 Natalie Geyer1
Natalie Geyer1 Rosalie Ridzewski1
Rosalie Ridzewski1 Julia Bauer1Maria Kuzyakova1
Julia Bauer1Maria Kuzyakova1 Kai Dittmann2Christian Dullin3
Kai Dittmann2Christian Dullin3 Albert Rosenberger4Hans-Ulrich Schildhaus5Anja Uhmann1
Albert Rosenberger4Hans-Ulrich Schildhaus5Anja Uhmann1 Simone Fulda6
Simone Fulda6 Heidi Hahn1*
Heidi Hahn1*- 1Institute for Human Genetics, University Medical Center Goettingen, Goettingen, Germany
- 2Institute for Celluar and Molecular Immunology, University Medical Center Goettingen, Goettingen, Germany
- 3Institute for Diagnostic and Interventional Radiology, University Medical Center Goettingen, Goettingen, Germany
- 4Department of Genetic Epidemiology, University Medical Center Goettingen, Goettingen, Germany
- 5Institute for Pathology, University Medical Center Goettingen, Goettingen, Germany
- 6Institute for Experimental Cancer Research in Pediatrics, Goethe-University Frankfurt, Frankfurt, Germany
Rhabdomyosarcoma (RMS) is the most common pediatric soft tissue sarcoma with poor prognosis. RMS frequently show Hedgehog (HH) pathway activity, which is predominantly seen in the embryonal subtype (ERMS). They also show activation of Phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) signaling. Here we compared the therapeutic effectiveness and the impact on HH target gene expression of Smoothened (SMO) antagonists with those of the PI3K inhibitor pictilisib in ERMS with and without mutations in the HH receptor Patched1 (PTCH). Our data demonstrate that growth of ERMS showing canonical Hh signaling activity due to Ptch germline mutations is efficiently reduced by SMO antagonists. This goes along with strong downregulation of the Hh target Gli1. Likewise Ptch mutant tumors are highly responsive toward the PI3K inhibitor pictilisib, which involves modulation of AKT and caspase activity. Pictilisib also modulates Hh target gene expression, which, however, is rather not correlated with its antitumoral effects. In contrast, sporadic ERMS, which usually express HH target genes without having PTCH mutation, apparently lack canonical HH signaling activity. Thus, stimulation by Sonic HE (SHH) or SAG (Smoothened agonist) or inhibition by SMO antagonists do not modulate HH target gene expression. In addition, SMO antagonists do not provoke efficient anticancer effects and rather exert off-target effects. In contrast, pictilisib and other PI3K/AKT/mTOR inhibitors potently inhibit cellular growth. They also efficiently inhibit HH target gene expression. However, of whether this is correlated with their antitumoral effects it is not clear. Together, these data suggest that PI3K inhibitors are a good and reliable therapeutic option for all ERMS, whereas SMO inhibitors might only be beneficial for ERMS driven by PTCH mutations.
Introduction
Hedgehog (HH) signaling plays a major role in a variety of human cancers. The main components of the canonical HH signaling pathway are HH ligands, the transmembrane proteins Patched (PTCH) and Smoothened (SMO) and GLI transcription factors. In the absence of the HH ligand, PTCH represses SMO. Upon binding of HH to PTCH this repression is released. This leads to accumulation of SMO in the primary cilium where it triggers the modulation of a variety of proteins, which finally results in translocation of the GLI2 and GLI3 transcription factors into the nucleus. This activates the expression of HH target genes, which include GLI1 that can amplify the response on transcriptional level. Indeed, GLI1 transcripts serve as the most reliable readout for active HH signaling. Alternative HH-target genes are HHIP and PTCH. However, in contrast to GLI1, the prediction of pathway activity from HHIP and PTCH levels is more difficult, because the respective proteins mediate a negative feedback by sequestering HH [for review see (1)].
The first link between HH signaling and cancer was the discovery of mutations in the HH receptor PTCH in the rare autosomal dominant inherited basal cell nevus syndrome (BCNS, also known as NBCCS or Gorlin Syndrome). Almost all BCNS patients develop basal cell carcinoma (BCC) that show high GLI1 expression and thus an active HH signaling cascade. In addition to BCC these patients are predisposed to other tumors, first of all to medulloblastoma (MB) and less frequently to rhabdomyoma and rhabdomyosarcoma (RMS) (1, 2). Therefore PTCH mutations are considered as driver mutations for these tumors. This is similar to heterozygous Ptch+/− mice, which develop BCC, MB, and RMS at a high frequency (3–5).
Today it is known that HH signaling is also active in many sporadic tumors. Whereas activation of this pathway by somatic mutations occurs only in some tumors (e.g., BCC and MB), other tumors show overexpression of HH ligands, which is seen e.g. in pancreatic, lung, and prostate cancers [for review see (6)]. In addition, the activity of GLI transcription factors and the expression of HH target genes can also be regulated in a non-canonical manner independently of HH, PTCH, and SMO, which can occur e.g., in melanoma and astrocytoma [for review see (7)].
Recently, we and others showed that sporadic RMS express HH ligands and the HH targets GLI1 and PTCH (8–10). This led to the hypothesis that the HH pathway in sporadic RMS may be activated in a canonical ligand-dependent manner.
RMS is the most common pediatric soft tissue sarcoma with poor prognosis for high-risk patients (11). In children, RMS is divided by histology in embryonal (ERMS) and alveolar RMS (ARMS). The latter one can be classified by PAX/FOXO1-gene fusion status as fusion-positive or fusion-negative. Fusion-positive ARMS are very aggressive, whereas fusion-negative ARMS are clinically and molecularly indistinguishable from the less aggressive ERMS subtype (12). Interestingly, ERMS and fusion-negative ARMS exhibit significantly higher HH target gene expression compared to fusion-positive ARMS (8, 9).
Because HH target gene expression in sporadic RMS may be driven by HH ligands i.e., via the canonical HH/PTCH/SMO axis, we recently tested SMO inhibitors including vismodegib and sonidegib for their antitumorigenic effects in human cell lines derived from sporadic RMS. However, the effects of the drugs and the response of the cells were very heterogeneous and did not necessarily correlate with inhibition of HH signaling. Thus, some drugs paradoxically induced cellular proliferation at certain concentrations or showed antiproliferative effects without reduction of HH signaling activity (13). Therefore we hypothesize that (i) reliable anticancer effects using SMO inhibitors may only be achieved in RMS cells harboring PTCH mutations, (ii) some of the above-mentioned results may reflect off-target effects of the SMO inhibitors, and (iii) the HH pathway in RMS is not only regulated via the canonical axis, but also in a HH/PTCH/SMO-independent manner e.g., by the PI3K/AKT/mTOR cascade. The latter assumption is based on the observation that the PI3K/AKT axis plays a crucial role in RMS (14–17) and, like in many other tumors or tumor cell lines [e.g., (18–20)], shows cooperation with HH signaling in RMS cells (21).
We here tested these 3 assumptions by analyzing ERMS cells harboring a Ptch mutation and ERMS cells without obvious PTCH mutations. We used the SMO inhibitors vismodegib, sonidegib (both have been approved for advanced BCC), or HhAntag alone or in combination with PI3K/AKT/mTOR-inhibitors (pictilisib, PI-103, MK-2206, rapamycin, or everolimus) and analyzed the impact on the HH and PI3K pathway and on growth behavior of ERMS cells.
Materials and Methods
Drugs
All used drugs, the respective provider, solvents, applications, and final concentrations are listed in Supplementary Table S1.
Cell Culture
The human ERMS cell lines RD and RUCH-2 were obtained from ATCC [for cell lines see (22)] and were cultured in DMEM, 10% FCS (Thermo Fisher Scientific Inc., Waltham, MA, United States of America) and 1% penicillin/streptomycin (PAN Biotech GmbH, Aidenbach, Germany; stock 10.000 U/ml).
