 Emilie Horvilleur
Emilie Horvilleur Lindsay A. Wilson
Lindsay A. Wilson Amandine Bastide
Amandine Bastide David Piñeiro
David Piñeiro Anne E. Willis
Anne E. Willis- Medical Research Council Toxicology Unit, Leicester, UK
Hematological malignancies are a heterogeneous group of diseases deriving from blood cells progenitors. Although many genes involved in blood cancers contain internal ribosome entry sites (IRESes), there has been only few studies focusing on the role of cap-independent translation in leukemia and lymphomas. Expression of IRES trans-acting factors can also be altered, and interestingly, BCL-ABL1 fusion protein expressed from “Philadelphia” chromosome, found in some types of leukemia, regulates several of them. A mechanism involving c-Myc IRES and cap-independent translation and leading to resistance to chemotherapy in multiple myeloma emphasize the contribution of cap-independent translation in blood cancers and the need for more work to be done to clarify the roles of known IRESes in pathology and response to chemotherapeutics.
Introduction
Hematopoietic malignancies, here referred to as blood cancers, are the fourth most diagnosed cancer type in the world. In 2012, they had an estimated incidence of almost one million per year (6.5% of all cancers diagnosed) and an estimated mortality of over half a million, thus being responsible for ~6.9% of all cancer deaths (1). Hematopoietic malignancies can be classed as three broad groups: lymphomas which form tumors in the lymph nodes; leukemia cells that accumulate in the blood; and myelomas which are cancers of plasmocytes. Blood cancers result from genetic alterations in the hematopoietic lineage, leading to uncontrolled proliferation and/or resistance to apoptosis.
Hematopoiesis is a complex and tightly regulated process. In response to various signals, hematopoietic stem cells mature into either lymphoid or myeloid progenitors. Myeloid progenitors ultimately differentiate into erythrocytes, platelets, monocytes, and granulocytes (2). The lymphoid progenitor undergoes several subsequent maturation steps in the bone marrow and secondary lymphoid organs to produce B-cells, T-cells, and natural killer cells (NK). Malignancies can derive from any of these lineages and from cells at various differentiation stages; hence, the types of blood cancers are completely different diseases. Most hematopoietic malignancies occur in the elderly, but Hodgkin lymphoma (HL) affects young adults, and leukemias are the most common pediatric cancers. While some of these malignancies are very aggressive like acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), and diffuse large B-cell lymphoma (DLBCL), others such as follicular lymphoma (FL), chronic myeloid leukemia (CML), or chronic lymphocytic leukemia (CLL) are relatively indolent and take several years to develop. They also have very different prognosis, e.g., 80% of HL can be cured while CLL remains incurable [for reviews, see Ref. (3–6)].
Despite the heterogeneity, many blood cancers share a common oncogenic mechanism. BCR and TCR genes undergo somatic recombinations to generate a broad range of receptors that would recognize various antigens. Off-target effects of these mechanisms can translocate proto-oncogenes to the highly transcribed BCR locus or lead to the expression of fusion proteins (7). While other mutations are found, the translocations are specific for each type of blood cancer and used for diagnosis and prognosis.
Differentiation and proliferation of blood cells is tightly controlled by an array of different extracellular signaling molecules, growth factors, and cytokines delivered by osteoblasts, osteoclasts, endothelial cells, stromal cells, mesenchymal progenitor cells, and adipocytes that populate the microenvironment of maturing hematopoietic cells: these components, together with the extracellular matrix, constitute the hematopoietic niche (8). While all hematopoietic malignancies interact with the hematopoietic niche, diseases in which no specific translocation is found (CLL and HL) are particularly dependent on their microenvironment and associated signaling pathways.
Various mechanisms deregulate cap-dependent translation in blood cancer, with the mTOR pathway being probably the most studied. mTOR is usually activated in aggressive diseases (ALL, AML, and DLBCL) resulting in an upregulation of translation. mTOR inhibitors, which should curtail translation, have been trialed in lymphomas, AML and CLL with mixed results (9). Protein synthesis can also be affected by deregulation of translation machinery components [e.g., eIF4E in leukemia (10) or eIF4B in DLBCL (11)], causing both global and gene specific effects. Finer gene-specific regulation of translation can be achieved with microRNAs, which can have either tumor-suppressor or pro-oncogenic effects. For example, miR146a expression is lost in lymphoma (12) and deletion of miR15/16 locus is common in CLL (13), while the miR17-92 cluster is a target of c-Myc and mediates its oncogenic effects [for review, see Ref. (14)].
When cap-dependent translation pathways have been downregulated, for example, through a change in microenvironment or presence of chemotherapeutics, cancer cells may still drive selective oncogene translation via “cap-independent” mechanisms. In such cases, an internal ribosome entry site (IRES) in the 5′ UTR of an mRNA interacts with a selection of canonical initiation factors and specific IRES trans-acting factors (ITAFs) to recruit ribosomes and translate protein independently of the 5′ cap. ITAFs are RNA-binding proteins that can either activate or inhibit IRES activity (15). Cap-independent translation could be of interest in hematopoietic malignancies since many drugs used for treatment, such as DNA-damaging agents or more recently mTOR inhibitors, shut down cap-dependent translation. By acting to maintain key survival pathways in the face of global translation shutdown, cap-independent translation has the potential to impede many chemotherapeutics’ action.
This review flags some instances in which IRESes are found in genes relevant to blood cancers, but in which their context is not understood. In many cases, these IRESes may be incidental to disease progression and response to treatment, since cap-dependent translation may be the only relevant mechanism. In other cases, particularly where upregulation of both ITAF and its cognate IRES are seen, both may be playing a critical role. Their potential to help or hinder treatment of hematological malignancies should not be overlooked. To underscore this point, we end this review by elaborating on the rare examples where the roles of cap-independent translation are better understood.
