 Renata Lyrio Rafael Baptista1*
Renata Lyrio Rafael Baptista1* Anna Cláudia Evangelista dos Santos2
Anna Cláudia Evangelista dos Santos2 Luciana Mayumi Gutiyama3
Luciana Mayumi Gutiyama3 Cristiana Solza1
Cristiana Solza1 Ilana Renault Zalcberg3
Ilana Renault Zalcberg3
- 1Departamento de Medicina Interna/Hematologia, Hospital Universitário Pedro Ernesto, Rio de Janeiro, Brazil
- 2Programa de Genética, Instituto Nacional do Câncer, Rio de Janeiro, Brazil
- 3Divisão de Laboratórios do Centro de Transplantes de Medula Óssea (CEMO), Instituto Nacional do Câncer, Rio de Janeiro, Brazil
Although most cases of myeloid neoplasms are sporadic, a small subset has been associated with germline mutations. The 2016 revision of the World Health Organization classification included these cases in a myeloid neoplasm group with a predisposing germline mutational background. These patients must have a different management and their families should get genetic counseling. Cases identification and outline of the major known syndromes characteristics will be discussed in this text.
Introduction
Most cases of myeloid neoplasms are sporadic; however, a small subset has been associated with germline mutations (1–3). The 2016 revision of the World Health Organization (WHO) classification included a group of myeloid neoplasms—such as myelodysplastic syndrome (MDS), MDS/myeloproliferative neoplasms, and acute myeloid leukemia (AML)—with a predisposing, germline mutational background. The presence of underlying genetic alterations or predisposition syndromes is crucial for diagnosing these familial cases (4).
In familial neoplasms, mutations are present in the heterozygous condition, most commonly resulting in loss of functional alleles and subsequent haploinsufficiency, although gain-of-function mutations have also been reported (5). It seems likely, although still unknown, that progression to hematologic malignancy requires the additional acquisition of somatic mutations in bone marrow stem or progenitor cells, probably in the same genes previously affected by germline mutations.
As many genes related to familial predisposition to myeloid neoplasms were also found to be recurrently mutated in sporadic cases, investigation of familial myeloid neoplasms may further provide insights into normal and malignant hematopoiesis and pathogenic mechanisms underlying hematologic malignancies. Moreover, the presence of germline genetic alterations associated to myeloid neoplasms should not be limited to the proband: family members might be at higher risk of developing myeloid neoplasms (6, 7).
Despite efforts to identify familial cases, only a minority with germline mutation can be explained by known genetic factors. The use of next-generation gene sequencing is allowing more cases of the syndrome to be diagnosed, including those without gene mutation (8). However, it is crucial for the germline material availability from the proband and affected family members for this analysis.
Therapy-related MDS/AML seems to be associated with germline mutations in familial cancer predisposition genes. This increases the possibility of these mutations being susceptibility factors for AML development (9, 10).
Given the above clinical conditions, physicians should be trained to identify highly suspicious cases of familial neoplasms. A detailed family medical history, collected in all cases of myeloid neoplasms, especially in younger patients, must be mandatory. Close collaboration between hematologists and experienced geneticists in suspected familial cases is crucial. In this review, we will discuss the specific germline alterations associated to familial myeloid malignancies aiming to provide hematologists with diagnosis tools (Figure 1).
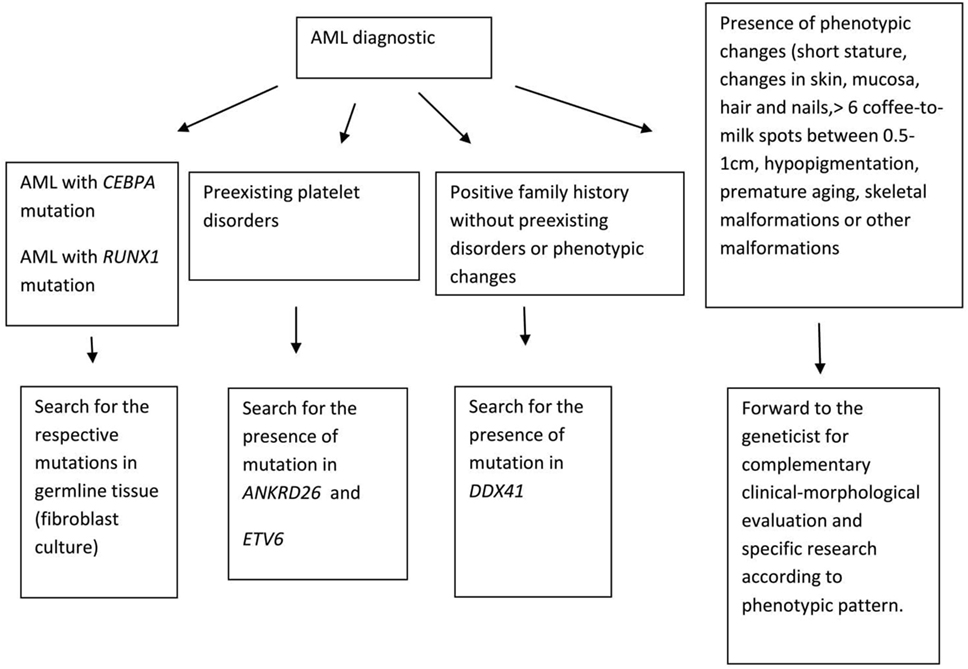
Figure 1. How to identify acute myeloid leukemia (AML) familial cases.
Myeloid Neoplasms with Germline Predisposition
The 2016 revision of the WHO classification included, in a new subset of hematological malignancies associated to germline mutations, the following conditions: (1) myeloid neoplasms with germline predisposition without a preexisting disorder or organ dysfunction, (2) myeloid neoplasms with germline predisposition and preexisting platelet disorders, and (3) myeloid neoplasms with germline predisposition and other organ dysfunction (Table 1) (4).

