 Anna S. Nikonova1†
Anna S. Nikonova1† Alexander Y. Deneka1,2†
Alexander Y. Deneka1,2† Louisa Eckman1
Louisa Eckman1 Meghan C. Kopp3
Meghan C. Kopp3 Harvey H. Hensley1
Harvey H. Hensley1 Brian L. Egleston1
Brian L. Egleston1 Erica A. Golemis1*
Erica A. Golemis1*
- 1Program in Molecular Therapeutics, Fox Chase Cancer Center, Philadelphia, PA, USA
- 2Kazan Federal University, Kazan, Russia
- 3Cancer Biology, Drexel University College of Medicine, Philadelphia, PA, USA
Aurora-A kinase (AURKA) overexpression in numerous tumors induces aneuploidy, in part because of cytokinetic defects. Alisertib and other small-molecule inhibitors targeting AURKA are effective in some patients as monotherapies or combination therapies. Epidermal growth factor receptor (EGFR) pro-proliferative signaling activity is commonly elevated in cancer, and the EGFR inhibitor erlotinib is commonly used as a standard of care agent for cancer. An erlotinib/alisertib combination therapy is currently under assessment in clinical trials, following pre-clinical studies that indicated synergy of these drugs in cancer. We were interested in further exploring the activity of this drug combination. Beyond well-established functions for AURKA in mitotic progression, additional non-mitotic AURKA functions include control of ciliary stability and calcium signaling. Interestingly, alisertib exacerbates the disease phenotype in mouse models for autosomal-dominant polycystic kidney disease (ADPKD), a common inherited syndrome induced by aberrant signaling from PKD1 and PKD2, cilia-localized proteins that have calcium channel activity. EGFR is also more active in ADPKD, making erlotinib also of potential interest in this disease setting. In this study, we have explored the interaction of alisertib and erlotinib in an ADPKD model. These experiments indicated erlotinib-restrained cystogenesis, opposing alisertib action. Erlotinib also interacted with alisertib to regulate proliferative signaling proteins, albeit in a complicated manner. Results suggest a nuanced role of AURKA signaling in different pathogenic conditions and inform the clinical use of AURKA inhibitors in cancer patients with comorbidities.
Introduction
In its role as a mitotic regulator, Aurora-A kinase (AURKA) accumulates through G2 at the centrosome, becomes active at G2/M transition, and remains active through M phase as it translocates along the mitotic spindle to the midzone, with the bulk of AURKA degraded at the midbody at cytokinesis. A large number of proteins have been identified that directly associate with AURKA either in its N-terminal unstructured domain or C-terminal kinase domain, and regulate AURKA activation, including the highly studied TPX2 (1–7), but in addition, the scaffolding factors NEDD9, nucleophosmin/B23, PAK kinases, CEP192, and others (8–12). Human AURKA is overexpressed in many tumors arising from breast, colon, ovary, and other tissues, and to function as an oncogene when exogenously expressed in numerous cell line models (13–18). AURKA overexpression is invariably associated with increased number of centrosomes and multipolar spindles, which arise as consequence of failed cytokinesis, and reflect failure to downregulate AURKA activity at the end of mitosis. Inhibitors designed to block AURKA mitotic activity are currently undergoing clinical assessment as cancer therapeutics, with MLN8237/alisertib in multiple late-stage trials (19, 20).
Although most work on AURKA focuses on the activity of this protein in mitotic cells, a number of studies have now identified additional functions of AURKA in non-mitotic cells. For example, AURKA activity is required for neurite extension, in a post-mitotic cell population (21, 22). AURKA is also transiently activated by elevated cytoplasmic calcium, which triggers calmodulin binding to the N-terminal unstructured domain of AURKA and enhances binding to some partners, such as NEDD9, and reciprocally, AURKA phosphorylation of the polycystin 2 (PC2) calcium channel can inhibit its activity in interphase cells (23, 24). In addition, growth factor stimulation of quiescent ciliated cells induces NEDD9 expression and AURKA activation, leading to resorption of the cilium (25). These latter activities were of particular interest, as they not only potentially informed some roles of AURKA relevant to cancer (26, 27) but also connected AURKA activity to another pathological condition, autosomal-dominant polycystic kidney disease (ADPKD).
Autosomal-dominant polycystic kidney disease arises from inactivating mutations in the genes PKD1 or PKD2, and currently has few treatment options (28). Formation of cysts is marked by multiple phenotypic changes in the cells lining renal tubules [reviewed in Ref. (28)]. These pleiotropic changes reflect the complex cellular action of the polycystins PC1 and PC2, products of the PKD1 and PKD2 genes. ADPKD is classified as a ciliopathy (29), based on the obligate functional heterodimerization of PC1 and PC2 on cell cilia, where PC1 normally acts as a flow sensor to trigger the calcium channel activity of PC2: calcium influx and other signaling interactions of the PC1/PC2 heterodimer act to restrain cell growth and govern the polarity of cell division in normal cells (30). Loss of cilia or defects in ciliary function can independently induce cyst formation (31).
As ADPKD signaling defects have become better understood, an unexpected feature has been the recognition that they possess extensive similarity to signaling defects seen in cancer (32). Exploiting these convergences, current research into the effective clinical management of ADPKD has been exploring the inhibition of signaling proteins, such as mTOR and SRC, that typically have elevated expression or activity in response to mutation of PC1/PC2 signaling, and actively contribute to cystic growth [reviewed in Ref. (30)]. Given the connections described above among AURKA, PC2, and cilia, and the identification that AURKA itself is elevated in cystic epithelia (23, 33), we previously explored efficacy of AURKA inhibition in controlling cyst growth in a mouse model of ADPKD (33). The initially surprising result of this study was that alisertib strongly exacerbated cyst formation. However, this outcome was compatible with an independent study that in the specific context of driver lesions in PKD1 or PKD2, genetic ablation of cilia reduces symptoms, suggesting the hypothesis that it is abnormal signaling rather than loss of signaling from the cilium that induces cyst formation (34). If so, then inhibiting signaling processes downstream of polycystins would potentially oppose the activity of alisertib. Epidermal growth factor receptor (EGFR) is activated in ADPKD (35, 36), and interacts with polycystins (37). In cancer, the combination of erlotinib and alisertib was first suggested by an siRNA screen that identified genes that influenced cellular response to inhibition of EGFR (38). In this work, AURKA inhibitors were shown to combine effectively with both small molecule and antibody inhibitors of EGFR in vitro and in vivo, providing the conceptual basis for two ongoing clinical trials (NCT01471964 and NCT01540682, clinicaltrials.gov). In the current study, to probe these novel actions of AURKA in ADPKD, we have evaluated the interaction of the EGFR inhibitor erlotinib with alisertib in control of cyst formation.
Materials and Methods
Mouse Strains and Drug Treatment
Conditional Pkd1−/− mice in which tamoxifen induction of the Cre-flox regulatory system permits targeted inactivation of the Pkd1 gene in vivo have been described (33, 39, 40). Pkd1fl/fl;Cre/Esr1+ (referred to as Pkd1−/−), and control mice lacking an intact Cre-flox system (Pkd1fl/fl;Cre/Esr1−) mice were injected intraperitoneally with tamoxifen [250 mg/kg body weight (BW), formulated in corn oil] on post-natal days P2 and P3 for the early cyst induction, or post-natal days P35 and P36 for late cyst induction, to induce Pkd1 deletion in the test group, as described (39). Alisertib (Millennium Pharmaceuticals, Inc., Cambridge, MA, USA) was formulated in 10% 2-hydroxypropyl-β-cyclodextrin (Sigma Aldrich, St. Louis, MO, USA) with 1% sodium bicarbonate and 20 mg/kg administered orally twice daily (BID), using a 5-day on/2-day off schedule. Erlotinib was formulated in 10% DMSO saline and 10 mg/kg administered orally once daily (QD), using a 5-day on/2-day off schedule. Treatment began at the age of 4 months and cyst growth monitored by magnetic resonance micro-imaging (MRI); mice were euthanized 10 weeks after the beginning of treatment to collect kidneys and liver for analysis. The Institutional Animal Care and Use Committee (IACUC) of Fox Chase Cancer Center approved all experiments involving mice.
