 Christiana Neophytou
Christiana Neophytou Panagiotis Boutsikos
Panagiotis Boutsikos Panagiotis Papageorgis
Panagiotis Papageorgis- 1Department of Biological Sciences, School of Pure and Applied Sciences, University of Cyprus, Nicosia, Cyprus
- 2Department of Life Sciences, European University Cyprus, Nicosia, Cyprus
Breast cancer represents a highly heterogeneous disease comprised by several subtypes with distinct histological features, underlying molecular etiology and clinical behaviors. It is widely accepted that triple-negative breast cancer (TNBC) is one of the most aggressive subtypes, often associated with poor patient outcome due to the development of metastases in secondary organs, such as the lungs, brain, and bone. The molecular complexity of the metastatic process in combination with the lack of effective targeted therapies for TNBC metastasis have fostered significant research efforts during the past few years to identify molecular “drivers” of this lethal cascade. In this review, the most current and important findings on TNBC metastasis, as well as its closely associated basal-like subtype, including metastasis-promoting or suppressor genes and aberrantly regulated signaling pathways at specific stages of the metastatic cascade are being discussed. Finally, the most promising therapeutic approaches and novel strategies emerging from these molecular targets that could potentially be clinically applied in the near future are being highlighted.
Introduction: Tumor Heterogeneity and Current Challenges in Triple-Negative Breast Cancer (TNBC) Treatment
Breast cancer is the most frequently diagnosed cancer among women in the United States and Europe (1, 2). Despite the relative improvement in patient survival rates, breast cancer remains the most commonly diagnosed cancer and the second leading cause of cancer deaths in women worldwide. One of major challenges for the effective treatment of breast cancer is its intertumoral and intratumoral heterogeneity (3). Breast cancer can be initially classified into three different types based on the presence or absence of estrogen receptors (ERs), progesterone receptors (PRs), and the human epidermal growth factor receptor 2 (Her2/neu) (4). Hormone receptor-positive breast cancers that express ER and/or PR constitute approximately 60% of all breast cancers (5). The Her2/neu receptor is overexpressed in approximately 20% of all breast cancer cases; while TNBC constitute approximately 20% of breast cancer cases and are negative for the expression of ER, PR, and Her2/neu (6, 7).
Based on their molecular profile, breast cancers may also be clustered into basal-like and luminal subsets. Luminal breast cancers are more heterogeneous compared to basal cancers in terms of gene expression, mutation spectrum, copy number changes, and patient outcomes and can be further subdivided into luminal A and B subtypes (8, 9). The luminal A subtype represents 50–60% of breast cancer cases and is characterized by low histological grade and good prognosis. Luminal A cancers express ER and PR and have a low frequency of P53 mutations (9). Luminal B represents 10–20% of all breast cancers; compared with the luminal A subtype, these cancers are more aggressive; they have a higher grade, worse prognosis, and worse proliferative index. Luminal B display an increased expression of proliferation genes; they are ER+, PR+/−, Her-2+/−, and EGFR+ and have a higher frequency of P53 mutation (9). Because luminal cancers have a high frequency of PIK3CA mutations, the gene that encodes the p110α catalytic subunit of the phosphatidylinositol 3-kinase (PI3K), agents targeting the PI3K/AKT/mammalian target of rapamycin pathway may be useful for their treatment (10).
The basal-like subtype represents 10–20% of breast cancer cases. They are characterized by high proliferation, high histological grade, and poor prognosis. Basal-like cancers can be triple negative and have a high frequency of P53 mutations combined with loss of Rb1 (9, 11). However, not all basal-like cancers are triple negative; studies have shown that 5–45% of basal-like cancers express ER while 14% express Her2/Neu (12, 13). TNBC is a diverse group of malignancies and can be further categorized to different subtypes. An analysis of 21 breast cancer data sets containing 587 TNBC cases identified seven subtypes based on differential expression of a set of 2,188 genes: two basal like (BL1 and BL2), a mesenchymal (M), a mesenchymal-stem cell-like, an immunomodulatory, a luminal androgen receptor/luminal-like, and an unclassified type (14).
The deregulation of adult mammary stem cells (aMaSC) during tumorigenesis is believed to contribute to the development of TNBC. aMaSCs give rise to common progenitor cells that can differentiate either to basal progenitors that develop mature basal cells, or luminal progenitors. Disruption in the homeostasis of luminal progenitor cells may lead to the development of TNBC. Contributors in the development of TNBC include aberrantly activated signaling pathways, such as Wnt/β-catenin and Notch, transcriptional factors, like Snail, and embryonic stem cell markers including Sox2, Nanog, and Oct4. These alterations allow the restoration of proliferation capacity as well as the de-differentiation of these progenitor cells, leading to the accumulation of mutations that give rise to TNBC (15).
Traditionally, due to the lack of ER, PR, and Her2/Neu expression, the ineffectiveness of current breast cancer targeted therapies as well as due to the challenges in identifying key molecular drivers of TNBC progression, chemotherapy has been the foundation of treatment for patients with this disease over the last decades. Despite its sensitivity to chemotherapy, TNBC is associated with a higher risk of distant recurrence, high rates of metastases, higher probability of relapse and worse overall survival (OS) compared to other subtypes (16, 17).
Complexity of TNBC Metastasis
The dissemination of breast cancer cells and eventual metastatic growth to distant organs—predominantly the bone, lungs, and brain—represents a significant clinical problem, as metastatic disease is incurable and is the primary cause of death for the vast majority of TNBC patients. Metastatic spread of tumor cells is a highly complex, yet poorly understood process, and consists of multiple steps, including acquisition of invasive properties through genetic and epigenetic alterations, angiogenesis, tumor–stroma interactions, intravasation through the basement membrane, survival in the circulation, and extravasation of some cancer cells to distal tissues (18). However, disseminated cells that survive pro-apoptotic signals in their new environment often remain quiescent in secondary organs undergoing long periods of latency, also known as the dormancy period (19). It is well established that the outgrowth of metastatic cells in a foreign tissue microenvironment is a highly inefficient process and is considered as the rate-limiting step of breast cancer metastasis (20) (Figure 1). During this stage, breast cancer cells are usually difficult to detect and exhibit resistance to chemotherapy due to lack of proliferation (19). This remains a major clinical problem since patients, often considered as “survivors,” can develop metastatic disease years later. Disseminated tumor cells (DTCs) can enter a state of dormancy in secondary organs by exiting the proliferative cycle for an indefinite period or by achieving a balanced state of proliferation and apoptosis. Successful emergence from dormancy is the result of further evolution of surviving DTCs, by accumulating molecular alterations as well as via permissive interactions with the tumor microenvironment (19). By acquiring these characteristics, metastatic populations can optimally adapt to the host microenvironment and initiate colonization. While significant progress has been made to highlight some of the specific processes required for the breast tumor initiation, efforts have recently been focused on elucidating the roles of critical genes, the underlying molecular mechanisms and signaling pathways involved in the fatal late stages of metastatic dissemination. These studies are of outmost importance for the development of novel effective treatments against metastasis of TNBC.
