 Danielle H. Lapin
Danielle H. Lapin Maria Tsoli
Maria Tsoli David S. Ziegler
David S. Ziegler- 1Children’s Cancer Institute, Lowy Cancer Research Centre, University of New South Wales, Randwick, NSW, Australia
- 2Kids Cancer Centre, Sydney Children’s Hospital, Randwick, NSW, Australia
Diffuse intrinsic pontine glioma (DIPG) is a highly aggressive pediatric brainstem tumor with a peak incidence in middle childhood and a median survival of less than 1 year. The dismal prognosis associated with DIPG has been exacerbated by the failure of over 250 clinical trials to meaningfully improve survival compared with radiotherapy, the current standard of care. The traditional practice to not biopsy DIPG led to a scarcity in available tissue samples for laboratory analysis that till recently hindered therapeutic advances. Over the past few years, the acquisition of patient derived tumor samples through biopsy and autopsy protocols has led to distinct breakthroughs in the identification of key oncogenic drivers implicated in DIPG development. Aberrations have been discovered in critical genetic drivers including histone H3, ACVR1, TP53, PDGFRA, and Myc. Mutations, previously not identified in other malignancies, highlight DIPG as a distinct biological entity. Identification of novel markers has already greatly influenced the direction of preclinical investigations and offers the exciting possibility of establishing biologically targeted therapies. This review will outline the current knowledge of the genomic landscape related to DIPG, overview preclinical investigations, and reflect how biological advances have influenced the focus of clinical trials toward targeted therapies.
Introduction
Diffuse intrinsic pontine glioma (DIPG) is a pediatric brainstem glioma that originates in the ventral pons, accounts for 75–80% of brainstem tumors in children and has a peak incidence in middle childhood (1–3). Histopathologically, DIPG have been classified as grade II–IV gliomas, namely, diffuse astrocytoma, anaplastic astrocytoma, or glioblastoma (GBM) (4). Under new recently defined WHO classification most would now be defined as diffuse midline gliomas with histone H3K27M mutation, as described below. Patients typically present with a neurological triad of cranial nerve deficits, ataxia, and long tract signs that have manifested over a short clinical history of less than 3 months (1, 5). Diagnosis is ascertained from clinical signs accompanied by the presence of characteristic radiological appearances (3, 5).
Due to the anatomical location of the tumor within the brainstem, they are unable to be resected, and in many centers are not biopsied. Palliative radiotherapy remains the sole standard therapy offered to patients albeit providing only transient improvements to neurological and radiological function (1, 5, 6). Chemotherapy as a neoadjuvant (7), combination (8–15), adjuvant (8–12, 14), and radiosensitizing (15, 16) agent has been extensively explored in over 250 clinical trials. Disappointedly, none of these studies have produced benefit meaningfully superior to radiotherapy, with the median OS ranging from 9 to 12 months (8, 12, 13, 16) and 1-year survival rates of 27–61% (12, 14, 16). The role of MRI in diagnosis, in addition to the concern that tissue biopsy may lead to increased morbidity, has till recently meant that tissue samples have rarely been available for preclinical research, thus hampering biologically driven therapeutic advances (3).
The recent establishment of stereotactic biopsy and autopsy protocols has allowed for acquisition of primary tumor samples that have subsequently facilitated extensive genomic profiling and crucial breakthroughs in key oncogenic drivers (17). Understanding the mutational processes underlying DIPG is of vital importance to identifying critical oncogenic pathways and defining high-frequency mutations with potential therapeutic relevance (18). The identification of biological markers with established targeted agents has transformed the design and direction being undertaken in preclinical and clinical trials with encouraging results already being discovered (19–21). This review will highlight the current state of knowledge of DIPG tumor biology and underlying genomic processes. It will subsequently outline how this understanding is beginning to guide research in both preclinical models and in clinical trials of novel targeted agents.