MRI Protocol and Image Analysis
Magnetic resonance micro-imaging was performed exactly as described in Ref. (33, 41, 42). Briefly, mice were anesthetized with 1–2% isoflurane in O2 and then imaged using a vertical bore 7-T magnet, Bruker DRX300 spectrometer, ParaVision 3.0.2 software (Bruker), and a single tuned 1H cylindrical radiofrequency coil. Kidney and cyst volume were quantified using Image J (43). For estimation of kidney volume, the kidney parenchyma was manually surrounded while excluding the renal pelvis, and summing up the products of area measurements of contiguous images and slice thickness, as in Ref. (44). Subsequently isolated kidney areas were prepared using defined settings for background subtraction and band passing, with a threshold set for each kidney based on the original images by targeting threshold values designating the transition between parenchyma and cyst at the border of the larger cysts in the kidneys. Cyst volume was estimated using a semi-automatic threshold approach (45, 46).
Tissue Preparation and Histology
All tissues were collected and fixed in 10% phosphate-buffered formaldehyde (formalin) for 24–48 h, dehydrated and embedded in paraffin. Hematoxylin and eosin (H&E) stained 5 μm sections were used for morphological evaluation.
Western Blotting
To analyze the expression levels of individual proteins, kidney tissues were lysed and resolved by SDS-PAGE. Western blotting was performed using standard procedures, and developed by chemiluminescence using Luminata Western HRP substrates (Classico, Crescendo and Forte) (EMD Millipore) and Immun-Star AP Substrate (Bio-Rad Laboratories). Quantification of signals on Western blots was done using the NIH ImageJ Imaging and Processing Analysis Software with signaling intensity normalized to loading control (β-actin or vinculin). Primary antibodies included anti-Src (Cell Signaling, #2110), anti-phospho-Src Tyr418 (Abcam, #ab4816), anti-S6 (Cell Signaling, #4858), anti-phospho-S6 S235/236 (Cell Signaling, #2317), anti-phospho-ERK Thr202/Tyr204 (Cell Signaling, #9101), anti-phospho-EGFR Y1068 (Cell Signaling, #3777), anti-phospho-EGFR Y1173 (Invitrogen, #44794G), anti-EGFR (Cell Signaling, #2646), anti-phospho-Akt S473 (Cell Signaling, #4060), anti-Akt (Cell Signaling, #2920), anti-Aurora-A (mouse, BD Transduction, #610939 and rabbit, Cell Signaling, #3092), anti-histone H3 (Cell Signaling, #3638S), anti-vinculin (Sigma, #V9131), and mouse anti-β-actin conjugated to HRP (Abcam, #ab49900). Secondary anti-mouse and anti-rabbit HRP-conjugated antibodies (GE Healthcare) were used at a dilution of 1:10,000 and secondary anti-mouse and anti-rabbit AP-conjugated antibodies (Jackson Immunoresearch Labs) were used at a dilution of 1:5,000.
Phosphorylation Assay
Histone H3 (Upstate, Charlottesville, VA, USA) was used as substrate for AURKA kinase activity, using standard methods. Parallel aliquots without [γ32P]ATP were processed for SDS-PAGE. To assess Aurora-A activation, we performed an in vitro kinase assay using AURKA immunoprecipitated from whole kidney lysates using beads conjugated with anti-Aurora A antibody (Bethyl Laboratories, S300-070-3). Immunoprecipitation samples were incubated overnight with antibody at 4°C, washed, and resolved by SDS-PAGE.
Statistical Analysis
Analyses were performed using STATA version 12. Data were analyzed using Wilcoxon rank-sum tests and generalized linear models with appropriate family and link functions (e.g., Gaussian or Gamma families with log or identity links). Where necessary, we estimated growth curves using generalized estimating equations (GEE) with exchangeable or Markov working correlation matrices to account for correlated data (46).
Results
Alisertib and Erlotinib Treatment of a Conditional Knockout Model for ADPKD: Modest Effect on Kidney Volume and Weight
We used a previously described Pkd1 conditional knockout mouse model in which tamoxifen induction of a Cre-flox regulatory system allows targeted inactivation of the Pkd1 gene in vivo (39, 40). In this system, the loss of Pkd1 at post-natal day 28 results in development of renal cysts at ~4.5–5 months of age, progressing to severe enlargement of the kidney and renal failure at 6–7 months of age. The experimental outline is shown in Figure 1A. We defined four cohorts of Pkd1−/− mice: Cohort 1 (n = 11), vehicle (10% cyclodextrin, 1% sodium hydrocarbonate, and 5% dextrose, with 10% DMSO mixed in 1:1 ratio) twice a day; Cohort 2 (n = 16), alisertib, 20 mg/kg, twice a day (40 mg/kg daily); Cohort 3 (n = 13), erlotinib, 10 mg/kg, once a day; and Cohort 4 (n = 14), alisertib 20 mg/kg, twice a day plus erlotinib, 10 mg/kg, once a day (2 h after the morning dose of alisertib). Parallel cohorts 5–8 were also run, with wild type mice that received the same dosing regimen: each of these cohorts contained 8–10 animals. Starting at the time of injection, mice were weighed weekly. Treatment with alisertib or alisertib plus erlotinib resulted in slower weight gain over 10 weeks in both wild type and Pkd1−/− groups, while erlotinib alone had no effect on weight gain (Figure 1B).
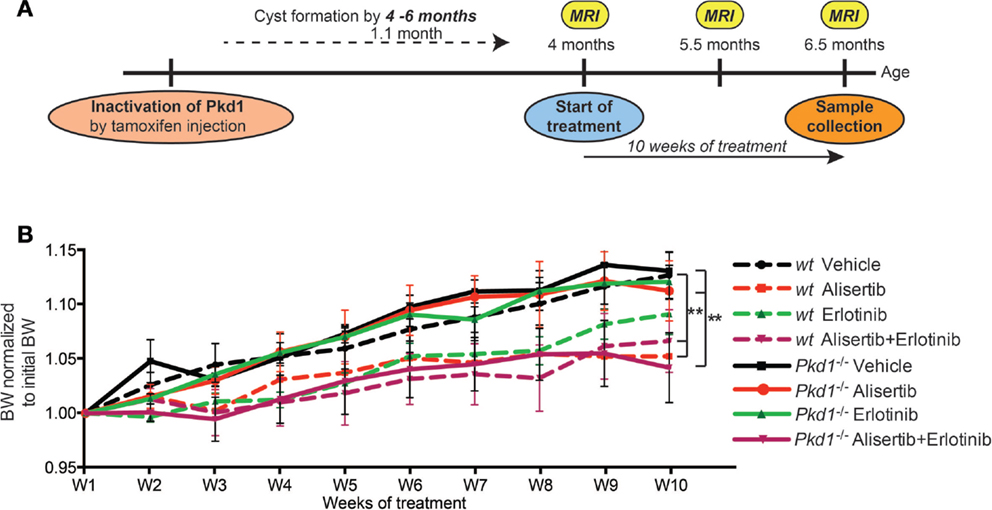
Figure 1. Alisertib and erlotinib treatment of a conditional knockout model for ADPKD. (A) Experimental design. In vivo experiments were performed using a conditional knockout mouse model for tamoxifen-induced, Cre-dependent ablation of Pkd1 (39). (B) Body weight (BW) dynamics for the course of the treatment (vehicle, alisertib, erlotinib, alisertib + erlotinib), measured weekly. Differences between genotypes and drug treatment groups at 10 weeks were statistically significant between alisertib and alisertib + erlotinib versus the vehicle-treated groups in both wt and Pkd1−/− groups. *P < 0.01 relative to vehicle.
