 Alan Dal Pra
Alan Dal Pra Jennifer A. Locke
Jennifer A. Locke Gerben Borst1,2§
Gerben Borst1,2§ Stephane Supiot
Stephane Supiot- 1Radiation Medicine Program, Ontario Cancer Institute, Princess Margaret Cancer Centre, University Health Network, Toronto, ON, Canada
- 2Department of Radiation Oncology, University of Toronto, Toronto, ON, Canada
- 3Integrated Center of Oncology (ICO) René Gauducheau, Nantes, France
Radiation therapy (RT) is one of the mainstay treatments for prostate cancer (PCa). The potentially curative approaches can provide satisfactory results for many patients with non-metastatic PCa; however, a considerable number of individuals may present disease recurrence and die from the disease. Exploiting the rich molecular biology of PCa will provide insights into how the most resistant tumor cells can be eradicated to improve treatment outcomes. Important for this biology-driven individualized treatment is a robust selection procedure. The development of predictive biomarkers for RT efficacy is therefore of utmost importance for a clinically exploitable strategy to achieve tumor-specific radiosensitization. This review highlights the current status and possible opportunities in the modulation of four key processes to enhance radiation response in PCa by targeting the: (1) androgen signaling pathway; (2) hypoxic tumor cells and regions; (3) DNA damage response (DDR) pathway; and (4) abnormal extra-/intracell signaling pathways. In addition, we discuss how and which patients should be selected for biomarker-based clinical trials exploiting and validating these targeted treatment strategies with precision RT to improve cure rates in non-indolent, localized PCa.
Introduction
The Role of RT in Localized Prostate Cancer
In 2014, it was estimated that over 233,000 men would be diagnosed with prostate cancer (PCa) in the North America leading to over 29,480 deaths (1). The prognosis and treatment of these men is currently determined by a number of different risk classification systems (2–5). All of these use combinations of the conventional risk stratifications: TNM staging, pathologic Gleason score (GS), and prostate specific antigen (PSA) level. Treatment options vary from active surveillance for indolent low-risk PCa (6) to different combinations of external beam radiotherapy (RT), brachytherapy, androgen deprivation therapy (ADT), and surgery. A comprehensive review of levels of evidence for the use of different types of treatment technologies, RT dose escalation, and the use of ADT is beyond the scope of this review, and the reader is pointed to several recent reviews in this area (7–15).
Despite the use of clinical prognostic factors and improved technological advances in radiation delivery and surgery, patients with localized PCa are at risk for local failure and occult metastases (not appreciated by current radiographic staging prior to treatment). Local recurrence after RT is thought to occur predominantly in regions bearing higher histological tumor burden (16, 17). Thus, strategies that improve both local control at the tumor site and eradicate occult metastases are required.
There is a pressing need to develop novel radiosensitizing strategies and agents to specifically target tumor cells to improve treatment outcome. Research exploiting the tumor-specific biology in relation to the normal tissue cells will reveal the Achilles heel of the most resistant tumor cells and regions. In this review we focus on approaches that combine RT with one or more agents to enhance the radiation response specifically for tumor cells. We focus on four important pathways that could influence RT outcome, including the: (1) androgen signaling pathway; (2) hypoxic tumor cells and regions; (3) DNA damage response (DDR) pathway; and (4) abnormal extra-/intracell signaling pathways. In addition, we provide an insight into which patients will benefit from this approach and how to select these patients by clinically feasible biomarkers.
Current Molecular Prognostic Factors and Combination Treatments
Patient selection and stratification over and above the current use of clinical prognostic factors is the cornerstone for an individualized treatment with local therapy alone or combinations of local and systemic therapies (including the use of novel molecular targeted drugs). To explore this, the Radiation Therapy Oncology Group (RTOG) has completed studies on a wide range of immunohistochemical (IHC) markers (18). Tissues from Phase III RT trial (with and without ADT) were evaluated within a variety of localized risk groups. IHC-based assessment of protein overexpression for p53, p16 INK4a, Ki-67, MDM2, CYP3A4, and BCL2 were associated with adverse clinical outcomes (18) but has not yet been validated in modern-day IGRT–IMRT cohorts. Another approach is to study the somatic tumor genetics of patients based on tissues derived from pretreatment biopsies and utilizes genomics to add prognostic power for personalized medicine approaches. Indeed, recent studies from our own laboratory have implicated allelic changes in c-MYC, NKX3.1, PTEN, StAR, and HSD17B2 as adverse prognostic factors following IGRT (19–21). Novel gene signatures reflective of the underlying biology of PCa progression are also being developed in biopsy material and radical prostatectomy specimens [i.e., Myriad Genetics Prolaris Score™, Genome Health OncotypeDx™ Genomic Prostate Score, GenomeDx Biosciences Decipher™, NF-kB-activated recurrence predictor 21 (NARP21)] (22–26). The ability to analyze RNA expression on routine, archived, formalin-fixed, paraffin-embedded tissue samples is currently being developed and may provide analysis on the small amounts of tissue available from prostate biopsy specimens to help prognosticate patients prior to precision RT.
Table 1 presents a summary of some of the current biomarkers tested in PCa RT. If these prognostic markers were also predictive of efficacy for targeted drugs directed against abnormalities in cellular pathways in cancer cells, then this could lead to combining such drugs with precision RT.
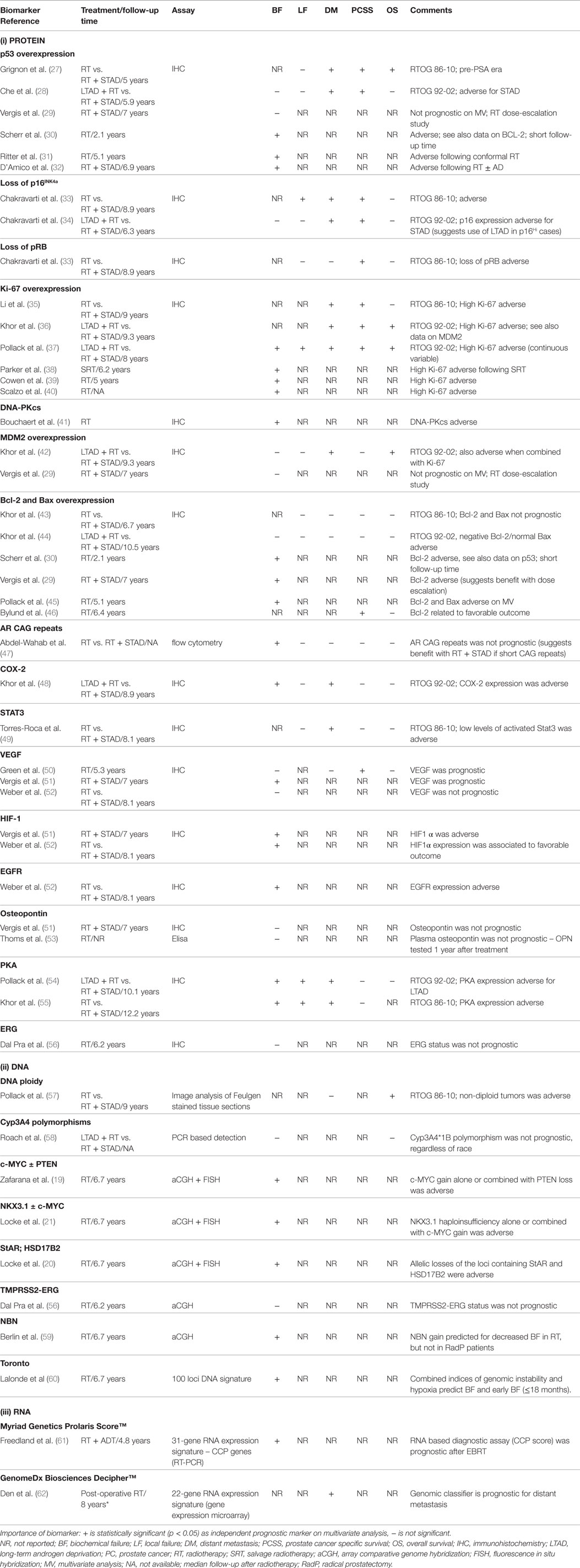
Table 1. Selected biomarkers tested with prostate cancer radiotherapy.
Although several of these gene markers and signatures have demonstrated prognostic roles in small patient cohorts, many have not been validated in large-scale clinical trials of specific groups of patients (i.e., low, intermediate and high-risk PCa). Furthermore, comparison between genetic signatures has been limited; thus the best gene signature has not been identified. Future clinical trial studies should further probe these prognostic markers in larger cohorts to help optimize therapy for the individual patient.
Furthermore, once prognosticated appropriately, the best combinational therapy with RT should be better specified for the individual patient. Although a number of preclinical PCa studies have tested novel targeted agents in combination with RT, a search of MEDLINE and EMBASE databases from 2000 to 2014 shows that few of these preclinical strategies have led to the clinical trials evaluating these combinations. Instead, many of the ongoing trials are testing the use of non-targeted chemotherapies with RT in high-risk groups (Table 2). Early evidence supports this approach mainly through cytotoxic effects to micrometastatic disease and possibly addressing androgen-resistant clones. Neoadjuvant setting chemotherapy would present a synergistic role by radiosensitizing tumor cells at the primary site (63–65). The RTOG 0521 is a Phase III trial that tested adjuvant combination of docetaxel, ADT, and RT in comparison to RT and ADT in patients with high-risk localized PCa. Four-year OS rates were 89% for men who received ADT and RT vs. 93% for men treated with ADT, RT, and docetaxel (HR = 0.70; 90% CI, 0.51–0.98; P = 0.04). Whether adding chemotherapy will become a standard of care for this population, especially considering toxicity outcomes, remains to be seen (66).
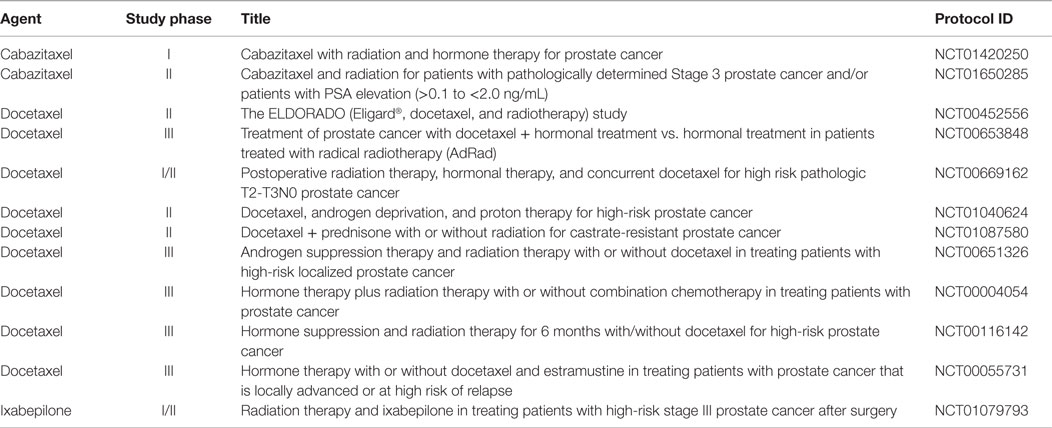
Table 2. Ongoing clinical trials testing radiotherapy combined with chemotherapy in non-indolent, localized prostate cancer.
Herein, we share insight as to how to move this area of research forward improving personalized medicine for PCa patients in this era of novel prognostic and predictive markers and targeted therapies.
Combining Molecular Targeting and RT in PCa
Androgen Depriving Associated Therapies and RT
Conventional ADT Plus RT
In the 1990s, ADT such as luteinizing hormone-releasing hormone (LH-RH) agonist or antiandrogens were tested as a combined modality therapy with RT (67). Phase III studies showed that ADT combined with RT allowed for better tumor control and survival as compared to RT alone in intermediate- and high-risk patients, and it is now considered as a standard treatment (68–70). However, despite ADT–RT combined treatments, long-term follow-up at 10 years shows that about 50% of patients relapse and eventually 10–25% die of PCa (68, 71, 72), which further strengthens the need for novel drugs especially in the high-risk category.
ADT Plus RT: Mechanistic Insight
The mechanism(s) of interaction between ADT and RT is still not completely clarified. An important in vitro study showed that different PCa cells lines lacked an overall radiosensitization by ADT (73) whereas in vivo data showed synergism with ADT and RT (fractionated vs. single-dose). This may be explained by the fact that the ADT effect was related to the tumor microenvironment and not to the tumor cells per se (74). ADT potentially affects tumor vascularization, and subsequently, tumor oxygenation. Testosterone was shown to act as a potent stimulator of prostatic endothelial cell growth (75, 76), and ADT induced a decrease in Mean Vessel Density (MVD) rapidly followed by an increase in MVD (76). Hypoxia is considered as an adverse predictive factor of RT response of prostate tumors (51, 77). ADT could decrease tumor hypoxia fraction in PCa, and this may represent a plausible explanation of the radiosensitizing properties of ADT (74). Moreover, it has been recently shown important new interactions between androgen signaling and DNA repair genes. In biopsies from patients with locally advanced PCa, androgen deprivation caused decreased levels of the Ku70 protein [responsible for non-homologous end-joining (NHEJ) repair of DNA double-strand breaks (DSBs)]; thus impairing DNA repair and possibly explaining increased radiosensitivity (78). Polkinghorn et al. (79) has recently shown that androgen receptor (AR) regulates a transcriptional program of DNA repair genes that promote PCa radioresistance. PCa cells treated with irradiation plus androgen demonstrated enhanced DNA repair and decreased DNA damage, whereas antiandrogen treatment caused increased DNA damage (also via decreased classical NHEJ) and decreased clonogenic survival. Careful monitoring of tumor vascularization, hypoxia, DNA damage markers (i.e., Ku70), the development of serum biomarkers of CYP17A1 (see below), and AR activity will be crucial to identify those patients likely to respond to ADT and RT as well as new combined modality combinations.