The Genomic Landscape of DIPG
Describing the genomic landscape underpinning DIPG tumorigenesis is vital to characterizing key oncogenic pathways and high-frequency mutations that represent actionable targets (18, 22). Pathway analysis of protein and mRNA profiles suggests that DIPG is a unique type of glioma while sharing some biological similarities with supratentorial high-grade gliomas (HGG) such as GBM (18, 23). Despite the highly malignant nature of DIPG, only a limited number of somatic mutations are yet to be implicated as disease defining. The identification of mutations such as histone H3, ACVR1, TP53, PDGFRA, PIK3CA, and MYC highlight, important somatic events associated with tumor evolution (18, 24, 25) (Table 1). However, intra- and inter-tumor heterogeneity has been observed within these disease-defining markers thereby reflecting the inherent barriers toward engendering effective therapeutic strategies (24). Additional somatic aberrations implicated in DIPG include gains in chromosomes 1q (H3F3A), 2q, 8q, and 9q, as well as repeated losses of chromosomes 11p, 17p13.1, 14q, 18p, and 22q (26–29). The discovery of histone mutations, present in up to 80% of DIPG tumors, has revolutionized our understanding of DIPG biology and introduces the potential for redefining clinical and therapeutic management (23, 30, 31). The histone mutation H3K27M is caused by the conversion of a lysine to methionine at residue 27, inducing unique gain-of-function mechanisms that lead to the loss of histone trimethylation (H3K27me3). Reduced H3K27me3 inhibits polycomb repressive complex 2 and promotes abnormal epigenetic silencing (26, 30). Functional analysis has highlighted the role of H3K27M as contributing to abnormal cell-cycle control, inhibition of autophagy and potentially augmenting tumor resistance to radiotherapy (23). However, the precise role of H3K27M in tumor initiation remains undefined as it is not sufficient on its own for tumorigenesis in vivo (32). However, the combination of H3K27M with additional mutational events, such as altered cell-cycle regulatory genes (TP53/PPM1D) and growth factor related pathways (ACVR1/PI3KR1), synergistically enhances tumorigenesis and together is thought to be early transformational event in DIPG (26, 32, 33). In vitro and in vivo models of developmentally relevant neural stem cells with combined histone H3.3, p53, and PDGFR mutations demonstrate increased tumor formation compared to non-mutated counterparts (32).
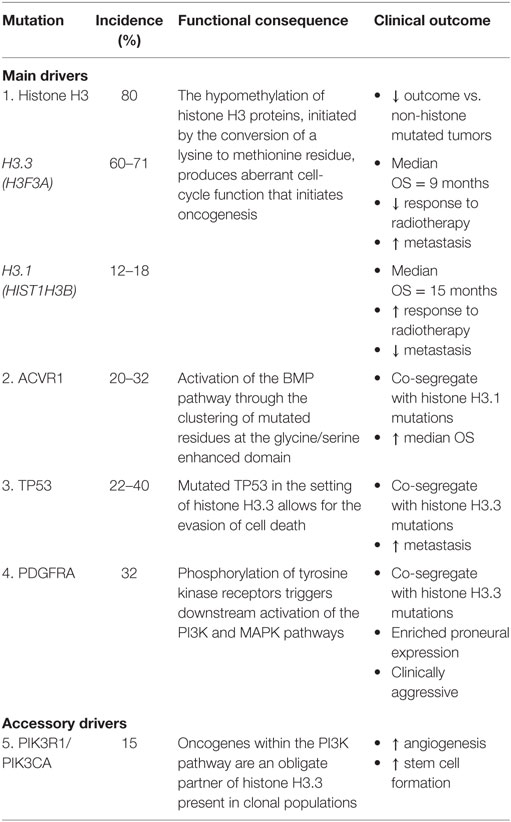
Table 1. Major genomic mutations identified in diffuse intrinsic pontine glioma.
Histone mutations are subclassified into H3.1 or H3.3 variants, encoded by the HIST1H3B and H3F3A genes, respectively, and include additional novel mutants in HIST2H3C and a lysine to isoleucine substitution (26, 30). H3.1 K27M mutations are exclusively linked to DIPG whereas H3.3 mutations in K27M and G34R/V (a glycine substitution of arginine or valine at position 34) are implicated in midline and supratentorial lateral tumors GBM, respectively (26, 34). The clinicopathological variation existing between the H3.1 and H3.3 subgroups, including differing median OS, phenotype, and responses to radiation, reflects previously unrecognized links between DIPG biology and clinical outcome (26, 31). Histone H3.1 is associated with a slight improved survival benefit with a median OS of 15 months and reduced presence of metastasis, whereas histone H3.3 has a median OS of 9 months and an inferior response to radiation therapy (17). Overall, tumors containing the presence of any H3 mutation are associated with poorer outcomes compared with non-histone mutated tumors (19, 26).
ACVR1 mutations have been identified in up to 32% of DIPG tumors, co-segregate with H3.1 mutations, and have been linked to increased median OS (18, 35, 36). Mutated residues cluster around the inhibitory glycine/serine enhanced domain or ATP-binding region and shift the kinase into an active conformation, subsequently leading to BMP pathway activation (18, 35). Somatic mutations in ACVR1 are almost exclusively limited to DIPG with reports by the Catalogue of Somatic Mutations in Cancer highlighting ACVR1 variants as present in only 0.3% of all tumor types (35). Indeed, this would seem highly suggestive of ACVR1 as potential oncogenic driver of tumorigenesis (18). However, germline mutations in ACRV1, including R206H, are present in patients with fibrodysplasia ossificans progressiva who do not proceed to develop DIPG, or other malignancies (35, 37). Nonetheless, as ACVR1 mutations facilitate early tumor propagation in conjunction with other molecular aberrations, they represent novel targets for future therapies (18).
