 Roberta Alfieri1
Roberta Alfieri1 Elisa Giovannetti
Elisa Giovannetti Mara Bonelli
Mara Bonelli Andrea Cavazzoni
Andrea Cavazzoni- 1Department of Medicine and Surgery, University of Parma, Parma, Italy
- 2Department of Medical Oncology, VU University Medical Center, Amsterdam, Netherlands
- 3Cancer Pharmacology Laboratory, AIRC Start Up Unit, University of Pisa, Pisa, Italy
Deep genetic studies revealed that phosphatase and tensin homolog (PTEN) mutations or loss of expression are not early events in cancer development but characterize tumor progression and invasion. Loss of PTEN function causes a full activation of the prosurvival phosphoinositide 3-kinase (PI3K)/AKT/mTOR pathway, but the treatment with specific inhibitors of PI3K/AKT/mTOR did not produce the expected results. One of the alternative targets of PTEN is the focal adhesion kinase (FAK) kinase, mainly involved in the control of cancer cell spread. The connection between PTEN and FAK has been demonstrated in different tumor types, with reduced PTEN activity often correlated with increased expression and phosphorylation of FAK. FAK inhibition may thus represent a promising strategy, and some clinical trials are testing FAK inhibitors alone or combined with other agents in a number of solid tumors. However, only few preclinical and clinical data described the effects of the combination of PI3K/AKT/mTOR and FAK inhibitors. Increasing knowledge on the PTEN/FAK connection could confirm PTEN as a good prognostic marker for a combination strategy based on concomitant inhibition of PI3K/AKT and FAK signaling, in advanced metastatic malignancies with altered or reduced PTEN expression.
Introduction
The development of new approaches directed to evaluate molecular alterations in cancer cells represents a key goal to better define the intrinsic characteristics of tumors and to explore molecular targets that are commonly deregulated in cancer cells. Novel insights regarding the second most commonly mutated tumor suppressor gene phosphatase and tensin homolog (PTEN) have been reported: loss of PTEN has been documented in a variety of solid tumors, including lung (1), prostate (2), colorectal (3), and brain (4) cancers. Typically, loss of function of PTEN causes aberrant activation of the phosphoinositide 3-kinase (PI3K)/AKT pathway, associated with increased malignant transformation and progression.
Recently, a series of new targets have been associated with PTEN phosphatase activity. PTEN is indeed a critical protein in the intracellular compartment, and controls, by its phosphatase activity, several important proteins, including signal transducers and activators of transcription (STAT) (5), Jun N-terminal kinase (JNK) (6), extracellular signal regulated kinase (ERK1/2) (7), AKT (8), and focal adhesion kinase (FAK) (9).
In this review, we will focus on the correlation between PTEN and one of its kinase targets, FAK.
Regulation and Role of PTEN
Phosphatase and tensin homolog is located on chromosome 10q23 and encodes for a lipid phosphatase that converts PtdIns(3,4,5)P3 to PtdIns(4,5)P2 (10), by inhibiting 3-phosphoinositide-dependent kinase (PDK1) and consequently protein kinase B (AKT) activation. Moreover, PTEN exerts a protein phosphatase activity toward different protein substrates (5–9). PTEN is a member of the type-I protein tyrosine phosphatase family, composed by 403 amino acids organized in five functional domains: a PtdIns (4,5)P2 binding domain, a phosphatase domain, a C2 domain involved in targeting protein to cell membrane, a carboxyterminal region and a protein interaction (PDZ) domain (11). PTEN loss causes accumulation of PtdIns (3,4,5)P3, with recruitment and activation of AKT mediated by PDK1; PDK1 phosphorylates AKT at the critical residue Thr308 (12), whereas the Ser473 residue of AKT is a substrate of mammalian target of rapamycin 2 complex (mTORC2) (13).
Phosphatase and tensin homolog function can be compromised by different mechanisms including genetic alterations, transcriptional repression, microRNA regulation, promoter hypermethylation, and post-translational modifications.
Germ line mutations are present in 60–80% of patients with hamartoma tumor syndromes and Cowden disease (14). Somatic alterations, such as mutations, insertions and deletions have been found throughout the full gene and are not specific for a peculiar cancer type. PTEN inactivation is also a consequence of promoter hypermethylation (15) and reduction of transcription: Sal-like protein 4 and SNAIL can bind PTEN promoter mediating its repression through the interaction with the Mi-2/NuRD complex (16), and competition with p53 (17), respectively.
At post-transcriptional level, PTEN is downregulated by small non-coding RNAs. Some miRNAs (a class of 20–25 nucleotide non-coding RNA) that partially match with the 3′UTR region of PTEN mRNA are able to abrogate PTEN expression: in particular, mir-21, mir-22, mir-25A, and mir-200 family member (miR-200a and miR-200b) stably reduce PTEN protein levels (18–20). More recently, miR-93 has also been reported to reduce PTEN expression in non-small cell lung cancer (NSCLC) by directly targeting PTEN mRNA (21).
Finally, PTEN can be regulated at post-translational level. PTEN phosphorylation at Ser380, Ser385, Thr382, and Thr383 reduces its phosphatase activity by moving PTEN from the intracellular membrane level to the cytosol (22, 23). This event prevents its ability to interact with PtdIns(3,4,5)P3, which is located to the internal side of the plasma membrane. Reduction of PTEN activity is also observed after phosphorylation at residues Ser362 and Thr366 by glycogen synthase kinase 3-beta (GSK3-β), an enzyme activated by constitutive stimulation of the PI3K/AKT pathway (24).
