 Clare M. Adams
Clare M. Adams Sean Clark-Garvey
Sean Clark-Garvey Pierluigi Porcu
Pierluigi Porcu Christine M. Eischen
Christine M. Eischen- 1Department of Cancer Biology, Sidney Kimmel Cancer Center, Thomas Jefferson University, Philadelphia, PA, United States
- 2Internal Medicine Residency Program, Department of Internal Medicine, Sidney Kimmel Medical College, Thomas Jefferson University, Philadelphia, PA, United States
- 3Division of Hematologic Malignancies and Hematopoietic Stem Cell Transplantation, Department of Medical Oncology, Sidney Kimmel Cancer Center, Thomas Jefferson University, Philadelphia, PA, United States
Although lymphoma is a very heterogeneous group of biologically complex malignancies, tumor cells across all B cell lymphoma subtypes share a set of underlying traits that promote the development and sustain malignant B cells. One of these traits, the ability to evade apoptosis, is essential for lymphoma development. Alterations in the Bcl-2 family of proteins, the key regulators of apoptosis, is a hallmark of B cell lymphoma. Significant efforts have been made over the last 30 years to advance knowledge of the biology, molecular mechanisms, and therapeutic potential of targeting Bcl-2 family members. In this review, we will highlight the complexities of the Bcl-2 family, including our recent discovery of overexpression of the anti-apoptotic Bcl-2 family member Bcl-w in lymphomas, and describe recent advances in the field that include the development of inhibitors of anti-apoptotic Bcl-2 family members for the treatment of B cell lymphomas and their performance in clinical trials.
Introduction
Acquiring resistance to apoptosis, a highly-regulated, evolutionarily conserved process, is a characteristic shared among all types of cancer, including B cell lymphomas (1). For a cell to divide and grow uncontrollably, as malignant cells do, it must not only hijack the normal cellular growth pathways, but it must also avoid cellular death signals. During lymphomagenesis, B cells encounter a broad range of stress stimuli, including oncogene activation, DNA damage, and oxygen and cytokine deprivation, all of which can elicit an apoptotic cell death response. Apoptosis is regulated by complex interactions between pro-apoptotic and anti-apoptotic members of the B cell lymphoma-2 (Bcl-2) protein family (2). Thus, a delicate balance between members of the Bcl-2 family dictate whether the B cell will live or die under stress conditions. As such, alterations that deregulate the apoptotic process lead to increased survival and facilitate lymphomagenesis (3). Moreover, these alterations can render lymphoma cells refractory to therapies that are designed to induce death (4). Specifically, overexpression of the anti-apoptotic Bcl-2 family members and/or reduced expression of specific pro-apoptotic members are a common feature shared among B cell lymphomas (4, 5). Much of what we know today about the Bcl-2 family and its role in B cells and B cell lymphoma comes from decades of research utilizing genetically engineered mice and a mouse model of Myc oncogene-induced B cell lymphoma [Eμ-myc transgenic mice, (6)]. However, recent discoveries and low complete response rates in clinical trials with targeted therapy against BCL-2 in lymphoma reveal significant gaps in knowledge remain (7–9). This review comprehensively examines each member of the Bcl-2 protein family, defining their contribution to B cell lymphomagenesis through mouse models and the alterations that occur in them in human B cell lymphomas, including our recent discovery of Bcl-w overexpression. In addition, this review also describes current therapeutic efforts to target specific anti-apoptotic Bcl-2 family members in lymphoma patients alone or in combinations to improve survival.
Bcl-2 Protein Family and apoptosis
B cells continuously monitor their environment and make decisions as to whether they should live or die. The Bcl-2 protein family are the central gatekeepers of the intrinsic or mitochondrial apoptotic response. The family is comprised of structurally-related proteins with opposing functions that either promote or inhibit apoptosis by interacting with one another (10). The Bcl-2 family is typically classified into three groups, including pro-apoptotic initiators, pro-apoptotic effectors, and anti-apoptotic proteins (Figure 1A). The apoptotic-promoting effects from the pro-apoptotic initiators and effectors are countered by their direct interaction with the anti-apoptotic family members. It is this delicate and dynamic balance between the pro- and anti-apoptotic Bcl-2 family members that governs whether a B cell undergoes apoptosis or survives. We discuss the consequences of alterations for each of the Bcl-2 family members in lymphoma in mouse models and make comparisons to what is observed in human lymphomas (see Table 1).
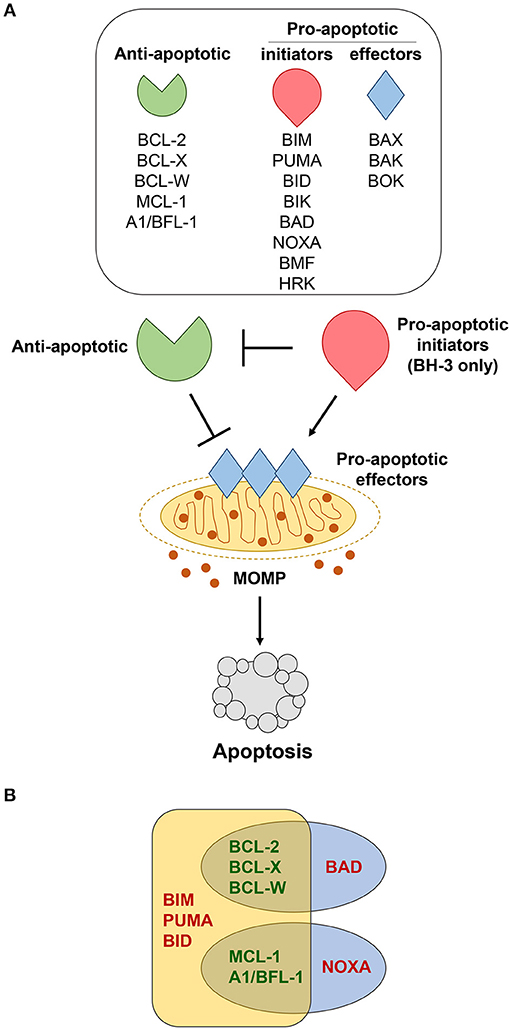
Figure 1. Bcl-2 family members regulate apoptosis. (A) Various cellular stressors induce apoptosis through the intrinsic, mitochondrial pathway, which is regulated by the Bcl-2 family of proteins. These stress signals activate pro-apoptotic BH-3 only initiators (red), which inhibit the anti-apoptotic proteins (green). This, in turn, allows the pro-apoptotic effectors (blue) to be activated. Activation of the effector proteins results in their oligomerization and subsequent mitochondrial outer membrane permeabilization (MOMP), enabling the release of apoptotic factors that initiate the caspase cascade and final stages of cellular destruction. (B) Pro-apoptotic BH-3 only proteins bind to anti-apoptotic Bcl-2 family members with different affinities. BIM, PUMA, and BID bind strongly to all anti-apoptotic Bcl-2 proteins, whereas BAD binds preferentially to BCL-2, BCL-X, and BCL-W, and NOXA binds preferentially to MCL-1 and A1/BFL-1.

Table 1. Alterations in Bcl-2 family members in mouse models and human lymphoma.
Pro-apoptotic Bcl-2 Family Members
Members of the Bcl-2 protein family share sequence homology within conserved regions known as Bcl-2 homology (BH) domains, which dictate structure and function (67, 68). All anti-apoptotic family members and a subset of pro-apoptotic members are multi-domain proteins, sharing sequence homology within three to four BH domains. A subset of pro-apoptotic Bcl-2 family members known as BH-3 only proteins only contain the BH-3 domain, which is known as the minimal death domain that is required for binding the multi-domain Bcl-2 family members (69).
BH-3 Only Proteins: Initiators of Apoptosis
The BH-3 only group of pro-apoptotic Bcl-2 proteins consists of Bim (BCL2L11), Puma/BBC3, Bad (Bcl-2/Bcl-x-associated death promoter), Bid (BH-3 interacting-domain death agonist), Bik (Bcl-2-interacting killer), Noxa/PMAIP1, Bmf (Bcl-2-modifying factor), and Hrk (Harakiri) [Figure 1A, (70)] and are essential for initiating the apoptotic cascade. While the BH-3 only proteins can initiate apoptotic signaling by binding directly to the anti-apoptotic Bcl-2 proteins, thereby freeing up Bax and Bak to undergo homo-dimerization, some have been reported to bind directly to and activate Bax and Bak (71). Years of studies using mouse models have revealed certain BH-3 only proteins are preferentially solicited in response to different apoptotic stimuli (72–76).
BH-3 only proteins serve as the first responders to cellular insults, including from dysregulation of oncogenes, such as Myc, and serve as a blockade against the development of B cell lymphoma. For example, loss of Bim or Puma accelerated Myc-driven B cell lymphoma development in a mouse model engineered to overexpress Myc in B cells (Eμ-myc transgenic) (11, 15, 16). Loss of BIM may also contribute to human lymphomas, as ~20% of mantle cell lymphomas (MCL) have deleted both alleles of BIM (12). In addition, single nucleotide polymorphisms in the BIM gene have been associated with risk of developing follicular lymphoma (FL), diffuse large B cell lymphoma (DLBCL), and chronic lymphocytic leukemia (CLL) (13). Furthermore, ~40% of human Burkitt lymphomas express very low levels of BIM or PUMA mRNA, which may be the result of epigenetic silencing (14, 15). In contrast to Bim and Puma, loss of Noxa had no effect on Myc-induced B cell lymphomagenesis in mice, but did increase the number of B lineage cells (16). It is unknown whether NOXA loss contributes to human B cell lymphoma. A quarter of mice with deletion of Bad develop DLBCL at old age, suggesting that it may have a tumor suppressive function in mature B cells (18). Deletion of Bad also accelerated Myc-induced B cell lymphoma (17). Despite the findings in mice, BAD loss has not been linked to DLBCL in humans. Deletion of Bid did not result in B cell lymphoma development in mice. Instead, chronic myelomonocytic leukemia emerged in Bid-null mice after a long latency period, indicating Bid function is critical for the myeloid lineage (19). It is unknown if alterations in BID contribute to human lymphoma. In mice, loss of Bik alone had no effect on hematopoietic cells and did not accelerate Myc-induced B cell lymphoma development, suggesting that it has no role in B cells (20, 21). However, somatic missense mutations have been observed in BIK in B cell lymphomas in humans, including FL, marginal zone (MZL), and DLBCL (22), suggesting that its loss may contribute to these lymphomas. Loss of Bmf in mice increased B cell numbers and cooperated with Myc overexpression to accelerate lymphomagenesis; preferentially developing an IgM+ B cell lymphoma (17). Reduced levels of BMF were observed in Burkitt lymphoma patient samples and cell lines (17). Together, the data indicate that each pro-apoptotic BH-3 only family member may facilitate tumor suppression in specific hematopoietic cells, but not all may have a role in B cells.
Bax, Bak, and Bok: Effectors of Apoptosis
Activation of Bax and Bak, and possibly the lesser-studied Bok, involves homo-dimerization followed by oligomerization within the outer mitochondrial membrane (71). This conformational change induces mitochondrial outer membrane permeabilization (MOMP) by creating a pore and triggering the release of apoptosis-inducing proteins, such as cytochrome c and second mitochondria-derived activator of caspase (Smac)/direct IAP-binding protein with low pI (DIABLO) from the mitochondria [Figure 1A, (77)]. Following this release, the caspase cascade is activated, ultimately resulting in the proteolysis of intra-cellular proteins and cellular destruction.
