 Praneeth R. Kuninty
Praneeth R. Kuninty Jonas Schnittert
Jonas Schnittert Gert Storm1,2
Gert Storm1,2 Jai Prakash
Jai Prakash- 1Targeted Therapeutics Section, Department of Biomaterials, Science and Technology, MIRA Institute for Biomedical Technology and Technical Medicine, University of Twente, Enschede, Netherlands
- 2Department of Pharmaceutics, Utrecht University, Utrecht, Netherlands
Communication between stromal cells and tumor cells initiates tumor growth, angiogenesis, invasion, and metastasis. Stromal cells include cancer-associated fibroblasts, tumor-associated macrophages, pericytes, endothelial cells, and infiltrating immune cells. MicroRNAs (miRNAs) in the tumor microenvironment have emerged as key players involved in the development of cancer and its progression. miRNAs are small endogenous non-protein-coding RNAs that negatively regulate the expression of multiple target genes at post-transcriptional level and thereby control many cellular processes. In this review, we provide a comprehensive overview of miRNAs dysregulated in different stromal cells and their impact on the regulation of intercellular crosstalk in the tumor microenvironment. We also discuss the therapeutic significance potential of miRNAs to modulate the tumor microenvironment. Since miRNA delivery is quite challenging and the biggest hurdle for clinical translation of miRNA therapeutics, we review various non-viral miRNA delivery systems that can potentially be used for targeting miRNA to stromal cells within the tumor microenvironment.
Introduction
The tumor microenvironment is composed of cancer cells and non-cancerous cells so-called stromal cells such as cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), pericytes, endothelial cells, and infiltrating immune cells (1, 2). Over the last decade, it was well established that stromal cells promote tumor growth, angiogenesis, invasion, and metastasis (3, 4). These effects are observed in breast, pancreatic, liver, brain, ovarian, and prostate cancer. Evidence suggests that tumor cells recruit stromal cells by secreting chemokines and growth factors, which educate them to create a tumor-favoring microenvironment (4). The “educated” stromal cells, such as CAFs, endothelial cells, pericytes, TAMs, and other immune cells, interact with tumor cells as well as among themselves to stimulate tumor growth, metastasis, and development of resistance to chemotherapy (3, 4). Intervening into these interactions within the tumor microenvironment is an interesting strategy to develop novel therapies for cancer treatment.
MicroRNAs (miRNAs) are represented as a novel class of therapeutics, regulating multiple signaling pathways within the tumor microenvironment (5). miRNAs, a class of small (17–25 nt) endogenous non-coding RNAs, regulate gene expression at the post-transcriptional level and thereby control cellular processes such as differentiation, proliferation, and migration (6). miRNAs have the ability to regulate not only one but also hundreds of target genes simultaneously and thereby control multiple signaling pathways (7). Gene silencing occurs through imperfect/perfect complementary base pairing between