 John Lynes
John Lynes Victoria Sanchez
Victoria Sanchez Gifty Dominah1
Gifty Dominah1 Edjah Nduom
Edjah Nduom- 1National Institute of Neurological Disorders and Stroke, Bethesda, MD, United States
- 2MedStar Georgetown University Hospital, Washington, DC, United States
Glioblastoma is in need of innovative treatment approaches. Immune therapy for cancer refers to the use of the body's immune system to target malignant cells in the body. Such immune therapeutics have recently been very successful in treating a diverse group of cancerous lesions. As a result, many new immune therapies have gained Food and Drug Administration approval for the treatment of cancer, and there has been an explosion in the study of immune therapeutics for cancer treatment over the past few years. However, the immune suppression of glioblastoma and the unique immune microenvironment of the brain make immune therapeutics more challenging to apply to the brain than to other systemic cancers. Here, we discuss the existing barriers to successful immune therapy for glioblastoma and the ongoing development of immune therapeutics. We will discuss the discovery and classification of immune suppressive factors in the glioblastoma microenvironment; the development of vaccine-based therapies; the use of convection-enhanced delivery to introduce tumoricidal viruses into the tumor microenvironment, leading to secondary immune responses; the emerging use of adoptive cell therapy in the treatment of glioblastoma; and future frontiers, such as the use of cerebral microdialysis for immune monitoring and the use of sequencing to develop patient-specific therapeutics. Armed with a better understanding of the challenges inherent in immune therapy for glioblastoma, we may soon see more successes in immune-based clinical trials for this deadly disease.
Introduction
GBM is an often-fatal brain malignancy that accounts for the majority of primary malignant brain tumors (1, 2) and has a recurrence rate of more than 90% (3). The current standard treatment for patients with GBM is maximal safe resection of the tumor followed by radiotherapy with temozolomide (TMZ), but survival is poor, with a median survival of just over 14 months (4). While other treatments, such as Gliadel wafers (5), bevacizumab (6), and tumor treatment fields (7), have been cleared by the Federal Drug Administration (FDA) for the treatment of glioblastoma, no other treatment has been accepted by the neuro-oncology community as standard of care, due to the inability of these treatments to significantly affect overall survival.
Immunotherapy is a rising field of study wherein one's own immune system is manipulated to target cancer antigens. Though the first report of a connection between tumor regression and infection was by Chekhov in 1884 (8), the concept of immune therapy for cancer is often attributed to the first use of Coley's toxin in 1893. William Coley, a “bone surgeon,” inoculated sarcoma patients with heat-inactivated streptococcus after observing a case of a patient having tumor regression after accidental infection (9). Over a century later, there have been several breakthroughs in the field of immune-oncology, leading to the FDA approval of several new agents, including checkpoint inhibitors.
Checkpoint inhibitors nivolumab, an anti-programmed death-1 (PD-1) antibody, and ipilimumab, an anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) antibody, demonstrated increased survival in untreated melanoma (10) and were FDA approved in 2015. Pembrolizumab, another anti-PD-1 antibody, has shown benefit in non-small cell lung cancer (11) and was FDA approved in 2017. Chimeric Antigen Receptor (CAR) T-cell therapy and blinatumomab, a targeted antibody against CD19, were approved for pediatric leukemias in 2017. In parallel with these advances, numerous groups have pursued strategies for immunotherapy in glioblastoma, given its recalcitrance in the face of traditional therapies. However, glioblastoma has remained a challenging disease to treat with immune therapeutics, as it has been a challenge with conventional therapeutics.
It was previously believed that the brain was immune privileged (12), because it could not induce an adequate immune response in the case of graft rejection. This led to understandable skepticism regarding the use of immune therapy for these lesions. However, new insight has revealed that the CNS, in communication with the rest of the body, can mount appropriate immune responses (13). Despite this, the success of immune therapy is not guaranteed. Immune therapy for glioblastoma is limited by the immunosuppressive mechanisms in the glioblastoma microenvironment (14). Therefore, scientists are working to determine the role that these different immunosuppressive factors play in tumor formation and progression.
This review aims to highlight the development of immune therapy for primary brain malignancies. Specifically, we will provide a detailed review of key players of immune suppression in the tumor microenvironment and outline the development of new immune treatments for glioblastoma. These new immune therapeutics include: checkpoint inhibition, tumor vaccines, adoptive cell therapies and convection enhanced delivery of tumoricidal viruses. Finally, we will discuss areas of future research for immune therapy, including advances in immune biomarker development.
Immunophenotyping the Tumor Microenvironment
Immunophenotyping, or the description of the immune system's form and functioning in the tumor microenvironment, has emerged as an important factor in understanding tumorigenesis, tumor survival, and potential for utilizing the immune system against glioblastoma. A variety of immune cell types are found in this environment with complex, still incompletely understood interactions (Figure 1).
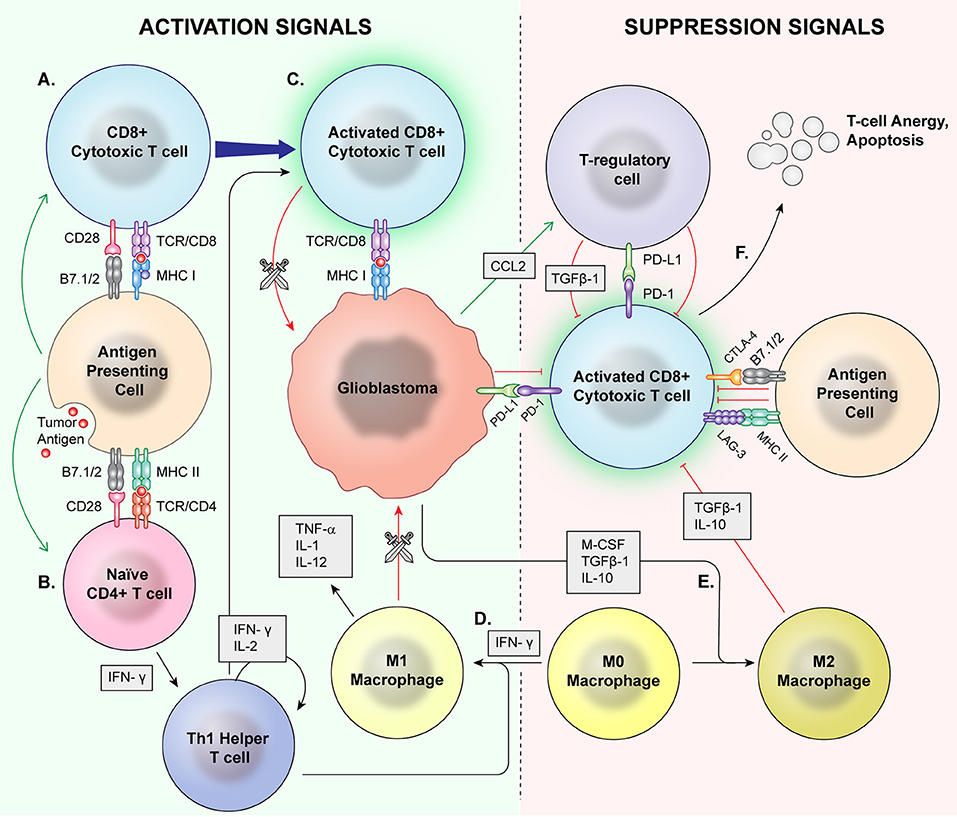
Figure 1. Normal Inflammation vs. Immunosuppression Mechanisms. Antigen presenting cells (APCs) phagocytose tumor antigens and present to cytotoxic T cells as well as naïve CD4+ cells. Via coactivation signals, the APCS activate the cytotoxic T cells (A) and skew helper T cells to a proinflammatory Th1 lineage (B). The activated cytotoxic T cells then recognize and attack malignant cells (C). T regulatory cells, M2 macrophages, and MDSCs are major mediators of immune suppression. M0 macrophages may be skewed toward a pro-inflammatory M1 phenotype by IFN-γ (D), which directly phagocytose target cells and release proinflammatory cytokines. (E) Glioblastoma cells also signal M0 macrophages to skew toward an M2 phenotype which release immunosuppressive cytokines. Immune checkpoints induce anergy and apoptosis of CD8+ cytotoxic T cells (F) and CD4+ cells.
Regulatory T Cells
Several cell types have been associated with the immunosuppressive glioblastoma microenvironment. Regulatory T Cells (Tregs), traditionally CD4+CD25+ FoxP3+ lymphocytes, help balance the immune system in a non-pathologic context, preventing injury from excessive activity and autoimmune disease (15). These cells induce a shift toward the T-Helper-2 (TH2) immune phenotype and immunosuppressive cytokine production. However, Tregs are found in the blood of glioblastoma patients at a higher ratio to CD4+ non-Tregs as compared to healthy controls (16). Glioblastoma cells have been found to release chemokines that attract Tregs to the tumor microenvironment (17) via the chemokine CCL2 (18). Most Tregs found in the tumor microenvironment are naturally occurring Tregs of thymic origin rather than induced Tregs (19). While CD4+ Tregs have been under extensive investigation for decades, the existence and role of CD8+ Tregs is less well studied and is not well understood in glioblastoma. First described by Damle in the 1980s, CD8+ Tregs are CD8+ T cells that are immunosuppressive (20), similar to CD4+ Tregs Kiniwa et al. would go on to describe CD8+ Tregs from an oncologic perspective in prostate cancer (21). Subsequent groups would describe these immunosuppressive cells in colorectal cancer (22), hepatocellular carcinoma (23), non-small cell lung cancer (24), and ovarian cancer (25). However, similar studies have not yet been performed in glioblastoma.
Macrophages and Myeloid Derived Suppressor Cells
Monocyte-derived macrophages or microglia native to the CNS can constitute as high as 12% of glioblastoma mass (26) and have been associated with poor outcome in non-CNS malignancies (27, 28). These tumor-associated macrophages can be nonpolarized M0 macrophages, classical (M1) lineage or M2 lineage. M1 macrophages upregulate cell surface molecules associated with antigen presentation and recognition; release proinflammatory cytokines such as TNF-α, IL-1β, and IL-12; and directly phagocytose targets (29). The M2 macrophages, however, help mediate immunosuppression and tumor invasion (30). Studies have suggested that the M2 lineage has been found in disproportionately high concentration in the glioblastoma environment (31) and their prevalence has been associated with glioma grade (32). This M2 immunosuppressive state is maintained by multiple signals, including the TGF-beta pathway and TH2 cytokines IL-4 and IL-10 (33). Conversely, interferon-gamma activates the inflammatory M1 phenotype (34). RNA sequencing of tumor associated macrophages in murine models and human samples suggests that a majority of macrophages found in glioblastoma are bone-marrow derived, rather than resident microglia, and that they have enriched proliferation and migration gene expression (35, 36). Recently, whole-genome analysis and microRNA expression profiling performed on human tumor tissue suggested that the macrophage population represents a spectrum rather than strict lineages, with the largest share most similar to M0 macrophages (37). Driving differentiation of TAMs to an M1 phenotype could be a new treatment approach. MiR-146, a microRNA found to direct hematopoietic differentiation (38), was found to be downregulated in glioblastoma macrophages and may direct the M0 phenotype toward a pro-inflammatory M1 lineage. Additionally, miR-142-3p is downregulated in glioma TAMs, and administration of miR-142-3p in a murine glioma model decreased infiltrating TAMs and extended median survival (39). A shift toward an M1 phenotype has been associated with response to therapy in a murine glioma model (40). However, iatrogenic sources may also affect balance of M1 and M2 macrophages, with one study finding that radiation therapy increased the proportion of M2 macrophages in in-vivo murine glioma models due to increased radiosensitivity of the M1 line (41).
Myeloid derived suppressor cells (MDSCs), precursors to both macrophage lineages, are found in higher levels in the serum of glioblastoma (42). This cell type is generally considered to have an overall immunosuppressive phenotype, though there have been reports of antitumor effects via nitrous oxide release (43) and potential M1 macrophage characteristics (44). However, in glioblastoma, these cells are immunosuppressive (45). MDSCs promote Treg proliferation (46), create oxidative stress that inhibits T cell proliferation (47), and deplete L-arginine, inhibiting CD3 production and T cell proliferation (48). Gielen et al describe a trend toward increased circulating MDSCs in glioma patients dependent on tumor grade, as well as increased arginase activity of these MDSCs compared to healthy controls (49). Additionally, immature monocytes have been found to express CTLA-4 (50) and PD-L1 (51), prominent immune checkpoints, which are membrane proteins associated with modulating T cell activation and anergy. Targeting MDSCs has shown benefit in pre-clinical models of non-CNS tumors (52), and a phase I trial is currently underway to evaluate if this strategy may be effective in glioblastoma (53).
Immune Checkpoints
In the physiologic state, immune checkpoints play an important role in the constant balance of immune modulation in order to prevent autoimmunity (54). In typical activation of T cells, coactivation of two receptor-ligand pairs is required. Major histocompatibility complex (MHC) I and II are key components of antigen presentation and recognition that make up one half of this co-stimulatory pathway. CD28/B7 interaction often acts as the second signal. Antigen presenting cells (APCs) activate cytotoxic T cells and T helper cells by presenting a tumor antigen on the MHC complex and providing co-stimulation (55). Cytotoxic T cells directly attack tumor cells that express a targeted antigen while helper T cells propagate an antitumor immune response via release of proinflammatory cytokines and induction of immune memory B and T cells (56). Immune checkpoints are regulators of the immune system that are expressed by T effector cells, APCs, and myeloid-derived cells in the normal immune system. When these checkpoints are engaged, they decrease immune activity by promoting T-cell anergy and apoptosis (57), preventing T-cell co-stimulation and activation by antigen presenting cells (58), and promoting Treg suppression of T effector cell functioning (59), among other mechanisms of immunosuppression. However, in the tumor microenvironment, tumor cells may utilize these pathways to suppress an effective immune response targeted toward the tumor (14).
Cytotoxic T-lymphocyte associated protein type 4 (CTLA-4) was described as an inhibitor of T cell activation by Walunas et al. (60). CTLA-4 on APCs prevents CD28/B7 co-stimulation by outcompeting CD28 to interact with B7, thus creating an inhibitory response in the T effector cell (61). However, it also exists on Tregs in a constitutively active form, potentially increasing this cell type's immunosuppressive potency (62). Since the discovery and characterization of CTLA-4, multiple other checkpoints have been discovered including PD-1 (63), LAG-3 (64), TIM-3 (65), CD137 (4-1BB) (66), GITR (67), and CD134 (OX40) (68). These molecular signals have been identified on infiltrating immune cells of many different malignancies, including glioblastoma, and the cancer cells often express the ligands for these checkpoints (69). Programmed cell death-1 (PD-1) is a cell surface immune checkpoint found on effector T cells, while its ligand, PD-L1, can be expressed by glioblastoma cells, with elevated PD-L1 being associated with poor overall survival in glioblastoma patients, independent of other factors (70–72). PD-L1 is additionally found on antigen presenting cells and immunosuppressive immune cells such as Tregs. One study found a correlation between PD-L1 expression and a marker of regulatory T-cells, FoxP3, as well as between FoxP3 expression and patient survival in patients with glioblastoma (73). Lymphocyte-activating gene-3 (LAG-3) is a cell membrane protein found on NK cells, APCs, and some T lymphocytes including Tregs (74). LAG-3 interacts with MHC class II molecules with greater affinity than their typical partner, CD4, and, in doing so, prevents CD4 T helper cell proliferation and cytokine release (60). Groups studying various malignancies have suggested that these checkpoints may work alone or in parallel for immune evasion (75).
Checkpoint Inhibition
Checkpoint inhibition describes the use of a treatment to interfere in the interaction between an immune checkpoint molecule and its target, receptor, or ligand (Figure 2). This is intended to cause a net immune stimulating effect by inhibiting an inhibitory signal. After the discovery of CTLA-4's immunosuppressive function, antibody-mediated CTLA-4 blockade caused tumor regression in murine models (54). Following several promising phase II trials (76– 78) the first phase III study of a checkpoint inhibitory showed anti-CTLA-4 improved survival in metastatic melanoma in 2010 (79). The Checkmate 067 trial demonstrated that anti-PD-1 and anti-CTLA-4 either alone or in combination imparted increased survival in untreated melanoma. Notably, patients whose tumors displayed increased PD-L1 expression had significantly improved survival on this therapy than those with low PD-L1 expression (10). Similar trends in response to anti-PD-1 therapy related to PD-L1 expression have been described by multiple groups in non-small cell lung cancer (80). However, PD-1 and CTLA-4 pathways may cause immune suppression in parallel, with one trial finding that treatment with either anti-PD-1 or anti-CTLA checkpoint inhibition in melanoma results in upregulation of the other pathway to continue immune evasion, suggesting a potential limitation to monotherapy (81).