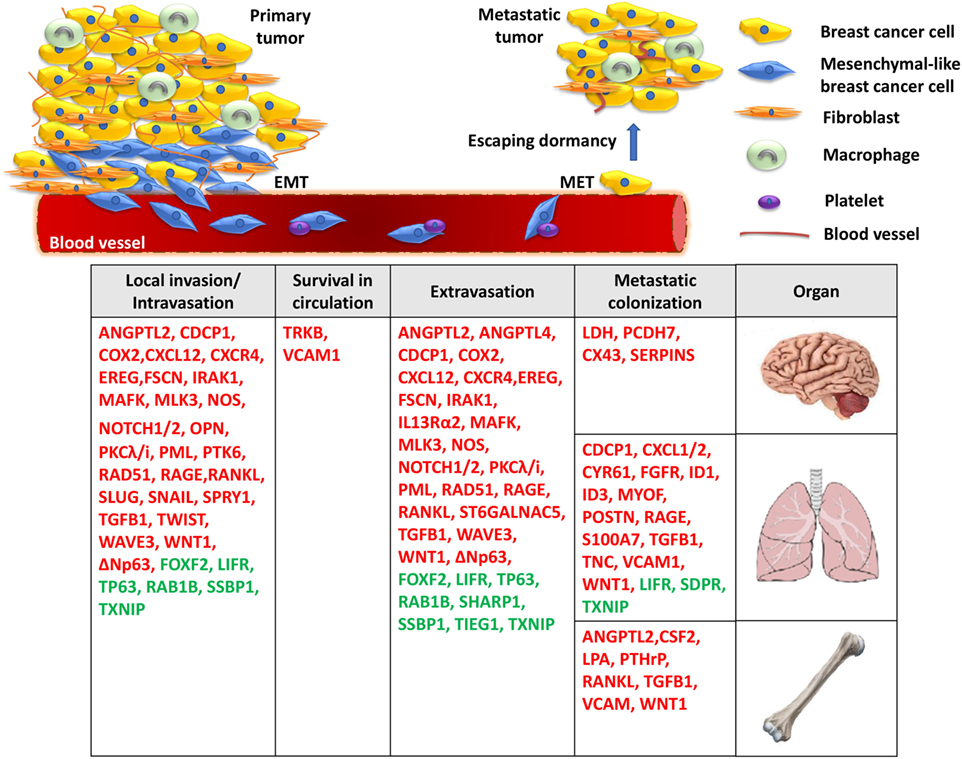
Figure 1. A model for the molecular basis of triple-negative breast cancer. During local invasion and intravasation, an epithelial-to-mesenchymal transition (EMT) transcriptional program is initiated along with the activation of matrix metalloproteases and pro-migratory signaling. Upon entering the circulation, breast cancer cells can interact with platelets, enable pro-survival pathways to suppress anoikis, and resist apoptotic signals. Then, migrated cancer cells extravasate through the endothelial blood vessel wall to a secondary organ where they enter a prolonged dormant state by forming micrometastases. Finally, the activation of metastasis-colonizing genes and the interaction with the local microenvironment create permissive conditions for macrometastatic outgrowth. Red: metastasis promoters, green: metastasis suppressors.
Genes Implicated in Multistep TNBC Metastasis
Local Invasion/Intravasation
Upon accumulation of genetic and/or epigenetic alterations, breast cancer cells at the primary tumor initially acquire properties, such as self-renewal, ability to migrate, and invade the surrounding normal tissues. During local invasion, breast cancer cells undergo epithelial-to-mesenchymal transition (EMT), a highly orchestrated transcriptional program, initially described during embryonic development, associated with dramatic remodeling of cytoskeleton, loss of apico-basolateral polarity, dissolution of cell–cell junctions, concomitant with downregulation of epithelial markers and upregulation of mesenchymal genes (21). This process is triggered by EMT-master regulators, such as the transcription factors Slug, Snail, and Twist to promote TNBC cell migration and intravasation in the circulation (22–24). The TGFβ pathway plays a critical role in regulating this early metastatic event. During intravasation, TGFβ promotes overexpression of musculoaponeurotic fibrosarcoma oncogene family protein K (MAFK) to induce EMT and enhance tumor formation and invasion in vivo (25). The TGFβ-Smad signaling axis controls the EMT step in the malignant progression of breast cancer cells either by inducing the expression of master transcriptional regulators of EMT, as described above, or by epigenetic silencing of epithelial genes, including CDH1 (26). The EMT program regulated by TGFβ/Smad signaling also involves WAVE3, a WASP/WAVE family actin-binding protein. In TNBC cells, depletion of WAVE3 expression prevented TGFβ-induced EMT phenotype (27). However, despite numerous studies using cell lines and animal models suggesting a functional role of EMT and EMT-inducing transcription factors in promoting breast cancer metastasis, the in vivo role and clinical relevance of this process remains controversial (28–31).
Moreover, the majority of genes implicated in TNBC metastasis have been reported to play a major role at the initial stages of cancer cell dissemination which include migration, invasion, and intravasation. This is not surprising given the fact that cancer cell dissemination is thought to be an early event during breast cancer evolution and that primary and metastatic tumor growth is likely to progress in parallel (32). For example, activation of CXCR4 receptor via its ligand CXCL12 or ANGPTL2 was found to induce MLK3 and Erk1/2 signaling and promote intravasation which leads to the development of lung and bone metastases (33–39). This hyperactive signaling axis may also function in multiple stages of the metastatic cascade, including angiogenesis, extravasation, and osteolysis at the secondary organ. At the same time, it is becoming increasingly clear that trans-endothelial migration and invasion of breast cancer cells in the vasculature is inhibited by metastasis suppressors, including TP63, LIFR, lysyl oxidase-like 4 (LOXL4), FOXF2, SSBP1, RAB1B, and TIEG1 (25, 40–47), suggesting that the migratory and invasive potential of breast cancer cells is ultimately determined by the balance in the activity of these molecules. The identification of numerous genes implicated in the initial stages of TNBC metastasis highlights the significant challenges for early molecular diagnosis and therapy.
Survival in Circulation
Upon entering the blood vessels, circulating tumor cells express proteins that have antiapoptotic and pro-survival functions which allow them to attach to and infiltrate specific secondary sites. Neurotrophic tyrosine kinase receptor TRKB was shown to inhibit anoikis, a form of cell death caused by lack of adhesion, via the PI3K/Akt pathway. These studies indicated that TRKB induces survival and proliferation of breast cancer cells to promote infiltration in the lymphatic and blood vessels and colonization in distant organs (48). In TNBC cells, brain-derived neurotrophic factor (BDNF) binds and activates TRKB receptor to regulate a network consisting of metalloproteases and calmodulin and thus modulate cancer–endothelial cells interaction. Importantly, Erk1/2 inhibitors were able to block the BDNF-induced phenotype, suggesting that blocking this pathway may be explored for therapeutic purposes against TNBC metastasis (49). In addition, the binding of platelets with circulating breast cancer cells has been shown to essential for their survival, evasion of pro-apoptotic signals, whereas interfering with this interaction inhibits the development of lung metastasis in TNBC mouse models (50, 51).