Table 1. Classification of myeloid neoplasms with germline predisposition (4).
Myeloid Neoplasms with Germline Predisposition without a Preexisting Disorder or Organ Dysfunction
AML with Germline CCAAT/Enhancer-Binding Protein-A (CEBPA) Mutation
The transcription factor CEBPA is allocated in 19q13.1. This gene, consisting of a single exon, is involved in myeloid differentiation. Familial AML with mutated CEBPA is an inherited autosomal dominant condition with complete or near-complete penetrance (11, 12). There is not a specific genotype–phenotype presentation. On the other hand, 10–15% of sporadic acute myeloid leukemia with normal karyotype (NK-AML) presents the somatic CEBPA mutations. Somatic, bi-allelic CEBPA mutations (CEBPAdm), found in 10–15% of NK-AML, confer a favorable prognosis, a reason why the identification of CEBPA mutation is currently incorporated in routine diagnosis (13, 14). The identification of the germline origin of CEBPA mutations in patients with CEBPAdm is recommended for discriminating between sporadic and familial cases. Family history is helpful since type or location differences between somatic and germline mutations are presently unknown.
Acute myeloid leukemia diagnosis may be difficult considering the lack of (i) specific clinical features preceding hematological history, (ii) anticipation, and (iii) genotype–phenotype correlation, thus making family history the only source of data for somatic versus familial cases distinction. Identification of familial cases may be impaired by occurrence of de novo mutations in a proband or early death of an affected parent without evident clinical AML manifestations. The replacement of the mutated allele in bone marrow can only be achieved by allogeneic stem cell transplantation from a previously tested related donor in whom the mutated allele has been excluded (15).
Finally, the recognition of familial AML with mutated CEBPA is essential since penetrance is nearly complete. Genetic counseling is key for managing these cases (16).
Myeloid Neoplasm with Germline DDX41 Mutation
DEAD/H-box helicase gene (DDX41), allocated in 5q35.3, contains 17 exons and encodes an RNA helicase protein apparently involved in RNA splicing. Its role in hematopoiesis and leukemogenesis remains unknown. Prevalence and penetrance of DDX41 mutations, as well as prognosis, are unclear (4). However, DDX41 mutations were present in 1.5% of patients with myeloid neoplasm in a cohort of 1,000 patients. Fifty percent of DDX41 mutations were germline suggesting that a germline analysis should be considered in these cases (17).
Familial AML with mutated DDX41 displays a pattern of autosomal dominant inheritance with the characteristics of other MDS/AML syndromes, including a long latency (18). Apart from the family history, there are no preceding clinical signs or symptoms suggesting increased risk for hematologic malignancy.
The majority of familial cases previously identified with this leukemia harbor a heterozygous germline frameshift mutation, DDX41 c.415_418dupGATG (p.D140Gfs*2), although missense and splice variants have also been described. Another mutation in the other DDX41 allele occurs in 50% of germline mutation carriers developing MDS or AML, thereby suggesting that DDX41 is a tumor suppressor gene (17). DDX41 may play a role in the pathogenesis of myeloid neoplasms with del(5q), since some of these deletions include DDX41 locus, leading to haploinsufficiency. The DDX41 defects in cases with del(5q) were associated with advanced disease and responsiveness to lenalidomide, a possible therapeutic intervention for otherwise poor-risk disease (19). The overall survival seems to be inferior in DDX41 mutations or deletions cases and a decreased DDX41 expression seems to be associated with worse outcomes (14).
Unfortunately, surveillance of unaffected individuals in the general population is not possible. Nevertheless, a bone marrow biopsy with cytogenetic analysis and blood count may be recommended at regular intervals to families with known DDX41 mutations or deletions and other predisposition syndromes (4).
Myeloid Neoplasms with Germline Predisposition and Preexisting Platelet Disorders
Myeloid Neoplasms with Germline RUNX1 Mutation
Runt-related transcription factor 1 (RUNX1) is a protein coding gene allocated in 21q22.12, which contains nine exons. It encodes the DNA-binding subunit of the core-binding factor transcription complex that is essential for normal hematopoiesis (20). Myeloid neoplasms with germline RUNX1 mutations result from monoallelic RUNX1 mutations occurring all along this gene, including missense, nonsense, frameshift, insertions, deletions, and, a recently reported, disrupting congenital translocation (21).
Monoallelic RUNX1 mutations carriers show a heterogeneous range of clinical manifestations: from moderate thrombocytopenia, bleeding, or myeloid neoplasm with frequent strong anticipation, to asymptomatic family members (22).
Management of patients with myeloid neoplasms and germline RUNX1 mutations depends on clinical presentation. Diagnosis of underlying germline mutations requires allogeneic stem cell transplantation as consolidation therapy provided that RUNX1 mutations are not carried by related donors (6). Management of asymptomatic RUNX1 mutation carriers is difficult because guidelines are not presently available for this recently described condition. Considering that myeloid neoplasms with germline RUNX1 mutations occur with strong anticipation, close follow-ups of the younger members of an affected family are necessary: a baseline blood count with annual checkups, and a bone marrow biopsy in the event of significant changes in peripheral blood counts (7). Finally, as RUNX1 mutations are found in 32% of sporadic AML (23, 24), translational studies might be relevant for clarifying leukemogenesis in familial platelet disorders (FDP) (23, 24).