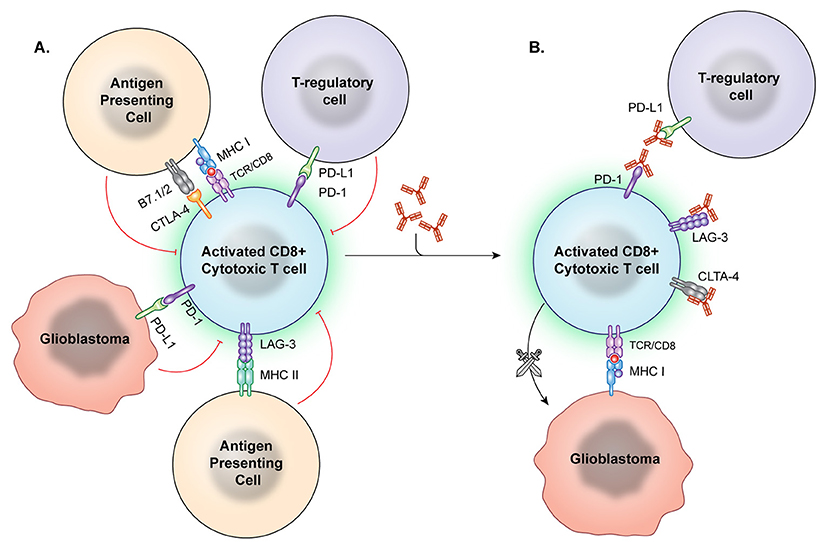
Figure 2. Immune checkpoint inhibition. (A) Immune checkpoints hinder T-cell activation and promote an immunosuppressive state. However, these checkpoint molecules can be neutralized by targeted antibodies. (B) After the checkpoint molecules are negated by these blocking antibodies, T effector cells are better able to recognize and attack tumor cells.
Work by groups led by Lesniak and Lim extended these positive findings to glioblastoma in murine models by showing survival benefit in glioma-implanted mice treated with PD-1 blockade in combination with CTLA-4 blockade (82) and with radiation therapy (83). In response to the promising results of checkpoint blockade in non-CNS cancer and what was believed, at the time, to be high expression of PD-L1 in glioblastoma (84) the Checkmate 143, Checkmate 498, and Checkmate 548 trials explored checkpoint inhibition in GBM. The checkmate 498 (85) and 548 (86) trials focused on the use of nivolumab on MGMT unmethylated and methylated newly diagnosed GBM, while Checkmate 143 investigated its use in recurrent GBM (87). Checkmate 143 has completed with no overall survival benefit of nivolumab treatment in this patient population (88). Filley et al. proposed that the negative result may be due to several different factors, including the profound immune suppressive microenvironment of glioblastoma, systemic immune suppression of glioblastoma patients and the antibody's inability to cross the blood-brain barrier (89). Additionally, they postulate that checkpoint inhibition may be less effective at reactivating immune cells rather than preventing immunosuppression in the immune environment in recurrent glioblastoma. Furthermore, steroid use at 4 mg of dexamethasone per day or higher was quite prevalent in the treated population, and the investigators did not obtain pathological confirmation of tumor recurrence prior to halting therapy. This could have led to premature discontinuation of immune therapy, given the difficulty in distinguishing inflammatory treatment response from tumor progression in glioblastoma patients (90). However, there are a variety of other trials investigating the combination of checkpoint inhibition with other therapies, such as vaccination, discussed below, which are still ongoing (85). Potential therapeutic benefit of LAG-3 blockade was shown in a phase I/II trial in metastatic breast cancer (91) and a phase I trial for renal cell carcinoma (92) with a study in pancreatic cancer terminated due to drug production difficulties (93). In response, several phase I studies of LAG-3 blockade are underway, including for a variety of solid tumors (94) and hematologic malignancies (95). One of these studies is currently investigating the use of anti-LAG3 checkpoint inhibition, along with PD-1 and CD137 blockade in recurrent glioblastoma, though results have not yet been released (96).
Tumor Vaccination
Vaccines have been extensively studied as a potential therapy for gliomas. In general, vaccines expose the immune system to a weakened or killed antigen to build immune memory against any future exposure to that antigen (97). Cancer vaccines work via similar mechanisms. They manipulate immune memory following a primary encounter with a cancer associated antigen to activate T-cells and induce an inflammatory response which is targeted against the tumor.
Dendritic Cell Vaccines
Dendritic cells (DCs) were first described as novel stellate cells found in lymphoid tissue in 1973 by Steinman and Nussenzweig (98). Our current understanding is that they are intermediate antigen presenting cells (APC) to both CD4+ and CD8+ T-cells as well as activators of natural killer cells (NK-cells) and NK-T-cells in the setting of MHC (96, 99). MHC expression can be downregulated in the glioblastoma microenvironment, reducing the efficiency of antigen presentation (100–102). This makes effective antigen presentation even more critical for successful immune therapy in the brain.
The general process of autologous DC vaccine development requires isolation of DCs from a subject's blood, pulsation of the immune cells with the cancer associated antigen for stimulation, and, finally, treatment of the subject with the newly formed vaccine (103) (Figure 3). Preclinical investigations in mice revealed that peripherally injected DC vaccines could induce cytotoxic T-lymphocyte (CTL) responses in the CNS without causing major adverse effects such as autoimmune responses (104). Further studies in glioma mouse models confirmed that DC vaccines could target and kill these tumors without significantly harming normal brain tissue (105, 106). In a Phase I clinical trial for DC vaccine use in glioma, Wheeler, Wu, and team pulsed immature DCs of 9 glioma patients with tumor peptides eluted from cultured autologous tumor cells (107). Patients underwent initial tumor resection followed by conventional radiotherapy (RT) prior to elution of MHC-I peptides from the tumor sample and DC extraction from host venous blood. They observed that 4 of the 7 patients that were given the vaccine had a positive cytotoxic T-cell response. After vaccination, 4 tumors were re-resected and 2 of them exhibited CD8+ T-cell and memory T-cell infiltration (103). This research illustrated the ability of ex-vivo educated DCs to augment the proinflammatory response against tumor.
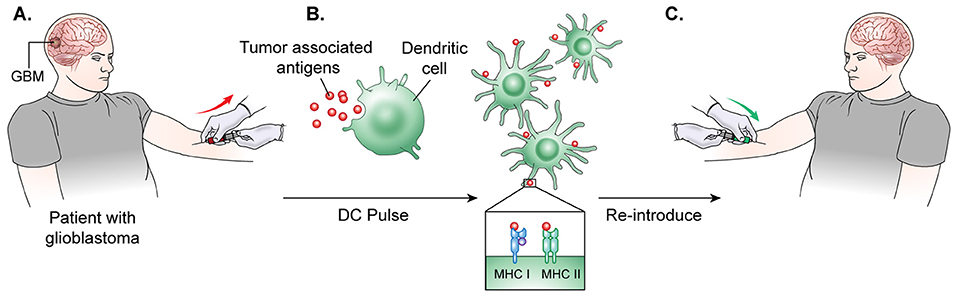
Figure 3. DC vaccine development.(A) The general process of DC vaccine development and immunization requires tumor lysate isolation. Patients first undergo resection of the tumor for production of lysate as well as patient leukapheresis to collect dendritic cells. (B) The tumor associated antigen, mRNA, or lysate is used to pulse mature or immature DCs obtained through patient leukopharesis. (C) Primed DCs are then administered as a vaccine to patients peripherally.
CD133, also known as prominin-1, is a transmembrane glycoprotein receptor that has been proposed to play a vital role in cell proliferation due its association with the WNT signaling pathway, but its overall function still remains undefined (108). CD133+ cells have been recognized in a study by Singh et al. as potential brain tumor initiating cells. The study showed that only the CD133+ human brain tumor cells possessed the ability to initiate phenotypically identical tumor growth in non-obese diabetic severe combined immunodeficient mouse brains with just 100 cells injected. The CD133- cells on the other hand were not able to cause tumor growth even with as many as 105 cells injected into the mouse brains (109). CD133 is now a common marker used to identify malignant cancer stem cells in GBM (110) as well as in endometrial (111), colon (112), lung (113), prostate (114), ovarian (115), skin (116), and breast (117) cancer. CD133+ cancer stem cells in GBM have been shown to be resistant to radiotherapy and chemotherapeutic drugs (118) as well as contribute to the recurrence of the tumor after radiation (119). A promising DC vaccine trial (120) targeting CD133 was reported in June 2017 at the American Society of Clinical Oncology annual meeting (121). This Phase 1 trial of the ICT-121 DC vaccine was carried out in patients with recurrent glioblastoma who express the HLA-A2 phenotype. In this trial, the patient's DCs were pulsed with CD133 to create the vaccine. The vaccine was then administered to the patient once a week for 4 weeks for the initial induction phase, followed by once every 2 months for the maintenance phase. They reported that the ICT-121 DC vaccine was considered safe and tolerable. Eight out of the 20 patients enrolled were surviving at the time of the report, and cytokine mRNA expression suggested the presence of an active immune response to the CD133 epitopes.
Multiple clinical trials have since been performed to evaluate the safety and efficacy of dendritic cell vaccines in patients. Liau et al. reported Phase I clinical trial results of the use of DCVax-L, a DC vaccine generated with autologous tumor lysate, in newly diagnosed and recurrent glioblastoma patients (122) and they found that the treatment was safe. De Vleeschouwer and team reported in their Phase I/II study feasibility study using a tumor lysate pulsed DC vaccine with RT and concomitant TMZ in 2010, and they demonstrated a 18.3 month median survival with this approach (123). Dr Liau's team has recently reported early results from a Phase III trial of DCVax-L, which completed enrollment in November of 2015. In this study, patients underwent tumor resection for vaccine preparation, they received standard concurrent radiation and chemotherapy with TMZ, and they were subsequently randomized into two groups: a group receiving DCVax-L, and a placebo group (124). Of note, all patients were given the opportunity to receive DCVax-L at the time of progression/recurrence without breaking the blind regarding their initial treatment. As a result, at the time of publication, 86.4% of patients enrolled in the study had received DCVax-L at initial diagnosis or recurrence. The early median survival results show that patients who received the vaccine survived 23.1 months, which does appear promising, but we will be unable to determine the true impact of the treatment until enough events of progression and/or death have occurred to report the unblinded randomized results.
To improve the therapeutic index of these trials, some groups are combining dendritic cell vaccines with other immunomodulatory therapies. Sampson and team in 2014, investigated DC migration to vaccine site-draining lymph nodes following tetanus diptheria toxoid (Td) pre-conditioning in mouse models as well as in patients with newly diagnosed GBM (125). This led to human CMV pp65-LAMP mRNA-pulsed autologous DCs now being used in the Phase II ELEVATE trial (126). Patients with newly diagnosed GBM who have undergone resection and standard TMZ and RT are separated into 3 groups to understand how pre-conditioning the body can affect migration of the pulsed DC vaccine. Groups are given unpulsed DCs, Td, or Td accompanied with the immunosuppressive drug, basiliximab as pre-conditioning for the CMV pp65-LAMP DC vaccine. Basiliximab is a chimeric CD25 monoclonal antibody that has been shown to decrease Treg expansion in transplant patients, and it is being added to the trial to attempt to prevent Treg expansion after TMZ therapy (127).
Peptide Vaccines
Although DC vaccines have the potential to be effective in patients with GBM, their development poses a challenge to those that may not have the facilities to safely extract and pulse DCs with tumor components, therefore limiting the ability to rapidly scale these therapies for broad utilization. Similar to DC vaccines, peptide vaccines are made from tumor associated antigens, but instead of creating the vaccine in a personalized manner, peptide vaccines are “off-the-shelf” therapies that can be centrally produced. Peptide vaccines are thus more rapidly available for distribution to various medical centers, making them an attractive approach for multicenter trials for glioma immune therapy (128).
A well-studied target for peptide vaccines is the epidermal growth factor receptor (EGFR), a receptor tyrosine kinase that is highly expressed in high grade gliomas compared to normal brain tissue. In GBM, there are frequent mutations in EGFR, with the most common being the EGFR variant III (EGFRvIII) truncated mutant. EGFRvIII does not have a ligand-binding domain like the wild-type receptor, and it is therefore constitutively active, driving tumorigenesis (129, 130). This makes EGFRvIII a very attractive target for immune therapy, as it is a tumor-specific target driver of the malignancy, and it is expressed on the cell surface.
EGFRvIII peptide vaccine was generated by the fusion of a synthetic peptide that represents a truncated amino acid chain of EGFRvIII with keyhole limpet hemocyanin (KLH), a highly immunogenic peptide (Figure 4). Early investigations of EGFRvIII peptide vaccines in murine models resulted in a CTL mediated immune response to the EGFRvIII antigen (131).
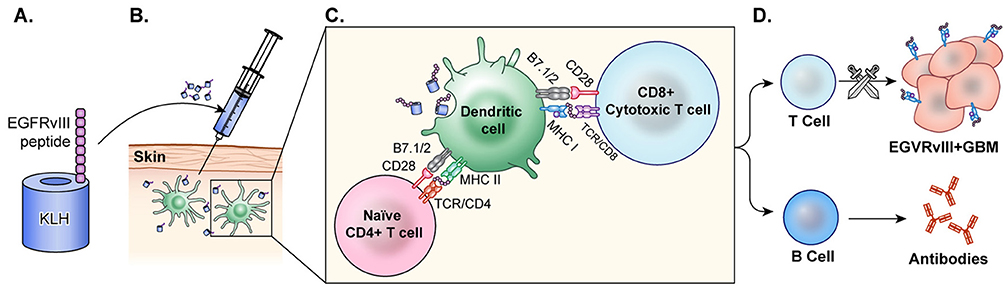
Figure 4. Peptide vaccine. (A) In the peptide vaccine, rindopepimut, EGFRvIII peptide is fused with highly immunogenic KLH (PEPvIII-KLH) for vaccine preparation. (B) The vaccine is administered intradermally and the antigen is recognized by APCs. (C) APCs present to T-cells and CTLs. (D) T-cells activate B-cells which then produce antibodies to EGFRvIII in the tumor. CTLs cross the blood brain barrier and target GBM cells with EGFRvIII on the surface. This activation of T-cells and CTLs results in anti-tumor response.
John Sampson, Amy Heimberger, and team conducted initial trials using rindopepimut, a peptide vaccine targeting EGFRvIII, in glioblastoma patients. They observed that the drug is tolerated at maximal doses with minimal adverse effects as determined by the Phase I VICTORI trial (132). In the Phase II ACTIVATE trial, they administered rindopepimut to newly diagnosed GBM patients who had undergone surgical resection and were receiving TMZ treatment. They showed a significant increase in both PFS and mOS at 14.2 and 26 months, respectively, with minor adverse reactions (133) which suggested that rindopepimut was both safe and beneficial to use with the standard GBM treatment of TMZ.
TMZ, however, can cause myelosuppression and lymphopenia in patients undergoing the treatment (134). ACT-II, a Phase II clinical trial of rindopepimut, revealed that EGFRvIII peptide vaccine could still be beneficial in conjunction with the standard treatment of TMZ in newly diagnosed GBM patients. However, they showed that at higher doses of TMZ, patients were at risk for greater toxic effects (135). Additionally, targeted tumor cells lost expression of EGFRvIII, demonstrating the specificity of the drug for its target (120).
The positive results observed in both the ACTIVATE and ACT-II clinical trials allowed for an expansion to a multicenter Phase II investigation of the drug known as ACT-III. Sixty-five newly diagnosed GBM patients were treated with rindopepimut and concomitant TMZ and the results continued to show increased PFS and mOS, as well as increased anti-EGFRvIII antibody titers in 85% of the patients (136). The vaccine then became part of a global, double-blind Phase III clinical trial in newly diagnosed GBM cases [ACT-IV 137).Unfortunately, ACT IV was terminated in 2016 as the control group had a higher mOS than the vaccinated group. It is believed that, in part, the heterogenous expression of EGFRvIII in GBM may have played a role in the failure of the trial (138).
The ReACT Phase II clinical trial used a combination of rindopepimut with the VEGF monoclonal antibody, bevacizumab, in patients with recurrent GBM, as bevacizumab has been shown to improve PFS in recurrent GBM (139). This trial showed a positive trend toward increased PFS in recurrent GBM patients (140).
