 Pau B. Esparza-Moltó
Pau B. Esparza-Moltó José M. Cuezva
José M. Cuezva- Departamento de Biología Molecular, Centro de Biología Molecular Severo Ochoa (CSIC-UAM), Centro de Investigación Biomédica en Red de Enfermedades Raras CIBERER-ISCIII, Instituto de Investigación Hospital 12 de Octubre (i+12), Universidad Autónoma de Madrid, Madrid, Spain
Cancer cells reprogram energy metabolism by boosting aerobic glycolysis as a main pathway for the provision of metabolic energy and of precursors for anabolic purposes. Accordingly, the relative expression of the catalytic subunit of the mitochondrial H+-ATP synthase—the core hub of oxidative phosphorylation—is downregulated in human carcinomas when compared with its expression in normal tissues. Moreover, some prevalent carcinomas also upregulate the ATPase inhibitory factor 1 (IF1), which is the physiological inhibitor of the H+-ATP synthase. IF1 overexpression, both in cells in culture and in tissue-specific mouse models, is sufficient to reprogram energy metabolism to an enhanced glycolysis by limiting ATP production by the H+-ATP synthase. Furthermore, the IF1-mediated inhibition of the H+-ATP synthase promotes the production of mitochondrial ROS (mtROS). mtROS modulate signaling pathways favoring cellular proliferation and invasion, the activation of antioxidant defenses, resistance to cell death, and modulation of the tissue immune response, favoring the acquisition of several cancer traits. Consistently, IF1 expression is an independent marker of cancer prognosis. By contrast, inhibition of the H+-ATP synthase by α-ketoglutarate and the oncometabolite 2-hydroxyglutarate, reduces mTOR signaling, suppresses cancer cell growth, and contributes to lifespan extension in several model organisms. Hence, the H+-ATP synthase appears as a conserved hub in mitochondria-to-nucleus signaling controlling cell fate. Unraveling the molecular mechanisms responsible for IF1 upregulation in cancer and the signaling cascades that are modulated by the H+-ATP synthase are of utmost interest to decipher the metabolic and redox circuits contributing to cancer origin and progression.
Overview of Metabolic Reprogramming in Cancer
Cancer cells experience a series of alterations during oncogenic transformation that confer them new features (1). Cellular metabolism is a central player in the acquisition of this new phenotype. Indeed, cancer cells are highly proliferative and readapt their metabolism to meet the demands imposed by the new phenotype, namely higher requirements of metabolic energy and of precursors for biosynthetic purposes (Figure 1) (2–5). One prominent feature of the metabolic reprogramming experienced by cancer cells is an enhanced glycolytic rate in the presence of oxygen, what is known as aerobic glycolysis (6) (Figure 1). Glycolysis provides cancer cells with various metabolic precursors that serve for the synthesis of amino acids, nucleotides, and lipids, as well as reducing power and ATP. In addition, mitochondrial function is readapted during oncogenic transformation and mutations in genes encoding mitochondrial proteins contribute to cancer development (7, 8). In particular, the relative contribution of mitochondria to energy provision is reduced, the organelles becoming mostly dedicated to produce anabolic precursors through the tricarboxylic acid (TCA) cycle (9) (Figure 1). Likewise, mitochondria, which are crucial hubs in intracellular signaling (10), readapt this function in cancer (11). Intermediates of the TCA cycle contribute to signaling tumorigenesis (12) and mtROS, which are key mediators in mitochondrial communication (13), activate signaling pathways that promote cell proliferation and tumorigenesis (14, 15). Moreover, mitochondrial dynamics is also reprogrammed in cancer (16).
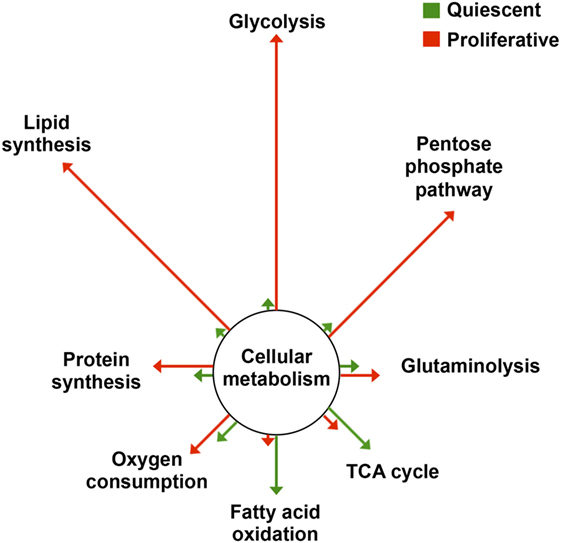
Figure 1. Metabolic reprogramming in proliferation. The changes in flux through the main pathways of cellular metabolism are depicted in proliferative cells (red) relative to that of quiescent cells (green). Arrows’ length is proportional to the relative change in flux. Data adapted from Ref. (2–5). Abbreviation: TCA cycle, tricarboxylic acid cycle.
The H+-ATP Synthase is Downregulated in Cancer
A critical enzyme complex within the mitochondria is the H+-ATP synthase, the rotatory engine of the inner mitochondrial membrane responsible for ATP synthesis by oxidative phosphorylation (OXPHOS) (17). The H+-ATP synthase consumes the proton electrochemical gradient generated across the inner mitochondrial membrane by the electron transport chain to drive ATP synthesis (17, 18). In addition, the H+-ATP synthase is a critical component of the mitochondrial permeability transition pore (PTP) whose prolonged opening triggers the execution of cell death (19–21). Although its mechanism of participation in PTP opening is currently debated (22–24), recent findings have mapped the residues in subunits of the H+-ATP synthase for Ca2+ activation (25) and pH inhibition (26) of the PTP, reinforcing the role of the H+-ATP synthase in PTP function and providing first evidence that single point mutations in the enzyme affect PTP modulation. Hence, the H+-ATP synthase integrates the bioenergetic and death-signaling functions of mitochondria, what makes it a relevant target for oncogenic transformation (27, 28). Actually, mutations in the mitochondrial-encoded subunit a of the H+-ATP synthase (MT-ATP6), which are found in different human carcinomas, promote tumor growth by restraining cell death (29, 30). Recent findings in yeast MT-ATP6 mutants have confirmed the role of these mutations in the PTP response to Ca2+ (31), providing additional genetic evidence that supports the involvement of mutations in the H+-ATP synthase in PTP functioning during carcinogenesis. However, it should be noted that the two mutations in MT-ATP6 impact the PTP response only when the function of the outer mitochondrial membrane porin complex is perturbed (i.e., OM45-GFP background) (31). In fact, permeability transition has been documented in rho0 cells that lack mtDNA (32), highlighting the relevance of the genetic background of the cancer cell for the desensitization of the PTP.