a miRNA guide strand and the 3′ UTR region of the mRNA, which leads to translational repression or mRNA degradation (6, 8). During cancer initiation and progression, the expression levels of multiple miRNAs are aberrantly up- or downregulated, resulting in an imbalance of cellular pathways ultimately leading to the attainment of a pathological state. In this article, we highlight dysregulated miRNAs in different tumor stromal cells and their functions in the regulation of the multifaceted tumor microenvironment. The expression of miRNAs can be controlled by administering either miRNA inhibitors (antagomiR) or miRNA agonists (miR mimics). We summarize various miRNA delivery approaches that have been or can potentially be applied to deliver miRNA as therapeutics into stromal cells.
MicroRNA in the Tumor Microenvironment
In recent years, many miRNAs have been identified in different stromal cells of the tumor microenvironment as illustrated in Figure 1. The Table 1 summarizes the miRNAs in the stromal cells from different cancer types with genes regulated by these miRNAs and their functions.
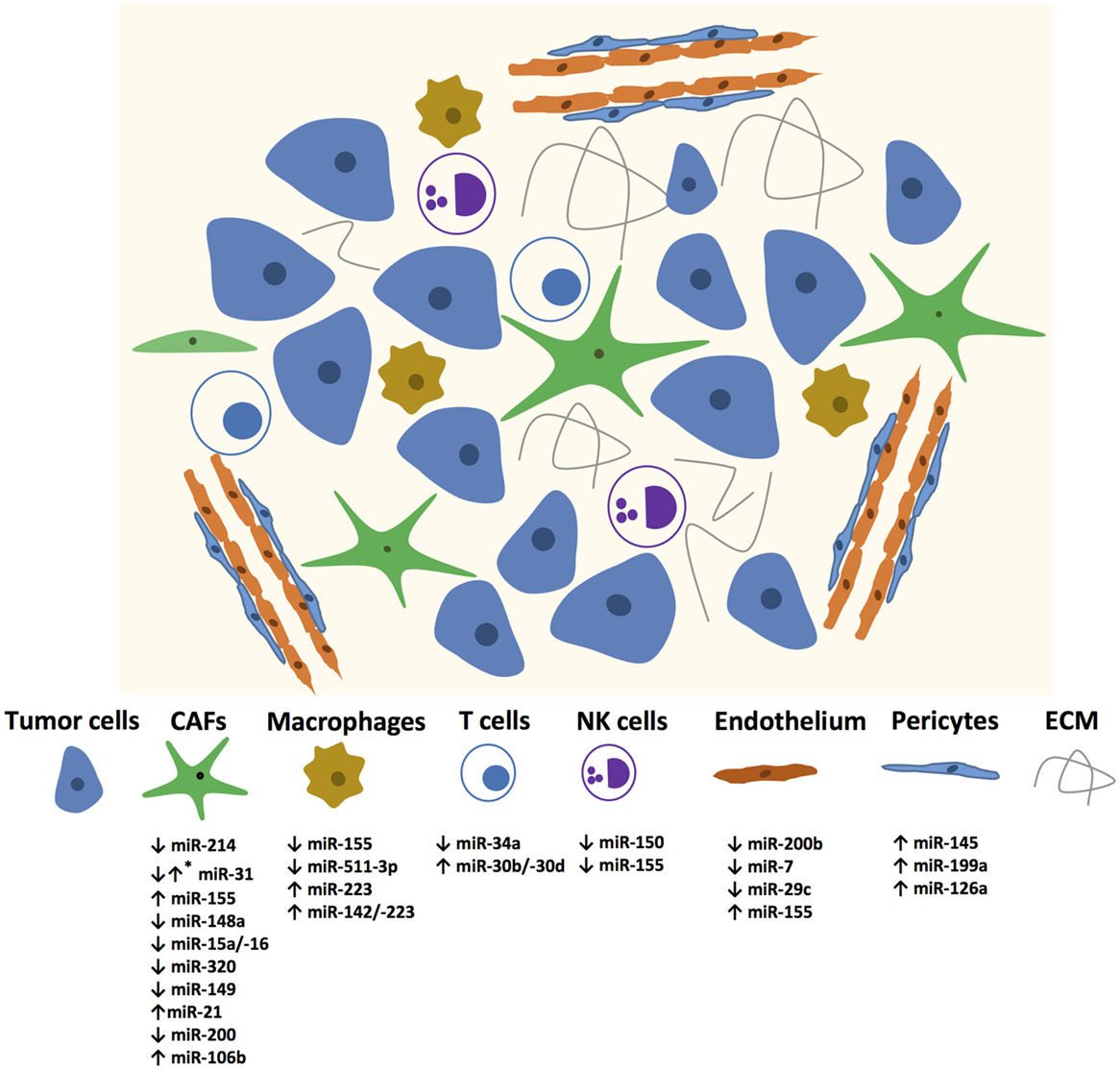
Figure 1. miRNAs mediating changes in tumor microenvironment components. Up and down regulated miRNAs are enlisted? *Mark denotes that the specific miRNA is expressed differentially in different CAFs.
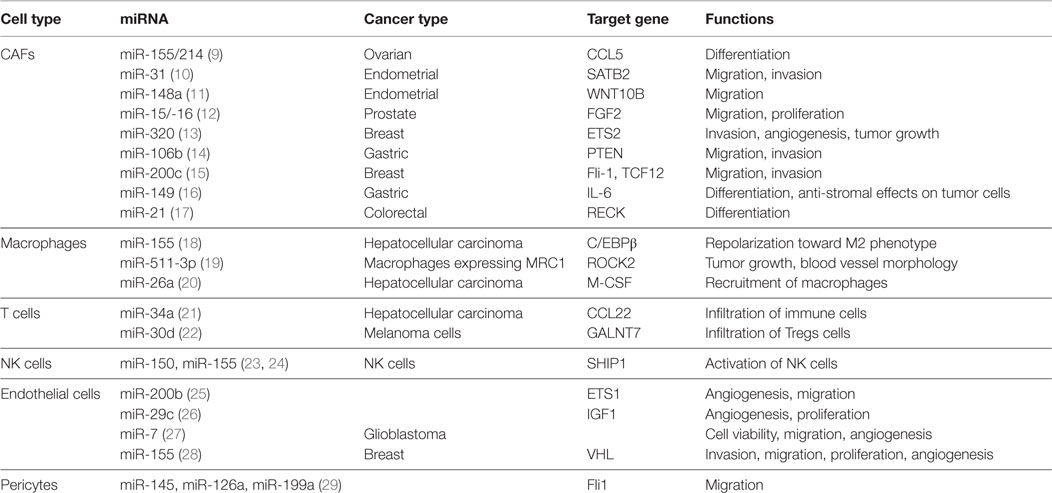
Table 1. List of miRNAs in various tumor stromal cells.
Cancer-Associated Fibroblasts
Cancer-Associated Fibroblasts are one of the most abundant cell types in the tumor microenvironment of many solid tumors (30, 31). In response to inflammatory stimuli, quiescent fibroblasts differentiate into activated myofibroblast (CAFs), expressing increased levels of α-smooth muscle actin (α-SMA). CAFs secrete numerous cytokines, chemokines, and ECM components, which actively participate in tumor progression, invasion, and metastasis (32–35). Differentiation of fibroblasts into a CAF phenotype has been proposed to be regulated at the post-transcriptional level by miRNAs (36).
Several studies have reported the importance of specific miRNAs in the activation and transdifferentiation of fibroblasts to CAFs and CAF-induced tumorigenic actions (9–11, 37). miRNA-21 is one of the most common miRNAs that is reported to be induced in tumor cells and CAFs of pancreatic and colorectal tumors (17, 37–40). Inhibition of miR-21 using antagomiR reduced the migration/invasion of CAFs (38). Not only upregulation but also downregulation of certain miRNAs can induce a CAF phenotype. Tang et al. identified downregulated miR-200 as a direct regulator of reprograming fibroblasts into CAFs (15). Downregulation of miR-200 in normal fibroblasts accelerated their migration and invasion potential similar to CAFs (15). Bronisz and coworkers identified miR-320 as a downstream regulator of the PTEN (Phosphatase and tensin homolog deleted on chromosome 10) gene that controls cell proliferation and migration in CAFs and was co-expressed in tumor stroma of breast tissue. Loss of PTEN and miR-320 has shown to be involved in the reprograming of the tumor microenvironment to promote tumor invasion and angiogenesis (13).