Aside from targeting EGFRvIII, ongoing clinical trials are utilizing vaccines targeting multiple antigens, such as the 11 tumor-associated peptides targeted by the IMA950 trial. In this study, the peptide vaccine is given together TMZ and either Poly-ICLC (141) a vaccine with broad innate and adaptive immune enhancing effects, or RT (142). This multi-peptide approach could overcome the antigen loss often seen when targeting a single tumor-associated antigen (120).
Heat Shock Protein Vaccination
Heat shock proteins (HSPs) can broadly activate both the innate and adaptive immune systems as well as enhance MHC-I and MHC-II presentation of antigens (143). Tumor cells have increased HSP expression because of their high metabolic rate, which can leave the cells riddled with misfolded and aberrant proteins, resulting in cellular stress (144). HSPs make complexes with cellular stressors such as antigens and traffic them to APCs, where they can ultimately induce an anti-tumor immune response, making them an attractive component in cancer vaccine development. The vaccine is generated following resection of a patient's tumor. HSPs released from the resected tumor cells are believed to form complexes with tumor associated antigens. The HSP-tumor peptide complexes are isolated from the tumor, ex vivo, verified via Western Blot, and purified to make the vaccine. Then, they are peripherally administered back to the patient, where it is hoped that these HSP complexes will help prime CTL against the tumor (145) (Figure 5). Initial investigations utilizing HSP peptide complex-96 (HSPPC-96), an HSP that can bind tumor-associated antigens, as a tumor vaccine revealed that this treatment was safe and there was significant peripheral immune response to the treatment (146). This phase I trial, led by Andrew Parsa and Orin Bloch, disclosed a significant immune response specific to the tumor site in 11 of the 12 recurrent GBM patients they treated with the vaccine with minimal adverse effects. The mOS for those 11 responders was 47 weeks post-surgery and vaccination compared to 16 weeks of the single non-responder (128). In a follow-up Phase II trial, the team revealed the safety and efficacy of the vaccine when used with concomitant standard TMZ therapy (147). The group further demonstrated that PD-L1 expression on migrating myeloid cells induced systemic immunosuppression that could diminish the effect of the vaccine. Their 2009 Phase II HeatShock trial in newly diagnosed GBM demonstrated that MGMT methylation, Karnofsky performance score (KPS), and PD-L1 expression were prognostic factors for vaccine effectiveness (148). Mark Gilbert and team are currently recruiting newly diagnosed glioblastoma patients for a Phase II clinical trial investigating the effect of HSPPC-96 vaccine treatment together with standard radiotherapy and TMZ as well as pembrolizumab, an anti-PD-1 checkpoint inhibitor, to elucidate whether the HSPs from these individuals can enhance pembrolizumab efficacy (149). Dr. Fangusaro is also investigating this approach in pediatric subjects diagnosed with either HGG or ependymoma. They aim to determine if the vaccine is both efficacious and safe in the pediatric population (150).
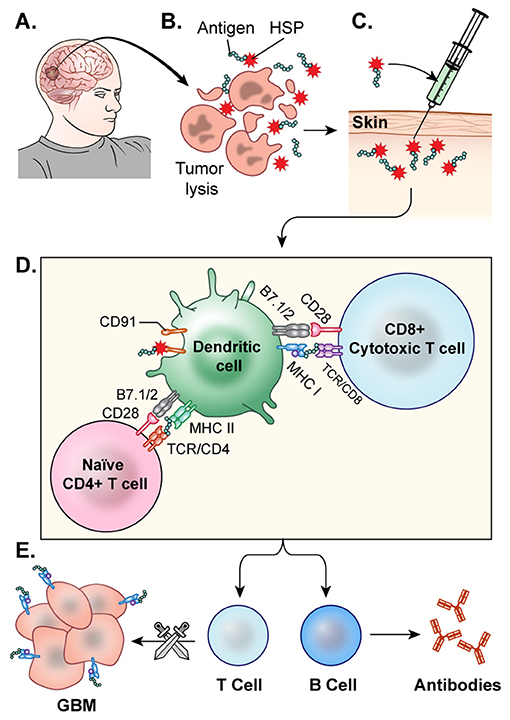
Figure 5. HSP vaccine. (A) The glioblastoma is resected. (B) HSPs bound to the tumor antigen are released by ex-vivo tumor cell lysis. (C) The desired HSPs are isolated and peripherally administered back to the patient as a vaccine. (D) Once injected, the HSP-tumor peptide complexes are taken up by antigen presenting cells, likely facilitated by CD91, and these peptide complexes are presented on MHC Class 1 molecules for recognition by CTLs (E) CTLs cross the blood brain barrier and target GBM cells. This activation of T-cells and CTLs results in anti-tumor response.
Adoptive Cell Therapy (ACT)
Adoptive cell therapy (ACT) has recently emerged as a promising immunotherapeutic regimen against various malignancies. ACT refers to the collection and manipulation of a patient's lymphocytes to target and kill cancer cells, first described when Delorme et al. observed that transferred lymphocytes could inhibit proliferating sarcomas in rat models (151).The first report of this approach for human use was in 1988 by Rosenburg et al. They used tumor-infiltrating lymphocytes (TILs) in combination with interleukin-2 to treat metastatic melanoma. In this study, they found objective regression in 60% of patient tumors. TIL treatment is now considered a highly effective therapy against this disease (152). Since then, ACT has advanced and now embodies a broad scope including several treatment modalities: tumor-infiltrating lymphocyte (TIL) immunotherapy, T-cell receptor (TCR) therapy, and chimeric antigen receptor (CAR) T-cell therapy.
TIL Therapy
The use of TILs, infiltrative lymphocytes with increased tumor-specificity that reside within the peritumoral space, for cancer therapy was introduced by the Surgery Branch at the NIH. This treatment involves TIL harvest at the time of tumor resection, expansion and stimulation with IL-2, and reinfusion of stimulated TILs into the body (153). Rosenburg et al. demonstrated the success of this treatment in metastatic melanoma (154). TIL therapy resulted in a complete response in approximately 20% of metastatic melanoma patients, with 40–50% of patients exhibiting transient response (155, 156). Solid tumor regression using TILs has also been reported in ovarian cancer and renal cell carcinoma (157). Additionally, TILs have recently gained popularity because these cells can be modified to recognize tumor-specific mutations creating neo-epitope reactive TILs (158). This treatment was applied in patients with colorectal cancers using TILs specifically targeting the Kras G12D mutant (159). Next generation sequencing to identify tumor-specific mutations shows promise in generating a genetically precise model of tumor-targeted TILs (160, 161) TIL therapy may be a promising mode of treatment for glioblastoma, but TIL therapy for the treatment of CNS malignancies has to be carefully investigated for its potential toxic effects and risk of cytokine release syndrome (162). Overall, recent technological advances in ACT expansion methods and preconditioning show progress for this field, but difficulties pertinent to treating central nervous system malignancies remain.
TCR Therapy
TCR therapy uses patient T-cells obtained from the peripheral blood which are modified to express tumor-specific α and β chains for enhanced antigen-recognition in treating solid tumors (163). TCRs express naturally occurring receptors that can recognize surface antigens as well as intracellular tumor antigens on antigen presenting cells by binding to the MCH complex (164, 165). The most notable clinical applications include the use of TCR therapy to target melanoma-associated antigen recognized by T-cells 1 (MART-1), melanoma-associated antigen A3 (MAGE-A3), and New York esophageal squamous cell carcinoma antigen (NY-ESO-1) (166–168). These trials demonstrated at least partial responses in patients with metastatic melanoma and synovial sarcoma and continue to show promise (148–150, 169). An ongoing trial is recruiting patients with diverse cancers including melanoma, synovial sarcoma, breast cancer, and non-small cell lung cancer patients for TCR directed NY-ESO-1 therapy (170, 171). While TCRs targeted to these specific antigens have not been tested in GBM, these antigens do present in gliomas as well, and due to their efficacy in other cancers warrant further investigation (172, 173).
CAR T-Cell Therapy
In CAR T-cell therapy, T-cells obtained from peripheral blood of patients are genetically engineered to express synthetic chimeric antigen receptors (CARs) on their cell surface which are specific for antigens expressed on a tumor's cell surface (174). These cells are then expanded in vitro and returned to the patient via infusion to subsequently proliferate in the body (175). Further modification and enhancement of CARs has generated a new generation of these synthetic T-cells. First-generation CARs are designed simply with an antigen recognition domain. However, second-generation CARs and beyond have additional co-stimulatory domains such as CD28 and 4-1BB which lower the barrier to activation and optimize receptor function (176) (Figure 6). CAR T-cell therapy has had success in treating various blood cancers by targeting an antigen, CD19, which is expressed among B-cells (177). Specifically, successful clinical trials led by Grupp et al. treating acute lymphoblastic leukemia (ALL) using CD19-targeted CAR T-cells led to its FDA approval in 2017 (178). However, disease recurrence has been observed in various clinical trials using CD19 CAR T-cell therapy to treat leukemias and lymphomas due to a phenomenon of antigen loss, where cancer cells no longer express CD19 (179). CD19-targeted CAR T-cell therapy has since been modified to address antigen loss by altering CARs to express antigen receptors that bind CD22 or CD123, which are antigens also expressed by these neoplastic B-cells (180). Additionally, Kochenderfer et al. developed a CAR T cell therapy targeted to B-cell maturation protein (BCMA), present in a majority of multiple myeloma (MM) cells, which resulted in a complete response in 50% of patients for this ongoing trial (181).
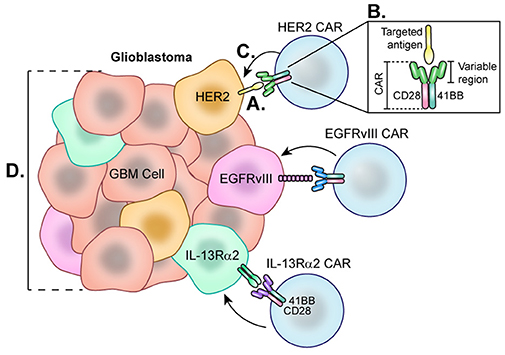
Figure 6. Chimeric Antigen Receptor (CAR) T-cells each directed at a specific GBM-specific tumor antigen. Each CAR T-cell therapy developed for the treatment of glioblastoma utilizes a CAR directed toward one antigen such as HER2, EGFRvIII, or IL-13Rα2. (A) Engagement of tumor-specific CAR T-cells with target cell surface antigen present on tumor cells causes CAR T-cell activation. (B) Second and third generation CAR T-cells are synthesized with co-stimulatory molecules such as 41BB, CD28, and CD3 which lower the CAR T-cell barrier to activation. (C) Fully activated CARs attack target cells causing tumor cell lysis. (D) Cells negative for the CAR T-cell target demonstrate the heterogeneity of the tumor and represent a barrier to treat as these cells not targeted continue to proliferate.
With the overt success of ACT treatment against blood cancers, questions have arisen about the potential efficacy of CAR-T-cells against solid tumors. Some in the field are skeptical that CAR T-cell therapy will demonstrate the same efficacy in solid tumors because the majority of solid tumor antigens exist inside the cell, away from CAR T-cell binding (182). However, investigators have made progress in generating CAR T-cells that target proteins overexpressed in many solid cancers with promising results in preclinical models of pancreatic cancer (183) neuroblastoma (184) and lymphoma (185) among others. As a result, phase I and phase I/II trials are underway targeting a variety of antigens in solid tumors (186).
For GBM, CAR T-cells generated to target one of three GBM-specific antigens—HER2, IL-13Rα2, or EGFRvIII—are being studied in clinical trials. HER2 is a tyrosine kinase receptor overexpressed in many cancers, including up to 80% of GBMs (187) (Figure 6). In 2010, a phase 1 trial was started at Baylor using CAR T-cells targeting HER2. Of note, these CARs were generated with a CD28 signaling domain and pre-selected for their ability to naturally recognize cytomegalovirus (CMV), which may augment therapeutic potency of these cells by also targeting CMV-related peptides in the glioblastoma microenvironment. A total of 16 recurrent HER2+ GBM patients were enrolled for treatment by this second generation HER2-CAR CMV-T cell which led to a partial response in 1 patient lasting more than 9 months. Of the 16 patients, 7 presented with stable disease for up to 29 months. Blood samples revealed that HER2-CARs were detected in only 7 patients at 6 weeks post-treatment, and levels continued to decline, suggesting these CARs were unable to expand in vivo (188). Although this therapy showed early signs of efficacy, one challenge is prolonging the life of these CAR T-cells. Beginning in 2018, City of Hope is leading a phase 1 clinical trial by Dr Badie and colleagues that aims to treat HGG patients with autologous memory-enriched T-cells transduced via lentivirus to express HER2 and 41-BB co-stimulant (189) as 41-BB has been reported to improve CAR T-cell persistence (190). The addition of this costimulatory signal has led to improved cytotoxicity to the target tumor cells (191, 192) as well as decreased T cell exhaustion (193) in in vitro models.
IL-13Rα2 is another promising antigen expressed in approximately 75% of GBMs but not at significant levels in normal brain cells (194) (Figure 6). From 2008 to 2011, Badie and others at City of Hope held a pilot safety and feasibility trial which enrolled 3 HGG patients for treatment using first generation CAR T-cells directed at IL13Rα2 via intracranial delivery directly to the resection cavity. Patients were not excluded based on lack of IL13Rα2 antigen positivity. Overall, all 3 HGG patients showed decreased IL13Rα2 tumor expression following therapy, and the post-relapse mean survival was 11 months (195). City of Hope then began using second generation IL-13Rα2-41BB co-stimulated CAR T-cells to treat recurrent or refractory HGG patients using intracavitary, intratumoral, or intraventricular infusions (196). While this study is still recruiting and final results have not been reported, a case study from this trial demonstrated complete response of recurrent multifocal GBM lasting for 7.5 months (197). This patient was treated with intracavitary infusion until leptomeningeal disease progression was found, at which point CAR T-cells were administered by intraventricular infusion which led to transient complete response, though notably, recurrence ultimately occurred in the form of distal metastases that expressed lower levels of IL13Rα2. The promising results of this work, though limited by small patient numbers, encourage further investigation.
Lastly, the EGFR variant, EGFRvIII, is a tumor-specific truncated version of the EGFR receptor, making it an attractive glioblastoma cell-surface target for CAR T cells. There are currently six ongoing trials that utilize CAR T-cells directed at EGFRvIII. Beginning in 2011, Rosenberg et al. of the National Cancer Institute led the first clinical trial directed at EGFRvIII, which is still ongoing (198). Early success has been reported in a phase I clinical trial of EGFRvIII-targeted CARs led by Donald O'Rourke and colleagues at the University of Pennsylvania. Of the 10 patients, 7 patients underwent reresection after therapy. Tissue from 3 of these patients demonstrated reduction of EGFRvIII expression, and 2 had complete elimination of detectable expression. Additionally, they noted that 3 patients demonstrated lymphocytic tumor infiltrates with broad T cell clonotype diversity. However, progression occurred in almost all cases despite antigen loss. As of 2017, one patient survived greater than 18 months with no further treatment while another two patients are alive, albeit with signs of disease progression. Importantly, the tumor microenvironment of surgical specimens from CAR T-Cell treated tumors in this trial displayed marked upregulation of PD-L1, IDO, and TGF-β, as well as FoxP3+ Tregs (199). This suggests that tumors adapt to treatment with this CAR T-cell and are capable of immune escape by activating various immunosuppressive pathways. Further investigations with in vivo animal models suggest that a durable anti-tumor response to CAR T cell therapy can be elicited by targeting this reciprocal immunosuppression with other immune-modulatory therapies (200–202) (Figure 6).
There are many complexities to treating GBM with CAR T-cells. Barriers to durable responses include the lack of long-term CAR T-cell persistence; ineffective delivery of cells to the infiltrating tumor; and GBM's characteristic immunosuppressive properties. Several of these barriers are being addressed in ongoing trials. For example, Memorial Sloan Kettering has created “Armored” CAR T-cells which constitutively secrete IL-12, a potent pro-inflammatory cytokine for cytotoxic T cells. In vitro work has shown that this IL-12 expression induces enhanced proliferation, persistent cytotoxicity, and decreased apoptosis of CAR T-cells (203).
Viral Therapeutics
Viral therapy has undergone extensive research over two decades, with some promising results in both pre-clinical and early clinical trials. Viral therapies use replication competent, albeit attenuated or genetically modified, viral species, taking advantage of both oncolytic and non-oncolytic mechanisms for high grade glioma targeting (Figure 7).