Regardless of oncogenic mutations on the H+-ATP synthase, it has been documented that the relative expression of the catalytic subunit of the complex (β-F1-ATPase) is downregulated in most prevalent human carcinomas when compared with the corresponding normal tissues (33, 34) [for review, see Ref. (2)]. The relative expression of β-F1-ATPase in the tissue provides a “bioenergetic signature” of the carcinoma that informs of the overall capacity of mitochondria. The bioenergetic signature [also known as the bioenergetic cellular (BEC) index (2)], is assessed as the protein ratio between β-F1-ATPase and GAPDH and has been shown to be significantly reduced in colon, lung, breast, gastric, and renal carcinomas (2, 33). Interestingly, the quantification of these two proteins in carcinomas derived from different tissues (lung, esophagus, and breast) show similar quantities irrespective of the large differences found in their content in normal tissues (35). These findings support that during oncogenic transformation the tissue-specific differences in energy metabolism are abolished to converge on a similar phenotype to support tumor growth (35). In addition, the BEC index is a biomarker for cancer prognosis and response to therapy. In fact, a higher BEC index predicts a better overall survival and/or disease-free survival in acute myeloid leukemia patients and in colon, lung, breast, and ovarian cancer patients (36–42). These findings thus support that an impaired bioenergetic function of mitochondria favors recurrence and progression of the disease. Moreover, the BEC index also provides a tool for predicting the therapeutic response to various chemotherapeutic strategies aimed at combating tumor progression (43–46).
From a mechanistic view point the control of β-F1-ATPase expression is essentially exerted at post-transcriptional levels (47). In this regard, the translation of β-F1-ATPase mRNA (β-mRNA) both during development and in oncogenesis requires the specific activity of a cis element in the 3′ untranslated region of the mRNA that tightly controls its translation by RNA binding proteins (48–53) and miRNAs (54).
The Diverse Role of Inhibitory Factor 1 (IF1) in Human Carcinomas
Besides the lower BEC index found in tumors, some prevalent human carcinomas also upregulate the expression of the ATPase IF1, which is the physiological inhibitor of the H+-ATP synthase (55, 56). Classically, IF1 was thought to function only to prevent mitochondrial ATP consumption by the reverse activity of the H+-ATP synthase (ATP hydrolase), which happens when mitochondria become de-energized such as in ischemia or in hypoxia (57, 58). However, more recent findings indicate that IF1 can bind to the H+-ATP synthase under normal phosphorylating conditions, hence inhibiting also the forward ATP synthetic activity of the enzyme (59). It should be noted that when arguing about the inhibition exerted by IF1 on the H+-ATP synthase it is important to take into consideration the tissue content of IF1 and the molar ratio that exists between IF1 and the H+-ATP synthase because the tissue availability of the inhibitor affects, among other factors, its interaction with the enzyme by the mass–action ratio. Unfortunately, the information of the tissue content of these two proteins in human and mouse tissues is presently missing.
In addition, it should be stressed that IF1 binding to the H+-ATP synthase, and hence its activity as an inhibitor of the enzyme, is subjected to a stringent posttranslational regulation of the protein by phosphorylation (59). In this regard, we have shown that IF1 is phosphorylated in S39 by a mitochondrial cAMP-dependent protein kinase that renders IF1 unable to bind to the H+-ATP synthase and hence inactive as an inhibitor of the enzyme (59). Regulation of IF1 phosphorylation depends on the cellular metabolic state to allow the fine tuning of ATP production to the cellular metabolic demand (56, 59). In this regard, we should stress that IF1 is found dephosphorylated, and hence active as an inhibitor of the enzyme in colon, lung, and breast carcinomas as well as in hypoxic cells and in cells progressing through the S/G2/M phases of the cell cycle (59).
In addition, IF1 is sharply upregulated in colon, lung, breast, and ovarian carcinomas, which are tissues that under normal physiological conditions are essentially devoid of the inhibitory protein (60–62). Not surprisingly, IF1 is an independent prognostic marker of disease progression for patients bearing these carcinomas. In non-small cell lung cancer (63), bladder carcinomas (64), and gliomas (65), a high expression level of IF1 in the tumor predicts a worse patient prognosis. On the contrary, in colon and breast cancer patients, a high level of IF1 expression predicts a better outcome (60, 66), especially in the bad prognosis group of triple-negative breast cancer patients (67). In the case of breast cancer, lymph node metastases show a lower expression level of IF1 when compared with the primary tumors (68). This finding suggested that breast cancer cells expressing low levels of IF1 may have a higher metastatic potential, which is in full agreement with our recent finding that low IF1 expression in triple-negative breast cancer cells confers a more invasive phenotype (67).
By contrast, human tissues that express high levels of IF1 under basal physiological conditions such as endometrium, kidney, liver, and stomach do not experience a relevant increase in IF1 expression by oncogenesis (60, 62). Nevertheless, in hepatocarcinomas (69) and in gastric carcinomas (70), a higher tumor content of IF1 predicts a worse prognosis for the patients. However, it should be mentioned that a higher IF1-mRNA expression level is correlated with a better prognosis in patients bearing the intestinal subtype of gastric cancer (66). This apparent discrepancy might arise from the histological type of gastric carcinomas analyzed in both studies and/or because the expression of IF1 in human carcinomas is primarily exerted at posttranscriptional levels (60). Overall, these findings support that IF1 plays a relevant role in cancer origin and progression. However, it remains to be elucidated the differential role played by IF1 in favoring or repressing cancer progression in different types of carcinomas, strongly emphasizing the need for specific studies in cellular, xenograft, and genetically modified mouse models in which to address these issues (67, 71).