SHH conditioned media (SHH-CM) or respective control-conditioned media (control-CM) were produced from HEK293 cells stably transfected with a SHH expression plasmid or from non-transfected HEK293, respectively, as described by Chen et al. (23). Shh-responsive B9 cells served as positive controls (24).
For preparation of primary murine RMS cultures the tumor was chopped with a razor blade and incubated with 3 mg/ml collagenase H (Roche, Mannheim, Germany) in DMEM for 60 min at 37°C while shaking with 1000 rpm. To release remaining cell aggregates the suspension was strained through a 40 μm nylon filter. Cells were sedimented at 300x g, 4°C for 5 min. The pellet was resuspended in DMEM containing 10% FCS and 1% penicillin/streptomycin and cells were seeded on collagen coated 96-well plates for proliferation analysis or 12-well plates for qRT-PCR analysis and western blot analysis. When 90% of cells were adherent, experiments were started. Cells were kept in culture for no longer than 4 days.
For analysis of cilia NIH/3T3 cells (control cells), RD and RUCH-2 were seeded at a density of 20.000 cells (standard cell culture condition) or 250,000 cells (highly confluent), respectively, per well of a 4 chamber CultureSlide (Falcon) and cultured for 48 h in 10% FCS (standard conditions). In another set of experiments the medium was replaced after 24 h by starvation medium (0.5% FCS). Then the cells were stained with antibodies against acetylated α-tubulin (Sigma Aldrich, T6793, clone 6-11B-1; 1:500) and Alexa488-conjugated anti-mouse secondary antibody (Dianova, 715-545-150, 1:400). Cells were mounted with ProLong Gold antifade reagent with DAPI (Life Technologies) and analyzed by fluorescence microscopy on a confocal laser scanning microscope equipped with software Fluoview FV100 (Olympus Corporation).
For BrdU incorporation assay with human ERMS cell lines 5,000 cells/well were seeded in 96-well-plates. After 24 h, the cells were incubated for 24 h with the respective drugs in the presence of BrdU. For BrdU incorporation and cell viability (WST-1) assay with the slow growing primary murine ERMS cells, cultures were grown to 90–100% confluency and were treated for 24 h with the respective drugs and for another 24 h with the respective drugs in the presence of BrdU. BrdU incorporation was measured using Cell Proliferation BrdU ELISA (Roche Diagnostics GmbH, Mannheim, Germany). The data are presented as the percentage of BrdU incorporation measured in time-matched solvent-treated controls that were set to 100%. Combination indices were calculated by CompuSyn software (25). Values >100% were not considered for calculation. Drug treatment was also 12 or 48 h for the cell viability assay. Afterwards WST-1 (Roche Diagnostics GmbH, Mannheim) was added to fresh culture medium and cells were incubated with this medium for 4 h. The colorimetric reaction was measured in a microplate reader at a wavelength of 450 nm.
For determination of apoptosis 100,000 RD cells/well were seeded in 6-well-plates. After treatment for 48 h with medium supplemented with drugs or solvent as indicated in the respective experiments, cells were stained with Annexin V-FITC (BD Biosciences, Heidelberg, Germany) and propidium iodide (PI, Miltenyi Biotec, Bergisch Gladbach, Germany) and apoptosis was determined by flow cytometry on a FACSCalibur (BD Biosciences, Heidelberg, Germany) equipped with FlowJo software (Tree Star Inc., Ashland, Oregon, United States of America).
If not stated otherwise, data shown summarize three independent experiments performed as triplicates.
Real-Time Quantitative RT-PCR-Analyses
For gene expression analysis 100,000 cells/well were seeded in 6-well-plates and allowed to attach for 24 h. After subsequent incubation of the cells for 24 h total RNA was isolated using TRIzol Reagent (Invitrogen GmbH, Karlsruhe, Germany). For RNA isolation from tissue samples, approximately 25 mg of tissue was chopped and homogenized on ice. RNA was isolated using TRIzol Reagent. cDNA was synthesized using Superscript II and random hexamers (Invitrogen, Karlsruhe, Germany). Gene expression was analyzed on the ABI Prism HT 7900 Detection System instrument and software (Thermo Fisher Scientific Inc., Waltham, United States of America) using SYBR-green-based qRT-PCR assays. Relative quantification was done by the standard curve method. All primer pairs except for 18S rRNA primers were intron-flanking and are shown in Supplementary Table S2.
Amplification of 18S rRNA served to normalize the amount of sample cDNA. Graphs represent mean value of three independent experiments measured in triplicates plus standard error of the mean (s.e.).
Immunohistochemistry
Isolated tumor samples were embedded in paraffin and sectioned for histological analyses. The identity of RMS was confirmed by examination of Haematoxylin and Eosin (H&E) stained sections. Paraffin sections were stained using the primary and secondary antibodies described in Supplementary Table S3. Supplementary Table S3 also gives the respective dilutions and the respective antigen retrieval methods. Diaminobenzidine chromogen was used as substrate.
Western Blot Analysis
Cells were lysed in RIPA buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.25% Na-deoxycholate, and a protease inhibitor cocktail (Merck KGaA, Darmstadt, Germany). For protein isolation from tissue samples, approximately 30 mg tissue was chopped and homogenized on ice. Protein concentrations were determined by the Pierce Protein BCA Assay Kit (Thermo Fisher Scientific Inc., Waltham, United States of America). Primary antibodies used to detect the individual target proteins and corresponding secondary antibodies are shown in Supplementary Table S3. Pierce ECL Plus Western Blot substrate (Thermo Fisher Scientific Inc., Waltham, United States of America) and the FluorChem Q detection system (Bio-Techne Corp., Minneapolis, United States of America) were used for visualization of protein signals. All Western blots shown are representative for at least two independent experiments.
Breeding of RMS-Bearing Mice, Drug Treatment, and Tumor Measurements
Ptchdel/+ mice on a C57BL/6N (B6) background, which harbor a heterozygous deletion of exon 8 and 9 within the Ptch gene (see Zibat et al. (26) for generation of Ptchdel/+ from Ptchflox/flox mice) were bred to Balb/c (Balb) wildtype (wt) mice to obtain a mixed B6/Balb genetic background that confers high susceptibility for RMS (27). The resulting B6xBalb-Ptchdel/+ mice are named Ptch+/− mice. Genotyping of the mice was done as described (26) (primers are additionally shown in Supplementary Table S2). RMS of Ptch+/− mice were detected upon weekly manual palpation.
RMS-bearing Ptch+/− mice were treated twice a day orally with vehicle/vehicle (controls), single drug/vehicle or drug/drug combinations as indicated in the respective experiments. Between the daily treatments we left an interjacent time span of 5 h to avoid potential drug-drug interactions or complexations, which could have lowered the intestinal resorption and hence the therapeutic efficacy of the combination treatment. The preparation of the drug suspensions and the dosing are given in Supplementary Table S1. The same volume of the vehicle methyl cellulose tween (MCT) was given to control mice.
Animals that received HhAntag and/or pictilisib were treated for a period of 35 days. Tumor size was monitored by low dose in vivo microCT before therapy onset, at day 21 and at the end of treatment. Since the tumors after combination treatment were too small to perform all molecular analyses, the treatment period was reduced to 14 days and tumors were isolated without subjection of the animals to microCT. In addition, the treatment period for vismodegib and sonidegib in combination with pictilisib was reduced to 21 days. The tumor size in these studies was monitored by microCT before therapy onset and at the end of treatment. In all settings, tumors for molecular analysis were isolated 1–4 h after the last treatment, which was either pictilisib or vehicle.