Novel Molecules Targeting Androgen Receptor Plus RT
Depicted in Figure 1A is a summary of targets of the androgen axis that are currently being exploited in PCa treatment. Many of these agents have shown efficacy in castration-resistant disease. We contend that a number of the newer targeted agents could be combined with RT in localized PCa to improve outcomes. Molecules targeting the AR pathway such as abiraterone (80), TAK700 (81), or enzalutamide (82) (formerly called MDV3100) were shown to induce tumor regression even in castration-resistant disease. As compared to LH-RH agonists that only reduce circulating testosterone levels, all of these second-generation androgen agents, except enzalutamide, inhibit also paracrine and intracrine intraprostatic testosterone production, which implies a possible direct effect on PCa cells leading to more pronounced effects on the tumor microenvironment (83). Additionally, new AR inhibitors such as enzalutamide have displayed higher potency and specificity for the AR than bicalutamide and flutamide in preclinical studies and may lead to decreased side effects (84–86).
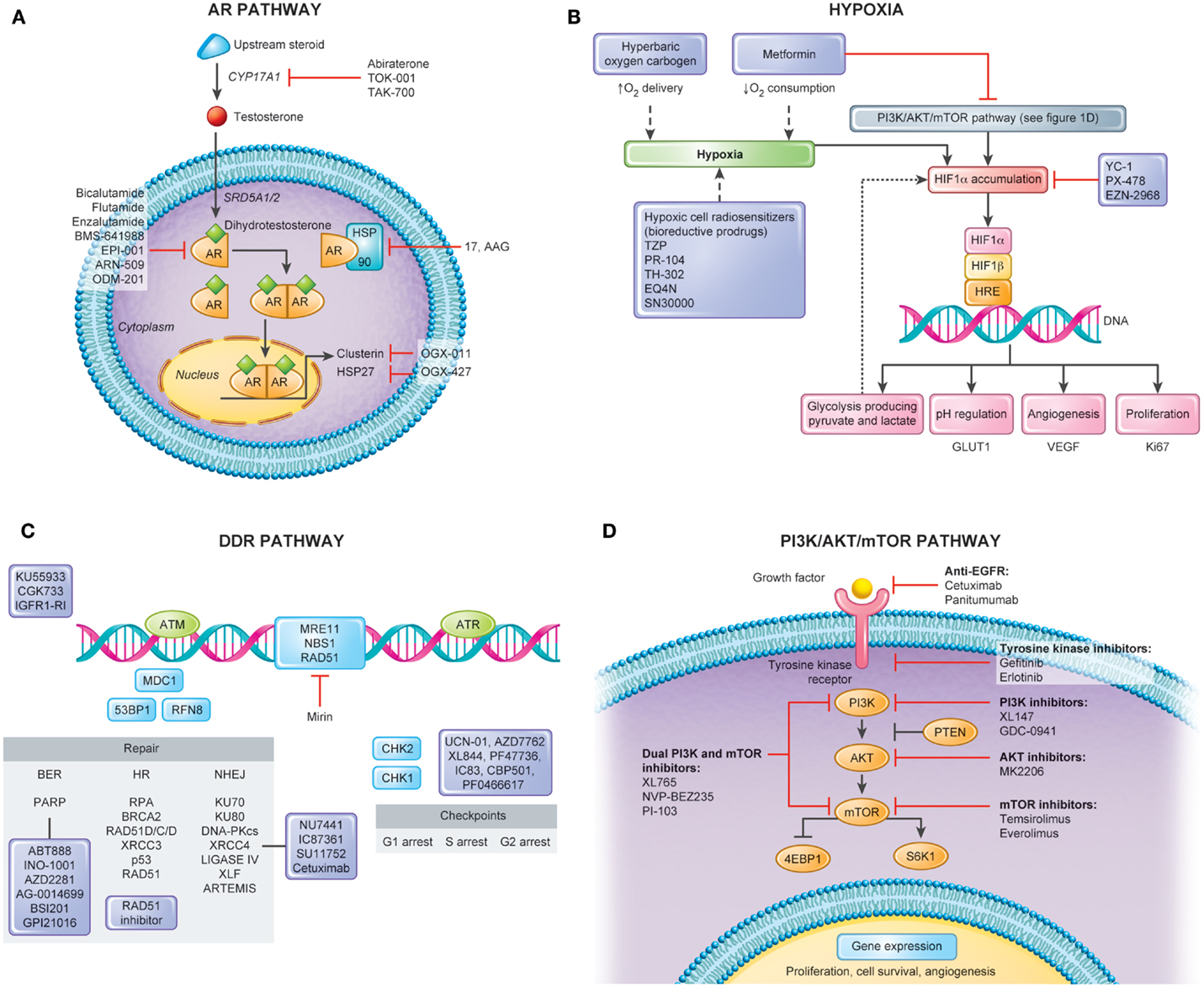
Figure 1. Pathways for molecular targeting in prostate cancer radiotherapy. Several pathways can serve as potential targets in attempt to modulate radiotherapy response and enhance clinical outcomes in non-indolent, localized prostate cancers. This figure depicts four important pathways involved in disease progression and radiation response along with its potential targets. (A) Androgen Receptor (AR) Pathway. AR has a central role in the transcription of several genes important in the survival and proliferation of prostate cancerous cells. Several new agents have been explored in castration-resistant prostate cancers with encouraging results. In localized disease, when combined with radiotherapy, these novel therapies also constitute a promising avenue for cure. (B) Hypoxia. Hypoxia modulation constitutes an important way to improve clinical outcomes following prostate cancer radiotherapy. Tumor hypoxia fraction can be targeted either by hypoxia cell radiosensitizers, enhancing oxygen delivery, or decreasing oxygen consumption. (C) DNA Damage Response (DDR) Pathway. Figure shows simplified DDR scheme with agents acting in different repair processes including Base Excision Repair (BER), Single Strand Break (SSB), Non-homologous End-Joining (NHEJ), and Homologous Recombination. Targeting cell cycle checkpoint can lead to cells with abrogated G1 and G2 checkpoints and accumulation of DNA breaks resulting in mitotic catastrophe. The use of these agents with RT would be expected to lead to increased residual SSBs and DSBs that can be tracked by DNA repair foci within irradiated tumor and normal tissues. In addition, the use of some of these inhibitors (e.g., PARP inhibitors) may lead to cytotoxicity as single agents based on the concept of synthetic lethality. (D) PI3K/AKT/mTOR Pathway. Proliferation of prostate cancer cells is under control of the PI3K/AKT/mTOR signaling. As major growth factor receptors (e.g., EGFR, VEGFR) require this downstream kinase pathway, it is also promising target for radiosensitization through inhibitors of mono- or multiple actions.
CYP17A1 Inhibitors Plus RT
The CYP17A1 inhibitor, abiraterone acetate, was shown to improve overall survival with minimal side effects in metastatic PCa (14.8 vs. 10.9 months, HR = 0.65) (80). CYP17A1 is an enzyme important in the synthesis of dihydrotestosterone (DHT) from cholesterol (Figure 1A) and may be targeted in the testes, adrenal glands, and prostate to reduce tumor burden in PCa. Wright et al. recently demonstrated that a SNP variant allele of CYP17A1 found in the serum of PCa patients is associated with survival (87). Furthermore, protein expression of CYP17A1 in the serum of patients with PCa is twofold higher than in the serum of healthy age-matched controls (88). These studies preclude the measurement of CYP17A1 in serum as a potential predictor for disease outcome and in light of the new CYP17A1 inhibitor, abiraterone acetate, a potential predictor for treatment response. The feasibility of circulating tumor cells (CTCs) as a biomarker of drug efficacy was recently tested and shown to be an easily obtained tissue for molecular analysis (89). It will be interesting to follow the current phase II trial of RT and ADT ± neoadjuvant or adjuvant abiraterone acetate to determine if this will be a useful means to cure intermediate to high-risk disease, and if serum CYP17A1 expression or maybe CTCs may be utilized for prediction of treatment response (NCT01023061 and NCT01780220).
TOK-001 (Galeterone) and TAK-700 (Orteronel) were shown in preclinical studies to antagonize the AR and CYP17A1 and decrease the overall expression of AR in PCa cells (90, 91). After positive results of TOK-001 in Phase II study in men with CRPC, a phase III study is planned to begin (91). TAK-700 advanced rapidly to Phase III trial in patients with CRPC; however, the study did not meet the primary endpoint of improved OS (92). Thus, further development of TAK-700 has been terminated. This affected the RTOG 1115, which was an ongoing Phase III study of dose-escalated RT with a LH-RH agonist ± TAK-700 in high-risk PCa (NCT01546987).
With the minimal side effects observed with CYP17A1 inhibitor abiraterone acetate (90), the use of these new compounds dually targeting CYP17A1 and AR are promising candidates to combine with RT.
Novel AR Inhibitors Plus RT
Enzalutamide is an AR antagonist with high affinity for the AR also inhibiting translocation of the AR to the nucleus and its binding to DNA. It was shown to improve OS in CRPC patients before and after chemotherapy (82, 93). In non-castrate-resistant disease, a significant biochemical response with minimal side effects was recently demonstrated in a phase II study (94). Due to important clinical response and low toxicity profile, enzalutamide is a promising drug to be utilized in the earlier stages of the disease. Possible biomarkers of enzalutamide response have been investigated and include CTCs, mutated AR, AR amplification, and AR splice variants that lack the ligand-binding domain (95–98). Detection of AR splice variant 7 messenger RNA (AR-V7) in CTCs from men with advanced disease was recently found to be associated with resistance to enzalutamide and abiraterone (98).
ODM-201 is a new generation inhibitor of the AR with superior preclinical efficacy compared to enzalutamide and bicalutamide. It does not enter the brain in preclinical studies and does not interact with cytochrome 3A4, therefore it may have lower toxicity as compared to other AR inhibitors (99).
There are several chaperone proteins associated with AR currently being targeted in CRPC including clusterin, HSP-27, and HSP-90. Serum levels of clusterin, an androgen-regulated chaperone protein, have been recently correlated with PCa outcome (100). Preclinical data has demonstrated that overexpression of clusterin decreases radiosensitivity in LNCaP cells (101) while clusterin knock-down has an effect to increase radiosensitivity of these cells (102). Moreover, a novel targeted agent to antisense clusterin (OGX-011) has been shown to be safe in men with PCa (103). Although trial data evaluating OGX-011 in patients with intermediate to high-risk PCa undergoing radical prostatectomy has been negative (104), the evaluation of OGX-011 with RT in patients with localized disease awaits investigation.
Androgen Depriving Associated Therapies and RT: How to Move Forward
To move forward in a personalized medicine setting, biomarker and mutation assays that reflect the functional status of the AR would help identify patients who may best benefit from these inhibitors. These assays are not in routine use in clinical RT practice, despite provocative data from the RTOG 86-10 trial that patients with short CAG repeats (which affect AR transcriptional activity) had better local control (47). The introduction of such AR biomarker methods into clinic may play an important role in the combination of current and future AR inhibitors and RT. With the introduction of such methods, second-generation antiandrogens represent interesting candidates to improve RT efficacy.
Hypoxia and RT
The biological effects of both chronic and acute/cycling tumor hypoxia are related to increased rates of genomic instability, systemic tumor spread, and resistance to RT, and several types of chemotherapy (105, 106). Hypoxic cells when compared to oxic cells show a twofold to threefold decrease in DNA damage and cell kill after RT. The increased resistance to chemotherapy is due to decreased perfusion of agents, decreased cell kill by proliferation-dependent drugs because of hypoxic cells arrest in G0–G1 state, and increased DNA damage repair (105, 106). An increased rate of metastases is due to multiple mechanisms including increased hypoxia-activated genes involved in metastasis and angiogenesis (e.g., VEGF, LOX) and selection of potential metastatic clones during tumor progression (106).
Clinically, hypoxia has been correlated with poor clinical outcomes in PCa following RT or surgery. Turaka et al. studied 57 patients with more than 8 years of follow-up. They showed that decreased prostate-to-muscle oxygen ratio was an important predictor of early biochemical recurrence following brachytherapy (107, 108) and suggested that hypoxia was a biomarker of occult metastases at the time of treatment. Using immunohistochemistry, Vergis et al. showed that increased expression of the hypoxic markers HIF1 and VEGF leads to rapid RT failure, independent of classical clinical-pathologic factors and RT dose (51). Milosevic et al. directly measured intra-prostatic O2 levels of 247 PCa patients using needle–electrode technique. This was the largest clinical study of PCa hypoxia with direct measurement of tumor oxygen levels, and showed that hypoxia is associated with early biochemical relapse and local recurrence in the prostate gland (77).
Drugs Dependent on Hypoxia Gradient in the Tumor
Targeting hypoxia in the clinical setting has been attempted for many years (Figure 1B). This includes increasing the oxygen delivery to the tumor by the blood (normo- or hyperbaric oxygen) or the use of hypoxic cell cytotoxins, or hypoxic cell radiosensitizers. The class of agents, N-oxides, such as tirapazamine (TZP), is a prodrug that under hypoxic conditions undergoes intracellular one-electron reduction to highly toxic radicals that cause DSBs and DNA base damage. This damage stalls and DNA collapse replication forks. In preclinical studies, TZP is 15- to 200-fold more cytotoxic under hypoxia compared to aerobic conditions (109). The randomized phase II and III TZP studies completed to date have shown mixed tumor responses while frequently having increased normal tissue toxicity. Dinitrobenzamide mustard (DNBM) is a new class of drugs that contain a latent nitrogen mustard moiety, which becomes activated when either of the nitro groups is reduced to the corresponding hydroxylamine or amine. This results in the selective generation of reactive nitrogen mustard metabolites causing DNA cross-linking in hypoxic cells (110). PR-104, a novel DNBM currently in clinical trials has shown great promise in preclinical studies and holds several advantages over other bioreductive drugs such as TZP. First its activation is confined to lower oxygen concentrations allowing for greater specificity, and second its activated metabolites are able to diffuse locally in tumor tissue, providing an efficient bystander effect. A recent study has shown that PR-104 can be selectively active in hypoxic cells within treated 22RV1 PCa xenograft models (111). TH-302, which has a hypoxia-generated DNA damaging warhead, has also been shown to sensitize LNCaP and DU145 cells under hypoxia (112).