IRESes are Present in Many Genes Relevant to Blood Cancers
Many genes of relevance to blood cancers contain an IRES element, including many involved in the characteristic translocations (see Table 1). Crucially, the contributions of these IRESes to transformation and progression, if any, are not yet known.
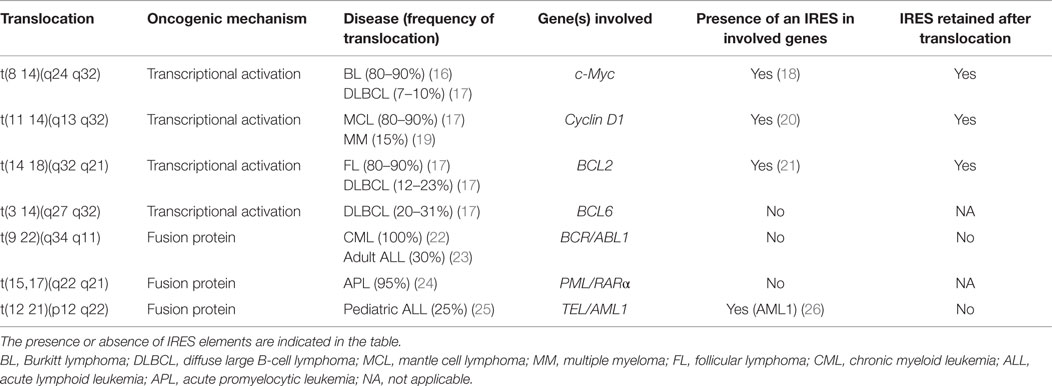
Table 1. Genes with the most common translocations observed in blood cancer.
Many other genetic rearrangements in hematological malignancies (e.g., less common translocations, deletions, duplications, and a wide range of mutations) involve IRES-containing genes. These include VEGF or b-FGF, which both are mutated in several cancers including CLL (27–30). p53 whose 5′ UTR contains two IRESes (31), is mutated in 5–50% of blood cancers [for review, see Ref. (32)], and mutations or deletions of p53 are particularly common in CLL (33).
ITAFs may also be Aberrantly Expressed in Blood Cancers
As discussed above, the ITAF proteins which can interact with specific IRESes are critical for cap-independent translation. Several ITAFs show expression changes in blood cancers. One such example is nucleolin which is overexpressed, and its subcellular localization altered, in CLL and myeloid leukemias (34, 35). SP1, whose IRES is targeted by nucleolin (36), is overexpressed and associated with bad prognosis in many cancers, including acute leukemia (37, 38), and it can drive drug resistance in leukemia stem cells (38, 39). In another example, HuR is overexpressed in subtypes of AML, CML, and ALL [reviewed in Ref. (40)]. HuR is an IRES-binding protein and has been reported to inhibit p27KIP1 IRES-mediated translation (41). Low expression of p27KIP1 is a bad prognosis factor in AML (42). To date, however, the relationship between the oncogenic properties of these two proteins and their cognate IRESes has not been explored.
Fusion proteins generated by translocations can also influence cap-independent translation regulation, e.g., signaling through the tyrosine kinase BCR-ABL1. BCR-ABL1 is a fusion gene that originates from a reciprocal translocation [t(9;22)(q34;q11)] between chromosomes 9 and 22, to generate an aberrant “Philadelphia” chromosome 22 (Table 1). It is present in >95% of CML cases and ~30% of cases of ALL, and sometimes also in AML. Studies suggest that BCR-ABL1 alone can be sufficient to cause CML [for a review, see Ref. (22)]. Because of this, the development of tyrosine kinase inhibitors, such as imatinib, has improved CML prognosis, although resistance can develop.
The BCR-ABL1 protein controls the transcription of several IRES-containing transcripts, including lymphoid enhancer factor-1 (LEF-1) (43). LEF-1 expression increases with CML progression (44, 45). Translation of full-length LEF-1 is partly controlled by an IRES in its 5′ UTR (43). Several LEF-1 ITAFs have been identified, including eIF4A1 (46), which is itself stimulated by BCR-ABL1 via the mTOR pathway. When both mTOR and eIF4A were inhibited, a reduction in the IRES-driven translation of LEF-1 was observed which correlated with a reduced proliferation in hematopoietic cell lines (46). Direct targeting of eIF4A1 activity, in combination with other chemotherapeutics, may therefore be of use in future treatments of CML in individuals who are resistant to tyrosine kinase inhibitors.
BCR-ABL1 also directly regulates transcription of several ITAFs including La/SSB (47), hnRNPA1 (48), hnRNPE2 (49), and hnRNPK (50). La/SSB has been shown to bind to the IRES of mRNA coding for the chaperone protein BIP (51). This protein is increased in cells expressing BRC/ABL1 fusion protein (52), and a cytosolic isoform of BIP has been described to activate PERK signaling and drive survival in leukemia cells (53). hnRNPA1, hnRNPE2, and hnRNPK have been shown to be important for BCR-ABL1-driven oncogenesis (54). Notari and colleagues also showed that, upon induction by BCR-ABL1, hnRNPK induced c-Myc IRES-dependant translation (50). Interestingly, hnRNPA1 has also been shown to associate with c-Myc IRES (55, 56). However, the role of the ITAF activity of these proteins in the context of CML has not been studied.
Overexpressed La/SSB, when induced by BCR/ABL1 or by JAK2 mutations, was also shown to bind to a 27 nucleotides sequence in the MDM2 5′ UTR and activate translation. Interestingly, MDM2 5′ UTR shares 70% identity with the 5′ UTR of BIP (47, 57). Regulation of MDM2 expression by La/SSB operates following DNA damage and consequent inhibition of cap-dependent translation, and altogether this would suggest that MDM2 5′ UTR does contain an IRES regulated by La/SSB. MDM2 is an ubiquitin ligase that targets p53, leading to its degradation and is overexpressed through various mechanisms in most blood cancer types (58). The potential impact on p53 means that further studies to explore cap-independent translation of MDM2 in hematological malignancies may be of value.