Myeloid Neoplasm with Germline ANKRD26 Mutation
The Ankyrin repeat domain-containing protein 26 gene (ANKRD26), located in 10p12.1, contains 34 exons. Mutations affecting this gene interfere with controlling mechanisms of ANKRD26 expression, impacting upon megakaryopoieses and platelet production (25). In 9 of 20 unrelated families with autosomal dominant non-syndromic thrombocytopenia-2 (THC2), 6 different mutations were localized in the 5′ promoter region of this gene (26). The overall incidence of these hematologic malignancies was 240/100,000, with acute leukemia accounting for 167/100,000, both above the expected incidences. THC2 has been characterized by platelet dysfunction. The thrombocytopenia is moderate, the mean platelet volume is normal with the presence of aggregation defect, and the bone marrow shows dysmegakaryopoiesis (27, 28). To date, the presence of familial cases of thrombocytopenia and predisposition to myeloid malignancies are the milestones for ANKRD26 mutations diagnosis (29).
The RUNX1 or ANKRD26 mutations should be suspected among patients with thrombocytopenia with a family history of bleeding and/or MDS/AML. If the presence of the mutations is confirmed, patients and family members should be referred to genetic counseling and evaluation of risk of developing myeloid malignancies (7).
Myeloid Neoplasm with Germline ETV6 Mutation
The ETS variant gene (ETV6), located in 12p13.2, contains eight exons and is essential in the development of the embryo and hematopoietic regulation (30). ETV6 mutations also play a role in hematologic malignancies (31, 32).
The hereditary syndrome of thrombocytopenia with normal platelet size has susceptibility to the development of diverse hematologic malignances. It is transmitted as a dominant trait associated with germline and missense ETV6 mutations (33). Patients present bleeding and thrombocytopenia and are often misdiagnosed with immune thrombocytopenia (34).
Three hotspot positions were identified in ETV6 accounting for p.A369G and p.A399C, at the highly conserved ETS, and p.Pro214L, thereby affecting DNA binding. These mutations were found to deter DNA binding, alter subcellular localization, and decrease the repression of transcription in a dominant-negative pattern, thereby impairing hematopoiesis (25). Most recently, five more possibly pathogenic variants were described: p.I358M, p.A377T, p.R396G, p.Y401N, and p.Y401H (35).
The ETV6 mutations, such as RUNX1 and ANKRD26 mutations, should be suspected among patients with thrombocytopenia with a family history of bleeding and/or hematologic malignances (36). If the presence of the mutations is confirmed, the patient and family members should be referred to genetic counseling. However, until now, there are not sufficient data in literature to suggest standardized treatment guidelines for these patients and their families.
Myeloid Neoplasms with Germline Predisposition Other Organ Dysfunction
Myeloid Neoplasms with Germline GATA2 Mutation
GATA-binding protein 2 gene (GATA2), located in 3q21.3, contains seven exons and encodes a GATA transcription factor family zinc finger that is essential in normal hematopoietic (37, 38).
In affected families, GATA2 mutations were transmitted as highly penetrant autosomal dominant traits with early MDS or AML onset. Onset at early age was reported in patients with syndromic presentations (39). Several families with GATA2 mutations have been described without any distinctive phenotypic or cytogenetic abnormality (2).
Some clinical syndromes, such as Emberger, MonoMAC, congenital neutropenia, and DCML (dendritic cell, monocyte, and lymphocyte deficiency), are associated with germline GATA2 mutations (40). Emberger syndrome is associated with predisposition to MDS/AL and the presence of systemic manifestations such as primary lymphedema confined to lower extremities and genitals, lymphopenia with low CD4/CD8 count ratio, cutaneous warts, and sensorineural deafness. Emberger syndrome also seems to be associated with eight independent GATA2 variants (41). The MonoMAC syndrome is connected to MDS/AL predisposition and immunologic defects—such as immunodeficiency, monocytopenia, NK cell, B cell, and macrophage deficiencies—that lead to predisposition to atypical infections and pulmonary alveolar proteinosis (42).
Pedigree analysis showed four different GATA2 mutations: two in familial AML (p.T354M and p.T355del, both in the second zinc finger domains) (21, 43) and two other in de novo AML (p.R308P and p.A350N351ins8) (43).
Transformation to MDS/AL rapidly occurs conferring an adverse prognosis, while indication of allogeneic hematopoietic stem cell transplantation appears to be the most adequate treatment (43).
Myeloid Neoplasms Associated with Bone Marrow Failure Syndromes
Inherited bone marrow failure syndromes (IBMFS) are rare genetic disorders with characteristic hematopoietic dysfunction and ensuing cytopenias, with high risk of transformation to clonal myeloid malignancies (CMMT) including MDS, AML, or isolated clonal cytogenetic abnormalities (44–48). Hematological neoplasms may occur as the initial manifestation of IBMF; approximately 25% of Fanconi anemia patients lack the typical disease phenotype such as short stature and radial ray anomalies (49, 50). Adult and pediatric de novo CMMT are not precisely defined, although diagnosis is based on peripheral blood cell counts and types, bone marrow blasts, cellularity, cytogenetic analysis, and dysplasia presence. A widely accepted definition of IBMFS-associated CMMT is, however, not presently available. Diagnosis of pediatric MDS is based on peripheral blood counts, marrow morphologic dysplasia, and blasts (51, 52). These are valuable indicators for defining MDS. Nevertheless, their applicability to IBMFS-associated MDS in the absence of transformation has not been tested. The risk of developing CMMT in patients with IBMFS has been estimated to be 2,284-fold higher than in general population (53).
The differential risk of developing CMMT among patients with various IBMFS types has not been precisely estimated due to dearth population-based data (54). Despite IBMFS types sharing several clinical and morphological phenotypes, IBMFS genes might be involved in different pathways, a reason why mutations in different IBMFS genes might have disparate malignant effects. This provides a rationale for routine leukemia surveillance in children with Fanconi anemia and Shwachman–Diamond syndrome.
