 Flora Guerra1†
Flora Guerra1† Nicoletta Guaragnella2†
Nicoletta Guaragnella2† Arnaldo A. Arbini3
Arnaldo A. Arbini3 Cecilia Bucci1
Cecilia Bucci1 Sergio Giannattasio2*
Sergio Giannattasio2* Loredana Moro2*
Loredana Moro2*
- 1Department of Biological and Environmental Sciences and Technologies (DiSTeBA), Università del Salento, Lecce, Italy
- 2Institute of Biomembranes, Bioenergetics and Molecular Biotechnologies, National Research Council, Bari, Italy
- 3Department of Pathology, NYU Langone Medical Center, New York, NY, United States
Epithelial-to-mesenchymal transition (EMT) allows epithelial cancer cells to assume mesenchymal features, endowing them with enhanced motility and invasiveness, thus enabling cancer dissemination and metastatic spread. The induction of EMT is orchestrated by EMT-inducing transcription factors that switch on the expression of “mesenchymal” genes and switch off the expression of “epithelial” genes. Mitochondrial dysfunction is a hallmark of cancer and has been associated with progression to a metastatic and drug-resistant phenotype. The mechanistic link between metastasis and mitochondrial dysfunction is gradually emerging. The discovery that mitochondrial dysfunction owing to deregulated mitophagy, depletion of the mitochondrial genome (mitochondrial DNA) or mutations in Krebs’ cycle enzymes, such as succinate dehydrogenase, fumarate hydratase, and isocitrate dehydrogenase, activate the EMT gene signature has provided evidence that mitochondrial dysfunction and EMT are interconnected. In this review, we provide an overview of the current knowledge on the role of different types of mitochondrial dysfunction in inducing EMT in cancer cells. We place emphasis on recent advances in the identification of signaling components in the mito-nuclear communication network initiated by dysfunctional mitochondria that promote cellular remodeling and EMT activation in cancer cells.
Introduction
Mitochondria are the cell powerhouse, on which amino acid, nucleic acid, lipid, and iron–sulfur cluster metabolic pathways converge. During the last decade, mitochondria have been recognized as key players in several aspects of cancer biology, including cancer development, metastasis, and drug resistance (1, 2), due to their central role as receivers, integrators, and transmitters of intracellular signals regulating various processes (3). Mitochondria are highly dynamic organelles whose biogenesis and functions, depending on cellular needs, is under tight nuclear control, through the so-called anterograde regulation, which allows mitochondria adaptation to the ever-changing cellular milieu (4). Only 1% of mitochondrial proteins are encoded by mitochondrial DNA (mtDNA), with all the others encoded by the nuclear genome, including proteins involved in mtDNA replication and transcription, such as mitochondrial single-stranded DNA-binding protein (mtSSB or SSBP1), transcription factor A of mitochondria (TFAM), and mitochondrial DNA polymerase γ (POLG) (5). When cells require enhanced mitochondrial function, anterograde transcriptional regulation of mitochondrial biogenesis is mediated by a set of transcription factors whose activity is regulated by the PPARγ co-activator 1 family members (4).
Epithelial-to-mesenchymal transition (EMT) is a complex transdifferentiation process that allows epithelial cancer cells to transiently acquire a predominantly mesenchymal phenotype (6, 7). EMT is characterized by loss of epithelial cell polarity and cell–cell/cell–extracellular matrix contacts, supported by concomitant changes in stromal cells, that enable some tumor cells to migrate out of the primary tumor, cross the basement membrane barriers, and intravasate into the blood stream (8, 9) (Figure 1A). These circulating tumor cells (CTCs) become sources of metastasis at distant sites as the “seeds” in Paget’s “seed and soil” theory (10). EMT requires a complex cellular reprogramming that may render the cells resistant to therapies designed against the primary tumor (11, 12) and has been connected with cancer cell stemness properties (6, 13, 14).
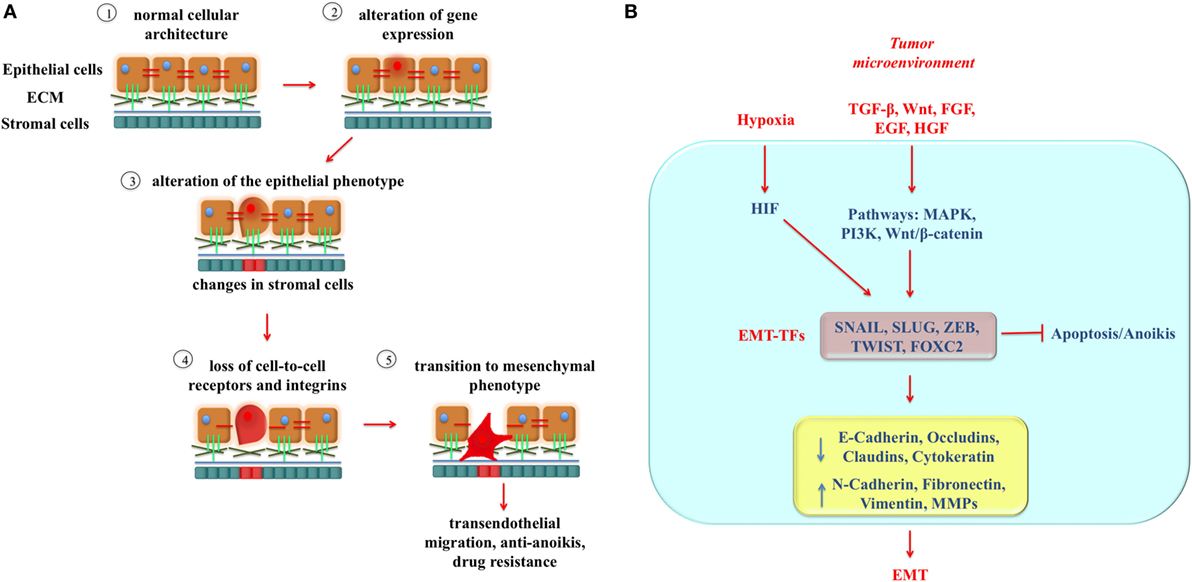
Figure 1. The mechanism of epithelial-to-mesenchymal transition (EMT). (A) Cellular changes associated with EMT. Epithelial tumor cells are shown in light brown, and stromal cells are shown in cyan. EMT begins with alterations in gene expression of epithelial cancer cells (step 2) that determine loss of the epithelial phenotype accompanied by alterations in nearby stromal cells (shown as a shift of stromal cell color from blue to red) (step 3). Loss of cell-to-cell attachment receptors and integrins occurs and continues to step 4 and beyond. EMT allows the cells to increase their invasiveness determining degradation of extracellular matrix (ECM) proteins, cytoskeleton reconstruction, extravasation, angiogenesis, as well as anoikis and drug resistance (step 5). (B) The regulatory network of EMT. Some important extracellular molecules in the tumor microenvironment, such as TGF-β, HGF, FGF, EGF, and Wnt bind to their respective receptors to induce activation of intracellular pathway, such as MAPK, PI3K, and Wnt/β-catenin. In turn, they regulate induction of EMT-inducing transcription factors (EMT-TFs), including SNAIL, SLUG, ZEB, TWIST, and FOXC2, which are responsible for molecular and physical changes occurring during EMT. Also hypoxia contributes to trigger EMT and participates in the EMT regulatory network through activation of HIFs.