For microCT mice were anesthetized with 1–2% isoflurane in 1:1 air:oxygen mix. In order to visualize the tumors 5 ml/kg of contrast agent Imeron 300 was injected into the tail vein approximately 30 sec prior imaging. The Quantum FX MicroCT (Perkin Elmer, Waltham, United States of America) was used for in vivo imaging of tumor bearing mice at 90 kVp tube voltage, 200 μA tube current and a 2 min total acquisition time. Data sets were reconstructed with a voxel size of 80 μm and analyzed using Scry v6.0 software (Kuchel & Sautter UG, Bad Teinach-Zavelstein, Germany).
All experiments using animals were performed in agreement with all relevant legal and ethical requirements and have been approved by the Lower Saxony State Office for Consumer Protection and Food Safety (file number 33.14-42502-04-13/1084).
Statistical Analysis
In vivo tumor growth was either considered as logarithmic tumor volume or classified into progressive disease (PD), stable disease (SD), or partial response (PR) according to the RECIST criteria (treatment response). The logarithmic tumor volume was considered for the analysis relative to the start of the treatment to avoid inter-mice variation. Treatment response was compared between treatment regimens or combinations thereof by two-sided Fisher's exact test. Relative logarithmic tumor volume was modeled in random effects mixed models with repeated volume measurements, adjusted for age at the start of treatment and sex. Multiple tumors of a mouse were considered as independent observations. The variance of tumor volume was allowed to vary between treatment regimes.
Multiple testing was addressed according to the method of Dunnett (differences between one control and several active treatment regimens) or Tukey-Kramer (differences among all treatment regimens), as appropriate. Spearman correlation was estimated between relative Gli1 expression and relative tumor size, and therefore being independent of scaling of these quantities.
Results are plotted as mean values plus one s.e. if not stated otherwise. The level of significance was set to α = 0.05. We used SAS 9.4 and GraphPad Prism 6 software to perform all statistical analyses.
Results
SMO or PI3K Inhibitors Inhibit Gli1 Expression in Cultured Ptch Mutant ERMS Cells
First, we analyzed the effectiveness of the SMO inhibitors vismodegib, sonidegib, or HhAntag and of the pure PI3K inhibitor pictilisib (also known as GDC-0941; used in the clinics) in PTCH mutant ERMS cells. PTCH mutations are driving RMS in BCNS patients whereas they are very rare in sporadic human RMS (2, 28–30). Since a siRNA-mediated knock-down of PTCH in a cell line derived from a sporadic ERMS would probably not adequately reflect this situation (i.e., it would not be a driver mutation), we tested the drugs in primary tumor cell cultures from Ptch mutant mice (Ptch+/−). As humans with PTCH mutations these mice develop ERMS-like tumors that show active HH signaling. In addition they show increased Akt activity (4, 31).
In general, the incubation of the primary murine tumor cells with vismodegib, sonidegib, or HhAntag significantly reduced the expression of the Hh target Gli1, but not of Hhip (Figure 1). Treatment with the pure PI3K inhibitor pictilisib (also known as GDC-0941; used in the clinics) also significantly reduced Gli1 levels (Figure 1), but at the same time significantly increased the expression of Hhip. Moreover, combined SMO inhibitor/pictilisib treatment resulted in a stronger downregulation of Gli1 than single treatments though it did not reach significance. Finally, the drugs sonidegib and HhAntag reversed the pictilisib-mediated upregulation of Hhip to basal levels (Figure 1).
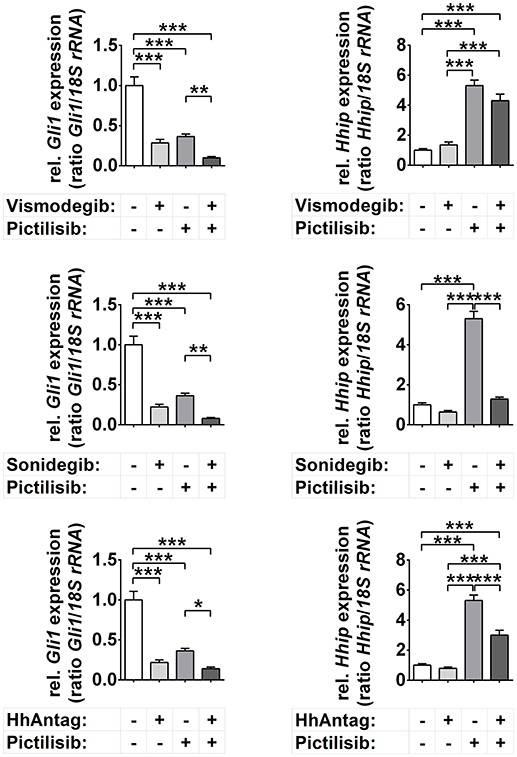
Figure 1. Effects of Smo and/or Pi3k inhibition on Hh target gene expression. Gli1 and Hhip expression in cells treated for 48 h with 10 μM vismodegib, 10 μM sonidegib, 10 μM HhAntag or 5 μM pictilisib. Bars represent the mean +s.e. of three independent experiments measured in duplicates. *P < 0.05, **P < 0.01, ***P < 0.001 when analyzed by Tukey's test for multiple comparisons.
The unchanged Hhip levels upon incubation with SMO inhibitors and the pictilisib mediated up- and downregulation of Hhip and Gli1, respectively, seem at the first glance puzzling. However, it is possible that Hhip is silenced in Ptch+/− ERMS by hypermethylation as described for other HH-associated cancer types (32, 33). In this case and because active PI3K/AKT signaling stabilizes DNA methyltransferase 1 (Dnmt1) (34), pictilisib may have decreased the level of Dnmt1, resulting in Hhip upregulation. Hypermethylation of the Hhip promotor also could explain why SMO inhibitors did not affect Hhip expression levels although they were in general functional. However, this is pure speculation and remains to be analyzed in the future. Together, these data show that vismodegib, sonidegib, HhAntag, and also pictilisib downregulate the expression of the major Hh target gene Gli1, whereas the expression of the Hh target Hhip is unchanged (SMO inhibitors) or upregulated (pictilisib).
Western blot analysis furthermore showed that pictilisib, but none of the SMO inhibitors, efficiently suppressed phosphorylation of Akt, which is the key component of the PI3K/AKT/mTOR pathway. Pictilisib also downregulated the total Akt level and enhanced caspase 3 cleavage in Ptch mutant ERMS cells in a concentration-dependent manner (Supplementary Figure S1A). These pictilisib-mediated changes were not affected by SMO inhibitors (Supplementary Figures S1A,B).
SMO and/or PI3K inhibitor treatment also resulted in significantly reduced BrdU incorporation of Ptch mutant ERMS cells after 48 h (data not shown). However, a cell viability assay revealed that the treatments became toxic for the cells with time (Supplementary Figure S2).
Together we can conclude that both SMO and PI3K inhibitors are involved in regulation of Hh target genes, at least of Gli1, in this murine ERMS model.
Strong Anticancer Effects of SMO or PI3K Inhibitors in ERMS-Bearing Ptch Mutant Mice
To evaluate the anticancer effects of SMO inhibitors and pictilisib in vivo, we treated ERMS-bearing Ptch mutant mice with vismodegib, sonidegib, HhAntag and/or pictilisib daily for 21 days (vismodegib and sonidegib cohorts) or 35 days (HhAntag cohort). All drugs were very well tolerated by the animals. Tumor growth was monitored by low dose in vivo microCT. For molecular analysis all tumors were isolated 1–4 h after the last vehicle or pictilisib treatment (see Materials and Methods section).