Myeloid Neoplasms Associated with Telomere Biology Disorders
Malignancies associated with telomere biology disorders result from mutations in nine different genes that induce an abnormal maintenance of telomeres leading to chromosome instability and apoptosis (55, 56). Dyskeratosis congenita (DKC), an X-linked recessive disease, is characterized by nail dystrophy, abnormal reticular skin pigmentation, and oral leukoplakia (57). The clinical presentation may vary, resulting in patients with constitutional defects while cancer and MDS predisposition are distinctive. Excessive telomere shortening in Xq28, where the X-linked gene DKC1 (Dyskerin) is located, leads to genetic instability and high cancer risk (58, 59). A high frequency of hematological malignancies is observed in DKC: approximately 200-fold for AML and 2,500-fold for MDS in relation to the normal population, a reason why the affected patient must be properly screened (60). Deleterious mutations affecting TERT (telomerase reverse transcriptase), a gene in 5p15.33, or TERC (telomerase RNA component), a gene in 3q26.2, are transmitted as autosomal dominant traits with heterogeneous phenotypes and incomplete penetrance (61). Phenotypes range from normal to severe hematological neoplasms, with variable age at onset and anticipation (62). This fact should not be ignored since children inheriting TERT/TERC mutations might present earlier clinical manifestations although their parents carrying the same mutations may not. Clinical presentation may include isolated idiopathic pulmonary fibrosis, hepatic cirrhosis, early-onset anogenital or head and neck cancer, and combinations of these features. The frequency of these associated manifestations is still unknown. These findings point out the need of TERC and TERT screening in families with more than one case of MDS/AL and/or patients with subtle blood abnormalities, failure to mobilize stem cells or clinical manifestations in other organs or systems (6).
Juvenile Myelomonocytic Leukemia (JMML) Associated with Neurofibromatosis, Noonan Syndrome, or Noonan Syndrome-Like Disorders
Juvenile myelomonocytic leukemia is an aggressive myelodysplastic/myeloproliferative malignancy. Most JMML cases are associated with somatic gain-of-function mutations in components of the RAS/MAPK signal transduction pathway (63). A minority of cases arise in young children with Noonan syndrome, a genetic disorder with increased RAS/MAPK signaling. Fifty percent of Noonan syndrome patients and 35% of JMML cases carry gain-of-function mutations in PTPN11 (protein-tyrosine phosphatase, non-receptor-type, 11), altering SHP-2, a tyrosine phosphatase involved in the regulation of the RAS/MAPK pathway (64).
In Noonan syndrome, JMML may occur due to PTPN11 germline mutations with similar clinical features to children with JMML arising from PTPN11 somatic mutations, although with a generally better outcome. Mutations in PTPN11, RAS, NF1, and CBL are exclusive in JMML indicating that one hit in the RAS/MAPK pathway is sufficient for leukemogenesis (65).
Neurofibromatosis
Approximately 10–15% of pediatric JMML occur in association with neurofibromatosis type I, disease resulting from mutations in the neurofibromatosis type I gene (NF1) that encodes neurofibromin. Neurofibromin is a molecule that regulates several intracellular processes, such as the RAS–MAPK pathway (66). Neurofibromatosis type I is an autosomal dominant disorder with a clinical presentation that includes café au lait spots, ocular Lisch nodules, and skin fibromatous tumors. The development of benign and malignant tumors is high in these individuals.
Noonan Syndrome-Like Disorders
Germline mutations affecting CBL (casitas-B-lineage lymphoma protooncogene), a gene located in 11q23.3, may result in variable Noonan syndrome-like phenotypes (OMIM#613563). In these patients, presence of neurologic features is relatively high, with predisposition to JMML, low prevalence of cardiac abnormalities, reduced growth, and cryptorchidism (67).
Finally, germline mutations affecting SHOC2 (suppressor of clear, C. elegans, homolog), a gene located in 10q25.2, usually result in Noonan syndrome-like phenotypes (OMIM#607721) and JMML, and a classic Noonan syndrome in a small proportion of affected individuals. A recurrent missense SHOC2 mutation (4A>G) has been identified in a NS subgroup with growth hormone deficiency, hyperactive behavior improving with age, hair anomalies, darkly pigmented skin with eczema or ichthyosis, hypernasal speech, and mitral valve dysplasia and septal defects respective with classic NS (68).
Myeloid Neoplasms Associated with Down Syndrome
The myeloid neoplasms associated with Down syndrome are Down syndrome transient abnormal myelopoiesis (DS-TAM) and myeloid leukemia Down syndrome (ML-DS). The GATA-binding protein 1 gene (GATA1), located in Xp11.23, encodes a zinc finger DNA-binding transcription factor that is critical for the normal development of hematopoietic cells. GATA1 mutations are a hallmark of DS-TAM and ML-DS (69). All GATA1 mutations have been allocated to exon 2 (or rarely exon 3) (70). Regardless of mutation types, all of them have been found to generate a premature stop codon, with transcription initiating from an in-frame ATG triplet in codon 84 resulting in a short GATA1 isoform (~40 kDa), called “GATA1s,” lacking an N-terminal transactivation domain. DS-TAM/ML is associated with a typically constitutional trisomy 21, although some patients have been shown to be mosaics for trisomy 21 or carriers of translocations involving chromosome 21. The lack of a typical DS phenotype cannot, therefore, exclude DS-TAM. DS-TAM is clinically and morphologically undistinguishable from AML, with blasts with morphologic and immunologic characteristics of the megakaryocytic lineage. It is unique to Down syndrome newborns, present in approximately 10% of DS but infrequent in phenotypically normal trisomy 21 mosaics (71).