Extravasation in Distal Sites
Many of the genetic alterations found to be involved in intravasation are also implicated in extravasation (Table 1) since, in large part, these two processes are considered “mirrored” to each other. The TGFβ pathway plays an important role in regulating both these metastatic steps. More specifically, TGFβ induces the assembly of a mutant-p53/Smad protein complex to inhibit the function of the metastasis suppressor TP63 and promote cell migration and invasion (40). During extravasation, TGFβ induces angiopoietin-like 4 (ANGPTL4) expression via the Smad signaling pathway; the increased levels of ANGPTL4 enhance the retention of cancer cells in the lungs by disrupting vascular endothelial cell–cell junctions, thus increasing the permeability of lung capillaries to facilitate trans-endothelial passage of breast cancer cells (52). Moreover, targeting the decoy interleukin-13 receptor alpha 2 (IL13Ra2) upregulates the metastasis suppressor TP63 in an IL13-mediated, STAT6-dependent manner and impairs extravasation of basal-like breast cancer cells to the lungs (41). Several reports also highlight the importance of the synergistic effects of genes in promoting metastasis by regulating specific stages of the process. For example, EREG, COX2, MMP1, and MMP2 can collectively promote metastatic extravasation to the lungs. These four genes were found to be overexpressed in TNBC cells independently of VEGF. Individual reduction of each gene or their silencing in different combinations produced limited effects on tumor growth in vivo while concurrent silencing of all four achieved nearly complete growth abrogation (53).
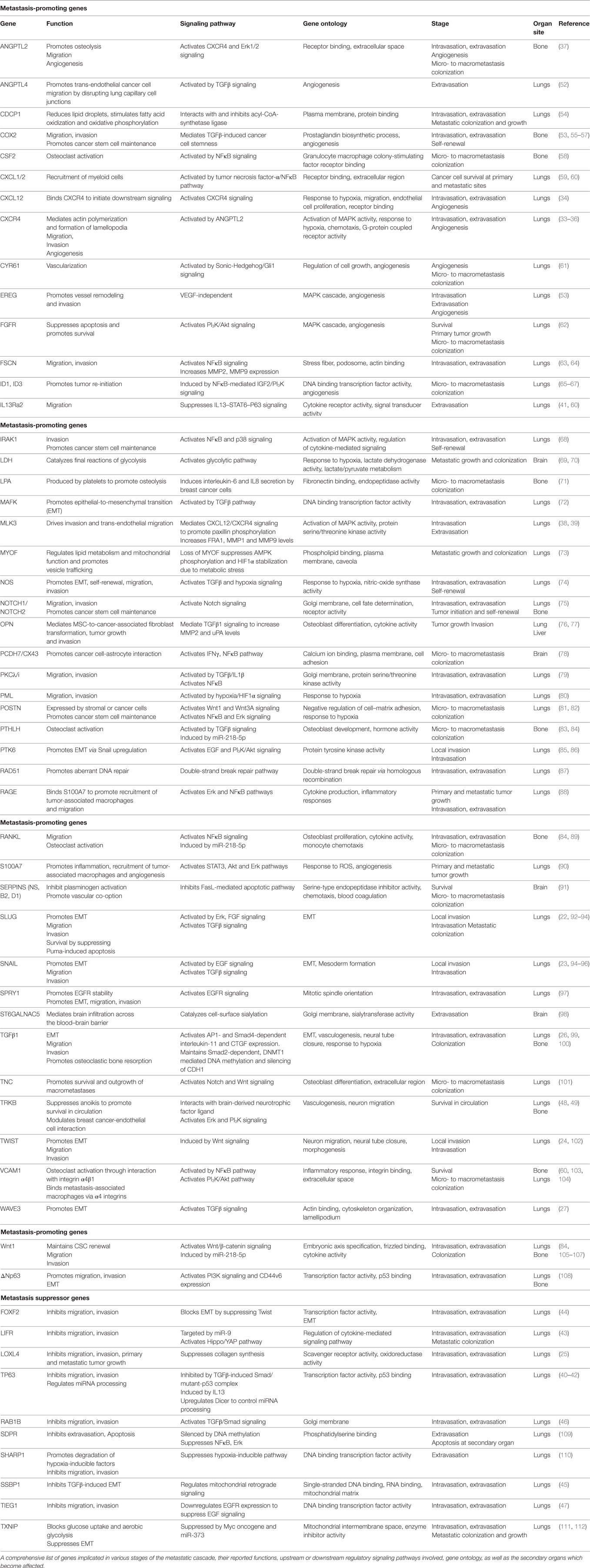
Table 1. List of genes involved triple-negative breast cancer metastasis.
Metastatic Colonization
Following extravasation and infiltration at the secondary site, a genetic program is initiated so that cancer cells can escape dormancy and form micro and macrometastatic tumors. Initially, EMT plasticity and the reversal to MET phenotype have been shown to be important for metastatic colonization (113). During this process, epithelial phenotype becomes re-established through miR-200-mediated downregulation of ZEB1, SIP1 to promote metastatic colonization (114, 115). Also, breast DTCs in the bone marrow gain the ability to form typical osteolytic metastases by producing parathyroid hormone-related protein (PTHLH), tumor necrosis factor-α (TNFα), interleukin-6 and/or interleukin-11. These factors stimulate the release of receptor activator of nuclear factor-κB ligand (RANKL) from osteoblasts which induces osteoclast formation (33, 58, 83, 116). Furthermore, inflammation in the lung microenvironment could also be responsible for triggering the escape of metastatic breast cancer cells from latency leading to metastatic colonization (117). A subset of genes contributing to primary tumor growth can also promote survival and growth at the secondary site. Chemokines CXCL1/2 mediate chemoresistance and lung metastasis by attracting myeloid cells into the tumor, which produce low molecular weight calcium-binding proteins S100A8/9 that enhance cancer cell survival by binding to the receptor for advanced glycation end products (RAGE) (59). Another calcium binding protein, S100A7 has been found to enhance tumor growth and metastasis, by binding to RAGE and activating Erk and NFκB signaling (88, 90). Furthermore, fibroblast growth factor receptor (FGFR) was shown to trigger pro-survival signals through PI3K/Akt signaling and promote outgrowth of metastatic breast cancer cells to the lungs (62). However, it needs to be highlighted that cellular and genetic context among cancers influences whether proteins act as tumor suppressors or metastasis promoters. One controversial example is LOXL4 which has been shown to recruit bone marrow-derived cells and facilitate colonization of TNBC to the lungs via a HIF1α-dependent mechanism (118). However, in another study, knockdown of LOXL4 expression in TNBC cells promoted primary tumor growth and lung metastasis which was associated with thickening of collagen bundles and remodeling of the extracellular matrix (ECM) within tumors (25). Overall, it is noteworthy that while some genes have been associated only with TNBC metastasis so far (i.e., TIEG1, MAFK, MLK3, SDPR), the majority is also involved in other tumor types, suggesting a more fundamental role in cancer progression.