The in vivo tumor growth analysis revealed an increase in mice with stable disease (SD) or partial response (PR) by mono-drug treatment with vismodegib, sonidegib, HhAntag, or pictilisib (p = 0.0003 considering any mono-drug treatment; p = 0.0152 for pictilisib and p = 0.0210 for sonidegib) (Figure 2A). Sonidegib mono-treatment led to PR in the majority of mice similar to the combination treatments sonidegib/pictilisib or vismodegib/pictilisib. However the most effective therapy seems to be the combination treatment HhAntag/pictilisib, since all mice reached a partial treatment response (Figure 2A). However, this ranking is done regardless to statistical uncertainty. Furthermore, animals treated with HhAntag/pictilisib have received the drugs for 35 days, whereas mice of the vismodegib/pictilisib and sonidegib/pictilisib study have been treated for 21 days (see material and methods). To mirror the duration of treatment, we fitted mixed random effects models to the growth in tumor volume (Supplementary Figure S3A; see material and methods). For a mono-drug treatment with vismodegib or HhAntag one can expect no essential tumor growth, since the tumor volume at the end of therapy was unchanged [for e.g., vismodegib relative tumor volume (rel.tv) = 0.96 95%-confidence interval (conf.I) [0.56–1.65]]. On the other hand sonidegib or pictilisib are expected to induce tumor regression, since the tumor volume was reduced at the end of therapy compared to vehicle treatment [for e.g., sonidegib rel.tv = 0.32 95%-conf.I [0.15–0.68]]. When vismodegib or sonidegib are combined with pictilisib tumor regression will be similar as for single pictilisib treatment [for e.g., sonidegib/pictilisib rel.tv = 0.49 95%-conf.I [0.26–0.94]], whereas the combination HhAntag/pictilisib will result in tumor regression that is more efficient than the single treatments (Supplementary Figure S3A).
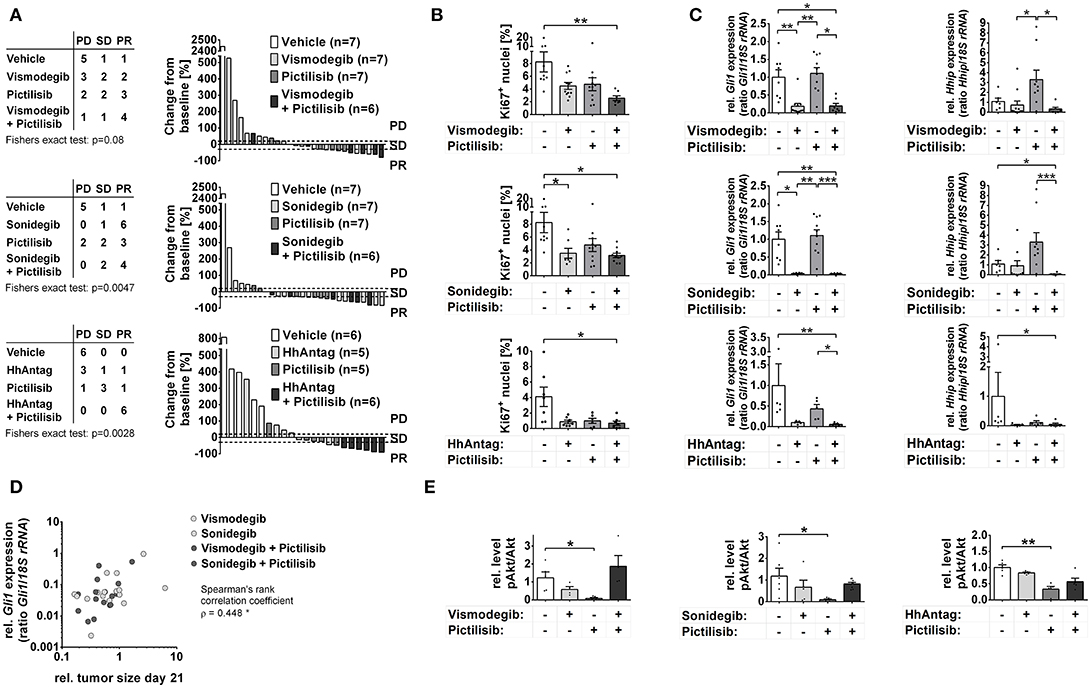
Figure 2. Effects of Smo and/or Pi3k inhibition on in vivo growth of Ptch mutant ERMS. Analysis of RMS of Ptch+/− mice that have been treated orally for 21 (vismodegib and/or pictilisib n = 27; sonidegib and/or pictilisib n = 27) or 35 days (HhAntag and/or pictilisib n = 22) as indicated. (A) Disease progression was classified according to RECIST criteria as progressive disease (PD), stable disease (PD) or partial response (PR). Right panel shows individual changes in tumor growth as measured by microCT at therapy end in comparison to the tumor size at therapy onset. (B) Quantification of Ki67 positive cells of tumors shown in A). (C) Gli1 and Hhip expression in tumor samples collected at day 21 (vismodegib and/or pictilisib study or sonidegib and/or pictilisib study) or day 14 (HhAntag and/or pictilisib study; see main text for explanation). (D) Correlation of tumor growth changes that have been treated with SMO inhibitors with Gli1 expression. (E) pAkt normalized to Akt expression levels as measured by semiquantitative densitometry of Western blot (Western blots and pAkt and Akt expression levels are shown in Supplementary Figures S3C,D, respectively). * P < 0.05, **P < 0.01, ***P < 0.001 when analyzed by Dunn's test for multiple comparisons.
In summary, these data demonstrate that in vivo treatment of Ptch mutant ERMS with SMO inhibitors and/or pictilisib either stop or reduce tumor growth and that sonidegib monotherapy or a HhAntag/pictilisib combination therapy show the strongest antitumor effects.
To substantiate these findings the proliferative activity of the tumors was analyzed by Ki67 immunhistochemical stainings. In agreement with the tumor growth analyses, all single and combination treatments strongly reduced the number of proliferating cells compared to vehicle-treated samples (Figure 2B). The reduction became significant in tumors that have been treated with sonidegib alone or with all of the drug combinations (Figure 2B).
Suppression of Gli1 Expression Correlates With Size Reduction of Ptch Mutant ERMS Upon Treatment With SMO Inhibitors, but Not With PI3K Inhibitors
Analyses of Hh signaling activity revealed extremely reduced Gli1 expression levels after treatment with SMO antagonists, whereas the expression of Hhip was unaltered. This is identical to the cell culture data. However, in contrast to the in vitro experiments pictilisib monotherapy did not inhibit Gli1 transcription in the tumors, whereas the pictilisib-mediated upregulation of Hhip was also seen in vivo (Figure 2C; please note the missing pictilisib-mediated upregulation in the HhAntag cohort. However, in order obtain enough material for RNA analysis, the tumors of this cohort have only been treated for 14 days whereas the others have been treated for 21 days. Therefore it is possible that pictilisib-mediated activation of Hhip expression requires more than 14 days).
Correlation analyses of Gli1 expression levels (see Figure 2C) and ERMS size (see Figure 2A) corroborates these results. Whereas Gli1 expression did not correlate with tumor size in the vehicle- or pictilisib-treated groups (Supplementary Figure S3B), a significant correlation was observed in the groups that have been treated with vismodegib, sonidegib, vismodegib/pictilisib, or sonidegib/pictilisib (Figure 2D; data was not available for HhAntag, see Material and Methods). Together these data show that the antitumor effects of SMO inhibitors in Ptch mutant ERMS are correlated with a decrease in Gli1 levels, whereas the antitumor effects of pictilisib are not.