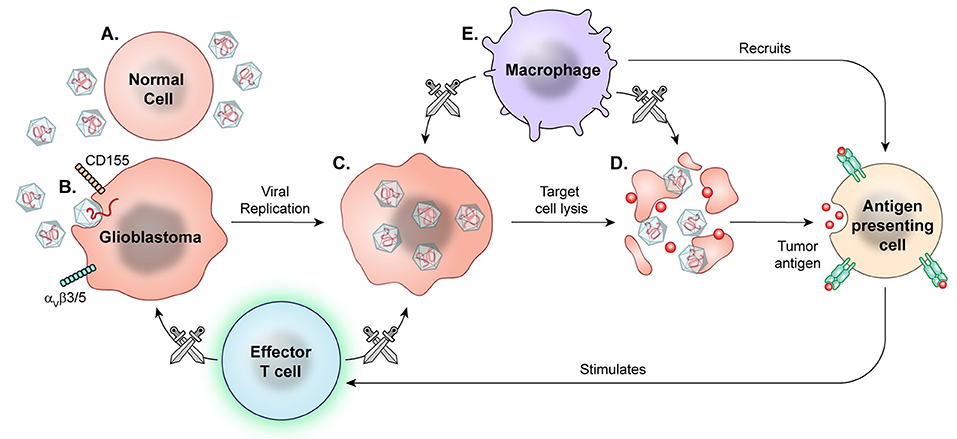
Figure 7. Prototypical mechanism of oncolytic viral therapy. The modified virus is infused into the tumor environment. (A) Normal cells exposed to viruses may have introduction of viral genetic information, but the viruses are modified to not replicate. (B) Viral particles then recognize enter cell based on specific surface proteins, such as CD155 in PVSRIPO and αVβ3/5 in Delta-24-RGD oncolytic adenovirus. (C) Oncolytic viral particles in tumors are replication-competent and recruit tumor cell replication machinery. (D) Viral replication results in cell lysis and release of viral particles to continue targeting tumor cells. (E) Macrophages detect and target virally infected cells, recruiting other APCs and effector T cells for secondary immune response against released tumor antigens.
Delivery of these viral therapeutic agents provides a practical challenge given the blood-brain barrier and the need to provide sufficient concentrations of agent at the site of action while minimizing risk of systemic toxicity. Convection enhanced delivery (CED) has been utilized to overcome these delivery challenges. CED, first described by Oldfield et al. in 1994, utilizes catheters to directly infuse a therapeutic agent to the central nervous system (204). This technique has been applied to glioblastoma treatment in a limited group of clinical trials, including the PRECISE trial. The PRECISE trial is, to date, the only completed phase III evaluation of convection enhanced delivery for the treatment of glioblastoma. The trial utilized cintredekin besudotox, a chimeric cytotoxin composed of a mutated Pseudomonas aeruginosa toxin attached to recombinant interleukin-13. Delivered by CED, this cytotoxin targets cells expressing the IL-13 receptor. They reported increased progression-free survival but not an increased overall survival compared to implanted Gliadel wafers in recurrent glioblastoma (205). This trial's failure may be attributed to less than 70% of catheters being placed according to protocol. Also, the statistical power study was designed such that although the CED group met the overall survival cutoff needed for significance, the control group had a significantly longer survival than expected, decreasing the difference in results of the two arms. These limitations highlight the difficulty of designing trials for CED in glioblastoma patients. However, other in vivo preclinical and early clinical studies have demonstrated feasibility of CED with other chemotherapeutic agents (206–209).
CED has been established as a viable technique for infusing viral therapeutics into the brain. Numerous groups have investigated a variety of genetically modified viruses in vitro and in non-human in vivo models via direct oncolytic activity or as a vector. Candidates include herpes-simplex virus (210) measles (211, 212) JC virus (213) Zika virus (214) adenovirus (192, 215–217) and pox virus (218) , among others. The combination of viral therapy with traditional chemoradiation is also under investigation (219, 220). Following the encouraging results of Delta-24-RGD oncolytic adenovirus in an immunocompetent murine glioma model (221) a phase I trial of the virus infused via CED into recurrent malignant gliomas was completed. The authors reported a subset of patients achieving long-term survival. This study was designed with two groups, one undergoing viral therapy alone, the other undergoing therapy followed by resection 2 weeks later in order to study the tumor tissue after undergoing treatment. They found that in the tumors resected 2 weeks after treatment, there was still active viral replication, while this was not seen in tumors resected at 6 weeks and 2.5 years post treatment. Additionally, at time of tumor resection, after receiving adenoviral therapy, the group resected 4 weeks after therapy displayed enhanced immune cell infiltration and decreased TIM-3 expression. As TIM-3 expression on T-cells is considered a marker for T cell exhaustion, this finding suggests improved T cell functioning in the treated tumor microenvironment. Based on the increased immune response at 4 weeks relative to 2 week samples in this trial, the investigators concluded that tumor regression reflects a delayed secondary immune response rather than direct oncolytic activity (222).
The recombinant polio-rhinovirus chimera (PVSRIPO), developed at Duke University, takes advantage of the poliovirus receptor CD155 found on many neoplastic cells. Preclinical studies demonstrated tumor regression in murine models of breast and prostate cancer treated by PVSRIPO (223). Similarly, treatment of preclinical murine glioma models with this viral therapy has led to tumor regression (224, 225). A phase I trial of PVSRIPO infused via CED in biopsy-confirmed recurrent glioblastoma showed a modest improvement in overall survival, but significant improvement in long term survival, with 21% of patients surviving at 24 and 36 months (226). While this study is limited by the use of historic controls, it warrants further study.
Future Directions and Biomarker Development
Even in the most successful trials of immune therapy for cancer, these treatments tend to be effective only in a subset of the treated patients. This highlights a need for better biomarkers that might predict which patients will be responders to immune therapy or could direct clinicians to other treatments that might augment the responses of predicted non-responders. In other cancers, there has been some limited progress in the identification of biomarkers of response to immune therapy. Wargo et al. illustrated the complexity of monitoring immune therapy response in melanoma patients, noting changing biomarker expression in longitudinal tissue samples over the course of treatment with serial checkpoint inhibitor monotherapy (77). Interferon-γ pathway loss has been associated with resistance to CTLA-4 blockade in melanoma (227) making it a potential biomarker of immune evasion and poor response to therapy. PD-L1, however, has had mixed reports as a tissue marker of potential therapeutic response. High expression has been associated with improved response to checkpoint inhibition in metastatic Merkel cell carcinoma (228) and squamous cell carcinoma (229). However, it does not appear to be predictive in non-small cell lung cancer (230) and metastatic urethral cancer (231). Additionally, Wolchok et al. reported from a phase III study of PD-1 and CTLA-4 blockade in melanoma that PD-L1 appeared to be prognostic of improved overall survival rather than predictive of checkpoint blockade therapeutic response (232). In all, current understanding of biomarkers is inconsistent and incomplete.
In glioblastoma patients, immunophenotyping has, thus far, been limited in that it typically relies on repeat tissue sampling. While stereotactic biopsies have relatively low morbidity, repeat biopsies are not considered standard practice in glioma clinical trials. This lack of sampling raises additional challenges, as it is quite difficult to determine if a glioblastoma patient has had response to immune therapy on imaging alone. Inflammatory responses to immune therapy often cause contrast enhancing lesions in the brain that can be difficult to distinguish from tumor progression on MRI (86). These limitations necessitate the identification of biomarkers to determine candidates for immune therapy and to track response reliably. The Checkmate 143 trial measured PD-L1 expression of patients entering the trial in an attempt to identify responders, but the full results of this trial have not yet been reported. Other trials have reported biomarkers in tissue pathology following tumor resection after treatment. Lang et al. reported increased TIM-3 expression on T-cells after treatment with Delta-24-RGD, suggesting improved T cell functioning (226). O'Rourke et al. reported upregulation of PD-L1, IDO, and TGF-β, as well as FoxP3+ Tregs in resected tumors following EGFRvIII CAR T-cells, potentially reflecting tumor adaptation and immune evasion (181). However, less invasive serum biomarkers have also been used effectively. In the phase 3 ACT-IV Trial for rindopepimut, investigators used serum titers for antibodies to EGFRvIII to monitor degree of host immune response to the vaccine after administration (119). In other studies, PD-L1 expression on circulating macrophages, and pathological response to therapy have been used to determine whether a response has occurred. In the phase II trial of autologous head shock protein peptide vaccination for glioblastoma, Bloch et al found that patients with low PD-L1 expression on peripheral myeloid cells had on overall survival of almost 45 months compared to 18 months in those with high expression (130). Non-invasive serum biomarkers such as soluble PD-L1 (233) cytokines (234) and peripheral mononuclear cells (235) have been described in monitoring response to immune therapy in non-CNS tumors (236) but similar studies have not been performed extensively in glioblastoma. However, additional biomarkers need to be developed to have a more complete understanding of host and tumor response.
The use of microdialysis in neuro-oncology is a promising addition to our arsenal for immune monitoring of glioma patients. Early use of cerebral microdialysis predominantly focused on patients with neurologic trauma (237–242) or subarachnoid hemorrhage (243–248) demonstrating safety and viability of the technology. More recently, this technique has been applied to brain tumor patients. Portnow et al. utilized microdialysis to monitor in real time if treatment with chemotherapy changed levels of 17 cytokines compared to craniotomy alone. This study showed that craniotomy induced an inflammatory response that dissipated over the next 96 h after surgery (249). Tabatabaei et al. utilized microdialysis catheters in peritumor tissue in high grade glioma patients undergoing radiation therapy, finding that treatment induces a strong inflammatory response via macrophages and monocytes (250). The development of checkpoint inhibitors, though demonstrably effective in metastatic melanoma and non-small cell lung carcinoma, has had mixed results human glioma studies thus far. Understanding the immune profile of the glioma microenvironment in patients undergoing this therapy may allow for patient stratification to determine those with the greatest potential for therapeutic benefit. The ongoing study “Cytokine Microdialysis for Immune Monitoring in Recurrent Glioblastoma Patients Undergoing Checkpoint Blockade” uses microdialysis for monitoring of immune functioning in the glioblastoma tumor environment following checkpoint inhibition. Concurrently, the study samples serum, CSF, and bone marrow for comprehensive analysis of potential biomarkers indicating response to therapy (251).
Sequencing and Biomarker Development
Advances in understanding the genetics of glioblastomas has led to discovery of prognostic and predictive factors as well as potential targets. Initially, the identification of 4 different molecular profiles (252) of GBM expanded understanding of pathology of the disease beyond histology. Subsequent identification of isocitrate dehydrogenase (IDH) mutation and MGMT methylation statuses and their association with survival (253, 254) as well as response to chemotherapy and radiation (255–257) have proven invaluable to patient care and stratification for appropriate therapy. As technology and methodology have improved, DNA and RNA sequencing allow further mutation identification (258) including biomarkers with associated survival implications (259, 260). Additionally, these techniques may now be able to move out of the laboratory into the clinical setting as sequencing technology becomes more accessible for use in patient care. Kazimierz has shown that whole-genome and RNA sequencing can be performed in a timely and efficient manner (261). As sequencing has become more efficient, its use in guiding clinical decision-making has become feasible, with Byron et al finding that results could consistently be obtained within 35 days of surgical resection (262). Pertinent to this review, sequencing may have implications for immune-based therapies. Early sequencing of common in-vitro glioblastoma cell lines identified which lines expressed HLA subtypes associated with improved antigen presentation and response to immunotherapy (263). Song et al. then expanded on similar sequencing work in 298 glioblastoma and control patients, identifying HLA subtypes associated with decreased tumor incidence (264).
Evidence from non-CNS malignancies has suggested an association between the number of somatic mutations identified via sequencing of malignant cells (mutational burden) and the cancer's relationship with the host immune system. Mutational burden is widely variable between malignancies (265) with subsequent groups demonstrating these mutations act as neoantigens to target in adoptive T-cell therapy (266) as well as checkpoint blockade (267). Cancers that have had promising results with checkpoint blockade in human trials, thus far, have been associated with high mutational burden, such as melanoma (268) and non-small cell lung cancer (269). However, further investigation suggests that degree of mutation burden has not had a strong association with response to checkpoint blockade across a broad cross section of malignancies (270). A pilot study reported two patients with glioblastomas due to bi-allelic mismatch repair deficiency, which results in high mutational burden. They found promising results in response to checkpoint inhibition, describing both clinical and radiographic improvement (271). However, despite the inter and intra-heterogeneity of the genomics of glioblastoma, Hodges et al found that only a minority of these tumors were found to have a high mutational burden, and the degree of burden did not correlate with immune cell infiltration into the tumor (272).
As mutational burden alone has not been a strong predictor, other groups have investigated specific somatic mutations, such as the interferon-gamma pathway that may portend a decreased response to checkpoint inhibition (273) in non-CNS tumors. Similarly, in glioblastoma, a diverse population of tumor-infiltrating lymphocytes, thought to more effectively combat a heterogeneous tumor, may not be dependent on intratumoral genomic heterogeneity (274) but rather in 23 genes in immune-related pathways whose expression was found to be significantly associated with prognosis via The Cancer Genome Atlas GBM dataset (275). Goodman et al analyzed a de-identified tumor database, finding that solid tumors with PD-L1 amplification in their genomic profile had almost 70% reported response to checkpoint inhibition, including in glioblastoma. This study again demonstrated that in these tumors, overall tumor mutational burden was only low-to-intermediate (276). As immune-relevant mutations have been identified, comprehensive testing of these mutations is being implemented in clinical decision-making. Peng et al utilized exome, whole genome, and RNA sequencing to identify glioblastoma patients that are least likely to respond to traditional therapy and may be candidates for immune therapy (277). Similarly, Chen et al. categorized multiple malignancies, including GBM, based on tumor microenvironment immune types, to identify genetic patterns that have the greatest potential for therapeutic response to immune therapy (278). While not yet described in GBM, sequencing of circulating tumor DNA in the serum has identified single nucleotide polymorphisms in PD-L1 genes that correlate with improved response to PD-1 blockade in NSCLC (279).
Sequencing and Development of Patient-Specific Therapeutics
Beyond identifying patients who may have the greatest benefit from immune therapy, Tran et al. reported using neoantigens identified via sequencing as targets for adopted T cell transfer with promising results in non-CNS cancers (141, 280). Monovalent and polyvalent vaccination, described previously, may incorporate these additional targets as more are identified (281). Dunn et al used whole exome and RNA sequencing to identify potential tumor neoantigens in murine glioma models, then use ELISPOT to screen for immunogenicity. These techniques allowed the identification of targets that infiltrating tumor CD8 cytotoxic T cells recognized and bound with high affinity. They suggest that this view into the function of the endogenous immune response to a tumor may provide guidance in the development of personalized tumor vaccination (282). As these lines of research continue, there is great potential for both the development of novel therapeutics as well as the identification of the patients who will most benefit.
Bispecific T-Cell Engagers (BITEs)
First described in 1961, bispecific antibodies have two variable segments allowing for binding two antigens (283). Nitta et al reported the use of bispecific antibodies to coat activated lymphocytes with partial response in a subset of 10 high-grade glioma patients (284). Bispecific antibodies would be the precursor to bispecific T-cell engagers (BITEs). In 1985, Staerz reported the use of bispecific antibodies in which one of the variable segments targeted the T-cell receptor with the goal of recruiting T-cell mediated immune response (285).
In 1995, Mack et al used BITEs to induce effective cytotoxicity in a cell line transfected to express the target antigen (286). The first drug of this type, blinatumomab, used segments specific for CD19 and CD3 to facilitate T cell targeting of leukemia. It was successful in several phase II trials for adult and pediatric acute lymphocytic leukemia (287–289) and in one phase III trial (290). It was subsequently given FDA approval in 2017. Blinatumomab has also had positive initial results in the treatment of refractory non-Hodgkin lymphoma (291).
These developments have led to study of other targeted BITEs in multiple myeloma (292) hepatocellular carcinoma (293) and other solid tumors in conjunction with oncolytic virus and CAR-T therapies (172, 294). The expanding use of BITEs has been of particular interest in solid cancers that are relatively non-immunogenic, which may be poor candidates for checkpoint inhibition (295). As the most common neoantigen found in glioblastoma, EGFRvIII has been one of the first targets for BITEs therapy. Initial in-vivo work in murine glioma models showed high cure rates. Interestingly, after BITE therapy, immunosuppressive Tregs changed their behavior to attack EGFRvIII positive tumor cells via the granzyme-perforin pathway (296). In addition to targeting the EGFRvIII antigen, BITEs have also been applied to enhance the cytotoxic T cell response to cells expressing CD133+, a marker of tumor initiating cells in GBM (113). A preclinical study by Prasad et al. showed that BITEs targeting CD133 increased T cell ability to eradicate patient derived CD133+ GBM stem cells in orthotopic models of brain tumors (297). They showed that the BITEs specific to CD133 and CD3 were able to inhibit tumor progression in 4 out of the 5 mice, compared to control BITEs targeting prostate-specific membrane antigen (PSMA). They also confirmed the antitumor activity of the CD133 BITE in established tumors in orthotopic xenograft models by administering the bispecific antibodies on day 14 after the tumor implantation. In the group treated with PSMA BITEs, the tumor progressed between day 20 and 35, whereas the group treated with CD133 BITEs inhibited further tumor growth. Despite promising pre-clinical results, there are no current human trials for BITEs in glioblastoma (120).