Multiple Myeloma as a Paradigm for the Importance of Cap-Independent Translation in Blood Cancers
The blood cancer for which we have the clearest evidence for the importance of cap-independent translation is multiple myeloma (MM). MM is a disease caused by clonal expansion of plasmocytes. Normal, mature plasmocytes differentiate from activated B cells and produce large quantities (up to 2000 molecules/s) of antibody, such that immunoglobulins normally occupy ~20% of total plasma protein. This massive protein production means that plasma cells must adapt to great ER stress during their development, sustaining unfolded protein response (UPR) and autophagy without inducing apoptosis (59–61).
The normal lifespan of plasmocytes can vary from a few days to several years. Differentiated plasma cells appear to live longest in specialized niches in the bone marrow (62). It is thought that such niches are limited, generating competition between plasma cells arising at different times (63–65). Overall, the nature of normal plasma cells predisposes them to longevity, resistance to apoptotic effects of ER stress, chemotactic movement to migrate and establish in competition-intense niches, and traits which also aid malignant cell survival. MM usually initiates from a B cell with somatic hypermutation (often of c-Myc) and translocations in the immunoglobulin genes (66, 67). While the disease is traditionally described as arising from a transformed differentiated plasma cell, the malignant population may instead derive from a small population of relatively chemoresistant, stem cell-like B cell precursors (68).
Broad spectrum chemotherapeutics, such as Melphalan, are used to treat MM, often in combinations that vary with stage and chemoresistance. Due to the generally greater protein load of MM cells, drugs which target the proteasome (such as bortezomib) are often effective (69). Unusually, thalidomide, a drug with immunomodulatory properties whose mechanism of action is not completely understood, is also increasingly used. This, and similar drugs such as Lenalidomide, appears to have diverse antitumor effects (70, 71). IRESes have the potential to impact upon MM drug response. For example, thalidomide may reduce the proliferative effects of MM cells in part by targeting the IRES of the b-FGF growth factor (72).
The greatest focus of research, however, has been upon c-Myc (summarized in Figure 1). MM cells are thought often to be addicted to c-Myc overexpression and c-Myc levels correlate with disease progression (73–75). c-Myc overexpression is known to have impact on translation in many ways, for example, by acting as a transcriptional activator of ribosomal proteins and translation initiation factors, such as eIF4E [for a review, see Ref. (76)]. Intriguingly, it is also able to modulate mTORC1 activity to enhance 4EBP1 phosphorylation (76).
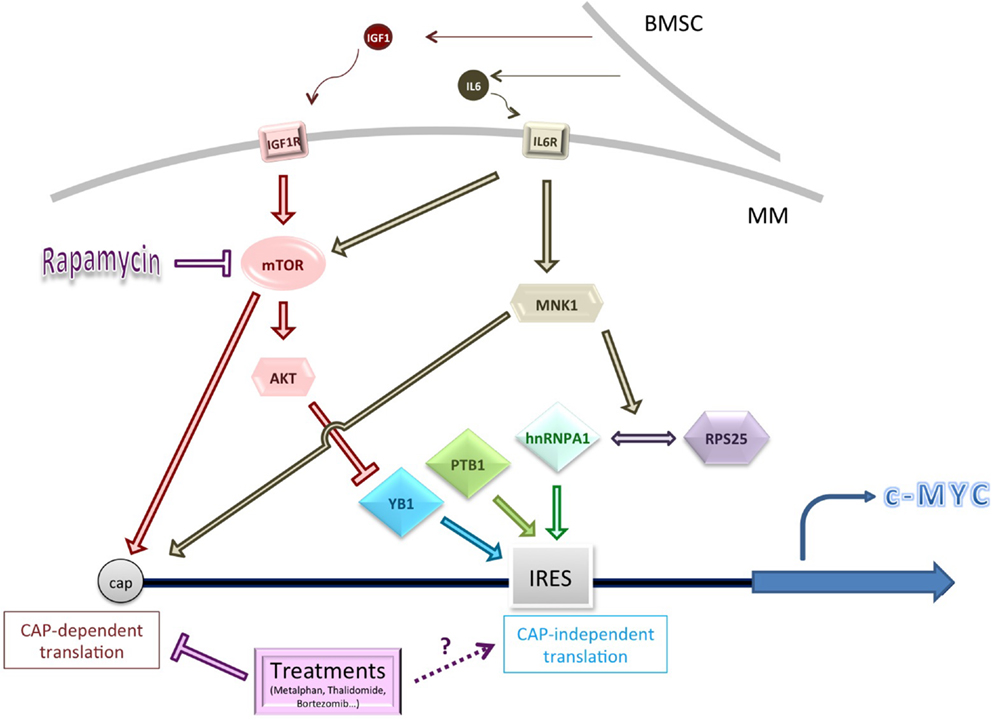
Figure 1. Schematic summarizing some of the genes and pathways known to influence c-Myc cap-independent translation in multiple myeloma (MM). Cytokine activation of key signaling pathways, or pathway dysregulation, may affect the levels or activity of IRES trans-activating factors (ITAFs), including YB1 (conditions of low Akt activity). MNK1-mediated phosphorylation of hnRNPA1 and/or RPS25 increases interaction between them and might improve ribosome loading on the c-Myc IRES (77). Conventional treatments for MM usually inhibit CAP-dependent translation but their impact on CAP-independent translation is unknown. BMSC, bone marrow stromal cell.
c-Myc contains an IRES in its 5′ UTR (18, 78). A C-T mutation at position 2796 of the IRES affects the secondary structure and correlates with increased c-Myc translation (79). The IRES mutation was found to be overrepresented in the bone marrow of patients with MM (80). Multiple proteins have been identified as Myc family ITAFs (55, 56, 81, 82). Of these, ITAFs PTB1 and YB1 showed a higher affinity for RNA containing the mutated IRES in vitro, compared to wild type, and acted synergistically to drive higher expression of a downstream luciferase in a reporter construct. A correlation was also observed between expression of ITAFs PTB1 and YB1 and c-Myc in two MM-derived cell lines carrying the mutation (83).