Mitra et al. found downregulation of miR-31 and miR-214 and upregulation of miR-155 in their miRNA profiling, and reversal of the activities of these miRNA in these patient-derived CAFs reversed their phenotype (9). This study suggested that miRNA reprograms fibroblasts into CAFs, and, therefore, targeting of miRNA in stromal cells could be a therapeutic approach to treat cancer (9). In other studies, miR-31 and miR-148a were shown to be downregulated in endometrial CAFs (10, 11). Overexpression of miR-31 or miR-148a in these CAFs impaired their ability to stimulate migration and invasion of endometrial cancer cells, which were linked to the direct targets SATB2 and WNT10B for miR-31 and miR-148a, respectively (10, 11).
Using miRNA microarray analysis, several dysregulated miRNAs have been identified in breast CAFs, e.g., miR-31-3p, 221-5p, and 221-3p were upregulated and miR-205, miR-200b, miR-200c, miR-141, miR-101, miR-342-3p, let-7g, and miR-26b were downregulated (41, 42). Furthermore, many miRNA-responsive target genes and signaling pathways were revealed that regulate different cellular processes such as cell differentiation, adhesion, migration, proliferation, and cell–cell interaction (41). It is important to note that miR-31 was found to be downregulated in CAFs derived from ovarian and endometrial tumors (9, 10) while it was upregulated in CAFs from breast tumor (41), indicating that the same miRNA can have dual activities, thereby acting as an oncogene in one tissue and as a tumor suppressor in another.
In prostate CAFs, miR-15a and miR-16 were shown to be downregulated in CAFs obtained from 23 patients (12). Downregulation of miR-15 and miR-16 in CAFs promoted tumor progression through the reduced post-transcriptional repression of Fgf-2 and its receptor Fgfr1. These pathways act on both stromal and tumor cells to enhance cancer cell survival, proliferation, and migration (12). Reconstitution of tumor-suppressive miR-15a and miR-16 in CAFs inhibited tumor-promoting ability of stromal cells as shown in a co-injection (tumor cells and CAFs) mouse model, proposing these miRNA as potential targets for the development of novel therapies (12).
Additionally, Li et al. demonstrated that miR-149 mediates the crosstalk between the tumor cell and CAFs in gastric cancer via prostaglandin E2 (PGE2) and interleukin (IL)-6 signaling pathways (16). While it remains unclear how PGE-2 modulates this crosstalk, it was demonstrated that by targeting IL-6, miR-149 inhibited fibroblast activation (16). The effects of CAFs on gastric cancer development were negatively regulated by miR-149 (16). Additionally, CAFs enhanced the epithelial to mesenchymal transition (EMT) and stem-like properties of gastric cancer cells in a miR-149-/IL-6-dependent manner (16). In another study, miR-106b has been identified as a marker of poor prognosis in gastric cancer (14). Downregulation of miR-106b expression in CAFs resulted in significantly inhibited CAF-induced gastric cancer cell migration and invasion mediated through PTEN pathway (14). These studies reveal the miRNA targets as diagnostic biomarkers and therapeutic targets for developing the anti-CAF therapy.
Tumor-Associated Macrophages and Immune Cells
Macrophages and other immune cells such as T-cells and natural killer (NK) cells are the major inflammatory cells infiltrating into the tumor microenvironment (3, 43). In the past, the infiltration of innate and adaptive immune cells into the tumor microenvironment was considered as an immune attack against cancer (43). However, now it is widely accepted that immune cells do also promote cancer initiation, progression, and metastasis (44). Macrophages within the tumor microenvironment can be polarized from antitumorigenic M1 macrophages to pro-tumorigenic M2 macrophages [TAMs, via changes in their metabolic pathways and the production of cytokines (CSF-1, IL-4, IL-13) by immune cells (43)]. TAMs promote tumor progression by stimulating angiogenesis, tumor cell migration, and extravasation at metastatic sites and suppressing antitumor immunity thereby reduce patient survival (43, 45, 46). Recent studies have unraveled the significance/dysregulation of miRNA in macrophages (47). In a study by Graff et al., miRNA expression profiles were determined in monocyte-derived macrophages differentially polarized into M1, M2a, M2b, and M2c phenotypes (48). They reveal several miRNAs to be uniquely regulated in human macrophages polarized into M1 (miR-125a-3p, miR-26a-2*), M2a (miR-193b), and M2b (miR-27a*, miR-29-b-1*, miR-132, miR-222*) (48). Herein, we report approaches through which dysregulated miRNAs have been targeted to reprogram miRNA expression in TAMs and thereby suppress their pro-tumorigenic properties.
Squadrito et al. showed that miR-511-3p, encoding for the macrophage mannose receptor, is upregulated in MRC1+ TAMs (19, 49). Enforcing miR-511-3p expression in MRC1+ TAMs resulted in the suppression of pro-tumoral genes and inhibited tumor growth with a change in blood vessel morphology. These effects were attributed to ROCK2, a direct target of miR-511-3p (19). The protein expression of transcription factor C/EBPβ showed elevated levels in TAMs as well as human hepatocellular carcinoma (HCC) tumor sections in situ (18). C/EBPβ expression was correlated with the production of cytokines in tumor-activated monocytes and shown to be regulated by sustained reduction of miR-155 (18). Overexpression of miRNA-155 was shown to attenuate the production of the cytokines (IL-6, IL-10, and TNF-α) by suppressing C/EBPβ expression, which led to inversion of pro-tumoral M2 into pro-inflammatory M1 macrophages, as demonstrated by upregulated M1 markers (TNF-α, NOS2, and IL-12) and downregulated M2 markers (Arg1, Ym1, Msr2, Fizz1, and IL-10) (50). More recently, ectopic expression of miR-26 in HCC cells suppressed the tumor growth, downregulated the expression of macrophage colony-stimulating factor (M-CSF) through the PI3K/Akt pathway, and suppressed the infiltration of macrophages in tumors (20). In addition, miR-26a expression was inversely correlated with M-CSF expression and the infiltration of macrophages into the tumor tissue of HCC patients (20).