For analysis of kidney enlargement over time, Pkd1−/− and wt mice were assessed at 4, 5.5, and 6.5 months of age using a MRI approach (41) (Figure 2A). In general, drug effects on rate of kidney growth did not rise to statistical significance. Following normalization to BW, alisertib slightly increased the rate of kidney growth versus vehicle-treated Pkd1−/− mice at all time points (Figure 2B). Erlotinib did not significantly affect growth, at all time periods. The alisertib/erlotinib combination initially resulted in a rate of kidney growth similar to vehicle or erlotinib-treated mice, but at latter time points, the ratio of kidney volume to BW indicated a phenotype more similar to alisertib. However, it is important to note that mice treated with this drug combination had a significantly lower BW (Figure 1B), which likely contributes to the difference. As a control, we established that no drug affected kidney volume increase in wild type mice (Figure 2C). After 10 weeks, mice were euthanized and kidney weight to BW ratio directly determined. This confirmed findings from MRI, with a non-statistically significantly trend toward elevated BW in Pkd1−/− mice treated with alisertib or alisertib/erlotinib, and all mice with a Pkd1−/− genotype having a statistically significant greater kidney weight than all wt mice (Figure 2D). Hepatic cysts are a common feature of ADPKD in humans, occurring in a significant number of patients. In a previous study, we showed that inhibition of HSP90 significantly reduced liver cyst burden (42), while also reducing the rate of development of kidney cysts (41). In the present study, the alisertib effect was specific to kidney tissue, and no effect was seen with any drug treatment in liver from wild type or Pkd1−/− mice (Figure 2E).
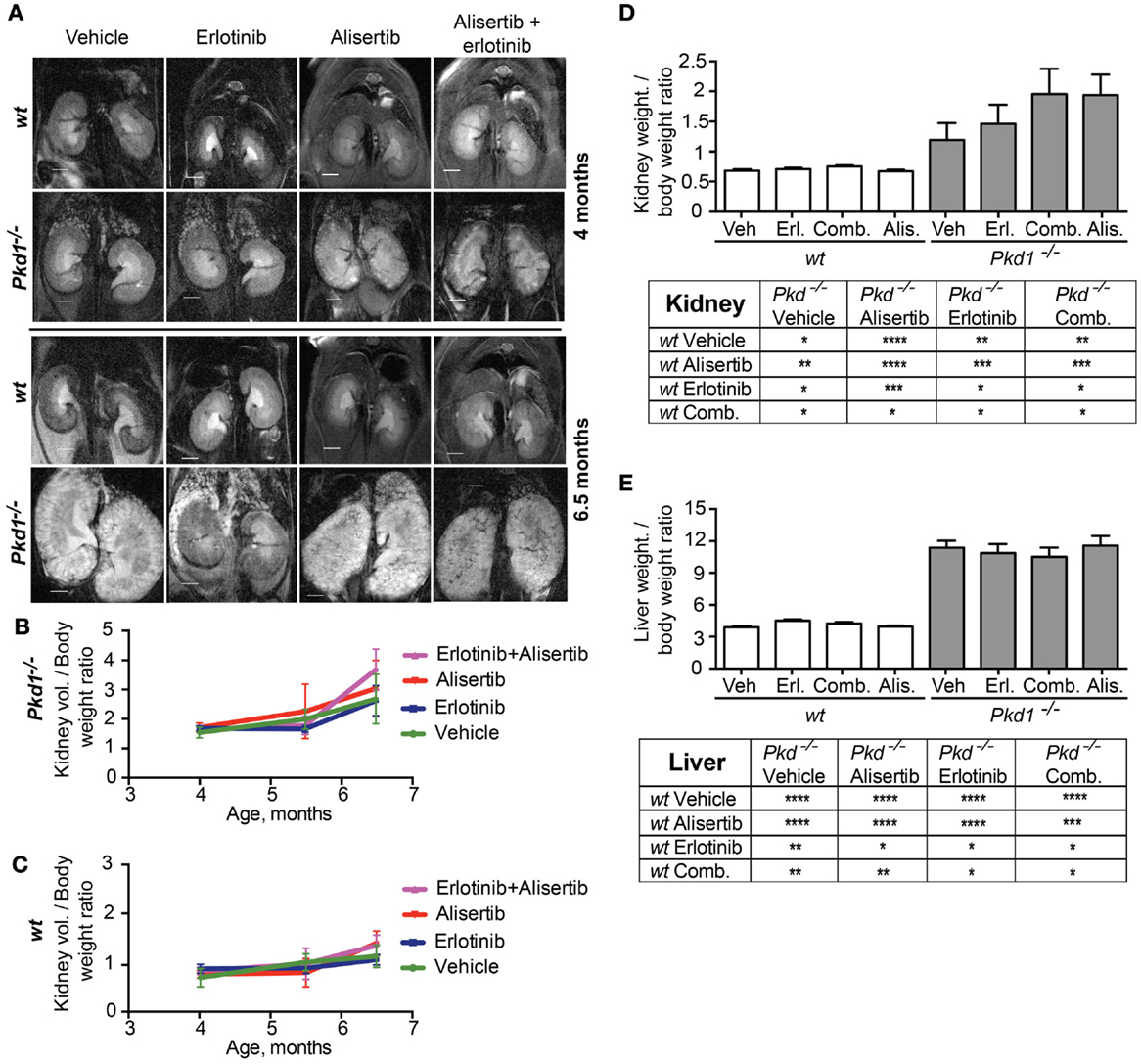
Figure 2. Effects of alisertib and erlotinib on kidney growth. (A) Representative images of murine kidneys acquired by using magnetic resonance micro-imaging (MRI) approach, at 4 (top) and 6.5 (bottom) months of age following treatment with vehicle, alisertib, erlotinib, and a combination of these drugs. (B,C) Quantification of the MRI imaging results for kidney volume, normalized to body weight for Pkd1−/−(B) and wt (C) mice. Differences at experimental endpoint (10 weeks of treatment) are not statistically significant. (D,E) Direct measurement of kidney (D) and liver (E) weight normalized to body weight for Pkd1−/− and wt mice at experimental endpoint (10 weeks of treatment). Top – graphs representing results; bottom – summary of P-values for the presented data. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Alisertib and Erlotinib Treatment of a Conditional Knockout Model for ADPKD: Drug Interactions in Control of Cyst Development
The development of cysts was analyzed by quantification of MRI imaging (Figures 2A and 3A), and subsequently confirmed by visual assessment of hematoxylin and eosin (H&E) stained tissues collected after 10 weeks of treatment (Figures 3B,C). No wild type mice developed cysts. Among the Pkd1−/− mice, there was some heterogeneity in cyst development between individual animals, in concordance with the basic biology of the disease and previous reports using the model (39–41). Notably, the erlotinib treatment strikingly reduced cystogenesis in most animals, at all time points, in a statistically significant effect (Figure 3A). Alisertib treatment elevated cyst growth early, and cystogenesis was much greater than in vehicle-treated animals by the experimental endpoint, as previously noted (33). Interestingly, the alisertib/erlotinib combination treatment caused an initial delay in the formation of cysts, similar to erlotinib. However, at later time points, the beneficial effect was lost, and at experimental endpoint, the overall phenotype resembled alisertib-treated mice. Mice treated with alisertib or erlotinib plus alisertib had an extremely heterogeneous phenotype at the experimental endpoint. Although the majority had highly cystic kidneys, some had only limited cysts, suggesting a stochastic effect in response to drug between individual animals, and accounting for the large error bars.