Drugs Targeting HIF-1
HIF-1 is an important transcription factor that is stabilized by low oxygen levels and is key in the expression of greater than 100 gene products following hypoxic stress. Cycling hypoxia strongly induces HIF-1, increases glucose uptake, and drives the Warburg effect. This is due in part to reoxygenation post-hypoxia increasing free radicals and thereby increasing HIF-1. HIF-1 could be a potential therapeutic target for PCa RT as it is also activated by oncogenic stress in addition to hypoxia (113). Drugs that inhibit glucose consumption by hypoxic tumor cells may be another strategy that explores the effects of hypoxia (114). This can be accomplished via HIF-1 inhibitors or by inhibition of MCT1 to force aerobic tumor cells to consume more glucose and less lactate and reduce glucose availability to the less well-perfused hypoxic cells. Lactate levels have been proposed to be a biomarker for HIF-1 inhibitors. As shown in Figure 1B, HIF-1 alpha can also be modulated by multiple upstream factors, including the PI3K/AKT/mTOR pathway (see PTEN/PI3K/AKT/mTOR) and downstream pathways, affecting gene expression, metabolism, cell survival, tumorigenesis, and tumor growth (115).
Hypoxia downregulation seems to relate to the effects of androgen deprivation in improving RT response. Al-Ubaidi et al. using pre- and posttreatment biopsies of patients treated with androgen deprivation have shown decreased HIF-1α levels by immunofluorescence (116). Many preclinical studies have tested HIF inhibition in PCa. Silencing HIF-1 alpha expression by small interfering RNA (siRNA) has shown increased radiosensitization of PC3 cells. HIF-1 alpha inhibition attenuated repair of radiation injury, with an increase in both interphase death and reproductive death after irradiation, apoptotic potential, and cell cycle arrest at the G2–M phase (more sensitive to radiation) (117). The use of dietary compounds, like soy isoflavones, has shown to improve radiation response both in PCa cell lines and xenograft models. It is believed that isoflavones inhibit the activation of the Src/STAT3 signaling pathway by radiation and radiation-induced HIF-1α expression thus contributing to increased response of cancer cells to radiation. These findings correlated with decreased expression of APE1/Ref-1 resulting in decreased DNA binding activity of HIF-1α and NF-κB, thereby inhibiting transcription of downstream genes essential for tumor growth and angiogenesis (118). Through HIF-1 abrogation and altered DNA damage repair, increased radiation response has been seen with nitric oxide donating non-steroidal anti-inflammatory drugs (NO-NSAIDs) (119). PX-478 is an oral agent that is currently under investigation in a phase I trial for advanced PCa. In vitro, it was shown to decrease HIF-1 alpha in PC3 and DU145 cells and enhance the radiosensitivity of PC3 cells under normoxic and hypoxic conditions (120).
Drugs Targeting Oxygen Consumption
While most strategies to modulate tumor hypoxia aim at increasing oxygen supply during RT through breathing of hyperbaric oxygen (121) or an oxygen-rich gas like carbogen (95% O2, 5% CO2) in combination with vasodilating agents (122), an alternative approach is decreasing oxygen consumption (Figure 1B). This is a logical choice given that oxygen gradients and “diffusion limited” hypoxia arise due to high cellular oxygen consumption (123). Mathematical modeling suggests that decreasing oxygen consumption is more efficient at promoting tumor oxygenation than increasing oxygen supply (124).
Our group has investigated metformin, a commonly prescribed anti-diabetic drug, as an effective and inexpensive means to improve RT outcome. Metformin inhibits complex I activity in the mitochondrial electron transport chain (ETC), therefore inhibiting cellular oxygen consumption (125). We showed through in vivo and in vitro models that metformin could improve tumor radiation response through inhibiting tumor cell oxygen consumption and transiently increasing tumor oxygenation. We also tested the impact of metformin use on the outcome of 504 PCa patients treated with curative-intent RT. Metformin was associated with an independent and significant decrease in early biochemical relapse rates (126). Others groups have confirmed the clinical benefit of metformin in PCa patients undergoing RT (127, 128).
Targeting Hypoxia: How to Move Forward
In order to personalize combined therapy approaches in PCa, clear and accurate documentation of preexisting and/or treatment-induced aggressive/adaptive tumor microenvironments is required to tailor such treatment to patients. Any trials with hypoxia-modifying agents will require biomarkers that measure hypoxic fraction before and after modification to place hypoxic patients into appropriate trials and prove that the drug is active in hypoxic tumor subpopulatons. Currently, different methods of hypoxia measurement have been used including pO2 microelectrodes, in situ markers including extrinsic markers (EF-5 and pimonidazole) or intrinsic markers (e.g., HIF1, VEGF, and GLUT-1) and imaging modalities involving functional PET and MRI (129). If hypoxia is to become a criterion for disease management in PCa, an agreement on invasive and/or non-invasive biomarkers is notably required (105, 130, 131).
Existing technologies can deliver a higher RT dose to specific regions in the tumor (i.e., dose painting) without increasing the dose in surrounding normal tissue. Dose painting as function of non-invasive hypoxia imaging modalities in combination with hypoxia-targeted systemic agents may be the way forward (132); as the prognostic value of low pO2 and increased expression of hypoxia-associated markers in situ was shown to be independent of radiation dose (51, 77), the hypoxic sub-fraction may therefore benefit from both dose-escalation and systemic treatment.
Targeting DNA Damage Responses and DNA Repair
DNA Damage Responses and DNA Repair in RT
Radiation therapy results in the production of a variety of ionizing radiation-induced lesion in DNA. Specific pathways of DNA repair are required to repair the variety of lesions, which include DNA single-strand breaks (SSBs), DSBs, DNA base alterations, and DNA–DNA or DNA–protein cross-links. Non-repaired DNA damage can lead to normal and tumor cell death via apoptosis, mitotic catastrophe, autophagy, or terminal growth arrest senescence. In PCa patients, RT was shown to induce ATM-p53 DNA damage-dependent proteins thereby leading to long-term activation of p21WAF associated with reduced cell proliferation, but no apoptosis (133). Precise molecular targeting of the sensing and repair of DNA damage in PCa cells over surrounding normal tissues (e.g., rectum, bladder, bowel) is a promising area of combination therapy in non-indolent, localized PCa.
DNA DSBs are the most damaging breaks resulting in cell death. DNA DSBs are primarily repaired through two different pathways: HR and NHEJ. HR repair is a template-guided, error-free pathway predominantly operating in the S and G2 phases of the cell cycle, which express many HR-related proteins, including Rad51, the Rad51 paralogs (XRCC3, RAD51B,C.D) XRCC2, RPA, BRCA2, and BLM proteins. In contrast, NHEJ is operational in all phases of the cell cycle and uses the KU 70/80, DNA-PKcs, Artemis, XLF, XRCC4, and DNA ligase IV proteins. The latter pathway would therefore only be operational in G0/G1-arrested slowly proliferating, late-reacting tissues (which limit the total dose of fractionated RT) (134).
Approaches to Target DNA Damage Responses
Using the Genetic Defects in Tumor Cells Involved in DNA Damage Response
Strategies that target DNA repair pathways that are dependent on DNA replication (i.e., HR during the S phase) may give rise to a therapeutic ratio when combined with fractionated RT (Figure 1C). We have shown that Rad51 expression and functional HR can be reduced using imatinib in PCa cells in vitro and in vivo during experimental RT (135). This combined imatinib–RT treatment increased prostate tumor cell radiosensitization without increased gut toxicity. Similar preclinical data exist in vitro for the targeting of the SSB and BER repair pathways [e.g., inhibiting the activity of poly (ADP-ribose) polymerase (PARP) or DNA polymeraseβ] whereby the increased levels of non-repaired SSB are converted to more lethal DSBs during replication. As such, the differential targeting of DNA repair in replicating tumor cells vs. non-replicating late-reacting normal tissues could be exploited in clinical treatment protocols.
Synthetic cell lethality defines a genetic interaction in which the combination of mutations in two or more genes (each mutation on its own being non-toxic) leads to cell death. A number of lethal combinations have been discovered using silencing RNA (siRNA) and chemical screens and, subsequently, validated in isogenic preclinical model systems and phase I and II clinical trials. From these screens, it was observed that certain DNA repair inhibitors may lead to tumor cell kill when used as single agents as they cause synthetic cell lethality when combined with a germ-line or somatic genetic defect in DNA repair. A remarkable example of this interaction is the results of recent trials that have observed tumor responses in chemoresistant breast and ovarian cancers with HR defects (BRCA1/2 deficient and HR defective) using inhibitors of the SSB repair protein, PARP1 without toxicity to repair-proficient normal tissues (136). Other approaches are to use PARP inhibitors with tumors deficient in phosphatase and tensin homolog (PTEN), Aurora A kinase, and HR- or cell cycle-related pathways or using DNA polymerase-β inhibitors in mismatch repair deficient tumors. These synthetic lethality approaches can be designed to decrease the number of PCa clonogens prior to RT if used in a neoadjuvant fashion and improve RT outcome (134).
DNA repair enzyme inhibition (e.g., PARP inhibition) may be prolonged in tumor tissues relative to normal tissues in vivo and recent data suggest that PARP inhibitors can “trap” the PARP1 and PARP2 enzymes at damaged DNA (137). In the latter scenario, the pharmacodynamics of an oral or intravenous inhibitor could determine when RT is administered during the period when tumor enzymes are still inhibited for DNA repair function, yet the pathway is no longer inhibited in normal tissues. Careful pharmacodynamic studies may lead to an increased therapeutic ratio based on differential scheduling of fractionated RT with a DNA repair inhibitor. Knowledge a prior of germ-line and somatic mutations in DNA damage and repair genes in RT patients could therefore be very helpful if the mutations lead to a functional loss of specific response pathways. We have used array comparative genomic hybridization to show that there can be allelic loss of PARP1, ATM, DNA-PKcs, p53, Rb, and RAD17 in PCa (59). If this leads to functional loss of DNA repair or damage signaling, then these patients may benefit from targeted therapies (e.g., inhibitors of PARP, ATM, DNA-PKcs, MTp53, and CHK1) in addition to the potential tumor cell radiosensitization based on inherently abnormal DNA repair (105).
Using the Difference of Cell Cycle Phase Stages between Tumor and Normal Cells
The radiosensitivity of human cells varies throughout the cell cycle (i.e., G1, S, G2, and M phases). S-phase cells are relatively more radioresistant than G1 and G2/M cells. Tumor cells have a shorter interval of subsequent cell cycles with a higher S-phase fraction correlated with base excision repair and/or homologous recombination (HR) compared to late acting G1-arrested normal tissue cells. Therefore the use of inhibitors of HR such as a recently described RAD51 inhibitor (138) may be selective for tumor cells over late reacting normal tissues.
Targeting Hypoxia Related Differences in DNA Damage Repair Pathway in Tumor Cells
Prolonged acute or chronic hypoxia can lead to decreased expression of HR genes, which decreases the radioresistance (e.g., reduced oxygen enhancement ratio); HR-deficient hypoxic cells can then be more radiosensitive when reoxygenated than even HR-proficient oxic cells (139). Thus, although acutely anoxic tumor cells that are repair proficient may be highly resistant to ionizing radiation, chronically hypoxic tumor sub-regions may contain cells with differential radio- and chemosensitivity. We observed that HR-defective hypoxic cells are more sensitive to radiation, mitomycin C, and cisplatin (140). Furthermore, these repair-deficient cells may also be more sensitive to PARP inhibitors; a phenomenon termed “contextual synthetic lethality” (141). Clinically useful functional assays of repair-proficient vs. repair-deficient oxic and hypoxic cells will be required to show the fraction of repair-deficient hypoxic cells in a given tumor. This could be useful to tackle advantage of contextual synthetic lethality using molecular targeted inhibitors with RT (142).
Abnormal Extra-/Intracell Signaling
PTEN/PI3K/AKT/mTOR
Proliferation of PCa cells is under control of the PTEN/PI3K/AKT protein pathways (143, 144) (Figure 1D), and this is also critical for PCa stem-like cell maintenance (145). Loss of PTEN, a common event in many human cancers, can be detected in more than 60% of PCa. This leads to constitutive activation of AKT and thereby activation of a host of downstream proteins that are involved in cell cycle progression, apoptosis suppression, and glucose uptake and metabolism (146). Permanent AKT activation is a major factor of radioresistance and is an important target to increase the RT response (147, 148). Also, interactions between different extra- and intra-cell signaling pathways play a significant role in radioresistance.
In addition, signal transduction modulation interferes with DNA repair mechanisms, particularly DSB repair by NHEJ (147) being an important alternative to increase radiosensitivity. PI3K-dependent AKT phosphorylation triggers a downstream cascade of events that are likely to interact with AR transcriptional activity. These include interaction of the AR with FKHR and FKHRL1 transcription factors, cross-talk of AR and AKT with NF-κβ, regulation of AR via coactivator Wnt/β-catenin, and activation of AR via the mTOR pathway (147). PI3K/AKT/mTOR downstream kinase pathways also regulate NF-κB which, in turn, regulates AR expression (149) and various other pathways implicated in cell survival, proliferation, invasion, angiogenesis, and metastasis (112). Numerous agents identified from natural sources can block the NF-κB pathway, including curcumin, resveratrol, ursolic acid, capsaicin, silymarin, guggulsterone, and plumbagin. Curcumin was shown to downregulate both the NF-κB and Stat3 pathways (149–153).