Besides TAMs, other immune cells such as myeloid-derived suppressor cells (MDSCs), NK cells, and T cells also express miRNAs that regulate their pro-tumorigenic potential. MDSCs negatively regulate immune responses by suppressing the antitumor functions of CD4+ and CD8+ T cells by inhibiting the activities of NK cells (51). miR-155 and miR-21 are reported as the most upregulated miRNAs in MDSCs from bone marrow cells, regulating PTEN and SHIP1, respectively (52). They promote STAT3 activity by inducing MDSC expansion that promotes tumor aggressiveness via immunosuppression (52). In HCC, positive for the hepatitis B virus, suppressed levels of miR-34a resulted in the enhanced production of chemokine CCL22, thereby recruiting regulatory T cells (Tregs) into the tumor microenvironment to facilitate immune escape (21). In human melanoma upregulation of miR-30b/-30d correlates with stage, metastatic potential, shorter time to recurrence and reduced overall survival (22). Upregulation of miR-30d in the immunocompetent mice triggered immunosuppressive properties at the lung metastatic site, shown by an enhanced infiltration of Tregs (22). Bezman et al. suggested that miR-150 differentially controls the development of NK and invariant NK T (iNKT) cells by targeting c-Myb. Mice with miR-150 deletion showed a defect in their ability to develop mature NK cells (23). Overexpression of miR-150 promotes the development of mature NK-cells, which were highly responsive to activation (23). Contrarily, the number of iNKT cells was reduced upon miR-150 upregulation (23). MiR-155 was found to regulate partly interferon-gamma production in human NK-cells by downregulating SHIP1, making it a potential target in neoplastic disease (24).
Tumor Vascular Cells
Endothelial cells together with pericytes are the major cellular components of tumor blood vessels, thus playing an important role in angiogenesis during tumor development (2–4). Chan et al. identified avian erythroblastosis virus E26 (v-ets) oncogene homolog-1 (Ets-1), an angiogenesis-related transcription factor, regulated by miR-200b (25). Ectopic expression of miR-200b reduced the tube formation and cell migration ability of human microvascular endothelial cells (HMECs) in vitro by targeting Ets-1 and its associated genes, matrix metalloproteinase-1 and VEGF receptor-2 (25). Interestingly, the authors demonstrated that miR-200b downregulation is hypoxia-induced and represses Ets-1 expression to promote angiogenesis in HMECs (25). Hypoxia stimulation also influences several miRNA expression levels in rat cortical pericytes compared to normoxic conditions (29). Real-time PCR data revealed changes in the expression of miRNAs associated with hypoxia-inducable factor-1α (HIF-1α) (miR-322 and miR-199a), TGF-β1 (miR-140, miR145, and miR-376b-3p), and VEGF (miR-126a, miR-297, miR-16, and miR-17-5p) (29).
In human umbilical vein endothelial cells (HUVECs), miR-29c was identified to regulate cell cycle, proliferation, and angiogenesis in vitro, likely mediated by suppressing Insulin-like growth factor-1 (26). miRNA-7 was identified as a negative regulator of angiogenesis, strongly reducing cell viability, tube formation, sprouting and migration in vitro (27). In an in vivo murine neuroblastoma tumor model, angiogenesis and tumor growth were significantly inhibited upon local administration of miR-7 (27). MiR-155, which is known to be upregulated in various human cancers, is also involved in the angiogenesis of breast cancer by targeting the von Hippel–Lindau (VHL) tumor suppressor gene (28). Mammary fat pad xenotransplantation of ectopically expressed miR-155 strongly induced angiogenesis, proliferation, tumor necrosis, and recruitment of pro-inflammatory TAMs (28). Moreover, miR-155 was identified as a marker for late-stage, lymph node metastasis and poor prognosis in breast cancer (28).
Recently, miRNA array profiling revealed that endothelial cells communicate with pericytes via miRNA-containing exosomes, which increased VEGF-B expression in pericytes at both gene and proteins levels (53). In addition, Lim et al showed that CXCL12-specific miRNAs are transported from bone marrow stroma to breast cancer cells via gap junctions and reduced CXCL12 levels as well as proliferation (54). Furthermore, mechanisms for the transfer of siRNA and miRNA through the gap junctions have been reviewed (55).
MicroRNA Delivery to the Tumor Stromal Cells
To turn miRNAs into therapeutics, it is essential to deliver antagomiRs or miRNA mimics cell specifically into the target cells. However, naked miRNAs are unable to pass through cell membranes due to their hydrophilicity, polyanionic nature, and high molecular weight. Intravenous administration of naked miRNA leads to poor tissue distribution due to rapid renal excretion (plasma half-life ~1 h) and degradation by serum RNases (56). In vivo application of miRNA either requires a chemical modification or formulation into delivery systems. There are several articles describing targeting strategies for miRNA delivery (57–64). Despite having many strategies reported, the number of successful delivery approaches in vivo is still limited.
Strategies for miRNA Delivery
Encapsulation of miRNAs into nanoparticles has been addressed to protect miRNAs from degradation by nucleases, resulting in an improved circulating half-life when administered systemically (65). Viral and non-viral encapsulation strategies have been investigated for this purpose. Viral-based delivery of nucleotides shows high transfection efficiencies, but will not be discussed in this review due to concerns regarding strong inflammatory side effects (66). The advantages of non-viral delivery systems over viral delivery systems are their well-defined molecular composition, simpler manufacturing and considerably lower immunogenicity (66). The advantages of non-viral delivery systems over viral delivery systems are their well-defined molecular composition, simpler manufacturing and considerably lower immunogenicity (67).