The mutual interplay between EMT and mitochondrial metabolism in cancer has been recently highlighted (15–17). In this relationship, mitochondrial metabolic alterations can drive EMT or, else, EMT activation can fine-tune cancer cell metabolism by affecting the expression of metabolic genes. Mitochondrial dysfunction has been widely implicated in cancer development and progression [for a recent review, see Ref. (2)]. The precise mechanisms underlying mitochondrial dysfunction are multiple and may involve deregulated autophagic processes, unbalance in reactive oxygen species (ROS) homeostasis, mutations in oxidative phosphorylation (OXPHOS) complexes, electron transport chain (ETC), or Krebs’ cycle (TCA) enzymes. Despite the heterogeneity of the mechanisms, EMT induction has been described as one of the endpoint phenotypes in many epithelial tumor cells affected by mitochondrial dysfunction. In this review, we describe how dysregulation of the mitochondrial metabolism and genetics may promote EMT in cancer cells.
EMT in Cancer
Epithelial-to-mesenchymal transition has been initially described as a physiological process occurring at different stages of the embryonic development (type I EMT) (18). Type II EMT occurs in wound healing and fibrosis (18). Type III EMT is associated with cancer progression (18) and is the focus of this review.
Epithelial-to-mesenchymal transition is a multistep process that involves several molecular changes, including downregulation of the epithelial markers E-cadherin, claudins, desmosomes, and occludins (key components of intercellular junctions) as well as upregulation of the mesenchymal markers N-cadherin, vimentin, and fibronectin, thus fostering motility and invasion (19) (Figure 1B). These changes are orchestrated by transcription factors known as EMT-inducing transcription factors (EMT-TFs), which include TWIST1 and TWIST2, SNAIL 1, SNAIL 2 (SLUG), ZEB1, and ZEB2 as well as non-canonical EMT-TFs such as KLF8, FOXC2, and GSC. EMT-TFs regulate directly or indirectly the expression of adhesive factors and can also induce the expression of matrix metalloproteinases (MMPs), which degrade the basement membrane facilitating invasion and intravasation. Some extracellular factors, such as Wnt, TGF-β, EGF, FGF, and HGF can drive EMT by activating different signaling pathways (MAPK, Wnt/β-catenin, and PI3K) thus promoting the expression of EMT-TFs (20). In addition, tumor hypoxia is considered one of the possible triggers of EMT by inducing hypoxia-inducible transcription factors, e.g., HIF-1α and HIF-2α, which regulate the hypoxic response by modulating the expression of EMT-TFs (21, 22) (Figure 1B).
The pro-metastatic role of EMT-TFs has been extensively demonstrated [for a review, see Ref. (23)]. For example, using genetic mouse models of breast cancer, Tran et al. (24) demonstrated that transient expression of SNAIL 1 in breast tumors was sufficient to increase metastasis. Ectopic expression of TWIST1 in Twist1-negative breast cancer cells also induces EMT and cancer stem cell-like features, including expression of the stem-cell marker CD44 (13, 25–27), suggesting that EMT and acquisition of stemness capacity may be part of the same pathway. Besides promoting migration, invasion and cancer stem-cell properties, EMT would also facilitate survival of CTCs in the peripheral system by inhibiting anoikis as well as apoptosis triggered by chemotherapy or radiotherapy (28, 29). Of note, EMT induction is also regulated by changes in the expression of splicing factors (30): suppression of epithelial-specific splicing proteins (ESPR) is an indicator of the EMT process (31). In addition, identification of epigenetic changes and microRNAs as potent EMT regulators adds further complexity to the regulatory network governing EMT (32, 33).
Mitochondrial Dysfunction and EMT
Mitochondrial dysfunction has been associated with increased invasiveness, metastatic potential, and drug resistance of cancer cells (2, 34–37). The mechanisms contributing to mitochondrial dysfunction may be multiple and may occur at the level of mtDNA- or nuclear-encoded mitochondrial proteins. In the next paragraphs, we will summarize current knowledge on factors promoting mitochondrial dysfunction that has been implicated in EMT induction in cancer cells.
Mutations/Changes in Expression of Nuclear-Encoded Mitochondrial Metabolic Enzymes
Mutations in the TCA cycle enzymes fumarate hydratase (FH), isocitrate dehydrogenase (IDH), and succinate dehydrogenase (SDH) have long been recognized as oncogenic but only recently, they have been associated with EMT activation.
Fumarate hydratase mutations suppress conversion of fumarate to malate and cause hereditary leiomyomatosis and highly aggressive renal cell cancer able to metastasize at an early stage even when the primary tumor is still very small (38). Accumulation of fumarate in FH-deficient cells would promote EMT through an epigenetic mechanism: fumarate suppresses the antimetastatic miRNA cluster mir-200ba429 by inhibiting demethylation of a regulatory region, thus resulting in expression of EMT-TFs (39). This novel mechanism provides a rationale to explain the aggressive nature of FH-mutated tumors.
Isocitrate dehydrogenase promotes oxidative decarboxylation of isocitrate to α-ketoglutarate. Mutations in IDH1/2 isoforms are common in oligodendrogliomas and astrocytomas and have been also found in leukemia, melanomas, prostate, colon, and lung cancers (40). Mutant IDHs are neomorphic and catalyze the transformation of α-ketoglutarate to 2-hydroxyglutarate, an oncometabolite that has been shown to induce EMT and to be associated with the presence of distant metastasis in colorectal cancer (41). The oncometabolite 2-hydroxyglutarate, an inhibitor of Jumonji-family histone demethylase, would induce EMT by increasing the trimethylation of H3K4 in the promoter of the ZEB1 gene, thus increasing the expression of ZEB1, a master regulator of EMT (41).