Inactivation or loss of PTEN expression has been reported in different solid tumors including NSCLC, breast, colorectal, endometrial, and ovarian cancers (Table 1): in NSCLCs, altered PTEN expression, has been detected in 8.2–59% and in 2.1–46% of squamous cell lung cancer (SCC, squamous hystotype) and adenocarcinoma (AD) hystotype, respectively (25). Breast cancer tissues show significant reduction of PTEN expression compared to non-tumor tissues, and a meta-analysis (26) reported a positive correlation between PTEN loss and later TNM stage, evidencing that PTEN loss is not an early event, but it is associated with tumor progression. Patients with colorectal cancer display increasing inactivation of PTEN expression, as a consequence of promoter hypermethylation, decreased DNA copy number, and a general reduction of protein expression. All these alterations are associated with increased stage of disease; 20% of PTEN loss has been detected in stage I, while up to 58.9% has been found in stage IV (27). In prostate cancer, a high frequency of PTEN loss (between 16 and 41% of tumor samples) (2) has been described, being homozygous deletion the most frequent event. The clinical trials considering PTEN alteration among the inclusion criteria in different solid tumors are reported in Table 1.
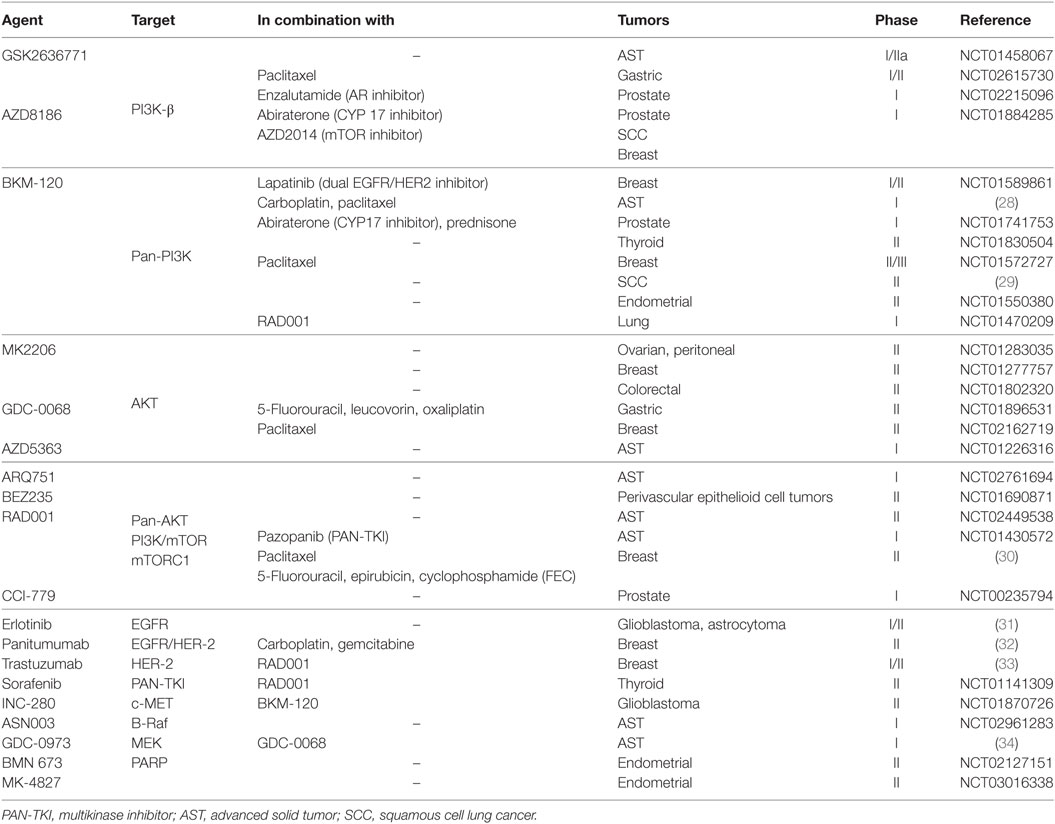
Table 1. Clinical trials with PTEN alteration as inclusion criteria (http://clinicaltrials.gov/).
As a regulator of the PI3K-AKT-mTOR pathway, PTEN controls many intracellular processes promoting cancer growth, cell metabolism, angiogenesis and cell motility (25, 35). In particular, it regulates the plasma membrane expression of GLUT1 either by its lipidic phosphatase activity on PtdIns(3,4,5)P3 or by direct dephosphorylation of AKT (8). In addition, PTEN decreases the levels of pyruvate kinase isozyme M2 and 6-phosphofructo-1-kinase/fructose-2,6-biphosphatase isoform 3, both involved in the glycolytic process, thus exerting an anti-Warburg effect (36).
Loss of PTEN also influences cell polarization and motility, e.g., increased PtdIns (4,5)P2 level in normal cells attracts CDC42 to the apical membrane establishing cellular polarization after binding with PAR-6/aPKC complex (37). Loss of PTEN prevents the acquisition of this epithelial characteristic, increasing the possibility of cells to undergo epithelial to mesenchymal transition (EMT) (38). Similar to the effects obtained by complete loss of PTEN, two point inactivating mutations (C124S and G129E) cause acquisition of a highly malignant phenotype, whereas single allelic loss produces an intermediate effect (39).
Recently, the loss of PTEN has been described as a mechanism of resistance to immune checkpoint blockade in solid tumors. In melanoma models the loss of PTEN induces immunoresistance prompting VEGF and other immunosuppressive cytokines expression (40); similarly, PTEN-null prostate tumors suppress antitumor immune response by activating the JAK2-STAT3 pathway (41). The correlation between PTEN-loss and immunotherapy resistance has been also confirmed by evaluating PTEN expression in patients with metastatic melanoma treated with anti–PD-1 antibodies: PTEN expression is generally related to a greater reduction of tumor size in respect to PTEN-negative tumors (40). Accordingly, in a case report of uterine leiomyosarcoma, immunotherapy resistance is associated with biallelic PTEN-loss (42).