TP53 mutations, corresponding to the 17p13.1 locus, have been identified in 22–40% of tumor samples and frequently occur in the setting of PDGFR amplification (25, 27, 35). TP53 mutations and allelic loss have been reported at comparable rates in both histone H3.3 mutation and wild-type subsets (38). TP53 and to a lesser extent PPM1D mutations represent obligate partners of H3.3 K27M that promote malignancy in DIPG (22). This partnership has specifically been described to evade cell death and senescence by possibly allowing H3.3 K27M to influence epigenetic regulation (22).
Recurring focal gains in receptor tyrosine kinases (RTKs) and regulatory cell-cycle genes in addition to phospho-mammalian target of rapamycin (mTOR) immunopositivity have been reported in DIPG and may also represent valid therapeutic targets (28, 29). PDGFRA is the most commonly observed amplification, present in approximately 32% of tumors, and highlights an important focal gain implicated in the RTK–Ras–PI3K–Akt signaling network, active in 69% of DIPG (18, 22, 27, 29). In pediatric gliomagenesis, PDGFRA strongly promotes phosphorylation at various phosphotyrosine domains, thereby initiating downstream signaling activation of PI3K and MAPK pathways (38). PDGFRA gains and amplifications co-segregate with histone H3.3 mutations have an enriched proneural gene expression profile and are clinically aggressive regardless of histological classification (26, 27, 39).
PIK3R1 and PIK3CA, genes involved in the PI3K pathway, have been recognized as oncogenes present in grade II–IV gliomas including DIPG (24, 25, 39). Mutations in PIK3R1 have been characterized as an obligatory partner in H3.3 K27M and are reported in clonal populations of DIPG. Interestingly, alterations in PIK3CA have been found in subclonal DIPG populations and are regarded as an accessory driver. While PIK3CA mutations are not crucial to DIPG development, they provide an important angiogenic advantage and promote cancer cell stemness (22). While the exact function of PIK3CA in DIPG in clonal and subclonal tumor evolution remains undefined, spatial tumor conservation of PIK3CA further supports the therapeutic advantage of targeting the RTK–PI3K–MAPK pathway (16).
MYC and MYCN aberrations are present in DIPG and constitute transcriptional regulators that specifically enhance gene expression across the whole genome (18). MYCN amplification is associated with hypermethylation, increased histological grade, and chromothripsis at chromosome 2p. The aberrant functionality of these transcription factors further highlights the critical impact of epigenetics within the DIPG mutational landscape (22). Additionally, G1 checkpoint regulators CCND1, 2, and 3, CDK4, and CDK6 have been identified as amplified in DIPG, indicating abnormalities in cell-cycle regulation (24).
In vivo models have highlighted anatomical and temporal associations between neural precursor cells of the pons and DIPG cells (22). Upregulation of sonic hedgehog (SHh), induces hyperplasia of the ventral pons and may cause precursor cell populations to undergo tumor transformation (22, 23). However, dysregulation of SHh alone cannot induce oncogenesis. Homeobox and HLH genes are associated with brainstem tumors and have a suspected role in reprogramming embryonic signaling organizers during tumor development (27).
Preclinical and Clinical Development of Targeted Therapies for DIPG
Revolutionary developments in our understanding of the biological processes underlying DIPG have led to the establishment of targeted therapeutic approaches with the aim of clinical translation (19–21, 35). The identification of histone mutations as disease-defining events has led to the investigation of epigenetic modifiers as potential therapies. Recently, panobinostat, an approved multi-histone deacetylase inhibitor, has been shown to have potent anti-DIPG activity (19, 40). Mechanism of action studies showed that panobinostat increased trimethylation and acetylation levels of H3K27M. Panobinostat has been shown to have antitumor efficacy on H3K27M expressing cells in vitro, reducing both cell proliferation and viability, and potent activity in some in vivo models, but not in others (19, 40). This may relate to a narrow therapeutic index, with dose limiting toxicities seen at the concentrations required to have an antitumor effect (40). A phase I trial exploring the use of panobinostat as a single agent in DIPG is currently underway in North America (http://ClinicalTrials.gov Identifier: NCT02717455).
GSKJ4, a H3K27 demethylase inhibitor, has also been shown to increase H3K27me3 in H3K27M expressing cells at inhibitory concentrations of 1.3–3.0 µM (20) and has demonstrated synergy with panobinostat (19).