Down syndrome transient abnormal myelopoiesis shows a heterogeneous clinical presentation and most patients are asymptomatic. This is the reason why it is incidentally diagnosed in peripheral blood checkups showing thrombocytopenia or thrombocytosis, high white blood cell counts, excess of blasts, and, frequently, nucleated red blood cells. Hepatomegaly is a common feature, while infrequent severe manifestations may include fetal hydrops, liver failure, jaundice, coagulation defects, bleeding diathesis, heart failure, pleural effusions, ascites, and/or respiratory failure. Symptoms may appear as early first 3 weeks of life. In most patients, DS-TAM undergoes spontaneous remission within the first 3 months of life (72). ML-DS is frequently preceded by a MDS-like phase that may last for months, characterized by decreasing thrombocytopenia, ineffective erythropoiesis with subsequent anemia, and dysplastic alterations in bone marrow (73).
While coexisting GATA1 mutations and trisomy 21 might account for DS-TAM, additional alterations in preexisting DS-TAM—GATA1 seem to be necessary for generating ML-DS. These include trisomy 8 and 21, partial or complete deletions of chromosome 5 and 7, dup(1q), del(16q) (74) and somatic mutations in JAK1, JAK2, JAK3 (75), TP53 (76), FLT3, and MPL in small subsets of cases.
Individuals with DS have a 50-fold increase in the incidence of acute leukemia during the first 5 years of life compared to non-DS individuals. The great majority of DS children with ML-DS are under 5 years of age (77). ML-DS occurs in 20–30% of children with prior history of TAM and leukemia usually occurs 1–3 years after TAM. ML is usually acute megakaryoblastic leukemia in 50% of cases. The clinical course in children with less than 20% blast cells in bone marrow appears to be indolent, with initial presentation of a period of thrombocytopenia. AML prognosis for infants with DS is more favorable than for patients with non-DS AMKL. Treatment with current chemotherapy protocols is associated with 80% rates of event-free survival. Despite the excellent response to therapy, death resulting from toxicity remains a problem, occurring in approximately 7% of cases. Gene expression signatures differ between ML-DS and non-DS AMKL (78). These differences cannot be attributed to the simple presence of an additional set of chromosome 21 genes. Overexpression of c-Kit, c-MYC, and GATA2, occurs in ML-DS, in contrast to non-DS AMKL cases, indicating that these malignancies are different entities (72).
Conclusion
Diagnosis of inherited forms of MDS/AML in adults has recently increased. The family history of young adults with MDS/AML should be investigated. Following diagnosis of genetic predisposition syndromes, critical clinical conducts must include therapy, HSCT, cancer surveillance, and genetic counseling.
Author Contributions
All authors contributed to the conception, drafting of the abstract, and its revision for important intellectual content.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Hector Nicolas Seuánez for reading and revising the manuscript.
References
1. Owen C, Barnett M, Fitzgibbon J. Familial myelodysplasia and acute myeloid leukaemia – a review. Br J Haematol (2008) 140(2):123. doi:10.1111/j.1365-2141.2007.06909
2. Hahn C, Chong C, Carmichael C, Wilkins E, Brautigan P, Li X, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet (2011) 43(10):1012. doi:10.1038/ng.913
3. Nickels EM, Soodalter J, Churpek JE, Godley LA. Recognizing familial myeloid leukemia in adults. Ther Adv Hematol (2013) 4(4):254. doi:10.1177/2040620713487399
4. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood (2016) 127(20):2391. doi:10.1182/blood-2016-03-643544
5. Godley LA. Inherited predisposition to acute myeloid leukemia. Semin Hematol (2014) 51(4):306. doi:10.1053/j.seminhematol.2014.08.001
6. West AH, Godley LA, Churpek JE. Familial myelodysplastic syndrome/acute leukemia syndromes: a review and utility for translational investigations. Ann N Y Acad Sci (2014) 1310(10):111. doi:10.1111/nyas.12346
7. Churpek JE, Lorenz R, Nedumgottil S, Onel K, Olopade OI, Sorrell A, et al. Proposal for the clinical detection and management of patients and their family members with familial myelodysplastic syndrome/acute leukemia predisposition syndromes. Leuk Lymphoma (2013) 54(1):28. doi:10.3109/10428194.2012.701738
8. Churpek JE, Pyrtel K, Kanchi K-L, Shao J, Koboldt D, Miller CA, et al. Genomic analyses of germline and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood (2015) 126:2484. doi:10.1182/blood-2015-04-641100
9. Churpek J, Marquez R, Neistadt B, Claussen K, Lee M, Churpek M, et al. Inherited mutations in cancer susceptibility genes are common among breast cancer survivors who develop therapy-related leukemia. Cancer (2016) 122:304. doi:10.1002/cncr.29615
10. Voso MT, Fabiani E, Zang Z, Fianchi L, Falcioni G, Padella A, et al. Fanconi anemia gene variants in therapy-related myeloid neoplasms. Blood Cancer J (2015) 5:e323. doi:10.1038/bcj.2015.44