The Impact of IF1-Mediated Inhibition of the H+-ATP Synthase in Cancer
The role of the H+-ATP synthase in cancer and in signaling has been studied by developing cellular and mouse models with regulated expression of IF1. The overexpression of IF1 both in cultured cells (55, 60, 61, 67) and in different tissues in vivo (71–73) is sufficient to promote metabolic reprogramming to an enhanced aerobic glycolysis (Figure 2). Upregulation of glycolysis results from the limitation of cellular ATP availability as a result of the inhibition of the H+-ATP synthase, supporting that the rate of ATP production by OXPHOS defines the rate of glucose consumption by aerobic glycolysis (74, 75). Likewise, this metabolic situation triggers the activation of the energy sensor AMPK (71–73) (Figure 2). Conversely, the silencing of IF1 in cells that express high levels of the protein has the opposite metabolic effect (55, 60, 61).
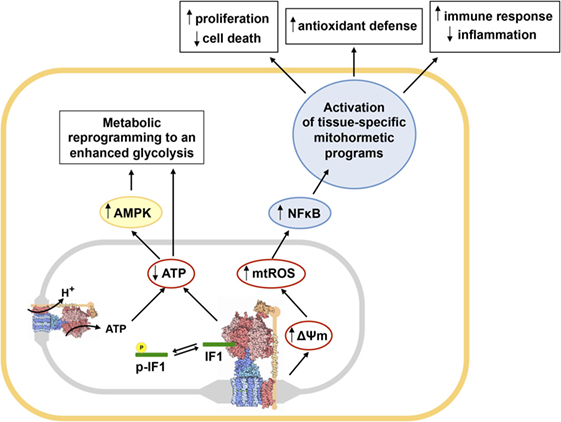
Figure 2. Main metabolic and redox circuits regulated by the inhibition of the H+-ATP synthase by inhibitory factor 1 (IF1). Dephosphorylated IF1 (green rod) inhibits the H+-ATP synthase (structure) when bound to the enzyme, while IF1 phosphorylation (yellow) prevents its binding and hence its inhibitory activity (59). The inhibition of a relevant pool of molecules of H+-ATP synthase by IF1 reduces cellular ATP availability, promoting metabolic reprogramming to an enhanced glycolysis and the activation of AMPK (71–73). Moreover, IF1 inhibition of the H+-ATP synthase triggers mitochondrial hyperpolarization because prevents H+ backflow through the enzyme enhancing the production of mitochondrial ROS (mtROS) that activate the canonical NFκB pathway (61, 72, 73). Activation of NFκB triggers the induction of different tissue-specific mitohormetic programs in the nucleus of the cell. In colon cancer cells overexpressing IF1, these programs favor proliferation, invasion, and resistance to cell death (61). In transgenic mice overexpressing IF1 in neurons or liver, promote the activation of survival pathways and antioxidant defenses (71, 72). In transgenic mice overexpressing IF1 in the intestine, the activated programs modulate the immune response of the tissue, favoring the development of an anti-inflammatory phenotype (73). Abbreviations: p-IF1, phosphorylated IF1; AMPK, AMP-activated protein kinase; NFκB, nuclear factor kappa B. The structure of the H+-ATP synthase was obtained from PDB.
The inhibition of the H+-ATP synthase by IF1 also reduces the backflow of protons into the mitochondrial matrix, triggering mitochondrial hyperpolarization and a mild increase in the production of mitochondrial ROS (mtROS) (Figure 2), both in vitro and in vivo (55, 60, 61, 67, 72, 73). In cells and tissues overexpressing IF1 mtROS trigger the carbonylation of some cellular proteins and signal the activation of the canonical nuclear factor kappa B (NFκB) pathway (71–73) (Figure 2). In colon cancer cells, NFκB induces a nuclear transcriptional program that favors cellular proliferation, invasion, and evasion of cell death (61). These results are in line with other findings reporting that mtROS are necessary for proliferation and tumorigenesis and that ROS scavenging with mitochondrial-targeted antioxidants reduces cancer cell growth and prosurvival pathways (14, 76, 77). A recent study also argues that IF1 overexpression in carcinomas might contribute to cancer progression by limiting the processing of the pro-fusion dynamin-related protein optic atrophy 1 and thus limiting cristae remodeling during apoptosis (78).
However, the phenotypic changes triggered by IF1 overexpression in colon cancer cells cannot be generalized to other cellular types. In fact, the transcriptional program triggered by IF1 overexpression in triple-negative breast cancer cells supports just the opposite, a less proliferative and invasive phenotype (67). This phenotype for breast cancer cells was confirmed by functional analysis illustrating that IF1 overexpression promotes cell adhesion and maintenance of the extracellular matrix hampering epithelial to mesenchymal transition (67). Accordingly, the results may explain why breast cancer patients with high IF1 expression in the carcinoma have a better prognosis (60, 67).
In the mouse model overexpressing IF1-H49K (a constitutively active mutant of IF1) in the liver, we have shown that downregulation of OXPHOS triggers the induction of AMPK rendering a liver phenotype that is prone to cancer development (71). Indeed, transgenic mice when challenged with the carcinogen diethylnitrosamine develop more and bigger tumors than control mice because there is more extensive proliferation and diminished apoptosis of liver cells (71). Remarkably, IF1 overexpression in human hepatocarcinomas also triggers the activation of NFκB (Figure 2), which drives the promotion of angiogenesis and epithelial to mesenchymal transition (69). Not surprisingly, the expression of IF1 in liver cancer predicts a bad overall prognosis and the recurrence of the disease in these patients (69).