Pharmacological mTOR inhibition has been demonstrated to block the induction of the proliferative, pro-survival, and oncogenic functions of mTOR (154), with important effects in PTEN-deficient tumors. mTOR signaling has been implicated as a determinant of cell survival in response to DNA damage (155). mTOR inhibitors have been shown to potentiate the effects DNA damaging agents, including ionizing radiation (156–159). As such, the mTOR-signaling pathway is a promising target for RT optimization in PCa.
Rapamycin, when administered in localized PCa patients before prostatectomy, attained high intra-prostatic levels with minimal adverse effects and effectively limited mTOR signaling. This was determined by inhibition of S6 kinase phosphorylation, which is a downstream target of mTOR activity involved in protein translation (160). Although some preliminary results with mTOR inhibitors (temsirolimus and everolimus) have been disappointing when administered as single agents in castration-resistant disease, they showed radiosensitizing effects independent of castration status (156). The combination of RAD001 with radiation has been tested in phase I and II trials (NCT00657982, NCT01548807, NCT00943956). Dual PI3K/mTOR inhibitors (BEZ235 or PI103) when combined with RT greatly improved treatment efficacy by repressing colony formation, inducing more apoptosis, leading to the arrest of the G2/M phase, increased double-strand break levels, and less inactivation of cell cycle check point, autophagy and NHEJ/ HR repair pathway proteins in PCa-radioresistant cells (161). BEZ235 has been shown to improve tumor sensitization by improving tumor oxygenation and vascular structure (162–164), and the radiosensitizing properties of BEZ235 seem to occur in normoxic and hypoxic PCa cells (164). Other PI3K inhibitors like XL147, GDC-0941, XL765, and small-molecule AKT inhibitors MK2206 are currently in Phase I trial (144) and are promising candidates for future studies with RT. Predictive biomarkers are essential for the clinical success of these agents targeting the PI3K/AKT/mTOR pathway. Possible biomarkers for mTOR inhibitors response may be phosphorylated p70S6K, pS6, AKT as well as VEGF, BCL2, and PTEN.
The Akt inhibitor Erufosine (ErPC3) was studied in PCa cell lines. It was shown to have a potential therapeutic benefit when used as monotherapy or in combination with RT (165). The Akt inhibitor P529 potentiates the effect of RT in PC3 cells mainly not only through the blockade of Akt activation but also through the alteration of other cancer-related pathways involving MMP-2, MMP-9, Id1, and VEGF. P529 also enhances the antitumor effect of RT in vivo by reducing the proliferation rates and promoting apoptosis. This ability to act at different pathway levels, all of them involved in the response to radiation, makes this compound an interesting agent for radiosensitization (166).
EGFR
EGFR for a long time has been considered an appealing target for monoclonal antibody (mAb) therapy. The treatment of head and neck cancers with EGFR inhibitors represents a model for the optimization of RT with molecular targets (167–169). In non-metastatic PCa, studies have reported high EGFR expression ranging from 18 to 41% (170, 171). There is evidence that the activation of EGFR and downstream signaling pathways are implicated in cell survival and proliferation following radiation (172–174) thus several studies have addressed potential mechanisms for radiosensitization by EGFR inhibitors. The prognostic role of EGFR expression in PCa is not clearly defined, although some studies have shown that an increased EGFR expression was associated with higher GS, early PSA relapse, and progression to CRPC (170, 175–178).
Exposure of tumor cells to radiation results in immediate activation of EGFR by autophosphorylation (179) and a secondary prolonged release of TGF-α (180). This creates an autocrine loop, which is important for proliferation and is thought to play a part in accelerated repopulation following radiation (181). EGFR activation of downstream pathways including the Ras/Raf/MAPK and STAT3 pathways results in protection from radiation induced cell death (182, 183). EGFR inhibition in different model systems has shown to affect proliferation, angiogenesis, and cell survival. Radiosensitization by EGFR inhibition seem to involve changes in cell-cycle arrest, endothelial cell sensitivity, apoptosis and DDR (184, 185).
Phase I and II trials tested the EGFR inhibitor gefitinib in combination with PCa RT (186). The toxicity profile of the combination appears to be acceptable, as less than 10% of patients had toxicity-related interruptions of RT. The preliminary efficacy seems promising compared to matched controls treated with a slightly higher biologically effective dose; however, further studies are required.
When cetuximab was tested in DU145 cells, it increased the radiosensitivity through antiproliferative effect, inhibition of clonal growth, G(0)/G(1) phase arrest, apoptosis induction, and inhibition of EGFR-signaling pathways by the downregulation of MAPK activation (187). The simultaneous blockade of EGFR and VEGFR (i.e., AEE788) has been tested with radiation and can lead to significant tumor growth delay in DU145 cells (188–190). Potential mechanisms of action could include: (1) enhanced tumor vasculature destruction and (2) decreased proliferation of tumor cells surviving cytotoxic effects of RT (191). Preclinical studies with PCa cells using coinhibitors of both EGFR and type 1 insulin-like growth factor receptor (IGF1R) significantly dampened cellular growth and DDR, therefore increasing radiosensitivity. The synergistic effect of the EGFR and IGF1R inhibitors was also confirmed in nude mouse xenograft assays, thus may provide a therapeutic rationale to be tested in future clinical trials (192).
Immune Checkpoint Inhibitors
Experimental data from multiple cancer models have provided cumulative evidence of an interaction of ionizing radiation with the systemic antitumor immunity, and this has created several opportunities in the field (193).
The combination of immunologic checkpoint inhibitors with RT offers an additional area to improve cancer cell kill in PCa (194). Based on preclinical data, manipulating immune response through checkpoint molecules using mAbs has thus gained interest (195). Early phase I and II clinical trials have demonstrated favorable safety profiles with cytotoxic lymphocyte antigen-4 (CTLA-4) blockade via the mAbs ipilimumab and tremelimumab (196–200). Recent phase I/II trials have been conducted combining single-dose RT concomitant or sequential to ipilimumab. These trials have confirmed the preclinical data that RT could help prime an immune response (200). A phase III study subsequently compared ipilimumab with a placebo following RT (8 Gy in one fraction) and demonstrated a significant PFS benefit but no benefit in terms of OS (201).
Future clinical trials are further investigating the ability of immunologic checkpoint inhibitors to enhance RT’s effect on tumor growth rate kinetics and cellular apoptosis on clinical endpoints such as PFS and OS. Clinical studies with novel immune strategies must include tissue and blood for interrogation of how and which immunologic populations can benefit from this approach.
Concluding Remarks and Future Perspectives
Despite all technological advances in RT delivery over the recent years, improvements in molecular characterization of PCa have not changed clinical practice. Decision-making in RT for PCa treatment is still guided by conventional clinical-pathological factors: PSA levels, GS and T category. In order to minimize RT failures (local and systemic) in non-indolent PCa, precision RT needs to exploit the rich molecular landscape of PCa.
Although the present review focused on four major pathways, other intricate and dynamic mechanisms related to intrinsic and/or acquired radioresistance contribute to the complexity of PCa radioresistance. Many groups have investigated the role of prostate cancer stem cells in providing a reservoir of cells resistant to radiation (202–205). The inhibition of signaling pathways in combination with RT may be a strategy to target PCa stem cells leading to better outcomes (205, 206) by providing improved local control and preventing the dissemination of resistant, proliferating stem-like PCa cells (207, 208). The specific role of prostate cancer stem cells has been recently reviewed (209). Another important feature in PCa radioresistance is the presence of neuroendocrine cells. Although rare, a neuroendocrine phenotype may be present at diagnosis and/or arise during the different stages of disease progression leading to a castrate-resistant state (neuroendocrine cells lack AR expression) and a lethal outcome. Although preclinical data have suggested that radiation can induce neuroendocrine transdifferentiation in PCa cell lines (210–212), an enhanced molecular identification of neuroendocrine cells with a better knowledge of the clinical impact of treatment-induced neuroendocrine differentiation are mostly warranted (211).
The optimization of PCa RT must take into consideration the importance of tumor heterogeneity. Elucidating those specific molecular processes of tumor progression in combination with a better characterization of tumor microenvironment and, most importantly, identifying and validating predictive biomarkers of treatment response are critical steps.
A significant progress has been made in the discovery of molecularly targeted therapies directly and indirectly involved in pathways of PCa progression and RT response. However, the lack of clinical trials combining RT and novel molecular agents faces several challenges. Enhancing the interest of pharmaceutical industry toward RT-based drug development and expediting the testing of these agents with RT are imperative to accelerate the field and improve patient outcomes (213).
The technological advances and the lower costs in genomic medicine should help standardize and validate assays for molecular characterizations. This is determining personalized PCa genomics and reflecting specific patterns of tumor progression. Incorporation of this knowledge into biomarker-based prospective clinical trials will enable us to marry precision RT, novel targeted agents, and biological endpoints in order to improve cure rates in non-indolent PCa.
Author Contributions
All authors contributed equally to researching data, discussing the content, writing the article, and performing review/editing of the manuscript before submission.
Conflict of Interest Statement
Alan Dal Pra has previously received honoraria or research grants from AstraZeneca, Astellas and Bayer. Robert G. Bristow has previously received honoraria or preclinical research grants from Abbott Pharmaceuticals, SuperGen, AstraZeneca, MERCK, and Sentinel. Stephane Supiot has previously received honoraria and research grants from Janssen, AstraZeneca, Novartis. The other co-authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Supported by a grant from the Ontario Institute for Cancer Research and Canadian Foundation for Innovation grant to the STTARR Innovation Facility. This research was funded in part by the Ontario Ministry of Health and Long Term Care. The views expressed do not necessarily reflect those of the Ontario Ministry of Health and Long Term Care. RB is a Canadian Cancer Society Research Scientist. Individual support is through a Canadian Cancer Society Research Scientist Award (RGB), Canadian Urology Oncology Group (CUOG) Fellowship (AD), “Région des Pays-de-Loire” young investigator award, Association pour la Recherche sur le Cancer, Ligue Contre le Cancer, comités Charente et Charente-Maritime, Programme d’Action Intégré de Recherche “PAIR prostate 2010” (SS) and the Comprehensive Research Experience for Medical Student (CREMS) program at the University of Toronto (JL).
References
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin (2014) 64:9–29. doi:10.3322/caac.21208
2. D’Amico AV, Whittington R, Malkowicz SB, Schultz D, Blank K, Broderick GA, et al. Biochemical outcome after radical prostatectomy, external beam radiation therapy, or interstitial radiation therapy for clinically localized prostate cancer. JAMA (1998) 280:969–74. doi:10.1001/jama.280.11.969
3. Mohler J, Bahnson RR, Boston B, Busby JE, D’Amico A, Eastham JA, et al. NCCN clinical practice guidelines in oncology: prostate cancer. J Natl Compr Canc Netw (2010) 8:162–200.
4. Cooperberg MR, Lubeck DP, Mehta SS, Carroll PR. Time trends in clinical risk stratification for prostate cancer: implications for outcomes (data from CaPSURE). J Urol (2003) 170:S21–5. doi:10.1097/01.ju.0000095025.03331.c6
5. Heidenreich A, Aus G, Bolla M, Joniau S, Matveev VB, Schmid HP, et al. EAU guidelines on prostate cancer. Eur Urol (2008) 53:68–80. doi:10.1016/j.eururo.2007.09.002
6. Cooperberg MR, Carroll PR, Klotz L. Active surveillance for prostate cancer: progress and promise. J Clin Oncol (2011) 29:3669–76. doi:10.1200/JCO.2011.34.9738
7. Wolff JM, Mason M. Drivers for change in the management of prostate cancer – guidelines and new treatment techniques. BJU Int (2012) 109(Suppl 6):33–41. doi:10.1111/j.1464-410X.2012.11218.x
8. Heidenreich A, Bellmunt J, Bolla M, Joniau S, Mason M, Matveev V, et al. EAU guidelines on prostate cancer. Part 1: screening, diagnosis, and treatment of clinically localised disease. Eur Urol (2011) 59:61–71. doi:10.1016/j.eururo.2010.10.039
9. Mohler JL, Armstrong AJ, Bahnson RR, Boston B, Busby JE, D’Amico AV, et al. Prostate cancer, version 3.2012 featured updates to the NCCN guidelines. J Natl Compr Canc Netw (2012) 10:1081–7.