11. Pabst T, Eyholzer M, Haefliger S, Schardt J, Mueller B. Somatic CEBPA mutations are a frequent second event in families with germline CEBPA mutations and familial acute myeloid leukemia. J Clin Oncol (2008) 26:5088. doi:10.1200/JCO.2008.16.5563
12. Renneville A, Mialou V, Philippe N, KagialisGirard S, Biggio V, Zabot MT, et al. Another pedigree with familial acute myeloid leukemia and germline CEBPA mutation. Leukemia (2009) 23(4):804. doi:10.1038/Ieu.2008.294
13. Taskesen E, Bullinger L, Corbacioglu A, Sanders MA, Erpelinck CA, Wouters BJ, et al. Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood (2011) 117(8):2469. doi:10.1182/blood-2010-09-307280
14. Dufour A, Schneider F, Metzeler KH, Hoster E, Schneider S, Zellmeier E, et al. Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J Clin Oncol (2010) 28(4):570. doi:10.1200/JCO.2008.21.6010
15. Stelljes M, Corbacioglu A, Schlenk R, Dohner K, Fruhwald M, Rossig C, et al. Allogeneic stem cell transplant to eliminate germline mutations in the gene for CCAAT-enhancer-binding protein alpha from hematopoietic cells in a family with AML. Leukemia (2011) 25:1209. doi:10.1038/leu.2011.64
16. Tawana K, Wang J, Renneville A, Bödör C, Hills R, Loveday C, et al. Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood (2015) 126(10):1214. doi:10.1182/blood-2015-05-647172
17. Polprasert C, Schulze I, Sekeres MA, Makishima H, Przychodzen B, Hosono N, et al. Inherited and somatic defects in DDX41 in myeloid neoplasms. Cancer (2015) 27(5):658. doi:10.1016/j.ccell.2015.03.017
18. Lewinsohn M, Brown AL, Weinel LM, Phung C, Rafidi G, Lee MK, et al. Novel germline DDX41 mutations define families with a longer age of MDS/AML onset and lymphoid malignances. Blood (2016) 127:1017. doi:10.1182/blood-2015-10-676098
19. Jerez A, Gondek LP, Jankowska AM, Makishima H, Przychodzen B, Tiu RV, et al. Topography, clinical, and genomic correlates of 5q myeloid malignancies revisited. J Clin Oncol (2012) 30:1343. doi:10.1200/JCO.2011.36.1824
20. Liew E, Owen C. Familial myelodysplastic syndromes: a review of the literature. Haematologica (2011) 96:1536. doi:10.3324/haematol.2011.043422
21. Michaud J, Wu F, Osato M, Cottles GM, Yanagida M, Asou N, et al. In vitro analyses of known and novel RUNX1/AML1 mutations in dominant familial platelet disorder with predisposition to acute myelogenous leukemia: implications for mechanisms of pathogenesis. Blood (2002) 99:1364. doi:10.1182/blood.V99.4.1364
22. Churpek JE, Garcia JS, Madzo J, Jackson SA, Onel K, Godley LA. Identification and molecular characterization of a novel 3’; mutation in RUNX1 in a family with familial platelet disorder. Leuk Lymphoma (2010) 51:1931. doi:10.3109/10428194.2010.503821
23. Holme H, Hossain U, Kirwan M, Walne A, Vulliamy T, Dokal I. Marked genetic heterogeneity in familial myelodysplasia/acute myeloid leukaemia. Br J Haematol (2012) 158:242. doi:10.1111/j.1365-2141.2012.09136.x
24. Mendler JH, Maharry K, Radmacher MD, Mrózek K, Becker H, Metzeler KA, et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and MicroRNA expression signatures. J Clin Oncol (2012) 30:3109. doi:10.1200/JCO.2011.40.6652
25. Hahn Y, Bera TK, Pastan IH, Lee B. Duplication and extensive remodeling shaped POTE family genes encoding proteins containing ankyrin repeat and coiled coil domains. Gene (2006) 366(2):238. doi:10.1016/j.gene.2005.07.045
26. Pippucci T, Savoia A, Perrotta S, Pujol-Moix N, Noris P, Castegnaro G, et al. Mutations in the 5-UTR of ANKRD26, the ankirin (sic) repeat domain 26 gene, cause an autosomal-dominant form of inherited thrombocytopenia, THC2. Am J Hum Genet (2011) 88:115. doi:10.1016/j.ajhg.2010.12.006
27. Noris P, Perrotta S, Seri M, Pecci A, Gnan C, Loffredo G, et al. Mutations in ANKRD26 are responsible for a frequent form of inherited thrombocytopenia: analysis of 78 patients from 21 families. Blood (2011) 117(24):6673. doi:10.1182/blood-2011-02-336537
28. Noris P, Favier R, Alessi MC, Geddis AE, Kunishima S, Heller PG, et al. ANKRD26-related thrombocytopenia and myeloid malignancies. Blood (2013) 122(11):1987. doi:10.1182/blood-2013-04-499319
29. Marquez R, Hantel A, Lorenz R, Neistadt B, Wong J, Churpek JE, et al. A new family with a germline ANKRD26 mutation and predisposition to myeloid malignances. Leuk Lymphoma (2014) 55(12):2945. doi:10.3109/10428194.2014.903476
30. Hock H, Meade E, Medeiros S, Schindler JW, Valk PJ, Fujiwara Y, et al. Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev (2004) 18(19):2336. doi:10.1101/gad.1239604
31. Bohlander SK. ETV6: a versatile player in leukemogenesis. Semin Cancer Biol (2005) 15(3):162. doi:10.1016/j.semcancer.2005.01.008
32. Wall M, Rayeroux KC, MacKinnon RN, Zordan A, Campbell LJ. ETV6 deletion is a common additional abnormality in patients with myelodysplastic syndromes or acute myeloid leukemia and monosomy 7. Haematologica (2012) 97(12):1933. doi:10.3324/haematol.2012.069716