In order to investigate whether antitumoral efficacy of pictilisib was rather associated with inhibition of Pi3k signaling, the Akt phosphorylation status and caspase 3 cleavage was investigated in the tumors via Western blot analyses. Unfortunately, caspase 3 cleavage was very hard to detect in the samples. However, pictilisib-treated tumors showed a downregulation of pAkt levels in comparison to the tumors that have been treated with SMO inhibitors (Figure 2E; Supplementary Figures S3C,D). Surprisingly, when pictilisib was combined with SMO inhibitors this effect was abrogated (Figure 2E;Supplementary Figures S3C,D).
In summary, these results demonstrate that SMO antagonists can efficiently block growth or even induce regression of Ptch mutant ERMS, which goes along with efficient reduction of Gli1 expression. Therefore, SMO antagonists seem to be appropriate anticancer drugs for RMS that show canonical Hh signaling activity due to Ptch mutations. Similarly, the PI3K inhibitor pictilisib stops growth of Ptch mutant tumors, which is accompanied by inhibition of Akt phosphorylation and most likely by downstream processes such as induction of apoptosis, but not by inhibition of Hh target genes.
PI3K/AKT/mTOR-Mediated Activation of HH Target Gene Expression in Ptch wt ERMS Cells
As demonstrated above, the antitumoral response of individual ERMS driven by Ptch mutations toward different SMO antagonists is rather homogeneous. This is different from cell lines derived from human sporadic ERMS, which are most likely all wt for PTCH and which show a highly diverse response to SMO antagonists (see introduction). Since human ERMS—regardless of the general lack of PTCH mutations (i.e., PTCH mutations in sporadic RMS are exceedingly rare)—show high GLI1 expression (8) and express HH ligands (10), we hypothesized that HH signaling activity in ERMS is caused in a ligand-dependent manner. However, when we tried to stimulate HH signaling activity in the ERMS cell lines RD, that is PTCH wt (35), or RUCH-2, neither SHH-conditioned medium (SHH-CM) nor the SMO agonist SAG uniquely induced the expression of GLI1, HHIP or PTCH (Supplementary Figures S4A–C). As the cells did not develop cilia under the respective experimental conditions (Supplementary Figure 4D), the results implicate that canonical HH signaling cannot be activated in ERMS cells that are wt for PTCH.
Since GLI1 and HHIP expression can be modulated by PI3K inhibitors in Ptch mutant ERMS cells (see above), we next incubated the human RMS cell lines RD and RUCH-2 with pictilisib. We also employed the dual PI3K/mTOR inhibitor PI-103, the pure AKT inhibitor MK-2206 or the mTOR inhibitors everolimus and rapamycin. These drugs were used alone or in combination with vismodegib, sonidegib or HhAntag.
Analyses of the GLI1 transcription levels revealed that only HhAntag, but not vismodegib or sonidegib, significantly inhibited GLI1 expression in RD cells (Figure 3A). However, HhAntag did not alter GLI1 expression in RUCH-2 cells (Figure 3A). In addition, HHIP levels were not affected by SMO inhibitors (at least not by Vismodegib; Figure 3A). In contrast, pictilisib efficiently inhibited GLI1 and HHIP expression in both cell lines (Figure 3A). This was similar for other PI3K, AKT and/or mTOR inhibitors, which potently inhibited GLI1 expression in both cell lines (Supplementary Figure S5A). We also combined the PI3K, AKT and/or mTOR inhibitors and SMO inhibitors to search for potential cooperative effects in inhibition of GLI1 expression. However, none of the combinations was able to significantly intensify the inhibition (Figure 3A, Supplementary Figure S5A).
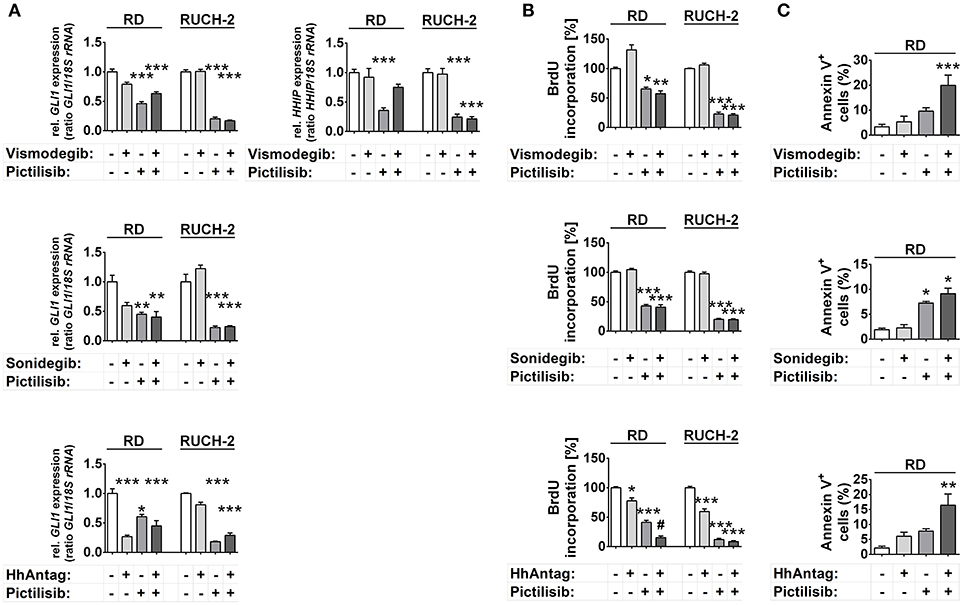
Figure 3. Effects of SMO and/or PI3K inhibition on HH target gene expression, proliferation and apoptosis in human ERMS cell lines. RD and RUCH-2 cells treated with 10 μM of SMO inhibitors and/or 10 μM of the PI3K inhibitor pictilisib. (A) GLI1 expression levels after treatment for 24 h. HHIP expression was investigated after treatment with vismodegib and/or pictilisib (B) BrdU incorporation after treatment for 24 h. BrdU incorporation of solvent treated cells was set to 100%. Bars represent the mean + s.e. of three independent experiments performed in triplicates. (C) Annexin V staining and subsequent FACS analysis of RD cells treated for 48 h with the drugs as indicated. Bars represent the mean number of Annexin V+ cells + s.e. of two independent experiments performed in duplicates.*P < 0.05, **P < 0.01, ***P < 0.01 compared to cells treated with solvent and analyzed by Tukey's test for multiple comparisons. #P < 0.05 compared to cells treated with either drug alone.
Together the data suggest that HH target gene expression in human ERMS cells is regulated by PI3K signaling and occurs downstream of PI3K at the level of AKT or mTOR.
HhAntag and the PI3K Inhibitor Pictilisib, but Not Vismodegib or Sonidegib, Can Induce Anticancer Effects in Ptch wt ERMS Cells
We next investigated the antitumoral potential of pictilisib and the other PI3K/AKT/mTOR inhibitors compared with that of SMO inhibitors. For this purpose we measured proliferation and apoptosis of RD and/or RUCH-2 cells after inhibitor treatment.
HhAntag and pictilisib and also PI-103 decreased proliferation of RD cells, whereas sonidegib had no effect and vismodegib revealed the already described paradoxical pro-proliferative effect (Figure 3B, Supplementary Figure S5B) (13). Similarly HhAntag, pictilisib, PI-103 and also MK-2206 had antiproliferative effects in RUCH-2 cells, whereas sonidegib and vismodegib had not (Figure 3B, Supplementary Figure S5B). Furthermore, a cooperative antiproliferative effect was induced in RD cells by the combination HhAntag (but not vismodegib or sonidegib) and pictilisib (Figure 3B). In RUCH-2 cells cooperative effects were detected by the combinations sonidegib/rapamycin, HhAntag/PI-103, HhAntag/MK-2206, and HhAntag/rapamycin (Supplementary Figure S5B).