It should be stressed that elevated YB1 protein levels in MM are likely to have impact widely on its establishment and progression, via both transcriptional and translational effects (84). YB1 effects may also vary with activity of the Ras-ERK and Ras-AKT pathways. High levels of AKT activity are observed in MM (85, 86), and other cancer studies have shown that YB1 is phosphorylated by AKT, rendering it less likely to enhance cap-independent translation (87). In support of this model in MM lines, when mTOR is inhibited by Rapamycin, and AKT activation is maintained, levels of c-Myc and Cyclin D proteins are not maintained by IRES-mediated translation and decline (88). This may be relevant to MM cells in the bone marrow, where malignant cells stimulate the bone marrow stromal cells (BMSCs) to produce the cytokines IL-6 and IGF1, which stimulates the IL6 and IGF1R receptors respectively, leading to activation of AKT (and ERK) signaling (89–91).
In contrast, low AKT activity allows cap-independent translation of c-Myc and Cyclin D to take place (20). It is possible that niche-responsive variation in AKT activity and expression of YB1 (and therefore also c-Myc) allows cells from the same clone, but in different environments, to plastically adopt a quiescent or proliferative phenotype that would result in differential sensitivity to chemotherapeutics (92, 93). A study of the phosphorylation status and relative levels and activity of nuclear and cytoplasmic YB1 and its proteolytically cleaved product within MM samples from different niches would be interesting to explore.
There also appears to be an important role for MNK1 in the activation of the c-Myc IRES under chemotherapeutic conditions. MNK1 is best known as a key player in cap-dependent translation; both p38 and ERK1/2 signaling pathways can catalyze its phosphorylation. This activated form can interact with scaffold protein eIF4G and subsequently activate eIF4E phosphorylation (94).
MNK1 can also phosphorylate eIF4B to activate translation [for a review, see Ref. (95)]. Activation of MNK1 via phosphorylation has also been implicated in cap-independent c-Myc translation to facilitate survival during treatment with Rapamycin (and also with common MM chemotherapeutics expected to inhibit cap-dependent translation, such as bortezomib) (77, 88). MNK1 is likely to be phosphorylated within the bone marrow microenvironment, since IL6 stimulates the activity of the p38 MAPK pathway (96).
MNK1 was found to be necessary for the interaction of the hnRNPA1 ITAF and RPS25 with the c-Myc IRES (77, 82). Both proteins have previously been established as having roles in cap-independent translation (which for RPS25 appears to outweigh any role in cap-dependent translation) (97). Recent work has identified a compound able to prevent hnRNPA1 binding to the c-Myc IRES; the compound was observed to reduce c-Myc expression only following induction of ER stress, when only IRES-dependent mechanisms would be expected to act (77). There is thus an exciting possibility that cap-independent translation inhibitors may be combined with chemotherapeutics that inhibit cap-dependent translation to block a possible survival route for cancer cells.
In a note of caution, the existence of multiple c-Myc ITAFs renders it possible that the c-Myc IRES could be stimulated in vivo even in the presence of an hnRNPA1 inhibitor, since it is currently unknown whether this particular ITAF is essential to c-Myc IRES function under all conditions. Other ITAFs, such as YB1, may be able to act independently of hnRNPA1 as c-Myc ITAFs (98). Detailed characterization of the key signaling pathways permitting survival in MM cells across different niches, and after different treatment regimens, would provide clarity on this subject.
Conclusion
Existing data illustrate that IRES function is relevant to several hematological malignancies and that specific IRESes or ITAFs may present druggable targets in the future. However, much work still needs to be done to clarify the roles of known IRESes in pathology and response to chemotherapeutics, in addition to finding further relevant examples of cap-independent translation. While acknowledging unusual examples such as LEF-1, one would expect that, in the majority of cases, IRES function is likely to be of most relevance when cap-dependent translation is compromised, e.g., following exposure to chemotherapeutic agents and in a hypoxic tumor cell environment. How IRESes mediate survival in such circumstances is likely to lead to a better understanding of which combinations of chemotherapeutics can be used to treat this very disparate and difficult to treat group of diseases. One way to address this question would be to ask if clones enriched for IRES-containing oncogenes arise, and persist, even as a small niche-specific subpopulation, following chemotherapy.