Concluding Remarks on Current and Future Perspectives on TNBC Metastasis Therapy
Due to their molecular heterogeneity, there are no drugs that can target the entire spectrum of TNBC tumors and each subtype is vulnerable to specific therapeutic approaches. Despite the lack of FDA-approved targeted therapies for TNBC to date, ongoing clinical trials are assessing the efficacy of single or combinatorial approaches that tackle different TNBC molecular alterations. Up to 20% of TNBC have been associated with germ-line mutations in BRCA1 (119). TNBC tumors with loss of function of BRCA1 or BRCA2 are sensitive to poly(ADP-ribose) polymerase inhibitors and alkylating agents that induce DNA double-strand breaks (120). Olaparib has been the most successful PARP inhibitor against BRCA-mutated TNBC, inducing partial responses in 54% of patients when administered as a single agent (121) and an overall response rate of 88% when combined with carboplatin (122). Anti-androgens as well as FGFR inhibitors have been tested in clinical trials against TNBCs that are androgen receptor-positive or harbor FGFR amplification, respectively (123, 124). Gamma-secretase inhibitors that block the NOTCH pathway are currently in clinical trials for TNBC patients with upregulated NOTCH signaling (125). All together clinical trials have shown that each agent alone provides small or no benefit in TNBC patients suggesting that further effort is needed to discover novel targets of TNBC and to identify each patient’s molecular profile that will lead to a more individualized treatment.
Toward this goal, some of the metastasis-promoting genes reported here could be further exploited for the future development of promising targeted therapies. Since local invasion, intravasation and possibly extravasation are thought to occur relatively early in the metastatic process (32), a plausible strategy would be to target dormancy and the outgrowth of macrometastatic tumors in distal organs. Since this final stage is considered the critical “rate-limiting” step of the “invasion-metastasis” cascade requiring even years to be completed, it provides a window of opportunity for effective therapy. Therefore, different approaches could aim against “druggable” molecules that facilitate metastatic colonization, such as overexpressed receptors or secreted molecules (i.e., CXCL1/2, FGFR, TGFβ1, WNT1, ANGPTL2, CSF2, RANKL), which target commonly deregulated signaling networks at this late-stage (Table 1). Ongoing clinical trials are evaluating the efficacy of the TGFβR1 inhibitor LY2157299 with paclitaxel (NCT02672475), whereas the FGFR inhibitor Lucitanib is also under testing (NCT02202746) for patients with metastatic TNBC. The ultimate goal would be, if not to completely eliminate dormant metastatic breast cancer cells, to prolong dormancy period and hopefully transform this stage into a chronic inactive cancer cell state.
Importantly, recent studies have shown that tumor cells are able to evade immune responses by activating negative regulatory pathways, also known as immune checkpoints, that block T-cell activation through cytotoxic T-lymphocyte protein 4 (CTLA4) or via binding of the programmed cell death protein 1 (PD1) receptor expressed on T-cell surface to the PDL1 ligand expressed by cancer cells in response to various cytokines (126). The recent development and FDA approval of anti-CTLA4, anti-PDL1, and anti-PDL1 monoclonal antibodies that elicit antitumor clinical responses in a variety of solid cancers created enthusiasm for cancer therapy (127). Currently, several clinical trials are underway to evaluate the efficacy of this approach in TNBC as well (128).
However, a major clinical problem is that breast cancer is considered one of the most desmoplastic tumor types due to the production of excessive amounts of ECM components, such as collagen and hyaluronan, which generate mechanical stresses within the growing tumor (129). This results in blood vessel compression, hypoperfusion, and hypoxia which promote cancer progression and metastasis as well as hinder drug delivery (130). Therefore, targeting components of the tumor microenvironment has also been recently proposed as another promising strategy for TNBC therapy by improving tumor penetration and delivery of cytotoxic drugs (131). For example, targeting of cancer-associated fibroblasts using pirfenidone, an FDA-approved drug for idiopathic pulmonary fibrosis, has been shown to suppress metastasis of TNBC in combination with doxorubicin (132). This effect is likely to be mediated through remodeling of tumor microenvironment which reduces ECM components through suppression of TGFβ signaling, improves perfusion and delivery of chemotherapy (133). Similar effects have also been demonstrated using the anti-fibrotic drug Tranilast or the anti-hypertensive drug Losartan in combination with chemotherapy or nanotherapy in mouse models for TNBC (134–136).
In conclusion, this evidence suggests that efforts in the near future should be focused toward the development and testing of novel anti-metastatic targeted therapies for late-stage TNBC that could be used in combination with existing chemotherapies, immunotherapies as well as with microenvironment-remodeling agents that can improve drug penetration and overall therapeutic efficacy.
Author Contributions
CN and PB wrote the paper and helped with illustrations. PP conceived the theme, wrote the paper, and prepared illustrations.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Cyprus Research Promotion Foundation (KOULTOURA/ΒΡ-ΝΕ/0415/03).
References
1. Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer (2013) 49(6):1374–403. doi:10.1016/j.ejca.2012.12.027
2. Siegel RL, Miller KD, Jemal A. cancer statistics, 2017. CA Cancer J Clin (2017) 67(1):7–30. doi:10.3322/caac.21387
3. Polyak K. Heterogeneity in breast cancer. J Clin Invest (2011) 121(10):3786–8. doi:10.1172/JCI60534
4. Carlson RW, Allred DC, Anderson BO, Burstein HJ, Carter WB, Edge SB, et al. Breast cancer. clinical practice guidelines in oncology. J Natl Compr Canc Netw (2009) 7(2):122–92. doi:10.6004/jnccn.2009.0012
5. Buzdar AU. Role of biologic therapy and chemotherapy in hormone receptor- and HER2-positive breast cancer. Ann Oncol (2009) 20(6):993–9. doi:10.1093/annonc/mdn739
6. Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med (2001) 344(11):783–92. doi:10.1056/NEJM200103153441101
7. Anders CK, Carey LA. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin Breast Cancer (2009) 9(Suppl 2):S73–81. doi:10.3816/CBC.2009.s.008
8. Hu Z, Fan C, Oh DS, Marron JS, He X, Qaqish BF, et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics (2006) 7:96. doi:10.1186/1471-2164-7-96
9. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature (2012) 490(7418):61–70. doi:10.1038/nature11412
10. De Abreu FB, Wells WA, Tsongalis GJ. The emerging role of the molecular diagnostics laboratory in breast cancer personalized medicine. Am J Pathol (2013) 183(4):1075–83. doi:10.1016/j.ajpath.2013.07.002
11. Perou CM. Molecular stratification of triple-negative breast cancers. Oncologist (2011) 16(Suppl 1):61–70. doi:10.1634/theoncologist.2011-S1-61
12. Rakha E, Ellis I, Reis-Filho J. Are triple-negative and basal-like breast cancer synonymous? Clin Cancer Res (2008) 14(2):618. doi:10.1158/1078-0432.CCR-07-1943
13. Rouzier R, Perou CM, Symmans WF, Ibrahim N, Cristofanilli M, Anderson K, et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin Cancer Res (2005) 11(16):5678–85. doi:10.1158/1078-0432.CCR-04-2421
14. Lehmann BD, Jovanovic B, Chen X, Estrada MV, Johnson KN, Shyr Y, et al. Refinement of triple-negative breast cancer molecular subtypes: implications for neoadjuvant chemotherapy selection. PLoS One (2016) 11(6):e0157368. doi:10.1371/journal.pone.0157368
15. Rangel MC, Bertolette D, Castro NP, Klauzinska M, Cuttitta F, Salomon DS. Developmental signaling pathways regulating mammary stem cells and contributing to the etiology of triple-negative breast cancer. Breast Cancer Res Treat (2016) 156(2):211–26. doi:10.1007/s10549-016-3746-7
16. Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet (2005) 365(9472):1687–717. doi:10.1016/S0140-6736(05)66544-0
17. Ovcaricek T, Frkovic SG, Matos E, Mozina B, Borstnar S. Triple negative breast cancer – prognostic factors and survival. Radiol Oncol (2011) 45(1):46–52. doi:10.2478/v10019-010-0054-4
18. Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nat Rev Genet (2007) 8(5):341–52. doi:10.1038/nrg2101
19. Giancotti FG. Mechanisms governing metastatic dormancy and reactivation. Cell (2013) 155(4):750–64. doi:10.1016/j.cell.2013.10.029
20. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell (2011) 147(2):275–92. doi:10.1016/j.cell.2011.09.024
21. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer (2013) 13(2):97–110. doi:10.1038/nrc3447
22. Ferrari-Amorotti G, Chiodoni C, Shen F, Cattelani S, Soliera AR, Manzotti G, et al. Suppression of invasion and metastasis of triple-negative breast cancer lines by pharmacological or genetic inhibition of slug activity. Neoplasia (2014) 16(12):1047–58. doi:10.1016/j.neo.2014.10.006
23. Tran HD, Luitel K, Kim M, Zhang K, Longmore GD, Tran DD. Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res (2014) 74(21):6330–40. doi:10.1158/0008-5472.CAN-14-0923
24. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell (2004) 117(7):927–39. doi:10.1016/j.cell.2004.06.006
25. Choi SK, Kim HS, Jin T, Moon WK. LOXL4 knockdown enhances tumor growth and lung metastasis through collagen-dependent extracellular matrix changes in triple-negative breast cancer. Oncotarget (2017) 8(7):11977–89. doi:10.18632/oncotarget.14450
26. Papageorgis P, Lambert AW, Ozturk S, Gao F, Pan H, Manne U, et al. Smad signaling is required to maintain epigenetic silencing during breast cancer progression. Cancer Res (2010) 70(3):968–78. doi:10.1158/0008-5472.CAN-09-1872
27. Taylor MA, Davuluri G, Parvani JG, Schiemann BJ, Wendt MK, Plow EF, et al. Upregulated WAVE3 expression is essential for TGF-beta-mediated EMT and metastasis of triple-negative breast cancer cells. Breast Cancer Res Treat (2013) 142(2):341–53. doi:10.1007/s10549-013-2753-1
28. Cheung SY, Boey YJ, Koh VC, Thike AA, Lim JC, Iqbal J, et al. Role of epithelial-mesenchymal transition markers in triple-negative breast cancer. Breast Cancer Res Treat (2015) 152(3):489–98. doi:10.1007/s10549-015-3485-1
29. Jang MH, Kim HJ, Kim EJ, Chung YR, Park SY. Expression of epithelial-mesenchymal transition-related markers in triple-negative breast cancer: ZEB1 as a potential biomarker for poor clinical outcome. Hum Pathol (2015) 46(9):1267–74. doi:10.1016/j.humpath.2015.05.010
30. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature (2015) 527(7579):472–6. doi:10.1038/nature15748
31. Ye X, Brabletz T, Kang Y, Longmore GD, Nieto MA, Stanger BZ, et al. Upholding a role for EMT in breast cancer metastasis. Nature (2017) 547(7661):E1–3. doi:10.1038/nature22816
32. Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer (2009) 9(4):302–12. doi:10.1038/nrc2627
33. Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell (2003) 3(6):537–49. doi:10.1016/S1535-6108(03)00132-6
34. Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature (2001) 410(6824):50–6. doi:10.1038/35065016
35. Liang Z, Yoon Y, Votaw J, Goodman MM, Williams L, Shim H. Silencing of CXCR4 blocks breast cancer metastasis. Cancer Res (2005) 65(3):967–71.
36. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell (2005) 121(3):335–48. doi:10.1016/j.cell.2005.02.034
37. Masuda T, Endo M, Yamamoto Y, Odagiri H, Kadomatsu T, Nakamura T, et al. ANGPTL2 increases bone metastasis of breast cancer cells through enhancing CXCR4 signaling. Sci Rep (2015) 5:9170. doi:10.1038/srep09170
38. Rattanasinchai C, Llewellyn BJ, Conrad SE, Gallo KA. MLK3 regulates FRA-1 and MMPs to drive invasion and transendothelial migration in triple-negative breast cancer cells. Oncogenesis (2017) 6(6):e345. doi:10.1038/oncsis.2017.44
39. Chen J, Gallo KA. MLK3 regulates paxillin phosphorylation in chemokine-mediated breast cancer cell migration and invasion to drive metastasis. Cancer Res (2012) 72(16):4130–40. doi:10.1158/0008-5472.CAN-12-0655
40. Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell (2009) 137(1):87–98. doi:10.1016/j.cell.2009.01.039
41. Papageorgis P, Ozturk S, Lambert AW, Neophytou CM, Tzatsos A, Wong CK, et al. Targeting IL13Ralpha2 activates STAT6-TP63 pathway to suppress breast cancer lung metastasis. Breast Cancer Res (2015) 17:98. doi:10.1186/s13058-015-0607-y
42. Su X, Chakravarti D, Cho MS, Liu L, Gi YJ, Lin YL, et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature (2010) 467(7318):986–90. doi:10.1038/nature09459