Regulation and Role of FAK Expression
Focal adhesion kinase is a key regulator of the focal adhesion complex, which controls various intracellular processes such as cell motility (43), invasion (44) cell growth, and survival (45, 46), by regulating signals from integrin-mediated cell–extracellular matrix (ECM) connection (47) and from membrane receptors with kinase activity (48). Moreover the role of FAK as a regulator of tumor-infiltrating immunosuppressive cells (TILs) has been recently demonstrated in a mouse model of human pancreatic ductal AD (49). FAK maps on chromosomal region 8q24.3 and gene amplification has been discovered in gastric cancer (50), whereas increased mRNA levels have been detected in ovarian, head and neck and metastatic breast carcinoma (51). Currently, a part from some polymorphisms revealed by DNA sequencing, no mutational activation has been detected in this gene. As a kinase, it is composed by different domains: a FERM domain (protein band 4.1-ezrin-radixin-moesin homology domain), a central kinase domain, three prolin-rich regions, and the focal adhesion targeting domain (51).
After FAK homodimerization, as a consequence of clustering of integrin receptors by cellular adhesion to ECM, an autophosphorylation reaction occurs at Tyr397 residue (52). With the recruitment of the SRC-family kinases at Tyr397, FAK is then phosphorylated at the residue Tyr925 of the kinase domain. The FAK-SRC complex activates a plethora of different substrates, including paxillin, Shc, p120RAsGDP, and PLCy (53). The main targets of FAK, p130Cas and paxillin, are involved in migration, by modulating the expression and activation of members of Rho family GTPases; after FAK activation, p130Cas and paxillin promote focal adhesion complex formation, maturation, and turnover (54). FAK involvement in invasiveness might be at least in part explained by the fact that FAK promotes expression and activation of metalloprotease-9 (MMP9), as reported in breast (55) and lung (56) cancers. MMP9 mediates matrix degradation, causing tumor cell spreading. In agreement with the acquisition of a malignant phenotype, FAK activation also controls EMT process. As reported for breast cancer (57), the insulin growth factor receptor 1 (IGF1R)-driven migration and invasion are predominantly mediated by FAK activation; moreover, transforming growth factor β, a well-known cytokine involved in EMT induction, causes FAK activation, triggers delocalization of E-cadherin from the plasma membrane, and increases levels of mesenchymal markers (58). Interestingly, some evidences concerning the connection between FAK and β-catenin are emerging: in colorectal cancer, a high expression of FAK activates the Wnt/β-catenin signaling by phosphorylating GSKβY216 (59) with subsequent stabilization of β-catenin protein. Recent data enforced the rationale of targeting FAK in a cohort of lung AD patients with KRAS mutation, which is typically detectable in the percentage of 15–25% of NSCLC patients. KRAS promotes tumor growth, by constitutive activation of RhoA, which in turn activates FAK, causing the acquisition of a more aggressive phenotype that could be reverted by pharmacological inhibition of FAK kinase (60).
The lack of NF2 tumor suppressor gene, coding for Merlin protein, is a frequent event in mesothelioma. Merlin negatively regulates some targets including Src-FAK complex (61), and its deficiency could represent a potential biomarker for the treatment with FAK inhibitors: specific FAK inhibition of Merlin-negative mesothelioma cancer cells causes a relevant reduction in cell viability, suggesting the potential benefit of this approach in NF2 negative mesothelioma cancer patients (62). FAK targeting strategies in different solid tumors are reported in Table 2.
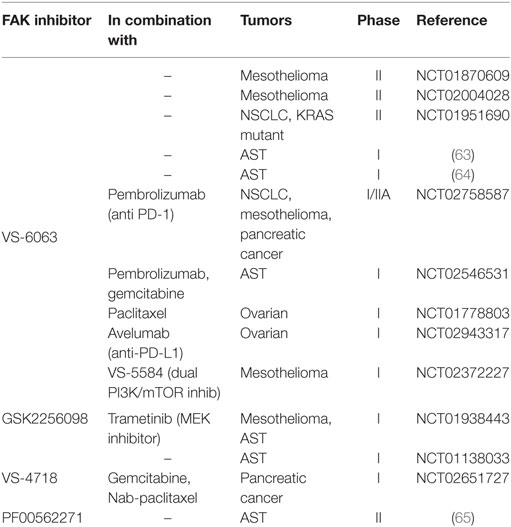
Table 2. Clinical trials involving specific focal adhesion kinase (FAK) inhibitors (http://clinicaltrials.gov/).
Several evidences demonstrated that FAK could be directly activated by PI3K/AKT pathway. In particular, the AKT-1 isoform is implicated in FAK activation, by directly phosphorylating three critical Serine residues (Ser517/601/695). These events promote FAKY397 autophosphorylation and thus enzyme activation. Notably, the cooperation between FAK and AKT is bidirectional: depletion of FAK reduces AKTS473 phosphorylation, evincing that a mutual cooperation could be responsible for cancer progression, at least in certain tumor types (66). Interestingly, novel data provided further insights on FAK and AKT correlation (67): in a preclinical model of ovarian cancer, the protein tyrosine phosphatase non-receptor type 12 (PTPN12) dephosphorylates FAK, shutting down the migratory properties. PTPN12 is downregulated after Her-2 activation, with reduction of PTEN protein and constitutive activation of PI3K/AKT pathway. In particular, AKT phosphorylates and inactivates GSK3 protein that in turn dephosphorylates FAK at the inhibitory target Ser722, promoting cell spread.