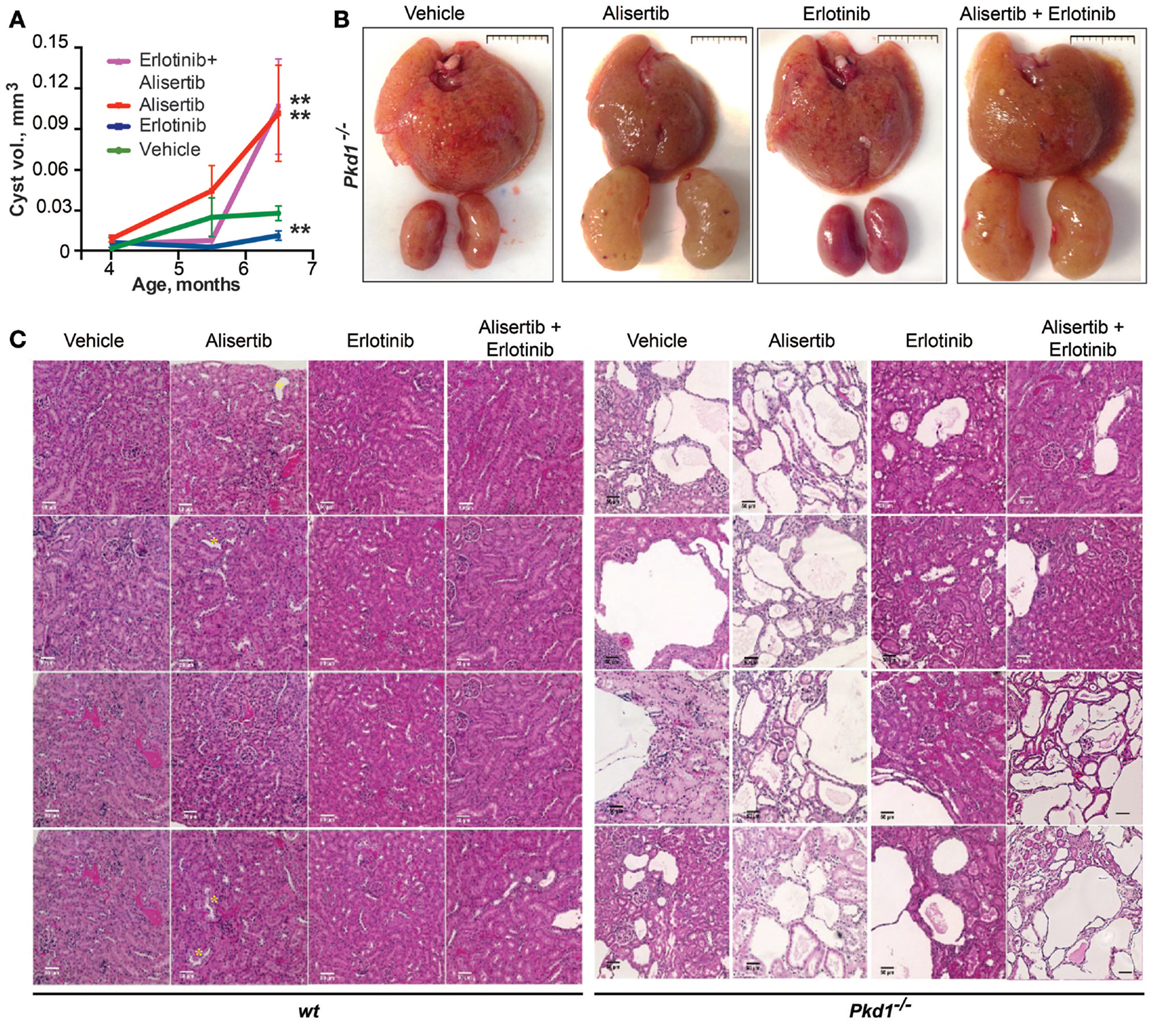
Figure 3. Cystogenesis during drug treatment in Pkd1−/− and wt mice. (A) Ratio of renal cyst volume to body weight in Pkd1−/− mice in the four drug treatment groups. *P < 0.05 and **P < 0.01 relative to vehicle treatment. (B) Representative images of collected livers (top) and kidneys (bottom) from each treatment cohort; scale bar, 1 cm. (C) Representative light microscopy images from H&E slides, reflecting cystic burden of Pkd1−/− and wt mice in all treatment groups at experimental endpoint (10 weeks of treatment). Images taken at 20× magnification; scale bar, 50 μm.
Signaling Consequences of Alisertib and Erlotinib Treatment Pkd1−/− and wt Kidneys
To better understand the functional interaction of inhibition of AURKA and EGFR, we analyzed activation of the signaling of these drug targets and of signaling pathways relevant to ADPKD in kidney tissue collected from Pkd1−/− and wt mice after 10 weeks of drug treatment.
Under conditions of vehicle treatment, AURKA expression was elevated in Pkd1−/− versus wt kidneys, as previously reported (33) (Figure 4A). In vitro kinase analysis of phosphorylation of the substrate histone H3 (HH3) by AURKA immunoprecipitated from kidney lysates, or the autophosphorylation of immunoprecipitated AURKA, normalized to total levels of immunoprecipitated kinase (Figure 4B), surprisingly indicated that drug treatments did not produce statistically significant effects on AURKA activity. However, parallel Western analysis (Figures 4C,D) indicated that total levels of AURKA were significantly depleted in tissue treated with each of the drugs, particularly in those treated with alisertib or alisertib plus erlotinib. Generally, similar effects of drug treatment were observed in wt kidneys. Hence, the primary consequence of alisertib treatment was to reduce overall AURKA activity by reducing total AURKA expression.
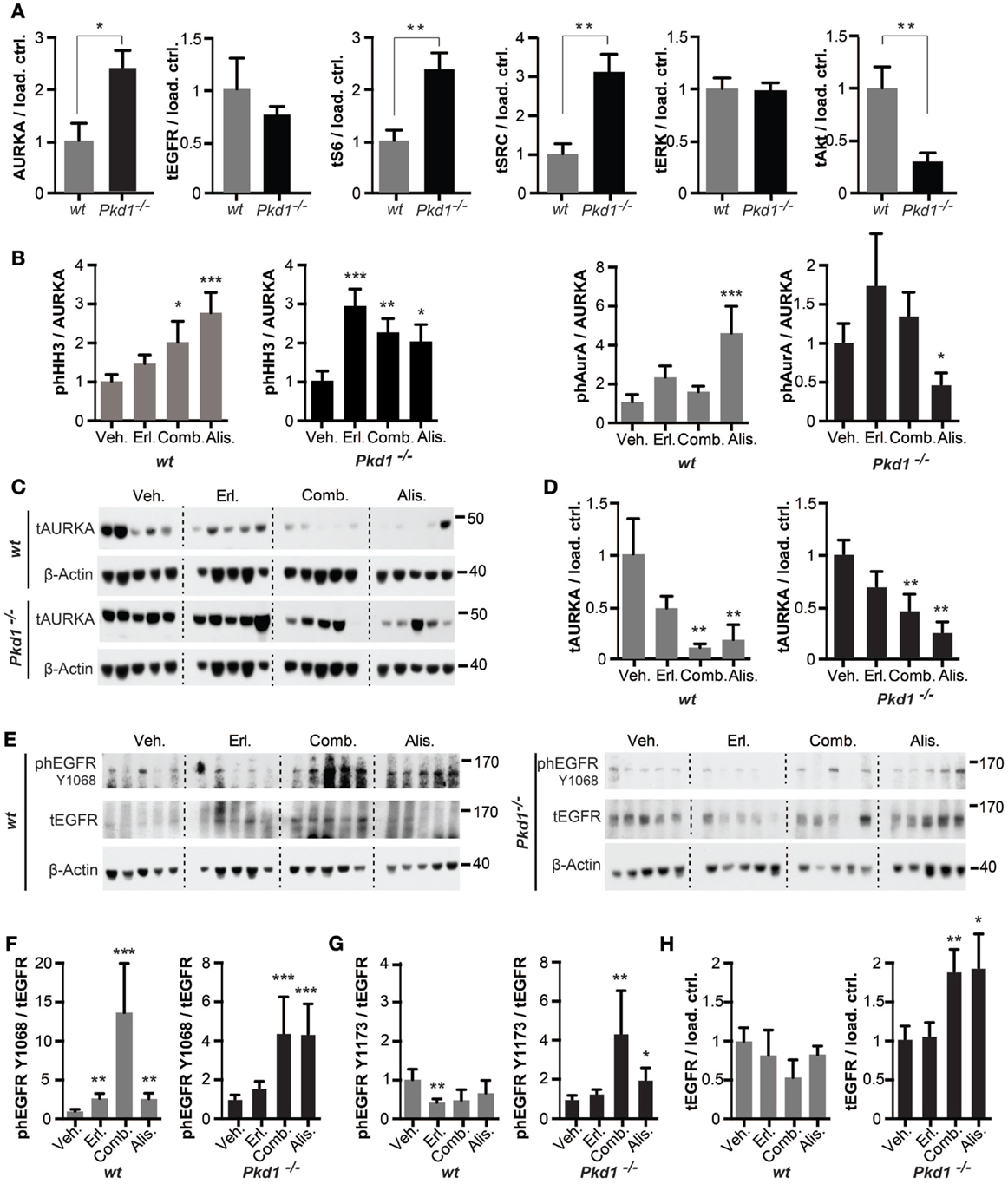
Figure 4. Drug inhibition of targets in Pkd1−/− and wt mice. (A) Quantification of Western data for indicated proteins from kidney lysates prepared from vehicle-treated wt or Pkd1−/− mice, normalized to β-actin or vinculin-loading control. *P < 0.05 and **P < 0.01 relative to vehicle treated. (B) Aurora-A (AURKA) was immunoprecipitated from kidney lysates and used for in vitro kinase with γ-32P-ATP to indicate autophosphorylation and phosphorylation of histone H3 kinase (HH3) (top two rows), with parallel blots probed by Western to allow normalization to total AURKA and HH3 in reaction. Quantitation of data from complete cohort of mice in each treatment group, indicating ratio of phosphorylated HH3 or AURKA to total immunoprecipitated AURKA in wt and Pkd1−/− mice following indicated drug treatments. *P < 0.05, **P < 0.01, and ***P < 0.001 relative to vehicle treated. (C) Western analysis showing representative expression of total Aurora-A (tAURKA) in kidney lysates after 10 weeks of treatment with indicated drugs, with β-actin loading control. (D) Quantitation of data from complete cohort of mice in each treatment group, indicating ratio of total AURKA to β-actin in wt and Pkd1−/− mice following indicated drug treatments. **P < 0.01 relative to vehicle treated. (E) Western blot with representative images of kidney lysates from each treatment cohort showing expression of EGFRphosphorylated (ph) at the indicated amino acids, total EGFR (tEGFR) or β-actin loading control. (F–H) Quantitation of data from complete cohort of mice in each treatment group, indicating ratio of phEGFR-Y1068 to tEGFR (F), phEGFR-Y1173 to tEGFR (G), or tEGFR to loading control (H). *P < 0.05, **P < 0.01, and ***P < 0.001, relative to vehicle treated.