The combined function of Bax and Bak is critical for development and mediating apoptosis. Mice lacking both Bak and Bax have severe developmental defects and have hematopoietic cells that are resistant to diverse stimuli that activate the intrinsic apoptotic pathway (23, 78). Mice lacking either Bak or Bax are phenotypically normal (23) or have mild lymphoid hyperplasia, respectively (24). Using the Eμ-myc mouse model of Myc-driven B cell lymphomagenesis, loss of Bax accelerated lymphoma development (25), but the effects of inactivation of Bak in Myc-induced B cell lymphoma have not been reported. Loss of Bok did not alter Myc-induced B cell lymphoma development (26). In humans, mutations in BAX that caused a frameshift were detected in cell lines derived from hematologic malignancies that did not include lymphoma and were associated with resistance to cell death and microsatellite instability (79–81). Alterations of BAX, BAK, or BOK have not been reported in human lymphoma; thus, loss/inactivation of BAX, BAK, or BOK either does not occur or is a rare event in human B cell lymphoma.
Anti-apoptotic Bcl-2 Family Members
Acquired resistance to apoptosis is regarded as one of the hallmarks of cancer (1). Accordingly, evidence continues to reveal that elevated expression of anti-apoptotic Bcl-2 family members (Bcl-2, Bcl-x, Bcl-w, Mcl-1, A1/Bfl-1) is one of the major contributing factors to B cell lymphomagenesis (2). Distinct biological roles for the individual anti-apoptotic Bcl-2 family members have been unveiled by genetically-engineered mouse models (3). Additionally, it has been hypothesized that levels of expression of individual anti-apoptotic Bcl-2 family proteins may be an indication of how dependent a cell is on the protein to maintain its survival (82). The mechanism by which the anti-apoptotic Bcl-2 family proteins inhibit apoptosis is predominately governed by their capacity to bind and sequester the pro-apoptotic BH-3-only proteins or Bax and Bak, ultimately preventing mitochondrial membrane permeabilization [Figure 1A, (83)]. The ability of the anti-apoptotic Bcl-2 proteins to bind pro-apoptotic Bcl-2 proteins does vary and depends, at least in part, on the apoptotic stimuli and which BH-3 only proteins are expressed and/or activated [Figure 1B, (10, 84, 85)]. We will discuss each of the anti-apoptotic Bcl-2 family members, focusing particular attention on Bcl-w to highlight its newly-exposed contributions to B cell survival and lymphomagenesis.
BCL-2
BCL-2 is translocated t(14;18)(q32;q21) to the immunoglobulin heavy chains, resulting in its constitutive expression in 90% of FL (29, 86–90). Bcl-2 knockout mice showed that Bcl-2 is required for normal B cells to survive (27), providing evidence for why B cell lymphomas would select for its overexpression. Somatic mutations in BCL-2 in FL are often associated with transformation of this indolent disease to more aggressive diffuse large B cell lymphoma (DLBCL) and decreased patient survival (31). Some of these mutations increased the affinity of BCL-2 to pro-apoptotic BH-3 only proteins and have also been detected in lymphoid cell lines (91, 92).
Approximately 20% of de novo DLBCL also harbor BCL-2 translocations (30). Increased BCL-2 expression has been linked to reduced survival of patients with DLBCL (93, 94). The first large-scale gene expression profiling studies classified DLBCL into two major molecular subgroups, germinal center B cell (GCB) and activated B cell (ABC) subtypes (95). GCB DLBCL that have increased levels of BCL-2 is most often due to a t(14;18) translocation (96), whereas in ABC DLBCL, amplification of the BCL-2 gene is more often observed (93, 97). Using gene expression profiling, Iqbal and colleagues reported a significant correlation between elevated BCL-2 mRNA expression and poor overall survival within the ABC subtype (93). We also observed elevated levels of BCL-2 in both subtypes of DLBCL, but BCL-2 was more highly expressed in the ABC subtype than the GCB subtype (9). More recently, new classifications of DLBCL subtypes have been reported by two groups (98, 99). Shipp and colleagues described a new classification of DLBCL subtypes based on genetic signatures of low-frequency alterations, recurrent mutations, somatic copy number alterations, and structural variants. One of their two distinct subtypes of GCB-DLBCL (cluster 3) has structural variants of BCL-2 and correlate to poor risk (98). Their ABC-DLBCL subtype (cluster 5) had amplification of BCL-2 (98), as previously described (93). Staudt and colleagues also reported new classifications of DLBCL that include four new genetic subtypes, one of which is EZB, which has EZH2 mutations and BCL-2 translocations (99).
DLBCL lymphomas that contain both rearrangements in BCL-2 and translocation of MYC are classified as “double hit lymphomas” (DHL) and represent ~10% of DLBCL cases (100, 101). DLBCLs that co-express high levels of MYC and BCL-2 proteins due to mechanisms other than chromosomal translocations are referred to as “dual expresser lymphomas” (DEL) and represent ~30% of DLBCL (102, 103). Both DHL and DEL tend to be clinically more aggressive and have a higher frequency of treatment failure than those that are non-DHL or non-DEL (102, 103). Because of this, these subtypes of DLBCL have become a new biomarker-defined subset, which illustrates the importance of knowing the status of MYC and BCL-2 to help guide treatment and monitoring of patients. These results also provide impetus to investigate the expression of other anti-apoptotic BCL-2 family members in lymphomas, as others may also alter prognosis.
Besides BCL-2 translocation and amplification, additional mechanisms are reported to contribute to its increased expression, including BCL-2 gene rearrangement (104), promoter hypomethylation (105), promoter hypermutation (106), and phosphorylation (107). A small subset of mantle cell lymphoma (MCL) have increased expression of BCL-2 (33). Additionally, BCL-2 can also become overexpressed in the indolent MZL (32). For patient samples of both MCL and MZL, BCL-2 overexpression is reportedly caused by non-genomic changes to BCL-2 (33, 108). BCL-2 expression can also be regulated by non-coding RNA. In B cells, BCL-2 expression is negatively regulated by the miR-15a/miR-16-1 cluster (109, 110). This region is deleted or inactivated by mutations in ~70% of CLL (110); however, no other B cell malignancy has been associated with loss of the region (111, 112).
Although the belief in the importance of BCL-2 in human B cell lymphomas is firmly embedded, two different transgenic mice generated ~30 years ago revealed that Bcl-2 is not a driver of B cell lymphoma, but increased levels in B cells did lead to their accumulation (113, 114). Bcl-2 overexpression did cooperate with Myc overexpression to accelerate B cell lymphomagenesis (28). The requirements of BCL-2 in the continued survival of human B cell lymphomas is just now being explored with some surprising results, as described below.
BCL-X
Shortly following the cloning of the BCL-2 gene, the gene coding for BCL-X (BCL2L1 for Bcl-2 like 1) was discovered due to its high level of sequence similarity to BCL-2 (115, 116). Similar to Bcl-2 transgenic mice, mice engineered to overexpress Bcl-x renders lymphoid cells resistant to numerous apoptotic stimuli and causes an abnormal accumulation of mature lymphocytes, but not overt lymphoma development (37). However, mice double-transgenic for Bcl-x and Myc in B cells developed lymphoproliferative disease and plasma cell malignancies (38). Knocking out Bcl-x revealed that it is not required for lymphocyte development, but is critical for erythropoiesis and platelets (34, 35, 117, 118). Loss of Bcl-x did delay Myc-induced B cell lymphoma development, suggesting that under conditions of oncogenic stress, B cells may rely on Bcl-x for survival (36).
Burkitt lymphomas can select to overexpress BCL-X (9). Elevated BCL-X expression has been detected in other B cell non-Hodgkin lymphomas, including FL and DLBCL, as well as T cell non-Hodgkin lymphomas (39). Moreover, it has been demonstrated that in DLBCL, elevated expression of BCL-X mRNA is associated with a chemoresistant, short-lived group of patients (119). Like BCL-2, selection for BCL-X overexpression occurs in a subset of MCL (40). Moderate levels of BCL-X protein were reported in several cases of CLL, FL, and MCL, but it was lowly expressed in MZL (33). In a large-scale analysis of gene expression profiling data, we reported that BCL-X mRNA was significantly elevated compared to normal human B cells in multiple types of non-Hodgkin B cell lymphoma, including Burkitt, DLBCL, FL, MCL, and MZL (9). Unlike BCL-2, which is lowly expressed in multiple myeloma (MM), levels of BCL-X are much higher and may therefore be a critical survival factor for MM (41–44). To date, no chromosomal translocation involving BCL-X has been reported in human tumor samples, but somatic copy number amplifications have been detected in hematopoietic malignancies, including non-Hodgkin lymphomas (120). High levels of BCL-X have also been attributed to the loss or silencing of the let-7 family of miRNA that targets BCL-X (109, 121, 122). Therefore, BCL-X likely contributes to B cell lymphomas.
MCL-1
Upregulation of the anti-apoptotic Bcl-2 family member Mcl-1 likely also contributes to lymphomagenesis. Mcl-1 was first identified in an immortalized myeloid leukemia-derived cell line, and consequently named myeloid cell leukemia 1 (123). Consistent with Bcl-2 and Bcl-x overexpression models, Mcl-1 overexpression in transgenic mice renders hematopoietic cells largely resistant to varying apoptotic stimuli and causes the accumulation of mature B and T lymphocytes (50, 51). In addition, half of the Mcl-1 transgenic mice develop B cell lymphomas within two years (52). Mcl-1 knockout mice are embryonic lethal, but conditional knockout of Mcl-1 in mice shows a requirement for Mcl-1 in hematopoietic stem cell and neutrophil survival (45–47). Furthermore, loss of Mcl-1 inhibited Myc-induced B cell lymphomagenesis in mice (48, 49).
A somatically acquired increase in MCL-1 copy number has been documented in a variety of non-hematopoietic malignancies, but only in a limited number of non-Hodgkin lymphomas (120). Gene amplification or chromosomal gains of MCL-1 occur in 20–25% of the activated B cell (ABC) subtype of DLBCL (53). Although individual lymphomas may select for overexpression of MCL-1, we determined MCL-1 expression was not elevated in a cohort of Burkitt lymphomas compared to normal human B cells, and that it was also not significantly different in patient samples of DLBCL, FL, MZL, and MCL compared to normal B cells (9). However, increased levels of MCL-1 mRNA are suggested to be essential for sustained growth, survival, and resistance to chemotherapeutics in multiple types of lymphoma as well as CLL (54, 55). Unlike BCL-2 and BCL-X, which are overexpressed in a subset of MCL, MCL-1 expression is typically low in this lymphoma (33). Increased levels of MCL-1 have been observed in multiple myeloma (56) and shown to correlate with disease progression (57).
The expression of Mcl-1 is tightly regulated at both the transcriptional and post-transcriptional level. In contrast to Bcl-2 and Bcl-x, Mcl-1 is a short-lived protein with a half-life of ~30 min compared to the >6 h half-lives observed for Bcl-2 and Bcl-x (124–126). Mcl-1 levels are also regulated by miRNA. Loss of miR-29 and decreased levels of miR-125b and miR-133b, miRNA that bind and negatively regulate MCL-1 expression (127–130), have been reported in many lymphomas, including Burkitt, anaplastic large cell, and DLBCL, which may also contribute to increased MCL-1 expression (131–133). Elevated expression of MCL-1 can also be the result of aberrant post-translational mechanisms. For instance, increased expression of the deubiquitinase USP9X, which is responsible for removing polyubiquitin chains that target MCL-1 protein for degradation, correlates with increased MCL-1 protein in FL, DLBCL, and multiple myeloma (134). In addition, increased protein stability of MCL-1 can also lead to protein overexpression as a result of genetic inactivation of FBW7, a ubiquitin ligase (135, 136).