Mechanistically, although dimers of H+-ATP synthase are critical components of the PTP (19), and the overexpression of IF1 in the liver in vivo favors the formation of dimers of the enzyme (71), we have observed that cell death protection is not related to differential opening and regulation of the PTP (71). Actually, we support that the cell death protection afforded by IF1 overexpression in the liver is related to mitohormetic signaling through the induction of an antioxidant response guided by Nrf2 [nuclear factor (erythroid-derived 2)-like 2] (Figure 2) (71) because the metabolically preconditioned hepatocytes are more resistant to acetaminophen induced toxicity (71). Interestingly, Nrf2 upregulation is also a strategy deployed by cancer cells to detoxify the higher ROS levels that are produced in these cells (79).
IF1-Mediated Inhibition of the H+-ATP Synthase Modulates Metabolic and Redox Circuits
Besides the liver, overexpression of IF1-H49K in neurons in vivo also reprograms energy metabolism to an enhanced glycolysis and affords metabolic preconditioning (72). In fact, mice overexpressing IF1-H49K in forebrain neurons are partially protected from excitotoxic damage induced by striatal administration of quinolinic acid because preconditioning partially protects neurons from death, reducing the lesion area in the brain and improving motor performance of the transgenic mice (72). Preconditioning in neurons also involves AMPK activation and mtROS-mediated signaling to implement a Bcl-xL-mediated protection of neurons from apoptosis (72) (Figure 2). Other findings also support the neuroprotective role of IF1 in ischemia reoxygenation by promoting autophagy and maintaining mitochondrial bioenergetics (80), although the contribution of IF1 to promotion of autophagy in neurons remains to be studied in the in vivo model.
Interestingly, the signaling pathways triggered by the IF1-mediated inhibition of the H+-ATP synthase are not limited to the cells overexpressing IF1 but also implicate non-cell autonomous processes. The transgenic mice overexpressing IF1 in enterocytes also show metabolic reprogramming to an enhanced glycolysis and activation of mtROS–NFκB signaling pathway (73) (Figure 2). Transgenic mice are partially protected from intestinal inflammation after the administration of the inflammatory agent dextran sodium sulfate (DSS), due to increased recruitment of regulatory T cells and macrophages that are mainly polarized to the M2 phenotype (73). These findings are consistent with the anti-inflammatory phenotype afforded by the overexpression of IF1 as revealed by the higher levels of anti-inflammatory cytokines present in plasma and intestine of the transgenic mice (73). Colonocytes of control mice induce the oncogenic Akt/mTOR/p70S6K and pro-inflammatory STAT3 pathways upon administration of DSS, something that is not observed in IF1-transgenic mice (73). Preconditioning and protection against stress in colon of IF1-transgenic mice clearly responds to the basal activation of the NFκB pathway due to mtROS production (73) (Figure 2), because protection from intestinal inflammation is blunted when an mtROS scavenger or an inhibitor of NFκB are administered (73).
Overall, transgenic mice overexpressing IF1 in liver, brain, or intestine reveal that by partial inhibition of OXPHOS and the production of mtROS the tissues acquire an advantageous phenotype against different forms of oxidative stress and inflammation (71–73) (Figure 2), stressing the role of the H+-ATP synthase as a therapeutic target in diverse human pathologies.
Longevity and the Inhibition of the H+-ATP Synthase
Besides IF1, the H+-ATP synthase can be inhibited by some mitochondrial metabolites produced in the TCA cycle, such as α-ketoglutarate (α-KG) (81). Therefore, partial inhibition of the enzyme by α-KG also reduces ATP availability and TOR signaling, promoting autophagy in Caenorhabditis elegans (81). In addition, the oncometabolite (R)-2-hydroxyglutarate (2-HG), which is structurally similar to α-KG, also inhibits the H+-ATP synthase both in C. elegans and in mammalian cell lines (82). 2-HG is highly accumulated in some gliomas and acute myeloid leukemias that harbor mutations in the genes encoding the cytosolic and mitochondrial isocitrate dehydrogenases (IDH1 and IDH2, respectively). These mutations result in neomorphic enzymes with higher affinity for α-KG that catalyze its conversion into 2-HG (83, 84). Interestingly, the inhibition of the H+-ATP synthase by 2-HG or α-KG in glioblastoma cells triggers cell growth arrest and cell death under conditions of limited glucose (82). These results are consistent with previous findings that indicate that brain cancer patients with IDH mutations have a longer median overall survival than patients without mutations (83, 85). The suppression of cell growth may be ascribed to reduced ATP levels and mTOR signaling in the tumors (82).
Interestingly, both α-KG and 2-HG, by inhibiting the H+-ATP synthase, also extend lifespan in C. elegans (81, 82), and α-KG might play a role in longevity induced by dietary restriction (81). Other interventions targeting the H+-ATP synthase have also been shown to extend lifespan in several model organisms, as recently reviewed (62). For instance, silencing of subunits of the H+-ATP synthase in C. elegans and Drosophila melanogaster promotes longevity (86, 87). No contribution to longevity has been reported so far in mammals for the modulation of the H+-ATP synthase. However, recent findings indicate that the specific inhibition of the enzyme by the small molecule J147 prevents the age-associated drift of the hippocampal transcriptome and plasma metabolome in mice and extends lifespan in D. melanogaster, providing an additional link between the activity of the H+-ATP synthase, aging and age-associated pathologies such as dementia (88).
Overall, these findings support the notion that the H+-ATP synthase is also a conserved hub in intracellular signaling that plays a key role in signaling mitohormesis contributing to cell fate decisions and longevity.