Epidermal growth factor receptor activation is reflected by phosphorylation of EGFR at Y1068, which allows it to associate with GRB2 to activate downstream signaling cascades (47), and at Y1173, which is important for SHC binding and EGFR internalization (48). Total EGFR expression was not elevated in wt versus Pkd1−/− kidneys (Figure 4A). However, in Pkd1−/− kidneys, Y1068 phosphorylation was significantly increased by treatment with alisertib, or alisertib plus erlotinib, and Y1173 phosphorylation was increased, albeit to a lesser degree (Figures 4E–G). Furthermore, in Pkd1−/− kidneys, total EGFR expression was also elevated by treatment with alisertib or the alisertib/erlotinib combination (Figures 4E,H). Together, these results emphasized the role of AURKA inhibition in potentiating proliferative signaling relevant to a severe cystic phenotype. By contrast, no drug treatment significantly affected EGFR expression or phosphorylation on Y1173 in relation to vehicle in wt kidneys, although interestingly, the alisertib/erlotinib combination significantly induced Y1068 phosphorylation in a subset of wt kidneys (Figures 4E–H).
We then analyzed the activity and expression of the ADPKD-related proteins S6, SRC, ERK1/2, and AKT (Figures 4 and 5). For S6, AKT, and SRC, total levels were elevated in Pkd1−/− versus wt kidneys (Figure 4A). The patterns of response to drug treatment were complicated for these downstream pro-proliferative proteins. Focusing first on alisertib in Pkd1−/− kidneys, this treatment significantly reduced levels of total S6 and SRC, and increased total levels of ERK1/2 (Figure 5). However, alisertib also resulted in a very significant increase in the ratio of active (phosphorylated) S6, leading to a net gain in S6 activity in kidney lysates, compatible with an increased cystic phenotype. By contrast, activity of SRC and ERK was reduced by alisertib in Pkd1−/− kidneys. These patterns of expression and activation were very different from those evoked by alisertib treatment of wt kidneys. In wt kidneys, alisertib very significantly reduced S6 and ERK1/2 activation, and reduced SRC expression. Alisertib treatment also resulted in a variable pattern of SRC activation, with three mice having very high levels of phosphorylated SRC, but most having SRC activity reduced. Whereas erlotinib or erlotinib plus alisertib effectively reduced ERK1/2 and S6 activity in wt kidneys, these treatments were less effective in Pkd1−/− kidneys. With the exception of effect on total ERK1/2 expression in Pkd1−/− kidneys, erlotinib, and erlotinib plus alisertib resulted in statistically non-distinct effects on the expression and activation of the signaling proteins analyzed. This was surprising, given the very different results of these treatments on cystic phenotype.
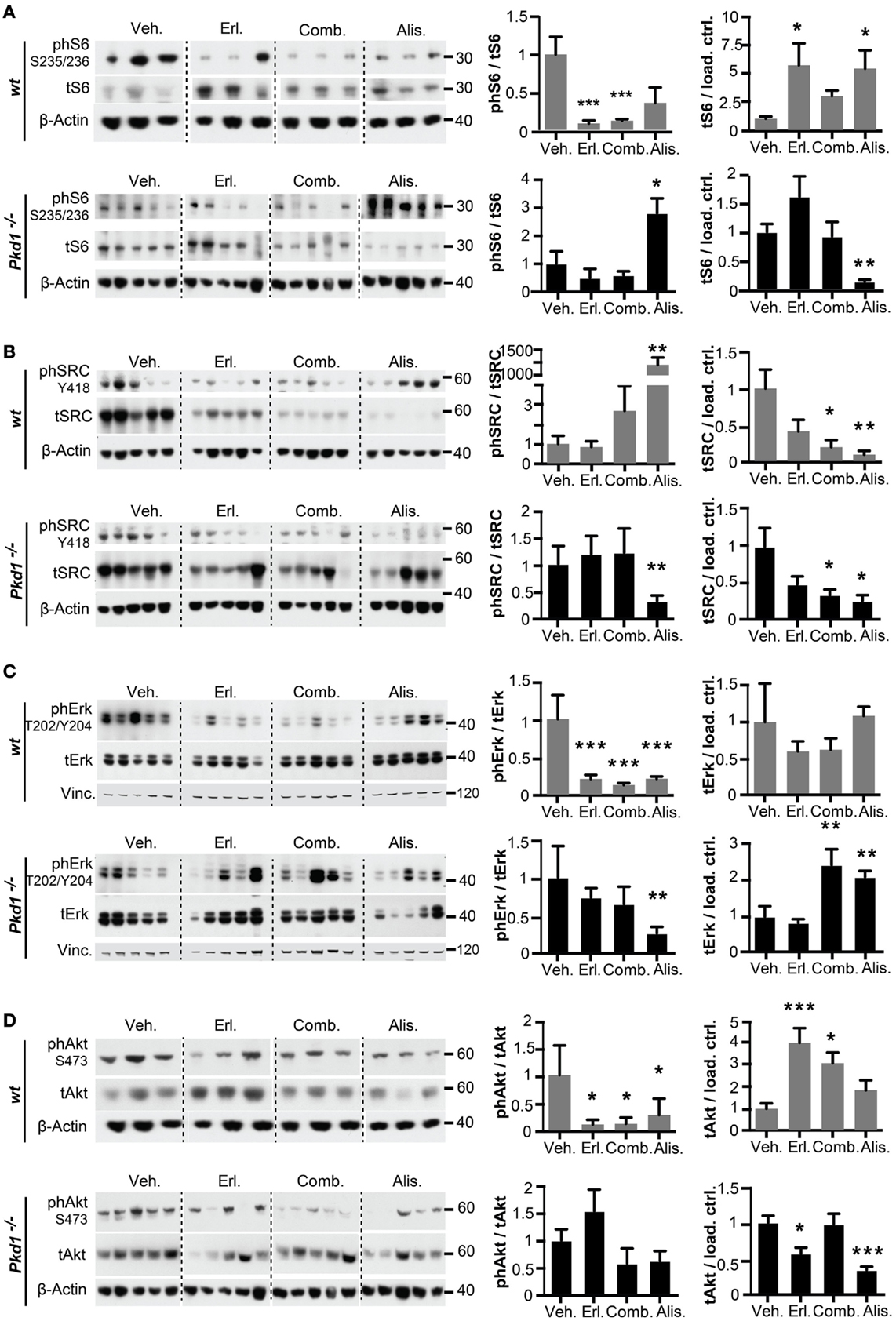
Figure 5. Drug inhibition of ADPKD-related signaling proteins in Pkd1−/− and wt mice. S6 (A), SRC (B), ERK1/2 (C), and AKT (D) in kidney lysates after 10 weeks of treatment with indicated drugs. Western analysis (left) showing representative expression of phosphorylated and total expression of proteins and quantitation of data (right) from complete cohort of mice in each treatment group with analysis, as described in Figure 4. (A–D) Ratios of phosphorylated to total protein for S6 (A), SRC (B), ERK1/2 (C), and AKT (D) are shown in the left graph, and ratios of total protein to β-actin or vinculin (vinc) loading control are shown in the right graph. *P < 0.05, **P < 0.01, and ***P < 0.001 relative to vehicle treated.