An emphasis on the aberrant activation of the RTK–PI3K–Akt pathway in DIPG has led to the investigation of therapies targeting this signaling network (25). In vitro use of dasatinib, a multi-tyrosine kinase inhibitor, has demonstrated reduced tumor proliferation due to the down regulation of PDGFRA activity and subsequent damaging of cell-cycle progression at G1 (21). The use of temsirolimus, an agent which inhibits mTOR which is a downstream protein of PI3K (41), has been explored in DIPG (42). A phase I study of the concurrent administration of temsirolimus and perifosine, an Akt inhibitor, in pediatric patients with solid tumors including DIPG has tested the concept of dual targeting of the PI3K–Akt–mTOR pathway and shown it to be safe and feasible (42). Currently, an open phase I study is examining the use of temsirolimus with vorinostat and radiation therapy in patients with newly diagnosed and progressive DIPG (http://ClinicalTrials.gov Identifier: NCT02420613). Another trial has been recently opened (BIOMEDE) that is stratifying patients to different RTK–PI3K–mTOR pathway inhibitors based on expression of EGFR and/or loss of PTEN established following stereotactic biopsy. Patients are assigned, and in some case randomized to different treatment arms with erlotinib, dasatinib, and everolimus (http://ClinicalTrials.gov Identifier: NCT02233049).
The use of PD-03332991 (PD), a CDK4/6 inhibitor, has been explored in preclinical models of DIPG where in vivo activity was demonstrated in combination with radiotherapy. PD was shown to have greater efficacy in DIPG tumor cells that exhibited Ink4a-ARF loss caused by cytostatic effects of halting progression through G0/G1 (43). Inhibition of WEE1 kinase, also expressed in DIPG, has been explored as a radiosensitizer and was demonstrated to have antitumor effects in vitro, but no activity in vivo (44). The use of temozolomide (TMZ), associated with MGMT inactivation and prolonged survival in adult GBM patients, has been investigated in DIPG patients but has failed to yield any therapeutic advantage (9, 10, 45, 46). The inefficacy of TMZ in DIPG patients may result from the poor association of K27M expressing tumor cells with MGMT methylation, present in only 3% of tissue samples (47). Thus, this disparity highlights the importance of biological rationale for driving clinical decision making (48). A recently closed multi-center phase II clinical study has implemented the use of biopsy and molecularly aided determination of treatment with TMZ and or erlotinib based on MGMT and EGFR status (http://ClinicalTrials.gov Identifier: NCT01182350). The trial has completed recruitment with results expected after completion of adequate patient follow-up.
One of the challenges around the development of effective therapies for DIPG relates to the impermeability of the blood–brain barrier (BBB). In fact, the BBB appears to be even more important in DIPG than other brain tumors, as DIPG exhibits reduced BBB permeability when compared to their cortical HGG counterparts (49). This has prompted the exploration of alternative novel delivery methods, such as the use of nanoparticles or distribution of chemotherapeutics via convection-enhanced delivery (CED) (19, 50). In the recent years, various approaches have been tested to improve delivery of therapies into brain tumors, such as, polymer/metal based nanoparticles and cell-mediated delivery methods (51). One exciting new delivery method has focused on the use of bacterial cell-derived vehicles to transport chemotherapeutic agents across the BBB (52). These vehicles are conjugated with bi-specific antibodies that can recognize polysaccharide moieties in the bacterial cell wall and EGFR moiety on the other side. Earlier studies have demonstrated by immunohistochemistry that DIPG express high levels of EGFR and reported this to be independent of gene amplification or mutations (28, 53). Preclinical experiments conducted in canine brain cancer models have shown that these EGFR-targeted vehicles loaded with doxorubicin exhibited highly significant tumor regression (54). A phase I study (ECREST) is currently open using mitoxantrone loaded vehicles in patients with solid and CNS tumors (including DIPG) that demonstrate EGFR expression (http://ClinicalTrials.gov Identifier: NCT02687386).
Convection-enhanced delivery represents a novel therapy modality that allows direct administration to local anatomical structures in order to reduce systemic toxicity and to bypass the BBB (40, 50). Administration of the chemotherapeutic agent carmustine indicated enhanced extension of survival in an orthotopic animal of DIPG (55). Small clinical studies and case reports of CED used in DIPG patients have suggested that it is safe and feasible. Anderson et al. reported on the treatment of two children with topotecan administered by CED. Both patients experienced neurological deficits following placement of bilateral catheters. While MRI findings exhibited a reduction in tumor size, the treatment was unable to prolong survival (56). Another study reported on a robotic navigated catheter implantation procedure to direct treatment with carboplatin. The procedure was well tolerated with an objective response initially seen on MRI. However, the patient died due to progression in regions beyond drug distribution (57). An additional study has been performed by the same research group treating a larger cohort of eight DIPG patients with CED of carboplatin. The investigators used multiple catheter implantations that were robotically directed and MRI guided. The patients experienced some neurological side effects following the first treatment but symptoms resolved before subsequent infusions. Preliminary results show that three of eight patients have survived beyond 15 months while seven of eight patients remained alive after a short follow-up interval (58). While these data suggest that the procedure is well tolerated, more advanced data are required to assess patient benefit. CED is also being employed to deliver novel therapies. For example, a currently open phase I trial is exploring the administration of a radioactively labeled antibody known as 124I-8H9 for DIPG patients (http://ClinicalTrials.gov Identifier: NCT01502917). 124I-8H9 is a chimeric toxin with demonstrated specificity toward B7-H3, a membrane protein that has been recognized as tumor selective in DIPG (50, 59). Conjugation of 124I to the anchoring antibody will allow the therapeutic effects of the radionucleotide against glioma cells to extend beyond B7-H3 tumor expressing cells (59). While the feasibility of CED has been established, ongoing studies are helping to improve the instrumentation to determine optimal flow levels and maximize safety and efficacy (50, 56).