Non-viral delivery systems that have been investigated for their usability as miRNA carrier systems are liposomes, lipoplexes, and polyplexes. Liposomes are composed of phospholipids possessing a hydrophilic head linked to a hydrophobic tail (68). This amphiphilic structure allows them to form vesicles with an inner aqueous compartment in which miRNAs can be encapsulated (68). Cationic lipids have been used to form complexes with negatively charged miRNA, called lipoplexes (69). Liposomes and lipoplexes have shown to protect miRNAs from degradation by nucleases, resulting in an improved circulating half-life when administered systemically (65). In addition, cationic polymers that are frequently used for intracellular delivery are polyethyleneimine (PEI) and polyamide amine dendrimers (PAMAM). PEI- and PAMAM-based polyplexes have been used successfully for the delivery of miRNA and in vitro and in vivo (70, 71). Systemic delivery of PAMAM had toxic effects on the liver and the kidney in mice (72). For PEI, clinical translation has been hampered by dose-dependent toxicity upon systemic administration (73). Similar toxicity issues can be expected from lipoplexes.
MicroRNA Delivery to Tumor Stroma Cells
The number of studies for the delivery of miRNAs to the compartments of the tumor stroma in vivo is highly limited. Polyplexes based on PEI delivered dicer substrate RNA duplexes, mimicking the structure of endogenous precursor miRNA-155 hairpin (Dmi155), into ID8-Defb29/Vegf-A tumors in mice (74). Increasing the levels of miRNA-155 in tumor-associated dendritic cells silenced multiple immunosuppressive mediators (74). These changes lead to the transformation of tumor-infiltrating dendritic cells from their immunosuppressive phenotype to immunostimulatory cells (74). Post-transformation of these cells were capable of triggering antitumor responses thereby inhibiting the progression of established ovarian cancers in mice (74).
Endothelial cells are the only cell type present in the tumor microenvironment, which have been actively targeted in vivo for miRNA delivery. Anti-angiogenic effects in tumors were achieved in a study performed by Ando et al. using lipoplexes modified with a PEG chain bearing the angiogenic vessel targeting peptide Ala–Pro–Arg–Pro–Gly (APRPG) (75). The APRPG–PEG-modified liposomes were used to form complexes with miR-499. The resulting lipoplexes were injected into mice bearing Colon 26 NL-17 xenografts (75). The lipoplexes accumulated in both angiogenic vessels and cancer cells resulting in downregulation of miR-499-regulated proteins and vascular endothelial growth factor (VEGF) (75). Following an injection of 0.5 mg/kg of the lipoplexes, tumor growth in mice was significantly inhibited (75).
In a study by Liu et al., anti-miR-296 antagomiR was delivered into HUVECs (76). PEGylated liposome-polycation-hyaluronic acid (LPH) nanoparticles conjugated with a cyclic RGD peptide (cRGD) were used as carrier systems for the specific targeting of αvβ3, a receptor present on endothelial cells in the tumor neovasculature (76). They reported inhibition of tube formation and endothelial cell migration, and the significant upregulation of hepatocyte growth factor-regulated tyrosine kinase substrate (HGS), one of the genes suppressed by miRNA-296 (76). A matrigel plug assay was performed to analyze the effect of anti-miR-296 delivery on in vivo angiogenesis (76). As a result, a decrease in microvessel formation by preventing CD31-positive endothelial cells from invading into the matrigel in combination with an induction of HGS in angiogenic endothelial cells was observed (76).
Anand et al. aimed to repress neovascularization in tumors by inhibiting miRNA-132 levels via the delivery of anti-miRNA-132 (77). Liposomal nanoparticles composed of distearoylphosphatidylcholine (DSPC), cholesterol, dioleoylphosphatidylethanolamine (DOPE), distearoylphosphatidylethanolamine (DSPE)-mPEG2000 modified with a DSPE–cRGD, targeting αvβ3 were used as a delivery system (77). miR-132, highly overexpressed in the endothelium of human tumors, mediated loss of p120RasGAP thereby inducing neovascularization (77). By restoring p120RasGAP levels using anti-miR-132 delivery, neovascularization was suppressed, and tumor burdens were decreased in an orthotopic xenograft mouse model of human breast carcinoma, and vessels were maintained in their non-pathological resting state (77). Systemically administered miRNA7 loaded peptide/polymer-based delivery systems modified with a cRGP ligand targeting the integrins αvβ3 and αvβ5 (78), targeted human glioblastoma xenografts in mice and strongly reduced angiogenesis and tumor proliferation.
Conclusion
In recent years, miRNAs have been extensively discovered in the tumor stromal cells either to be used as biomarkers or to show their potential for inhibiting cellular processes. Undoubtedly, the miRNA field has a high potential to develop novel therapeutics against cancer; however, at the same time development of technologies to deliver miRNAs to specific cells are highly essential to utilize them for therapeutic purposes. With this review, we bring together the fields of tumor biology and miRNA delivery, which will surely benefit both biologists and technology developers.
Author Contributions
JP designed the structure of the review, wrote, and edited the review article. PK and JS wrote the review article. GS read the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was financially supported by Swedish Research Council, Stockholm, Sweden under the project nr. K7/60501283 (JP). Authors sincerely thank Ruchi Bansal for proof reading this review.