Discussion
As significant findings, this study has established that the cystogenic activity of the AURKA inhibitor in ADPKD can be partially reversed by treatment with erlotinib, and for the first time showed that erlotinib itself has a potent activity in limiting cystic growth. Second, it also demonstrated that erlotinib and alisertib elicited distinct response profiles in the kidneys of wt versus Pkd1−/− mice, potentially reflecting differences in signaling landscape associated with the replacement of the normal renal parenchyma with cysts. Third, it also demonstrated that in the context of drug treatment, changes in degree of cystogenesis in Pkd1−/− mice could not be aligned with specific changes in expression or activity of AKT, S6, ERK1/2, or SRC, in spite of the common association of elevated activity of these signaling proteins with disease etiology. Finally, as discussed below, this work emphasized some significant differences between the interactions of alisertib and erlotinib in the context of ADPKD versus cancer, such that this treatment is potentially beneficial in cancer but not ADPKD.
These findings confirmed earlier reports that AURKA activity was associated with cyst formation, and that inhibition of AURKA exacerbated cystogenesis in the context of genetic loss of Pkd1 (23, 24, 33). A particularly interesting finding was that treatment with alisertib alone or in combination with erlotinib was more strongly associated with loss of AURKA protein, rather than inhibition of AURKA activity. There are two potential explanations for this observation. First, AURKA expression is highly regulated by protein degradation, and the protein is more susceptible to degradation when catalytically inactive (49, 50). Hence, alisertib treatment may be elevating the rate of AURKA destruction, and in this context, the elevated activity of the remaining AURKA may represent a sub-population effectively protected by interaction partners (49, 50). Second, AURKA expression has been reported as most abundant and active in the renal epithelia of early cysts (23, 24, 33). However, in the tissue isolated at the end of this experiment and used for signaling analysis, the majority of tissue in alisertib- and alisertib/erlotinib-treated cells reflects loss of normal renal structure and replacement with enlarged late-stage cysts that have in many cases lost epithelial lining, and fibrotic tissue. In this interpretation, a difference in tissue composition explains the loss of AURKA activity. Potentially, both explanations contribute to the observed phenotypes.
In analysis of signaling consequences of AURKA inhibition, alisertib reduction of SRC expression and activity was notable. One previous study has shown direct AURKA phosphorylation of SRC enhances SRC activity (51); it is possible that this phosphorylation also contributes to SRC stability, so that alisertib destabilizes SRC. The significant effect of AURKA inhibition on S6 phosphorylation is compatible with the enhanced cystogenesis seen in alisertib-treated mice. However, comparison of alisertib versus alisertib/erlotinib-treated mice confounds simple interpretation of specific signaling changes as relevant to the experimental endpoint, given the cystogenic phenotype of the combination resembles alisertib, whereas the signaling phenotype resembles erlotinib. This discordance suggests that while proteins such as S6 and SRC may have elevated expression and activity in ADPKD, their contribution to disease pathogenesis is not an essential driver of disease progression. This interpretation would be compatible with the limited activity of inhibitors of the S6 activator mTOR (52–54), or inhibitors of SRC (55), in assessment for treatment of polycystic kidney disease.
Although erlotinib has not previously been assessed, some prior studies have used alternative inhibitors of EGFR (56–58) or related ERBB-family kinases (59) in various models for cyst formation. Typically although not invariably, these studies have found inhibition of EGFR/HER2 signaling reduced cystic burden. In this study, we show that erlotinib is well tolerated and effective in controlling cyst growth in the context of loss of Pkd1, and that erlotinib can delay the growth of cysts that alisertib promotes. These results suggest that erlotinib may be useful for clinical evaluation in ADPKD. It is also interesting and important to note that erlotinib treatment reduces the level of AURKA in wt but not Pkd1−/− kidneys (Figure 4). In this case, one possible explanation is that erlotinib treatment causes wt cells to accumulate in the G1 phase of cell cycle by inhibiting multiple mitogenic effector pathways (60), hence reducing the population of G2/M phase cells in which AURKA is most abundant, whereas this inhibition is partially overcome in ADPKD tissue. This interpretation would suggest that small pool of AURKA at the basal body of cilia (61) – the pool potentially most relevant to the control of cystic severity, based on the model developed by Ma and colleagues (34) – might not be affected by erlotinib, as it is active in G0/G1. Test of this idea requires development of antibodies or probes suitable for analysis of AURKA activity by immunofluorescence in mouse tissue, currently a technical limitation on performing this work (50). Additional analyses of future interest would be the profiling of renal function (rather than cystic burden) following treatment with alisertib, erlotinib, or the combination, as well as broader profiling of gene expression changes following such treatments.
Finally, understanding the basis of AURKA and EGFR activity in ADPKD is of considerable interest for the field of oncology. One in 500 individuals suffers from ADPKD, many of whom will ultimately develop cancer and could potentially be treated with alisertib or erlotinib, given the common use of these agents. This work emphasizes not only the importance of avoiding alisertib but also suggests these patients would safely receive erlotinib. This study also emphasizes the different functional and signaling interactions of targeted inhibitors in distinct cellular contexts. Typically, AURKA inhibitors are used in therapeutic combinations with cytotoxic agents or other targeted agents (62): there is great interest in identifying productive therapeutic combinations. The positive interaction of alisertib and erlotinib in the context of oncogenic drivers (38), versus the opposing activity in the context of lesions in Pkd1, emphasizes the dynamic nature of signaling networks. Given the large number of comorbidities that are commonly experienced by cancer patients, including diabetes, cardiovascular disease, and other conditions that can affect cell signaling, there is clearly a need for further study.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors were supported by NCI Core Grant P30 CA006927 (to Fox Chase Cancer Center) and funding from the PKD Foundation and DOD PRMRP W81XWH-12-1-0437/PR110518 (to EG); and by a project of the Russian Government to support the Program for Competitive Growth of Kazan Federal University (to AD).