A1/BFL-1
Protein-based structural analyses indicate, A1 (BCL2A1), the mouse homolog of human BFL-1, is most highly related to the anti-apoptotic Mcl-1 (137). Because A1/Bfl-1-null mice are embryonic lethal, recently, an A1/Bfl-1-conditional knockout mouse was generated that was viable, fertile, and only showed minor defects in the hematopoietic system (58, 59). A1/Bfl-1 transgenic mice demonstrated that overexpression of A1/Bfl-1 did not cooperate with Myc to drive B cell lymphomagenesis (60). However, overexpression of A1/BFL-1 in lymphoma cell lines provided protection from apoptosis induced by a number of stimuli, including ligation of the B cell receptor (138), cytokine deprivation (139), and treatment with staurosporine or etoposide (139, 140). In contrast, knocking down A1/BFL-1 resulted in increased sensitivity of B cell lymphoma cells to cell death caused by CD20 cross-linking and DNA-damaging drugs (141).
Increased levels of A1/BFL-1 mRNA have been reported in DLBCL and CLL (61–63). However, our analysis of public gene expression data showed that compared to normal B cells, A1/BFL-1 was not overexpressed in DLBCL, FL, MZL, and MCL (9). We also observed reduced A1/BFL-1 expression in Burkitt lymphoma patient samples compared to normal human B cells (9). A1/BFL-1 protein can be post-translationally regulated by ubiquitin-mediated proteasomal degradation (139, 140). While regulatory mechanisms, including ubiquitination/proteasomal degradation (139, 140) and direct transcriptional activation by NF-κB (nuclear factor-kappaB) (142) have been reported, the contributions of these mechanisms to increased levels of A1/BFL-1 in B cell lymphomas have not been evaluated. Based on the available data, the contribution of A1/BFL-1 to B cell lymphomas does not appear to be significant.
BCL-W
The gene encoding BCL-W (BCL2L2 for Bcl-2 like 2) was initially discovered via a PCR-based strategy while searching for additional BCL-2 related genes (143). Mice lacking Bcl-w were determined to be essentially normal, except for a profound block in male spermatogenesis (64, 65). Several observations have pointed to a potential role for Bcl-w in tumorigenesis. For example, in a mouse model of Myc-driven myeloid leukemogenesis, Bcl-w overexpression cooperated with Myc to accelerate leukemia development (66). In addition, high levels of BCL-W were present in cell lines derived from human lymphomas, leukemias, and multiple solid organ cancers (66). Similar to its anti-apoptotic relatives, overexpression of Bcl-w in mouse B and T lymphocytes imparted resistance to cytotoxic agents (143, 144). Recently, we discovered Bcl-w has a critical, previously unexplored function in B cell survival and lymphomagenesis (8, 9). We demonstrated with mouse models that loss of Bcl-w profoundly delayed Myc-induced B cell lymphoma development and sensitized B cells to Myc-induced apoptosis (8). We also evaluated the importance of BCL-W in human lymphomas known to be driven by or reliant on MYC expression, specifically Burkitt and DLBCL. The vast majority of Burkitt lymphoma patient samples examined overexpressed BCL-W at both the mRNA and protein levels (8). When BCL-W-specific shRNA was introduced into human Burkitt lymphoma cell lines, they rapidly underwent apoptosis, indicating BCL-W is essential for their continued survival. Additionally, we determined BCL-W was frequently overexpressed in DLBCL. We detected BCL-W mRNA and protein were as often and as highly expressed as BCL-2 in DLBCL, where BCL-2 has long served as the hallmark of a prognostically unfavorable subset (94). Notably, we also observed that increased BCL-W mRNA expression correlated with poor patient survival when levels of BCL-2 mRNA were lower in DLBCL (8).
More recently, we expanded our analyses to explore the additional contributions of BCL-W in other non-Hodgkin B cell lymphomas. We performed an unprecedented analysis of all anti-apoptotic BCL-2 family members across different B cell lymphomas, including Burkitt, DLBCL, FL, MZL, and MCL (9). In all five types of B cell lymphomas, BCL-W was overexpressed compared to normal B cell controls. Increased levels of BCL-W mRNA and protein mirrored those of BCL-2 in FL, which was unexpected, as FL is historically recognized to be driven by a BCL-2 translocation (9, 86–90). Of note, and consistent with previous reports, as the grade of FL increased, levels of BCL-2 decreased (9, 145), whereas BCL-W expression remained elevated in both low and high grade FL (9). Taken together, BCL-W appears to have a critically important, and previously unrecognized, anti-apoptotic role in B cell lymphoma.
Multiple avenues to regulate levels of Bcl-w have been proposed. Overexpression of BCL-W can result from increased activity at the BCL-W promoter (146). A number of studies using different tumor types have documented that elevated expression of BCL-W may be attributed to the downregulation of miRNA that target BCL-W mRNA (128, 147, 148). We recently demonstrated that tumor suppressive miRNA target BCL-W, BCL-2, and BCL-X as a novel, miRNA-mediated mechanism of apoptosis induced by the oncogenic transcription factor Myc (8, 109, 121). Specifically, in normal cells, Myc transcriptionally activates the miR-15 family and let-7a, which bind and negatively regulate the expression of BCL-W, BCL-2, and BCL-X, causing cells to undergo apoptosis (Figure 2). However, cancer cells, including lymphoma, inactivate this mechanism. By means that have not been fully elucidated, but do involve histone deacetylase (HDAC) enzymes, Myc switches from a transcriptional activator to a transcriptional repressor of the miR-15 family and let-7a in lymphoma. This, in turn, allows the expression of anti-apoptotic BCL-W, BCL-2, and BCL-X to increase, thereby facilitating tumorigenesis (Figure 2). We established a link between regulation of BCL-W expression and Myc. Identifying this novel mechanism of miRNA-mediated apoptosis also answered a question that has remained largely unexplored. Previously, it was shown that Myc suppressed the expression of Bcl-2 and Bcl-x through an unknown, indirect mechanism (149, 150). These recent studies provide direct evidence of a mechanism whereby Myc transcriptionally modulates miRNA expression leading to altered expression of Bcl-w as well as Bcl-2 and Bcl-x (8, 109, 121). Therefore, increased expression of anti-apoptotic Bcl-2 family members, through a variety of mechanisms, are essential survival factors to B cell lymphoma.
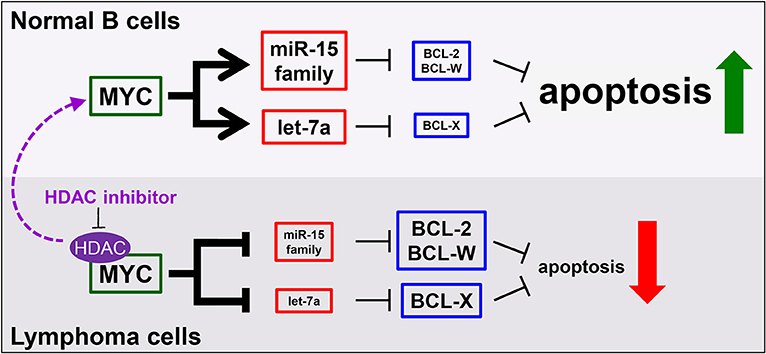
Figure 2. MicroRNA mediate a novel mechanism of Myc-induced apoptosis that is inactivated in malignant cells, but re-activated by HDAC inhibition. In normal B cells (top), MYC transcriptionally upregulates the miR-15 family and let-7a to target and reduce BCL-2, BCL-W, and BCL-X levels, thereby promoting apoptosis. However, in lymphoma cells (bottom), MYC, in complex with histone deacetylases (HDAC), transcriptionally repress the same miRNA, causing increased levels of anti-apoptotic proteins and reduced apoptosis. This mechanism can be re-activated in lymphoma cells by inhibiting HDACs (purple arrow).
Targeting Anti-Apoptotic Bcl-2 Family Members in Lymphoma
Biological Rationale, Drug Development, and Early Clinical Studies for Targeting BCL-2
Due to the persuasive in vitro and in vivo evidence that anti-apoptotic BCL-2 family members confer a survival advantage to neoplastic cells, and contribute to chemotherapeutic resistance in different types of hematologic malignancies, including B cell lymphomas, several strategies have been developed to target them. Since BCL-2 was considered the most important anti-apoptotic BCL-2 family member in B cell lymphoma, it was targeted first. Initial attempts to pharmacologically target BCL-2 consisted of decreasing the intracellular levels of BCL-2 with the delivery of RNA antisense molecules (Figure 3). Several of these antisense molecules had encouraging preclinical efficacy and entered clinical trials, including Oblimersen (G3139/Genasense) (151, 152), PNT2258 (NCT02226965) (153, 154), and SPC2996 (NCT00285103) (155). Of the three anti-BCL-2 antisense oligonucleotides, the best characterized was Oblimersen sodium (G3139, Genasense; Genta Inc.). Oblimersen was the first drug to demonstrate proof-of-principle specific down-regulation of BCL-2 protein in human tumors (156, 157) and provided initial preclinical and clinical evidence of synergy with cytotoxic drugs, biological agents, and steroids in a variety of human cancers, including non-Hodgkin lymphoma, multiple myeloma and acute myeloid leukemia (AML) (156–159).
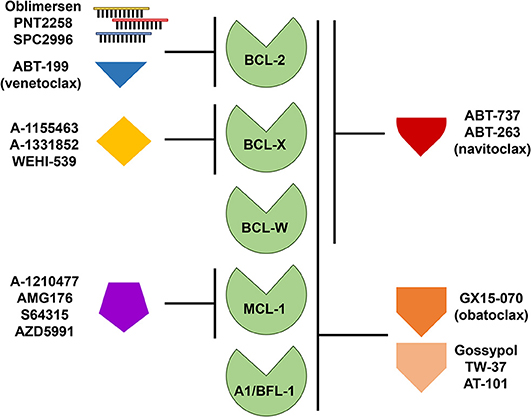
Figure 3. Targeting anti-apoptotic BCL-2 family members for therapy. Therapeutic strategies directed at targeting single or multiple anti-apoptotic BCL-2 family members include the use of antisense oligonucleotides and small molecule inhibitors or mimetics. Like the pro-apoptotic BH-3 only proteins, these small molecules display varying affinities for anti-apoptotic BCL-2 family members as indicated.
The limited efficacy of Oblimersen in AML (158) and other hematologic malignancies was attributed, in part, to inefficient intracellular delivery and led to the search for small molecules that could target the anti-apoptotic proteins themselves. Each of the anti-apoptotic proteins has a hydrophobic groove where they interact with the BH-3 domains of other BCL-2 family members. With the advances made by structural biology and improved knowledge of the protein-protein interactions within the Bcl-2 family, small molecule inhibitors, known as BH-3 mimetics were developed. BH-3 mimetics, based on their functional mimicry of the BH-3 only pro-apoptotic proteins, can bind with high affinity to the same hydrophobic pocket of one or more anti-apoptotic Bcl-2 family members. The high affinity and specificity of the BH-3 mimetics displaces already-bound or prevents binding of newly synthesized pro-apoptotic BH-3 only proteins from their anti-apoptotic partners, leaving the cells in a “primed” state for apoptosis. This has proven particularly important in combination treatments, where the addition of BH-3 mimetics sensitizes cells to a variety of anti-cancer compounds and aids in circumventing intrinsic or acquired resistance (160).