Succinate dehydrogenase is another TCA cycle enzyme involved in EMT. It catalyzes the conversion of succinate to fumarate and loss-of-function SDH mutations predispose to hereditary pheochromocytoma, paraganglioma, gastrointestinal stromal tumor, and renal cell carcinoma (42). In metastatic pheochromocytomas and paragangliomas, mutations in the SDHB subunit are associated with activation of SNAIL and SLUG as a result of epigenetic remodeling due to hypermethylation of promoter CpG islands (43, 44). Focal deletions of SDHB have been also identified in serous ovarian (45) and colorectal (46) cancer and have been shown to promote EMT through an epigenetic mechanism.
Finally, a combined RNAseq and metabolomics profiling of different solid cancers has shown that downregulation of mitochondrial proteins, particularly those involved in OXPHOS, correlates with poor clinical prognosis across different cancer types and is associated with an EMT gene signature (47). Consistently, loss of OXPHOS genes was observed in metastatic cancer cell lines and in metastatic melanoma and renal cancer specimens. OXPHOS was downregulated in about 60% of low-survival patients, with subunits of Complex I and IV of the ETC being the most affected. In cancers exhibiting OXPHOS downregulation, EMT was the most upregulated cellular program, suggesting a causal role of mitochondrial dysfunction in EMT induction, and, consequently, in cancer aggressiveness and poor outcome.
mtDNA Modifications
Mutations in mtDNA-encoded proteins also contribute to mitochondrial dysfunction by directly affecting the ETC/OXPHOS system. Until a few years ago, mtDNA was believed to be very susceptible to damage because of absence of DNA repair systems. Nowadays, it is widely accepted that both yeast and mammalian mitochondria are equipped with almost all known nuclear DNA repair pathways, including base excision repair, mismatch repair, single-strand break repair, and possibly non-homologous end joining and homologous recombination [for details, see Ref. (48, 49)]. Despite the presence of DNA repair systems, the mtDNA mutation rate is considerably higher than nuclear DNA, due also to the close proximity of mtDNA to ROS-generating sites. Accumulation of mtDNA mutations has been detected in several cancer types and has been associated with metastatic progression and/or chemoresistance (2, 50–52). In 2008, Ishikawa et al. (53) demonstrated that the mtDNA mutation G13997A in the NADH dehydrogenase (ND) subunit 6 gene promotes metastasis through an ROS-dependent mechanism. Other mtDNA mutations, such as C12084T and A13966G affecting ND4 and ND5, respectively, confer a metastatic phenotype to breast cancer cells but in an ROS-independent manner (54). Another mtDNA mutation affecting ND3 (A10398G) has been detected selectively in bone metastasis of 7/10 prostate cancer patients, suggesting that the A10398G mtDNA mutation may confer a selective advantage to prostate cancer cells to colonize the bone metastatic sites (55). Frequent mtDNA mutations in Complex I genes have been detected in both benign and malignant oncocytic thyroid tumors (56, 57). Intriguingly, oncocytic thyroid carcinomas, also known as Hurthle cell carcinomas, are more aggressive than non-oncocytic thyroid cancers (58, 59), suggesting a potential role of mtDNA mutations in acquisition of the aggressive phenotype. However, despite several evidences showing a link between certain mtDNA point mutations and metastasis, it remains to be investigated whether the mechanism involves EMT activation.
Besides single mtDNA mutations, reduction in mtDNA copy number has been reported in several cancer types and has been associated with metabolic reprogramming, increased metastatic potential, chemoresistance, and EMT activation. Different mechanisms have been proposed to explain reduction of mtDNA in cancer cells. Guo et al. (60) reported frequent truncating mutations in the mitochondrial transcription factor TFAM in colorectal cancer cells, which induced mtDNA depletion and apoptosis resistance. A recent study has shown that methylation of the mitochondrial polymerase POLG may also regulate the mtDNA copy number in cancer cells (61). Besides methylation, POLG mutations have been associated with mtDNA depletion in breast cancer tissues (62). Expression changes in other nuclear genes have been reported to affect mtDNA content and induce EMT: for instance, reduced β-catenin levels in basal ErbB2-positive breast cancer cells promote an EMT program through reduction of the mtDNA content, correlated with downregulation of mitochondrial biogenesis transcription factors TFAM and PGC-1α (63). A recent study performed on 207 primary breast tumor specimens shows a direct correlation between low mtDNA content and presence of distant metastasis: patients with ≤350 mtDNA molecules per cell showed a poorer 10-year distant metastasis-free survival compared with patients with> 350 mtDNA molecules per cell (64), suggesting that low mtDNA content might be a prognostic marker for distant metastasis in breast cancer. Reduced mtDNA content has been associated with aggressive features also in other cancer types, including prostate (35, 65, 66) and colorectal (60) cancers, and it has been directly correlated with induction of EMT through activation of mitochondria-to-nucleus signaling (retrograde signaling; Figure 2).
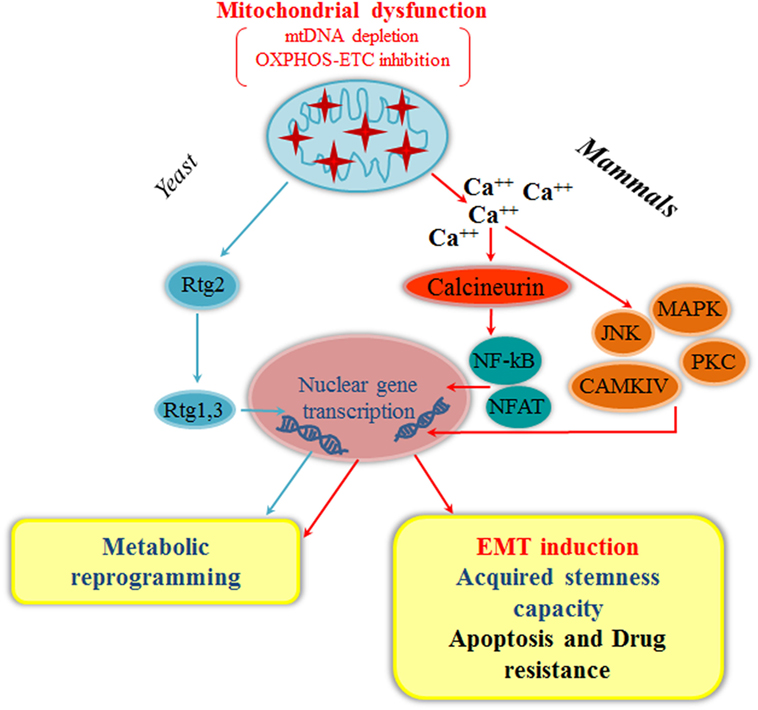
Figure 2. Mitochondrial retrograde signaling and epithelial-to-mesenchymal transition (EMT). Mitochondrial dysfunction, such as mitochondrial DNA (mtDNA) depletion or oxidative phosphorylation (OXPHOS) inhibition, triggers mitochondrial retrograde signaling, which is evolutionary conserved from yeast to mammals. In yeast, Rtg2 regulates the Rtg1,3 translocation into the nucleus eliciting a metabolic reprogramming through the upregulation of specific genes involved in anaplerotic reactions (cyan arrows). In mammals, deregulation in calcium homeostasis due to mitochondrial stress [mtDNA depletion, OXPHOS/electron transport chain (ETC) inhibition] can activate a Ca++-dependent retrograde signaling that converges on two possible branches: one mediated by calcineurin for the nuclear translocation of NF-κB or NFAT, and the other directly dependent on activation of Ca++-dependent protein kinases, such as PKC, JNK, MAPK, and CAMKIV. These pathways culminate with the activation of different transcription factors that lead to metabolic reprogramming, EMT induction, acquired stemness capacity, apoptosis resistance, and drug resistance (red arrows). Alternative RTG signaling pathways in yeast, Caenorhabditis elegans, and mammals are discussed in the text.