References
1. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell (2012) 21:309–22. doi:10.1016/j.ccr.2012.02.022
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
3. Mueller MM, Fusenig NE. Friends or foes – bipolar effects of the tumour stroma in cancer. Nat Rev Cancer (2004) 4:839–49. doi:10.1038/nrc1477
4. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19:1423–37. doi:10.1038/nm.3394
5. Suzuki HI, Katsura A, Matsuyama H, Miyazono K. MicroRNA regulons in tumor microenvironment. Oncogene (2015) 34:3085–94. doi:10.1038/onc.2014.254
6. Melo SA, Kalluri R. Molecular pathways: microRNAs as cancer therapeutics. Clin Cancer Res (2012) 18:4234–9. doi:10.1158/1078-0432.CCR-11-2010
7. Esquela-Kerscher A, Slack FJ. Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer (2006) 6:259–69. doi:10.1038/nrc1840
8. Bader AG, Brown D, Stoudemire J, Lammers P. Developing therapeutic microRNAs for cancer. Gene Ther (2011) 18:1121–6. doi:10.1038/gt.2011.79
9. Mitra AK, Zillhardt M, Hua Y, Tiwari P, Murmann AE, Peter ME, et al. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov (2012) 2:1100–8. doi:10.1158/2159-8290.CD-12-0206
10. Aprelikova O, Yu X, Palla J, Wei BR, John S, Yi M, et al. The role of miR-31 and its target gene SATB2 in cancer-associated fibroblasts. Cell Cycle (2010) 9:4387–98. doi:10.4161/cc.9.21.13674
11. Aprelikova O, Palla J, Hibler B, Yu X, Greer YE, Yi M, et al. Silencing of miR-148a in cancer-associated fibroblasts results in WNT10B-mediated stimulation of tumor cell motility. Oncogene (2013) 32:3246–53. doi:10.1038/onc.2012.351
12. Musumeci M, Coppola V, Addario A, Patrizii M, Maugeri-Saccà M, Memeo L, et al. Control of tumor and microenvironment cross-talk by miR-15a and miR-16 in prostate cancer. Oncogene (2011) 30:4231–42. doi:10.1038/onc.2011.140
13. Bronisz A, Godlewski J, Wallace JA, Merchant AS, Nowicki MO, Mathsyaraja H, et al. Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-320. Nat Cell Biol (2012) 14:159–67. doi:10.1038/ncb2396
14. Yang TS, Yang XH, Chen X, Wang XD, Hua J, Zhou DL, et al. MicroRNA-106b in cancer-associated fibroblasts from gastric cancer promotes cell migration and invasion by targeting PTEN. FEBS Lett (2014) 588:2162–9. doi:10.1016/j.febslet.2014.04.050
15. Tang X, Hou Y, Yang G, Wang X, Tang S, Du YE, et al. Stromal miR-200s contribute to breast cancer cell invasion through CAF activation and ECM remodeling. Cell Death Differ (2015) 23(1):132–45. doi:10.1038/cdd.2015.78
16. Li P, Shan JX, Chen XH, Zhang D, Su LP, Huang XY, et al. Epigenetic silencing of microRNA-149 in cancer-associated fibroblasts mediates prostaglandin E2/interleukin-6 signaling in the tumor microenvironment. Cell Res (2015) 25:588–603. doi:10.1038/cr.2015.51
17. Bullock MD, Pickard KM, Nielsen BS, Sayan AE, Jenei V, Mellone M, et al. Pleiotropic actions of miR-21 highlight the critical role of deregulated stromal microRNAs during colorectal cancer progression. Cell Death Dis (2013) 4:e684. doi:10.1038/cddis.2013.213
18. He M, Xu Z, Ding T, Kuang DM, Zheng L. MicroRNA-155 regulates inflammatory cytokine production in tumor-associated macrophages via targeting C/EBPbeta. Cell Mol Immunol (2009) 6:343–52. doi:10.1038/cmi.2009.45
19. Squadrito ML, Pucci F, Magri L, Moi D, Gilfillan GD, Ranghetti A, et al. miR-511-3p modulates genetic programs of tumor-associated macrophages. Cell Rep (2012) 1:141–54. doi:10.1016/j.celrep.2011.12.005
20. Chai ZT, Zhu XD, Ao JY, Wang WQ, Gao DM, Kong J, et al. microRNA-26a suppresses recruitment of macrophages by down-regulating macrophage colony-stimulating factor expression through the PI3K/Akt pathway in hepatocellular carcinoma. J Hematol Oncol (2015) 8:56. doi:10.1186/s13045-015-0150-4
21. Yang P, Li QJ, Feng Y, Zhang Y, Markowitz GJ, Ning S, et al. TGF-beta-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell (2012) 22:291–303. doi:10.1016/j.ccr.2012.07.023
22. Gaziel-Sovran A, Segura MF, Di Micco R, Collins MK, Hanniford D, Vega-Saenz de Miera E, et al. miR-30b/30d regulation of GalNAc transferases enhances invasion and immunosuppression during metastasis. Cancer Cell (2011) 20:104–18. doi:10.1016/j.ccr.2011.05.027
23. Bezman NA, Chakraborty T, Bender T, Lanier LL. miR-150 regulates the development of NK and iNKT cells. J Exp Med (2011) 208:2717–31. doi:10.1084/jem.20111386
24. Trotta R, Chen L, Ciarlariello D, Josyula S, Mao C, Costinean S, et al. miR-155 regulates IFN-gamma production in natural killer cells. Blood (2012) 119:3478–85. doi:10.1182/blood-2011-12-398099
25. Chan YC, Khanna S, Roy S, Sen CK. miR-200b targets Ets-1 and is down-regulated by hypoxia to induce angiogenic response of endothelial cells. J Biol Chem (2011) 286:2047–56. doi:10.1074/jbc.M110.158790
26. Hu Y, Deng F, Song J, Lin J, Li X, Tang Y, et al. Evaluation of miR-29c inhibits endotheliocyte migration and angiogenesis of human endothelial cells by suppressing the insulin like growth factor 1. Am J Transl Res (2015) 7:866–77.