The first synthetic BH-3 mimetic was ABT-737, a small-molecule that binds with high affinity to BCL-2, BCL-X, and BCL-W, but not MCL-1 or A1/BFL-1 (Figure 3), and was shown to efficiently induce apoptosis at sub-micromolar concentrations in a variety of non-Hodgkin lymphoma cell lines (161, 162) and in primary CLL cells (163, 164). An orally bioavailable derivative of ABT-737, ABT-263 (navitoclax), was later developed. ABT-263 (navitoclax) proved to be efficacious in B cell lymphoma xenograft models as a single agent or in combination with a variety of cytotoxic drugs, including rituximab (165). Of note, ectopic overexpression of BCL-2 in B cell lymphoma cell lines protects from ABT-263 (navitoclax)-induced death in vitro (8, 166). We demonstrated that, similar to BCL-2 overexpression, BCL-W overexpression in Burkitt lymphoma cell lines conferred resistance to ABT-737 and ABT-263 (8). Also, ectopic Bcl-w overexpression in primary murine precursor and mature B and T cells induced resistance to ABT-737 (144). In early phase clinical trials, navitoclax showed antitumor activity in indolent B cell malignancies (CLL and FL) (167–169). However, the drug was found to induce rapid and severe, dose-limiting, thrombocytopenia and further studies were stopped. Navitoclax-induced thrombocytopenia is believed to be due to an on-target effect on BCL-X, which is a key factor for platelet survival (165, 170, 171). The hematologic toxicity of navitoclax led to a significant effort to develop a BH-3 mimetic that selectively targeted BCL-2. This effort was successful and produced ABT-199 (venetoclax), an orally bioavailable BCL-2-specific BH-3 mimetic [Figure 3, (172)]. Venetoclax (ABT-199) showed promising efficacy in vitro and in xenografts in vivo for a variety of hematologic malignancies (e.g., leukemia, CLL, DLBCL) without inducing severe thrombocytopenia (172–174).
Based on these encouraging preclinical data, further clinical development of venetoclax was launched in 2011 (Table 2), with a primary focus on relapsed/refractory (R/R) CLL and NHL (M12-175). There were high expectations of clinical benefit with venetoclax in low grade lymphomas, which historically have served as the canonical disease model for BCL-2 overexpression. The first subjects treated on the M12-175 venetoclax study, at daily doses of 100 or 200 mg, were R/R CLL patients with high nodal and blood tumor burden. These patients experienced very rapid (< 12 h) and dramatic reductions in lymphocyte count and lymph node size, but they also developed severe, life-threatening, tumor lysis syndrome (TLS). In all cases, the TLS was effectively managed, and the patients were able to resume therapy on study. These early data demonstrated the rapid pro-apoptotic effect and therapeutic potential of venetoclax in R/R CLL, but required the development of a mitigating strategy to prevent life-threatening TLS. The strategy included selecting a lower venetoclax starting dose (50 mg) and developing a weekly intra-patient dose ramp-up scheme, in addition to careful prophylaxis and management of TLS. Despite the implementation of this strategy, additional episodes of severe TLS were observed, leading to the death of one patient at the highest dose level (1,200 mg) in a large (N = 56) cohort of patients with CLL (175). After the starting dose of venetoclax was further decreased to 20 mg and with even more stringent TLS prophylaxis and monitoring, 60 additional CLL patients were treated in an expansion cohort, with ramp-up dosing up to 400 mg daily. No additional events of severe TLS were observed in this group. The most common Grade 3–4 non-TLS toxicity was Grade 3–4 neutropenia (~40% of patients) (175), which is believed to be an on-target effect of BCL-2 inhibition in neutrophil progenitor cells (180). Other common toxicities included Grade 1–2 gastrointestinal symptoms, such as nausea and diarrhea (~50% or patients). From the standpoint of anti-tumor responses, venetoclax showed highly encouraging efficacy in R/R CLL, with an overall response rate (ORR) of 79%, and a complete response rate (CRR) of 20%, in 116 patients. Most notably, responses were observed in high-risk CLL patients, including those with del(17p). These important early observations in high-risk CLL led to a landmark phase II study where 107 patients with del(17p) CLL were treated with venetoclax with the proven ramp-up dosing to 400 mg daily, with an ORR of 79% and a CRR of 8%. Based on the data from these phase I and II studies, venetoclax received accelerated FDA approval on April 11, 2016 for the treatment of patients with del(17p) CLL who have relapsed after, or are refractory to, ≥1 prior line of therapy. Based primarily on data from 389 R/R CLL patients enrolled in the MURANO clinical trial (NCT02005471), on June 8, 2018 the FDA expanded the approval of venetoclax, in combination with rituximab, to include patients with CLL who progressed after at least one prior therapy, regardless of the presence of del(17p) (181).
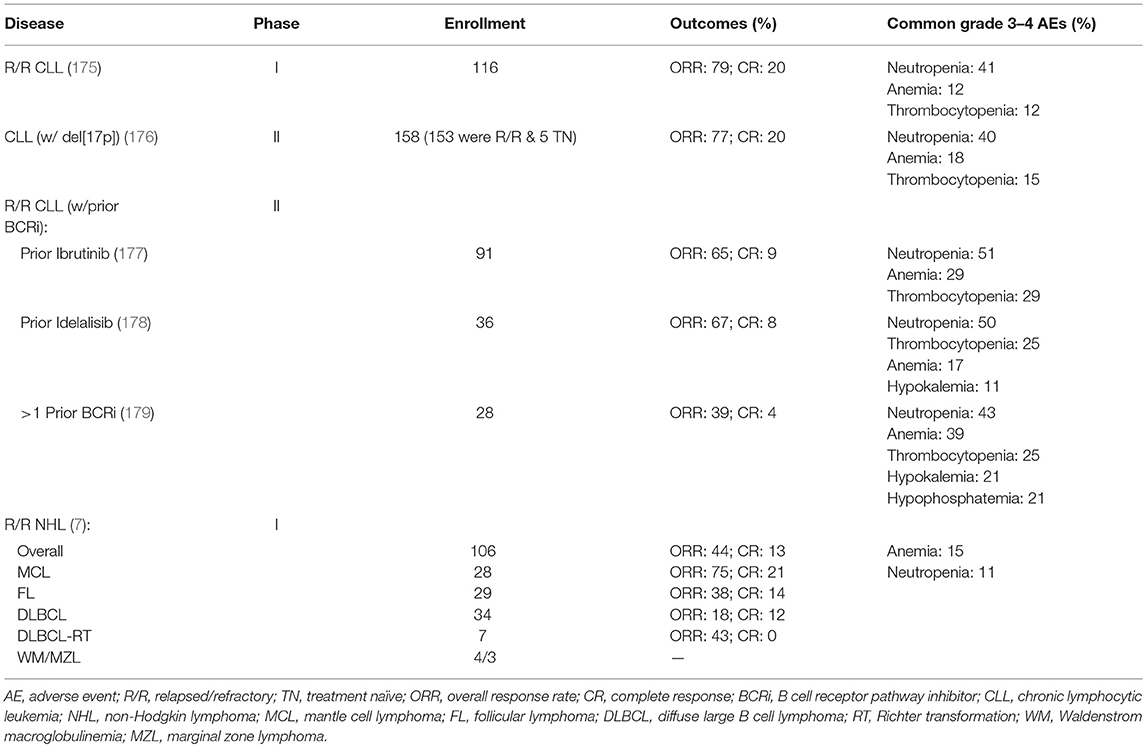
Table 2. Venetoclax as monotherapy in CLL and B cell lymphoma.
Clinical Trials With Single Agent Venetoclax in B Cell Lymphomas
In parallel with the clinical development of venetoclax in CLL, the drug's profile was also evaluated in patients with a broad variety of B cell lymphomas, including DLBCL, FL, MCL, lymphoplasmacytic lymphoma/Waldenström macroglobulinemia (LPL/WM), and MZL [Table 2, (7)]. The observed toxicity and the efficacy were significantly different compared to those in CLL. TLS did not occur and severe neutropenia was only observed in 11% of patients. The ORR for the entire cohort was 44%, with the best responses seen in MCL (75% ORR; 21% CRR). Remarkably, despite the well-characterized overexpression of BCL-2 in FL, ORR in FL was only 38%. A slightly higher ORR of 44% was noted in patients with FL treated at ≥1,200 mg compared with 27% in those treated at ≤ 900 mg, suggesting that higher doses could lead to better efficacy in the nodal disease, which is the primary disease site in FL. Results in DLBCL were even more disappointing, with ORR of 18% and no clear association between BCL-2 protein expression levels and response.
Combination Treatments With Venetoclax in Clinical Trials With CLL and B Cell Lymphoma
In light of the low single agent activity of venetoclax in B cell lymphomas, combination regimens are now being evaluated, mostly in the context of the general strategy of adding venetoclax to well-recognized chemo-immunotherapy combination with established efficacy (bendamustine and rituximab; CHOP and rituximab; CHOP and obinutuzumab; EPOCH and rituximab), or highly promising new drugs (ibrutinib) (Tables 3, 4). A phase II trial comparing a venetoclax/rituximab doublet to the three-drug combination of venetoclax/bendamustine/rituximab is ongoing in patients with R/R FL (NCT02187861), with preliminary evidence of superior efficacy with the three drug combination vs. the doublet (ORR 64 vs. 33%) (194). A combination of venetoclax with obinutuzumab in previously untreated FL is also currently underway (NCT02877550). As in CLL, the combination of ibrutinib and venetoclax is being evaluated in a phase II study in relapsed/refractory MCL (NCT02471391). The early experience in this trial suggests good tolerability and promising efficacy of the combination (196). There are also ongoing studies exploring whether venetoclax may act as a chemosensitizing agent, such as a study of venetoclax administered in combination with R-CHOP or obinutuzumab-CHOP in previously untreated DLBCL (NCT02055820). A preliminary report indicated that, as expected, CR rates were high, but follow-up was too short to assess progression-free survival (PFS). The toxicity profile was acceptable, but venetoclax dosing had to be reduced to administration only on days 1 to 10 (rather than continuous dosing) to mitigate the rate of Grade 3–4 neutropenia. A phase II study of venetoclax and dose-adjusted R-EPOCH in patients with Richter transformation to DLBCL has begun (NCT03054896), in addition to a study using a similar regimen for patients with de novo aggressive B cell lymphomas, including double-hit DLBCL (NCT03036904). A phase I study is also ongoing to examine the combination of venetoclax and the SYK tyrosine kinase inhibitor TAK-659 for patients with R/R DLBCL and FL (NCT03357627).
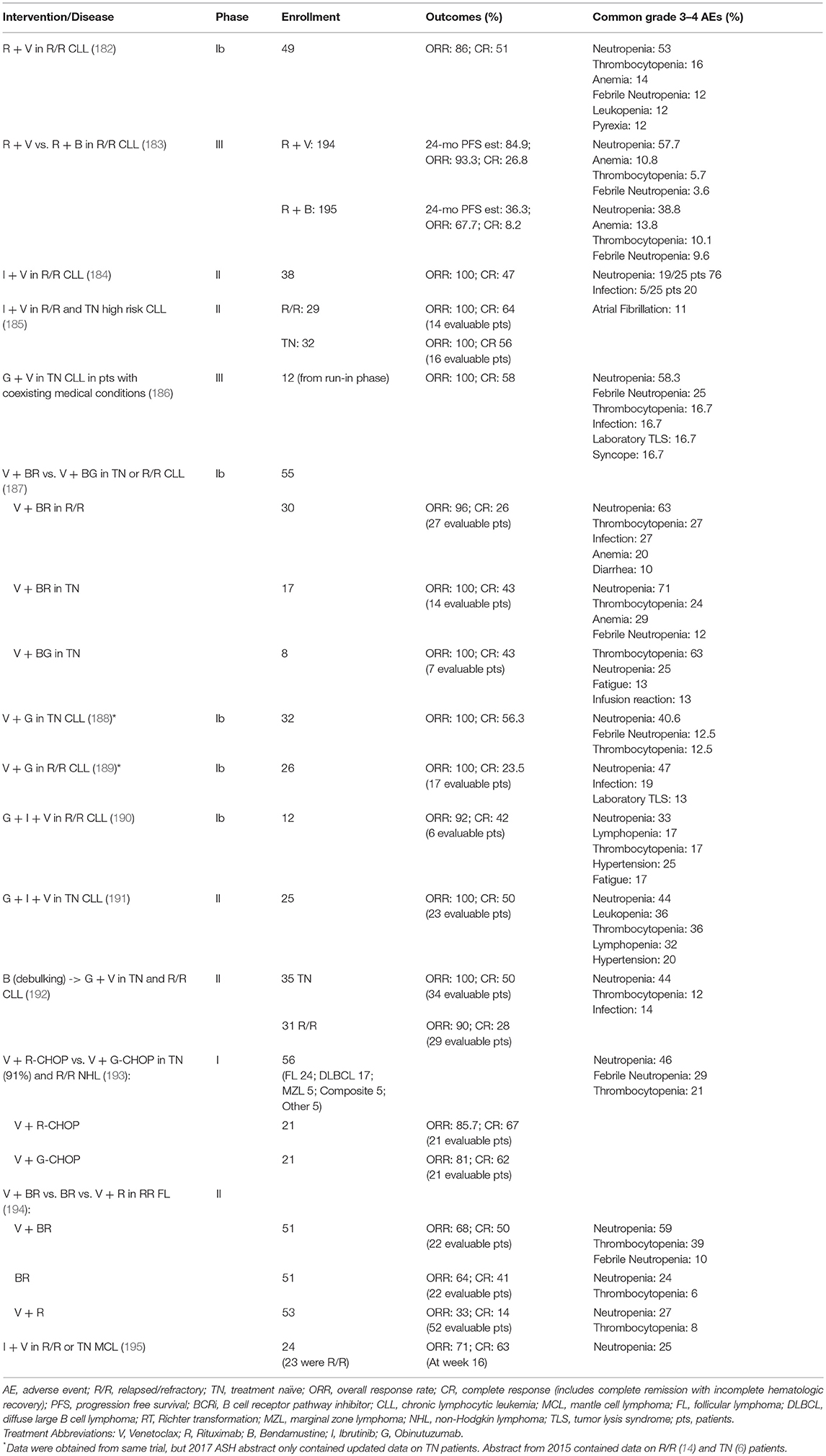
Table 3. Venetoclax in combination with anti-neoplastic agents in CLL and B cell lymphoma.