Mitophagy
Autophagy is the master mechanism of cell homeostasis through which destruction of unnecessary or dysfunctional molecules and organelles occur (67, 68). Withdrawal of nutrients and various stress conditions, such as alterations in glucose metabolism (69, 70), mitochondrial dysfunction, and oxidative stress (71, 72), induce autophagy with the aim of removing damaged macromolecules and organelles and/or to digest cell components to help the cell’s own maintenance (73–76). Being a homeostatic process, autophagy may have a double and opposite role in cancer, behaving as both tumor-promoter and tumor-suppressor depending on cancer cell type and tumorigenic context (77, 78). Cancer cells may indeed activate autophagy to overcome microenvironmental (nutrient deprivation, cell detachment, and hypoxia) or therapeutic (radiotherapy and chemotherapy) stress, thus promoting cancer progression (79, 80).
Mitophagy is a selective form of autophagy that specifically removes dysfunctional mitochondria from the cells. Besides traditional autophagy-related (ATG) proteins, such as LC3 (ATG8) and Beclin1 (ATG6), mitophagy relies upon specific proteins, including the E3 ubiquitin ligase Parkin (PARK2) and mitochondrially targeted PTEN-induced kinase-1 (81, 82). In yeast cells, Atg32, an outer mitochondrial membrane protein, is essential for mitophagy (83–86). Recently, Bcl2-L-13 has been identified as the mammalian homolog of Atg32: it induces mitophagy in Parkin-deficient cells (87), but its role in cancer remains to be investigated. Impaired Parkin activity in mammals has been correlated with cancer progression, suggesting that mitophagy may represent a tumor suppression mechanism (82). On the other hand, Whelan et al. (88) have recently reported that mitophagy supports EMT-mediated conversion of low CD44- to high CD44-expressing keratinocytes through modulation of oxidative stress and Parkin-dependent mitochondrial clearance. In this model, mitophagy was associated with mtDNA depletion, an event known to induce EMT and high-CD44 cell generation in mammary epithelial cells (89). It remains to be established if mitophagy drives EMT-mediated high-CD44 cell generation or is a permissive factor during this process. An independent recent study confirmed a positive role of mitophagy during EMT: Marín-Hernández et al. (90) reported that simultaneous exposure of cancer cells to hypoxia and hypoglycemia results in EMT activation and increased invasiveness, accompanied by activation of mitophagy and impaired mitochondrial functionality.
Taken together, these studies indicate a possible dichotomous nature of the relationship between EMT and mitophagy, which may be ascribed to cell type- and context-dependent factors, but much remains to be investigated.
Mitochondrial Retrograde Signaling and EMT
Dysfunctional mitochondria can generate a wide range of retrograde responses, i.e., intracellular signals relayed from mitochondria to the nucleus, leading to changes in the expression of nuclear genes for metabolic adjustments and cytoprotection (91–93). The first mitochondrial retrograde signaling was discovered by Butow (94) in yeast Saccharomyces cerevisiae. The main positive regulators of mitochondria-to-nucleus in yeast are three retrograde response (RTG) genes: RTG1 and RTG3, encoding for a heterodimeric transcription factor activating RTG target gene expression (95). RTG2, coding for a cytoplasmic protein with an N-terminal ATP-binding domain, acts as a sensor of the mitochondrial dysfunction and regulates Rtg1/3p localization (96). RTG genes dynamically interact with other regulators and signaling pathways to elicit a metabolic reprogramming through activation of anaplerotic reactions, supplying intermediates in response to respiratory defects initiated by mtDNA depletion/mutations or disruption of ETC/OXPHOS (97) (Figure 2). Interestingly, AUP1 encoding for a conserved mitochondrial protein phosphatase required for mitophagy in yeast has been shown to induce the RTG3-dependent retrograde signaling pathway (98), suggesting a possible interplay between mitophagy and mitochondrial retrograde signaling.
Another mitochondrial retrograde pathway, induced by mitochondrial proteotoxic stress, was discovered in mammalian cells by the pioneering work of Hoogenraad (99), but its detailed regulation has recently been elucidated in Caenorhabditis elegans (100). Disturbance of mitochondrial protein homeostasis and/or an increase in unassembled components initiates an retrograde response named mitochondrial unfolded-protein response (UPRmt). The current paradigm suggests that peptides resulting from proteolytic degradation of improperly folded mitochondrial proteins are released from mitochondria. However, mitochondrial import efficiency is reduced during mitochondrial dysfunction, causing ATFS-1, a pivotal transcription factor of the UPRmt, to accumulate in the cytosol and subsequently be imported into the nucleus. ATFS-1 in the nucleus regulates a transcriptional response to recover mitochondrial function including induction of mitochondrial proteases and chaperones, ROS detoxifying genes, and metabolic regulators leading to metabolic reprogramming (93, 100). The transcription factor ATF5 was recently identified as the mammalian ortholog of ATFS-1 (101). While a body of literature is already present on the function of ATF5 in cancer biology, notably in the regulation of survival and apoptosis (102, 103), it will be interesting to explore the role of ATF5 in the context of UPRmt and cancer, particularly in EMT regulation and metastasis.