References
1. Eyers PA, Maller JL. Regulation of Xenopus Aurora A activation by TPX2. J Biol Chem (2004) 279(10):9008–15. doi: 10.1074/jbc.M312424200
2. Giubettini M, Asteriti IA, Scrofani J, De Luca M, Lindon C, Lavia P, et al. Control of Aurora-A stability through interaction with TPX2. J Cell Sci (2011) 124(Pt 1):113–22. doi:10.1242/jcs.075457
3. Eyers PA, Erikson E, Chen LG, Maller JL. A novel mechanism for activation of the protein kinase Aurora A. Curr Biol (2003) 13(8):691–7. doi:10.1016/S0960-9822(03)00166-0
4. Kufer TA, Sillje HH, Korner R, Gruss OJ, Meraldi P, Nigg EA. Human TPX2 is required for targeting Aurora-A kinase to the spindle. J Cell Biol (2002) 158(4):617–23. doi:10.1083/jcb.200204155
5. Tsai MY, Wiese C, Cao K, Martin O, Donovan P, Ruderman J, et al. A Ran signalling pathway mediated by the mitotic kinase Aurora A in spindle assembly. Nat Cell Biol (2003) 5(3):242–8. doi:10.1038/ncb936
6. Dodson CA, Bayliss R. Activation of Aurora-A kinase by protein partner binding and phosphorylation are independent and synergistic. J Biol Chem (2012) 287(2):1150–7. doi:10.1074/jbc.M111.312090
7. Xu X, Wang X, Xiao Z, Li Y, Wang Y. Two TPX2-dependent switches control the activity of Aurora A. PLoS One (2011) 6(2):e16757. doi:10.1371/journal.pone.0016757
8. Pugacheva EN, Golemis EA. The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nat Cell Biol (2005) 7(10):937–46. doi:10.1038/ncb1309
9. Clark MA, Acharya RA, Arico-Muendel CC, Belyanskaya SL, Benjamin DR, Carlson NR, et al. Design, synthesis and selection of DNA-encoded small-molecule libraries. Nat Chem Biol (2009) 5(9):647–54. doi:10.1038/nchembio.211
10. Zhao ZS, Lim JP, Ng YW, Lim L, Manser E. The GIT-associated kinase PAK targets to the centrosome and regulates Aurora-A. Mol Cell (2005) 20(2):237–49. doi:10.1016/j.molcel.2005.08.035
11. Hirota T, Kunitoku N, Sasayama T, Marumoto T, Zhang D, Nitta M, et al. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell (2003) 114(5):585–98. doi:10.1016/S0092-8674(03)00642-1
12. Reboutier D, Troadec MB, Cremet JY, Fukasawa K, Prigent C. Nucleophosmin/B23 activates Aurora A at the centrosome through phosphorylation of serine 89. J Cell Biol (2012) 197(1):19–26. doi:10.1083/jcb.201107134
13. Meraldi P, Honda R, Nigg EA. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. EMBO J (2002) 21(4):483–92. doi:10.1093/emboj/21.4.483
14. Tatsuka M, Katayama H, Ota T, Tanaka T, Odashima S, Suzuki F, et al. Multinuclearity and increased ploidy caused by overexpression of the aurora- and Ipl1-like midbody-associated protein mitotic kinase in human cancer cells. Cancer Res (1998) 58(21):4811–6.
15. Zhang D, Hirota T, Marumoto T, Shimizu M, Kunitoku N, Sasayama T, et al. Cre-loxP-controlled periodic Aurora-A overexpression induces mitotic abnormalities and hyperplasia in mammary glands of mouse models. Oncogene (2004) 23(54):8720–30. doi:10.1038/sj.onc.1208153
16. Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell (2003) 3(1):51–62. doi:10.1016/S1535-6108(02)00235-0
17. Mehra R, Serebriiskii IG, Burtness B, Astsaturov I, Golemis EA. Aurora kinases in head and neck cancer. Lancet Oncol (2013) 14(10):e425–35. doi:10.1016/S1470-2045(13)70128-1
18. Dotan E, Meropol NJ, Zhu F, Zambito F, Bove B, Cai KQ, et al. Relationship of increased aurora kinase A gene copy number, prognosis and response to chemotherapy in patients with metastatic colorectal cancer. Br J Cancer (2012) 106(4):748–55. doi:10.1038/bjc.2011.587
19. Friedberg JW, Mahadevan D, Cebula E, Persky D, Lossos I, Agarwal AB, et al. Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J Clin Oncol (2014) 32(1):44–50. doi:10.1200/JCO.2012.46.8793
20. Kelly KR, Shea TC, Goy A, Berdeja JG, Reeder CB, McDonagh KT, et al. Phase I study of MLN8237 – investigational Aurora A kinase inhibitor – in relapsed/refractory multiple myeloma, non-Hodgkin lymphoma and chronic lymphocytic leukemia. Invest New Drugs (2014) 32(3):489–99. doi:10.1007/s10637-013-0050-9
21. Mori D, Yamada M, Mimori-Kiyosue Y, Shirai Y, Suzuki A, Ohno S, et al. An essential role of the aPKC-Aurora A-NDEL1 pathway in neurite elongation by modulation of microtubule dynamics. Nat Cell Biol (2009) 11(9):1057–68. doi:10.1038/ncb1919
22. Yamada M, Hirotsune S, Wynshaw-Boris A. The essential role of LIS1, NDEL1 and Aurora-A in polarity formation and microtubule organization during neurogenesis. Cell Adh Migr (2010) 4(2):180–4. doi:10.4161/cam.4.2.10715
23. Plotnikova OV, Pugacheva E, Golemis EA. Aurora-A kinase activity influences calcium signaling in kidney cells. J Cell Biol (2011) 193:1021–32. doi:10.1083/jcb.201012061
24. Plotnikova OV, Pugacheva EN, Dunbrack RL, Golemis EA. Rapid calcium-dependent activation of Aurora-A kinase. Nat Commun (2010) 1(6):64. doi:10.1038/ncomms1061
25. Plotnikova OV, Nikonova AS, Loskutov YV, Kozyulina PY, Pugacheva EN, Golemis EA. Calmodulin activation of Aurora-A kinase (AURKA) is required during ciliary disassembly and in mitosis. Mol Biol Cell (2012) 23(14):2658–70. doi:10.1091/mbc.E11-12-1056
26. Plotnikova OV, Golemis EA, Pugacheva EN. Cell cycle-dependent ciliogenesis and cancer. Cancer Res (2008) 68(7):2058–61. doi:10.1158/0008-5472.CAN-07-5838
27. Plotnikova OV, Pugacheva EN, Golemis EA. Primary cilia and the cell cycle. Methods Cell Biol (2009) 94:137–60. doi:10.1016/S0091-679X(08)94007-3
28. Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med (2009) 60:321–37. doi:10.1146/annurev.med.60.101707.125712
29. Patel V, Chowdhury R, Igarashi P. Advances in the pathogenesis and treatment of polycystic kidney disease. Curr Opin Nephrol Hypertens (2009) 18(2):99–106. doi:10.1097/MNH.0b013e3283262ab0
30. Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int (2009) 76(2):149–68. doi:10.1038/ki.2009.128
31. Lee JE, Gleeson JG. A systems-biology approach to understanding the ciliopathy disorders. Genome Med (2011) 3(9):59. doi:10.1186/gm275
32. Seeger-Nukpezah T, Geynisman DM, Nikonova AS, Benzing T, Golemis EA. The hallmarks of cancer: relevance to the pathogenesis of polycystic kidney disease. Nat Rev Nephrol (2015) 11:515–34. doi:10.1038/nrneph.2015.46
33. Nikonova AS, Plotnikova OV, Serzhanova V, Efimov A, Bogush I, Cai KQ, et al. Nedd9 restrains renal cystogenesis in Pkd1-/- mice. Proc Natl Acad Sci U S A (2014) 111(35):12859–64. doi:10.1073/pnas.1405362111
34. Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet (2013) 45(9):1004–12. doi:10.1038/ng.2715
35. Du J, Wilson PD. Abnormal polarization of EGF receptors and autocrine stimulation of cyst epithelial growth in human ADPKD. Am J Physiol (1995) 269(2 Pt 1):C487–95.