Measurement of apoptotic RD cells showed a moderate increase of apoptosis after pictilisib treatment, but not after treatment with SMO inhibitors (Figure 3C). Similarly, PI-103 induced apoptosis. However, MK-2206, everolimus and rapamycin had no effect (Figure 3C, Supplementary Figure S5C). The combination treatments with vismodegib/pictilisib, HhAntag/pictilisib (Figure 3C) or HhAntag/PI-103 (Supplementary Figure S5C) enhanced the apoptosis rate, however it was not a cooperative effect (please again note that combination treatments were considered cooperative when data were significant compared to either single treatment and to control). A significant cooperative proapoptotic effect was observed for HhAntag/MK-2206 (up to 45%; Supplementary Figure S5C). Apoptosis measurement of RUCH-2 cells was not possible due to the strong adherence of the cells to the culture dish, which would require aggressive trypsinization of the cells leading to unreliable apoptotic assay results.
To evaluate if all drugs were functional and the observed antitumoral effects were due to differences in the activation status of proteins involved in the PI3K/AKT/mTOR axis, caspase 3 cleavage, the phosphorylation status of AKT and of the mTOR downstream target S6 were investigated. The results are shown and described in detail in Supplementary Figure S6. In short, all PI3K/AKT/mTOR inhibitors were functional as shown by downregulation of pAKT and pS6 levels. As already described by our lab (13), HhAntag reduced pAKT in RD and RUCH-2 cells. We also found that vismodegib and sonidegib, but not HhAntag, enhanced the total level of AKT in RUCH-2 cells, but not in RD cells.
HhAntag significantly inhibited cellular proliferation of both RD and RUCH-2 cells. This went along with significant inhibition of HH signaling in RD but not in RUCH-2 cells (compare Figures 3A–C). Therefore it is tempting to speculate that the reduced pAKT levels, and not GLI1 levels, are responsible for the antiproliferative effects of HhAntag in comparison to vismodegib or sonidegib. However, this is pure speculation and remains to be elucidated in the future. Contrarily, a more obvious correlation is the enhanced cleavage of caspase 3 and the significant cooperative proapoptotic effect in RD cells that is seen upon treatment with HhAntag/MK-2206.
In summary, SMO antagonists do not show strong antitumoral effects in cultured PTCH wt ERMS cells. Indeed, their efficacy in terms of inhibition of proliferation or induction of apoptosis is much weaker compared to PI3K/AKT inhibitors. Finally, the results suggest off-target effects of SMO antagonists, because they can act on other signaling pathways (e.g., on AKT signaling).
Discussion
We here tested whether reliable anticancer effects using SMO inhibitors may only be achieved in RMS cells harboring PTCH mutations. Indeed, the three SMO inhibitors vismodegib, sonidegib, and HhAntag strongly downregulated Gli1 expression in Ptch mutant ERMS cells both in vitro and in vivo and strongly induced antiproliferative effects in vivo. Furthermore, tumor growth inhibition correlated with Gli1 expression levels, which suggests that Gli1 is a perfect biomarker for the antitumor effects of SMO inhibitors in Ptch mutant ERMS.
This is different in ERMS without PTCH mutations (RD cells) or in which we were not able to activate HH target gene expression by SHH or SAG (RD and RUCH-2 cells). In addition, SMO inhibitors are rather ineffective with respect to inhibition of HH target expression and proliferation. This strongly argues against canonical HH signaling activity in these cells and is similar to other reports showing that SMO inhibitors are mainly effective in medulloblastoma of the SHH subgroup (36) and in basal cell carcinoma that most frequently are driven by PTCH mutations (37), whereas they are of no benefit in e.g. pancreatic cancer or lung cell cancer, in which SHH overexpression was thought to be responsible for HH signaling activity in the tumors [for review see (38, 39)]. Thus our data support the recent hypothesis that SMO inhibitors are effective only in tumors driven by mutations in the HH pathway, whereas they in general lack efficacy in other tumors1 (39). Nevertheless, there is a chance that these drugs might have worked in RD or RUCH-2 xenografts, because the tumor microenvironment plays an important role in canonical HH signaling activity [for review see (39)]. Indeed, a recent paper shows that SMO-deficient RD cells do rarely form palpable tumors. However, as stated by the authors, this effect is not due to changes in cell viability, cell cycle or proliferation, but to unsuccessful tumor initiation (40). Thus, it is rather unlikely that SMO inhibitors may induce stable disease or even regression of ERMS that are wt for PTCH.
We also tried to elucidate whether SMO inhibitors display off-target effects. When used in targeted therapy, vismodegib, sonidegib, and HhAntag only should hit SMO and thus should block activity of GLI transcription factors. However, vismodegib and sonidegib can increase total AKT level and HhAntag can decrease pAKT levels. Although this observation is not surprising [i.e., canonical HH activity is known to regulate AKT (41–43)], it strongly argues for off-target effects of the drugs. Indeed, off-target effects can occur when SMO inhibitors are used at high concentrations [for review see (39)]. Finally, whereas a drug-induced decrease of pAKT may be beneficial in tumor therapy, an increase of total AKT may be deleterious for therapy outcome (44).
In addition, our data show that vice versa PI3K/AKT/mTOR signaling regulates HH/Hh target gene expression in ERMS.
In Ptch mutant ERMS we found that pictilisib upregulated Hhip expression both in vitro and in vivo. We also found that pictilisib reduced Gli1 levels in culture, but not when applied orally to the mouse. Whereas the pictilisib-mediated Hhip upregulation can be explained by epigenetic events (please see Results section), the different effects on Gli1 expression in the in vitro vs. the in vivo situation could be due to e.g. drug pharmacokinetics or the tumor microenvironment. However, since pictilisib potently reduced growth of Ptch mutant tumors in vivo without modulation of Gli1, its strong antitumoral effects in Ptch+/− mice are most likely not related to inhibition of Hh signaling.
In Ptch mutant ERMS, pictilisib also intensified antitumoral effects of some SMO antagonists. In this case, intensification may indeed rely on inhibition of HH signaling mediated by SMO inhibitors. However, it is apparently independent from the phosphorylation status of AKT. Thus, the pictilisib-mediated inhibition of AKT phosphorylation is abolished when pictilisib is applied in combination with SMO inhibitors.
Pictilisib and other PI3K/AKT/mTOR inhibitors also inhibited GLI1 expression in PTCH wt RD cells. We also found that pictilisib downregulated HHIP. Because PI3K signaling regulates the activity of GLI transcription factors in many other tumor entities [e.g., (19, 20, 45, 46)], it is now tempting to speculate that ERMS follows a similar scenario, in which the HH pathway in ERMS is regulated in a non-canonical manner. However, it is possible that PI3K/AKT/mTOR signaling modulates the expression independently of HH pathway components.
Furthermore, the downregulation of HH target gene expression was accompanied by inhibition of cellular proliferation and apoptosis induction. Thus, it is possible that the antitumoral effects of PI3K/AKT/mTOR inhibitors in PTCH wt ERMS is partly mediated by HH target inhibition. However, the fact that MK-2206, everolimus or rapamycin reduce GLI1 expression levels while not affecting cell proliferation or apoptosis, argues against this hypothesis.