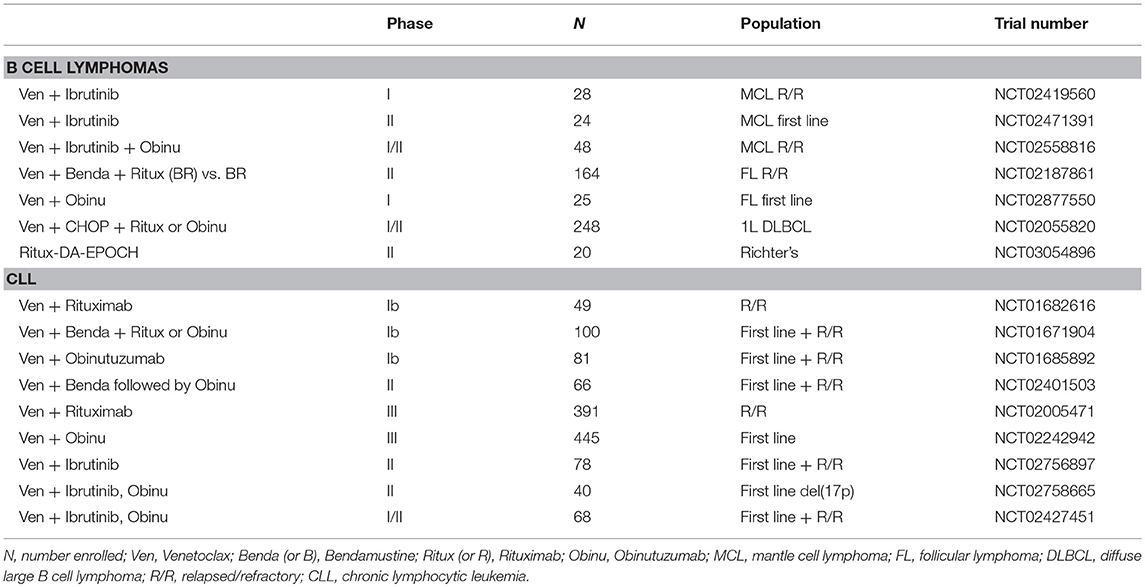
Table 4. Ongoing venetoclax combination studies in B cell lymphomas and CLL.
Inhibiting Other Anti-apoptotic BCL-2 Family Members
The accompanying toxicities that come with targeting multiple BCL-2 family members have fueled the development of new inhibitors to selectively target specific anti-apoptotic BCL-2 family members. Furthermore, individual lymphomas may be differentially reliant on one or more of the anti-apoptotic BCL-2 family members, which can be exploited therapeutically. In addition to the BCL-2 specific inhibitors developed, BCL-X and MCL-1 inhibitors have also been generated (5). There are currently no known inhibitors specific for BCL-W or A1/BFL-1.
BCL-X Inhibitors
While compounds selective for BCL-2 over BCL-X have shown anti-tumor effects in vivo with limited platelet toxicities (175, 180), not all cancers express BCL-2 or require BCL-2 for their continued survival (5, 197). The first reported BCL-X selective inhibitor with sub-nanomolar affinity and selectivity was WEHI-539, which antagonized the anti-apoptotic activity of BCL-X (198). However, WEHI-539 has been limited to in vitro studies as a tool compound due to its unfavorable chemical properties for use in vivo (198). Other efforts using high-throughput screening with nuclear magnetic resonance and structure-based medicinal chemistry led to the development of the BCL-X selective inhibitors A-1155463 and its orally bioavailable analog A-1331852 [Figure 3, (199, 200)]. Cell lines from a variety of malignancies, including AML showed sensitivity to the BCL-X inhibitors. In vivo studies in mice indicated inhibition of BCL-X with A-1331852 as a single agent reduced the volume of a T-cell leukemia, whereas venetoclax did not (180). In addition, the mice tolerated the BCL-X inhibitor. These data suggest that some cancers appear to be dependent on BCL-X and not on BCL-2 and that targeting one over the other may be beneficial.
MCL-1 Inhibitors
Multiple approaches have been explored to selectively target MCL-1. Initially, stabilized alpha-helices of BH-3 only proteins [known as “stapled” peptides (201)] and alpha- or beta-peptide foldamers (202) were designed to target MCL-1. Several compounds with affinity for MCL-1 were identified by screening libraries of natural compounds and small molecules. However, many of these have reported off-target or minimal effects (203, 204). Although obatoclax (GX15-070) targets other anti-apoptotic BCL-2 family members, it was one of the first BH-3 mimetics reported to inhibit MCL-1 [Figure 3, (205)]. Obatoclax has completed phase I/II clinical trials for a number of malignancies, including CLL, but with modest efficacy and neutropenia as a common toxicity (206). At this time, no further clinical trials with obatoclax are ongoing. Similar to obatoclax, TW-37, a gossypol derivative, has shown some toxicity, but has demonstrated potency as a single agent in the treatment of DLBCL cell lines and synergized with other chemotherapies in xenograft models [Figure 3, (207)]. Phase I clinical trials for AT-101, a more refined variant of the gossypol family of pan-BCL-2 family inhibitors, show that it is well-tolerated with treatable neutropenia (208). Two clinical trials of AT-101 as a single agent in R/R B cell lymphomas (NCT00275431) and in combination with rituximab in CLL (NCT00286780) have been completed, but final results have not been published.
A-1210477 was the first selective MCL-1 inhibitor to demonstrate picomolar binding to MCL-1 while maintaining selectivity for MCL-1 over other anti-apoptotic family members [Figure 3, (209)]. A-1210477 forms complexes with MCL-1, thereby disrupting endogenous MCL-1:BIM complexes. As a single agent, this compound induced apoptosis in MCL-1-dependent multiple myeloma cell lines (210). However, A-1210477 does not demonstrate the necessary pharmacokinetics for in vivo use. AMG176 was the first putative MCL-1 inhibitor to undergo clinical evaluation (NCT02675452) (211), but no data in humans has been reported. In addition, phase I trials are actively recruiting or soon to recruit patients for testing the MCL-1 specific inhibitors S64315 (MIK665) in patients with R/R DLBCL and multiple myeloma, AML, and myelodysplastic syndrome (NCT02992483, NCT02979366, and NCT03672695) and AZD5991 in patients with R/R hematologic malignancies, CLL, T-cell lymphoma, and multiple myeloma (NCT03218683). A question that remains largely unanswered is whether normal cells will tolerate MCL-1 inhibition at the level necessary for therapeutic benefit. Potential on-target toxicities may include cardiac (212, 213), hepatic (214, 215), and hematological (45–47), which are based on those observed in Mcl-1 knockout mouse models (45–47, 212–215).
Combination Treatments With Other Anti-apoptotic BCL-2 Family Inhibitors
At present, there are a limited number of clinical trials dedicated to evaluating the effects of anti-apoptotic BCL-2 family inhibitors aside from venetoclax either alone or in combination with other therapies. Navitoclax, which inhibits BCL-2, BCL-X, and BCL-W, was being tested in combination with bendamustine and rituximab in patients with relapsed DLBCL as part of the NAVIGATE study, but recruitment was terminated due to non-safety-related reasons (NCT01423539). A study of navitoclax together with venetoclax and chemotherapy (including peg-asparaginase, vincristine, dexamethasone, and tyrosine kinase inhibitor) is ongoing for patients with R/R ALL or R/R lymphoblastic lymphoma (NCT03181126). A phase I study evaluating the safety of navitoclax administered in combination with rituximab is active for patients with CD20-positive lymphoproliferative disorders and CLL (NCT00788684). In addition, obatoclax mesylate (GX15-070MS), which inhibits BCL-2, BCL-X, BCL-W, and MCL-1 (Figure 3) has been evaluated as a single agent followed by a combination with rituximab for treatment naive patients with FL (NCT00427856). Obatoclax has also been combined with bortezomib for the treatment of R/R MCL (NCT00407303). A phase I/II trial was initiated to study the side effects and the dose of obatoclax when given together with rituximab and bendamustine in treating patients with R/R non-Hodgkin lymphoma including MZL, FL, and MCL; however, the study was withdrawn due to lack of patient accrual (NCT01238146). A phase I dose escalation study of the MCL-1 selective inhibitor S64315 in combination with venetoclax (estimated start date: December 3, 2018) will be testing the safety and tolerability in patients with AML (NCT03672695). The continued efforts to develop novel anti-apoptotic BCL-2 family protein inhibitors will continue to pave the way for new clinical trials combining current inhibitors with both conventional and other novel agents in various lymphomas.
Resistance Mechanisms for BCL-2 Family Protein Inhibitors
Given that venetoclax selectively inhibits BCL-2, this compound should be effective in cancer cells that express BCL-2; however, this does not always occur. For example, FL expresses high levels of BCL-2 due to its t(14;18) translocation; yet, the clinical response rate to venetoclax is quite low in FL patients (7). This suggests BCL-2 expression alone is insufficient to predict BCL-2 dependence. In a recent study, a subset of lymphoma cell lines expressing BCL-2 protein were resistant to venetoclax, resulting from acquired mutations in BCL-2 and the pro-apoptotic protein BAX or a phosphorylation event on BCL-2 that prevented venetoclax from binding, thereby blocking apoptosis (216–218). Similarly, upon comparing venetoclax-resistant FL cells with their parental cell lines, the resistant FL cells had significantly higher levels of ERK1/2 and BIM phosphorylation at serine 69, targeting BIM for proteasomal degradation; thus, reducing the pro-apoptotic nature of the cells (219, 220). Another study showed increased phospho-ATK levels in a venetoclax-resistant B cell lymphoma line, suggesting activation of the PI3K pathway (221). Whole-exome sequencing and methylation profiling of serial CLL samples from eight patients before venetoclax treatment and at the time of venetoclax resistance did not show genetic alterations in BCL-2 (222). However, most patients acquired mutations in cancer-related genes, including BRAF, NOTCH1, RB1, and TP53, and had homozygous deletion of CDKN2A/B.