Connection Between PTEN and FAK Signaling Pathways
The connection between PTEN and FAK has been explored for few years but only recently this has emerged as a potential druggable signaling axis.
The interaction between PTEN and FAK is clearly demonstrated in MKN28 cells, a preclinical model of gastric cancer (68) which shows a significant reduction of FAK expression and activation when overexpressing PTEN, with inhibition of cell motility, invasiveness and in vivo tumor growth.
A negative correlation between PTEN and FAK is also detected in patients with multiple myeloma (MM) (69) and urologic malignancies (70) in advanced stage. Moreover, it has been reported that Notch1 controls PTEN expression in hepatocellular carcinoma: the abrogation of Notch1 by siRNA approach increases PTEN expression and phosphorylation, with inhibition of both AKT and FAK activity (71).
Loss of PTEN function, a frequent event reported in endometrial cancer (72), with percentage near to 60%, makes this mutation an attractive molecular target in this type of cancer. In a preclinical study, cell lines with wild-type or mutated PTEN show different sensitivity to the specific FAK inhibitor GSK2256028 given alone or combined with chemotherapy. In an in vivo model, the combined treatment shows a dramatic effect only in cells carrying mutated PTEN, with apoptosis induction, reduced cell growth and neo-angiogenesis. To further sustain this correlation, a cohort of 91 patients was analyzed for PTEN and FAK expression and phosphorylation at Tyr397. Patients with poor prognosis show reduced PTEN levels associated with increased FAK expression and Tyr397 phosphorylation, confirming that PTEN could be considered a prognostic biomarker and suggesting its role for predicting the response to anti-FAK targeted agents (73).
Phosphatase and tensin homolog mutations are detected in 15–25% of patients with acute lymphoblastic leukemia (T-ALL). However, while in vitro experiments demonstrated that the pharmacological inhibition of PI3K/AKT/mTOR pathway in PTEN null-T-ALL cells dramatically reduces cell growth and viability, the efficacy of this treatment is less pronounced in an in vivo system. As previously reported for MM, gastric and endometrial cancers, PTEN null-T-ALL cells display increased FAK activity. Genetic deletion or pharmacological inhibition of FAK, associated with inhibition of PI3K pathway, produce a more significant effect than single monotherapy (74) in this cellular model.
In our recent article (75), we analyzed the correlation between PTEN loss and FAK activation in squamous NSCLC patients. As previously reported (76), PTEN levels are reduced in 70 and 77% of patients with lung squamous (SCC) or AD histology, respectively. This event, observed in metastatic patients, has been confirmed by our analysis in a cohort of 51 patients with SCC, at different stage (I–IV). In particular, we demonstrated that loss of PTEN expression is not an early event, at least in SCC, but it is associated with cancer progression and acquisition of high malignancy: in fact, the majority of metastatic stage IV patients presents low PTEN expression associated with increased FAKY397 phosphorylation. Cell clones, expressing low level of PTEN, demonstrated increased FAKY397 and AKTS473 phosphorylation, high miR-21 levels, and the acquisition of a mesenchymal phenotype.
The acquisition of mesenchymal markers caused by PTEN abrogation can be a consequence of increased onco-miR levels: in particular, the miR-130 family, highly expressed in bladder cancer, shuts down PTEN protein, with increase of migration and invasiveness, MMP9 production, AKT, and FAK phosphorylation (77). Similarly, miR-301a is upregulated in tumor tissue from melanoma patients (78), and modulation of this miRNA in cancer cells revealed lower PTEN expression, with activation of both AKT and FAK.
Notwithstanding this connection is reported for different tumor types, it cannot be extended to all cancers: for example, esophageal squamous cell carcinoma cells depleted for PTEN (79) show increased malignancy, but the mechanism of metastatic spread was not correlated with FAK activation.
Finally, in most studies the connection between PTEN and FAK is based on the phosphatase activity of PTEN, but recent data (80) demonstrated that FAK could in turn phosphorylate PTENY336, increasing protein stability and phosphatase activity. This result shows a novel mechanism of PTEN regulation providing new insights for the role of FAK.
Targeting PTEN/FAK Signaling
Taking into account the relevance of genetic PTEN aberrations such as loss or reduced expression, point mutations or post-translational events, and the critical role of PTEN enzyme as lipid and protein phosphatase, it is of particular interest to approach new targeted strategies based on PTEN status evaluation (Table 1). PTEN, as tumor suppressor gene, cannot be considered a classical oncogenic driver such as EGFR or EML4/ALK in NSCLC patients or BRAF in melanoma patients, but it can constitutively regulate some intracellular oncogenic signaling pathways, such as the PI3K/AKT/mTOR axis. A high number of specific or pan inhibitors of this pathway have been developed, and novel molecules are continuously synthesized, but to date, no specific therapies are approved for patients with PTEN deficiency. This could be a consequence of the pleiotropic effects exerted by PTEN depletion; as reported, abrogation of this protein causes not only activation of PI3K/AKT pathway, but increases JNK, MAPK, STAT, and FAK activity, making problematic targeting this multiple molecular alterations. Moreover, PTEN alterations are often acquired during the last phase of tumor progression; in particular loss of PTEN can be associated with the development of metastatic disease and it can be considered a marker of advanced tumor stage.