Conclusion
Glioblastoma is a highly malignant disease particularly resistant to the current armament of chemoradiation. As a result, the need for novel therapeutic strategies has been paramount. Over decades, research has made enormous strides in defining the pathogenesis of GBM and its relationship with its human host. A greater understanding of the immune function both systemically and within the tumor microenvironment has provided new therapeutic targets. In addition, in-depth knowledge of each patient's particular disease may allow for improved patient stratification to determine the best emerging therapy to use. The ongoing development of multiple immunotherapeutic strategies has ever-increasing potential to fundamentally change the way patients with glioblastoma are treated and, hopefully, make meaningful improvements in outcome.
Author Contributions
JL provided the largest writing contribution to the manuscript and edited all sections of the manuscript. He was primarily responsible for assembling the manuscript and addressing comments by reviewers. VS performed literature and wrote multiple sections of the manuscript. She additionally assisted in creating figures and worked with medical arts to develop final figures. GD performed literature and wrote multiple sections of the manuscript and assisted in creating figures. AN provided additional literature review and writing of sections added to manuscript at time of review. EN provided extensive guidance, supervision, and editorial input to manuscript development.
Conflict of Interest Statement
The authors refer to an ongoing clinical trial being performed by several of the authors. This trial receives funding from the Intramural Research Program at the National Institutes of Health and non-financial drug support from Bristol-Myers Squibb. Otherwise, the authors declare no potential conflicts of interest.
Acknowledgments
The authors would like to acknowledge Rogelio Medina for assistance in proofreading.
References
1. Gedeon PC, Riccione KA, Fecci PE, Sampson JH. Antibody-based immunotherapy for malignant glioma. Seminars Oncol. (2014) 41:496–510. doi: 10.1053/j.seminoncol.2014.06.004
2. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab plus Radiotherapy–Temozolomide for newly diagnosed glioblastoma. N Engl J Med. (2014) 370:709–22. doi: 10.1056/NEJMoa1308345
3. Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008-2012. Neuro Oncol. (2015) 17(Suppl. 4):iv1–iv62. doi: 10.1093/neuonc/nov189
4. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. (2005) 352:987–96. doi: 10.1056/NEJMoa043330
5. Ashby LS, Smith KA, Stea B. Gliadel wafer implantation combined with standard radiotherapy and concurrent followed by adjuvant temozolomide for treatment of newly diagnosed high-grade glioma: a systematic literature review. World J Surg Oncol. (2016) 14:225. doi: 10.1186/s12957-016-0975-5
6. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. (2014) 370:699–708. doi: 10.1056/NEJMoa1308573
7. Mittal S, Klinger NV, Michelhaugh SK, Barger GR, Pannullo SC, Juhász C. Alternating electric tumor treating fields for treatment of glioblastoma: rationale, preclinical, and clinical studies. J Neurosurg. (2018) 128:414–21. doi: 10.3171/2016.9.JNS16452
9. McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J. (2006) 26:154–8.
10. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. (2015) 373:23–34. doi: 10.1056/NEJMoa1504030
11. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. (2015) 372:2018–28. doi: 10.1056/NEJMoa1501824
12. Medawar P. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br J Exp Pathol. (1948) 29:58–69.
13. Louveau A, Harris TH, Kipnis J. Revisiting the mechanisms of CNS immune privilege. Trends Immunology (2015) 36:569–77. doi: 10.1016/j.it.2015.08.006
14. Nduom EK, Weller M, Heimberger AB. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol. (2015) 17(Suppl. 7):vii9–14. doi: 10.1093/neuonc/nov151
15. Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. (2010) 11:7–13. doi: 10.1038/ni.1818
16. Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. (2006) 66:3294–302. doi: 10.1158/0008-5472.CAN-05-3773
17. Jordan JT, Sun W, Hussain SF, DeAngulo G, Prabhu SS, Heimberger AB. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol Immunother. (2008) 57:123–31. Erratum in: Cancer Immunol Immunother. (2008) 57:133. doi: 10.1007/s00262-007-0336-x
18. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, et al. CCL2 Produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. (2016) 76:5671–82. doi: 10.1158/0008-5472.CAN-16-0144
19. Wainwright DA, Sengupta S, Han Y, Lesniak MS. Thymus-derived rather than tumor-induced regulatory T cells predominate in brain tumors. Neuro Oncol. (2011) 13:1308–23. doi: 10.1093/neuonc/nor134
20. Damle NK, Childs AL, Doyle LV. Immunoregulatory T lymphocytes in man. Soluble antigen-specific suppressor-inducer T lymphocytes are derived from the CD4+CD45R-p80+ subpopulation. J Immunol. (1987) 139:1501–8. Erratum in: J Immunol (1987) 139:3911.
21. Kiniwa Y, Miyahara Y, Wang HY, Peng W, Peng G, Wheeler TM, et al. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin Cancer Res. (2007) 13:6947–58. doi: 10.1158/1078-0432.CCR-07-0842
22. Chaput N, Louafi S, Bardier A, Charlotte F, Vaillant JC, Ménégaux F, et al. Identification of CD8+CD25+Foxp3+ suppressive T cells in colorectal cancer tissue. Gut (2009) 58:520–9. doi: 10.1136/gut.2008.158824
23. Wang Q, Yu T, Yuan Y, Zhuang H, Wang Z, Liu X, et al. Sorafenib reduces hepatic infiltrated regulatory T cells in hepatocellular carcinoma patients by suppressing TGF-beta signal. J Surg Oncol. (2013) 107:422–7. doi: 10.1002/jso.23227
24. Chen C, Chen D, Zhang Y, Chen Z, Zhu W, Zhang B, et al. Changes of CD4+CD25+FOXP3+ and CD8+CD28- regulatory T cells in non-small cell lung cancer patients undergoing surgery. Int Immunopharmacol. (2014) 18:255–61. doi: 10.1016/j.intimp.2013.12.004
25. Zhang S, Ke X, Zeng S, Wu M, Lou J, Wu L, et al. Analysis of CD8+ Treg cells in patients with ovarian cancer: a possible mechanism for immune impairment. Cell Mol Immunol. (2015) 12:580–91. doi: 10.1038/cmi.2015.57
26. Badie B, Schartner JM. Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery (2000) 46:957–61; discussion 961–2. doi: 10.1097/00006123-200004000-00035
27. Tsutsui S, Yasuda K, Suzuki K, Tahara K, Higashi H, Era S. Macrophage infiltration and its prognostic implications in breast cancer: the relationship with VEGF expression and microvessel density. Oncol Rep. (2005) 14:425–31. doi: 10.3892/or.14.2.425
28. Komohara Y, Niino D, Saito Y, Ohnishi K, Horlad H, Ohshima K, et al. Clinical significance of CD163? tumor-associated macrophages in patients with adult T-cell leukemia/lymphoma. Cancer Sci. (2013) 104:945–51. doi: 10.1111/cas.12167
29. Kennedy BC, Showers CR, Anderson DE, Anderson L, Canoll P, Bruce JN, et al. Tumor-associated macrophages in glioma: friend or foe? J Oncol. (2013) 2013:486912. doi: 10.1155/2013/486912
30. Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. (2010) 12:1113–25. doi: 10.1093/neuonc/noq082
31. Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol. (2008) 216:15–24. doi: 10.1002/path.2370
32. Prosniak M, Harshyne LA, Andrews DW, Kenyon LC, Bedelbaeva K, Apanasovich TV, et al. Glioma grade is associated with the accumulation and activity of cells bearing M2 monocyte markers. Clin Cancer Res. (2013) 19:3776–86. doi: 10.1158/1078-0432.CCR-12-1940
33. Mia S, Warnecke A, Zhang XM, Malmström V, Harris RA. An optimized protocol for human M2 macrophages using M-CSF and IL-4/IL-10/TGF-β yields a dominant immunosuppressive phenotype. Scand J Immunol. (2014) 79:305–14. doi: 10.1111/sji.12162
34. Lisi L, Stigliano E, Lauriola L, Navarra P, Dello Russo C. Proinflammatory-activated glioma cells induce a switch in microglial polarization and activation status, from a predominant M2b phenotype to a mixture of M1 and M2a/B polarized cells. ASN Neuro. (2014) 6:171–83. doi: 10.1042/AN20130045
35. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. (2017) 77:2266–78. doi: 10.1158/0008-5472.CAN-16-2310
36. Szulzewsky F, Arora S, de Witte L, Ulas T, Markovic D, Schultze JL, et al. Human glioblastoma-associated microglia/monocytes express a distinct RNA profile compared to human control and murine samples. Glia (2016) 64:1416–36. doi: 10.1002/glia.23014
37. Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight (2016) 1:e85841. doi: 10.1172/jci.insight.85841
38. Fallah P, Arefian E, Naderi M, Aghaee-Bakhtiari SH, Atashi A, Ahmadi K, et al. miR-146a and miR-150 promote the differentiation of CD133± cells into T-lymphoid lineage. Mol Biol Rep. (2013) 40:4713–9. doi: 10.1007/s11033-013-2567-6
39. Xu S, Wei J, Wang F, Kong LY, Ling XY, Nduom E, et al. Effect of miR-142-3p on the M2 macrophage and therapeutic efficacy against murine glioblastoma. J Natl Cancer Inst. (2014) 106:dju162. doi: 10.1093/jnci/dju162
40. Saha D, Martuza RL, Rabkin SD. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell (2017) 32:253–267.e5. doi: 10.1016/j.ccell.2017.07.006
41. Leblond MM, Pérès EA, Helaine C, Gérault AN, Moulin D, Anfray C, et al. M2 macrophages are more resistant than M1 macrophages following radiation therapy in the context of glioblastoma. Oncotarget (2017) 8:72597–612. doi: 10.18632/oncotarget.19994
42. Rodrigues JC, Gonzalez GC, Zhang L, Ibrahim G, Kelly JJ, Gustafson MP, et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro Oncol. (2010) 12:351–65. doi: 10.1093/neuonc/nop023
43. Angulo I, Rullas J, Campillo JA, Obregón E, Heath A, Howard M, et al. Early myeloid cells are high producers of nitric oxide upon CD40 plus IFN-gamma stimulation through a mechanism dependent on endogenous TNF-alpha and IL-1alpha. Eur J Immunol. (2000) 30:1263–71. doi: 10.1002/(SICI)1521-4141(200005)30:5<1263::AID-IMMU1263>3.0.CO;2-5
44. Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol. (2008) 83:1136–44. doi: 10.1189/jlb.0907611
45. Gielen PR, Schulte BM, Kers-Rebel ED, Verrijp K, Petersen-Baltussen HM, ter Laan M, et al. Increase in both CD14-positive and CD15-positive myeloid-derived suppressor cell subpopulations in the blood of patients with glioma but predominance of CD15-positive myeloid-derived suppressor cells in glioma tissue. J Neuropathol Exp Neurol. (2015) 74:390–400. doi: 10.1097/NEN.0000000000000183
46. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1±CD115± immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. (2006) 66:1123–31. doi: 10.1158/0008-5472.CAN-05-1299
47. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8± T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. (2004) 172:989–99. Erratum in: J Immunol. (2004) 172:4647. doi: 10.4049/jimmunol.172.2.989
48. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. (2004) 64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465
49. Gielen PR, Schulte BM, Kers-Rebel ED, Verrijp K, Bossman SA, Ter Laan M, et al. Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro Oncol. (2016) 18:1253–64. doi: 10.1093/neuonc/now034
50. Yang R, Cai Z, Zhang Y, Yutzy WH IV, Roby KF, Roden RB. CD80 in immune suppression by mouse ovarian carcinoma-associated Gr-1±CD11b± myeloid cells. Cancer Res. (2006) 66:6807–15. doi: 10.1158/0008-5472.CAN-05-3755
51. Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res. (2013) 19:3165–75. doi: 10.1158/1078-0432.CCR-12-3314
52. Najjar YG, Finke JH. Clinical perspectives on targeting of myeloid derived suppressor cells in the treatment of cancer. Front Oncol. (2013) 3:49. doi: 10.3389/fonc.2013.00049
53. US National Library of Medicine. ClinicalTrials.gov. (2016). Available online at:https://clinicaltrials.gov/ct2/show/NCT02669173
54. Ceeraz S, Nowak EC, Burns CM, Noelle RJ. Immune checkpoint receptors in regulating immune reactivity in rheumatic disease. Arthrit Res Ther. (2014) 16:469. doi: 10.1186/s13075-014-0469-1
55. Frauwirth KA, Thompson CB. Activation and inhibition of lymphocytes by costimulation. J Clin Invest. (2002) 109:295–9. doi: 10.1172/JCI0214941
57. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity (2007) 27:111–22. doi: 10.1016/j.immuni.2007.05.016
58. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (1996) 271:1734–6.
59. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
60. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity (1994) 1:405–13.
61. Carreno BM, Bennett F, Chau TA, Ling V, Luxenberg D, Jussif J, et al. CTLA-4 (CD152) can inhibit T cell activation by two different mechanisms depending on its level of cell surface expression. J Immunol. (2000) 165:1352–6. doi: 10.4049/jimmunol.165.3.1352
62. Thompson CB, Allison JP. The emerging role of CTLA-4 as an immune attenuator. Immunity (1997) 7:445–50.
63. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. (2002) 8:793–800. Erratum in: Nat Med 2002 8:1039. doi: 10.1038/nm730
64. Huard B, Prigent P, Pagès F, Bruniquel D, Triebel F. T cell major histocompatibility complex class II molecules down-regulate CD4+ T cell clone responses following LAG-3 binding. Eur J Immunol. (1996) 26:1180–6.
65. Su EW, Lin JY, Kane LP. TIM-1 and TIM-3 proteins in immune regulation. Cytokine (2008) 44:9–13. doi: 10.1016/j.cyto.2008.06.013
66. Shuford WW, Klussman K, Tritchler DD, Loo DT, Chalupny J, Siadak AW, et al. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J Exp Med. (1997) 186:47–55.
67. Schaer DA, Murphy JT, Wolchok JD. Modulation of GITR for cancer immunotherapy. Curr Opin Immunol. (2012) 24:217–24. doi: 10.1016/j.coi.2011.12.011
68. Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, Brogdon JL. OX40 engagement depletes intratumoral Tregs via activating FcγRs, leading to antitumor efficacy. Immunol Cell Biol. (2014) 92:475–80. doi: 10.1038/icb.2014.26
69. Hung AL, Garzon-Muvdi T, Lim M. Biomarkers and Immunotherapeutic Targets in Glioblastoma. World Neurosurg. (2017) 102:494–506. doi: 10.1016/j.wneu.2017.03.011
70. Lee KS, Lee K, Yun S, Moon S, Park Y, Han JH, et al. Prognostic relevance of programmed cell death ligand 1 expression in glioblastoma. J Neurooncol. (2018) 136:453–61. doi: 10.1007/s11060-017-2675-6
71. Nduom EK, Wei J, Yaghi NK, Huang N, Kong LY, Gabrusiewicz K, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. (2016) 18:195–205. doi: 10.1093/neuonc/nov172
72. Pratt D, Dominah G, Lobel G, Obungu A, Lynes J, Sanchez V, et al. Programmed Death Ligand 1 is a negative prognostic marker in recurrent isocitrate dehydrogenase-wildtype glioblastoma. Neurosurgery (2018). doi: 10.1093/neuros/nyy268. [Epub ahead of print].