Author Contributions
All authors contributed to writing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was funded by BBSRC grant numbers for AB (BB/H024980/1) and DP (BB/M006700/1) and MRC programme grant funding for EH, LW, TP, and AW.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer (2015) 136:E359–86. doi: 10.1002/ijc.29210
2. Chitteti BR, Cheng YH, Poteat B, Rodriguez-Rodriguez S, Goebel WS, Carlesso N, et al. Impact of interactions of cellular components of the bone marrow microenvironment on hematopoietic stem and progenitor cell function. Blood (2010) 115:3239–48. doi:10.1182/blood-2009-09-246173
3. Hallek M. Chronic lymphocytic leukemia: 2015 update on diagnosis, risk stratification, and treatment. Am J Hematol (2015) 90:446–60. doi:10.1002/ajh.23979
4. Estey EH. Acute myeloid leukemia: 2014 update on risk-stratification and management. Am J Hematol (2014) 89:1063–81. doi:10.1002/ajh.23834
5. Hunger SP, Mullighan CG. Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood (2015) 125:3977–87. doi:10.1182/blood-2015-02-580043
6. Hennessy BT, Hanrahan EO, Daly PA. Non-Hodgkin lymphoma: an update. Lancet Oncol (2004) 5:341–53. doi:10.1016/S1470-2045(04)01490-1
7. Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer (2007) 7:233–45. doi:10.1038/nrc2091
8. Raaijmakers MH, Scadden DT. Evolving concepts on the microenvironmental niche for hematopoietic stem cells. Curr Opin Hematol (2008) 15:301–6. doi:10.1097/MOH.0b013e328303e14c
9. Eyre TA, Collins GP, Goldstone AH, Cwynarski K. Time now to TORC the TORC? New developments in mTOR pathway inhibition in lymphoid malignancies. Br J Haematol (2014) 166:336–51. doi:10.1111/bjh.12945
10. Pettersson F, Del Rincon SV, Miller WH Jr. Eukaryotic translation initiation factor 4E as a novel therapeutic target in hematological malignancies and beyond. Expert Opin Ther Targets (2014) 18:1035–48. doi:10.1517/14728222.2014.937426
11. Horvilleur E, Sbarrato T, Hill K, Spriggs RV, Screen M, Goodrem PJ, et al. A role for eukaryotic initiation factor 4B overexpression in the pathogenesis of diffuse large B-cell lymphoma. Leukemia (2014) 28(5):1092–102. doi:10.1038/leu.2013.295
12. Paik JH, Jang JY, Jeon YK, Kim WY, Kim TM, Heo DS, et al. MicroRNA-146a downregulates NFkappaB activity via targeting TRAF6 and functions as a tumor suppressor having strong prognostic implications in NK/T cell lymphoma. Clin Cancer Res (2011) 17:4761–71. doi:10.1158/1078-0432.CCR-11-0494
13. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A (2002) 99:15524–9. doi:10.1073/pnas.242606799
14. Dal Bo M, Bomben R, Hernandez L, Gattei V. The MYC/miR-17-92 axis in lymphoproliferative disorders: a common pathway with therapeutic potential. Oncotarget (2015) 6:19381–92. doi:10.18632/oncotarget.4574
15. Martinez-Salas E, Lozano G, Fernandez-Chamorro J, Francisco-Velilla R, Galan A, Diaz R. RNA-binding proteins impacting on internal initiation of translation. Int J Mol Sci (2013) 14:21705–26. doi:10.3390/ijms141121705
16. Molyneux EM, Rochford R, Griffin B, Newton R, Jackson G, Menon G, et al. Burkitt’s lymphoma. Lancet (2012) 379:1234–44. doi:10.1016/S0140-6736(11)61177-X
17. Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer (2005) 5:251–62. doi:10.1038/nrc1589
18. Nanbru C, Lafon I, Audigier S, Gensac MC, Vagner S, Huez G, et al. Alternative translation of the proto-oncogene c-myc by an internal ribosome entry site. J Biol Chem (1997) 272:32061–6. doi:10.1074/jbc.272.51.32061
19. Kuehl WM, Bergsagel PL. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J Clin Invest (2012) 122:3456–63. doi:10.1172/JCI61188
20. Shi Y, Sharma A, Wu H, Lichtenstein A, Gera J. Cyclin D1 and c-myc internal ribosome entry site (IRES)-dependent translation is regulated by AKT activity and enhanced by rapamycin through a p38 MAPK- and ERK-dependent pathway. J Biol Chem (2005) 280:10964–73. doi:10.1074/jbc.M407874200
21. Sherrill KW, Byrd MP, Van Eden ME, Lloyd RE. BCL-2 translation is mediated via internal ribosome entry during cell stress. J Biol Chem (2004) 279:29066–74. doi:10.1074/jbc.M402727200
22. Chereda B, Melo JV. Natural course and biology of CML. Ann Hematol (2015) 94(Suppl 2):S107–21. doi:10.1007/s00277-015-2325-z
23. Buchner M, Swaminathan S, Chen Z, Muschen M. Mechanisms of pre-B-cell receptor checkpoint control and its oncogenic subversion in acute lymphoblastic leukemia. Immunol Rev (2015) 263:192–209. doi:10.1111/imr.12235
24. Melnick A, Licht JD. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood (1999) 93:3167–215.
25. Golub TR, Barker GF, Lovett M, Gilliland DG. Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell (1994) 77:307–16. doi:10.1016/0092-8674(94)90322-0
26. Pozner A, Goldenberg D, Negreanu V, Le SY, Elroy-Stein O, Levanon D, et al. Transcription-coupled translation control of AML1/RUNX1 is mediated by cap- and internal ribosome entry site-dependent mechanisms. Mol Cell Biol (2000) 20:2297–307. doi:10.1128/MCB.20.7.2297-2307.2000
27. Stein I, Itin A, Einat P, Skaliter R, Grossman Z, Keshet E. Translation of vascular endothelial growth factor mRNA by internal ribosome entry: implications for translation under hypoxia. Mol Cell Biol (1998) 18:3112–9. doi:10.1128/MCB.18.6.3112
28. Vagner S, Touriol C, Galy B, Audigier S, Gensac MC, Amalric F, et al. Translation of CUG- but not AUG-initiated forms of human fibroblast growth factor 2 is activated in transformed and stressed cells. J Cell Biol (1996) 135:1391–402. doi:10.1083/jcb.135.5.1391
29. Galy B, Maret A, Prats AC, Prats H. Cell transformation results in the loss of the density-dependent translational regulation of the expression of fibroblast growth factor 2 isoforms. Cancer Res (1999) 59:165–71.
30. Sella O, Gerlitz G, Le SY, Elroy-Stein O. Differentiation-induced internal translation of c-sis mRNA: analysis of the cis elements and their differentiation-linked binding to the hnRNP C protein. Mol Cell Biol (1999) 19:5429–40. doi:10.1128/MCB.19.8.5429
31. Ray PS, Grover R, Das S. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep (2006) 7:404–10. doi:10.1038/sj.embor.7400623
32. Peller S, Rotter V. TP53 in hematological cancer: low incidence of mutations with significant clinical relevance. Hum Mutat (2003) 21:277–84. doi:10.1002/humu.10190
33. el Rouby S, Thomas A, Costin D, Rosenberg CR, Potmesil M, Silber R, et al. p53 gene mutation in B-cell chronic lymphocytic leukemia is associated with drug resistance and is independent of MDR1/MDR3 gene expression. Blood (1993) 82:3452–9.