43. Chen D, Sun Y, Wei Y, Zhang P, Rezaeian AH, Teruya-Feldstein J, et al. LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat Med (2012) 18(10):1511–7. doi:10.1038/nm.2940
44. Wang QS, Kong PZ, Li XQ, Yang F, Feng YM. FOXF2 deficiency promotes epithelial-mesenchymal transition and metastasis of basal-like breast cancer. Breast Cancer Res (2015) 17:30. doi:10.1186/s13058-015-0531-1
45. Jiang HL, Sun HF, Gao SP, Li LD, Huang S, Hu X, et al. SSBP1 suppresses TGFbeta-driven epithelial-to-mesenchymal transition and metastasis in triple-negative breast cancer by regulating mitochondrial retrograde signaling. Cancer Res (2016) 76(4):952–64. doi:10.1158/0008-5472.CAN-15-1630
46. Jiang HL, Sun HF, Gao SP, Li LD, Hu X, Wu J, et al. Loss of RAB1B promotes triple-negative breast cancer metastasis by activating TGF-beta/SMAD signaling. Oncotarget (2015) 6(18):16352–65. doi:10.18632/oncotarget.3877
47. Jin W, Chen BB, Li JY, Zhu H, Huang M, Gu SM, et al. TIEG1 inhibits breast cancer invasion and metastasis by inhibition of epidermal growth factor receptor (EGFR) transcription and the EGFR signaling pathway. Mol Cell Biol (2012) 32(1):50–63. doi:10.1128/MCB.06152-11
48. Douma S, Van Laar T, Zevenhoven J, Meuwissen R, Van Garderen E, Peeper DS. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature (2004) 430(7003):1034–9. doi:10.1038/nature02765
49. Tsai YF, Tseng LM, Hsu CY, Yang MH, Chiu JH, Shyr YM. Brain-derived neurotrophic factor (BDNF) -TrKB signaling modulates cancer-endothelial cells interaction and affects the outcomes of triple negative breast cancer. PLoS One (2017) 12(6):e0178173. doi:10.1371/journal.pone.0178173
50. Wenzel J, Zeisig R, Fichtner I. Inhibition of metastasis in a murine 4T1 breast cancer model by liposomes preventing tumor cell-platelet interactions. Clin Exp Metastasis (2010) 27(1):25–34. doi:10.1007/s10585-009-9299-y
51. Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer (2011) 11(2):123–34. doi:10.1038/nrc3004
52. Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell (2008) 133(1):66–77. doi:10.1016/j.cell.2008.01.046
53. DeJong RJ, Miller LM, Molina-Cruz A, Gupta L, Kumar S, Barillas-Mury C. Reactive oxygen species detoxification by catalase is a major determinant of fecundity in the mosquito Anopheles gambiae. Proc Natl Acad Sci U S A (2007) 104(7):2121–6. doi:10.1073/pnas.0608407104
54. Wright HJ, Hou J, Xu B, Cortez M, Potma EO, Tromberg BJ, et al. CDCP1 drives triple-negative breast cancer metastasis through reduction of lipid-droplet abundance and stimulation of fatty acid oxidation. Proc Natl Acad Sci U S A (2017) 114(32):E6556–65. doi:10.1073/pnas.1703791114
55. Larkins TL, Nowell M, Singh S, Sanford GL. Inhibition of cyclooxygenase-2 decreases breast cancer cell motility, invasion and matrix metalloproteinase expression. BMC Cancer (2006) 6:181. doi:10.1186/1471-2407-6-181
56. Singh B, Berry JA, Shoher A, Ayers GD, Wei C, Lucci A. COX-2 involvement in breast cancer metastasis to bone. Oncogene (2007) 26(26):3789–96. doi:10.1038/sj.onc.1210154
57. Tian J, Hachim MY, Hachim IY, Dai M, Lo C, Raffa FA, et al. Cyclooxygenase-2 regulates TGFbeta-induced cancer stemness in triple-negative breast cancer. Sci Rep (2017) 7:40258. doi:10.1038/srep40258
58. Park BK, Zhang H, Zeng Q, Dai J, Keller ET, Giordano T, et al. NF-kappaB in breast cancer cells promotes osteolytic bone metastasis by inducing osteoclastogenesis via GM-CSF. Nat Med (2007) 13(1):62–9. doi:10.1038/nm1519
59. Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell (2012) 150(1):165–78. doi:10.1016/j.cell.2012.04.042
60. Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes that mediate breast cancer metastasis to lung. Nature (2005) 436(7050):518–24. doi:10.1038/nature03799
61. Harris LG, Pannell LK, Singh S, Samant RS, Shevde LA. Increased vascularity and spontaneous metastasis of breast cancer by hedgehog signaling mediated upregulation of cyr61. Oncogene (2012) 31(28):3370–80. doi:10.1038/onc.2011.496
62. Dey JH, Bianchi F, Voshol J, Bonenfant D, Oakeley EJ, Hynes NE. Targeting fibroblast growth factor receptors blocks PI3K/AKT signaling, induces apoptosis, and impairs mammary tumor outgrowth and metastasis. Cancer Res (2010) 70(10):4151–62. doi:10.1158/0008-5472.CAN-09-4479
63. Chen L, Yang S, Jakoncic J, Zhang JJ, Huang XY. Migrastatin analogues target fascin to block tumour metastasis. Nature (2010) 464(7291):1062–6. doi:10.1038/nature08978
64. Al-Alwan M, Olabi S, Ghebeh H, Barhoush E, Tulbah A, Al-Tweigeri T, et al. Fascin is a key regulator of breast cancer invasion that acts via the modification of metastasis-associated molecules. PLoS One (2011) 6(11):e27339. doi:10.1371/journal.pone.0027339
65. Gupta GP, Perk J, Acharyya S, de Candia P, Mittal V, Todorova-Manova K, et al. ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc Natl Acad Sci U S A (2007) 104(49):19506–11. doi:10.1073/pnas.0709185104
66. Fong S, Itahana Y, Sumida T, Singh J, Coppe JP, Liu Y, et al. Id-1 as a molecular target in therapy for breast cancer cell invasion and metastasis. Proc Natl Acad Sci U S A (2003) 100(23):13543–8. doi:10.1073/pnas.2230238100
67. Tominaga K, Shimamura T, Kimura N, Murayama T, Matsubara D, Kanauchi H, et al. Addiction to the IGF2-ID1-IGF2 circuit for maintenance of the breast cancer stem-like cells. Oncogene (2017) 36(9):1276–86. doi:10.1038/onc.2016.293
68. Wee ZN, Yatim SM, Kohlbauer VK, Feng M, Goh JY, Bao Y, et al. IRAK1 is a therapeutic target that drives breast cancer metastasis and resistance to paclitaxel. Nat Commun (2015) 6:8746. doi:10.1038/ncomms9746
69. Dong T, Liu Z, Xuan Q, Wang Z, Ma W, Zhang Q. Tumor LDH-A expression and serum LDH status are two metabolic predictors for triple negative breast cancer brain metastasis. Sci Rep (2017) 7(1):6069. doi:10.1038/s41598-017-06378-7
70. Vergara D, Simeone P, del Boccio P, Toto C, Pieragostino D, Tinelli A, et al. Comparative proteome profiling of breast tumor cell lines by gel electrophoresis and mass spectrometry reveals an epithelial mesenchymal transition associated protein signature. Mol Biosyst (2013) 9(6):1127–38. doi:10.1039/c2mb25401h
71. Boucharaba A, Serre CM, Gres S, Saulnier-Blache JS, Bordet JC, Guglielmi J, et al. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J Clin Invest (2004) 114(12):1714–25. doi:10.1172/JCI22123