10. Bauman G, Rumble RB, Chen J, Loblaw A, Warde P. Intensity-modulated radiotherapy in the treatment of prostate cancer. Clin Oncol (R Coll Radiol) (2012) 24:461–73. doi:10.1016/j.clon.2012.05.002
11. Zumsteg ZS, Zelefsky MJ. Short-term androgen deprivation therapy for patients with intermediate-risk prostate cancer undergoing dose-escalated radiotherapy: the standard of care? Lancet Oncol (2012) 13:e259–69. doi:10.1016/S1470-2045(12)70084-0
12. Yamada Y, Rogers L, Demanes DJ, Morton G, Prestidge BR, Pouliot J, et al. American brachytherapy society consensus guidelines for high-dose-rate prostate brachytherapy. Brachytherapy (2012) 11:20–32. doi:10.1016/j.brachy.2011.09.008
13. Jaffray DA. Image-guided radiotherapy: from current concept to future perspectives. Nat Rev Clin Oncol (2012) 9:688–99. doi:10.1038/nrclinonc.2012.194
14. Zaorsky NG, Harrison AS, Trabulsi EJ, Gomella LG, Showalter TN, Hurwitz MD, et al. Evolution of advanced technologies in prostate cancer radiotherapy. Nat Rev Urol (2013) 10:565–79. doi:10.1038/nrurol.2013.185
15. Locke JA, Dal Pra A, Supiot S, Warde P, Bristow RG. Synergistic action of image-guided radiotherapy and androgen deprivation therapy. Nat Rev Urol (2015) 12:193–204. doi:10.1038/nrurol.2015.50
16. Chopra S, Toi A, Taback N, Evans A, Haider MA, Milosevic M, et al. Pathological predictors for site of local recurrence after radiotherapy for prostate cancer. Int J Radiat Oncol Biol Phys (2012) 82:e441–8. doi:10.1016/j.ijrobp.2011.05.035
17. Arrayeh E, Westphalen AC, Kurhanewicz J, Roach M III, Jung AJ, Carroll PR, et al. Does local recurrence of prostate cancer after radiation therapy occur at the site of primary tumor? Results of a longitudinal MRI and MRSI study. Int J Radiat Oncol Biol Phys (2012) 82:e787–93. doi:10.1016/j.ijrobp.2011.11.030
18. Roach M III, Waldman F, Pollack A. Predictive models in external beam radiotherapy for clinically localized prostate cancer. Cancer (2009) 115:3112–20. doi:10.1002/cncr.24348
19. Zafarana G, Ishkanian AS, Malloff CA, Locke JA, Sykes J, Thoms J, et al. Copy number alterations of c-MYC and PTEN are prognostic factors for relapse after prostate cancer radiotherapy. Cancer (2012) 118(16):4053–62. doi:10.1002/cncr.26729
20. Locke JA, Zafarana G, Malloff CA, Lam WL, Sykes J, Pintilie M, et al. Allelic loss of the loci containing the androgen synthesis gene, StAR, is prognostic for relapse in intermediate-risk prostate cancer. Prostate (2011) 72(12):1295–305. doi:10.1002/pros.22478
21. Locke JA, Zafarana G, Ishkanian AS, Milosevic M, Thoms J, Have CL, et al. NKX3.1 haploinsufficiency is prognostic for prostate cancer relapse following surgery or image-guided radiotherapy. Clin Cancer Res (2012) 18:308–16. doi:10.1158/1078-0432.CCR-11-2147
22. Abdueva D, Wing M, Schaub B, Triche T, Davicioni E. Quantitative expression profiling in formalin-fixed paraffin-embedded samples by affymetrix microarrays. J Mol Diagn (2010) 12:409–17. doi:10.2353/jmoldx.2010.090155
23. Bishoff JT, Freedland SJ, Gerber L, Tennstedt P, Reid J, Welbourn W, et al. Prognostic utility of the cell cycle progression score generated from biopsy in men treated with prostatectomy. J Urol (2014) 192:409–14. doi:10.1016/j.juro.2014.02.003
24. Cuzick J, Swanson GP, Fisher G, Brothman AR, Berney DM, Reid JE, et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. Lancet Oncol (2011) 12:245–55. doi:10.1016/S1470-2045(10)70295-3
25. Wu CL, Schroeder BE, Ma XJ, Cutie CJ, Wu S, Salunga R, et al. Development and validation of a 32-gene prognostic index for prostate cancer progression. Proc Natl Acad Sci USA (2013) 110:6121–6. doi:10.1073/pnas.1215870110
26. Erho N, Crisan A, Vergara IA, Mitra AP, Ghadessi M, Buerki C, et al. Discovery and validation of a prostate cancer genomic classifier that predicts early metastasis following radical prostatectomy. PLoS One (2013) 8:e66855. doi:10.1371/journal.pone.0066855
27. Grignon DJ, Caplan R, Sarkar FH, Lawton CA, Hammond EH, Pilepich MV, et al. p53 status and prognosis of locally advanced prostatic adenocarcinoma: a study based on RTOG 8610. J Natl Cancer Inst (1997) 89(2):158–65. doi:10.1093/jnci/89.2.158
28. Che M, DeSilvio M, Pollack A, Grignon DJ, Venkatesan VM, Hanks GE, et al. Prognostic value of abnormal p53 expression in locally advanced prostate cancer treated with androgen deprivation and radiotherapy: a study based on RTOG 9202. Int J Radiat Oncol Biol Phys (2007) 69(4):1117–23. doi:10.1016/j.ijrobp.2007.04.070
29. Vergis R, Corbishley CM, Thomas K, Horwich A, Huddart R, Khoo V, et al. Expression of Bcl-2, p53, and MDM2 in localized prostate cancer with respect to the outcome of radical radiotherapy dose escalation. Int J Radiat Oncol Biol Phys (2010) 78(1):35–41. doi:10.1016/j.ijrobp.2009.07.1728
30. Scherr DS, Vaughan ED Jr, Wei J, Chung M, Felsen D, Allbright R, et al. BCL-2 and p53 expression in clinically localized prostate cancer predicts response to external beam radiotherapy. J Urol (1999) 162(1):12–6; discussion 6–7. doi:10.1097/00005392-199907000-00003
31. Ritter MA, Gilchrist KW, Voytovich M, Chappell RJ, Verhoven BM. The role of p53 in radiation therapy outcomes for favorable-to-intermediate-risk prostate cancer. Int J Radiat Oncol Biol Phys (2002) 53(3):574–80. doi:10.1016/S0360-3016(02)02781-5
32. D’Amico AV, Halabi S, Vollmer R, Loffredo M, McMahon E, Sanford B, et al. p53 Protein expression status and recurrence in men treated with radiation and androgen suppression therapy for higher-risk prostate cancer: a prospective phase II Cancer and Leukemia Group B Study (CALGB 9682). Urology (2008) 71(5):933–7. doi:10.1016/j.urology.2007.11.005
33. Chakravarti A, Heydon K, Wu CL, Hammond E, Pollack A, Roach M, et al. Loss of p16 expression is of prognostic significance in locally advanced prostate cancer: an analysis from the Radiation Therapy Oncology Group protocol 86-10. J Clin Oncol (2003) 21(17):3328–34. doi:10.1200/JCO.2003.12.151
34. Chakravarti A, DeSilvio M, Zhang M, Grignon D, Rosenthal S, Asbell SO, et al. Prognostic value of p16 in locally advanced prostate cancer: a study based on Radiation Therapy Oncology Group Protocol 9202. J Clin Oncol (2007) 25(21):3082–9. doi:10.1200/JCO.2006.08.4152
35. Li R, Heydon K, Hammond ME, Grignon DJ, Roach M III, Wolkov HB, et al. Ki-67 staining index predicts distant metastasis and survival in locally advanced prostate cancer treated with radiotherapy: an analysis of patients in radiation therapy oncology group protocol 86-10. Clin Cancer Res (2004) 10(12 Pt 1):4118–24. doi:10.1158/1078-0432.CCR-1052-03
36. Khor LY, Bae K, Paulus R, Al-Saleem T, Hammond ME, Grignon DJ, et al. MDM2 and Ki-67 predict for distant metastasis and mortality in men treated with radiotherapy and androgen deprivation for prostate cancer: RTOG 92-02. J Clin Oncol (2009) 27(19):3177–84. doi:10.1200/JCO.2008.19.8267
37. Pollack A, DeSilvio M, Khor LY, Li R, Al-Saleem TI, Hammond ME, et al. Ki-67 staining is a strong predictor of distant metastasis and mortality for men with prostate cancer treated with radiotherapy plus androgen deprivation: Radiation Therapy Oncology Group Trial 92-02. J Clin Oncol (2004) 22(11):2133–40. doi:10.1200/JCO.2004.09.150
38. Parker AS, Heckman MG, Wu KJ, Crook JE, Hilton TW, Pisansky TM, et al. Evaluation of ki-67 staining levels as an independent biomarker of biochemical recurrence after salvage radiation therapy for prostate cancer. Int J Radiat Oncol Biol Phys (2009) 75(5):1364–70. doi:10.1016/j.ijrobp.2008.12.061
39. Cowen D, Troncoso P, Khoo VS, Zagars GK, von Eschenbach AC, Meistrich ML, et al. Ki-67 staining is an independent correlate of biochemical failure in prostate cancer treated with radiotherapy. Clin Cancer Res (2002) 8(5):1148–54.
40. Scalzo DA, Kallakury BV, Gaddipati RV, Sheehan CE, Keys HM, Savage D, et al. Cell proliferation rate by MIB-1 immunohistochemistry predicts postradiation recurrence in prostatic adenocarcinomas. Am J Clin Pathol (1998) 109(2):163–8.
41. Bouchaert P, Guerif S, Debiais C, Irani J, Fromont G. DNA-PKcs expression predicts response to radiotherapy in prostate cancer. Int J Radiat Oncol Biol Phys (2012) 84(5):1179–85. doi:10.1016/j.ijrobp.2012.02.014
42. Khor LY, Desilvio M, Al-Saleem T, Hammond ME, Grignon DJ, Sause W, et al. MDM2 as a predictor of prostate carcinoma outcome: an analysis of Radiation Therapy Oncology Group Protocol 8610. Cancer (2005) 104(5):962–7. doi:10.1002/cncr.21261
43. Khor LY, Desilvio M, Li R, McDonnell TJ, Hammond ME, Sause WT, et al. Bcl-2 and bax expression and prostate cancer outcome in men treated with radiotherapy in Radiation Therapy Oncology Group protocol 86-10. Int J Radiat Oncol Biol Phys (2006) 66(1):25–30. doi:10.1016/j.ijrobp.2006.03.056
44. Khor LY, Moughan J, Al-Saleem T, Hammond EH, Venkatesan V, Rosenthal SA, et al. Bcl-2 and Bax expression predict prostate cancer outcome in men treated with androgen deprivation and radiotherapy on radiation therapy oncology group protocol 92-02. Clin Cancer Res (2007) 13(12):3585–90. doi:10.1158/1078-0432.CCR-06-2972
45. Pollack A, Cowen D, Troncoso P, Zagars GK, von Eschenbach AC, Meistrich ML, et al. Molecular markers of outcome after radiotherapy in patients with prostate carcinoma: Ki-67, bcl-2, bax, and bcl-x. Cancer (2003) 97(7):1630–8. doi:10.1002/cncr.11230
46. Bylund A, Stattin P, Widmark A, Bergh A. Predictive value of bcl-2 immunoreactivity in prostate cancer patients treated with radiotherapy. Radiother Oncol (1998) 49(2):143–8. doi:10.1016/S0167-8140(98)00111-X
47. Abdel-Wahab M, Berkey BA, Krishan A, O’Brien T, Hammond E, Roach M III, et al. Influence of number of CAG repeats on local control in the RTOG 86-10 protocol. Am J Clin Oncol (2006) 29(1):14–20. doi:10.1097/01.coc.0000195085.34162.88
48. Khor LY, Bae K, Pollack A, Hammond ME, Grignon DJ, Venkatesan VM, et al. COX-2 expression predicts prostate-cancer outcome: analysis of data from the RTOG 92-02 trial. Lancet Oncol (2007) 8(10):912–20. doi:10.1016/S1470-2045(07)70280-2
49. Torres-Roca JF, DeSilvio M, Mora LB, Khor LY, Hammond E, Ahmad N, et al. Activated STAT3 as a correlate of distant metastasis in prostate cancer: a secondary analysis of Radiation Therapy Oncology Group 86-10. Urology (2007) 69(3):505–9. doi:10.1016/j.urology.2006.11.006
50. Green MM, Hiley CT, Shanks JH, Bottomley IC, West CM, Cowan RA, et al. Expression of vascular endothelial growth factor (VEGF) in locally invasive prostate cancer is prognostic for radiotherapy outcome. Int J Radiat Oncol Biol Phys (2007) 67(1):84–90. doi:10.1016/j.ijrobp.2006.08.077
51. Vergis R, Corbishley CM, Norman AR, Bartlett J, Jhavar S, Borre M, et al. Intrinsic markers of tumour hypoxia and angiogenesis in localised prostate cancer and outcome of radical treatment: a retrospective analysis of two randomised radiotherapy trials and one surgical cohort study. Lancet Oncol (2008) 9(4):342–51. doi:10.1016/S1470-2045(08)70076-7
52. Weber DC, Tille JC, Combescure C, Egger JF, Laouiti M, Hammad K, et al. The prognostic value of expression of HIF1alpha, EGFR and VEGF-A, in localized prostate cancer for intermediate- and high-risk patients treated with radiation therapy with or without androgen deprivation therapy. Radiat Oncol (2012) 7:66. doi:10.1186/1748-717X-7-66
53. Thoms JW, Dal Pra A, Anborgh PH, Christensen E, Fleshner N, Menard C, et al. Plasma osteopontin as a biomarker of prostate cancer aggression: relationship to risk category and treatment response. Br J Cancer (2012) 107(5):840–6. doi:10.1038/bjc.2012.345
54. Pollack A, Bae K, Khor LY, Al-Saleem T, Hammond ME, Venkatesan V, et al. The importance of protein kinase A in prostate cancer: relationship to patient outcome in Radiation Therapy Oncology Group trial 92-02. Clin Cancer Res (2009) 15(17):5478–84. doi:10.1158/1078-0432.CCR-08-2704
55. Khor LY, Bae K, Al-Saleem T, Hammond EH, Grignon DJ, Sause WT, et al. Protein kinase A RI-alpha predicts for prostate cancer outcome: analysis of radiation therapy oncology group trial 86-10. Int J Radiat Oncol Biol Phys (2008) 71(5):1309–15. doi:10.1016/j.ijrobp.2007.12.010
56. Dal Pra A, Lalonde E, Sykes J, Warde F, Ishkanian A, Meng A, et al. TMPRSS2-ERG status is not prognostic following prostate cancer radiotherapy: implications for fusion status and DSB repair. Clin Cancer Res (2013) 19(18):5202–9. doi:10.1158/1078-0432.CCR-13-1049
57. Pollack A, Grignon DJ, Heydon KH, Hammond EH, Lawton CA, Mesic JB, et al. Prostate cancer DNA ploidy and response to salvage hormone therapy after radiotherapy with or without short-term total androgen blockade: an analysis of RTOG 8610. J Clin Oncol (2003) 21(7):1238–48. doi:10.1200/JCO.2003.02.025
58. Roach M III, De Silvio M, Rebbick T, Grignon D, Rotman M, Wolkov H, et al. Racial differences in CYP3A4 genotype and survival among men treated on Radiation Therapy Oncology Group (RTOG) 9202: a phase III randomized trial. Int J Radiat Oncol Biol Phys (2007) 69(1):79–87. doi:10.1016/j.ijrobp.2007.03.008
59. Berlin A, Lalonde E, Sykes J, Zafarana G, Chu KC, Ramnarine VR, et al. NBN gain is predictive for adverse outcome following image-guided radiotherapy for localized prostate cancer. Oncotarget. (2014) 5(22):11081–90. doi:10.18632/oncotarget.2404
60. Lalonde E, Ishkanian AS, Sykes J, Fraser M, Ross-Adams H, Erho N, et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: a retrospective cohort study. Lancet Oncol (2014) 15(13):1521–32. doi:10.1016/S1470-2045(14)71021-6
61. Freedland SJ, Gerber L, Reid J, Welbourn W, Tikishvili E, Park J, et al. Prognostic utility of cell cycle progression score in men with prostate cancer after primary external beam radiation therapy. Int J Radiat Oncol Biol Phys (2013) 86(5):848–53. doi:10.1016/j.ijrobp.2013.04.043
62. Den RB, Yousefi K, Trabulsi EJ, Abdollah F, Choeurng V, Feng FY, et al. Genomic classifier identifies men with adverse pathology after radical prostatectomy who benefit from adjuvant radiation therapy. J Clin Oncol (2015) 33(8):944–51. doi:10.1200/JCO.2014.59.0026
63. Hennequin C, Giocanti N, Favaudon V. Interaction of ionizing radiation with paclitaxel (Taxol) and docetaxel (Taxotere) in HeLa and SQ20B cells. Cancer Res (1996) 56:1842–50.