36. Zheleznova NN, Wilson PD, Staruschenko A. Epidermal growth factor-mediated proliferation and sodium transport in normal and PKD epithelial cells. Biochim Biophys Acta (2011) 1812(10):1301–13. doi:10.1016/j.bbadis.2010.10.004
37. Ma R, Li WP, Rundle D, Kong J, Akbarali HI, Tsiokas L. PKD2 functions as an epidermal growth factor-activated plasma membrane channel. Mol Cell Biol (2005) 25(18):8285–98. doi:10.1128/MCB.25.18.8285-8298.2005
38. Astsaturov I, Ratushny V, Sukhanova A, Einarson MB, Bagnyukova T, Zhou Y, et al. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci Signal (2010) 3(140):ra67. doi:10.1126/scisignal.2001083
39. Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med (2007) 13(12):1490–5. doi:10.1038/nm1675
40. Piontek KB, Huso DL, Grinberg A, Liu L, Bedja D, Zhao H, et al. A functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J Am Soc Nephrol (2004) 15(12):3035–43. doi:10.1097/01.ASN.0000144204.01352.86
41. Seeger-Nukpezah T, Proia DA, Egleston BL, Nikonova AS, Kent T, Cai KQ, et al. Inhibiting the HSP90 chaperone slows cyst growth in a mouse model of autosomal dominant polycystic kidney disease. Proc Natl Acad Sci U S A (2013) 110:12786–91. doi:10.1073/pnas.1301904110
42. Smithline ZB, Nikonova AS, Hensley HH, Cai KQ, Egleston BL, Proia DA, et al. Inhibiting heat shock protein 90 (HSP90) limits the formation of liver cysts induced by conditional deletion of Pkd1 in mice. PLoS One (2014) 9(12):e114403. doi:10.1371/journal.pone.0114403
43. Rasband WS. ImageJ. Bethesda, MD: U. S. National Institutes of Health (1997-2014). Available from: http://imagej.nih.gov/ij/
44. Reichardt W, Romaker D, Becker A, Buechert M, Walz G, von Elverfeldt D. Monitoring kidney and renal cyst volumes applying MR approaches on a rapamycin treated mouse model of ADPKD. MAGMA (2009) 22(3):143–9. doi:10.1007/s10334-008-0158-7
45. Lee YR, Lee KB. Reliability of magnetic resonance imaging for measuring the volumetric indices in autosomal-dominant polycystic kidney disease: correlation with hypertension and renal function. Nephron Clin Pract (2006) 103(4):c173–80. doi:10.1159/000092915
46. Shults J, Ratcliffe SJ, Leonard M. Improved generalized estimating equation analysis via xtqls for implementation of quasi-least squares in STATA. STATA J (2007) 7:147–66.
47. Batzer AG, Rotin D, Urena JM, Skolnik EY, Schlessinger J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol Cell Biol (1994) 14(8):5192–201.
48. Tanos B, Pendergast AM. Abl tyrosine kinase regulates endocytosis of the epidermal growth factor receptor. J Biol Chem (2006) 281(43):32714–23. doi:10.1074/jbc.M603126200
49. Ice RJ, McLaughlin SL, Livengood RH, Culp MV, Eddy ER, Ivanov AV, et al. NEDD9 depletion destabilizes Aurora A kinase and heightens the efficacy of Aurora A inhibitors: implications for treatment of metastatic solid tumors. Cancer Res (2013) 73(10):3168–80. doi:10.1158/0008-5472.CAN-12-4008
50. Shagisultanova E, Dunbrack RL Jr, Golemis EA. Issues in interpreting the in vivo activity of Aurora-A. Exp Opin Ther Targets (2015) 19(2):187–200. doi:10.1517/14728222.2014.981154
51. Ratushny V, Pathak HB, Beeharry N, Tikhmyanova N, Xiao F, Li T, et al. Dual inhibition of SRC and Aurora kinases induces postmitotic attachment defects and cell death. Oncogene (2012) 31(10):1217–27. doi:10.1038/onc.2011.314
52. Braun WE, Schold JD, Stephany BR, Spirko RA, Herts BR. Low-dose rapamycin (sirolimus) effects in autosomal dominant polycystic kidney disease: an open-label randomized controlled pilot study. Clin J Am Soc Nephrol (2014) 9(5):881–8. doi:10.2215/CJN.02650313
53. Walz G, Budde K, Mannaa M, Nurnberger J, Wanner C, Sommerer C, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med (2010) 363(9):830–40. doi:10.1056/NEJMoa1003491
54. Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med (2010) 363(9):820–9. doi:10.1056/NEJMoa0907419
55. Sweeney WE Jr, von Vigier RO, Frost P, Avner ED. Src inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol (2008) 19(7):1331–41. doi:10.1681/ASN.2007060665
56. Sweeney WE, Chen Y, Nakanishi K, Frost P, Avner ED. Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int (2000) 57(1):33–40. doi:10.1046/j.1523-1755.2000.00829.x
57. Torres VE, Sweeney WE Jr, Wang X, Qian Q, Harris PC, Frost P, et al. EGF receptor tyrosine kinase inhibition attenuates the development of PKD in Han:SPRD rats. Kidney Int (2003) 64(5):1573–9. doi:10.1046/j.1523-1755.2003.00256.x
58. Torres VE, Sweeney WE Jr, Wang X, Qian Q, Harris PC, Frost P, et al. Epidermal growth factor receptor tyrosine kinase inhibition is not protective in PCK rats. Kidney Int (2004) 66(5):1766–73. doi:10.1111/j.1523-1755.2004.00952.x
59. Wilson SJ, Amsler K, Hyink DP, Li X, Lu W, Zhou J, et al. Inhibition of HER-2(neu/ErbB2) restores normal function and structure to polycystic kidney disease (PKD) epithelia. Biochim Biophys Acta (2006) 1762(7):647–55. doi:10.1016/j.bbadis.2006.04.006
60. Hopper-Borge EA, Nasto RE, Ratushny V, Weiner LM, Golemis EA, Astsaturov I. Mechanisms of tumor resistance to EGFR-targeted therapies. Expert Opin Ther Targets (2009) 13(3):339–62. doi:10.1517/14712590902735795
61. Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell (2007) 129:1351–63. doi:10.1016/j.cell.2007.04.035
Keywords: PKD1, Aurora-A kinase, mouse models, renal cyst, EGFR, SRC
Citation: Nikonova AS, Deneka AY, Eckman L, Kopp MC, Hensley HH, Egleston BL and Golemis EA (2015) Opposing effects of inhibitors of Aurora-A and EGFR in autosomal-dominant polycystic kidney disease. Front. Oncol. 5:228. doi: 10.3389/fonc.2015.00228
Received: 08 June 2015; Accepted: 01 October 2015;
Published: 16 October 2015
Edited by:
Mar Carmena, The University of Edinburgh, UKReviewed by:
Bernd E. R. Nuernberg, University of Tuebingen Hospitals and Clinics, GermanyDeilson Elgui De Oliveira, Faculdade de Medicina de Botucatu – UNESP, Brazil
Copyright: © 2015 Nikonova, Deneka, Eckman, Kopp, Hensley, Egleston and Golemis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erica A. Golemis, ZXJpY2EuZ29sZW1pc0BmY2NjLmVkdQ==
†Anna S. Nikonova and Alexander Y. Deneka have contributed equally to this work.