Resistance of FL and DLBCL to single agent venetoclax may be attributed, in part, to elevated levels of other anti-apoptotic family members. For example, we recently reported that BCL-W was elevated in both FL and DLBCL at a similar frequency as BCL-2 (9). Moreover, our recent large-scale evaluation of all anti-apoptotic BCL-2 family members in multiple non-Hodgkin lymphomas revealed that many selected for the overexpression of more than one anti-apoptotic family member, simultaneously (9). These data provide a potential explanation into why just targeting BCL-2 with venetoclax did not result in high complete response rates for FL or DLBCL (7). Moreover, levels of BCL-X and MCL-1 were upregulated in venetoclax-resistant DLBCL cell lines (221). Furthermore, in lymphoma cell lines that have become resistant to navitoclax, increased levels of anti-apoptotic A1/BFL-1 and MCL-1 are observed (223).
With the increasing focus on MCL-1 as an important regulator of apoptosis in leukemia and lymphoma (224) and potential mediator of venetoclax resistance, a number of efforts are in progress to better define the role of MCL-1 in venetoclax resistance and to develop strategies to downregulate MCL-1 levels as a possible way to overcome it. While MCL-1 specific inhibitors are in early phase of development (225, 226), several studies have already shown that downregulation of MCL-1 mRNA and/or protein levels in BH-3 mimetic-resistant cells increases the sensitivity to navitoclax and venetoclax (54, 227). For example, the pan CDK inhibitor dinaciclib restored sensitivity to navitoclax- and venetoclax-mediated apoptosis in resistant lymphoma cells via inhibition of MCL-1 phosphorylation by CDK2/Cyclin E, which in turn led to the destabilization of MCL-1 protein and release of BIM from MCL-1 (228). The combination of dinaciclib and venetoclax resulted in robust synergistic cell death in DLBCL cell lines and in primary CLL patient samples. Additional ongoing strategies to enhance the therapeutic efficacy of venetoclax in B cell neoplasms (increasing response rate, depth of response, overcoming primary and secondary resistance), include combinations with BTK inhibitors (229), dual delta- and gamma-PI3 kinase inhibitors (230), SYK inhibitors (231), and BET inhibitors (232). While all these studies are of very high interest, and many of the preclinical concepts are being evaluated in clinical trials, there is a need to better understand the specific mechanistic functions of the BCL-2 family members in each of these pro-apoptotic combinations, where they are redundant and where they may lead to synthetic lethality.
With clinical trials on-going and many more being developed using anti-apoptotic BCL-2 family inhibitors for treatment of B cell lymphoma, as well as other hematologic malignancies and solid-organ cancers, there remains a significant lack of knowledge of these proteins and their requirements in non-Hodgkin lymphomas. Aside from FL and DLBCL, alterations in specific anti-apoptotic BCL-2 family members have not been well-characterized or associated with other B cell lymphomas, which is likely due to the lack of a comprehensive analysis prior to the one we recently published (9). Therefore, it is likely necessary to measure and monitor the levels of BCL-2 family members when enrolling patients onto clinical trials testing selective anti-apoptotic inhibitors. Inhibiting one anti-apoptotic BCL-2 family member that is not expressed in that lymphoma should have no effect on the lymphoma and be unnecessary treatment for that patient. Additionally, targeting one anti-apoptotic BCL-2 family member may lead to the dependency on another, ultimately leading to therapeutic resistance.
Non-apoptotic Effects of BCL-2 Family Inhibition
There have been reports suggesting that inhibition of anti-apoptotic BCL-2 family members may activate cell signaling pathways, in addition to apoptosis, to further promote survival and resistance to cell death. For example, a recent proteomic analysis following treatment of DLBCL and MCL cell lines with venetoclax showed venetoclax treatment (both short- and long-term) altered the levels of proteins involved in apoptosis, but also the expression levels and phosphorylation status of proteins involved in the DNA damage response (i.e., γH2AX, CHK2, ATM), growth/survival signaling pathways (i.e., PTEN, Src, MAPK, AKT), and cellular metabolism (i.e., HK2, PDK1, PKM2, GCLM) were also affected (233). However, further studies are necessary to determine whether these effects are directly attributable to venetoclax-mediated BCL-2 inhibition and the resulting apoptosis or effects that are independent of BCL-2. The BCL-2 family of proteins has also been reported to function in maintaining calcium homeostasis (234). The early BH-3-only protein mimetic HA14-1 was shown to dysregulate intracellular calcium signaling in platelets, reportedly due to off-target effects, but neither navitoclax nor venetoclax disrupted intracellular calcium transport mechanisms (235, 236). Furthermore, investigations have shown that BCL-2 family proteins may be involved in autophagy (237). Specifically, studies have reported that navitoclax and the BH-3-mimetic HA14-1 can block the interaction between BCL-2/BCL-X and Beclin1, a protein important for the localization of autophagic proteins, leading to the activation of Beclin1-dependent autophagy (237–240). BCL-2 family BH-3-only mimetics can also regulate cell autophagy through activation of the unfolded protein response signaling pathway PERK-eIF2α-ATF4, which upregulates expression of the autophagy gene Atg12 (241–243).
Conclusion
Defects in apoptosis are universal to all B cell lymphomas and defects in apoptotic signaling is frequently associated with resistance to chemotherapy (2, 4, 244). In this review, we have highlighted the Bcl-2 family network of proteins in lymphoma, including the recent discovery of BCL-W overexpression in B cell lymphomas, and described current clinical strategies to inhibit anti-apoptotic Bcl-2 family proteins that aim to develop more effective therapies for B cell lymphoma. Despite decades of significant progress in identifying the molecular underpinnings of apoptotic cell death and their contributions to the pathogenesis, survival, and resistance to treatment of individual B cell lymphomas, recent efforts have revealed that several critical factors have been significantly underestimated. The identification of BCL-W as a previously unrecognized key contributor to B cell lymphoma has substantially aided in increasing our understanding of the BCL-2 family and the alterations in their expression that contribute to B cell lymphoma survival and therapy resistance. This new knowledge has opened the door to the development of additional selective cancer therapeutics and combination therapies that may redefine the treatment of B cell lymphoma. Revealing the involvement of BCL-W in many types of B cell lymphoma has also opened the door to studying its possible role in treatment resistance. To fully exploit the potential of selective inhibitors of anti-apoptotic BCL-2 proteins for the treatment of B cell lymphoma, we must know which inhibitors should be given to which patients. To guide the use of specific inhibitors in individual patients, or molecularly defined patient subsets, we must know which anti-apoptotic BCL-2 protein(s) are the most relevant target(s). For example, while one malignancy may have requirements for BCL-2, another might require BCL-X and/or BCL-W, highlighting the importance of using the right biomarkers to evaluate each lymphoma. In the era of personalized medicine, these recent advances attest to the power of discoveries in basic science being directly translated into the clinic to improve targeted treatment strategies for individual lymphomas.
Author Contributions
CA, SC-G, PP, and CE contributed to the literature review, and to the writing and editing of the material presented in this manuscript.
Funding
This work was supported by the NCI Cancer Center support grant P30CA056036 and the Sidney Kimmel Cancer Center.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Eischen laboratory for their thoughtful discussions and technical efforts that have contributed to any of our studies highlighted in this review.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
2. Adams JM, Cory S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. (2018) 25:27–36. doi: 10.1038/cdd.2017.161
3. Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer (2016) 16:99–109. doi: 10.1038/nrc.2015.17
4. Vogler M, Walter HS, Dyer MJS. Targeting anti-apoptotic BCL2 family proteins in haematological malignancies–from pathogenesis to treatment. Br J Haematol. (2017) 178:364–79. doi: 10.1111/bjh.14684
5. Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov. (2017) 16:273–84. doi: 10.1038/nrd.2016.253
6. Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature (1985) 318:533–8.
7. Davids MS, Roberts AW, Seymour JF, Pagel JM, Kahl BS, Wierda WG, et al. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-Hodgkin lymphoma. J Clin Oncol. (2017) 35:826–33. doi: 10.1200/JCO.2016.70.4320
8. Adams CM, Kim AS, Mitra R, Choi JK, Gong JZ, Eischen CM. BCL-W has a fundamental role in B cell survival and lymphomagenesis. J Clin Invest. (2017) 127:635–50. doi: 10.1172/JCI89486
9. Adams CM, Mitra R, Gong JZ, Eischen CM. Non-Hodgkin and Hodgkin lymphomas select for overexpression of BCLW. Clin Cancer Res. (2017) 23:7119–29. doi: 10.1158/1078-0432.CCR-17-1144
10. Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. (2018) 25:65–80. doi: 10.1038/cdd.2017.186
11. Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci USA. (2004) 101:6164–9. doi: 10.1073/pnas.0401471101
12. Tagawa H, Karnan S, Suzuki R, Matsuo K, Zhang X, Ota A, et al. Genome-wide array-based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene (2005) 24:1348–58. doi: 10.1038/sj.onc.1208300
13. Kelly JL, Novak AJ, Fredericksen ZS, Liebow M, Ansell SM, Dogan A, et al. Germline variation in apoptosis pathway genes and risk of non-Hodgkin's lymphoma. Cancer Epidemiol Biomarkers Prev. (2010) 19:2847–58. doi: 10.1158/1055-9965.EPI-10-0581
14. Richter-Larrea JA, Robles EF, Fresquet V, Beltran E, Rullan AJ, Agirre X, et al. Reversion of epigenetically mediated BIM silencing overcomes chemoresistance in Burkitt lymphoma. Blood (2010) 116:2531–42. doi: 10.1182/blood-2010-02-268003
15. Garrison SP, Jeffers JR, Yang C, Nilsson JA, Hall MA, Rehg JE, et al. Selection against PUMA gene expression in Myc-driven B-cell lymphomagenesis. Mol Cell Biol. (2008) 28:5391–402. doi: 10.1128/MCB.00907-07
16. Michalak EM, Jansen ES, Happo L, Cragg MS, Tai L, Smyth GK, et al. Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ. (2009) 16:684–96. doi: 10.1038/cdd.2008.195
17. Frenzel A, Labi V, Chmelewskij W, Ploner C, Geley S, Fiegl H, et al. Suppression of B-cell lymphomagenesis by the BH3-only proteins Bmf and Bad. Blood (2010) 115:995–1005. doi: 10.1182/blood-2009-03-212670
18. Ranger AM, Zha J, Harada H, Datta SR, Danial NN, Gilmore AP, et al. Bad-deficient mice develop diffuse large B cell lymphoma. Proc Natl Acad Sci USA. (2003) 100:9324–9. doi: 10.1073/pnas.1533446100
19. Zinkel SS, Ong CC, Ferguson DO, Iwasaki H, Akashi K, Bronson RT, et al. Proapoptotic BID is required for myeloid homeostasis and tumor suppression. Genes Dev. (2003) 17:229–39. doi: 10.1101/gad.1045603
20. Happo L, Phipson B, Smyth GK, Strasser A, Scott CL. Neither loss of Bik alone, nor combined loss of Bik and Noxa, accelerate murine lymphoma development or render lymphoma cells resistant to DNA damaging drugs. Cell Death Dis. (2012) 3:e306. doi: 10.1038/cddis.2012.42
21. Coultas L, Bouillet P, Stanley EG, Brodnicki TC, Adams JM, Strasser A. Proapoptotic BH3-only Bcl-2 family member Bik/Blk/Nbk is expressed in hemopoietic and endothelial cells but is redundant for their programmed death. Mol Cell Biol. (2004) 24:1570–81. doi: 10.1128/MCB.24.4.1570-1581.2004
22. Arena V, Martini M, Luongo M, Capelli A, Larocca LM. Mutations of the BIK gene in human peripheral B-cell lymphomas. Genes Chromosomes Cancer (2003) 38:91–6. doi: 10.1002/gcc.10245
23. Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell (2000) 6:1389–99. doi: 10.1016/S1097-2765(00)00136-2
24. Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science (1995) 270:96–9.