The failure of conventional cytotoxic therapies in DIPG has prompted the exploration of alternative therapeutic strategies, such as immunotherapies, that target glioma-associated antigens (GAAs), which are preferentially expressed by tumor cells (60, 61). Immunohistochemical (IHC) evaluation of IL-13Rα2, EphA2, and survivin proteins, three GAAs previously recognized in adult gliomas, has demonstrated expression of at least one GAA in 87% of DIPG samples (60). A small phase I trial evaluating subcutaneous administration of IL-13Rα2, EphA2, and survivin peptide-based vaccinations was well tolerated with preliminary indications of immunologic activity and clinical response (61). The targeting of multiple GAAs is therapeutically advantageous to overcome mixed GAA expression patterns among tumor samples and combat possible immunoediting that may occur within subclonal populations (60, 61). An additional phase I trial is currently exploring the delivery of IL13-PE38QQR, a recombinant mutated Pseudomonas aeruginosa toxin, via CED for pediatric patients with DIPG and HGG (50, 62). The presence of IL-13Rα2 alone has been recognized in 61–67% of DIPG tumors and is virtually non-detectable in normal brain tissue, therefore making it a suitable candidate for targeted immunotherapy (60, 62). Future trials will likely require pretreatment biopsies to identify biomarkers by IHC in order to appropriately stratify patients based on the expression of GAAs (62).
Conclusion
The lack of progress of over three decades of clinical trials means that DIPG remains an almost universally fatal pediatric tumor. Improved access to tumor samples for preclinical investigations has led to substantial breakthroughs and the identification of important genomic mutations responsible for tumorigenesis. Moreover, the significant progress toward uncovering targetable mutations has already vastly transformed the preclinical and clinical landscape. Further elucidation of the pathways involved in the growth and development of DIPG will improve our understanding of the biological landscape and by extension provide rationales for novel treatment protocols.
Author Contributions
DL, MT, and DZ contributed to the initial and subsequent drafts of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This publication was supported by grants from National Health and Medical Research Council (NHMRC) of Australia (1065117, 1085411), Cure Brain Cancer Foundation, The Cure Starts Now, and The Kids Cancer Alliance.
References
1. Lewis J, Lucraft H, Gholkar A. UKCCSG study of accelerated radiotherapy for paediatric brain stem gliomas. Int J Radiat Oncol Biol Phys (1997) 30(5):5.
2. Fisher PG, Breiter SN, Carson BS, Wharam MD, Williams JA, Weingart JD, et al. A clinicopathologic reappraisal of brain stem tumor classification. Identification of pilocytic astrocytoma and fibrillary astrocytoma as distinct entities. Cancer (2000) 89(7):1569–76. doi: 10.1002/1097-0142(20001001)89:7<1569::AID-CNCR22>3.0.CO;2-0
3. Warren KE. Diffuse intrinsic pontine glioma: poised for progress. Front Oncol (2012) 2:205. doi:10.3389/fonc.2012.00205
4. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol (2016) 131(6):803–20. doi:10.1007/s00401-016-1545-1
5. Hankinson TC, Patibandla MR, Green A, Hemenway M, Foreman N, Handler M, et al. Hypofractionated radiotherapy for children with diffuse intrinsic pontine gliomas. Pediatr Blood Cancer (2016) 63(4):716–8. doi:10.1002/pbc.25836
6. Janssens GO, Jansen MH, Lauwers SJ, Nowak PJ, Oldenburger FR, Bouffet E, et al. Hypofractionation vs conventional radiation therapy for newly diagnosed diffuse intrinsic pontine glioma: a matched-cohort analysis. Int J Radiat Oncol Biol Phys (2013) 85(2):315–20. doi:10.1016/j.ijrobp.2012.04.006
7. Jennings MT, Sposto R, Boyett JM, Vezina LG, Holmes E, Berger MS, et al. Preradiation chemotherapy in primary high-risk brainstem tumors: phase II study CCG-9941 of the children’s cancer group. J Clin Oncol (2002) 20(16):3431–7. doi:10.1200/JCO.2002.04.109