27. Babae N, Bourajjaj M, Liu Y, Van Beijnum JR, Cerisoli F, Scaria PV, et al. Systemic miRNA-7 delivery inhibits tumor angiogenesis and growth in murine xenograft glioblastoma. Oncotarget (2014) 5:6687–700. doi:10.18632/oncotarget.2235
28. Kong W, He L, Richards EJ, Challa S, Xu CX, Permuth-Wey J, et al. Upregulation of miRNA-155 promotes tumour angiogenesis by targeting VHL and is associated with poor prognosis and triple-negative breast cancer. Oncogene (2014) 33:679–89. doi:10.1038/onc.2012.636
29. Truettner JS, Katyshev V, Esen-Bilgin N, Dietrich WD, Dore-Duffy P. Hypoxia alters microRNA expression in rat cortical pericytes. Microrna (2013) 2:32–44. doi:10.2174/2211536611302010005
30. Adegboyega PA, Mifflin RC, DiMari JF, Saada JI, Powell DW. Immunohistochemical study of myofibroblasts in normal colonic mucosa, hyperplastic polyps, and adenomatous colorectal polyps. Arch Pathol Lab Med (2002) 126:829–36.
31. De Wever O, Mareel M. Role of tissue stroma in cancer cell invasion. J Pathol (2003) 200:429–47. doi:10.1002/path.1398
32. Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev (2015). doi:10.1016/j.addr.2015.07.007
33. Madar S, Goldstein I, Rotter V. ‘Cancer associated fibroblasts’ – more than meets the eye. Trends Mol Med (2013) 19:447–53. doi:10.1016/j.molmed.2013.05.004
34. Xing F, Saidou J, Watabe K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front Biosci (Landmark Ed) (2010) 15:166–79. doi:10.2741/3613
35. Strell C, Rundqvist H, Ostman A. Fibroblasts – a key host cell type in tumor initiation, progression, and metastasis. Ups J Med Sci (2012) 117:187–95. doi:10.3109/03009734.2012.654859
36. Aprelikova O, Green JE. MicroRNA regulation in cancer-associated fibroblasts. Cancer Immunol Immunother (2012) 61:231–7. doi:10.1007/s00262-011-1139-7
37. Yao Q, Cao S, Li C, Mengesha A, Kong B, Wei M. Micro-RNA-21 regulates TGF-beta-induced myofibroblast differentiation by targeting PDCD4 in tumor-stroma interaction. Int J Cancer (2011) 128:1783–92. doi:10.1002/ijc.25506
38. Ali S, Suresh R, Banerjee S, Bao B, Xu Z, Wilson J, et al. Contribution of microRNAs in understanding the pancreatic tumor microenvironment involving cancer associated stellate and fibroblast cells. Am J Cancer Res (2015) 5:1251–64.
39. Kadera BE, Li L, Toste PA, Wu N, Adams C, Dawson DW, et al. MicroRNA-21 in pancreatic ductal adenocarcinoma tumor-associated fibroblasts promotes metastasis. PLoS One (2013) 8:e71978. doi:10.1371/journal.pone.0071978
40. Li Q, Zhang D, Wang Y, Sun P, Hou X, Larner J, et al. MiR-21/Smad 7 signaling determines TGF-beta1-induced CAF formation. Sci Rep (2013) 3:2038. doi:10.1038/srep02038
41. Zhao L, Sun Y, Hou Y, Peng Q, Wang L, Luo H, et al. MiRNA expression analysis of cancer-associated fibroblasts and normal fibroblasts in breast cancer. Int J Biochem Cell Biol (2012) 44:2051–9. doi:10.1016/j.biocel.2012.08.005
42. Verghese ET, Drury R, Green CA, Holliday DL, Lu X, Nash C, et al. MiR-26b is down-regulated in carcinoma-associated fibroblasts from ER-positive breast cancers leading to enhanced cell migration and invasion. J Pathol (2013) 231:388–99. doi:10.1002/path.4248
43. Fang H, Declerck YA. Targeting the tumor microenvironment: from understanding pathways to effective clinical trials. Cancer Res (2013) 73:4965–77. doi:10.1158/0008-5472.CAN-13-0661
44. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer (2006) 6:24–37. doi:10.1038/nrc1782
45. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141:39–51. doi:10.1016/j.cell.2010.03.014
46. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) (2014) 6:1670–90. doi:10.3390/cancers6031670
47. Squadrito ML, Etzrodt M, De Palma M, Pittet MJ. MicroRNA-mediated control of macrophages and its implications for cancer. Trends Immunol (2013) 34:350–9. doi:10.1016/j.it.2013.02.003
48. Graff JW, Dickson AM, Clay G, McCaffrey AP, Wilson ME. Identifying functional microRNAs in macrophages with polarized phenotypes. J Biol Chem (2012) 287:21816–25. doi:10.1074/jbc.M111.327031
49. Martinez-Pomares L. The mannose receptor. J Leukoc Biol (2012) 92:1177–86. doi:10.1189/jlb.0512231
50. Cai X, Yin Y, Li N, Zhu D, Zhang J, Zhang CY, et al. Re-polarization of tumor-associated macrophages to pro-inflammatory M1 macrophages by microRNA-155. J Mol Cell Biol (2012) 4:341–3. doi:10.1093/jmcb/mjs044
51. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12:253–68. doi:10.1038/nri3175
52. Li L, Zhang J, Diao W, Wang D, Wei Y, Zhang CY, et al. MicroRNA-155 and microRNA-21 promote the expansion of functional myeloid-derived suppressor cells. J Immunol (2014) 192:1034–43. doi:10.4049/jimmunol.1301309
53. Yamamoto S, Niida S, Azuma E, Yanagibashi T, Muramatsu M, Huang TT, et al. Inflammation-induced endothelial cell-derived extracellular vesicles modulate the cellular status of pericytes. Sci Rep (2015) 5:8505. doi:10.1038/srep08505