64. Milas L, Milas MM, Mason KA. Combination of taxanes with radiation: preclinical studies. Semin Radiat Oncol (1999) 9:12–26.
65. Mason KA, Hunter NR, Milas M, Abbruzzese JL, Milas L. Docetaxel enhances tumor radioresponse in vivo. Clin Cancer Res (1997) 3:2431–8.
66. Sandler HM, Hu C, Rosenthal SA. A phase III protocol of androgen suppression (AS) and 3DCRT/IMRT vs AS and 3DCRT/IMRT followed by chemotherapy (CT) with docetaxel and prednisone for localized, high-risk prostate cancer (RTOG 0521). J Clin Oncol (2015) 33(15):LBA5002.
67. Martin JM, Supiot S, Berthold DR. Pharmacotherapeutic management of locally advanced prostate cancer: current status. Drugs (2011) 71:1019–41. doi:10.2165/11591500-000000000-00000
68. Bolla M, Van Tienhoven G, Warde P, Dubois JB, Mirimanoff RO, Storme G, et al. External irradiation with or without long-term androgen suppression for prostate cancer with high metastatic risk: 10-year results of an EORTC randomised study. Lancet Oncol (2010) 11:1066–73. doi:10.1016/S1470-2045(10)70223-0
69. Warde P, Mason M, Ding K, Kirkbride P, Brundage M, Cowan R, et al. Combined androgen deprivation therapy and radiation therapy for locally advanced prostate cancer: a randomised, phase 3 trial. Lancet (2011) 378:2104–11. doi:10.1016/S0140-6736(11)61095-7
70. Dal Pra A, Cury FL, Souhami L. Radiation therapy and androgen deprivation in the management of high risk prostate cancer. Int Braz J Urol (2011) 37:161–79. doi:10.1590/S1677-55382011000200003
71. D’Amico AV, Chen MH, Renshaw AA, Loffredo M, Kantoff PW. Androgen suppression and radiation vs radiation alone for prostate cancer: a randomized trial. JAMA (2008) 299:289–95. doi:10.1001/jama.299.3.289
72. Bolla M, Hannoun-Levi JM, Ferrero JM, Maingon P, Buffet-Miny J, Bougnoux A, et al. Concurrent and adjuvant docetaxel with three-dimensional conformal radiation therapy plus androgen deprivation for high-risk prostate cancer: preliminary results of a multicentre phase II trial. Radiother Oncol (2010) 97:312–7. doi:10.1016/j.radonc.2010.08.012
73. Pollack A, Salem N, Ashoori F, Hachem P, Sangha M, von Eschenbach AC, et al. Lack of prostate cancer radiosensitization by androgen deprivation. Int J Radiat Oncol Biol Phys (2001) 51:1002–7. doi:10.1016/S0360-3016(01)01750-3
74. Milosevic M, Chung P, Parker C, Bristow R, Toi A, Panzarella T, et al. Androgen withdrawal in patients reduces prostate cancer hypoxia: implications for disease progression and radiation response. Cancer Res (2007) 67:6022–5. doi:10.1158/0008-5472.CAN-07-0561
75. Godoy A, Watts A, Sotomayor P, Montecinos VP, Huss WJ, Onate SA, et al. Androgen receptor is causally involved in the homeostasis of the human prostate endothelial cell. Endocrinology (2008) 149:2959–69. doi:10.1210/en.2007-1078
76. Godoy A, Montecinos VP, Gray DR, Sotomayor P, Yau JM, Vethanayagam RR, et al. Androgen deprivation induces rapid involution and recovery of human prostate vasculature. Am J Physiol Endocrinol Metab (2011) 300:E263–75. doi:10.1152/ajpendo.00210.2010
77. Milosevic M, Warde P, Ménard C, Chung P, Toi A, Ishkanian A, et al. Tumor hypoxia predicts biochemical failure following radiotherapy for clinically localized prostate cancer. Clin Cancer Res (2012) 18:2108–14. doi:10.1158/1078-0432.CCR-11-2711
78. Al-Ubaidi FL, Schultz N, Loseva O, Egevad L, Granfors T, Helleday T. Castration therapy results in decreased Ku70 levels in prostate cancer. Clin Cancer Res (2013) 19:1547–56. doi:10.1158/1078-0432.CCR-12-2795
79. Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, et al. Androgen Receptor Signaling Regulates DNA Repair in Prostate Cancers. Cancer Discov (2013) 3(11):1245–53. doi:10.1158/2159-8290.CD-13-0172
80. de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med (2011) 364:1995–2005. doi:10.1056/NEJMoa1014618
81. Kaku T, Hitaka T, Ojida A, Matsunaga N, Adachi M, Tanaka T, et al. Discovery of orteronel (TAK-700), a naphthylmethylimidazole derivative, as a highly selective 17, 20-lyase inhibitor with potential utility in the treatment of prostate cancer. Bioorg Med Chem (2011) 19:6383–99. doi:10.1016/j.bmc.2011.08.066
82. Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med (2012) 367(13):1187–97. doi:10.1056/NEJMoa1207506
83. Molina A, Belldegrun A. Novel therapeutic strategies for castration resistant prostate cancer: inhibition of persistent androgen production and androgen receptor mediated signaling. J Urol (2011) 185:787–94. doi:10.1016/j.juro.2010.10.042
84. Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science (2009) 324:787–90. doi:10.1126/science.1168175
85. Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell (2010) 17:535–46. doi:10.1016/j.ccr.2010.04.027
86. Rathkopf D, Liu G, Carducci MA, Eisenberger MA, Anand A, Morris MJ, et al. Phase I dose-escalation study of the novel antiandrogen BMS-641988 in patients with castration-resistant prostate cancer. Clin Cancer Res (2011) 17:880–7. doi:10.1158/1078-0432.CCR-10-2955
87. Wright JL, Kwon EM, Lin DW, Kolb S, Koopmeiners JS, Feng Z, et al. CYP17 polymorphisms and prostate cancer outcomes. Prostate (2010) 70:1094–101. doi:10.1002/pros.21143
88. Locke JA, Fazli L, Adomat H, Smyl J, Weins K, Lubik AA, et al. A novel communication role for CYP17A1 in the progression of castration-resistant prostate cancer. Prostate (2009) 69:928–37. doi:10.1002/pros.20940
89. Bjartell A. Circulating tumour cells as surrogate biomarkers in castration-resistant prostate cancer trials. Eur Urol (2011) 60:905–7. doi:10.1016/j.eururo.2011.08.024
90. Vasaitis TS, Bruno RD, Njar VC. CYP17 inhibitors for prostate cancer therapy. J Steroid Biochem Mol Biol (2011) 125:23–31. doi:10.1016/j.jsbmb.2010.11.005
91. Njar VC, Brodie AM. Discovery and development of Galeterone (TOK-001 or VN/124-1) for the treatment of all stages of prostate cancer. J Med Chem (2015) 58:2077–87. doi:10.1021/jm501239f
92. Saad F, Fizazi K, Jinga V, Efstathiou E, Fong PC, Hart LL, et al. Orteronel plus prednisone in patients with chemotherapy-naive metastatic castration-resistant prostate cancer (ELM-PC 4): a double-blind, multicentre, phase 3, randomised, placebo-controlled trial. Lancet Oncol (2015) 16:338–48. doi:10.1016/S1470-2045(15)70027-6
93. Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med (2014) 371:424–33. doi:10.1056/NEJMoa1405095
94. Tombal B, Borre M, Rathenborg P, Werbrouck P, Van Poppel H, Heidenreich A, et al. Enzalutamide monotherapy in hormone-naive prostate cancer: primary analysis of an open-label, single-arm, phase 2 study. Lancet Oncol (2014) 15:592–600. doi:10.1016/S1470-2045(14)70129-9
95. Hoffman-Censits J, Kelly WK. Enzalutamide: a novel antiandrogen for patients with castrate-resistant prostate cancer. Clin Cancer Res (2013) 19:1335–9. doi:10.1158/1078-0432.CCR-12-2910
96. Yap TA, Zivi A, Omlin A, de Bono JS. The changing therapeutic landscape of castration-resistant prostate cancer. Nat Rev Clin Oncol (2011) 8:597–610. doi:10.1038/nrclinonc.2011.117
97. Kohli M, Qin R, Jimenez R, Dehm SM. Biomarker-based targeting of the androgen-androgen receptor axis in advanced prostate cancer. Adv Urol (2012) 2012:781459. doi:10.1155/2012/781459
98. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med (2014) 371:1028–38. doi:10.1056/NEJMoa1315815
99. Fizazi K, Massard C, Bono P, Jones R, Kataja V, James N, et al. Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): an open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial. Lancet Oncol (2014) 15:975–85. doi:10.1016/S1470-2045(14)70240-2
100. Miyake H, Muramaki M, Furukawa J, Kurahashi T, Fujisawa M. Serum level of clusterin and its density in men with prostate cancer as novel biomarkers reflecting disease extension. Urology (2010) 75:454–9. doi:10.1016/j.urology.2009.08.029
101. Zellweger T, Kiyama S, Chi K, Miyake H, Adomat H, Skov K, et al. Overexpression of the cytoprotective protein clusterin decreases radiosensitivity in the human LNCaP prostate tumour model. BJU Int (2003) 92:463–9. doi:10.1046/j.1464-410X.2003.04349.x
102. Zellweger T, Chi K, Miyake H, Adomat H, Kiyama S, Skov K, et al. Enhanced radiation sensitivity in prostate cancer by inhibition of the cell survival protein clusterin. Clin Cancer Res (2002) 8:3276–84.
103. Chi KN, Goldenberg L, Kollmannsberger C, Murray N, Fazli L, Gleave ME. A phase II neoadjuvant study of OGX-011, a 2’methoxyethyl phosphorothioate antisense to clusterin, in patients with prostate cancer prior to prostatectomy. J Clin Oncol (2008) 14:833–9.