34. Shen N, Yan F, Pang J, Wu LC, Al-Kali A, Litzow MR, et al. A nucleolin-DNMT1 regulatory axis in acute myeloid leukemogenesis. Oncotarget (2014) 5:5494–509. doi:10.18632/oncotarget.2131
35. Otake Y, Soundararajan S, Sengupta TK, Kio EA, Smith JC, Pineda-Roman M, et al. Overexpression of nucleolin in chronic lymphocytic leukemia cells induces stabilization of bcl2 mRNA. Blood (2007) 109:3069–75. doi:10.1182/blood-2006-08-043257
36. Hung CY, Yang WB, Wang SA, Hsu TI, Chang WC, Hung JJ. Nucleolin enhances internal ribosomal entry site (IRES)-mediated translation of Sp1 in tumorigenesis. Biochim Biophys Acta (2014) 1843:2843–54. doi:10.1016/j.bbamcr.2014.08.009
37. Beishline K, Azizkhan-Clifford J. Sp1 and the ‘hallmarks of cancer’. FEBS J (2015) 282:224–58. doi:10.1111/febs.13148
38. Liu S, Wu LC, Pang J, Santhanam R, Schwind S, Wu YZ, et al. Sp1/NFkappaB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukemia. Cancer Cell (2010) 17:333–47. doi:10.1016/j.ccr.2010.03.008
39. Zhang Y, Chen HX, Zhou SY, Wang SX, Zheng K, Xu DD, et al. Sp1 and c-Myc modulate drug resistance of leukemia stem cells by regulating survivin expression through the ERK-MSK MAPK signaling pathway. Mol Cancer (2015) 14:56. doi:10.1186/s12943-015-0326-0
40. Baou M, Norton JD, Murphy JJ. AU-rich RNA binding proteins in hematopoiesis and leukemogenesis. Blood (2011) 118:5732–40. doi:10.1182/blood-2011-07-347237
41. Kullmann M, Gopfert U, Siewe B, Hengst L. ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5’UTR. Genes Dev (2002) 16:3087–99. doi:10.1101/gad.248902
42. Yokozawa T, Towatari M, Iida H, Takeyama K, Tanimoto M, Kiyoi H, et al. Prognostic significance of the cell cycle inhibitor p27Kip1 in acute myeloid leukemia. Leukemia (2000) 14:28–33. doi:10.1038/sj.leu.2401640
43. Jimenez J, Jang GM, Semler BL, Waterman ML. An internal ribosome entry site mediates translation of lymphoid enhancer factor-1. RNA (2005) 11:1385–99. doi:10.1261/rna.7226105
44. Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med (2004) 351:657–67. doi:10.1056/NEJMoa040258
45. Tsai BP, Wang X, Huang L, Waterman ML. Quantitative profiling of in vivo-assembled RNA-protein complexes using a novel integrated proteomic approach. Mol Cell Proteomics (2011) 10:M110007385. doi:10.1074/mcp.M110.007385
46. Tsai BP, Jimenez J, Lim S, Fitzgerald KD, Zhang M, Chuah CT, et al. A novel Bcr-Abl-mTOR-eIF4A axis regulates IRES-mediated translation of LEF-1. Open Biol (2014) 4:140180. doi:10.1098/rsob.140180
47. Trotta R, Vignudelli T, Candini O, Intine RV, Pecorari L, Guerzoni C, et al. BCR/ABL activates mdm2 mRNA translation via the La antigen. Cancer Cell (2003) 3:145–60. doi:10.1016/S1535-6108(03)00020-5
48. Iervolino A, Santilli G, Trotta R, Guerzoni C, Cesi V, Bergamaschi A, et al. hnRNP A1 nucleocytoplasmic shuttling activity is required for normal myelopoiesis and BCR/ABL leukemogenesis. Mol Cell Biol (2002) 22:2255–66. doi:10.1128/MCB.22.7.2255-2266.2002
49. Perrotti D, Cesi V, Trotta R, Guerzoni C, Santilli G, Campbell K, et al. BCR-ABL suppresses C/EBPalpha expression through inhibitory action of hnRNP E2. Nat Genet (2002) 30:48–58. doi:10.1038/ng791
50. Notari M, Neviani P, Santhanam R, Blaser BW, Chang JS, Galietta A, et al. /HNRPK pathway controls BCR/ABL oncogenic potential by regulating MYC mRNA translation. Blood (2006) 107:2507–16. doi:10.1182/blood-2005-09-3732
51. Buchkovich NJ, Yu Y, Pierciey FJ Jr, Alwine JC. Human cytomegalovirus induces the endoplasmic reticulum chaperone BiP through increased transcription and activation of translation by using the BiP internal ribosome entry site. J Virol (2010) 84:11479–86. doi:10.1128/JVI.01330-10
52. Tanimura A, Yujiri T, Tanaka Y, Hatanaka M, Mitani N, Nakamura Y, et al. The anti-apoptotic role of the unfolded protein response in Bcr-Abl-positive leukemia cells. Leuk Res (2009) 33:924–8. doi:10.1016/j.leukres.2009.01.027
53. Ni M, Zhou H, Wey S, Baumeister P, Lee AS. Regulation of PERK signaling and leukemic cell survival by a novel cytosolic isoform of the UPR regulator GRP78/BiP. PLoS One (2009) 4:e6868. doi:10.1371/journal.pone.0006868
54. Perrotti D, Calabretta B. Post-transcriptional mechanisms in BCR/ABL leukemogenesis: role of shuttling RNA-binding proteins. Oncogene (2002) 21:8577–83. doi:10.1038/sj.onc.1206085
55. Jo OD, Martin J, Bernath A, Masri J, Lichtenstein A, Gera J. Heterogeneous nuclear ribonucleoprotein A1 regulates cyclin D1 and c-myc internal ribosome entry site function through Akt signaling. J Biol Chem (2008) 283:23274–87. doi:10.1074/jbc.M801185200