Concluding Remarks
Reprogramming cellular metabolism is a hallmark of cancer that is necessary to fulfill the metabolic demands imposed by the oncogenic process. In this context, the mitochondrial H+-ATP synthase is a main hub in rewiring energy metabolism and in retrograde signaling to the nucleus programs required for cancer progression. In this regard, most prevalent carcinomas show reduced expression of the catalytic subunit of the H+-ATP synthase (β-F1-ATPase) relative to the glycolytic GAPDH, what provides a protein signature of energy metabolism of clinical relevance in oncogenesis. Moreover, some prevalent carcinomas also show an increased expression of IF1, the physiological inhibitor of the enzyme. As revealed in different in vitro and in vivo systems, IF1 overexpression is sufficient to rewire energy metabolism to an enhanced glycolysis and to trigger an mtROS signal that promotes nuclear reprogramming. IF1-mediated reprogramming is mainly geared by the activation of AMPK and NFκB pathways resulting in the induction of tissue-specific programs aimed at preventing cell death, oxidative damage or inflammation. The precise molecular events that lead to the upregulation of IF1 in cancer and its role in cancer progression in different carcinomas remain to be established. The H+-ATP synthase, engine of OXPHOS, is also a crucial hub in mitohormetic signaling to modulate cytoprotective defenses that contribute to longevity in several organisms. Therefore, deciphering the metabolic and redox circuits controlled by the H+-ATP synthase and IF1 are of utmost importance to understand how they contribute to oncogenesis and thus providing new targets for cancer and age-associated diseases.
Author Contributions
PE-M and JC wrote the paper. All the authors read, contributed, and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
PE-M was supported by a predoctoral fellowship from Fundación La Caixa (Obra Social La Caixa). The work was supported by grants from MINECO (SAF2016-75916-R), CIBERER-ISCIII, and Fundación Ramón Areces, Spain.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
2. Cuezva JM, Ortega AD, Willers I, Sanchez-Cenizo L, Aldea M, Sanchez-Arago M. The tumor suppressor function of mitochondria: translation into the clinics. Biochim Biophys Acta (2009) 1792(12):1145–58. doi:10.1016/j.bbadis.2009.01.006
3. Wang R, Green DR. Metabolic reprogramming and metabolic dependency in T cells. Immunol Rev (2012) 249(1):14–26. doi:10.1111/j.1600-065X.2012.01155.x
4. Vander Heiden MG, Lunt SY, Dayton TL, Fiske BP, Israelsen WJ, Mattaini KR, et al. Metabolic pathway alterations that support cell proliferation. Cold Spring Harb Symp Quant Biol (2013) 76:325–34. doi:10.1101/sqb.2012.76.010900
5. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol (2011) 27:441–64. doi:10.1146/annurev-cellbio-092910-154237
7. Gaude E, Frezza C. Defects in mitochondrial metabolism and cancer. Cancer Metab (2014) 2:10. doi:10.1186/2049-3002-2-10
8. Sciacovelli M, Goncalves E, Johnson TI, Zecchini VR, Da Costa AS, Gaude E, et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature (2016) 537(7621):544–7. doi:10.1038/nature19353
9. Zong WX, Rabinowitz JD, White E. Mitochondria and cancer. Mol Cell (2016) 61(5):667–76. doi:10.1016/j.molcel.2016.02.011
10. Chandel NS. Evolution of mitochondria as signaling organelles. Cell Metab (2015) 22(2):204–6. doi:10.1016/j.cmet.2015.05.013
11. Sassano ML, Van Vliet AR, Agostinis P. Mitochondria-associated membranes as networking platforms and regulators of cancer cell fate. Front Oncol (2017) 7:174. doi:10.3389/fonc.2017.00174
12. Raimundo N, Baysal BE, Shadel GS. Revisiting the TCA cycle: signaling to tumor formation. Trends Mol Med (2011) 17(11):641–9. doi:10.1016/j.molmed.2011.06.001
13. Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell (2015) 163(3):560–9. doi:10.1016/j.cell.2015.10.001
14. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A (2010) 107(19):8788–93. doi:10.1073/pnas.1003428107
15. Woo DK, Green PD, Santos JH, D’souza AD, Walther Z, Martin WD, et al. Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APC(Min/+) mice. Am J Pathol (2012) 180(1):24–31. doi:10.1016/j.ajpath.2011.10.003
16. Pendin D, Filadi R, Pizzo P. The concerted action of mitochondrial dynamics and positioning: new characters in cancer onset and progression. Front Oncol (2017) 7:102. doi:10.3389/fonc.2017.00102
17. Walker JE. The ATP synthase: the understood, the uncertain and the unknown. Biochem Soc Trans (2013) 41(1):1–16. doi:10.1042/BST20110773
18. Saita E, Suzuki T, Kinosita K Jr, Yoshida M. Simple mechanism whereby the F1-ATPase motor rotates with near-perfect chemomechanical energy conversion. Proc Natl Acad Sci U S A (2015) 112(31):9626–31. doi:10.1073/pnas.1422885112
19. Giorgio V, Von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A (2013) 110(15):5887–92. doi:10.1073/pnas.1217823110
20. Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A (2014) 111(29):10580–5. doi:10.1073/pnas.1401591111