Another important aspect emerging from preclinical studies is the pivotal role of PI3K-β as an enzyme often overexpressed in the presence of PTEN loss (2, 81). This enzyme can be targeted by specific (GSK-2636771, AZD8186 or TGX-221), pan (GDC-0032, XL-147, GDC-0941, NVP-BKM120, PX-866, BAY 80-6946), or dual PI3K/mTOR (NVP-BEZ235, XL-765) inhibitors. In a preclinical in vivo model characterized by PTEN loss, the selective inhibition of the β subunit of PI3K caused tumor shrinkage (82). Recently, a correlation between PTEN loss and high PI3K-β was reported in a cohort of NSCLC patients, with prevalence of squamous histology (83). Currently, four clinical trials (Table 1) (NCT01458067, NCT02215096, NCT02615730, and NCT01884285) are evaluating the role of specific PI3K-β inhibitors in patients with altered PTEN expression.
In view of the fact that PTEN is one of the most important regulators of FAK kinase, loss of PTEN can be regarded as a good biomarker for a combined therapy with PI3K and FAK inhibitors. Currently, this strategy (74) provides a better treatment for PTEN-null T-ALL cells compared to the inhibition of PI3K/AKT alone. The same approach has been tested by our group in a preclinical model on SCC with stable PTEN abrogation, demonstrating that a simultaneous inhibition of both targets induces synergistic reduction of cell growth and invasiveness (75).
This new combination deserves further investigations in the clinical practice. However, to date there is only a phase I clinical trial (NCT02372227) exploring the safety of the PI3K/AKT/mTOR inhibitor VS-5584 combined with the FAK inhibitor VS-6063 in patients with relapsed malignant mesothelioma (Table 2). Remarkably, since PTEN loss is typically both in advanced primary tumors and in metastatic sites, the development of a combined strategy directed to inhibiting PI3K aberrant activation caused by PTEN loss and FAK kinase could represent a promising strategy to target metastases, which are the leading cause of death in cancer patients.
Author Contributions
RA wrote the manuscript. EG revised the manuscript. MB performed bibliographic research. AC performed bibliographic research, planned, and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Stjernstrom A, Karlsson C, Fernandez OJ, Soderkvist P, Karlsson MG, Thunell LK. Alterations of INPP4B, PIK3CA and pAkt of the PI3K pathway are associated with squamous cell carcinoma of the lung. Cancer Med (2014) 3(2):337–48. doi:10.1002/cam4.191
2. Wise HM, Hermida MA, Leslie NR. Prostate cancer, PI3K, PTEN and prognosis. Clin Sci (Lond) (2017) 131(3):197–210. doi:10.1042/CS20160026
3. Perrone F, Lampis A, Orsenigo M, Di Bartolomeo M, Gevorgyan A, Losa M, et al. PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann Oncol (2009) 20(1):84–90. doi:10.1093/annonc/mdn541
4. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science (1997) 275(5308):1943–7. doi:10.1126/science.275.5308.1943
5. Wu R, Sun S, Steinberg BM. Requirement of STAT3 activation for differentiation of mucosal stratified squamous epithelium. Mol Med (2003) 9(3–4):77–84. doi:10.2119/2003-00001.Wu
6. Vivanco I, Palaskas N, Tran C, Finn SP, Getz G, Kennedy NJ, et al. Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer Cell (2007) 11(6):555–69. doi:10.1016/j.ccr.2007.04.021
7. Senol N, Ceyhan BM, Ersoy IH, Senol A, Acarturk G, Sutcu R. Aspirin increases NMDA receptor subunit 2A concentrations in rat hippocampus. J Recept Signal Transduct Res (2012) 32(1):17–21. doi:10.3109/10799893.2011.641975
8. Phadngam S, Castiglioni A, Ferraresi A, Morani F, Follo C, Isidoro C.PTEN dephosphorylates AKT to prevent the expression of GLUT1 on plasmamembrane and to limit glucose consumption in cancer cells. Oncotarget (2016) 7(51):84999–5020. doi:10.18632/oncotarget.13113
9. Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science (1998) 280(5369):1614–7. doi:10.1126/science.280.5369.1614
10. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem (1998) 273(22):13375–8. doi:10.1074/jbc.273.22.13375
11. Wang X, Jiang X. PTEN: a default gate-keeping tumor suppressor with a versatile tail. Cell Res (2008) 18(8):807–16. doi:10.1038/cr.2008.83
12. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell (2007) 129(7):1261–74. doi:10.1016/j.cell.2007.06.009
13. Bozulic L, Hemmings BA. PIKKing on PKB: regulation of PKB activity by phosphorylation. Curr Opin Cell Biol (2009) 21(2):256–61. doi:10.1016/j.ceb.2009.02.002
15. Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer (2011) 11(4):289–301. doi:10.1038/nrc3037
16. Lu J, Jeong HW, Kong N, Yang Y, Carroll J, Luo HR, et al. Stem cell factor SALL4 represses the transcriptions of PTEN and SALL1 through an epigenetic repressor complex. PLoS One (2009) 4(5):e5577. doi:10.1371/journal.pone.0005577
17. Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, et al. Regulation of PTEN transcription by p53. Mol Cell (2001) 8(2):317–25. doi:10.1016/S1097-2765(01)00323-9
18. Huse JT, Brennan C, Hambardzumyan D, Wee B, Pena J, Rouhanifard SH, et al. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev (2009) 23(11):1327–37. doi:10.1101/gad.1777409
19. Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology (2007) 133(2):647–58. doi:10.1053/j.gastro.2007.05.022
20. Yoneyama K, Ishibashi O, Kawase R, Kurose K, Takeshita T. miR-200a, miR-200b and miR-429 are onco-miRs that target the PTEN gene in endometrioid endometrial carcinoma. Anticancer Res (2015) 35(3):1401–10.