73. DiDomenico J, Lamano JB, Oyon D, Li Y, Veliceasa D, Kaur G, et al. The immune checkpoint protein PD-L1 induces and maintains regulatory T cells in glioblastoma. Oncoimmunology (2018) 7:e1448329. doi: 10.1080/2162402X.2018.1448329
74. He Y, Rivard CJ, Rozeboom L, Yu H, Ellison K, Kowalewski A, et al. Lymphocyte-activation gene-3, an important immune checkpoint in cancer. Cancer Sci. (2016) 107:1193–7. doi: 10.1111/cas.12986
75. Simonelli M, Persico P, Perrino M, Zucali PA, Navarria P, Pessina F, et al. Checkpoint inhibitors as treatment for malignant gliomas: “A long way to the top”. Cancer Treat Rev. (2018) 69:121–31. doi: 10.1016/j.ctrv.2018.06.016
76. Weber J, Thompson JA, Hamid O, Minor D, Amin A, Ron I, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. (2009) 15:5591–8. doi: 10.1158/1078-0432.CCR-09-1024
77. Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. (2010) 11:155–64. doi: 10.1016/S1470-2045(09)70334-1
78. O'Day SJ, Maio M, Chiarion-Sileni V, Gajewski TF, Pehamberger H, Bondarenko IN, et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single-arm phase II study. Ann Oncol. (2010) 21:1712–7. doi: 10.1093/annonc/mdq013
79. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
80. Brody R, Zhang Y, Ballas M, Siddiqui MK, Gupta P, Barker C, et al. PD-L1 expression in advanced NSCLC: Insights into risk stratification and treatment selection from a systematic literature review. Lung Cancer (2017) 112:200–15. doi: 10.1016/j.lungcan.2017.08.005
81. Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. (2016) 6:827–37. doi: 10.1158/2159-8290.CD-15-1545
82. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. (2014) 20:5290–301. doi: 10.1158/1078-0432.CCR-14-0514
83. Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. (2013) 86:343–9. doi: 10.1016/j.ijrobp.2012.12.025
84. Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wöhrer A, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. (2015) 17:1064–75. doi: 10.1093/neuonc/nou307
85. US National Library of Medicine. ClinicalTrials.gov. (2015). Available online at: https://clinicaltrials.gov/ct2/show/NCT02617589
86. US National Library of Medicine. ClinicalTrials.gov. (2016). Available online at: https://clinicaltrials.gov/ct2/show/NCT02667587
87. US National Library of Medicine. ClinicalTrials.gov. (2013). Available online at: https://clinicaltrials.gov/ct2/show/NCT02017717
88. Reardon DA, Omuro A, Brandes AA, Rieger J, Wick A, Sepulveda J, et al. OS10.3 randomized phase 3 study evaluating the efficacy and safety of Nivolumab vs. Bevacizumab in patients with recurrent glioblastoma: CheckMate 143, Neuro Oncol. (2017) 19 (Suppl. 3):iii21. doi: 10.1093/neuonc/nox036.071
89. Filley AC, Henriquez M, Dey M. Recurrent glioma clinical trial, CheckMate-143: the game is not over yet. Oncotarget (2017) 8:91779–94. doi: 10.18632/oncotarget.21586
90. Ranjan S, Quezado M, Garren N, Boris L, Siegel C, Lopes Abath Neto O, et al. Clinical decision making in the era of immunotherapy for high grade-glioma: report of four cases. BMC Cancer (2018) 8:239. doi: 10.1186/s12885-018-4131-1
91. Brignone C, Gutierrez M, Mefti F, Brain E, Jarcau R, Cvitkovic F, et al. First-line chemoimmunotherapy in metastatic breast carcinoma: combination of paclitaxel and IMP321 (LAG-3Ig) enhances immune responses and antitumor activity. J Transl Med. (2010) 8:71. doi: 10.1186/1479-5876-8-71
92. Brignone C, Escudier B, Grygar C, Marcu M, Triebel F. A phase I pharmacokinetic and biological correlative study of IMP321, a novel MHC class II agonist, in patients with advanced renal cell carcinoma. Clin Cancer Res. (2009) 15:6225–31. doi: 10.1158/1078-0432.CCR-09-0068
93. US National Library of Medicine. ClinicalTrials.gov (2008). Available online at: https://clinicaltrials.gov/ct2/show/NCT00732082
94. US National Library of Medicine. ClinicalTrials.gov. (2013). Available online at: https://clinicaltrials.gov/ct2/show/NCT01968109
95. US National Library of Medicine. ClinicalTrials.gov. (2014). Available online at: https://clinicaltrials.gov/ct2/show/NCT02061761
96. US National Library of Medicine. ClinicalTrials.gov. (2016). Available online at: https://clinicaltrials.gov/ct2/show/NCT02658981
97. Plotkin SA. Vaccines, vaccination, and vaccinology. J Infect Dis. (2003) 187:1349–59. doi: 10.1086/374419
98. Steinman RM, Nussenzweig MC. Dendritic cells: features and functions. Immunol Rev. (1980) 53:127–47.
99. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, et al. Immunobiology of dendritic cells. Ann Rev Immunol. (2000) 18:767–811. doi: 10.1146/annurev.immunol.18.1.767
100. Fong L, Hou Y, Rivas A, Benike C, Yuen A, Fisher GA, et al. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc Natl Acad Sci USA. (2001) 98:8809–14. doi: 10.1073/pnas.141226398
101. Morse MA, Nair S, Fernandez-Casal M, Deng Y, St Peter M, Williams R, et al. Preoperative mobilization of circulating dendritic cells by Flt3 ligand administration to patients with metastatic colon cancer. J Clin Oncol. (2000) 18:3883–93. doi: 10.1200/JCO.2000.18.23.3883
102. Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL, et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med. (1998) 4:321–7.
103. Weller M, Roth P, Preusser M, Wick W, Reardon DA, Platten M, et al. Vaccine-based immunotherapeutic approaches to gliomas and beyond. Nat Rev Neurol. (2017) 13:363. doi: 10.1038/nrneurol.2017.64
104. Ashley DM, Faiola B, Nair S, Hale LP, Bigner DD, Gilboa E. Bone marrow–generated dendritic cells pulsed with tumor extracts or tumor RNA induce antitumor immunity against central nervous system tumors. J Exp Med. (1997) 186:1177–82.
105. Heimberger AB, Crotty LE, Archer GE, McLendon RE, Friedman A, Dranoff G, et al. Bone marrow-derived dendritic cells pulsed with tumor homogenate induce immunity against syngeneic intracerebral glioma. J Neuroimmunol. (2000) 103:16–25. doi: 10.1016/S0165-5728(99)00172-1
106. Kuwashima N, Nishimura F, Eguchi J, Sato H, Hatano M, Tsugawa T, et al. Delivery of dendritic cells engineered to secrete IFN-α into central nervous system tumors enhances the efficacy of peripheral tumor cell vaccines: dependence on apoptotic pathways. J Immunol. (2005) 175:2730–40. doi: 10.4049/jimmunol.175.4.2730
107. Wheeler CJ, Zeltzer PM, Ying H, Finger DN, Lee PK, Yong WH, et al. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. (2001) 61:842–847.
108. Mak AB, Nixon AM, Kittanakom S, Stewart JM, Chen GI, Curak J, et al. Regulation of CD133 by HDAC6 promotes β-catenin signaling to suppress cancer cell differentiation. Cell Rep. (2012) 2:951–63. doi: 10.1016/j.celrep.2012.09.016
109. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature (2004) 432:396–401. doi: 10.1038/nature03128
110. Okamoto OK, Oba-Shinjo SM, Lopes L, Nagahashi Marie SK. Expression of HOXC9 and E2F2 are up-regulated in CD133(±) cells isolated from human astrocytomas and associate with transformation of human astrocytes. Biochim Biophys Acta. (2007) 1769:437–42. doi: 10.1016/j.bbaexp.2007.05.002
111. Ding D-C, Liu H-W, Chang Y-H, Chu T-Y. Expression of CD133 in endometrial cancer cells and its implications. J Cancer (2017) 8:2142–53. doi: 10.7150/jca.18869
112. Yang Z-L, Zheng Q, Yan J, Pan Y, Wang Z-G. Upregulated CD133 expression in tumorigenesis of colon cancer cells. World J Gastroenterol. (2011) 17:932–937. doi: 10.3748/wjg.v17.i7.932
113. Alama A, Gangemi R, Ferrini S, Barisione G, Orengo AM, Truini M, et al. CD133-Positive Cells from Non-Small Cell Lung Cancer Show Distinct Sensitivity to Cisplatin and Afatinib. Arch Immunol Ther Exp (Warsz). (2015) 63:207–14. doi: 10.1007/s00005-015-0330-5
114. Pellacani D, Oldridge EE, Collins AT, Maitland NJ. Prominin-1 (CD133) expression in the prostate and prostate cancer: a marker for quiescent stem cells. Adv Exp Med Biol. (2013) 777:167–84. doi: 10.1007/978-1-4614-5894-4_11
115. Kryczek I, Liu S, Roh M, Vatan L, Szeliga W, Wei S, et al. Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int J Cancer. (2012) 130:29–39. doi: 10.1002/ijc.25967
116. Nam-Cha SH, Serrano-Vargas R, Escario E, Azaña JM, Calero-Oliver R, Martín AG, et al. CD133 expression in normal skin and in epithelial cutaneous tumors. BioMed Res. Int. (2013) 2013:385604. doi: 10.1155/2013/385604
117. Luis T, Karen P, Roberto U-I, Jeel M. CD133 in breast cancer cells and in breast cancer stem cells as another target for immunotherapy. Gaceta Mexicana de Oncología (2016) 15:22–30. doi: 10.1016/j.gamo.2016.01.003
118. Kang MK, Kang SK. Tumorigenesis of chemotherapeutic drug-resistant cancer stem-like cells in brain glioma. Stem Cells Dev. (2007) 16:837–47. doi: 10.1089/scd.2007.0006
119. Lubensky IA, Vortmeyer AO, Kim S, Lonser RR, Park DM, Ikejiri B, et al. Identification of tumor precursor cells in the brains of primates with radiation-induced de novo glioblastoma multiforme. Cell Cycle (2006) 5:452–6. doi: 10.4161/cc.5.4.2482
120. US National Library of Medicine. ClinicalTrials.gov. (2014) Available online at: https://clinicaltrials.gov/ct2/show/NCT02049489
121. Rudnick JD, Fink KL, Landolfi JC, Markert J, Piccioni DE, Glantz MJ, et al. Immunological targeting of CD133 in recurrent glioblastoma: A multi-center phase I translational and clinical study of autologous CD133 dendritic cell immunotherapy. J Clin Oncol. (2017) 35(Suppl 15):2059. doi: 10.1200/JCO.2017.35.15_suppl.2059
122. Liau LM, Prins RM, Kiertscher SM, Odesa SK, Kremen TJ, Giovannone AJ, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. (2005) 11:5515–25. doi: 10.1158/1078-0432.CCR-05-0464
123. Ardon H, Van Gool SW, Verschuere T, Maes W, Fieuws S, Sciot R, et al. Integration of autologous dendritic cell-based immunotherapy in the standard of care treatment for patients with newly diagnosed glioblastoma: results of the HGG-2006 phase I/II trial. Cancer Immunol Immunother. (2012) 61:2033–44. doi: 10.1007/s00262-012-1261-1
124. Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Translat Med. (2018) 16:142. doi: 10.1186/s12967-018-1507-6
125. Mitchell DA, Batich KA, Gunn MD, Huang M-N, Sanchez-Perez L, Nair SK, et al. Tetanus toxoid and CCL3 improve DC vaccines in mice and glioblastoma patients. Nature (2015) 519:366–9. doi: 10.1038/nature14320
126. US National Library of Medicine. ClinicalTrials.gov (2015). Available online at: https://clinicaltrials.gov/ct2/show/NCT02366728?term=NCT02366728&rank=1
127. Krystufkova E, Sekerkova A, Striz I, Brabcova I, Girmanova E, Viklicky O. Regulatory T cells in kidney transplant recipients: the effect of induction immunosuppression therapy. Nephrol Dial Transp. (2012) 27:2576–82. doi: 10.1093/ndt/gfr693
128. Kirkwood JM, Butterfield LH, Tarhini AA, Zarour H, Kalinski P, Ferrone S. Immunotherapy of cancer in 2012. CA Cancer J Clin. (2012) 62:309–35. doi: 10.3322/caac.20132
129. Huang HJS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji XD, et al. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem. (1997) 272:2927–35.
130. Guo G, Gong K, Wohlfeld B, Hatanpaa KJ, Zhao D, Habib AA. Ligand-independent EGFR signaling. Cancer Res. (2015) 75:3436–41. doi: 10.1158/0008-5472.CAN-15-0989
131. Moscatello DK, Ramirez G, Wong AJ. A naturally occurring mutant human epidermal growth factor receptor as a target for peptide vaccine immunotherapy of tumors. Cancer Res. (1997) 57:1419–24.
132. Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Herndon JE, Lally-Goss D, et al. An epidermal growth factor receptor variant III–targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Therapeut. (2009) 8:2773–9. doi: 10.1158/1535-7163.MCT-09-012
133. Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. (2010) 28:4722. doi: 10.1200/JCO.2010.28.6963
134. Sengupta S, Marrinan J, Frishman C, Sampath P. Impact of temozolomide on immune response during malignant glioma chemotherapy. Clin Develop Immunol (2012) 2012:831090. doi: 10.1155/2012/831090
135. Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. (2011) 13:324–33. doi: 10.1093/neuonc/noq157
136. Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol. (2015) 17:854–61. doi: 10.1093/neuonc/nou348
137. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. ACT IV trial investigators. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. (2017) 18:1373–85. doi: 10.1016/S1470-2045(17)30517-X
138. Yang J, Yan J, Liu B. Targeting EGFRvIII for glioblastoma multiforme. Cancer Lett. (2017) 403:224–30. doi: 10.1016/j.canlet.2017.06.024
139. Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. (2009) 27:740. doi: 10.1200/JCO.2008.16.3055
140. Reardon DA, Schuster J, Tran DD, Fink KL, Nabors LB, Li G, et al. ReACT: Overall survival from a randomized phase II study of rindopepimut (CDX-110) plus bevacizumab in relapsed glioblastoma. (2015) 33(Suppl):2009. doi: 10.1227/01.neu.0000467069.86811.3f
141. US National Library of Medicine. ClinicalTrials.gov (2013). Available online at: https://clinicaltrials.gov/ct2/show/NCT01920191?term=NCT01920191&rank=1
142. US National Library of Medicine. ClinicalTrials.gov. (2010) Available online at: https://clinicaltrials.gov/ct2/show/NCT01222221?term=NCT01222221&rank=1
143. Fecci PE, Heimberger AB, Sampson JH. Immunotherapy for primary brain tumors: no longer a matter of privilege. Clin Cancer Res. (2014) 20:5620–9. doi: 10.1158/1078-0432.CCR-14-083
144. Ampie L, Choy W, Lamano JB, Fakurnejad S, Bloch O, Parsa AT. Heat shock protein vaccines against glioblastoma: from bench to bedside. J Neur Oncol. (2015) 123:441–8. doi: 10.1007/s11060-015-1837-7
145. di Pietro A, Tosti G, Ferrucci PF, Testori A. Oncophage®(vitespen®)-Heat shock protein peptide complex 96-based vaccines in melanoma: how far we are, how far we can get? Hum Vaccin. (2009) 5:727–37. doi: 10.4161/hv.5.11.9881
146. Crane CA, Han SJ, Ahn B, Oehlke J, Kivett V, Fedoroff A, et al. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin Cancer Res. (2012) 19:205–14. doi: 10.1158/1078-0432.CCR-11-3358
147. Bloch O, Crane CA, Fuks Y, Kaur R, Aghi MK, Berger MS, et al. Heat-shock protein peptide complex−96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro Oncol. (2013) 16:274–9. doi: 10.1093/neuonc/not203
148. Bloch O, Lim M, Sughrue ME, Komotar RJ, Abrahams JM, O'Rourke DM, et al. Autologous heat shock protein peptide vaccination for newly diagnosed glioblastoma: impact of peripheral PD-L1 expression on response to therapy. Clin Cancer Res. (2017) 23:3575–84. doi: 10.1158/1078-0432.CCR-16-1369
149. US National Library of Medicine. ClinicalTrials.gov (2017). Available online at: https://clinicaltrials.gov/ct2/show/NCT03018288?term=NCT03018288&rank=1
150. US National Library of Medicine. ClinicalTrials.gov (2016). Available online at: https://clinicaltrials.gov/ct2/show/NCT02722512?term=NCT02722512&rank=1
151. Delorme EJ, Alexander P. Treatment of primary fibrosarcoma in the rat with immune lymphocytes. Lancet (1964) 284:117–20.
152. Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocyts and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J. Med. (1988) 319:1676–80.
153. Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med. (1985) 313:1485–92.
154. Rosenberg SA, Yang JC, White DE, Steinberg SM. Durability of complete responses in patients with metastatic cancer treated with high-dose interleukin-2: identification of the antigens mediating response. Ann Surg. (1998) 228:307.