21. Bernardi P, Rasola A, Forte M, Lippe G. The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol Rev (2015) 95(4):1111–55. doi:10.1152/physrev.00001.2015
22. He J, Carroll J, Ding S, Fearnley IM, Walker JE. Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc Natl Acad Sci U S A (2017) 114(34):9086–91. doi:10.1073/pnas.1711201114
23. He J, Ford HC, Carroll J, Ding S, Fearnley IM, Walker JE. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc Natl Acad Sci U S A (2017) 114(13):3409–14. doi:10.1073/pnas.1702357114
24. Giorgio V, Guo L, Bassot C, Petronilli V, Bernardi P. Calcium and regulation of the mitochondrial permeability transition. Cell Calcium (2017) 70:56–63. doi:10.1016/j.ceca.2017.05.004
25. Giorgio V, Burchell V, Schiavone M, Bassot C, Minervini G, Petronilli V, et al. Ca(2+) binding to F-ATP synthase beta subunit triggers the mitochondrial permeability transition. EMBO Rep (2017) 18(7):1065–76. doi:10.15252/embr.201643354
26. Antoniel M, Jones K, Antonucci S, Spolaore B, Fogolari F, Petronilli V, et al. The unique histidine in OSCP subunit of F-ATP synthase mediates inhibition of the permeability transition pore by acidic pH. EMBO Rep (2018) 19(2):257–68. doi:10.15252/embr.201744705
27. Sanchez-Arago M, Formentini L, Cuezva JM. Mitochondria-mediated energy adaption in cancer: the H(+)-ATP synthase-geared switch of metabolism in human tumors. Antioxid Redox Signal (2013) 19(3):285–98. doi:10.1089/ars.2012.4883
28. Martinez-Reyes I, Cuezva JM. The H(+)-ATP synthase: a gate to ROS-mediated cell death or cell survival. Biochim Biophys Acta (2014) 1837(7):1099–112. doi:10.1016/j.bbabio.2014.03.010
29. Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A (2005) 102(3):719–24. doi:10.1073/pnas.0408894102
30. Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, Manfredi G, et al. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res (2005) 65(5):1655–63. doi:10.1158/0008-5472.CAN-04-2012
31. Niedzwiecka K, Tisi R, Penna S, Lichocka M, Plochocka D, Kucharczyk R. Two mutations in mitochondrial ATP6 gene of ATP synthase, related to human cancer, affect ROS, calcium homeostasis and mitochondrial permeability transition in yeast. Biochim Biophys Acta (2018) 1865(1):117–31. doi:10.1016/j.bbamcr.2017.10.003
32. Masgras I, Rasola A, Bernardi P. Induction of the permeability transition pore in cells depleted of mitochondrial DNA. Biochim Biophys Acta (2012) 1817(10):1860–6. doi:10.1016/j.bbabio.2012.02.022
33. Cuezva JM, Krajewska M, De Heredia ML, Krajewski S, Santamaria G, Kim H, et al. The bioenergetic signature of cancer: a marker of tumor progression. Cancer Res (2002) 62(22):6674–81.
34. Isidoro A, Martinez M, Fernandez PL, Ortega AD, Santamaria G, Chamorro M, et al. Alteration of the bioenergetic phenotype of mitochondria is a hallmark of breast, gastric, lung and oesophageal cancer. Biochem J (2004) 378(Pt 1):17–20. doi:10.1042/bj20031541
35. Acebo P, Giner D, Calvo P, Blanco-Rivero A, Ortega AD, Fernandez PL, et al. Cancer abolishes the tissue type-specific differences in the phenotype of energetic metabolism. Transl Oncol (2009) 2(3):138–45. doi:10.1593/tlo.09106
36. Cuezva JM, Chen G, Alonso AM, Isidoro A, Misek DE, Hanash SM, et al. The bioenergetic signature of lung adenocarcinomas is a molecular marker of cancer diagnosis and prognosis. Carcinogenesis (2004) 25(7):1157–63. doi:10.1093/carcin/bgh113
37. Isidoro A, Casado E, Redondo A, Acebo P, Espinosa E, Alonso AM, et al. Breast carcinomas fulfill the Warburg hypothesis and provide metabolic markers of cancer prognosis. Carcinogenesis (2005) 26(12):2095–104. doi:10.1093/carcin/bgi188
38. Lopez-Rios F, Sanchez-Arago M, Garcia-Garcia E, Ortega AD, Berrendero JR, Pozo-Rodriguez F, et al. Loss of the mitochondrial bioenergetic capacity underlies the glucose avidity of carcinomas. Cancer Res (2007) 67(19):9013–7. doi:10.1158/0008-5472.CAN-07-1678
39. Lin PC, Lin JK, Yang SH, Wang HS, Li AF, Chang SC. Expression of beta-F1-ATPase and mitochondrial transcription factor A and the change in mitochondrial DNA content in colorectal cancer: clinical data analysis and evidence from an in vitro study. Int J Colorectal Dis (2008) 23(12):1223–32. doi:10.1007/s00384-008-0539-4
40. Aldea M, Clofent J, Nunez De Arenas C, Chamorro M, Velasco M, Berrendero JR, et al. Reverse phase protein microarrays quantify and validate the bioenergetic signature as biomarker in colorectal cancer. Cancer Lett (2011) 311(2):210–8. doi:10.1016/j.canlet.2011.07.022
41. Hjerpe E, Egyhazi Brage S, Carlson J, Frostvik Stolt M, Schedvins K, Johansson H, et al. Metabolic markers GAPDH, PKM2, ATP5B and BEC-index in advanced serous ovarian cancer. BMC Clin Pathol (2013) 13(1):30. doi:10.1186/1472-6890-13-30
42. Xiao X, Yang J, Li R, Liu S, Xu Y, Zheng W, et al. Deregulation of mitochondrial ATPsyn-beta in acute myeloid leukemia cells and with increased drug resistance. PLoS One (2013) 8(12):e83610. doi:10.1371/journal.pone.0083610