104. Chi KN, Zoubeidi A, Gleave ME. Custirsen (OGX-011): a second-generation antisense inhibitor of clusterin for the treatment of cancer. Expert Opin Investig Drugs (2008) 17:1955–62. doi:10.1517/13543780802528609
105. Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer (2008) 8:180–92. doi:10.1038/nrc2344
106. Chan N, Milosevic M, Bristow RG. Tumor hypoxia, DNA repair and prostate cancer progression: new targets and new therapies. Future Oncol (2007) 3:329–41. doi:10.2217/14796694.3.3.329
107. Movsas B, Chapman JD, Hanlon AL, Horwitz EM, Greenberg RE, Stobbe C, et al. Hypoxic prostate/muscle pO2 ratio predicts for biochemical failure in patients with prostate cancer: preliminary findings. Urology (2002) 60:634–9. doi:10.1016/S0090-4295(02)01858-7
108. Turaka A, Buyyounouski MK, Hanlon AL, Horwitz EM, Greenberg RE, Movsas B. Hypoxic prostate/muscle PO2 ratio predicts for outcome in patients with localized prostate cancer: long-term results. Int J Radiat Oncol Biol Phys (2012) 82:e433–9. doi:10.1016/j.ijrobp.2011.05.037
109. Zeman EM, Brown JM, Lemmon MJ, Hirst VK, Lee WW. SR-4233: a new bioreductive agent with high selective toxicity for hypoxic mammalian cells. Int J Radiat Oncol Biol Phys (1986) 12:1239–42. doi:10.1016/0360-3016(86)90267-1
110. Helsby NA, Wheeler SJ, Pruijn FB, Palmer BD, Yang S, Denny WA, et al. Effect of nitroreduction on the alkylating reactivity and cytotoxicity of the 2,4-dinitrobenzamide-5-aziridine CB 1954 and the corresponding nitrogen mustard SN 23862: distinct mechanisms of bioreductive activation. Chem Res Toxicol (2003) 16:469–78. doi:10.1021/tx025662b
111. Patterson AV, Ferry DM, Edmunds SJ, Gu Y, Singleton RS, Patel K, et al. Mechanism of action and preclinical antitumor activity of the novel hypoxia-activated DNA cross-linking agent PR-104. Clin Cancer Res (2007) 13:3922–32. doi:10.1158/1078-0432.CCR-07-0478
112. Meng F, Evans JW, Bhupathi D, Banica M, Lan L, Lorente G, et al. Molecular and cellular pharmacology of the hypoxia-activated prodrug TH-302. Mol Cancer Ther (2012) 11:740–51. doi:10.1158/1535-7163.MCT-11-0634
113. Chan DA, Giaccia AJ. Hypoxia, gene expression, and metastasis. Cancer Metastasis Rev (2007) 26:333–9. doi:10.1007/s10555-007-9063-1
114. Jordan BF, Sonveaux P. Targeting tumor perfusion and oxygenation to improve the outcome of anticancer therapy. Front Pharmacol (2012) 3:94. doi:10.3389/fphar.2012.00094
115. Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer (2008) 8:851–64. doi:10.1038/nrc2501
116. Al-Ubaidi FL, Schultz N, Egevad L, Granfors T, Helleday T. Castration therapy of prostate cancer results in downregulation of HIF-1alpha levels. Int J Radiat Oncol Biol Phys (2012) 82:1243–8. doi:10.1016/j.ijrobp.2011.10.038
117. Huang Y, Yu J, Yan C, Hou J, Pu J, Zhang G, et al. Effect of small interfering RNA targeting hypoxia-inducible factor-1alpha on radiosensitivity of PC3 cell line. Urology (2012) 79(744):e17–24. doi:10.1016/j.urology.2011.10.024
118. Singh-Gupta V, Zhang H, Banerjee S, Kong D, Raffoul JJ, Sarkar FH, et al. Radiation-induced HIF-1alpha cell survival pathway is inhibited by soy isoflavones in prostate cancer cells. Int J Cancer (2009) 124:1675–84. doi:10.1002/ijc.24015
119. Stewart GD, Nanda J, Katz E, Bowman KJ, Christie JG, Brown DJ, et al. DNA strand breaks and hypoxia response inhibition mediate the radiosensitisation effect of nitric oxide donors on prostate cancer under varying oxygen conditions. Biochem Pharmacol (2011) 81:203–10. doi:10.1016/j.bcp.2010.09.022
120. Palayoor ST, Mitchell JB, Cerna D, Degraff W, John-Aryankalayil M, Coleman CN. PX-478, an inhibitor of hypoxia-inducible factor-1alpha, enhances radiosensitivity of prostate carcinoma cells. Int J Cancer (2008) 123:2430–7. doi:10.1002/ijc.23807
121. Bennett MH, Feldmeier J, Smee R, Milross C. Hyperbaric oxygenation for tumour sensitisation to radiotherapy. Cochrane Database Syst Rev (2012) 4:CD005007. doi:10.1002/14651858.CD005007.pub3
122. Kaanders JH, Bussink J, van der Kogel AJ. ARCON: a novel biology-based approach in radiotherapy. Lancet Oncol (2002) 3:728–37. doi:10.1016/S1470-2045(02)00929-4
123. Durand RE, Biaglow JE. Radiosensitization of hypoxic cells of an in vitro tumor model by respiratory inhibitors. Radiat Res (1977) 69:359–66. doi:10.2307/3574443
124. Secomb TW, Hsu R, Ong ET, Gross JF, Dewhirst MW. Analysis of the effects of oxygen supply and demand on hypoxic fraction in tumors. Acta Oncol (1995) 34:313–6. doi:10.3109/02841869509093981
125. Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) (2012) 122:253–70. doi:10.1042/CS20110386
126. Zannella VE, Dal Pra A, Muaddi H, McKee TD, Stapleton S, Sykes J, et al. Reprogramming Metabolism with metformin improves tumor oxygenation and radiotherapy response. Clin Cancer Res (2013) 19(24):6741–50. doi:10.1158/1078-0432.CCR-13-1787
127. Spratt DE, Zhang C, Zumsteg ZS, Pei X, Zhang Z, Zelefsky MJ. Metformin and prostate cancer: reduced development of castration-resistant disease and prostate cancer mortality. Eur Urol (2013) 63:709–16. doi:10.1016/j.eururo.2012.12.004
128. Margel D, Urbach DR, Lipscombe LL, Bell CM, Kulkarni G, Austin PC, et al. Metformin use and all-cause and prostate cancer-specific mortality among men with diabetes. J Clin Oncol (2013) 31:3069–75. doi:10.1200/JCO.2012.46.7043
129. Chopra S, Foltz WD, Milosevic MF, Toi A, Bristow RG, Ménard C, et al. Comparing oxygen-sensitive MRI (BOLD R2*) with oxygen electrode measurements: a pilot study in men with prostate cancer. Int J Radiat Biol (2009) 85:805–13. doi:10.1080/09553000903043059
130. Dewhirst MW, Cao Y, Li CY, Moeller B. Exploring the role of HIF-1 in early angiogenesis and response to radiotherapy. Radiother Oncol (2007) 83:249–55. doi:10.1016/j.radonc.2007.05.016
131. Dal Pra A, Milosevic M, Hill R, Wouters B, Warde P, Bristow RG. Hypoxia, androgen deprivation and systemic metastases in prostate cancer (in response to “Antivascular effects of neoadjuvant androgen deprivation for prostate cancer: an in vivo human study using susceptibility and relaxitivity dynamic MRI”: in regard to Alonzi R et al. (Int J Radiat Oncol Biol Phys 2011;80(3):721-727)). Int J Radiat Oncol Biol Phys (2012) 82:1319. doi:10.1016/j.ijrobp.2011.11.017
132. Wang JZ, Li XA, Mayr NA. Dose escalation to combat hypoxia in prostate cancer: a radiobiological study on clinical data. Br J Radiol (2006) 79:905–11. doi:10.1259/bjr/18700614
133. Supiot S, Shubbar S, Fleshner N, Warde P, Hersey K, Wallace K, et al. A phase I trial of pre-operative radiotherapy for prostate cancer: clinical and translational studies. Radiother Oncol (2008) 88:53–60. doi:10.1016/j.radonc.2008.03.019
134. Thoms J, Bristow RG. DNA repair targeting and radiotherapy: a focus on the therapeutic ratio. Semin Radiat Oncol (2010) 20:217–22. doi:10.1016/j.semradonc.2010.06.003
135. Choudhury A, Zhao H, Jalali F, Al Rashid S, Ran J, Supiot S, et al. Targeting homologous recombination using imatinib results in enhanced tumor cell chemosensitivity and radiosensitivity. Mol Cancer Ther (2009) 8:203–13. doi:10.1158/1535-7163.MCT-08-0959
136. Luoto KR, Kumareswaran R, Bristow RG. Tumor hypoxia as a driving force in genetic instability. Genome Integr (2013) 4:5. doi:10.1186/2041-9414-4-5
137. Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res (2012) 72:5588–99. doi:10.1158/0008-5472.CAN-12-2753
138. Zhu J, Chen H, Guo XE, Qiu XL, Hu CM, Chamberlin AR, et al. Synthesis, molecular modeling, and biological evaluation of novel RAD51 inhibitors. Eur J Med Chem (2015) 96:196–208. doi:10.1016/j.ejmech.2015.04.021
139. Chan N, Bristow RG. “Contextual” synthetic lethality and/or loss of heterozygosity: tumor hypoxia and modification of DNA repair. Clin Cancer Res (2010) 16:4553–60. doi:10.1158/1078-0432.CCR-10-0527
140. Chan N, Koritzinsky M, Zhao H, Bindra R, Glazer PM, Powell S, et al. Chronic hypoxia decreases synthesis of homologous recombination proteins to offset chemoresistance and radioresistance. Cancer Res (2008) 68:605–14. doi:10.1158/0008-5472.CAN-07-5472
141. Chan N, Pires IM, Bencokova Z, Coackley C, Luoto KR, Bhogal N, et al. Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res (2010) 70:8045–54. doi:10.1158/0008-5472.CAN-10-2352
142. Liu SK, Olive PL, Bristow RG. Biomarkers for DNA DSB inhibitors and radiotherapy clinical trials. Cancer Metastasis Rev (2008) 27:445–58. doi:10.1007/s10555-008-9137-8
143. Majumder PK, Sellers WR. Akt-regulated pathways in prostate cancer. Oncogene (2005) 24:7465–74. doi:10.1038/sj.onc.1209096
144. Sarker D, Reid AH, Yap TA, de Bono JS. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin Cancer Res (2009) 15:4799–805. doi:10.1158/1078-0432.CCR-08-0125
145. Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira SM, García-Echeverría C, et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci U S A (2009) 106:268–73. doi:10.1073/pnas.0810956106
146. Chow LM, Baker SJ. PTEN function in normal and neoplastic growth. Cancer Lett (2006) 241:184–96. doi:10.1016/j.canlet.2005.11.042
147. Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer (2011) 11:239–53. doi:10.1038/nrc3007
148. Riesterer O, Tenzer A, Zingg D, Hofstetter B, Vuong V, Pruschy M, et al. Novel radiosensitizers for locally advanced epithelial tumors: inhibition of the PI3K/Akt survival pathway in tumor cells and in tumor-associated endothelial cells as a novel treatment strategy? Int J Radiat Oncol Biol Phys (2004) 58:361–8. doi:10.1016/j.ijrobp.2003.09.050
149. Wang Y, Kreisberg JI, Ghosh PM. Cross-talk between the androgen receptor and the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer. Curr Cancer Drug Targets (2007) 7:591–604. doi:10.2174/156800907781662248
150. Kunnumakkara AB, Anand P, Aggarwal BB. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett (2008) 269:199–225. doi:10.1016/j.canlet.2008.03.009
151. Chendil D, Ranga RS, Meigooni D, Sathishkumar S, Ahmed MM. Curcumin confers radiosensitizing effect in prostate cancer cell line PC-3. Oncogene (2004) 23:1599–607. doi:10.1038/sj.onc.1207284
152. Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y. Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res (2004) 24:2783–840.
153. Adhami VM, Syed DN, Khan N, Mukhtar H. Dietary flavonoid fisetin: a novel dual inhibitor of PI3K/Akt and mTOR for prostate cancer management. Biochem Pharmacol (2012) 84:1277–81. doi:10.1016/j.bcp.2012.07.012
154. Hidalgo M, Rowinsky EK. The rapamycin-sensitive signal transduction pathway as a target for cancer therapy. Oncogene (2000) 19:6680–6. doi:10.1038/sj.onc.1204091
155. Shen C, Lancaster CS, Shi B, Guo H, Thimmaiah P, Bjornsti MA. TOR signaling is a determinant of cell survival in response to DNA damage. Mol Cell Biol (2007) 27:7007–17. doi:10.1128/MCB.00290-07
156. Schiewer MJ, Den R, Hoang DT, Augello MA, Lawrence YR, Dicker AP, et al. mTOR is a selective effector of the radiation therapy response in androgen receptor-positive prostate cancer. Endocr Relat Cancer (2012) 19:1–12. doi:10.1530/ERC-11-0072
157. Braunstein S, Badura ML, Xi Q, Formenti SC, Schneider RJ. Regulation of protein synthesis by ionizing radiation. Mol Cell Biol (2009) 29:5645–56. doi:10.1128/MCB.00711-09
158. Saunders P, Cisterne A, Weiss J, Bradstock KF, Bendall LJ. The mammalian target of rapamycin inhibitor RAD001 (everolimus) synergizes with chemotherapeutic agents, ionizing radiation and proteasome inhibitors in pre-B acute lymphocytic leukemia. Haematologica (2011) 96:69–77. doi:10.3324/haematol.2010.026997
159. Murphy JD, Spalding AC, Somnay YR, Markwart S, Ray ME, Hamstra DA. Inhibition of mTOR radiosensitizes soft tissue sarcoma and tumor vasculature. Clin Cancer Res (2009) 15:589–96. doi:10.1158/1078-0432.CCR-08-1019
160. Armstrong AJ, Netto GJ, Rudek MA, Halabi S, Wood DP, Creel PA, et al. A pharmacodynamic study of rapamycin in men with intermediate- to high-risk localized prostate cancer. Clin Cancer Res (2010) 16:3057–66. doi:10.1158/1078-0432.CCR-10-0124
161. Chang L, Graham PH, Hao J, Ni J, Bucci J, Cozzi PJ, et al. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell death & disease (2014) 5:e1437. doi:10.1038/cddis.2014.415
162. Fokas E, Im JH, Hill S, Yameen S, Stratford M, Beech J, et al. Dual inhibition of the PI3K/mTOR pathway increases tumor radiosensitivity by normalizing tumor vasculature. Cancer Res (2012) 72:239–48. doi:10.1158/0008-5472.CAN-11-2263
163. Fokas E, Yoshimura M, Prevo R, Higgins G, Hackl W, Maira SM, et al. NVP-BEZ235 and NVP-BGT226, dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitors, enhance tumor and endothelial cell radiosensitivity. Radiat Oncol (2012) 7:48. doi:10.1186/1748-717X-7-48
164. Potiron VA, Abderrahmani R, Giang E, Chiavassa S, Di Tomaso E, Maira SM, et al. Radiosensitization of prostate cancer cells by the dual PI3K/mTOR inhibitor BEZ235 under normoxic and hypoxic conditions. Radiother Oncol (2013) 106:138–46. doi:10.1016/j.radonc.2012.11.014
165. Rudner J, Ruiner CE, Handrick R, Eibl HJ, Belka C, Jendrossek V. The Akt-inhibitor erufosine induces apoptotic cell death in prostate cancer cells and increases the short term effects of ionizing radiation. Radiat Oncol (2010) 5:108. doi:10.1186/1748-717X-5-108
166. Diaz R, Nguewa PA, Diaz-Gonzalez JA, Hamel E, Gonzalez-Moreno O, Catena R, et al. The novel Akt inhibitor Palomid 529 (P529) enhances the effect of radiotherapy in prostate cancer. Br J Cancer (2009) 100:932–40. doi:10.1038/sj.bjc.6604938
167. Eke I, Sandfort V, Mischkus A, Baumann M, Cordes N. Antiproliferative effects of EGFR tyrosine kinase inhibition and radiation-induced genotoxic injury are attenuated by adhesion to fibronectin. Radiother Oncol (2006) 80:178–84. doi:10.1016/j.radonc.2006.07.028
168. Baumann M, Krause M, Dikomey E, Dittmann K, Dörr W, Kasten-Pisula U, et al. EGFR-targeted anti-cancer drugs in radiotherapy: preclinical evaluation of mechanisms. Radiother Oncol (2007) 83:238–48. doi:10.1016/j.radonc.2007.04.006
169. Bonner JA, Harari PM, Giralt J, Cohen RB, Jones CU, Sur RK, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol (2010) 11:21–8. doi:10.1016/S1470-2045(09)70311-0
170. Di Lorenzo G, Tortora G, D’Armiento FP, De Rosa G, Staibano S, Autorino R, et al. Expression of epidermal growth factor receptor correlates with disease relapse and progression to androgen-independence in human prostate cancer. Clin Cancer Res (2002) 8:3438–44.