155. Crystal JS, Prickett TD, Gartner JJ, Parkhurst MR, Lu YC, Gros A, et al. Diversity of mutated antigen recognition by tumor infiltrating lymphocytes from patients with metastatic melanoma. J Immunother Cancer (2015) 3:P9. doi: 10.1186/2051-1426-3-S2-P9
156. Merhavi-Shoham E, Itzhaki O, Markel G, Schachter J, Besser MJ. Adoptive cell therapy for metastatic melanoma. Cancer J. (2017) 23:48–53. doi: 10.1097/PPO.0000000000000240
157. Andersen R, Donia M, Westergaard MC, Pedersen M, Hansen M, Svane IM. Tumor infiltrating lymphocyte therapy for ovarian cancer and renal cell carcinoma. Hum Vaccin Immunother (2015) 11:2790–5. doi: 10.1080/21645515.2015.1075106
158. Forget MA, Tavera RJ, Haymaker C, Ramachandran R, Malu S, Zhang M, et al. A novel method to generate and expand clinical-grade, genetically modified, tumor-infiltrating lymphocytes. Front Immunol. (2017) 8:908. doi: 10.3389/fimmu.2017.00908
159. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. (2016) 375:2255–62.
160. Weisman PS, Ng CKY, Brogi E, Eisenberg RE, Won HH, Piscuoglio S, et al. Genetic Alterations of Triple Negative Breast Cancer By Targeted Next Generation Sequencing And Correlation With Tumor Morphology. Modern Pathol. (2016) 29:476–88. doi: 10.1038/modpathol.2016.39
161. Wein L, Savas P, Luen SJ, Virassamy B, Salgado R, Loi S. Clinical Validity and Utility of Tumor-Infiltrating Lymphocytes in Routine Clinical Practice for Breast Cancer Patients: Current and Future Directions. Front Oncol. (2017) 7:156. doi: 10.3389/fonc.2017.00156
162. Dunn IF, Black PM. The neurosurgeon as local oncologist: cellular and molecular neurosurgery in malignant glioma therapy. Neurosurgery (2003) 52:1411–24. doi: 10.1227/01.NEU.0000064808.27512.CF
163. Hughes MS, Yu YY, Dudley ME, Zheng Z, Robbins PF, Li Y, et al. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum Gene Ther. (2005) 16:457–72. doi: 10.1089/hum.2005.16.457
164. Robbins PF, Li YF, El-Gamil M, Zhao Y, Wargo JA, Zheng Z, et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol. (2008) 180:6116–31. doi: 10.4049/jimmunol.180.9.6116
165. Draper LM, Kwong ML, Gros A, Stevanovic S, Tran E, Kerkar S, et al. Targeting of HPV-16+ Epithelial Cancer Cells by TCR Gene Engineered T Cells Directed against E6. Clin Cancer Res. (2015) 21:4431–9. doi: 10.1158/1078-0432.CCR-14-3341
166. Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY-ESO-1–specific TCR–engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. (2015) 21:914. doi: 10.1038/nm.3910
167. Morgan RA, Chinnasamy N, Abate-Daga DD, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurologic toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother (2013) 36:133–51. doi: 10.1097/CJI.0b013e3182829903
168. US National Library of Medicine. ClinicalTrials.gov (2007). Available online at: https://clinicaltrials.gov/ct2/show/study/NCT00509288
169. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. (2011) 29:917–24. doi: 10.1200/JCO.2010.32.2537
170. Coccoris M, Straetemans T, Govers C, Lamers C, Sleijfer S, Debets R. T cell receptor (TCR) gene therapy to treat melanoma: lessons from clinical and preclinical studies. Expert Opin Biol Ther. (2010) 10:547–62. doi: 10.1517/14712591003614756
171. Abate-Daga D, Hanada KI, Davis JL, Yang JC, Rosenberg SA, Morgan RA. Expression profiling of TCR-engineered T cells demonstrates over-expression of multiple inhibitory receptors in persisting lymphocytes. Blood (2013) 122:1399–410. doi: 10.1182/blood-2013-04-49553
172. Everson RG, Antonios JP, Lisiero DN, Soto H, Scharnweber R, Garrett MC, et al. Efficacy of systemic adoptive transfer immunotherapy targeting NY-ESO-1 for glioblastoma. Neuro Oncol. (2015) 18:368–78. doi: 10.1093/neuonc/nov153
173. Sasaki M, Nakahira K, Kawano Y, Katakura H, Yoshimine T, Shimizu K, et al. MAGE-E1, a new member of the melanoma-associated antigen gene family and its expression in human glioma. Cancer Res. (2001) 61:4809–14.
174. Dudley ME, Rosenberg SA. Adoptive cell transfer therapy. Semin Oncol. (2007) 34:524–31. doi: 10.1053/j.seminoncol.2007.09.002
175. Hollyman D, Stefanski J, Przybylowski M, Bartido S, Borquez-Ojeda O, Taylor C, et al. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother. (2009) 32:169–80. doi: 10.1097/CJI.0b013e318194a6e8
176. Abate-Daga D, Davila ML. CAR models: next-generation CAR modifications for enhanced T-cell function. Mol Ther Oncolyt. (2016) 3:16014.doi: 10.1038/mto.2016.14
177. Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor–transduced T cells. Blood (2012) 119:2709–20. doi: 10.1182/blood-2011-10-384388
178. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood (2015) 125:4017–23. doi: 10.1182/blood-2014-12-580068
179. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-CAR T Cells Induce Remissions in CD19-CAR Naïve and Resistant B-ALL. Nat Med. (2018) 24:20–8. doi: 10.1038/nm.4441
180. Tsukahara T, Iwase N, Kawakami K, Iwasaki M, Yamamoto C, Ohmine K, et al. The Tol2 transposon system mediates the genetic engineering of T-cells with CD19-specific chimeric antigen receptors for B-cell malignancies. Gene Ther (2015) 22:209–15. doi: 10.1038/gt.2014.104
181. Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T Cells Expressing an Anti-B-Cell-Maturation-Antigen Chimeric Antigen Receptor Cause Remissions of Multiple Myeloma (2016) Available online at: http://www.bloodjournal.org/content/early/2016/07/13/blood-2016-04-711903?sso-checked=true
182. Mirzaei HR, Rodriguez A, Shepphird J, Brown CE, Badie B. Chimeric Antigen Receptors T cell therapy in solid tumor: challenges and clinical applications. Front Immunol. (2017) 8:1850. doi: 10.3389/fimmu.2017.01850
183. Watanabe K, Luo Y, Da T, Guedan S, Ruella M, Scholler J, et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight (2018) 3:e99573. doi: 10.1172/jci.insight.99573
184. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. (2010) 33:780–8. doi: 10.1097/CJI.0b013e3181ee6675
185. Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood (2009) 113:6392–402. doi: 10.1182/blood-2009-03-209650
186. Newick K, O'Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Ann Rev Med. (2017) 68:139–52. doi: 10.1146/annurev-med-062315-120245
187. Liu G, Ying H, Zeng G, Wheeler CJ, Black KL, Yu JS. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res. (2004) 64:4980–6. 10.1158/0008-5472.CAN-03-3504
188. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-Specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. (2017) 3:1094–101. doi: 10.1001/jamaoncol.2017.0184
189. Memory-EnrichedT Cells in Treating Patients With Recurrent or Refractory Grade III-IV Glioma. (n.d.). Available online at: https://clinicaltrials.gov/ct2/show/NCT03389230?term=city_of_hope_her2_car_t&rank = 1
190. Wing A, Fajardo CA, Posey Jr AD, Shaw C, Da T, Young RM, et al. Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus-Driven Production of a Bispecific T-cell Engager. Cancer Immunol Res. (2018) 6:605–16. doi: 10.1158/2326-6066.CIR-17-0314
191. Zhang H, Snyder KM, Suhoski MM, Maus MV, Kapoor V, June CH, et al. 4-1BB is superior to CD28 costimulation for generating CD8+ cytotoxic lymphocytes for adoptive immunotherapy. J Immunol. (2007) 179:4910–8. doi: 10.4049/jimmunol.179.7.4910
192. Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia (2004) 18:676–84. doi: 10.1038/sj.leu.2403302
193. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. (2015) 21:581–90. doi: 10.1038/nm.3838
194. Mintz A, Gibo DM, Slagle-Webb B, Christensen ND, Debinski W. IL-13Ralpha2 is a glioma-restricted receptor for interleukin-13. Neoplasia (2002) 4:388–99. doi: 10.1038/sj.neo.7900234
195. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and Safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. (2015) 21:4062–72. doi: 10.1158/1078-0432.CCR-15-0428
196. Brown CE, Aguilar B, Starr R, Yang X, Chang WC, Weng L, et al. Optimization of IL13Rα2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol Ther. (2018) 26:31–44. doi: 10.1016/j.ymthe.2017.10.002
197. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa161049
198. US National Library of Medicine. ClinicalTrials.gov (2011). Available online at: https://clinicaltrials.gov/ct2/show/NCT01454596?term=CAR+T+Cell+Therapy&cond=Glioblastoma&rank=2
199. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
200. Suarez ER, Chang de K, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget (2016) 7:34341–55. doi: 10.18632/oncotarget.9114
201. John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. (2013) 19:5636–46. doi: 10.1158/1078-0432.CCR-13-0458
202. Miao H, Choi BD, Suryadevara CM, Sanchez-Perez L, Yang S, De Leon G, et al. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLoS ONE (2014) 9:e94281. doi: 10.1371/journal.pone.0094281
203. Yeku OO, Purdon TJ, Koneru M, Spriggs D, Brentjens RJ. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci Reports (2017) 7:10541. doi: 10.1038/s41598-017-10940-8
204. Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci USA. (1994) 91:2076–80.
205. Kunwar S, Chang S, Westphal M, Vogelbaum M, Sampson J, Barnett G, et al. Phase III randomized trial of CED of IL13-PE38QQR vs. Gliadel wafers for recurrent glioblastoma. Neuro Oncol. (2010) 12:871–81. doi: 10.1093/neuonc/nop054
206. Zhang C, Nance EA, Mastorakos P, Chisholm J, Berry S, Eberhart C, et al. Convection enhanced delivery of cisplatin-loaded brain penetrating nanoparticles cures malignant glioma in rats. J Control Release. (2017) 263:112–9. doi: 10.1016/j.jconrel.2017.03.007
207. Nordling-David MM, Yaffe R, Guez D, Meirow H, Last D, Grad E, et al. Liposomal temozolomide drug delivery using convection enhanced delivery. J Control Rel. (2017) 261:138–46. doi: 10.1016/j.jconrel.2017.06.028
208. Young JS, Bernal G, Polster SP, Nunez L, Larsen GF, Mansour N, et al. Convection-enhanced delivery of polymeric nanoparticles encapsulating chemotherapy in canines with spontaneous supratentorial tumors. World Neurosurg. (2018) 117:e698–704. doi: 10.1016/j.wneu.2018.06.114
209. Bruce JN, Fine RL, Canoll P, Yun J, Kennedy BC, Rosenfeld SS, et al. Regression of recurrent malignant gliomas with convection-enhanced delivery of topotecan. Neurosurgery (2011) 69:1272–80. doi: 10.1227/neu.0b013e3182233e24
210. White E, Bienemann A, Megraw L, Bunnun C, Gill S. Evaluation and optimization of the administration of a selectively replicating herpes simplex viral vector to the brain by convection-enhanced delivery. Cancer Gene Ther. (2011) 18:358–69. doi: 10.1038/cgt.2011.2
211. Allen C, Opyrchal M, Aderca I, Schroeder MA, Sarkaria JN, Domingo E, et al. Oncolytic measles virus strains have significant antitumor activity against glioma stem cells. Gene Therapy (2012) 20:444–9. doi: 10.1038/gt.2012.62
212. Allen C, Vongpunsawad S, Nakamura T, James CD, Schroeder M, Cattaneo R, et al. EGFRvIII retargeted oncolytic measles virus strains have significant antitumor activity against gliomas. Mol Ther. (2005) 11:S279. doi: 10.1016/j.ymthe.2005.07.260
213. Chao C, Yang Y, Wu M, Chou M, Fang C, Lin M, et al. Gene therapy for human glioblastoma using neurotropic JC virus-like particles as a gene delivery vector. Sci Reports (2018) 8:2213. doi: 10.1038/s41598-018-19825-w
214. Lubin JA, Zhang RR, Kuo JS. Zika Virus has oncolytic activity against glioblastoma stem cells. Neurosurgery (2018) 82:E113–4. doi: 10.1093/neuros/nyy047
215. Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, Mcdonnell TJ, Mitlianga P, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene (2000) 19:2–12. doi: 10.1038/sj.onc.1203251
216. The Oncolytic Adenovirus DNX-2401 has antitumor activity in Glioblastoma. Cancer Discovery (2018) 8:382. doi: 10.1158/2159–8290.cd-rw2018-031
217. Barua N, Woolley M, Bienemann A, Johnson D, Wyatt M, Irving C, et al. Convection-enhanced delivery of AAV2 in white matter—A novel method for gene delivery to cerebral cortex. J Neurosci Methods, (2013) 220:1–8. doi: 10.1016/j.jneumeth.2013.08.011
218. Lun X, Yang W, Alain T, Shi ZQ, Muzik H, Barrett JW, et al. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res. (2005) 65:9982–9990. doi: 10.1158/0008-5472.CAN-05-1201
219. Kanai R, Wakimoto H, Martuza RL, Rabkin SD. Combining oncolytic herpes simplex virus and chemotherapy: G47Δ and temozolomide synergize in killing glioma stem cells. Mol Ther. (2010) 18:S320. doi: 10.1016/s1525-0016(16)38270-3
220. Hadjipanayis CG, Fellows-Mayle W, Deluca NA. Therapeutic efficacy of a herpes simplex virus with radiation or temozolomide for intracranial glioblastoma after convection-enhanced delivery. Mol Ther. (2008) 16:1783–8. doi: 10.1038/mt.2008.185
221. Jiang H, Clise-Dwyer K, Ruisaard KE, Fan X, Tian W, Gumin J, et al. Delta-24-RGD oncolytic adenovirus elicits anti-glioma immunity in an immunocompetent mouse model. PLoS ONE (2014) 9:e97407. doi: 10.1371/journal.pone.0097407
222. Lang FF, Conrad C, Gomez-Manzano C, Yung WA, Sawaya R, Weinberg JS, et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J Clin Oncol. (2018) 36:1419–27. doi: 10.1200/jco.2017.75.8219
223. Holl EK, Brown MC, Boczkowski D, McNamara MA, George DJ, Bigner DD, et al. Recombinant oncolytic poliovirus, PVSRIPO, has potent cytotoxic and innate inflammatory effects, mediating therapy in human breast and prostate cancer xenograft models. Oncotarget (2016) 7:79828–41. doi: 10.18632/oncotarget.12975
224. Gromeier M, Lachmann S, Rosenfeld MR, Gutin PH, Wimmer E. Intergeneric poliovirus recombinants for the treatment of malignant glioma. Proc Natl Acad Sci USA. (2000) 97:6803–8. doi: 10.1073/pnas.97.12.6803
225. Goetz C, Everson RG, Zhang LC, Gromeier M. MAPK signal-integrating kinase controls cap-independent translation and cell type-specific cytotoxicity of an oncolytic poliovirus. Mol Ther. (2010) 18:1937–46. doi: 10.1038/mt.2010.145
226. Desjardins A, Gromeier M, Herndon JE II Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. (2018) 379:150–61. doi: 10.1056/NEJMoa1716435
227. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to Anti-CTLA-4 therapy. Cell (2016) 167:397–404.e9. doi: 10.1016/j.cell.2016.08.069
228. Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D'Angelo SP, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. (2016) 17:1374–85. doi: 10.1016/S1470-2045(16)30364-3
229. Chow LQM, Haddad R, Gupta S, Mahipal A, Mehra R, Tahara M, et al. Antitumor activity of pembrolizumab in biomarker-unselected patients with recurrent and/or metastatic head and neck squamous cell carcinoma: results from the phase Ib KEYNOTE-012 Expansion Cohort. J Clin Oncol. (2016) 34:3838–45. doi: 10.1200/JCO.2016.68.1478
230. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. (2015) 373:123–35. doi: 10.1056/NEJMoa1504627
231. Sharma P, Retz M, Siefker-Radtke A, Baron A, Necchi A, Bedke J, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. (2017) 18:312–22. doi: 10.1016/S1470-2045(17)30065-7
232. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Lance Cowey C, et al. Updated results from a phase III trial of nivolumab (NIVO) combined with ipilimumab (IPI) in treatment-naive patients (pts) with advanced melanoma (MEL) (CheckMate 067). In: 2016 ASCO Annual Meeting. Chicago, IL (2016).