21. Li C, Lyu J, Meng QH. MiR-93 promotes tumorigenesis and metastasis of non-small cell lung cancer cells by activating the PI3K/Akt pathway via inhibition of LKB1/PTEN/CDKN1A. J Cancer (2017) 8(5):870–9. doi:10.7150/jca.17958
22. Vazquez F, Grossman SR, Takahashi Y, Rokas MV, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J Biol Chem (2001) 276(52):48627–30. doi:10.1074/jbc.C100556200
23. Liang K, Esteva FJ, Albarracin C, Stemke-Hale K, Lu Y, Bianchini G, et al. Recombinant human erythropoietin antagonizes trastuzumab treatment of breast cancer cells via Jak2-mediated Src activation and PTEN inactivation. Cancer Cell (2010) 18(5):423–35. doi:10.1016/j.ccr.2010.10.025
24. Jang HD, Noh JY, Shin JH, Lin JJ, Lee SY. PTEN regulation by the Akt/GSK-3beta axis during RANKL signaling. Bone (2013) 55(1):126–31. doi:10.1016/j.bone.2013.02.005
25. Fumarola C, Bonelli MA, Petronini PG, Alfieri RR. Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochem Pharmacol (2014) 90(3):197–207. doi:10.1016/j.bcp.2014.05.011
26. Li S, Shen Y, Wang M, Yang J, Lv M, Li P, et al. Loss of PTEN expression in breast cancer: association with clinicopathological characteristics and prognosis. Oncotarget (2017) 8(19):32043–54. doi:10.18632/oncotarget.16761
27. Lin PC, Lin JK, Lin HH, Lan YT, Lin CC, Yang SH, et al. A comprehensive analysis of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) loss in colorectal cancer. World J Surg Oncol (2015) 13:186. doi:10.1186/s12957-015-0601-y
28. Smyth LM, Monson KR, Jhaveri K, Drilon A, Li BT, Abida W, et al. A phase 1b dose expansion study of the pan-class I PI3K inhibitor buparlisib (BKM120) plus carboplatin and paclitaxel in PTEN deficient tumors and with dose intensified carboplatin and paclitaxel. Invest New Drugs (2017). doi:10.1007/s10637-017-0445-0
29. Vansteenkiste JF, Canon JL, Braud FD, Grossi F, De Pas T, Gray JE, et al. Safety and efficacy of buparlisib (BKM120) in patients with PI3K pathway-activated non-small cell lung cancer: results from the phase II BASALT-1 study. J Thorac Oncol (2015) 10(9):1319–27. doi:10.1097/JTO.0000000000000607
30. Gonzalez-Angulo AM, Akcakanat A, Liu S, Green MC, Murray JL, Chen H, et al. Open-label randomized clinical trial of standard neoadjuvant chemotherapy with paclitaxel followed by FEC versus the combination of paclitaxel and everolimus followed by FEC in women with triple receptor-negative breast cancerdagger. Ann Oncol (2014) 25(6):1122–7. doi:10.1093/annonc/mdu124
31. Kesavabhotla K, Schlaff CD, Shin B, Mubita L, Kaplan R, Tsiouris AJ, et al. Phase I/II study of oral erlotinib for treatment of relapsed/refractory glioblastoma multiforme and anaplastic astrocytoma. J Exp Ther Oncol (2012) 10(1):71–81.
32. Yardley DA, Ward PJ, Daniel BR, Eakle JF, Lamar RE, Lane CM, et al. Panitumumab, gemcitabine, and carboplatin as treatment for women with metastatic triple-negative breast cancer: a Sarah Cannon Research Institute phase II trial. Clin Breast Cancer (2016) 16(5):349–55. doi:10.1016/j.clbc.2016.05.006
33. Morrow PK, Wulf GM, Ensor J, Booser DJ, Moore JA, Flores PR, et al. Phase I/II study of trastuzumab in combination with everolimus (RAD001) in patients with HER2-overexpressing metastatic breast cancer who progressed on trastuzumab-based therapy. J Clin Oncol (2011) 29(23):3126–32. doi:10.1200/JCO.2010.32.2321
34. Nannini MA, Hong R, Lee BB, Arrazate A, Kassees R, Lin J, et al. Combined targeting of Akt and MEK with the Akt inhibitor GDC-0068 and MEK inhibitor GDC-0973 demonstrates synergistic anti-tumor effects. Cancer Res (2012) 72(8 Suppl):873. doi:10.1158/1538-7445.AM2012-873
35. Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol (2012) 13(5):283–96. doi:10.1038/nrm3330
36. Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, de Boer VC, et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell (2012) 149(1):49–62. doi:10.1016/j.cell.2012.02.030
37. Martin-Belmonte F, Gassama A, Datta A, Yu W, Rescher U, Gerke V, et al. PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through CDC42. Cell (2007) 128(2):383–97. doi:10.1016/j.cell.2006.11.051
38. Mulholland DJ, Kobayashi N, Ruscetti M, Zhi A, Tran LM, Huang J, et al. PTEN loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res (2012) 72(7):1878–89. doi:10.1158/0008-5472.CAN-11-3132
39. Papa A, Wan L, Bonora M, Salmena L, Song MS, Hobbs RM, et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell (2014) 157(3):595–610. doi:10.1016/j.cell.2014.03.027
40. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov (2016) 6(2):202–16. doi:10.1158/2159-8290.CD-15-0283
41. Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M, et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep (2014) 9(1):75–89. doi:10.1016/j.celrep.2014.08.044