25. Eischen CM, Roussel MF, Korsmeyer SJ, Cleveland JL. Bax loss impairs Myc-induced apoptosis and circumvents the selection of p53 mutations during Myc-mediated lymphomagenesis. Mol Cell Biol. (2001) 21:7653–62. doi: 10.1128/MCB.21.22.7653-7662.2001
26. Ke F, Voss A, Kerr JB, O'Reilly LA, Tai L, Echeverry N, et al. BCL-2 family member BOK is widely expressed but its loss has only minimal impact in mice. Cell Death Differ. (2012) 19:915–25. doi: 10.1038/cdd.2011.210
27. Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell (1993) 75:229–40.
28. Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature (1990) 348:331–3. doi: 10.1038/348331a0
29. Kahl BS, Yang DT. Follicular lymphoma: evolving therapeutic strategies. Blood (2016) 127:2055–63. doi: 10.1182/blood-2015-11-624288
30. Schuetz JM, Johnson NA, Morin RD, Scott DW, Tan K, Ben-Nierah S, et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia (2012) 26:1383–90. doi: 10.1038/leu.2011.378
31. Correia C, Schneider PA, Dai H, Dogan A, Maurer MJ, Church AK, et al. BCL2 mutations are associated with increased risk of transformation and shortened survival in follicular lymphoma. Blood (2015) 125:658–67. doi: 10.1182/blood-2014-04-571786
32. Meda BA, Frost M, Newell J, Bohling SD, Huebner-Chan DR, Perkins SL, et al. BCL-2 is consistently expressed in hyperplastic marginal zones of the spleen, abdominal lymph nodes, and ileal lymphoid tissue. Am J Surg Pathol. (2003) 27:888–94. doi: 10.1097/00000478-200307000-00003
33. Agarwal B, Naresh KN. Bcl-2 family of proteins in indolent B-cell non-Hodgkin's lymphoma: study of 116 cases. Am J Hematol. (2002) 70:278–82. doi: 10.1002/ajh.10139
34. Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Nakayama K, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science (1995) 267:1506–10.
35. Motoyama N, Kimura T, Takahashi T, Watanabe T, Nakano T. bcl-x prevents apoptotic cell death of both primitive and definitive erythrocytes at the end of maturation. J Exp Med. (1999) 189:1691–8.
36. Kelly PN, Grabow S, Delbridge AR, Strasser A, Adams JM. Endogenous Bcl-xL is essential for Myc-driven lymphomagenesis in mice. Blood (2011) 118:6380–6. doi: 10.1182/blood-2011-07-367672
37. Fang W, Weintraub BC, Dunlap B, Garside P, Pape KA, Jenkins MK, et al. Self-reactive B lymphocytes overexpressing Bcl-xL escape negative selection and are tolerized by clonal anergy and receptor editing. Immunity (1998) 9:35–45.
38. Linden M, Kirchhof N, Carlson C, Van Ness B. Targeted overexpression of Bcl-XL in B-lymphoid cells results in lymphoproliferative disease and plasma cell malignancies. Blood (2004) 103:2779–86. doi: 10.1182/blood-2003-10-3399
39. Xerri L, Parc P, Brousset P, Schlaifer D, Hassoun J, Reed JC, et al. Predominant expression of the long isoform of Bcl-x (Bcl-xL) in human lymphomas. Br J Haematol. (1996) 92:900–6.
40. Rummel MJ, de Vos S, Hoelzer D, Koeffler HP, Hofmann WK. Altered apoptosis pathways in mantle cell lymphoma. Leuk Lymphoma (2004) 45:49–54. doi: 10.1080/1042819031000151112
41. Krajewski S, Krajewska M, Shabaik A, Wang HG, Irie S, Fong L, et al. Immunohistochemical analysis of in vivo patterns of Bcl-X expression. Cancer Res. (1994) 54:5501–7.
42. Puthier D, Derenne S, Barille S, Moreau P, Harousseau JL, Bataille R, et al. Mcl-1 and Bcl-xL are co-regulated by IL-6 in human myeloma cells. Br J Haematol. (1999) 107:392–5.
43. Tu Y, Renner S, Xu F, Fleishman A, Taylor J, Weisz J, et al. BCL-X expression in multiple myeloma: possible indicator of chemoresistance. Cancer Res. (1998) 58:256–62.
44. Gauthier ER, Piche L, Lemieux G, Lemieux R. Role of bcl-X(L) in the control of apoptosis in murine myeloma cells. Cancer Res. (1996) 56:1451–6.
45. Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature (2003) 426:671–6. doi: 10.1038/nature02067
46. Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, et al. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science (2005) 307:1101–4. doi: 10.1126/science.1106114
47. Dzhagalov I, St John A, He YW. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood (2007) 109:1620–6. doi: 10.1182/blood-2006-03-013771
48. Grabow S, Delbridge AR, Aubrey BJ, Vandenberg CJ, Strasser A. Loss of a single Mcl-1 allele inhibits MYC-driven lymphomagenesis by sensitizing pro-B cells to apoptosis. Cell Rep. (2016) 14:2337–47. doi: 10.1016/j.celrep.2016.02.039
49. Kelly GL, Grabow S, Glaser SP, Fitzsimmons L, Aubrey BJ, Okamoto T, et al. Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev. (2014) 28:58–70. doi: 10.1101/gad.232009.113
50. Campbell KJ, Bath ML, Turner ML, Vandenberg CJ, Bouillet P, Metcalf D, et al. Elevated Mcl-1 perturbs lymphopoiesis, promotes transformation of hematopoietic stem/progenitor cells, and enhances drug resistance. Blood (2010) 116:3197–207. doi: 10.1182/blood-2010-04-281071
51. Zhou P, Qian L, Bieszczad CK, Noelle R, Binder M, Levy NB, et al. Mcl-1 in transgenic mice promotes survival in a spectrum of hematopoietic cell types and immortalization in the myeloid lineage. Blood (1998) 92:3226–39.
52. Zhou P, Levy NB, Xie H, Qian L, Lee CY, Gascoyne RD, et al. MCL1 transgenic mice exhibit a high incidence of B-cell lymphoma manifested as a spectrum of histologic subtypes. Blood (2001) 97:3902–9. doi: 10.1182/blood.V97.12.3902
53. Wenzel SS, Grau M, Mavis C, Hailfinger S, Wolf A, Madle H, et al. MCL1 is deregulated in subgroups of diffuse large B-cell lymphoma. Leukemia (2013) 27:1381–90. doi: 10.1038/leu.2012.367
54. Phillips DC, Xiao Y, Lam LT, Litvinovich E, Roberts-Rapp L, Souers AJ, et al. Loss in MCL-1 function sensitizes non-Hodgkin's lymphoma cell lines to the BCL-2-selective inhibitor venetoclax (ABT-199). Blood Cancer J. (2015) 5:e368. doi: 10.1038/bcj.2015.88
55. Al-Harbi S, Hill BT, Mazumder S, Singh K, Devecchio J, Choudhary G, et al. An antiapoptotic BCL-2 family expression index predicts the response of chronic lymphocytic leukemia to ABT-737. Blood (2011) 118:3579–90. doi: 10.1182/blood-2011-03-340364
56. Zhang B, Gojo I, Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood (2002) 99:1885–93. doi: 10.1182/blood.V99.6.1885
57. Quinn BA, Dash R, Azab B, Sarkar S, Das SK, Kumar S, et al. Targeting Mcl-1 for the therapy of cancer. Expert Opin Investig Drugs (2011) 20:1397–411. doi: 10.1517/13543784.2011.609167
58. Tuzlak S, Schenk RL, Vasanthakumar A, Preston SP, Haschka MD, Zotos D, et al. The BCL-2 pro-survival protein A1 is dispensable for T cell homeostasis on viral infection. Cell Death Differ. (2017) 24:523–33. doi: 10.1038/cdd.2016.155
59. Schenk RL, Tuzlak S, Carrington EM, Zhan Y, Heinzel S, Teh CE, et al. Characterisation of mice lacking all functional isoforms of the pro-survival BCL-2 family member A1 reveals minor defects in the haematopoietic compartment. Cell Death Differ. (2017) 24:534–45. doi: 10.1038/cdd.2016.156
60. Mensink M, Anstee NS, Robati M, Schenk RL, Herold MJ, Cory S, et al. Anti-apoptotic A1 is not essential for lymphoma development in Emicro-Myc mice but helps sustain transplanted Emicro-Myc tumour cells. Cell Death Differ. (2018) 25:795–806. doi: 10.1038/s41418-017-0045-8
61. Park IC, Lee SH, Whang DY, Hong WS, Choi SS, Shin HS, et al. Expression of a novel Bcl-2 related gene, Bfl-1, in various human cancers and cancer cell lines. Anticancer Res. (1997) 17:4619–22.
62. Morales AA, Olsson A, Celsing F, Osterborg A, Jondal M, Osorio LM. High expression of bfl-1 contributes to the apoptosis resistant phenotype in B-cell chronic lymphocytic leukemia. Int J Cancer (2005) 113:730–7. doi: 10.1002/ijc.20614
63. Olsson A, Norberg M, Okvist A, Derkow K, Choudhury A, Tobin G, et al. Upregulation of bfl-1 is a potential mechanism of chemoresistance in B-cell chronic lymphocytic leukaemia. Br J Cancer (2007) 97:769–77. doi: 10.1038/sj.bjc.6603951
64. Print CG, Loveland KL, Gibson L, Meehan T, Stylianou A, Wreford N, et al. Apoptosis regulator bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc Natl Acad Sci USA. (1998) 95:12424–31.
65. Ross AJ, Waymire KG, Moss JE, Parlow AF, Skinner MK, Russell LD, et al. Testicular degeneration in Bclw-deficient mice. Nat Genet. (1998) 18:251–6. doi: 10.1038/ng0398-251
66. Beverly LJ, Varmus HE. MYC-induced myeloid leukemogenesis is accelerated by all six members of the antiapoptotic BCL family. Oncogene (2009) 28:1274–9. doi: 10.1038/onc.2008.466
67. Moldoveanu T, Follis AV, Kriwacki RW, Green DR. Many players in BCL-2 family affairs. Trends Biochem Sci. (2014) 39:101–11. doi: 10.1016/j.tibs.2013.12.006
68. Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta (2004) 1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012
69. Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. (1999) 13:1899–911.
70. Huang DC, Strasser A. BH3-Only proteins-essential initiators of apoptotic cell death. Cell (2000) 103:839–42. doi: 10.1016/S0092-8674(00)00187-2
71. Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. (2008) 9:47–59. doi: 10.1038/nrm2308
72. Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science (2003) 302:1036–8. doi: 10.1126/science.1090072
73. Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell (2003) 4:321–8. doi: 10.1016/S1535-6108(03)00244-7
74. Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science (1999) 286:1735–8.
75. Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell (2007) 129:1337–49. doi: 10.1016/j.cell.2007.04.027
76. Michalak EM, Villunger A, Adams JM, Strasser A. In several cell types tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ. (2008) 15:1019–29. doi: 10.1038/cdd.2008.16
77. Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. (2008) 18:157–64. doi: 10.1016/j.tcb.2008.01.007
78. Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. (2001) 15:1481–6. doi: 10.1101/gad.897601
79. Meijerink JP, Smetsers TF, Sloetjes AW, Linders EH, Mensink EJ. Bax mutations in cell lines derived from hematological malignancies. Leukemia (1995) 9:1828–32.
80. Meijerink JP, Mensink EJ, Wang K, Sedlak TW, Sloetjes AW, de Witte T, et al. Hematopoietic malignancies demonstrate loss-of-function mutations of BAX. Blood (1998) 91:2991–7.