233. Zhu X, Lang J. Soluble PD-1 and PD-L1: predictive and prognostic significance in cancer. Oncotarget. (2017) 8:97671–82. doi: 10.18632/oncotarget.18311
234. Yamazaki N, Kiyohara Y, Uhara H, Iizuka H, Uehara J, Otsuka F, et al. Cytokine biomarkers to predict antitumor responses to nivolumab suggested in a phase 2 study for advanced melanoma. Cancer Sci. (2017) 108:1022–31. doi: 10.1111/cas.13226
235. Akyüz N, Brandt A, Stein A, Schliffke S, Mährle T, Quidde J, et al. Binder M. T-cell diversification reflects antigen selection in the blood of patients on immune checkpoint inhibition and may be exploited as liquid biopsy biomarker. Int J Cancer (2017) 140:2535–44. doi: 10.1002/ijc.30549
236. Mitsuhashi A, Okuma Y. Perspective on immune oncology with liquid biopsy, peripheral blood mononuclear cells, and microbiome with non-invasive biomarkers in cancer patients. Clin Transl Oncol. (2018) 20:966–74. doi: 10.1007/s12094-017-1827-7
237. Winter CD, Iannotti F, Pringle AK, Trikkas C, Clough GF, Church MK. A microdialysis method for the recovery of IL-1β, IL-6 and nerve growth factor from human brain in vivo. J Neurosci Methods (2002) 119:45–50. doi: 10.1016/s0165-0270(02)00153-x
238. Winter CD. Raised parenchymal interleukin-6 levels correlate with improved outcome after traumatic brain injury. Brain (2004) 127:315–20. doi: 10.1093/brain/awh039
239. Helmy A, Carpenter KL, Menon DK, Pickard JD, Hutchinson PJ. The cytokine response to human traumatic brain injury: temporal profiles and evidence for cerebral parenchymal production. J Cerebl Blood Flow Metabolism (2010) 31:658–70. doi: 10.1038/jcbfm.2010.142
240. Hutchinson PJ, Oconnell MT, Rothwell NJ, Hopkins SJ, Nortje J, Carpenter KL, et al. Inflammation in human brain injury: intracerebral concentrations of IL-1α, IL-1β, and their endogenous inhibitor IL-1ra. J Neurotrauma (2007) 24:1545–57. doi: 10.1089/neu.2007.0295
241. Mellergård P, Åneman O, Sjögren F, Pettersson P, Hillman J. Changes in extracellular concentrations of some cytokines, chemokines, and neurotrophic factors after insertion of intracerebral microdialysis catheters in neurosurgical patients. Neurosurgery (2008) 62:151–8. doi: 10.1227/01.neu.0000311072.33615.3a
242. Zeiler FA, Thelin EP, Czosnyka M, Hutchinson PJ, Menon DK, Helmy A. Cerebrospinal fluid and microdialysis cytokines in severe traumatic brain injury: a scoping systematic review. Front Neurol. (2017) 8:331. doi: 10.3389/fneur.2017.00331
243. Zeiler FA, Thelin EP, Czosnyka M, Hutchinson PJ, Menon DK, Helmy A. Cerebrospinal fluid and microdialysis cytokines in aneurysmal subarachnoid hemorrhage: a scoping systematic review. Front Neurol. (2017) 8:379. doi: 10.3389/fneur.2017.00379
244. Helbok R, Schiefecker AJ, Beer R, Dietmann A, Antunes AP, Sohm F, et al. Early brain injury after aneurysmal subarachnoid hemorrhage: a multimodal neuromonitoring study. Critical Care (2015) 19:2–9. doi: 10.1186/s13054–015-0809-9
245. Helbok R, Kofler M, Schiefecker AJ, Gaasch M, Rass V, Pfausler B, et al. Clinical use of cerebral microdialysis in patients with aneurysmal subarachnoid hemorrhage—state of the art. Front Neurol. (2017) 8:565. doi: 10.3389/fneur.2017.00565
246. Sarrafzadeh A, Schlenk F, Gericke C, Vajkoczy P. Relevance of cerebral interleukin-6 after aneurysmal subarachnoid hemorrhage. Neurocritical Care (2010) 13:339–46. doi: 10.1007/s12028-010-9432-4
247. Hanafy KA, Stuart RM, Khandji AG, Connolly ES, Badjatia N, Mayer SA, et al. Relationship between brain interstitial fluid tumor necrosis factor-α and cerebral vasospasm after aneurysmal subarachnoid hemorrhage. J Clin Neurosci. (2010) 17:853–856. doi: 10.1016/j.jocn.2009.11.041
248. Hillman J, Åneman O, Persson M, Andersson C, Dabrosin C, Mellergård P. Variations in the response of interleukins in neurosurgical intensive care patients monitored using intracerebral microdialysis. J Neurosurg. (2007) 106:820–5. doi: 10.3171/jns.2007.106.5.820
249. Portnow J, Badie B, Liu X, Frankel P, Mi S, Chen M, et al. A pilot microdialysis study in brain tumor patients to assess changes in intracerebral cytokine levels after craniotomy and in response to treatment with a targeted anti-cancer agent. J Neuro Oncol. (2014) 118:169–77. doi: 10.1007/s11060-014-1415-4
250. Tabatabaei P, Visse E, Bergström P, Brännström T, Siesjö P, Bergenheim AT. Radiotherapy induces an immediate inflammatory reaction in malignant glioma: A clinical microdialysis study. J Neuro Oncol. (2016) 131:83–92. doi: 10.1007/s11060-016-2271-1
251. US National Library of Medicine. ClinicalTrials.gov (2018). Available online at: https://clinicaltrials.gov/ct2/show/NCT03493932
252. Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. (2004) 64:6892–9. doi: 10.1158/0008-5472.CAN-04-1337
253. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science (2008) 321:1807–12. doi: 10.1126/science.1164382
254. Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. (2009) 27:4150–4. doi: 10.1200/JCO.2009.21.9832
255. Li S, Chou AP, Chen W, Chen R, Deng Y, Phillips HS, et al. Overexpression of isocitrate dehydrogenase mutant proteins renders glioma cells more sensitive to radiation. Neuro Oncol. (2013) 15:57–68. doi: 10.1093/neuonc/nos261
256. Li H, Li J, Cheng G, Zhang J, Li X. IDH mutation and MGMT promoter methylation are associated with the pseudoprogression and improved prognosis of glioblastoma multiforme patients who have undergone concurrent and adjuvant temozolomide-based chemoradiotherapy. Clin Neurol Neurosurg. (2016) 151:31–6. doi: 10.1016/j.clineuro.2016.10.004
257. Hegi ME, Diserens A, Gorlia T, Hamou M, Tribolet ND, Weller M, et al. MGMTGene Silencing and Benefit from Temozolomide in Glioblastoma. New Engl J Med. (2005) 352:997–1003. doi: 10.1056/nejmoa043331
258. Tabone T, Abuhusain HJ, Nowak AK, Erber WN, Mcdonald KL. Multigene profiling to identify alternative treatment options for glioblastoma: a pilot study. J Clin Pathol. (2014) 67:550–5. doi: 10.1136/jclinpath-2014-202173
259. Huang Y, Hsu T, Kelsey KT, Lin C. Integrative analysis of micro-RNA, gene expression, and survival of glioblastoma multiforme. Genetic Epidemiol. (2014) 39:134–43. doi: 10.1002/gepi.21875
260. Song YC, Lu GX, Zhang HW, Zhong XM, Cong XL, Xue SB, et al. Proteogenomic characterization and integrative analysis of glioblastoma multiforme. Oncotarget (2017) 8:97304–12. doi: 10.18632/oncotarget.21937
261. Wrzeszczynski KO, Frank MO, Koyama T, Rhrissorrakrai K, Robine N, Utro F, et al. Comparing sequencing assays and human-machine analyses in actionable genomics for glioblastoma. Neurol Genet. (2017) 3:e164. doi: 10.1212/nxg.0000000000000164
262. Byron SA, Tran NL, Halperin RF, Phillips JJ, Kuhn JG, Groot JF, et al. Prospective Feasibility Trial for Genomics-Informed Treatment in Recurrent and Progressive Glioblastoma. Clin Cancer Res (2017) 24:295–305. doi: 10.1158/1078-0432.ccr-17-0963
263. Wu AH, Hall WA, Low WC. Identification of HLA a*0201 glioblastoma multiforme cell lines for immunotherapy by PCR-SSP and DNA sequencing. J Neurooncol. (2004) 66:1–8. doi: 10.1023/B:NEON.0000013460.53527.28
264. Song W, Ruder AM, Hu L, Li Y, Ni R, Shao W, et al. Genetic epidemiology of glioblastoma multiforme: confirmatory and new findings from analyses of human leukocyte antigen alleles and motifs. PLoS ONE (2009) 4:e7157. doi: 10.1371/journal.pone.0007157
265. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500:415–21. doi: 10.1038/nature12477
266. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. (2013) 19:747–52. doi: 10.1038/nm.3161
267. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature (2014) 515:577–81. doi: 10.1038/nature13988
268. van Rooij N, van Buuren MM, Philips D, Toebes M, Heemskerk B, van Dijk LJ, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. (2013) 31:e439–42. doi: 10.1200/JCO.2012.47.7521
269. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348:124–8. doi: 10.1126/science.aaa1348
270. Dijkstra KK, Voabil P, Schumacher TN, Voest EE. Genomics- and Transcriptomics-Based Patient Selection for Cancer Treatment With Immune Checkpoint Inhibitors: A Review. JAMA Oncol. (2016) 2:1490–5. doi: 10.1001/jamaoncol.2016.2214
271. Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. (2016) 34:2206–11. doi: 10.1200/JCO.2016.66.6552
272. Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S, et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol. (2017) 19:1047–57. doi: 10.1093/neuonc/nox026
273. Budczies J, Bockmayr M, Klauschen F, Endris V, Fröhling S, Schirmacher P, et al. Mutation patterns in genes encoding interferon signaling and antigen presentation: a pan-cancer survey with implications for the use of immune checkpoint inhibitors. Genes Chromosomes Cancer (2017) 56:651–9. doi: 10.1002/gcc.22468
274. Mahlokozera T, Vellimana AK, Li T, Mao DD, Zohny ZS, Kim DH, et al. Biological and therapeutic implications of multisector sequencing in newly diagnosed glioblastoma. Neuro Oncol. (2017) 20:472–83. doi: 10.1093/neuonc/nox232
275. Feng L, Qian H, Yu X, Liu K, Xiao T, Zhang C, et al. Heterogeneity of tumor-infiltrating lymphocytes ascribed to local immune status rather than neoantigens by multi-omics analysis of glioblastoma multiforme. Sci Rep. (2017) 7:6968. doi: 10.1038/s41598-017-05538-z
276. Goodman AM, Piccioni D, Kato S, Boichard A, Wang HY, Frampton G, et al. Prevalence of PDL1 amplification and preliminary response to immune checkpoint blockade in solid tumors. JAMA Oncol. (2018) 4:1237–44. doi: 10.1001/jamaoncol.2018.1701
277. Peng S, Dhruv H, Armstrong B, Salhia B, Legendre C, Kiefer J, et al. Integrated genomic analysis of survival outliers in glioblastoma. Neuro Oncol. (2017) 19:833–44. doi: 10.1093/neuonc/now269
278. Chen YP, Zhang Y, Lv JW, Li YQ, Wang YQ, He QM, et al. Genomic analysis of tumor microenvironment immune types across 14 solid cancer types: immunotherapeutic implications. Theranostics (2017) 7:3585–94. doi: 10.7150/thno.21471
279. Nomizo T, Ozasa H, Tsuji T, Funazo T, Yasuda Y, Yoshida H, et al. Clinical impact of single nucleotide polymorphism in PD-L1 on response to nivolumab for advanced non-small-cell lung cancer patients. Sci Rep. (2017) 7:45124. doi: 10.1038/srep45124
280. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science (2014) 344:641–5. doi: 10.1126/science.1251102
281. Li L, Goedegebuure SP, Gillanders WE. Preclinical and clinical development of neoantigen vaccines. Ann Oncol. (2017) 28(Suppl. 12):xii11–7. doi: 10.1093/annonc/mdx681
282. Johanns TM, Ward JP, Miller CA, Wilson C, Kobayashi DK, Bender D, et al. Endogenous neoantigen-specific CD8 T cells identified in two glioblastoma models using a cancer immunogenomics approach. Cancer Immunol Res. (2016) 4:1007–15. doi: 10.1158/2326-6066.CIR-16-0156
283. Nisonoff A, Rivers MM. Recombination of a mixture of univalent antibody fragments of different specificity. Arch Biochem Biophys. (1961) 93:460–2.
284. Nitta T, Sato K, Yagita H, Okumura K, Ishii S. Preliminary trial of specific targeting therapy against malignant glioma. Lancet (1990) 335:368–71.
285. Staerz UD, Kanagawa O, Bevan MJ. Hybrid antibodies can target sites for attack by T cells. Nature (1985) 314:628–31.
286. Mack M, Riethmüller G, Kufer P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc Natl Acad Sci USA. (1995) 92:7021–5.
287. Topp MS, Kufer P, Gk¨buget N, Goebeler M, Klinger M, Neumann S, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-re- fractory minimal residual disease in B-lineage ALL patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol. (2011) 29:2493–8. doi: 10.1200/JCO.2010.32.7270
288. Martinelli G, Boissel N, Chevallier P, Ottmann O, Gkbuget N, Topp MS, et al. Complete haematologic and molecular response in adult patients with relapsed/refractory Philadelphia chromosome-Positive B-precursor ALL following treatment with blinatumomab: results from a Phase II, Single-Arm, Multicenter Study. J Clin Oncol. (2017) 35:1795–802. doi: 10.1200/JCO.2016.69.3531
289. Topp MS, G¨kbuget N, Zugmaier G, Klappers P, Stelljes M, Neumann S, et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows haemato- logic and molecular remissions in patients with relapsed or refractory B-precursor ALL. J Clin Oncol. (2014) 32:4134–40. doi: 10.1200/JCO.2014.56.3247
290. Kantarjian H, Stein A, Gökbuget N, Fielding AK, Schuh AC, Ribera JM, et al. Blinatumomab versus chemotherapy for advanced ALL. N Engl J Med. (2017) 376:836–47. doi: 10.1056/NEJMoa1609783
291. Sanders S, Stewart DA. Targeting non-Hodgkin lymphoma with blinatumomab. Expert Opin Biol Ther. (2017) 17:1013–17. doi: 10.1080/14712598.2017.1334053
292. Hipp S, Tai YT, Blanset D, Deegen P, Wahl J, Thomas O, et al. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia (2017) 31:2278. doi: 10.1038/leu.2017.219
293. Bi Y, Jiang H, Wang P, Song B, Wang H, Kong X, et al. Treatment of hepatocellular carcinoma with a GPC3-targeted bispecific T cell engager. Oncotarget (2017) 8:52866–76. doi: 10.18632/oncotarget.17905
294. Scott EM, Duffy MR, Freedman JD, Fisher KD, Seymour LW. Solid tumor immunotherapy with T cell engager-armed oncolytic viruses. Macromol Biosci. (2018) 18:3–6. doi: 10.1002/mabi.201700187
295. Ishiguro T, Sano Y, Komatsu SI, Kamata-Sakurai M, Kaneko A, Kinoshita Y, et al. An anti-glypican 3/CD3 bispecific T cell-redirecting antibody for treatment of solid tumors. Sci Transl Med. (2017) 9:eaal4291. doi: 10.1126/scitranslmed.aal4291
296. Choi BD, Gedeon PC, Sanchez-Perez L, Bigner DD, Sampson JH. Regulatory T cells are redirected to kill glioblastoma by an EGFRvIII-targeted bispecific antibody. Oncoimmunology (2013) 2:e26757. doi: 10.4161/onci.26757
Keywords: glioblastoma, immunotherapy, virus, vaccination, checkpoint, sequencing, cell therapy
Citation: Lynes J, Sanchez V, Dominah G, Nwankwo A and Nduom E (2018) Current Options and Future Directions in Immune Therapy for Glioblastoma. Front. Oncol. 8:578. doi: 10.3389/fonc.2018.00578
Received: 28 August 2018; Accepted: 19 November 2018;
Published: 05 December 2018.
Edited by:
Marcos Vinicius Calfat Maldaun, Hospital Sírio-Libanês, BrazilReviewed by:
Lincoln A. Edwards, Cornell University, United StatesJustin Lathia, Cleveland Clinic Lerner College of Medicine, United States
Copyright © 2018 Lynes, Sanchez, Dominah, Nwankwo and Nduom. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edjah Nduom, ZWRqYWgubmR1b21AbmloLmdvdg==