56. Evans JR, Mitchell SA, Spriggs KA, Ostrowski J, Bomsztyk K, Ostarek D, et al. Members of the poly (rC) binding protein family stimulate the activity of the c-myc internal ribosome entry segment in vitro and in vivo. Oncogene (2003) 22:8012–20. doi:10.1038/sj.onc.1206645
57. Nakatake M, Monte-Mor B, Debili N, Casadevall N, Ribrag V, Solary E, et al. JAK2(V617F) negatively regulates p53 stabilization by enhancing MDM2 via La expression in myeloproliferative neoplasms. Oncogene (2012) 31:1323–33. doi:10.1038/onc.2011.313
58. Quesnel B, Preudhomme C, Oscier D, Lepelley P, Collyn-d’Hooghe M, Facon T, et al. Over-expression of the MDM2 gene is found in some cases of haematological malignancies. Br J Haematol (1994) 88:415–8. doi:10.1111/j.1365-2141.1994.tb05044.x
59. Cenci S, Mezghrani A, Cascio P, Bianchi G, Cerruti F, Fra A, et al. Progressively impaired proteasomal capacity during terminal plasma cell differentiation. EMBO J (2006) 25:1104–13. doi:10.1038/sj.emboj.7601009
60. Auner HW, Cenci S. Recent advances and future directions in targeting the secretory apparatus in multiple myeloma. Br J Haematol (2015) 168:14–25. doi:10.1111/bjh.13172
61. Auner HW, Beham-Schmid C, Dillon N, Sabbattini P. The life span of short-lived plasma cells is partly determined by a block on activation of apoptotic caspases acting in combination with endoplasmic reticulum stress. Blood (2010) 116:3445–55. doi:10.1182/blood-2009-10-250423
62. Hargreaves DC, Hyman PL, Lu TT, Ngo VN, Bidgol A, Suzuki G, et al. A coordinated change in chemokine responsiveness guides plasma cell movements. J Exp Med (2001) 194:45–56. doi:10.1084/jem.194.1.45
63. Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity (1998) 8:363–72. doi:10.1016/S1074-7613(00)80541-5
64. Ahuja A, Anderson SM, Khalil A, Shlomchik MJ. Maintenance of the plasma cell pool is independent of memory B cells. Proc Natl Acad Sci U S A (2008) 105:4802–7. doi:10.1073/pnas.0800555105
65. Manz RA, Thiel A, Radbruch A. Lifetime of plasma cells in the bone marrow. Nature (1997) 388:133–4. doi:10.1038/40540
66. Bergsagel PL, Kuehl WM. Chromosome translocations in multiple myeloma. Oncogene (2001) 20:5611–22. doi:10.1038/sj.onc.1204641
67. Bakkus MH, Heirman C, Van Riet I, Van Camp B, Thielemans K. Evidence that multiple myeloma Ig heavy chain VDJ genes contain somatic mutations but show no intraclonal variation. Blood (1992) 80:2326–35.
68. Matsui W, Wang Q, Barber JP, Brennan S, Smith BD, Borrello I, et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res (2008) 68:190–7. doi:10.1158/0008-5472.CAN-07-3096
69. Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci U S A (2003) 100:9946–51. doi:10.1073/pnas.1334037100
70. Olson KB, Hall TC, Horton J, Khung CL, Hosley HF. Thalidomide (N-Phthaloylglutamimide) in the treatment of advanced cancer. Clin Pharmacol Ther (1965) 6:292–7.
71. Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med (1999) 341:1565–71. doi:10.1056/NEJM199911183412102
72. Lien IC, Horng LY, Hsu PL, Wu CL, Sung HC, Wu RT. Internal ribosome entry site of bFGF is the target of thalidomide for IMiDs development in multiple myeloma. Genes Cancer (2014) 5:127–41.
73. Holien T, Vatsveen TK, Hella H, Waage A, Sundan A. Addiction to c-MYC in multiple myeloma. Blood (2012) 120:2450–3. doi:10.1182/blood-2011-08-371567
74. Chng WJ, Huang GF, Chung TH, Ng SB, Gonzalez-Paz N, Troska-Price T, et al. Clinical and biological implications of MYC activation: a common difference between MGUS and newly diagnosed multiple myeloma. Leukemia (2011) 25:1026–35. doi:10.1038/leu.2011.53
75. Skopelitou A, Hadjiyannakis M, Tsenga A, Theocharis S, Alexopoulou V, Kittas C, et al. Expression of C-myc p62 oncoprotein in multiple myeloma: an immunohistochemical study of 180 cases. Anticancer Res (1993) 13:1091–5.
76. van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer (2010) 10:301–9. doi:10.1038/nrc2819
77. Shi Y, Yang Y, Hoang B, Bardeleben C, Holmes B, Gera J, et al. Therapeutic potential of targeting IRES-dependent c-myc translation in multiple myeloma cells during ER stress. Oncogene (2015). doi:10.1038/onc.2015.156
78. Stoneley M, Paulin FE, Le Quesne JP, Chappell SA, Willis AE. C-Myc 5’ untranslated region contains an internal ribosome entry segment. Oncogene (1998) 16:423–8. doi:10.1038/sj.onc.1201763
79. Paulin FE, West MJ, Sullivan NF, Whitney RL, Lyne L, Willis AE. Aberrant translational control of the c-myc gene in multiple myeloma. Oncogene (1996) 13:505–13.