The mitochondrial retrograde signaling is conserved in mammals both in response to energy metabolism impairment and to proteotoxic stress (93, 104). Of the multiple retrograde signaling pathways activated in mammals by mitochondrial dysfunction (91, 105), Ca++/calcineurin-mediated retrograde signaling has been involved in EMT activation (105) (Figure 2). Ca++ homeostasis strictly depends on mitochondria and its deregulation due to different mitochondrial stresses, such as mtDNA depletion or ETC/OXPHOS inhibition, can elicit an increase in cytosolic Ca++ that activates a Ca++-dependent retrograde signaling. Depending on cell type and conditions, there are essentially two branches in this pathway: (i) a Ca++-calcineurin-mediated retrograde signaling, through the nuclear translocations of transcription factors, NF-κB, NFAT, CREB, and HnRNPA2; (ii) a direct activation of Ca++-dependent protein kinases, such as PKC, JNK, MAPK, and CAMKIV (94, 104). Activation of these signaling pathways in epithelial cells converge on the upregulation of genes affecting several cellular functions, including apoptosis resistance, multidrug resistance, invasion, and EMT (66, 89, 106). Mitochondrial dysfunction induced by mtDNA depletion promotes EMT in breast epithelial cells through a calcineurin A-mediated mitochondrial retrograde signaling that triggers transcriptional activation of SLUG, SNAIL, and TWIST, the MMP-9 metalloproteinase, and the mesenchymal markers fibronectin, vimentin, and N-cadherin, with a corresponding decrease in the epithelial marker E-cadherin. In addition, mtDNA-depleted breast cells exhibited loss of the ESPR such as ESPR1, indicative of their mesenchymal phenotype, and expressed stem-cell markers, suggesting generation of cancer stem cells (13) (Figure 2). Of note, mtDNA-depleted cells exhibit also unorganized trajectory and higher mitochondrial fission, characteristic of cells with high metastatic ability (105). The potential link between mitochondrial dysfunction and EMT was also reported in prostate and breast adenocarcinoma cell lines depleted of mtDNA, which acquired a mesenchymal phenotype and showed TGF-β overexpression (107). More recently, mtDNA depletion was shown to induce EMT in hepatocellular carcinoma cells through TGF-β/SMAD/SNAIL signaling (108). In addition, suppression of SSBP1 promoted triple-negative breast cancer cell metastasis through mtDNA depletion, which triggered calcineurin A-mediated mitochondrial retrograde signaling resulting in c-Rel/p50 translocation to the nucleus, increased levels of TGF-β and TGF-β-driven EMT (109).
Concluding Remarks
Epithelial-to-mesenchymal transition endows cancer cells with the ability to detach from the primary tumor bulk and survive during invasion, dissemination, and metastasis. The observation that mitochondrial dysfunction can drive EMT is important as it unfolds novel therapeutic scenarios: EMT could be potentially blocked by targeting mitochondrial stress-specific EMT marker genes, effectors of the mitochondrial retrograde signaling, specific metabolic enzymes, or metabolism-dependent epigenetic reprogramming, with the aim to limit or prevent cancer metastasis. Several questions, however, remain to be answered. For instance, how and why different types of mitochondrial dysfunction converge on EMT remains a puzzle. It is possible that transient transition to a mesenchymal phenotype may confer a survival advantage to epithelial cancer cells under nutrient or oxygen stress, or in the presence of genetic defects in metabolic enzymes. In this context, EMT would represent a strategy to equip cancer cells with the necessary “armor” (increased survival) and “skills” (increased motility, invasion) to strive while exploring more advantageous metabolic microenvironments. Further studies aimed at understanding the interplay between mitochondrial retrograde signaling pathways and changing microenvironments as well as identifying the molecular determinants of the mito-nuclear network linking mitochondrial dysfunction with EMT activation may provide useful therapeutic targets for treatment and prevention of metastatic cancer.
Author Contributions
LM and SG designed and outlined structure and contents of the review. FG, NG, AA, CB, SG, and LM contributed to the literature analysis, interpretation, and writing of the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the FCRP project “Identificazione di molecole attive per lo sviluppo di nuovi farmaci antitumorali contro il carcinoma di prostata” (to LM) and by AIRC (IG2016 N. 19068 to CB).
References
1. Vyas S, Zaganjor E, Haigis MC. Mitochondria and cancer. Cell (2016) 166(3):555–66. doi:10.1016/j.cell.2016.07.002
2. Guerra F, Arbini AA, Moro L. Mitochondria and cancer chemoresistance. Biochim Biophys Acta (2017) 1858(8):686–99. doi:10.1016/j.bbabio.2017.01.012
3. Ryan MT, Hoogenraad NJ. Mitochondrial-nuclear communications. Annu Rev Biochem (2007) 76:701–22. doi:10.1146/annurev.biochem.76.052305.091720
4. Hock MB, Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annu Rev Physiol (2009) 71:177–203. doi:10.1146/annurev.physiol.010908.163119
5. Bogenhagen DF. Mitochondrial DNA nucleoid structure. Biochim Biophys Acta (2012) 1819(9–10):914–20. doi:10.1016/j.bbagrm.2011.11.005
6. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol (2017) 14(10):611–29. doi:10.1038/nrclinonc.2017.44
7. Yeung KT, Yang J. Epithelial-mesenchymal transition in tumor metastasis. Mol Oncol (2017) 11(1):28–39. doi:10.1002/1878-0261.12017
8. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer (2009) 9(4):265–73. doi:10.1038/nrc2620
9. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell (2009) 139(5):871–90. doi:10.1016/j.cell.2009.11.007
10. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell (2006) 127(4):679–95. doi:10.1016/j.cell.2006.11.001
11. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature (2015) 527(7579):472–6. doi:10.1038/nature15748
12. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature (2015) 527(7579):525–30. doi:10.1038/nature16064
13. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell (2008) 133(4):704–15. doi:10.1016/j.cell.2008.03.027
14. Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports (2014) 2(1):78–91. doi:10.1016/j.stemcr.2013.11.009
15. Payen VL, Porporato PE, Baselet B, Sonveaux P. Metabolic changes associated with tumor metastasis, part 1: tumor pH, glycolysis and the pentose phosphate pathway. Cell Mol Life Sci (2016) 73(7):1333–48. doi:10.1007/s00018-015-2098-5
16. Porporato PE, Payen VL, Baselet B, Sonveaux P. Metabolic changes associated with tumor metastasis, part 2: mitochondria, lipid and amino acid metabolism. Cell Mol Life Sci (2016) 73(7):1349–63. doi:10.1007/s00018-015-2100-2
17. Sciacovelli M, Frezza C. Oncometabolites: unconventional triggers of oncogenic signalling cascades. Free Radic Biol Med (2016) 100:175–81. doi:10.1016/j.freeradbiomed.2016.04.025
18. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest (2009) 119(6):1420–8. doi:10.1172/JCI39104
19. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15(3):178–96. doi:10.1038/nrm3758
20. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer (2013) 13(2):97–110. doi:10.1038/nrc3447
21. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal (2014) 7(344):re8. doi:10.1126/scisignal.2005189
22. Cannito S, Novo E, Compagnone A, Valfre di Bonzo L, Busletta C, Zamara E, et al. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis (2008) 29(12):2267–78. doi:10.1093/carcin/bgn216
23. Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol (2014) 16(6):488–94. doi:10.1038/ncb2976
24. Tran HD, Luitel K, Kim M, Zhang K, Longmore GD, Tran DD. Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res (2014) 74(21):6330–40. doi:10.1158/0008-5472.CAN-14-0923
25. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell (2004) 117(7):927–39. doi:10.1016/j.cell.2004.06.006
26. Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res (2007) 67(5):1979–87. doi:10.1158/0008-5472.CAN-06-1479
27. Xu Y, Qin L, Sun T, Wu H, He T, Yang Z, et al. Twist1 promotes breast cancer invasion and metastasis by silencing Foxa1 expression. Oncogene (2017) 36(8):1157–66. doi:10.1038/onc.2016.286
28. Cao Z, Livas T, Kyprianou N. Anoikis and EMT: lethal “Liaisons” during cancer progression. Crit Rev Oncog (2016) 21(3–4):155–68. doi:10.1615/CritRevOncog.2016016955
29. Frisch SM, Schaller M, Cieply B. Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J Cell Sci (2013) 126(Pt 1):21–9. doi:10.1242/jcs.120907
30. Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, et al. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet (2011) 7(8):e1002218. doi:10.1371/journal.pgen.1002218
31. Warzecha CC, Jiang P, Amirikian K, Dittmar KA, Lu H, Shen S, et al. An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. EMBO J (2010) 29(19):3286–300. doi:10.1038/emboj.2010.195
32. Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med (2013) 19(11):1438–49. doi:10.1038/nm.3336
33. Lamouille S, Subramanyam D, Blelloch R, Derynck R. Regulation of epithelial-mesenchymal and mesenchymal-epithelial transitions by microRNAs. Curr Opin Cell Biol (2013) 25(2):200–7. doi:10.1016/j.ceb.2013.01.008
34. Moro L, Arbini AA, Marra E, Greco M. Mitochondrial DNA depletion reduces PARP-1 levels and promotes progression of the neoplastic phenotype in prostate carcinoma. Cell Oncol (2008) 30(4):307–22.
35. Moro L, Arbini AA, Yao JL, di Sant’Agnese PA, Marra E, Greco M. Mitochondrial DNA depletion in prostate epithelial cells promotes anoikis resistance and invasion through activation of PI3K/Akt2. Cell Death Differ (2009) 16(4):571–83. doi:10.1038/cdd.2008.178
36. Chen EI. Mitochondrial dysfunction and cancer metastasis. J Bioenerg Biomembr (2012) 44(6):619–22. doi:10.1007/s10863-012-9465-9
37. Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep (2014) 8(3):754–66. doi:10.1016/j.celrep.2014.06.043
38. Schmidt LS, Linehan WM. Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Renovasc Dis (2014) 7:253–60. doi:10.2147/IJNRD.S42097
39. Sciacovelli M, Goncalves E, Johnson TI, Zecchini VR, da Costa AS, Gaude E, et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature (2016) 537(7621):544–7. doi:10.1038/nature19353
40. Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov (2013) 3(7):730–41. doi:10.1158/2159-8290.CD-13-0083
41. Colvin H, Nishida N, Konno M, Haraguchi N, Takahashi H, Nishimura J, et al. Oncometabolite D-2-hydroxyglurate directly induces epithelial-mesenchymal transition and is associated with distant metastasis in colorectal cancer. Sci Rep (2016) 6:36289. doi:10.1038/srep36289
42. Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochim Biophys Acta (2011) 1807(11):1432–43. doi:10.1016/j.bbabio.2011.07.003
43. Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell (2013) 23(6):739–52. doi:10.1016/j.ccr.2013.04.018
44. Loriot C, Domingues M, Berger A, Menara M, Ruel M, Morin A, et al. Deciphering the molecular basis of invasiveness in Sdhb-deficient cells. Oncotarget (2015) 6(32):32955–65. doi:10.18632/oncotarget.5106
45. Aspuria PP, Lunt SY, Varemo L, Vergnes L, Gozo M, Beach JA, et al. Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab (2014) 2:21. doi:10.1186/2049-3002-2-21
46. Wang H, Chen Y, Wu G. SDHB deficiency promotes TGFbeta-mediated invasion and metastasis of colorectal cancer through transcriptional repression complex SNAIL1-SMAD3/4. Transl Oncol (2016) 9(6):512–20. doi:10.1016/j.tranon.2016.09.009
47. Gaude E, Frezza C. Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat Commun (2016) 7:13041. doi:10.1038/ncomms13041
48. Copeland WC, Longley MJ. Mitochondrial genome maintenance in health and disease. DNA Repair (Amst) (2014) 19:190–8. doi:10.1016/j.dnarep.2014.03.010
49. Stein A, Sia EA. Mitochondrial DNA repair and damage tolerance. Front Biosci (Landmark Ed) (2017) 22:920–43. doi:10.2741/4525