33. Zhang MY, Churpek JE, Keel SB, Walsh T, Lee MK, Loeb KR, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet (2015) 47:180. doi:10.1038/ng.3177
34. Noetzli L, Lo RW, Lee-Sherick AB, Callaghan M, Noris P, Savoia A, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet (2015) 47:535. doi:10.1038/ng.3253
35. Poggi M, Canault M, Favier M, Turro E, Saultier P, Ghalloussi D, et al. Germline variants in ETV6 underlie reduced platelet formation, platelet dysfunction and increased levels of circulating CD34+ progenitors. Haematologica (2017) 102:282. doi:10.3324/haematol.2016.147694
36. Babushok DV, Bessler M, Alson TS. Genetic predisposition to myelodysplastic syndrome and acute myeloid leukemia in children and young adults. Leuk Lymphoma (2016) 57:520. doi:10.3109/10428194.2015.1115041
37. Rodrigues NP, Janzen V, Forkert R, Dombkowski DM, Boyd AS, Orkin SH, et al. Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood (2005) 106:477. doi:10.1182/blood-2004-08-2989
38. Kazenwadel J, Secker GA, Liu YJ, Rosenfeld JA, Wildin RS, Cuellar-Rodriguez J. Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood (2012) 119:1283. doi:10.1182/blood-2011-08-374363
39. Pasquet M, Bellanné-Chantelot C, Tavitian S, Prade N, Beaupain B, Larochelle O, et al. High frequency of GATA2 mutations in patients with mild chronic neutropenia evolving to MonoMac syndrome, myelodysplasia, and acute myeloid leukemia. Blood (2013) 121:8229. doi:10.1182/blood-2012-08-447367
40. Mir MA, Kochuparambil ST, Abraham RS, Rodriguez V, Howard M, Hsu AP, et al. Spectrum of myeloid neoplasms and immune deficiency associated with germline GATA2 mutations. Cancer Med (2015) 4(4):490. doi:10.1002/cam4.384
41. Ostergaard P, Simpson M, Connell F, Steward C, Brice G, Woollard W, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet (2011) 43:929. doi:10.1038/ng.923
42. Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, Patel SY, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood (2011) 118:2653. doi:10.1182/blood-2011-05-356352
43. Bodor C, Renneville A, Smith M, Charazac A, Iqbal S, Etancelin P, et al. Germ-line GATA2 p.THR354MET mutation in familial myelodysplastic syndrome with acquired monosomy 7 and ASXL1 mutation demonstrating rapid onset and poor survival. Haematologica (2012) 97:890. doi:10.3324/haematol.2011.054361
44. Dror Y, Durie P, Ginzberg H, Herman R, Banerjee A, Champagne M, et al. Clonal evolution in marrows of patients with Shwachman-Diamond syndrome: a prospective 5-year follow-up study. Exp Hematol (2002) 30(7):659. doi:10.1016/S0301-472X(02)00815-9
45. Göhring G, Karow A, Steinemann D, Wilkens L, Lichter P, Zeidler C, et al. Chromosomal aberrations in congenital bone marrow failure disorders-an early indicator for leukemogenesis? Ann Hematol (2007) 86(10):733. doi:10.1007/s00277-007-0337-z
46. Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood (2009) 113(26):6549. doi:10.1182/blood-2008-12-192880
47. Alter BP. Fanconi anemia and development of leukemia. Best Pract Res Clin Haematol (2014) 27(0):214. doi:10.1016/j.beha.2014.10.002
48. Aul C, Giagounidis A, Germing U, Ganser A. Evaluating the prognosis of patients with myelodysplastic syndromes. Ann Hematol (2002) 81(9):485. doi:10.1007/s00277-002-0530-z
49. Kee Y, D’Andrea AD. Molecular pathogenesis and clinical management of Fanconi anemia. J Clin Invest (2012) 122:3799. doi:10.1172/JCI58321
50. Alter BP. Diagnosis, genetics, and management of inherited bone marrow failure syndromes. Hematology Am Soc Hematol Educ Program (2007) 2007(1):29–39. doi:10.1182/asheducation-2007.1.29
51. Mandel K, Dror Y, Poon A, Freedman MH. A practical, comprehensive classification for pediatric myelodysplastic syndromes: the CCC system. J Pediatr Hematol Oncol (2002) 24(7):596. doi:10.1007/s00277-009-0745-3
52. Hasle H, Niemeyer CM, Chessells JM, Baumann I, Bennett JM, Kerndrup G, et al. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia (2003) 17(2):277. doi:10.1038/sj.leu.2402765
53. Cada M, Segbefia CI, Klaassen R, Fernandez CV, Yanofsky RA, Wu J, et al. The impact of category, cytopathology and cytogenetics on development and progression of clonal and malignant myeloid transformation in inherited bone marrow failure syndromes. Haematologica (2015) 100(5):633. doi:10.3324/haematol.2014.117457
54. Hashmi SK, Allen C, Klaassen R, Fernandez CV, Yanofsky R, Shereck E, et al. Comparative analysis of Shwachman Diamond syndrome to other inherited bone marrow failure syndromes and genotype phenotype correlation. Clin Genet (2011) 79(5):448. doi:10.1111/j.1399-0004.2010.01468.x
55. Ballew BJ, Savage SA. Updates on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev Hematol (2013) 6:327. doi:10.1586/ehm.13.23
56. Ballew BJ, Yeager M, Jacobs K, Giri N, Boland J, Burdett L, et al. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in dyskeratosis congenita. Hum Genet (2013) 132:473. doi:10.1007/s00439-013-1265-8
57. Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, et al. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet (1998) 19:32. doi:10.1038/ng0598-32