54. Lim PK, Bliss SA, Patel SA, Taborga M, Dave MA, Gregory LA, et al. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res (2011) 71:1550–60. doi:10.1158/0008-5472.CAN-10-2372
55. Brink PR, Valiunas V, Gordon C, Rosen MR, Cohen IS. Can gap junctions deliver? Biochim Biophys Acta (2012) 1818:2076–81. doi:10.1016/j.bbamem.2011.09.025
56. Wang J, Lu Z, Wientjes MG, Au JL. Delivery of siRNA therapeutics: barriers and carriers. AAPS J (2010) 12:492–503. doi:10.1208/s12248-010-9210-4
57. Gandhi NS, Tekade RK, Chougule MB. Nanocarrier mediated delivery of siRNA/miRNA in combination with chemotherapeutic agents for cancer therapy: current progress and advances. J Control Release (2014) 194:238–56. doi:10.1016/j.jconrel.2014.09.001
58. Li Z, Rana TM. Therapeutic targeting of microRNAs: current status and future challenges. Nat Rev Drug Discov (2014) 13:622–38. doi:10.1038/nrd4359
59. Kortylewski M, Nechaev S. How to train your dragon: targeted delivery of microRNA to cancer cells in vivo. Mol Ther (2014) 22:1070–1. doi:10.1038/mt.2014.73
60. Kwekkeboom RF, Lei Z, Doevendans PA, Musters RJ, Sluijter JP. Targeted delivery of miRNA therapeutics for cardiovascular diseases: opportunities and challenges. Clin Sci (2014) 127:351–65. doi:10.1042/CS20140005
61. Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature (2005) 438:685–9. doi:10.1038/nature04303
62. Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet (2011) 43:371–8. doi:10.1038/ng.786
63. Lennox KA, Behlke MA. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther (2011) 18:1111–20. doi:10.1038/gt.2011.100
64. Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S. Inhibition of microRNA function by antimiR oligonucleotides. Silence (2012) 3:1. doi:10.1186/1758-907X-3-1
65. Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov (2010) 9:775–89. doi:10.1038/nrd3179
66. Choi YS, Lee JY, Suh JS, Kwon YM, Lee SJ, Chung JK, et al. The systemic delivery of siRNAs by a cell penetrating peptide, low molecular weight protamine. Biomaterials (2010) 31:1429–43. doi:10.1016/j.biomaterials.2009.11.001
67. Park TG, Jeong JH, Kim SW. Current status of polymeric gene delivery systems. Adv Drug Deliv Rev (2006) 58:467–86. doi:10.1016/j.addr.2006.03.007
68. Tros de Ilarduya C, Sun Y, Duzgunes N. Gene delivery by lipoplexes and polyplexes. Eur J Pharm Sci (2010) 40:159–70. doi:10.1016/j.ejps.2010.03.019
69. Ben-Shushan D, Markovsky E, Gibori H, Tiram G, Scomparin A, Satchi-Fainaro R. Overcoming obstacles in microRNA delivery towards improved cancer therapy. Drug Deliv Transl Res (2014) 4:38–49. doi:10.1007/s13346-013-0160-0
70. Ren Y, Kang CS, Yuan XB, Zhou X, Xu P, Han L, et al. Co-delivery of as-miR-21 and 5-FU by poly(amidoamine) dendrimer attenuates human glioma cell growth in vitro. J Biomater Sci Polym Ed (2010) 21:303–14. doi:10.1163/156856209X415828
71. Zhang Y, Wang Z, Gemeinhart RA. Progress in microRNA delivery. J Control Release (2013) 172:962–74. doi:10.1016/j.jconrel.2013.09.015
72. Chauhan AS, Jain NK, Diwan PV. Pre-clinical and behavioural toxicity profile of PAMAM dendrimers in mice. Proc R Soc A (2010) 466(2117):1535–50. doi:10.1098/rspa.2009.0448
73. Zhang Y, Satterlee A, Huang L. In vivo gene delivery by nonviral vectors: overcoming hurdles? Mol Ther (2012) 20:1298–304. doi:10.1038/mt.2012.79
74. Cubillos-Ruiz JR, Baird JR, Tesone AJ, Rutkowski MR, Scarlett UK, Camposeco-Jacobs AL, et al. Reprogramming tumor-associated dendritic cells in vivo using miRNA mimetics triggers protective immunity against ovarian cancer. Cancer Res (2012) 72:1683–93. doi:10.1158/0008-5472.CAN-11-3160
75. Ando H, Asai T, Koide H, Okamoto A, Maeda N, Tomita K, et al. Advanced cancer therapy by integrative antitumor actions via systemic administration of miR-499. J Control Release (2014) 181:32–9. doi:10.1016/j.jconrel.2014.02.019
76. Liu XQ, Song WJ, Sun TM, Zhang PZ, Wang J. Targeted delivery of antisense inhibitor of miRNA for antiangiogenesis therapy using cRGD-functionalized nanoparticles. Mol Pharm (2011) 8:250–9. doi:10.1021/mp100315q
77. Anand S, Majeti BK, Acevedo LM, Murphy EA, Mukthavaram R, Scheppke L, et al. MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat Med (2010) 16:909–14. doi:10.1038/nm.2186
Keywords: tumor microenvironment, tumor stroma, microRNA, gene delivery, cancer-associated fibroblasts, tumor-associated macrophages
Citation: Kuninty PR, Schnittert J, Storm G and Prakash J (2016) MicroRNA Targeting to Modulate Tumor Microenvironment. Front. Oncol. 6:3. doi: 10.3389/fonc.2016.00003
Received: 19 October 2015; Accepted: 03 January 2016;
Published: 19 January 2016
Edited by:
Ruggero De Maria, Istituto Superiore di Sanità, ItalyReviewed by:
Justin Lathia, Cleveland Clinic Lerner Research Institute, USAMin Hee Kang, Texas Tech University Health Sciences Center School of Medicine, USA
Copyright: © 2016 Kuninty, Schnittert, Storm and Prakash. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jai Prakash, ai5wcmFrYXNoQHV0d2VudGUubmw=