8. Hummel TR, Salloum R, Drissi R, Kumar S, Sobo M, Goldman S, et al. A pilot study of bevacizumab-based therapy in patients with newly diagnosed high-grade gliomas and diffuse intrinsic pontine gliomas. J Neurooncol (2016) 127(1):53–61. doi:10.1007/s11060-015-2008-6
9. Cohen KJ, Heideman RL, Zhou T, Holmes EJ, Lavey RS, Bouffet E, et al. Temozolomide in the treatment of children with newly diagnosed diffuse intrinsic pontine gliomas: a report from the Children’s Oncology Group. Neuro Oncol (2011) 13(4):410–6. doi:10.1093/neuonc/noq205
10. Bailey S, Howman A, Wheatley K, Wherton D, Boota N, Pizer B, et al. Diffuse intrinsic pontine glioma treated with prolonged temozolomide and radiotherapy – results of a United Kingdom phase II trial (CNS 2007 04). Eur J Cancer (2013) 49(18):3856–62. doi:10.1016/j.ejca.2013.08.006
11. Zaky W, Wellner M, Brown RJ, Bluml S, Finlay JL, Dhall G. Treatment of children with diffuse intrinsic pontine gliomas with chemoradiotherapy followed by a combination of temozolomide, irinotecan, and bevacizumab. Pediatr Hematol Oncol (2013) 30(7):623–32. doi:10.3109/08880018.2013.829895
12. Korones DN, Fisher PG, Kretschmar C, Zhou T, Chen Z, Kepner J, et al. Treatment of children with diffuse intrinsic brain stem glioma with radiotherapy, vincristine and oral VP-16: a Children’s Oncology Group phase II study. Pediatr Blood Cancer (2008) 50(2):227–30. doi:10.1002/pbc.21154
13. Packer RJ, Krailo M, Mehta M, Warren K, Allen J, Jakacki R, et al. Phase 1 study of concurrent RMP-7 and carboplatin with radiotherapy for children with newly diagnosed brainstem gliomas. Cancer (2005) 104(6):1281–7. doi:10.1002/cncr.21403
14. Pollack IF, Stewart CF, Kocak M, Poussaint TY, Broniscer A, Banerjee A, et al. A phase II study of gefitinib and irradiation in children with newly diagnosed brainstem gliomas: a report from the pediatric brain tumor consortium. Neuro Oncol (2011) 13(3):290–7. doi:10.1093/neuonc/noq199
15. Allen J, Siffert J, Donahue B, Nirenberg A, Jakacki R, Robertson P, et al. A phase I/II study of carboplatin combined with hyperfractionated radiotherapy for brainstem gliomas. Cancer (1999) 86(6):1064–9. doi:10.1002/(SICI)1097-0142(19990915)86:6<1064::AID-CNCR24>3.0.CO;2-1
16. Porkholm M, Valanne L, Lonnqvist T, Holm S, Lannering B, Riikonen P, et al. Radiation therapy and concurrent topotecan followed by maintenance triple anti-angiogenic therapy with thalidomide, etoposide, and celecoxib for pediatric diffuse intrinsic pontine glioma. Pediatr Blood Cancer (2014) 61(9):1603–9. doi:10.1002/pbc.25045
17. Jones C, Baker SJ. Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat Rev Cancer (2014) 14(10): 651–61. doi:10.1038/nrc3811
18. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet (2014) 46(5):444–50. doi:10.1038/ng.2938
19. Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily MA, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med (2015) 21(7):827. doi:10.1038/nm0715-827a
20. Hashizume R, Andor N, Ihara Y, Lerner R, Gan H, Chen X, et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med (2014) 20(12):1394–6. doi:10.1038/nm.3716
21. Truffaux N, Philippe C, Paulsson J, Andreiuolo F, Guerrini-Rousseau L, Cornilleau G, et al. Preclinical evaluation of dasatinib alone and in combination with cabozantinib for the treatment of diffuse intrinsic pontine glioma. Neuro Oncol (2015) 17(7):953–64. doi:10.1093/neuonc/nou330
22. Monje M, Mitra SS, Freret ME, Raveh TB, Kim J, Masek M, et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci U S A (2011) 108(11):4453–8. doi:10.1073/pnas.1101657108
23. Saratsis AM, Kambhampati M, Snyder K, Yadavilli S, Devaney JM, Harmon B, et al. Comparative multidimensional molecular analyses of pediatric diffuse intrinsic pontine glioma reveals distinct molecular subtypes. Acta Neuropathol (2014) 127(6):881–95. doi:10.1007/s00401-013-1218-2
24. Hoffman LM, DeWire M, Ryall S, Buczkowicz P, Leach J, Miles L, et al. Spatial genomic heterogeneity in diffuse intrinsic pontine and midline high-grade glioma: implications for diagnostic biopsy and targeted therapeutics. Acta Neuropathol Commun (2016) 4(1):1. doi:10.1186/s40478-015-0269-0
25. Grill J, Puget S, Andreiuolo F, Philippe C, MacConaill L, Kieran MW. Critical oncogenic mutations in newly diagnosed pediatric diffuse intrinsic pontine glioma. Pediatr Blood Cancer (2012) 58(4):489–91. doi:10.1002/pbc.24060