50. Giannattasio S, Guaragnella N, Arbini AA, Moro L. Stress-related mitochondrial components and mitochondrial genome as targets of anticancer therapy. Chem Biol Drug Des (2013) 81(1):102–12. doi:10.1111/cbdd.12057
51. Guaragnella N, Giannattasio S, Moro L. Mitochondrial dysfunction in cancer chemoresistance. Biochem Pharmacol (2014) 92(1):62–72. doi:10.1016/j.bcp.2014.07.027
52. Girolimetti G, Guerra F, Iommarini L, Kurelac I, Vergara D, Maffia M, et al. Platinum-induced mitochondrial DNA mutations confer lower sensitivity to paclitaxel by impairing tubulin cytoskeletal organization. Hum Mol Genet (2017) 26(15):2961–74. doi:10.1093/hmg/ddx186
53. Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science (2008) 320(5876):661–4. doi:10.1126/science.1156906
54. Imanishi H, Hattori K, Wada R, Ishikawa K, Fukuda S, Takenaga K, et al. Mitochondrial DNA mutations regulate metastasis of human breast cancer cells. PLoS One (2011) 6(8):e23401. doi:10.1371/journal.pone.0023401
55. Arnold RS, Fedewa SA, Goodman M, Osunkoya AO, Kissick HT, Morrissey C, et al. Bone metastasis in prostate cancer: recurring mitochondrial DNA mutation reveals selective pressure exerted by the bone microenvironment. Bone (2015) 78:81–6. doi:10.1016/j.bone.2015.04.046
56. Gasparre G, Porcelli AM, Bonora E, Pennisi LF, Toller M, Iommarini L, et al. Disruptive mitochondrial DNA mutations in complex I subunits are markers of oncocytic phenotype in thyroid tumors. Proc Natl Acad Sci U S A (2007) 104(21):9001–6. doi:10.1073/pnas.0703056104
57. Zimmermann FA, Mayr JA, Neureiter D, Feichtinger R, Alinger B, Jones ND, et al. Lack of complex I is associated with oncocytic thyroid tumours. Br J Cancer (2009) 100(9):1434–7. doi:10.1038/sj.bjc.6605028
58. Goffredo P, Roman SA, Sosa JA. Hurthle cell carcinoma: a population-level analysis of 3311 patients. Cancer (2013) 119(3):504–11. doi:10.1002/cncr.27770
59. De Luise M, Girolimetti G, Okere B, Porcelli AM, Kurelac I, Gasparre G. Molecular and metabolic features of oncocytomas: seeking the blueprints of indolent cancers. Biochim Biophys Acta (2017) 1858(8):591–601. doi:10.1016/j.bbabio.2017.01.009
60. Guo J, Zheng L, Liu W, Wang X, Wang Z, Wang Z, et al. Frequent truncating mutation of TFAM induces mitochondrial DNA depletion and apoptotic resistance in microsatellite-unstable colorectal cancer. Cancer Res (2011) 71(8):2978–87. doi:10.1158/0008-5472.CAN-10-3482
61. Lee W, Johnson J, Gough DJ, Donoghue J, Cagnone GL, Vaghjiani V, et al. Mitochondrial DNA copy number is regulated by DNA methylation and demethylation of POLGA in stem and cancer cells and their differentiated progeny. Cell Death Dis (2015) 6:e1664. doi:10.1038/cddis.2015.34
62. Singh KK, Ayyasamy V, Owens KM, Koul MS, Vujcic M. Mutations in mitochondrial DNA polymerase-gamma promote breast tumorigenesis. J Hum Genet (2009) 54(9):516–24. doi:10.1038/jhg.2009.71
63. Vergara D, Stanca E, Guerra F, Priore P, Gaballo A, Franck J, et al. beta-Catenin knockdown affects mitochondrial biogenesis and lipid metabolism in breast cancer cells. Front Physiol (2017) 8:544. doi:10.3389/fphys.2017.00544
64. Weerts MJ, Sieuwerts AM, Smid M, Look MP, Foekens JA, Sleijfer S, et al. Mitochondrial DNA content in breast cancer: impact on in vitro and in vivo phenotype and patient prognosis. Oncotarget (2016) 7(20):29166–76. doi:10.18632/oncotarget.8688
65. Koochekpour S, Marlowe T, Singh KK, Attwood K, Chandra D. Reduced mitochondrial DNA content associates with poor prognosis of prostate cancer in African American men. PLoS One (2013) 8(9):e74688. doi:10.1371/journal.pone.0074688
66. Arbini AA, Guerra F, Greco M, Marra E, Gandee L, Xiao G, et al. Mitochondrial DNA depletion sensitizes cancer cells to PARP inhibitors by translational and post-translational repression of BRCA2. Oncogenesis (2013) 2:e82. doi:10.1038/oncsis.2013.45
67. Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell (2010) 40(2):280–93. doi:10.1016/j.molcel.2010.09.023
68. Palikaras K, Tavernarakis N. Mitochondrial homeostasis: the interplay between mitophagy and mitochondrial biogenesis. Exp Gerontol (2014) 56:182–8. doi:10.1016/j.exger.2014.01.021
69. Hoffman WH, Shacka JJ, Andjelkovic AV. Autophagy in the brains of young patients with poorly controlled T1DM and fatal diabetic ketoacidosis. Exp Mol Pathol (2012) 93(2):273–80. doi:10.1016/j.yexmp.2011.10.007
70. Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov (2014) 4(8):914–27. doi:10.1158/2159-8290.CD-14-0363
71. Kiffin R, Bandyopadhyay U, Cuervo AM. Oxidative stress and autophagy. Antioxid Redox Signal (2006) 8(1–2):152–62. doi:10.1089/ars.2006.8.152
72. Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev (2013) 27(13):1447–61. doi:10.1101/gad.219642.113
73. Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature (2004) 432(7020):1032–6. doi:10.1038/nature03029
74. Suzuki SW, Onodera J, Ohsumi Y. Starvation induced cell death in autophagy-defective yeast mutants is caused by mitochondria dysfunction. PLoS One (2011) 6(2):e17412. doi:10.1371/journal.pone.0017412
75. Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, et al. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci (2004) 117(Pt 20):4837–48. doi:10.1242/jcs.01370
76. Guerra F, Bucci C. Multiple roles of the small GTPase Rab7. Cells (2016) 5(3):34. doi:10.3390/cells5030034
77. White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res (2009) 15(17):5308–16. doi:10.1158/1078-0432.CCR-07-5023
78. Gugnoni M, Sancisi V, Manzotti G, Gandolfi G, Ciarrocchi A. Autophagy and epithelial-mesenchymal transition: an intricate interplay in cancer. Cell Death Dis (2016) 7(12):e2520. doi:10.1038/cddis.2016.415
79. Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J (2015) 34(7):856–80. doi:10.15252/embj.201490784
80. Kenific CM, Debnath J. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol (2015) 25(1):37–45. doi:10.1016/j.tcb.2014.09.001
81. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol (2008) 183(5):795–803. doi:10.1083/jcb.200809125