80. Chappell SA, LeQuesne JP, Paulin FE, deSchoolmeester ML, Stoneley M, Soutar RL, et al. A mutation in the c-myc-IRES leads to enhanced internal ribosome entry in multiple myeloma: a novel mechanism of oncogene de-regulation. Oncogene (2000) 19:4437–40. doi:10.1038/sj.onc.1203791
81. Cobbold LC, Spriggs KA, Haines SJ, Dobbyn HC, Hayes C, de Moor CH, et al. Identification of internal ribosome entry segment (IRES)-trans-acting factors for the Myc family of IRESs. Mol Cell Biol (2008) 28:40–9. doi:10.1128/MCB.01298-07
82. Shi YJ, Frost PJ, Hoang BQ, Benavides A, Sharma S, Gera JF, et al. IL-6-induced stimulation of c-Myc translation in multiple myeloma cells is mediated by Myc internal ribosome entry site function and the RNA-binding protein, hnRNP A1. Cancer Res (2008) 68:10215–22. doi:10.1158/0008-5472.CAN-08-1066
83. Cobbold LC, Wilson LA, Sawicka K, King HA, Kondrashov AV, Spriggs KA, et al. Upregulated c-myc expression in multiple myeloma by internal ribosome entry results from increased interactions with and expression of PTB-1 and YB-1. Oncogene (2010) 29:2884–91. doi:10.1038/onc.2010.31
84. Chatterjee M, Rancso C, Stuhmer T, Eckstein N, AndruliS M, Gerecke C, et al. The Y-box binding protein YB-1 is associated with progressive disease and mediates survival and drug resistance in multiple myeloma. Blood (2008) 111:3714–22. doi:10.1182/blood-2007-05-089151
85. Hsu JH, Shi YJ, Krajewski S, Renner S, Fisher M, Reed JC, et al. The AKT kinase is activated in multiple myeloma tumor cells. Blood (2001) 98:2853–5. doi:10.1182/blood.V98.9.2853
86. Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell (2009) 137:873–86. doi:10.1016/j.cell.2009.03.046
87. Evdokimova V, Ovchinnikov LP, Sorensen PHB. Y-box binding protein 1 – providing a new angle on translational regulation. Cell Cycle (2006) 5:1143–7. doi:10.4161/cc.5.11.2784
88. Shi Y, Frost P, Hoang B, Yang Y, Fukunaga R, Gera J, et al. MNK kinases facilitate c-myc IRES activity in rapamycin-treated multiple myeloma cells. Oncogene (2013) 32:190–7. doi:10.1038/onc.2012.43
89. Bataille R, Robillard N, Avet-Loiseau H, Harousseau JL, Moreau P. CD221 (IGF-1R) is aberrantly expressed in multiple myeloma, in relation to disease severity. Haematologica (2005) 90:706–7.
90. Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene (2007) 26:1932–40. doi:10.1038/sj.onc.1209990
91. Wegiel B, Bjartell A, Culig Z, Persson JL. Interleukin-6 activates PI3K/Akt pathway and regulates cyclin A1 to promote prostate cancer cell survival. Int J Cancer (2008) 122:1521–9. doi:10.1002/ijc.23261
92. Evdokimova V, Tognon C, Ng T, Ruzanov P, Melnyk N, Fink D, et al. Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell (2009) 15:402–15. doi:10.1016/j.ccr.2009.03.017
93. Kim ER, Selyutina AA, Buldakov IA, Evdokimova V, Ovchinnikov LP, Sorokin AV. The proteolytic YB-1 fragment interacts with DNA repair machinery and enhances survival during DNA damaging stress. Cell Cycle (2013) 12:3791–803. doi:10.4161/cc.26670
94. Shveygert M, Kaiser C, Bradrick SS, Gromeier M. Regulation of eukaryotic initiation factor 4E (eIF4E) phosphorylation by mitogen-activated protein kinase occurs through modulation of Mnk1-eIF4G interaction. Mol Cell Biol (2010) 30:5160–7. doi:10.1128/MCB.00448-10
95. Blagden SP, Willis AE. The biological and therapeutic relevance of mRNA translation in cancer. Nat Rev Clin Oncol (2011) 8:280–91. doi:10.1038/nrclinonc.2011.16
96. Hideshima T, Akiyama M, Hayashi T, Richardson P, Schlossman R, Chauhan D, et al. Targeting p38 MAPK inhibits multiple myeloma cell growth in the bone marrow milieu. Blood (2003) 101:703–5. doi:10.1182/blood-2002-06-1874
97. Hertz MI, Landry DM, Willis AE, Luo G, Thompson SR. Ribosomal protein S25 dependency reveals a common mechanism for diverse internal ribosome entry sites and ribosome shunting. Mol Cell Biol (2013) 33:1016–26. doi:10.1128/MCB.00879-12
98. Frost P, Shi Y, Hoang B, Gera J, Lichtenstein A. Regulation of D-cyclin translation inhibition in myeloma cells treated with mammalian target of rapamycin inhibitors: rationale for combined treatment with extracellular signal-regulated kinase inhibitors and rapamycin. Mol Cancer Ther (2009) 8:83–93. doi:10.1158/1535-7163.MCT-08-0254
Keywords: leukemia, lymphoma, myeloma, IRES, cap-independent, translation
Citation: Horvilleur E, Wilson LA, Bastide A, Piñeiro D, Pöyry TAA and Willis AE (2015) Cap-Independent Translation in Hematological Malignancies. Front. Oncol. 5:293. doi: 10.3389/fonc.2015.00293
Received: 22 September 2015; Accepted: 08 December 2015;
Published: 21 December 2015
Edited by:
Sunnie R. Thompson, University of Alabama at Birmingham, USAReviewed by:
Giovanni Porta, University of Insubria, ItalyAnna Rita Migliaccio, Mount Sinai School of Medicine, USA
Jian Lu, Johns Hopkins University, USA
Copyright: © 2015 Horvilleur, Wilson, Bastide, Piñeiro, Pöyry and Willis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anne E. Willis, YWV3NUBsZS5hYy51aw==
†Emilie Horvilleur and Lindsay A. Wilson have contributed equally to this work.