26. Castel D, Philippe C, Calmon R, Le Dret L, Truffaux N, Boddaert N, et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol (2015) 130(6):815–27. doi:10.1007/s00401-015-1478-0
27. Puget S, Philippe C, Bax DA, Job B, Varlet P, Junier MP, et al. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS One (2012) 7(2):e30313. doi:10.1371/journal.pone.0030313
28. Zarghooni M, Bartels U, Lee E, Buczkowicz P, Morrison A, Huang A, et al. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J Clin Oncol (2010) 28(8):1337–44. doi:10.1200/JCO.2009.25.5463
29. Paugh BS, Broniscer A, Qu C, Miller CP, Zhang J, Tatevossian RG, et al. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J Clin Oncol (2011) 29(30):3999–4006. doi:10.1200/JCO.2011.35.5677
30. Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science (2013) 340(6134):857–61. doi:10.1126/science.1232245
31. Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol (2012) 124(3):439–47. doi:10.1007/s00401-012-0998-0
32. Funato K, Major T, Lewis PW, Allis CD, Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science (2014) 346(6216):1529–33. doi:10.1126/science.1253799
33. Nikbakht H, Panditharatna E, Mikael LG, Li R, Gayden T, Osmond M, et al. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat Commun (2016) 7:11185. doi:10.1038/ncomms11185
34. Liu X, McEachron TA, Schwartzentruber J, Wu G. Histone H3 mutations in pediatric brain tumors. Cold Spring Harb Perspect Biol (2014) 6(4):a018689. doi:10.1101/cshperspect.a018689
35. Taylor KR, Mackay A, Truffaux N, Butterfield YS, Morozova O, Philippe C, et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet (2014) 46(5):457–61. doi:10.1038/ng.2925
36. Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet (2014) 46(5):451–6. doi:10.1038/ng.2936
37. Shen Q, Little SC, Xu M, Haupt J, Ast C, Katagiri T, et al. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J Clin Invest (2009) 119(11):3462–72. doi:10.1172/JCI37412
38. Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J, et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res (2013) 73(20):6219–29. doi:10.1158/0008-5472.CAN-13-1491
39. Buczkowicz P, Bartels U, Bouffet E, Becher O, Hawkins C. Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications. Acta Neuropathol (2014) 128(4):573–81. doi:10.1007/s00401-014-1319-6
40. Hennika T, Hu G, Olaciregui NG, Barton KL, Ehteda A, Chitranjan A, et al. Pre-clinical study of panobinostat in xenograft and genetically engineered murine diffuse intrinsic pontine glioma models. PLoS One (2017) 12(1):e0169485. doi:10.1371/journal.pone.0169485
41. Holmes D. PI3K pathway inhibitors approach junction. Nat Rev Drug Discov (2011) 10(8):563–4. doi:10.1038/nrd3527
42. Becher OJ, Gilheeney SW, Khakoo Y, Lyden DC, Haque S, De Braganca KC, et al. A phase I study of perifosine with temsirolimus for recurrent pediatric solid tumors. Pediatr Blood Cancer (2016) 1–9. doi:10.1002/pbc.26409
43. Barton KL, Misuraca K, Cordero F, Dobrikova E, Min HD, Gromeier M, et al. PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma. PLoS One (2013) 8(10):e77639. doi:10.1371/journal.pone.0077639
44. Caretti V, Hiddingh L, Lagerweij T, Schellen P, Koken PW, Hulleman E, et al. WEE1 kinase inhibition enhances the radiation response of diffuse intrinsic pontine gliomas. Mol Cancer Ther (2013) 12(2):141–50. doi:10.1158/1535-7163.MCT-12-0735
45. Tolcher AW, Gerson SL, Denis L, Geyer C, Hammond LA, Patnaik A, et al. Marked inactivation of O6-alkylguanine-DNA alkyltransferase activity with protracted temozolomide schedules. Br J Cancer (2003) 88(7):1004–11. doi:10.1038/sj.bjc.6600827
46. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352(10):987–96. doi:10.1056/NEJMoa043330
47. Korshunov A, Ryzhova M, Hovestadt V, Bender S, Sturm D, Capper D, et al. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol (2015) 129(5):669–78. doi:10.1007/s00401-015-1405-4
48. Puget S, Beccaria K, Blauwblomme T, Roujeau T, James S, Grill J, et al. Biopsy in a series of 130 pediatric diffuse intrinsic pontine gliomas. Childs Nerv Syst (2015) 31(10):1773–80. doi:10.1007/s00381-015-2832-1