43. Shin YK, Yoo BC, Chang HJ, Jeon E, Hong SH, Jung MS, et al. Down-regulation of mitochondrial F1F0-ATP synthase in human colon cancer cells with induced 5-fluorouracil resistance. Cancer Res (2005) 65(8):3162–70. doi:10.1158/0008-5472.CAN-04-3300
44. Li RJ, Zhang GS, Chen YH, Zhu JF, Lu QJ, Gong FJ, et al. Down-regulation of mitochondrial ATPase by hypermethylation mechanism in chronic myeloid leukemia is associated with multidrug resistance. Ann Oncol (2010) 7:1506–14. doi:10.1093/annonc/mdp569
45. Sanchez-Arago M, Cuezva JM. The bioenergetic signature of isogenic colon cancer cells predicts the cell death response to treatment with 3-bromopyruvate, iodoacetate or 5-fluorouracil. J Transl Med (2011) 9:19. doi:10.1186/1479-5876-9-19
46. Yizhak K, Le Devedec SE, Rogkoti VM, Baenke F, De Boer VC, Frezza C, et al. A computational study of the Warburg effect identifies metabolic targets inhibiting cancer migration. Mol Syst Biol (2014) 10:744. doi:10.15252/msb.20134993
47. Willers IM, Cuezva JM. Post-transcriptional regulation of the mitochondrial H(+)-ATP synthase: a key regulator of the metabolic phenotype in cancer. Biochim Biophys Acta (2011) 1807(6):543–51. doi:10.1016/j.bbabio.2010.10.016
48. Izquierdo JM, Cuezva JM. Control of the translational efficiency of beta-F1-ATPase mRNA depends on the regulation of a protein that binds the 3’ untranslated region of the mRNA. Mol Cell Biol (1997) 17(9):5255–68. doi:10.1128/MCB.17.9.5255
49. Izquierdo JM, Cuezva JM. Internal-ribosome-entry-site functional activity of the 3’-untranslated region of the mRNA for the beta subunit of mitochondrial H+-ATP synthase. Biochem J (2000) 346(Pt 3):849–55. doi:10.1042/bj3460849
50. De Heredia ML, Izquierdo JM, Cuezva JM. A conserved mechanism for controlling the translation of beta-F1-ATPase mRNA between the fetal liver and cancer cells. J Biol Chem (2000) 275(10):7430–7. doi:10.1074/jbc.275.10.7430
51. Willers IM, Isidoro A, Ortega AD, Fernandez PL, Cuezva JM. Selective inhibition of beta-F1-ATPase mRNA translation in human tumours. Biochem J (2010) 426(3):319–26. doi:10.1042/BJ20091570
52. Ortega AD, Sala S, Espinosa E, Gonzalez-Baron M, Cuezva JM. HuR and the bioenergetic signature of breast cancer: a low tumor expression of the RNA-binding protein predicts a higher risk of disease recurrence. Carcinogenesis (2008) 29(11):2053–61. doi:10.1093/carcin/bgn185
53. Ortega AD, Willers IM, Sala S, Cuezva JM. Human G3BP1 interacts with beta-F1-ATPase mRNA and inhibits its translation. J Cell Sci (2010) 123(Pt 16):2685–96. doi:10.1242/jcs.065920
54. Willers IM, Martínez-Reyes I, Martínez-Diez M, Cuezva J. miR-127-5p targets the 3’UTR of human β-F1-ATPase mRNA and inhibits its translation. Biochim Biophys Acta Bioenergetics (2012) 1817(5):838–48. doi:10.1016/j.bbabio.2012.03.005
55. Sanchez-Cenizo L, Formentini L, Aldea M, Ortega AD, Garcia-Huerta P, Sanchez-Arago M, et al. Up-regulation of the ATPase inhibitory factor 1 (IF1) of the mitochondrial H+-ATP synthase in human tumors mediates the metabolic shift of cancer cells to a Warburg phenotype. J Biol Chem (2010) 285(33):25308–13. doi:10.1074/jbc.M110.146480
56. Garcia-Bermudez J, Cuezva JM. The ATPase inhibitory factor 1 (IF1): a master regulator of energy metabolism and of cell survival. Biochim Biophys Acta (2016) 1857:1167–82. doi:10.1016/j.bbabio.2016.02.004
57. Gledhill JR, Montgomery MG, Leslie AG, Walker JE. How the regulatory protein, IF(1), inhibits F(1)-ATPase from bovine mitochondria. Proc Natl Acad Sci U S A (2007) 104(40):15671–6. doi:10.1073/pnas.0707326104
58. Campanella M, Casswell E, Chong S, Farah Z, Wieckowski MR, Abramov AY, et al. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab (2008) 8(1):13–25. doi:10.1016/j.cmet.2008.06.001
59. Garcia-Bermudez J, Sanchez-Arago M, Soldevilla B, Del Arco A, Nuevo-Tapioles C, Cuezva JM. PKA phosphorylates the ATPase inhibitory factor 1 and inactivates its capacity to bind and inhibit the mitochondrial H-ATP synthase. Cell Rep (2015) 12:2143–55. doi:10.1016/j.celrep.2015.08.052
60. Sanchez-Arago M, Formentini L, Martinez-Reyes I, Garcia-Bermudez J, Santacatterina F, Sanchez-Cenizo L, et al. Expression, regulation and clinical relevance of the ATPase inhibitory factor 1 in human cancers. Oncogenesis (2013) 2:e46. doi:10.1038/oncsis.2013.9
61. Formentini L, Sánchez-Aragó M, Sánchez-Cenizo L, Cuezva JM. The mitochondrial ATPase inhibitory factor 1 (IF1) triggers a ROS-mediated retrograde pro-survival and proliferative response. Mol Cell (2012) 45:731–42. doi:10.1016/j.molcel.2012.01.008
62. Esparza-Molto PB, Nuevo-Tapioles C, Cuezva JM. Regulation of the H+-ATP synthase by IF1: a role in mitohormesis. Cell Mol Life Sci (2017) 74(12):2151–66. doi:10.1007/s00018-017-2462-8
63. Gao YX, Chen L, Hu XG, Wu HB, Cui YH, Zhang X, et al. ATPase inhibitory factor 1 expression is an independent prognostic factor in non-small cell lung cancer. Am J Cancer Res (2016) 6(5):1141–8.