42. George S, Miao D, Demetri GD, Adeegbe D, Rodig SJ, Shukla S, et al. Loss of PTEN is associated with resistance to anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity (2017) 46(2):197–204. doi:10.1016/j.immuni.2017.02.001
43. Tomar A, Lim ST, Lim Y, Schlaepfer DD. A FAK-p120RasGAP-p190RhoGAP complex regulates polarity in migrating cells. J Cell Sci (2009) 122(Pt 11):1852–62. doi:10.1242/jcs.046870
44. Schneider GB, Kurago Z, Zaharias R, Gruman LM, Schaller MD, Hendrix MJ. Elevated focal adhesion kinase expression facilitates oral tumor cell invasion. Cancer (2002) 95(12):2508–15. doi:10.1002/cncr.10992
45. Hochgrafe F, Zhang L, O’Toole SA, Browne BC, Pinese M, Porta Cubas A, et al. Tyrosine phosphorylation profiling reveals the signaling network characteristics of basal breast cancer cells. Cancer Res (2010) 70(22):9391–401. doi:10.1158/0008-5472.CAN-10-0911
46. Gilmore AP, Romer LH. Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Mol Biol Cell (1996) 7(8):1209–24. doi:10.1091/mbc.7.8.1209
47. Schaller MD. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J Cell Sci (2010) 123(Pt 7):1007–13. doi:10.1242/jcs.045112
48. Long W, Yi P, Amazit L, LaMarca HL, Ashcroft F, Kumar R, et al. SRC-3Delta4 mediates the interaction of EGFR with FAK to promote cell migration. Mol Cell (2010) 37(3):321–32. doi:10.1016/j.molcel.2010.01.004
49. Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med (2016) 22(8):851–60. doi:10.1038/nm.4123
50. Park JH, Lee BL, Yoon J, Kim J, Kim MA, Yang HK, et al. Focal adhesion kinase (FAK) gene amplification and its clinical implications in gastric cancer. Hum Pathol (2010) 41(12):1664–73. doi:10.1016/j.humpath.2010.06.004
51. Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer (2014) 14(9):598–610. doi:10.1038/nrc3792
52. Brami-Cherrier K, Gervasi N, Arsenieva D, Walkiewicz K, Boutterin MC, Ortega A, et al. FAK dimerization controls its kinase-dependent functions at focal adhesions. EMBO J (2014) 33(4):356–70. doi:10.1002/embj.201386399
53. Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol (1999) 71(3–4):435–78. doi:10.1016/S0079-6107(98)00052-2
54. Brown MC, Turner CE. Paxillin: adapting to change. Physiol Rev (2004) 84(4):1315–39. doi:10.1152/physrev.00002.2004
55. Mitra SK, Lim ST, Chi A, Schlaepfer DD. Intrinsic focal adhesion kinase activity controls orthotopic breast carcinoma metastasis via the regulation of urokinase plasminogen activator expression in a syngeneic tumor model. Oncogene (2006) 25(32):4429–40. doi:10.1038/sj.onc.1209482
56. Meng XN, Jin Y, Yu Y, Bai J, Liu GY, Zhu J, et al. Characterisation of fibronectin-mediated FAK signalling pathways in lung cancer cell migration and invasion. Br J Cancer (2009) 101(2):327–34. doi:10.1038/sj.bjc.6605154
57. Taliaferro-Smith L, Oberlick E, Liu T, McGlothen T, Alcaide T, Tobin R, et al. FAK activation is required for IGF1R-mediated regulation of EMT, migration, and invasion in mesenchymal triple negative breast cancer cells. Oncotarget (2015) 6(7):4757–72. doi:10.18632/oncotarget.3023
58. Cicchini C, Laudadio I, Citarella F, Corazzari M, Steindler C, Conigliaro A, et al. TGFbeta-induced EMT requires focal adhesion kinase (FAK) signaling. Exp Cell Res (2008) 314(1):143–52. doi:10.1016/j.yexcr.2007.09.005
59. Gao C, Chen G, Kuan SF, Zhang DH, Schlaepfer DD, Hu J. FAK/PYK2 promotes the Wnt/beta-catenin pathway and intestinal tumorigenesis by phosphorylating GSK3beta. Elife (2015) 4. doi:10.7554/eLife.10072
60. Konstantinidou G, Ramadori G, Torti F, Kangasniemi K, Ramirez RE, Cai Y, et al. RHOA-FAK is a required signaling axis for the maintenance of KRAS-driven lung adenocarcinomas. Cancer Discov (2013) 3(4):444–57. doi:10.1158/2159-8290.CD-12-0388
61. Li W, Cooper J, Karajannis MA, Giancotti FG. Merlin: a tumour suppressor with functions at the cell cortex and in the nucleus. EMBO Rep (2012) 13(3):204–15. doi:10.1038/embor.2012.11
62. Shapiro IM, Kolev VN, Vidal CM, Kadariya Y, Ring JE, Wright Q, et al. Merlin deficiency predicts FAK inhibitor sensitivity: a synthetic lethal relationship. Sci Transl Med (2014) 6(237):237ra68. doi:10.1126/scitranslmed.3008639
63. Shimizu T, Fukuoka K, Takeda M, Iwasa T, Yoshida T, Horobin J, et al. A first-in-Asian phase 1 study to evaluate safety, pharmacokinetics and clinical activity of VS-6063, a focal adhesion kinase (FAK) inhibitor in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol (2016) 77(5):997–1003. doi:10.1007/s00280-016-3010-1
64. Jones SF, Siu LL, Bendell JC, Cleary JM, Razak AR, Infante JR, et al. A phase I study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Invest New Drugs (2015) 33(5):1100–7. doi:10.1007/s10637-015-0282-y
65. Infante JR, Camidge DR, Mileshkin LR, Chen EX, Hicks RJ, Rischin D, et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J Clin Oncol (2012) 30(13):1527–33. doi:10.1200/JCO.2011.38.9346