72. Okita Y, Kimura M, Xie R, Chen C, Shen LT, Kojima Y, et al. The transcription factor MAFK induces EMT and malignant progression of triple-negative breast cancer cells through its target GPNMB. Sci Signal (2017) 10(474):eaak9397. doi:10.1126/scisignal.aak9397
73. Blomme A, Costanza B, de Tullio P, Thiry M, Van Simaeys G, Boutry S, et al. Myoferlin regulates cellular lipid metabolism and promotes metastases in triple-negative breast cancer. Oncogene (2017) 36(15):2116–30. doi:10.1038/onc.2016.369
74. Granados-Principal S, Liu Y, Guevara ML, Blanco E, Choi DS, Qian W, et al. Inhibition of iNOS as a novel effective targeted therapy against triple-negative breast cancer. Breast Cancer Res (2015) 17:25. doi:10.1186/s13058-015-0527-x
75. Azzam DJ, Zhao D, Sun J, Minn AJ, Ranganathan P, Drews-Elger K, et al. Triple negative breast cancer initiating cell subsets differ in functional and molecular characteristics and in gamma-secretase inhibitor drug responses. EMBO Mol Med (2013) 5(10):1502–22. doi:10.1002/emmm.201302558
76. Weber CE, Kothari AN, Wai PY, Li NY, Driver J, Zapf MA, et al. Osteopontin mediates an MZF1-TGF-beta1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene (2015) 34(37):4821–33. doi:10.1038/onc.2014.410
77. Mi Z, Guo H, Russell MB, Liu Y, Sullenger BA, Kuo PC. RNA aptamer blockade of osteopontin inhibits growth and metastasis of MDA-MB231 breast cancer cells. Mol Ther (2009) 17(1):153–61. doi:10.1038/mt.2008.235
78. Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature (2016) 533(7604):493–8. doi:10.1038/nature18268
79. Paul A, Gunewardena S, Stecklein SR, Saha B, Parelkar N, Danley M, et al. PKClambda/iota signaling promotes triple-negative breast cancer growth and metastasis. Cell Death Differ (2014) 21(9):1469–81. doi:10.1038/cdd.2014.62
80. Ponente M, Campanini L, Cuttano R, Piunti A, Delledonne GA, Coltella N, et al. PML promotes metastasis of triple-negative breast cancer through transcriptional regulation of HIF1A target genes. JCI Insight (2017) 2(4):e87380. doi:10.1172/jci.insight.87380
81. Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, Lehr HA, Delaloye JF, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature (2011) 481(7379):85–9. doi:10.1038/nature10694
82. Lambert AW, Wong CK, Ozturk S, Papageorgis P, Raghunathan R, Alekseyev Y, et al. Tumor cell-derived periostin regulates cytokines that maintain breast cancer stem cells. Mol Cancer Res (2016) 14(1):103–13. doi:10.1158/1541-7786.MCR-15-0079
83. Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, et al. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest (1999) 103(2):197–206. doi:10.1172/JCI3523
84. Taipaleenmaki H, Farina NH, van Wijnen AJ, Stein JL, Hesse E, Stein GS, et al. Antagonizing miR-218-5p attenuates Wnt signaling and reduces metastatic bone disease of triple negative breast cancer cells. Oncotarget (2016) 7(48):79032–46. doi:10.18632/oncotarget.12593
85. Kamalati T, Jolin HE, Fry MJ, Crompton MR. Expression of the BRK tyrosine kinase in mammary epithelial cells enhances the coupling of EGF signalling to PI 3-kinase and Akt, via erbB3 phosphorylation. Oncogene (2000) 19(48):5471–6. doi:10.1038/sj.onc.1203931
86. Ito K, Park SH, Nayak A, Byerly JH, Irie HY. PTK6 inhibition suppresses metastases of triple-negative breast cancer via SNAIL-dependent E-cadherin regulation. Cancer Res (2016) 76(15):4406–17. doi:10.1158/0008-5472.CAN-15-3445
87. Wiegmans AP, Al-Ejeh F, Chee N, Yap PY, Gorski JJ, Da Silva L, et al. Rad51 supports triple negative breast cancer metastasis. Oncotarget (2014) 5(10):3261–72. doi:10.18632/oncotarget.1923
88. Nasser MW, Wani NA, Ahirwar DK, Powell CA, Ravi J, Elbaz M, et al. RAGE mediates S100A7-induced breast cancer growth and metastasis by modulating the tumor microenvironment. Cancer Res (2015) 75(6):974–85. doi:10.1158/0008-5472.CAN-14-2161
89. Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, Sarosi I, et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature (2006) 440(7084):692–6. doi:10.1038/nature04524
90. Nasser MW, Qamri Z, Deol YS, Ravi J, Powell CA, Trikha P, et al. S100A7 enhances mammary tumorigenesis through upregulation of inflammatory pathways. Cancer Res (2012) 72(3):604–15. doi:10.1158/0008-5472.CAN-11-0669
91. Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ, et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell (2014) 156(5):1002–16. doi:10.1016/j.cell.2014.01.040
92. Savagner P, Yamada KM, Thiery JP. The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J Cell Biol (1997) 137(6):1403–19. doi:10.1083/jcb.137.6.1403
93. Kim S, Yao J, Suyama K, Qian X, Qian BZ, Bandyopadhyay S, et al. Slug promotes survival during metastasis through suppression of puma-mediated apoptosis. Cancer Res (2014) 74(14):3695–706. doi:10.1158/0008-5472.CAN-13-2591
94. Dhasarathy A, Phadke D, Mav D, Shah RR, Wade PA. The transcription factors snail and slug activate the transforming growth factor-beta signaling pathway in breast cancer. PLoS One (2011) 6(10):e26514. doi:10.1371/journal.pone.0026514
95. Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol (2000) 2(2):76–83. doi:10.1038/35000025
96. Kim J, Kong J, Chang H, Kim H, Kim A. EGF induces epithelial-mesenchymal transition through phospho-Smad2/3-Snail signaling pathway in breast cancer cells. Oncotarget (2016) 7(51):85021–32. doi:10.18632/oncotarget.13116
97. He Q, Jing H, Liaw L, Gower L, Vary C, Hua S, et al. Suppression of Spry1 inhibits triple-negative breast cancer malignancy by decreasing EGF/EGFR mediated mesenchymal phenotype. Sci Rep (2016) 6:23216. doi:10.1038/srep23216
98. Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature (2009) 459(7249):1005–9. doi:10.1038/nature08021
99. Tang B, Vu M, Booker T, Santner SJ, Miller FR, Anver MR, et al. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest (2003) 112(7):1116–24. doi:10.1172/JCI18899
100. Kang Y, He W, Tulley S, Gupta GP, Serganova I, Chen CR, et al. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci U S A (2005) 102(39):13909–14. doi:10.1073/pnas.0506517102
101. Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med (2011) 17(7):867–74. doi:10.1038/nm.2379
102. Howe LR, Watanabe O, Leonard J, Brown AM. Twist is up-regulated in response to Wnt1 and inhibits mouse mammary cell differentiation. Cancer Res (2003) 63(8):1906–13.
103. Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell (2011) 20(6):701–14. doi:10.1016/j.ccr.2011.11.002
104. Chen Q, Zhang XH, Massague J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell (2011) 20(4):538–49. doi:10.1016/j.ccr.2011.08.025
105. Jang GB, Kim JY, Cho SD, Park KS, Jung JY, Lee HY, et al. Blockade of Wnt/beta-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci Rep (2015) 5:12465. doi:10.1038/srep12465
106. Dey N, Barwick BG, Moreno CS, Ordanic-Kodani M, Chen Z, Oprea-Ilies G, et al. Wnt signaling in triple negative breast cancer is associated with metastasis. BMC Cancer (2013) 13:537. doi:10.1186/1471-2407-13-537
107. Geyer FC, Lacroix-Triki M, Savage K, Arnedos M, Lambros MB, MacKay A, et al. beta-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod Pathol (2011) 24(2):209–31. doi:10.1038/modpathol.2010.205
108. Di Franco S, Turdo A, Benfante A, Colorito ML, Gaggianesi M, Apuzzo T, et al. DeltaNp63 drives metastasis in breast cancer cells via PI3K/CD44v6 axis. Oncotarget (2016) 7(34):54157–73. doi:10.18632/oncotarget.11022
109. Ozturk S, Papageorgis P, Wong CK, Lambert AW, Abdolmaleky HM, Thiagalingam A, et al. SDPR functions as a metastasis suppressor in breast cancer by promoting apoptosis. Proc Natl Acad Sci U S A (2016) 113(3):638–43. doi:10.1073/pnas.1514663113
110. Montagner M, Enzo E, Forcato M, Zanconato F, Parenti A, Rampazzo E, et al. SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia-inducible factors. Nature (2012) 487(7407):380–4. doi:10.1038/nature11207
111. Shen L, O’Shea JM, Kaadige MR, Cunha S, Wilde BR, Cohen AL, et al. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc Natl Acad Sci U S A (2015) 112(17):5425–30. doi:10.1073/pnas.1501555112
112. Chen D, Dang BL, Huang JZ, Chen M, Wu D, Xu ML, et al. MiR-373 drives the epithelial-to-mesenchymal transition and metastasis via the miR-373-TXNIP-HIF1alpha-TWIST signaling axis in breast cancer. Oncotarget (2015) 6(32):32701–12. doi:10.18632/oncotarget.4702
113. Gunasinghe NP, Wells A, Thompson EW, Hugo HJ. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev (2012) 31(3–4):469–78. doi:10.1007/s10555-012-9377-5
114. Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol (2008) 10(5):593–601. doi:10.1038/ncb1722
115. Dykxhoorn DM, Wu Y, Xie H, Yu F, Lal A, Petrocca F, et al. miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS One (2009) 4(9):e7181. doi:10.1371/journal.pone.0007181
116. Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer (2002) 2(8):584–93. doi:10.1038/nrc867
117. De Cock JM, Shibue T, Dongre A, Keckesova Z, Reinhardt F, Weinberg RA. Inflammation triggers Zeb1-dependent escape from tumor latency. Cancer Res (2016) 76(23):6778–84. doi:10.1158/0008-5472.CAN-16-0608
118. Wong CC, Gilkes DM, Zhang H, Chen J, Wei H, Chaturvedi P, et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc Natl Acad Sci U S A (2011) 108(39):16369–74. doi:10.1073/pnas.1113483108
119. Bosch A, Eroles P, Zaragoza R, Vina JR, Lluch A. Triple-negative breast cancer: molecular features, pathogenesis, treatment and current lines of research. Cancer Treat Rev (2010) 36(3):206–15. doi:10.1016/j.ctrv.2009.12.002
120. Okuma HS, Yonemori K. BRCA Gene Mutations and Poly(ADP-Ribose) Polymerase Inhibitors in Triple-Negative Breast Cancer. Adv Exp Med Biol (2017) 1026:271–86. doi:10.1007/978-981-10-6020-5_13
121. Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet (2010) 376(9737):235–44. doi:10.1016/S0140-6736(10)60892-6
122. Dent RA, Lindeman GJ, Clemons M, Wildiers H, Chan A, McCarthy NJ, et al. Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast Cancer Res (2013) 15(5):R88. doi:10.1186/bcr3484
123. Gucalp A, Tolaney S, Isakoff SJ, Ingle JN, Liu MC, Carey LA, et al. Cancer Research: Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic breast cancer. Clin Cancer Res (2013) 19(19):5505–12. doi:10.1158/1078-0432.CCR-12-3327
124. Gelmon K, Dent R, Mackey JR, Laing K, McLeod D, Verma S. Targeting triple-negative breast cancer: optimising therapeutic outcomes. Ann Oncol (2012) 23(9):2223–34. doi:10.1093/annonc/mds067
125. Locatelli MA, Aftimos P, Dees EC, LoRusso PM, Pegram MD, Awada A, et al. Phase I study of the gamma secretase inhibitor PF-03084014 in combination with docetaxel in patients with advanced triple-negative breast cancer. Oncotarget (2017) 8(2):2320–8. doi:10.18632/oncotarget.13727
126. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature (2017) 541(7637):321–30. doi:10.1038/nature21349
127. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med (2016) 8(328):328rv4. doi:10.1126/scitranslmed.aad7118
128. Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol (2016) 13(11):674–90. doi:10.1038/nrclinonc.2016.66
129. Walker RA. The complexities of breast cancer desmoplasia. Breast Cancer Res (2001) 3(3):143–5. doi:10.1186/bcr287
130. Jain RK, Martin JD, Stylianopoulos T. The role of mechanical forces in tumor growth and therapy. Annu Rev Biomed Eng (2014) 16:321–46. doi:10.1146/annurev-bioeng-071813-105259
131. Gkretsi V, Stylianou A, Papageorgis P, Polydorou C, Stylianopoulos T. Remodeling components of the tumor microenvironment to enhance cancer therapy. Front Oncol (2015) 5:214. doi:10.3389/fonc.2015.00214
132. Takai K, Le A, Weaver VM, Werb Z. Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget (2016) 7(50):82889–901. doi:10.18632/oncotarget.12658
133. Polydorou C, Mpekris F, Papageorgis P, Voutouri C, Stylianopoulos T. Pirfenidone normalizes the tumor microenvironment to improve chemotherapy. Oncotarget (2017) 8(15):24506–17. doi:10.18632/oncotarget.15534
134. Papageorgis P, Polydorou C, Mpekris F, Voutouri C, Agathokleous E, Kapnissi-Christodoulou CP, et al. Tranilast-induced stress alleviation in solid tumors improves the efficacy of chemo- and nanotherapeutics in a size-independent manner. Sci Rep (2017) 7:46140. doi:10.1038/srep46140
135. Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun (2013) 4:2516. doi:10.1038/ncomms3516
Keywords: triple-negative breast cancer, metastasis, targeted therapy, tumor microenvironment, dormancy
Citation: Neophytou C, Boutsikos P and Papageorgis P (2018) Molecular Mechanisms and Emerging Therapeutic Targets of Triple-Negative Breast Cancer Metastasis. Front. Oncol. 8:31. doi: 10.3389/fonc.2018.00031
Received: 28 November 2017; Accepted: 30 January 2018;
Published: 22 February 2018
Edited by:
Scott Bultman, University of North Carolina at Chapel Hill, United StatesReviewed by:
Saraswati Sukumar, Johns Hopkins University, United StatesDaniele Vergara, University of Salento, Italy
Copyright: © 2018 Neophytou, Boutsikos and Papageorgis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Panagiotis Papageorgis, cC5wYXBhZ2Vvcmdpc0BldWMuYWMuY3k=