64. Wei S, Fukuhara H, Kawada C, Kurabayashi A, Furihata M, Ogura S, et al. Silencing of ATPase inhibitory factor 1 inhibits cell growth via cell cycle arrest in bladder cancer. Pathobiology (2015) 82(5):224–32. doi:10.1159/000439027
65. Wu J, Shan Q, Li P, Wu Y, Xie J, Wang X. ATPase inhibitory factor 1 is a potential prognostic marker for the migration and invasion of glioma. Oncol Lett (2015) 10(4):2075–80. doi:10.3892/ol.2015.3548
66. Zhang C, Min L, Liu J, Tian W, Han Y, Qu L, et al. Integrated analysis identified an intestinal-like and a diffuse-like gene sets that predict gastric cancer outcome. Tumour Biol (2016) 37(12):16317–35. doi:10.1007/s13277-016-5454-7
67. Garcia-Ledo L, Nuevo-Tapioles C, Cuevas-Martin C, Martinez-Reyes I, Soldevilla B, Gonzalez-Llorente L, et al. Overexpression of the ATPase inhibitory factor 1 favors a non-metastatic phenotype in breast cancer. Front Oncol (2017) 7:69. doi:10.3389/fonc.2017.00069
68. Kurbasic E, Sjostrom M, Krogh M, Folkesson E, Grabau D, Hansson K, et al. Changes in glycoprotein expression between primary breast tumour and synchronous lymph node metastases or asynchronous distant metastases. Clin Proteomics (2015) 12(1):13. doi:10.1186/s12014-015-9084-7
69. Song R, Song H, Liang Y, Yin D, Zhang H, Zheng T, et al. Reciprocal activation between ATPase inhibitory factor 1 and NF-kappaB drives hepatocellular carcinoma angiogenesis and metastasis. Hepatology (2014) 60:1659–73. doi:10.1002/hep.27312
70. Yin T, Lu L, Xiong Z, Wei S, Cui D. ATPase inhibitory factor 1 is a prognostic marker and contributes to proliferation and invasion of human gastric cancer cells. Biomed Pharmacother (2015) 70:90–6. doi:10.1016/j.biopha.2014.12.036
71. Santacatterina F, Sanchez-Cenizo L, Formentini L, Mobasher MA, Casas E, Rueda CB, et al. Down-regulation of oxidative phosphorylation in the liver by expression of the ATPase inhibitory factor 1 induces a tumor-promoter metabolic state. Oncotarget (2016) 7(1):490–508. doi:10.18632/oncotarget.6357
72. Formentini L, Pereira MP, Sanchez-Cenizo L, Santacatterina F, Lucas JJ, Navarro C, et al. In vivo inhibition of the mitochondrial H+-ATP synthase in neurons promotes metabolic preconditioning. EMBO J (2014) 33(7):762–78. doi:10.1002/embj.201386392
73. Formentini L, Santacatterina F, Nunez De Arenas C, Stamatakis K, Lopez-Martinez D, Logan A, et al. Mitochondrial ROS production protects the intestine from inflammation through functional M2 macrophage polarization. Cell Rep (2017) 19(6):1202–13. doi:10.1016/j.celrep.2017.04.036
75. Sanchez-Arago M, Chamorro M, Cuezva JM. Selection of cancer cells with repressed mitochondria triggers colon cancer progression. Carcinogenesis (2010) 31(4):567–76. doi:10.1093/carcin/bgq012
76. Nazarewicz RR, Dikalova A, Bikineyeva A, Ivanov S, Kirilyuk IA, Grigor’ev IA, et al. Does scavenging of mitochondrial superoxide attenuate cancer prosurvival signaling pathways? Antioxid Redox Signal (2013) 19(4):344–9. doi:10.1089/ars.2013.5185
77. Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep (2014) 8(3):754–66. doi:10.1016/j.celrep.2014.06.043
78. Faccenda D, Nakamura J, Gorini G, Dhoot GK, Piacentini M, Yoshida M, et al. Control of mitochondrial remodeling by the ATPase inhibitory factor 1 unveils a pro-survival relay via OPA1. Cell Rep (2017) 18(8):1869–83. doi:10.1016/j.celrep.2017.01.070
79. Denicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature (2011) 475(7354):106–9. doi:10.1038/nature10189
80. Matic I, Cocco S, Ferraina C, Martin-Jimenez R, Florenzano F, Crosby J, et al. Neuroprotective coordination of cell mitophagy by the F1Fo-ATPase inhibitory factor 1 (IF). Pharmacol Res (2015) 103:56–68. doi:10.1016/j.phrs.2015.10.010
81. Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature (2014) 510(7505):397–401. doi:10.1038/nature13264
82. Fu X, Chin RM, Vergnes L, Hwang H, Deng G, Xing Y, et al. 2-hydroxyglutarate inhibits ATP synthase and mTOR signaling. Cell Metab (2015) 22(3):508–15. doi:10.1016/j.cmet.2015.06.009
83. Yan H, Parsons DW, Jin G, Mclendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med (2009) 360(8):765–73. doi:10.1056/NEJMoa0808710
84. Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med (2010) 207(2):339–44. doi:10.1084/jem.20092506
85. Van Den Bent MJ, Dubbink HJ, Marie Y, Brandes AA, Taphoorn MJ, Wesseling P, et al. IDH1 and IDH2 mutations are prognostic but not predictive for outcome in anaplastic oligodendroglial tumors: a report of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Clin Cancer Res (2010) 16(5):1597–604. doi:10.1158/1078-0432.CCR-09-2902
86. Hansen M, Hsu AL, Dillin A, Kenyon C. New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet (2005) 1(1):119–28. doi:10.1371/journal.pgen.0010017
87. Sun X, Wheeler CT, Yolitz J, Laslo M, Alberico T, Sun Y, et al. A mitochondrial ATP synthase subunit interacts with TOR signaling to modulate protein homeostasis and lifespan in Drosophila. Cell Rep (2014) 8(6):1781–92. doi:10.1016/j.celrep.2014.08.022
Keywords: oxidative phosphorylation, ATPase inhibitory factor 1, mitohormesis, metabolic reprogramming, hepatocarcinogenesis, inflammation
Citation: Esparza-Moltó PB and Cuezva JM (2018) The Role of Mitochondrial H+-ATP Synthase in Cancer. Front. Oncol. 8:53. doi: 10.3389/fonc.2018.00053
Received: 15 January 2018; Accepted: 20 February 2018;
Published: 07 March 2018
Edited by:
Andrea Rasola, Università degli Studi di Padova, ItalyReviewed by:
Giovanna Lippe, University of Udine, ItalyValentina Giorgio, Institute of Neuroscience (CNR), Italy
Copyright: © 2018 Esparza-Moltó and Cuezva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José M. Cuezva, am1jdWV6dmFAY2JtLmNzaWMuZXM=