66. Wang S, Basson MD. Akt directly regulates focal adhesion kinase through association and serine phosphorylation: implication for pressure-induced colon cancer metastasis. Am J Physiol Cell Physiol (2011) 300(3):C657–70. doi:10.1152/ajpcell.00377.2010
67. Villa-Moruzzi E. PTPN12 controls PTEN and the AKT signalling to FAK and HER2 in migrating ovarian cancer cells. Mol Cell Biochem (2013) 375(1–2):151–7. doi:10.1007/s11010-012-1537-y
68. Zhang LL, Liu J, Lei S, Zhang J, Zhou W, Yu HG. PTEN inhibits the invasion and metastasis of gastric cancer via downregulation of FAK expression. Cell Signal (2014) 26(5):1011–20. doi:10.1016/j.cellsig.2014.01.025
69. Wang SY, Hao HL, Deng K, Li Y, Cheng ZY, Lv C, et al. Expression levels of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) and focal adhesion kinase in patients with multiple myeloma and their relationship to clinical stage and extramedullary infiltration. Leuk Lymphoma (2012) 53(6):1162–8. doi:10.3109/10428194.2011.647311
70. Haber T, Jockel E, Roos FC, Junker K, Prawitt D, Hampel C, et al. Bone metastasis in renal cell carcinoma is preprogrammed in the primary tumor and caused by AKT and integrin alpha5 signaling. J Urol (2015) 194(2):539–46. doi:10.1016/j.juro.2015.01.079
71. Hu YJ, Li HY, Qiu KJ, Li DC, Zhou JH, Hu YH, et al. Downregulation of Notch1 inhibits the invasion of human hepatocellular carcinoma HepG2 and MHCC97H cells through the regulation of PTEN and FAK. Int J Mol Med (2014) 34(4):1081–6. doi:10.3892/ijmm.2014.1889
72. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, et al. Integrated genomic characterization of endometrial carcinoma. Nature (2013) 497(7447):67–73. doi:10.1038/nature12113
73. Thanapprapasr D, Previs RA, Hu W, Ivan C, Armaiz-Pena GN, Dorniak PL, et al. PTEN expression as a predictor of response to focal adhesion kinase inhibition in uterine cancer. Mol Cancer Ther (2015) 14(6):1466–75. doi:10.1158/1535-7163.MCT-14-1077
74. You D, Xin J, Volk A, Wei W, Schmidt R, Scurti G, et al. FAK mediates a compensatory survival signal parallel to PI3K-AKT in PTEN-null T-ALL cells. Cell Rep (2015) 10(12):2055–68. doi:10.1016/j.celrep.2015.02.056
75. Cavazzoni A, La Monica S, Alfieri R, Ravelli A, Van Der Steen N, Sciarrillo R, et al. Enhanced efficacy of AKT and FAK kinase combined inhibition in squamous cell lung carcinomas with stable reduction in PTEN. Oncotarget (2017). doi:10.18632/oncotarget.18087
76. Beck JT, Ismail A, Tolomeo C. Targeting the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway: an emerging treatment strategy for squamous cell lung carcinoma. Cancer Treat Rev (2014) 40(8):980–9. doi:10.1016/j.ctrv.2014.06.006
77. Egawa H, Jingushi K, Hirono T, Ueda Y, Kitae K, Nakata W, et al. The miR-130 family promotes cell migration and invasion in bladder cancer through FAK and Akt phosphorylation by regulating PTEN. Sci Rep (2016) 6:20574. doi:10.1038/srep20574
78. Cui L, Li Y, Lv X, Li J, Wang X, Lei Z, et al. Expression of MicroRNA-301a and its functional roles in malignant melanoma. Cell Physiol Biochem (2016) 40(1–2):230–44. doi:10.1159/000452540
79. Sun Z, Ji N, Bi M, Zhang Z, Liu X, Wang Z. Negative expression of PTEN identifies high risk for lymphatic-related metastasis in human esophageal squamous cell carcinoma. Oncol Rep (2015) 33(6):3024–32. doi:10.3892/or.2015.3928
80. Tzenaki N, Aivaliotis M, Papakonstanti EA. Focal adhesion kinase phosphorylates the phosphatase and tensin homolog deleted on chromosome 10 under the control of p110delta phosphoinositide-3 kinase. FASEB J (2015) 29(12):4840–52. doi:10.1096/fj.15-274589
81. Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, et al. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A (2008) 105(35):13057–62. doi:10.1073/pnas.0802655105
82. Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature (2008) 454(7205):776–9. doi:10.1038/nature07091
83. Cumberbatch M, Tang X, Beran G, Eckersley S, Wang X, Ellston RP, et al. Identification of a subset of human non-small cell lung cancer patients with high PI3Kbeta and low PTEN expression, more prevalent in squamous cell carcinoma. Clin Cancer Res (2014) 20(3):595–603. doi:10.1158/1078-0432.CCR-13-1638
Keywords: phosphatase and tensin homolog, focal adhesion kinase, targeted therapy, kinase, drug combination
Citation: Alfieri R, Giovannetti E, Bonelli M and Cavazzoni A (2017) New Treatment Opportunities in Phosphatase and Tensin Homolog (PTEN)-Deficient Tumors: Focus on PTEN/Focal Adhesion Kinase Pathway. Front. Oncol. 7:170. doi: 10.3389/fonc.2017.00170
Received: 06 June 2017; Accepted: 26 July 2017;
Published: 09 August 2017
Edited by:
Barbara Zavan, University of Padua, ItalyReviewed by:
Ciro Isidoro, Università degli Studi del Piemonte Orientale, ItalyJason Roszik, University of Texas MD Anderson Cancer Center, United States
Copyright: © 2017 Alfieri, Giovannetti, Bonelli and Cavazzoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Cavazzoni, YW5kcmVhLmNhdmF6em9uaUB1bmlwci5pdA==