Finally, a treatment of PTCH wt RD cells with the combinations vismodegib/pictilisib, HhAntag/pictilisib or HhAntag/PI-103 enhanced apoptosis. These results are similar to our recent data showing that concomitant inhibition of HH and PI3K/AKT/mTOR signaling using the GLI1/2 inhibitor GANT61 and the PI3K/mTOR inhibitor PI-103 synergistically induces apoptosis and growth reduction of cell lines derived from sporadic RMS. However, in contrast to the data shown here, this was associated with strong inhibition of GLI1 expression and a cooperative effect on caspase-dependent apoptosis via the mitochondrial pathway (21). Indeed, GANT61 can attenuate the proliferation of both embryonal and alveolar RMS cells-derived xenograft tumors thereby blocking their growth (47). Since GANT61 blocks the HH pathway at the level of GLI these data again demonstrate the importance of these transcription factors in PTCH wt ERMS. Unfortunately, GANT61 is not very stable and therefore is currently not used in the clinics (48).
Together, this work highlights that the use of SMO antagonists in ERMS is a double-edged sword. Thus, our data suggest that these drugs are very effective in PTCH mutant ERMS that show canonical HH signaling activity, whereas they are in all likelihood ineffective in PTCH wt ERMS that show HH target gene expression due to activity of other signaling pathways. Therefore, treatment with these drugs requires patient selection by either pre-testing of SMO antagonists in patient-derived xenograft cultures or screening of patients for PTCH mutations.
In contrast, PI3K inhibition by e.g., pictilisib may be a superior and more general option in ERMS treatment. PI3K inhibition evokes strong and stable anticancer effects in both PTCH mutant and PTCH wt ERMS. Although pictilisib and other PI3K/AKT/mTOR inhibitors can modulate HH target gene expression, the role of this effect in ERMS response is not clear and remains to be established in the future.
Author Contributions
NG, RR, and HH contributed to conception and design of the study. JB, MK, KD, CD, and AU contributed to data acquisition and helped with data interpretation. AR performed the statistical analysis; H-US contributed to data interpretation. SF provided reagents and ideas; HH and NG wrote the manuscript. All authors read and approved the manuscript.
Funding
This work was supported by the DKH grant Translational Oncology 111537 (to SF and HH) and partly by the DFG grant HA 2197/9-1 (to HH).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank the animal caretakers of the ZTE and Dr. Verena Reupke for excellent animal care and Christina Malowsky, Anke Frommhold and Ina Hess for technical assistance. We are grateful to the companies Genentech/Roche or Novartis for providing protocols for preparing suspensions/solutions of the drugs vismodegib, HhAntag, pictilisib, or sonidegib for in vivo application.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2018.00396/full#supplementary-material
Footnotes
References
1. Scales SJ, De Sauvage FJ. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci. (2009) 30:303–12. doi: 10.1016/j.tips.2009.03.007
2. Hettmer S, Teot LA, Kozakewich H, Werger AM, Davies KJ, Fletcher CD, et al. Myogenic tumors in nevoid Basal cell carcinoma syndrome. J Pediatr Hematol Oncol. (2015) 37:147–9. doi: 10.1097/MPH.0000000000000115
3. Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science (1997) 277:1109–13. doi: 10.1126/science.277.5329.1109
4. Hahn H, Wojnowski L, Zimmer AM, Hall J, Miller G, Zimmer A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nature Med. (1998) 4:619–22. doi: 10.1038/nm0598-619
5. Aszterbaum M, Epstein J, Oro A, Douglas V, Leboit PE, Scott MP, et al. Ultraviolet and ionizing radiation enhance the growth of BCCs and trichoblastomas in patched heterozygous knockout mice [In Process Citation]. Nat Med. (1999) 5:1285–91. doi: 10.1038/15242
6. Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers (2016) 8:20022. doi: 10.3390/cancers8020022
7. Aberger F, Kern D, Greil R, Hartmann TN. Canonical and noncanonical Hedgehog/GLI signaling in hematological malignancies. Vitam Horm. (2012) 88:25–54. doi: 10.1016/B978-0-12-394622-5.00002-X
8. Zibat A, Missiaglia E, Rosenberger A, Pritchard-Jones K, Shipley J, Hahn H, et al. Activation of the hedgehog pathway confers a poor prognosis in embryonal and fusion gene-negative alveolar rhabdomyosarcoma. Oncogene (2010) 29:6323–30. doi: 10.1038/onc.2010.368
9. Pressey JG, Anderson JR, Crossman DK, Lynch JC, Barr FG. Hedgehog pathway activity in pediatric embryonal rhabdomyosarcoma and undifferentiated sarcoma: a report from the Children's Oncology Group. Pediatr Blood Cancer (2011) 57:930–8. doi: 10.1002/pbc.23174
10. Almazan-Moga A, Zarzosa P, Molist C, Velasco P, Pyczek J, Simon-Keller K, et al. Ligand-dependent Hedgehog pathway activation in Rhabdomyosarcoma: the oncogenic role of the ligands. Br J Cancer (2017) 117:1314–25. doi: 10.1038/bjc.2017.305
11. Hawkins DS, Spunt SL, Skapek SX. Children's Oncology Group's 2013 blueprint for research: Soft tissue sarcomas. Pediatr Blood Cancer (2013) 60:1001–8. doi: 10.1002/pbc.24435
12. Williamson D, Missiaglia E, De Reynies A, Pierron G, Thuille B, Palenzuela G, et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol. (2010) 28:2151–8. doi: 10.1200/JCO.2009.26.3814
13. Ridzewski R, Rettberg D, Dittmann K, Cuvelier N, Fulda S, Hahn H. Hedgehog inhibitors in rhabdomyosarcoma: a comparison of four compounds and responsiveness of four cell lines. Front Oncol. (2015) 5:130. doi: 10.3389/fonc.2015.00130
14. Cen L, Hsieh FC, Lin HJ, Chen CS, Qualman SJ, Lin J. PDK-1/AKT pathway as a novel therapeutic target in rhabdomyosarcoma cells using OSU-03012 compound. Br J Cancer (2007) 97:785–91. doi: 10.1038/sj.bjc.6603952
15. Petricoin EF III, Espina V, Araujo RP, Midura B, Yeung C, Wan X, et al. Phosphoprotein pathway mapping: Akt/mammalian target of rapamycin activation is negatively associated with childhood rhabdomyosarcoma survival. Cancer Res. (2007) 67:3431–40. doi: 10.1158/0008-5472.CAN-06-1344
16. Renshaw J, Taylor KR, Bishop R, Valenti M, De Haven Brandon A, Gowan S, et al. Dual blockade of the PI3K/AKT/mTOR (AZD8055) and RAS/MEK/ERK (AZD6244) pathways synergistically inhibits rhabdomyosarcoma cell growth in vitro and in vivo. Clin Cancer Res. (2013) 19:5940–51. doi: 10.1158/1078-0432.CCR-13-0850
17. Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature (2018) 555:321–7. doi: 10.1038/nature25480
18. Agarwal NK, Qu C, Kunkalla K, Liu Y, Vega F. Transcriptional regulation of serine/threonine protein kinase (AKT) genes by glioma-associated oncogene homolog 1. J Biol Chem. (2013) 288:15390–401. doi: 10.1074/jbc.M112.425249
19. Sharma N, Nanta R, Sharma J, Gunewardena S, Singh KP, Shankar S, et al. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget (2015) 6:32039–60. doi: 10.18632/oncotarget.5055