58. Streffer C. Strong association between cancer and genomic instability. Radiat Environ Biophys (2010) 49:125. doi:10.1007/s00411-009-0258-4
59. Raynaud CM, Sabatier L, Philipot O, Olaussen KA, Soria JC. Telomere length, telomeric proteins and genomic instability during the multistep carcinogenic process. Crit Rev Oncol Hematol (2008) 66:99. doi:10.1016/j.critrevonc.2007.11.006
60. Gramatges MM, Alison AB. Short telomeres: from dyskeratosis congenita to sporadic aplastic anemia and malignancy. Transl Res (2013) 162(6):1. doi:10.1016/j.trsl.2013.05.003
61. Young NS. Bone marrow failure and the new telomere diseases: practice and research. Hematology (2012) 17(Suppl 1):S18. doi:10.1179/102453312X13336169155132
62. Armanios M. Syndromes of telomere shortening. Annu Rev Genomics Hum Genet (2009) 10:45. doi:10.1146/annurev-genom-082908-150046
63. Yoshida N, Doisaki S, Kojima S. Current management of juvenile myelomonocytic leukemia and the impact of RAS mutations. Paediatr Drugs (2012) 14:157. doi:10.2165/11631360-000000000-00000
64. Mulero-Navarro S, Sevilla A, Roman AC, Lee DF, D’Souza SL, Pardo S, et al. Myeloid dysregulation in a human induced pluripotent stem cell model of PTPN11-associated juvenile myelomonocytic leukemia. Cell Rep (2015) 13(3):504–15. doi:10.1016/j.celrep.2015.09.019
65. Perez B, Mechinaud F, Galambrun C, Ben Romdhane N, Isidor B, Philip N, et al. Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J Med Genet (2010) 47:686. doi:10.1136/jmg.2010.076836
66. Shen MH, Harper PS, Upadhyaya M. Molecular genetics of neurofibromatosis type 1 (NF1). J Med Genet (1996) 33:2. doi:10.1136/jmg.33.1.2
67. Martinelli S, De Luca A, Stellacci E, Rossi C, Checquolo S, Lepri F, et al. Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am J Hum Genet (2010) 87:250. doi:10.1016/j.ajhg.2010.06.015
68. Cordeddu V, Di Schiavi E, Pennacchio LA, Ma’ayan A, Sarkozy A, Fodale V, et al. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet (2009) 41:1022. doi:10.1038/ng.425
69. Roberts I, Alford K, Hall G, Juban G, Richmond H, Norton A, et al. GATA1-mutant clones are frequent and often unsuspected in babies with Down syndrome: identification of a population at risk of leukemia. Blood (2013) 122:3908. doi:10.1182/blood-2013-07-515148
70. Alford KA, Reinhardt K, Garnett C, Norton A, Bohmer K, von Neuhoff C, et al. Analysis of GATA1 mutations in Down syndrome transient myeloproliferative disorder and myeloid leukemia. Blood (2011) 118:2222. doi:10.1182/blood-2011-03-342774
71. Klusmann JH, Creutzig U, Zimmermann M, Dworzak M, Jorch N, Langebrake C, et al. Treatment and prognostic impact of transient leukemia in neonates with Down syndrome. Blood (2008) 111:2991. doi:10.1182/blood-2007-10-118810
72. Gamis AS, Alonzo TA, Gerbing RB, Hilden JM, Sorrell AD, Sharma M, et al. Natural history of transient myeloproliferative disorder clinically diagnosed in Down syndrome neonates: a report from the Children’s Oncology Group Study A2971. Blood (2011) 118:6752. doi:10.1182/blood-2011-04-350017
73. Zipursky A, Poon A, Doyle J. Leukemia in Down syndrome: a review. Pediatr Hematol Oncol (1992) 9:139. doi:10.3109/08880019209018329
74. Blink M, Zimmermann M, von Neuhoff C, Reinhardt D, de Haas V, Hasle H, et al. Normal karyotype is a poor prognostic factor in myeloid leukemia of Down syndrome: a retrospective, international study. Haematologica (2014) 99:299. doi:10.3324/haematol.2013.089425
75. Malinge S, Ragu C, Della-Valle V, Pisani D, Constantinescu SN, Perez C, et al. Activating mutations in human acute megakaryoblastic leukemia. Blood (2008) 112:4220. doi:10.1182/blood-2008-01-136366
76. Malkin D, Brown EJ, Zipursky A. The role of p53 in megakaryocyte differentiation and the megakaryocytic leukemias of Down syndrome. Cancer Genet Cytogenet (2000) 116:15. doi:10.1016/S0165-4608(99)00072-2
77. Rao A, Hills RK, Stiller C, Gibson BE, de Graaf SSN, Hann IM, et al. Treatment of myeloid leukaemia of Down syndrome: population-based experience in the UK and results from the Medical Research Council AML 10 and AML 12 trials. Br J Haematol (2006) 132:576. doi:10.1111/j.1365-2141.2005.05906.x
Keywords: familial, leukemia, myeloid neoplasms, germline mutations, WHO classification, GATA2, RUNX1, CEBPA
Citation: Baptista RLR, dos Santos ACE, Gutiyama LM, Solza C and Zalcberg IR (2017) Familial Myelodysplastic/Acute Leukemia Syndromes—Myeloid Neoplasms with Germline Predisposition. Front. Oncol. 7:206. doi: 10.3389/fonc.2017.00206
Received: 09 June 2017; Accepted: 23 August 2017;
Published: 12 September 2017
Edited by:
Massimo Breccia, Sapienza Università di Roma, ItalyReviewed by:
Gianni Binotto, University of Padua, ItalyMaria Teresa Voso, Università degli Studi di Roma Tor Vergata, Italy
Copyright: © 2017 Baptista, dos Santos, Gutiyama, Solza and Zalcberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renata Lyrio Rafael Baptista, cmVuYXRhLmx5cmlvQG91dGxvb2suY29t