171. Schlomm T, Kirstein P, Iwers L, Daniel B, Steuber T, Walz J, et al. Clinical significance of epidermal growth factor receptor protein overexpression and gene copy number gains in prostate cancer. Clin Cancer Res (2007) 13:6579–84. doi:10.1158/1078-0432.CCR-07-1257
172. Chen DJ, Nirodi CS. The epidermal growth factor receptor: a role in repair of radiation-induced DNA damage. Clin Cancer Res (2007) 13:6555–60. doi:10.1158/1078-0432.CCR-07-1610
173. Rodemann HP, Dittmann K, Toulany M. Radiation-induced EGFR-signaling and control of DNA-damage repair. Int J Radiat Biol (2007) 83:781–91. doi:10.1080/09553000701769970
174. Dent P, Reardon DB, Park JS, Bowers G, Logsdon C, Valerie K, et al. Radiation-induced release of transforming growth factor alpha activates the epidermal growth factor receptor and mitogen-activated protein kinase pathway in carcinoma cells, leading to increased proliferation and protection from radiation-induced cell death. Mol Biol Cell (1999) 10:2493–506.
175. Bostwick DG, Grignon DJ, Hammond ME, Amin MB, Cohen M, Crawford D, et al. Prognostic factors in prostate cancer. College of American Pathologists Consensus Statement 1999. Arch Pathol Lab Med (2000) 124:995–1000.
176. Shah RB, Ghosh D, Elder JT. Epidermal growth factor receptor (ErbB1) expression in prostate cancer progression: correlation with androgen independence. Prostate (2006) 66:1437–44. doi:10.1002/pros.20460
177. Zellweger T, Ninck C, Bloch M, Mirlacher M, Koivisto PA, Helin HJ, et al. Expression patterns of potential therapeutic targets in prostate cancer. Int J Cancer (2005) 113:619–28. doi:10.1002/ijc.20615
178. Cho KS, Lee JS, Cho NH, Park K, Ham WS, Choi YD. Gene amplification and mutation analysis of epidermal growth factor receptor in hormone refractory prostate cancer. Prostate (2008) 68:803–8. doi:10.1002/pros.20743
179. Olapade-Olaopa EO, Moscatello DK, MacKay EH, Horsburgh T, Sandhu DP, Terry TR, et al. Evidence for the differential expression of a variant EGF receptor protein in human prostate cancer. Br J Cancer (2000) 82:186–94. doi:10.1054/bjoc.1999.0898
180. Schmidt-Ullrich RK, Mikkelsen RB, Dent P, Todd DG, Valerie K, Kavanagh BD, et al. Radiation-induced proliferation of the human A431 squamous carcinoma cells is dependent on EGFR tyrosine phosphorylation. Oncogene (1997) 15:1191–7. doi:10.1038/sj.onc.1201275
181. Schmidt-Ullrich RK, Valerie K, Fogleman PB, Walters J. Radiation-induced autophosphorylation of epidermal growth factor receptor in human malignant mammary and squamous epithelial cells. Radiat Res (1996) 145:81–5. doi:10.2307/3579199
182. Carter S, Auer KL, Reardon DB, Birrer M, Fisher PB, Valerie K, et al. Inhibition of the mitogen activated protein (MAP) kinase cascade potentiates cell killing by low dose ionizing radiation in A431 human squamous carcinoma cells. Oncogene (1998) 16:2787–96. doi:10.1038/sj.onc.1201802
183. Grandis JR, Drenning SD, Chakraborty A, Zhou MY, Zeng Q, Pitt AS, et al. Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor- mediated cell growth in vitro. J Clin Invest (1998) 102:1385–92. doi:10.1172/JCI3785
184. Bandyopadhyay D, Mandal M, Adam L, Mendelsohn J, Kumar R. Physical interaction between epidermal growth factor receptor and DNA-dependent protein kinase in mammalian cells. J Biol Chem (1998) 273:1568–73. doi:10.1074/jbc.273.3.1568
185. Sartor CI. Mechanisms of disease: radiosensitization by epidermal growth factor receptor inhibitors. Nat Clin Pract Oncol (2004) 1:80–7. doi:10.1038/ncponc0048
186. Joensuu G, Joensuu T, Nokisalmi P, Reddy C, Isola J, Ruutu M, et al. A phase I/II trial of gefitinib given concurrently with radiotherapy in patients with nonmetastatic prostate cancer. Int J Radiat Oncol Biol Phys (2010) 78:42–9. doi:10.1016/j.ijrobp.2009.07.1731
187. Liu F, Wang JJ, You ZY, Zhang YD, Zhao Y. Radiosensitivity of prostate cancer cells is enhanced by EGFR inhibitor C225. Urol Oncol (2010) 28:59–66. doi:10.1016/j.urolonc.2008.07.001
188. Goudar RK, Shi Q, Hjelmeland MD, Keir ST, McLendon RE, Wikstrand CJ, et al. Combination therapy of inhibitors of epidermal growth factor receptor/vascular endothelial growth factor receptor 2 (AEE788) and the mammalian target of rapamycin (RAD001) offers improved glioblastoma tumor growth inhibition. Mol Cancer Ther (2005) 4:101–12.
189. Traxler P, Allegrini PR, Brandt R, Brueggen J, Cozens R, Fabbro D, et al. AEE788: a dual family epidermal growth factor receptor/ErbB2 and vascular endothelial growth factor receptor tyrosine kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res (2004) 64:4931–41. doi:10.1158/0008-5472.CAN-03-3681
190. Younes MN, Yigitbasi OG, Park YW, Kim SJ, Jasser SA, Hawthorne VS, et al. Antivascular therapy of human follicular thyroid cancer experimental bone metastasis by blockade of epidermal growth factor receptor and vascular growth factor receptor phosphorylation. Cancer Res (2005) 65:4716–27. doi:10.1158/0008-5472.CAN-04-4196
191. Huamani J, Willey C, Thotala D, Niermann KJ, Reyzer M, Leavitt L, et al. Differential efficacy of combined therapy with radiation and AEE788 in high and low EGFR-expressing androgen-independent prostate tumor models. Int J Radiat Oncol Biol Phys (2008) 71:237–46. doi:10.1016/j.ijrobp.2007.12.049
192. Wang Y, Yuan JL, Zhang YT, Ma JJ, Xu P, Shi CH, et al. Inhibition of both EGFR and IGF1R sensitized prostate cancer cells to radiation by synergistic suppression of DNA homologous recombination repair. PLoS One (2013) 8:e68784. doi:10.1371/journal.pone.0068784
193. Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst (2013) 105:256–65. doi:10.1093/jnci/djs629
194. Pilones KA, Vanpouille-Box C, Demaria S. Combination of radiotherapy and immune checkpoint inhibitors. Semin Radiat Oncol (2015) 25:28–33. doi:10.1016/j.semradonc.2014.07.004
195. Bracarda S, Altavilla A, Hamzaj A, Sisani M, Marrocolo F, Del Buono S, et al. Immunologic checkpoints blockade in renal cell, prostate, and urothelial malignancies. Semin Oncol (2015) 42:495–505. doi:10.1053/j.seminoncol.2015.02.004
196. Small EJ, Tchekmedyian NS, Rini BI, Fong L, Lowy I, Allison JP. A pilot trial of CTLA-4 blockade with human anti-CTLA-4 in patients with hormone-refractory prostate cancer. Clin Cancer Res (2007) 13:1810–5. doi:10.1158/1078-0432.CCR-06-2318
197. Fong L, Kwek SS, O’Brien S, Kavanagh B, McNeel DG, Weinberg V, et al. Potentiating endogenous antitumor immunity to prostate cancer through combination immunotherapy with CTLA4 blockade and GM-CSF. Cancer Res (2009) 69:609–15. doi:10.1158/0008-5472.CAN-08-3529
198. Harzstark AL, Fong L, Weinberg VK, Ryan CJ, Lin AM, Sun J, et al. Final results of a phase I study of CTLA-4 blockade in combination with GM-CSF for metastatic castration resistant prostate cancer (mCRPC). J Clin Oncol (2010) 28:15s.
199. McNeel DG, Smith HA, Eickhoff JC, Lang JM, Staab MJ, Wilding G, et al. Phase I trial of tremelimumab in combination with short-term androgen deprivation in patients with PSA-recurrent prostate cancer. Cancer Immunol Immunother (2012) 61:1137–47. doi:10.1007/s00262-011-1193-1
200. Slovin SF, Higano CS, Hamid O, Tejwani S, Harzstark A, Alumkal JJ, et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: results from an open-label, multicenter phase I/II study. Ann Oncol (2013) 24:1813–21. doi:10.1093/annonc/mdt107
201. Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol (2014) 15:700–12. doi:10.1016/S1470-2045(14)70189-5
202. Baumann M, Krause M, Hill R. Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer (2008) 8(7):545–54. doi:10.1038/nrc2419
203. Peitzsch C, Kurth I, Kunz-Schughart L, Baumann M, Dubrovska A. Discovery of the cancer stem cell related determinants of radioresistance. Radiother Oncol (2013) 108(3):378–87. doi:10.1016/j.radonc.2013.06.003
204. Cho YM, Kim YS, Kang MJ, Farrar WL, Hurt EM. Long-term recovery of irradiated prostate cancer increases cancer stem cells. Prostate (2012) 72(16):1746–56. doi:10.1002/pros.22527
205. Frame FM, Pellacani D, Collins AT, Simms MS, Mann VM, Jones GD, et al. HDAC inhibitor confers radiosensitivity to prostate stem-like cells. Br J Cancer (2013) 109:3023–33. doi:10.1038/bjc.2013.691
206. Cojoc M, Mäbert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Semin Cancer Biol (2015) 31(16):27. doi:10.1016/j.semcancer.2014.06.004
207. Blagosklonny MV. Target for cancer therapy: proliferating cells or stem cells. Leukemia (2006) 20(3):385–91. doi:10.1038/sj.leu.2404075
208. Trumpp A, Wiestler OD. Mechanisms of disease: cancer stem cells – targeting the evil twin. Nat Clin Pract Oncol (2008) 5(6):337–47. doi:10.1038/ncponc1110
209. Rybak A, Bristow RG, Kapoor A. Prostate cancer stem cells: deciphering the origins and pathways involved in prostate tumorigenesis and aggression. Oncotarget. (2014) 6(4):1900–19. doi:10.18632/oncotarget.2953
210. Deng X, Liu H, Huang J, Cheng L, Keller ET, Parsons SJ, et al. Ionizing radiation induces prostate cancer neuroendocrine differentiation through interplay of CREB and ATF2: implications for disease progression. Cancer Res (2008) 68(23):9663–70. doi:10.1158/0008-5472.CAN-08-2229
211. Deng X, Elzey BD, Poulson JM, Morrison WB, Ko SC, Hahn NM, et al. Ionizing radiation induces neuroendocrine differentiation of prostate cancer cells in vitro, in vivo and in prostate cancer patients. Am J Cancer Res. (2011) 1(7):834–44.
212. Hu C-D, Choo R, Huang J. Neuroendocrine differentiation in prostate cancer: a mechanism of radioresistance and treatment failure. Front Oncol (2015) 5:90. doi:10.3389/fonc.2015.00090
Keywords: prostate cancer, radiotherapy, biomarkers, genomics, targeted therapies, molecular oncology, combined modality
Citation: Dal Pra A, Locke JA, Borst G, Supiot S and Bristow RG (2016) Mechanistic Insights into Molecular Targeting and Combined Modality Therapy for Aggressive, Localized Prostate Cancer. Front. Oncol. 6:24. doi: 10.3389/fonc.2016.00024
Received: 24 October 2015; Accepted: 22 January 2016;
Published: 16 February 2016
Edited by:
John Varlotto, University of Massachusetts Medical Center, USACopyright: © 2016 Dal Pra, Locke, Borst, Supiot and Bristow. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert G. Bristow, cm9iLmJyaXN0b3dAcm1wLnVobi5vbi5jYQ==
†Alan Dal Pra and Jennifer A. Locke contributed equally to this work.
‡Stephane Supiot and Robert G. Bristow are both co-senior authors.
§Present address: Alan Dal Pra, Department of Radiation Oncology, Bern University Hospital, Bern, Switzerland;
Jennifer A. Locke, Department of Urologic Sciences, University of British Columbia, Vancouver, BC, Canada;
Gerben Borst, Netherlands Cancer Institute – Antoni van Leeuwenhoek Hospital, Amsterdam, Netherlands