20. Zhou J, Zhu G, Huang J, Li L, Du Y, Gao Y, et al. Non-canonical GLI1/2 activation by PI3K/AKT signaling in renal cell carcinoma: a novel potential therapeutic target. Cancer Lett. (2016) 370:313–23. doi: 10.1016/j.canlet.2015.11.006
21. Graab U, Hahn H, Fulda S. Identification of a novel synthetic lethality of combined inhibition of hedgehog and PI3K signaling in rhabdomyosarcoma. Oncotarget (2015) 6:8722–35. doi: 10.18632/oncotarget.2726
22. Hinson AR, Jones R, Crose LE, Belyea BC, Barr FG, Linardic CM. Human rhabdomyosarcoma cell lines for rhabdomyosarcoma research: utility and pitfalls. Front Oncol. (2013) 3:183. doi: 10.3389/fonc.2013.00183
23. Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci USA. (2002) 99:14071–6. doi: 10.1073/pnas.182542899
24. Uhmann A, Niemann H, Lammering B, Henkel C, Hess I, Nitzki F, et al. Antitumoral effects of calcitriol in basal cell carcinomas involve inhibition of hedgehog signaling and induction of vitamin D receptor signaling and differentiation. Mol Cancer Ther. (2011) 10:2179–88. doi: 10.1158/1535-7163.MCT-11-0422
25. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. (2006) 58:621–81. doi: 10.1124/pr.58.3.10
26. Zibat A, Uhmann A, Nitzki F, Wijgerde M, Frommhold A, Heller T, et al. Time-point and dosage of gene inactivation determine the tumor spectrum in conditional Ptch knockouts. Carcinogenesis (2009) 30:918–26. doi: 10.1093/carcin/bgp068
27. Hahn H, Nitzki F, Schorban T, Hemmerlein B, Threadgill D, Rosemann M. Genetic mapping of a Ptch1-associated rhabdomyosarcoma susceptibility locus on mouse chromosome 2. Genomics (2004) 84:853–8. doi: 10.1016/j.ygeno.2004.07.002
28. Calzada-Wack J, Schnitzbauer U, Walch A, Wurster KH, Kappler R, Nathrath M, et al. Analysis of the PTCH coding region in human rhabdomyosarcoma. Hum Mutat. (2002) 20:233–4. doi: 10.1002/humu.9056
29. Tostar U, Malm CJ, Meis-Kindblom JM, Kindblom LG, Toftgard R, Unden AB. Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J Pathol. (2006) 208:17–25. doi: 10.1002/path.1882
30. Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. (2014) 4:216–31. doi: 10.1158/2159-8290.CD-13-0639
31. Kappler R, Calzada-Wack J, Schnitzbauer U, Koleva M, Herwig A, Piontek G, et al. Molecular characterization of Patched-associated rhabdomyosarcoma. J Pathol. (2003) 200:348–56. doi: 10.1002/path.1361
32. Tada M, Kanai F, Tanaka Y, Tateishi K, Ohta M, Asaoka Y, et al. Down-regulation of hedgehog-interacting protein through genetic and epigenetic alterations in human hepatocellular carcinoma. Clin Cancer Res. (2008) 14:3768–76. doi: 10.1158/1078-0432.CCR-07-1181
33. Shahi MH, Zazpe I, Afzal M, Sinha S, Rebhun RB, Melendez B, et al. Epigenetic regulation of human hedgehog interacting protein in glioma cell lines and primary tumor samples. Tumour Biol. (2015) 36:2383–91. doi: 10.1007/s13277-014-2846-4
34. Sun L, Zhao H, Xu Z, Liu Q, Liang Y, Wang L, et al. Phosphatidylinositol 3-kinase/protein kinase B pathway stabilizes DNA methyltransferase I protein and maintains DNA methylation. Cell Signal. (2007) 19:2255–63. doi: 10.1016/j.cellsig.2007.06.014
35. Tostar U, Toftgard R, Zaphiropoulos PG, Shimokawa T. Reduction of human embryonal rhabdomyosarcoma tumor growth by inhibition of the hedgehog signaling pathway. Genes Cancer (2010) 1:941–51. doi: 10.1177/1947601910385449
36. Robinson GW, Orr BA, Wu G, Gururangan S, Lin T, Qaddoumi I, et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC-025B and PBTC-032. J Clin Oncol. (2015) 33:2646–54. doi: 10.1200/JCO.2014.60.1591
37. Von Hoff DD, Lorusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. (2009) 361:1164–72. doi: 10.1056/NEJMoa0905360
38. Atwood SX, Oro AE. “Atypical” regulation of Hedgehog-dependent cancers. Cancer Cell. (2014) 25:133–4. doi: 10.1016/j.ccr.2014.01.027
39. Curran T. Reproducibility of academic preclinical translational research: lessons from the development of Hedgehog pathway inhibitors to treat cancer. Open Biol. (2018) 8:180098. doi: 10.1098/rsob.180098
40. Satheesha S, Manzella G, Bovay A, Casanova EA, Bode PK, Belle R, et al. Targeting hedgehog signaling reduces self-renewal in embryonal rhabdomyosarcoma. Oncogene (2016) 35:2020–30. doi: 10.1038/onc.2015.267
41. Fu JR, Liu WL, Zhou JF, Sun HY, Xu HZ, Luo L, et al. Sonic hedgehog protein promotes bone marrow-derived endothelial progenitor cell proliferation, migration and VEGF production via PI 3-kinase/Akt signaling pathways. Acta Pharmacol Sin. (2006) 27:685–93. doi: 10.1111/j.1745-7254.2006.00335.x
42. Riobo NA, Lu K, Ai X, Haines GM, Emerson CP Jr. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc Natl Acad Sci USA. (2006) 103:4505–10. doi: 10.1073/pnas.0504337103
43. Morton JP, Mongeau ME, Klimstra DS, Morris JP, Lee YC, Kawaguchi Y, et al. Sonic hedgehog acts at multiple stages during pancreatic tumorigenesis. Proc Natl Acad Sci USA. (2007) 104:5103–8. doi: 10.1073/pnas.0701158104
44. Liu Z, Zhu G, Getzenberg RH, Veltri RW. The upregulation of PI3K/Akt and MAP kinase pathways is associated with resistance of microtubule-targeting drugs in prostate cancer. J Cell Biochem. (2015) 116:1341–9. doi: 10.1002/jcb.25091
45. Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, et al. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc Natl Acad Sci USA. (2007) 104:5895–900. doi: 10.1073/pnas.0700776104
46. Gotschel F, Berg D, Gruber W, Bender C, Eberl M, Friedel M, et al. Synergism between Hedgehog-GLI and EGFR Signaling in Hedgehog-Responsive Human Medulloblastoma Cells Induces Downregulation of Canonical Hedgehog-Target Genes and Stabilized Expression of GLI1. PLoS ONE (2013) 8:e0065403. doi: 10.1371/journal.pone.0065403
47. Srivastava RK, Kaylani SZ, Edrees N, Li C, Talwelkar SS, Xu J, et al. GLI inhibitor GANT-61 diminishes embryonal and alveolar rhabdomyosarcoma growth by inhibiting Shh/AKT-mTOR axis. Oncotarget (2014) 5:12151–65. doi: 10.18632/oncotarget.2569
Keywords: ERMS, HH, PTCH, PI3K, vismodegib, sonidegib, HhAntag, pictilisib
Citation: Geyer N, Ridzewski R, Bauer J, Kuzyakova M, Dittmann K, Dullin C, Rosenberger A, Schildhaus H-U, Uhmann A, Fulda S and Hahn H (2018) Different Response of Ptch Mutant and Ptch Wildtype Rhabdomyosarcoma Toward SMO and PI3K Inhibitors. Front. Oncol. 8:396. doi: 10.3389/fonc.2018.00396
Received: 16 June 2018; Accepted: 31 August 2018;
Published: 25 September 2018.
Edited by:
Anat Erdreich-Epstein, Children's Hospital of Los Angeles, United StatesReviewed by:
Marcin Wysoczynski, University of Louisville, United StatesAykut Üren, Georgetown University, United States
Copyright © 2018 Geyer, Ridzewski, Bauer, Kuzyakova, Dittmann, Dullin, Rosenberger, Schildhaus, Uhmann, Fulda and Hahn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heidi Hahn, aGhhaG5AZ3dkZy5kZQ==