49. Subashi E, Cordero FJ, Halvorson KG, Qi Y, Nouls JC, Becher OJ, et al. Tumor location, but not H3.3K27M, significantly influences the blood-brain-barrier permeability in a genetic mouse model of pediatric high-grade glioma. J Neurooncol (2016) 126(2):243–51. doi:10.1007/s11060-015-1969-9
50. Zhou Z, Singh R, Souweidane M. Convection-enhanced delivery for diffuse intrinsic pontine glioma treatment. Curr Neuropharmacol (2017) 15:116–28. doi:10.2174/1570159X14666160614093615
51. Zhang F, Xu CL, Liu CM. Drug delivery strategies to enhance the permeability of the blood-brain barrier for treatment of glioma. Drug Des Devel Ther (2015) 9:2089–100. doi:10.2147/DDDT.S79592
52. MacDiarmid JA, Mugridge NB, Weiss JC, Phillips L, Burn AL, Paulin RP, et al. Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell (2007) 11(5):431–45. doi:10.1016/j.ccr.2007.03.012
53. Ballester LY, Wang Z, Shandilya S, Miettinen M, Burger PC, Eberhart CG, et al. Morphologic characteristics and immunohistochemical profile of diffuse intrinsic pontine gliomas. Am J Surg Pathol (2013) 37(9):1357–64. doi:10.1097/PAS.0b013e318294e817
54. MacDiarmid JA, Langova V, Bailey D, Pattison ST, Pattison SL, Christensen N, et al. Targeted doxorubicin delivery to brain tumors via minicells: proof of principle using dogs with spontaneously occurring tumors as a model. PLoS One (2016) 11(4):e0151832. doi:10.1371/journal.pone.0151832
55. Sewing AC, Caretti V, Lagerweij T, Schellen P, Jansen MH, van Vuurden DG, et al. Convection enhanced delivery of carmustine to the murine brainstem: a feasibility study. J Neurosci Methods (2014) 238:88–94. doi:10.1016/j.jneumeth.2014.09.020
56. Anderson RC, Kennedy B, Yanes CL, Garvin J, Needle M, Canoll P, et al. Convection-enhanced delivery of topotecan into diffuse intrinsic brainstem tumors in children. J Neurosurg Pediatr (2013) 11(3):289–95. doi:10.3171/2012.10.PEDS12142
57. Barua NU, Lowis SP, Woolley M, O’Sullivan S, Harrison R, Gill SS. Robot-guided convection-enhanced delivery of carboplatin for advanced brainstem glioma. Acta Neurochir (Wien) (2013) 155(8):1459–65. doi:10.1007/s00701-013-1700-6
58. Singleton WGB, Barua NU, Morgan J, Bienemann AS, Killick-Cole CL, Asby DJ, et al., editors. Multi-Catheter Intermittent Convection-Enhanced Delivery of Carboplatin as a Treatment for Diffuse Intrinsic Pontine Glioma (DIPG): Pre-Clinical Rationale and Early Clinical Experience. MULTI-. 17th International Symposium on Pediatric Neuro-Oncology (ISPNO). Liverpool, UK: Neurooncology (2016).
59. Zhou Z, Luther N, Ibrahim GM, Hawkins C, Vibhakar R, Handler MH, et al. B7-H3, a potential therapeutic target, is expressed in diffuse intrinsic pontine glioma. J Neurooncol (2013) 111(3):257–64. doi:10.1007/s11060-012-1021-2
60. Okada H, Low KL, Kohanbash G, McDonald HA, Hamilton RL, Pollack IF. Expression of glioma-associated antigens in pediatric brain stem and non-brain stem gliomas. J Neurooncol (2008) 88(3):245–50. doi:10.1007/s11060-008-9566-9
61. Pollack IF, Jakacki RI, Butterfield LH, Hamilton RL, Panigrahy A, Potter DM, et al. Antigen-specific immune responses and clinical outcome after vaccination with glioma-associated antigen peptides and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in children with newly diagnosed malignant brainstem and nonbrainstem gliomas. J Clin Oncol (2014) 32(19):2050–8. doi:10.1200/JCO.2013.54.0526
Keywords: diffuse intrinsic pontine glioma, pediatric brainstem gliomas, histone H3K27M, ACVR1, PDGFR, preclinical studies, targeted therapies
Citation: Lapin DH, Tsoli M and Ziegler DS (2017) Genomic Insights into Diffuse Intrinsic Pontine Glioma. Front. Oncol. 7:57. doi: 10.3389/fonc.2017.00057
Received: 12 December 2016; Accepted: 14 March 2017;
Published: 28 March 2017
Edited by:
Katherine Warren, National Cancer Institute, USAReviewed by:
Matthias Karajannis, Memorial Sloan Kettering Cancer Center, USAJaume Mora, Hospital Sant Joan de Déu Barcelona, Spain
Copyright: © 2017 Lapin, Tsoli and Ziegler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David S. Ziegler, ZC56aWVnbGVyQHVuc3cuZWR1LmF1