82. Bernardini JP, Lazarou M, Dewson G. Parkin and mitophagy in cancer. Oncogene (2017) 36(10):1315–27. doi:10.1038/onc.2016.302
83. Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell (2009) 17(1):87–97. doi:10.1016/j.devcel.2009.06.013
84. Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell (2009) 17(1):98–109. doi:10.1016/j.devcel.2009.06.014
85. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol (2011) 12(1):9–14. doi:10.1038/nrm3028
86. Kanki T, Furukawa K, Yamashita S. Mitophagy in yeast: molecular mechanisms and physiological role. Biochim Biophys Acta (2015) 1853(10 Pt B):2756–65. doi:10.1016/j.bbamcr.2015.01.005
87. Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun (2015) 6:7527. doi:10.1038/ncomms8527
88. Whelan KA, Chandramouleeswaran PM, Tanaka K, Natsuizaka M, Guha M, Srinivasan S, et al. Autophagy supports generation of cells with high CD44 expression via modulation of oxidative stress and Parkin-mediated mitochondrial clearance. Oncogene (2017) 36(34):4843–58. doi:10.1038/onc.2017.102
89. Guha M, Srinivasan S, Ruthel G, Kashina AK, Carstens RP, Mendoza A, et al. Mitochondrial retrograde signaling induces epithelial-mesenchymal transition and generates breast cancer stem cells. Oncogene (2014) 33(45):5238–50. doi:10.1038/onc.2013.467
90. Marín-Hernández A, Gallardo-Perez JC, Hernandez-Resendiz I, Del Mazo-Monsalvo I, Robledo-Cadena DX, Moreno-Sanchez R, et al. Hypoglycemia enhances epithelial-mesenchymal transition and invasiveness, and restrains the Warburg phenotype, in hypoxic HeLa cell cultures and microspheroids. J Cell Physiol (2017) 232(6):1346–59. doi:10.1002/jcp.25617
91. Quiros PM, Mottis A, Auwerx J. Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol (2016) 17(4):213–26. doi:10.1038/nrm.2016.23
92. Eisenberg-Bord M, Schuldiner M. Mitochatting – if only we could be a fly on the cell wall. Biochim Biophys Acta (2017) 1864(9):1469–80. doi:10.1016/j.bbamcr.2017.04.012
93. Arnould T, Michel S, Renard P. Mitochondria retrograde signaling and the UPR mt: where are we in mammals? Int J Mol Sci (2015) 16(8):18224–51. doi:10.3390/ijms160818224
94. Butow RA, Avadhani NG. Mitochondrial signaling: the retrograde response. Mol Cell (2004) 14(1):1–15. doi:10.1016/S1097-2765(04)00179-0
95. Jia Y, Rothermel B, Thornton J, Butow RA. A basic helix-loop-helix-leucine zipper transcription complex in yeast functions in a signaling pathway from mitochondria to the nucleus. Mol Cell Biol (1997) 17(3):1110–7. doi:10.1128/MCB.17.3.1110
96. Liu Z, Sekito T, Spirek M, Thornton J, Butow RA. Retrograde signaling is regulated by the dynamic interaction between Rtg2p and Mks1p. Mol Cell (2003) 12(2):401–11. doi:10.1016/S1097-2765(03)00285-5
97. Liu Z, Butow RA. Mitochondrial retrograde signaling. Annu Rev Genet (2006) 40:159–85. doi:10.1146/annurev.genet.40.110405.090613
98. Journo D, Mor A, Abeliovich H. Aup1-mediated regulation of Rtg3 during mitophagy. J Biol Chem (2009) 284(51):35885–95. doi:10.1074/jbc.M109.048140
99. Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J (2002) 21(17):4411–9. doi:10.1093/emboj/cdf445
100. Qureshi MA, Haynes CM, Pellegrino MW. The mitochondrial unfolded protein response: signaling from the powerhouse. J Biol Chem (2017) 292(33):13500–6. doi:10.1074/jbc.R117.791061
101. Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr Biol (2016) 26(15):2037–43. doi:10.1016/j.cub.2016.06.002
102. Sheng Z, Li L, Zhu LJ, Smith TW, Demers A, Ross AH, et al. A genome-wide RNA interference screen reveals an essential CREB3L2-ATF5-MCL1 survival pathway in malignant glioma with therapeutic implications. Nat Med (2010) 16(6):671–7. doi:10.1038/nm.2158
103. Deng P, Haynes CM. Mitochondrial dysfunction in cancer: potential roles of ATF5 and the mitochondrial UPR. Semin Cancer Biol (2017) 47:43–9. doi:10.1016/j.semcancer.2017.05.002
104. Guha M, Tang W, Sondheimer N, Avadhani NG. Role of calcineurin, hnRNPA2 and Akt in mitochondrial respiratory stress-mediated transcription activation of nuclear gene targets. Biochim Biophys Acta (2010) 1797(6–7):1055–65. doi:10.1016/j.bbabio.2010.02.008
105. Srinivasan S, Guha M, Kashina A, Avadhani NG. Mitochondrial dysfunction and mitochondrial dynamics – the cancer connection. Biochim Biophys Acta (2017) 1858(8):602–14. doi:10.1016/j.bbabio.2017.01.004
106. Guha M, Avadhani NG. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion (2013) 13(6):577–91. doi:10.1016/j.mito.2013.08.007
107. Naito A, Cook CC, Mizumachi T, Wang M, Xie CH, Evans TT, et al. Progressive tumor features accompany epithelial-mesenchymal transition induced in mitochondrial DNA-depleted cells. Cancer Sci (2008) 99(8):1584–8. doi:10.1111/j.1349-7006.2008.00879.x
108. Yi EY, Park SY, Jung SY, Jang WJ, Kim YJ. Mitochondrial dysfunction induces EMT through the TGF-beta/Smad/Snail signaling pathway in Hep3B hepatocellular carcinoma cells. Int J Oncol (2015) 47(5):1845–53. doi:10.3892/ijo.2015.3154
Keywords: epithelial-to-mesenchymal transition, mitochondrial dysfunction, mitochondrial DNA, mitochondrial retrograde signaling, metastasis
Citation: Guerra F, Guaragnella N, Arbini AA, Bucci C, Giannattasio S and Moro L (2017) Mitochondrial Dysfunction: A Novel Potential Driver of Epithelial-to-Mesenchymal Transition in Cancer. Front. Oncol. 7:295. doi: 10.3389/fonc.2017.00295
Received: 10 October 2017; Accepted: 17 November 2017;
Published: 01 December 2017
Edited by:
Michael Breitenbach, University of Salzburg, AustriaReviewed by:
Johannes A. Mayr, Paracelsus Private Medical University of Salzburg, AustriaValentina Tosato, International Centre for Genetic Engineering and Biotechnology, Italy
Copyright: © 2017 Guerra, Guaragnella, Arbini, Bucci, Giannattasio and Moro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sergio Giannattasio, cy5naWFubmF0dGFzaW9AaWJpb20uY25yLml0;
Loredana Moro, bC5tb3JvQGliaW9tLmNuci5pdA==
†These authors have contributed equally to this work.