81. Brimmell M, Mendiola R, Mangion J, Packham G. BAX frameshift mutations in cell lines derived from human haemopoietic malignancies are associated with resistance to apoptosis and microsatellite instability. Oncogene (1998) 16:1803–12. doi: 10.1038/sj.onc.1201704
82. Rooswinkel RW, van de Kooij B, de Vries E, Paauwe M, Braster R, Verheij M, et al. Antiapoptotic potency of Bcl-2 proteins primarily relies on their stability, not binding selectivity. Blood (2014) 123:2806–15. doi: 10.1182/blood-2013-08-519470
83. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. (2014) 15:49–63. doi: 10.1038/nrm3722
84. Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell (2006) 9:351–65. doi: 10.1016/j.ccr.2006.03.027
85. Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. (2005) 19:1294–305. doi: 10.1101/gad.1304105
86. Fukuhara S, Rowley JD. Chromosome 14 translocations in non-Burkitt lymphomas. Int J Cancer (1978) 22:14–21.
87. Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science (1984) 226:1097–9.
88. Cleary ML, Sklar J. Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proc Natl Acad Sci USA (1985) 82:7439–43.
89. Cleary ML, Meeker TC, Levy S, Lee E, Trela M, Sklar J, et al. Clustering of extensive somatic mutations in the variable region of an immunoglobulin heavy chain gene from a human B cell lymphoma. Cell (1986) 44:97–106.
90. Bakhshi A, Jensen JP, Goldman P, Wright JJ, McBride OW, Epstein AL, et al. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell (1985) 41:899–906.
91. Dai H, Meng XW, Lee SH, Schneider PA, Kaufmann SH. Context-dependent Bcl-2/Bak interactions regulate lymphoid cell apoptosis. J Biol Chem. (2009) 284:18311–22. doi: 10.1074/jbc.M109.004770
92. Smith AJ, Dai H, Correia C, Takahashi R, Lee SH, Schmitz I, et al. Noxa/Bcl-2 protein interactions contribute to bortezomib resistance in human lymphoid cells. J Biol Chem. (2011) 286:17682–92. doi: 10.1074/jbc.M110.189092
93. Iqbal J, Neppalli VT, Wright G, Dave BJ, Horsman DE, Rosenwald A, et al. BCL2 expression is a prognostic marker for the activated B-cell-like type of diffuse large B-cell lymphoma. J Clin Oncol. (2006) 24:961–8. doi: 10.1200/JCO.2005.03.4264
94. Tsuyama N, Sakata S, Baba S, Mishima Y, Nishimura N, Ueda K, et al. BCL2 expression in DLBCL: reappraisal of immunohistochemistry with new criteria for therapeutic biomarker evaluation. Blood (2017) 130:489–500. doi: 10.1182/blood-2016-12-759621
95. Sandoval-Sus JD, Chavez J, Dalia S. A new therapeutic era in GCB and ABC diffuse large B-cell lymphoma molecular subtypes: a cell of origin-driven review. Curr Cancer Drug Targets (2016) 16:305–22. doi: 10.2174/1568009615666151030102539
96. Iqbal J, Sanger WG, Horsman DE, Rosenwald A, Pickering DL, Dave B, et al. BCL2 translocation defines a unique tumor subset within the germinal center B-cell-like diffuse large B-cell lymphoma. Am J Pathol. (2004) 165:159–66. doi: 10.1016/S0002-9440(10)63284-1
97. Monni O, Joensuu H, Franssila K, Klefstrom J, Alitalo K, Knuutila S. BCL2 overexpression associated with chromosomal amplification in diffuse large B-cell lymphoma. Blood (1997) 90:1168–74.
98. Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. (2018) 24:679–90. doi: 10.1038/s41591-018-0016-8
99. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. (2018) 378:1396–407. doi: 10.1056/NEJMoa1801445
100. Dunleavy K. Double-hit lymphomas: current paradigms and novel treatment approaches. Hematology Am Soc Hematol Educ Program (2014) 2014:107–12. doi: 10.1182/asheducation-2014.1.107
101. Lindsley RC, LaCasce AS. Biology of double-hit B-cell lymphomas. Curr Opin Hematol. (2012) 19:299–304. doi: 10.1097/MOH.0b013e328353bbbd
102. Johnson NA, Slack GW, Savage KJ, Connors JM, Ben-Neriah S, Rogic S, et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol. (2012) 30:3452–9. doi: 10.1200/JCO.2011.41.0985
103. Green TM, Young KH, Visco C, Xu-Monette ZY, Orazi A, Go RS, et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol. (2012) 30:3460–7. doi: 10.1200/JCO.2011.41.4342
104. Rao PH, Houldsworth J, Dyomina K, Parsa NZ, Cigudosa JC, Louie DC, et al. Chromosomal and gene amplification in diffuse large B-cell lymphoma. Blood (1998) 92:234–40.
105. Hanada M, Delia D, Aiello A, Stadtmauer E, Reed JC. bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood (1993) 82:1820–8.
106. Saito M, Novak U, Piovan E, Basso K, Sumazin P, Schneider C, et al. BCL6 suppression of BCL2 via Miz1 and its disruption in diffuse large B cell lymphoma. Proc Natl Acad Sci USA. (2009) 106:11294–9. doi: 10.1073/pnas.0903854106
107. Breitschopf K, Haendeler J, Malchow P, Zeiher AM, Dimmeler S. Posttranslational modification of Bcl-2 facilitates its proteasome-dependent degradation: molecular characterization of the involved signaling pathway. Mol Cell Biol. (2000) 20:1886–96. doi: 10.1128/MCB.20.5.1886-1896.2000
108. Davids MS. Targeting BCL-2 in B-cell lymphomas. Blood (2017) 130:1081–8. doi: 10.1182/blood-2017-04-737338
109. Adams CM, Hiebert SW, Eischen CM. Myc induces miRNA-mediated apoptosis in response to HDAC inhibition in hematologic malignancies. Cancer Res. (2016) 76:736–48. doi: 10.1158/0008-5472.CAN-15-1751
110. Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. (2005) 102:13944–9. doi: 10.1073/pnas.0506654102
111. Di Lisio L, Sanchez-Beato M, Gomez-Lopez G, Rodriguez ME, Montes-Moreno S, Mollejo M, et al. MicroRNA signatures in B-cell lymphomas. Blood Cancer J. (2012) 2:e57. doi: 10.1038/bcj.2012.1
112. Di Lisio L, Martinez N, Montes-Moreno S, Piris-Villaespesa M, Sanchez-Beato M, Piris MA. The role of miRNAs in the pathogenesis and diagnosis of B-cell lymphomas. Blood (2012) 120:1782–90. doi: 10.1182/blood-2012-05-402784
113. McDonnell TJ, Deane N, Platt FM, Nunez G, Jaeger U, McKearn JP, et al. bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell (1989) 57:79–88.
114. Strasser A, Whittingham S, Vaux DL, Bath ML, Adams JM, Cory S, et al. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc Natl Acad Sci USA. (1991) 88:8661–5.
115. Gonzalez-Garcia M, Perez-Ballestero R, Ding L, Duan L, Boise LH, Thompson CB, et al. bcl-XL is the major bcl-x mRNA form expressed during murine development and its product localizes to mitochondria. Development (1994) 120:3033–42.
116. Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, et al. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell (1993) 74:597–608.
117. Gregoli PA, Bondurant MC. The roles of Bcl-X(L) and apopain in the control of erythropoiesis by erythropoietin. Blood (1997) 90:630–40.
118. Wagner KU, Claudio E, Rucker EB III Riedlinger G, Broussard C, Schwartzberg PL, et al. Conditional deletion of the Bcl-x gene from erythroid cells results in hemolytic anemia and profound splenomegaly. Development (2000) 127:4949–58. doi: 10.1016/S0925-4773(01)00549-4
119. Bairey O, Zimra Y, Shaklai M, Okon E, Rabizadeh E. Bcl-2, Bcl-X, Bax, and Bak expression in short- and long-lived patients with diffuse large B-cell lymphomas. Clin Cancer Res. (1999) 5:2860–6.
120. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature (2010) 463:899–905. doi: 10.1038/nature08822
121. Adams CM, Eischen CM. Histone deacetylase inhibition reveals a tumor-suppressive function of MYC-regulated miRNA in breast and lung carcinoma. Cell Death Differ. (2016) 23:1312–21. doi: 10.1038/cdd.2016.9
122. Shimizu S, Takehara T, Hikita H, Kodama T, Miyagi T, Hosui A, et al. The let-7 family of microRNAs inhibits Bcl-xL expression and potentiates sorafenib-induced apoptosis in human hepatocellular carcinoma. J Hepatol. (2010) 52:698–704. doi: 10.1016/j.jhep.2009.12.024
123. Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci USA. (1993) 90:3516–20.
124. Morel C, Carlson SM, White FM, Davis RJ. Mcl-1 integrates the opposing actions of signaling pathways that mediate survival and apoptosis. Mol Cell Biol. (2009) 29:3845–52. doi: 10.1128/MCB.00279-09
125. Stewart DP, Koss B, Bathina M, Perciavalle RM, Bisanz K, Opferman JT. Ubiquitin-independent degradation of antiapoptotic MCL-1. Mol Cell Biol. (2010) 30:3099–110. doi: 10.1128/MCB.01266-09
126. Mojsa B, Lassot I, Desagher S. Mcl-1 ubiquitination: unique regulation of an essential survival protein. Cells (2014) 3:418–37. doi: 10.3390/cells3020418
127. Crawford M, Batte K, Yu L, Wu X, Nuovo GJ, Marsh CB, et al. MicroRNA 133B targets pro-survival molecules MCL-1 and BCL2L2 in lung cancer. Biochem Biophys Res Commun. (2009) 388:483–9. doi: 10.1016/j.bbrc.2009.07.143
128. Gong J, Zhang JP, Li B, Zeng C, You K, Chen MX, et al. MicroRNA-125b promotes apoptosis by regulating the expression of Mcl-1, Bcl-w and IL-6R. Oncogene (2013) 32:3071–9. doi: 10.1038/onc.2012.318
129. Mott JL, Kobayashi S, Bronk SF, Gores GJ. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene (2007) 26:6133–40. doi: 10.1038/sj.onc.1210436
130. Steele R, Mott JL, Ray RB. MBP-1 upregulates miR-29b that represses Mcl-1, collagens, and matrix-metalloproteinase-2 in prostate cancer cells. Genes Cancer (2010) 1:381–7. doi: 10.1177/1947601910371978
131. Desjobert C, Renalier MH, Bergalet J, Dejean E, Joseph N, Kruczynski A, et al. MiR-29a down-regulation in ALK-positive anaplastic large cell lymphomas contributes to apoptosis blockade through MCL-1 overexpression. Blood (2011) 117:6627–37. doi: 10.1182/blood-2010-09-301994
132. Mazzoccoli L, Robaina MC, Apa AG, Bonamino M, Pinto LW, Queiroga E, et al. MiR-29 silencing modulates the expression of target genes related to proliferation, apoptosis and methylation in Burkitt lymphoma cells. J Cancer Res Clin Oncol. (2018) 144:483–97. doi: 10.1007/s00432-017-2575-3
133. Malumbres R, Sarosiek KA, Cubedo E, Ruiz JW, Jiang X, Gascoyne RD, et al. Differentiation stage-specific expression of microRNAs in B lymphocytes and diffuse large B-cell lymphomas. Blood (2009) 113:3754–64. doi: 10.1182/blood-2008-10-184077
134. Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature (2010) 463:103–7. doi: 10.1038/nature08646
135. Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature (2011) 471:104–9. doi: 10.1038/nature09732
136. Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature (2011) 471:110–4. doi: 10.1038/nature09779
137. Lin EY, Orlofsky A, Berger MS, Prystowsky MB. Characterization of A1, a novel hemopoietic-specific early-response gene with sequence similarity to bcl-2. J Immunol. (1993) 151:1979–88.
138. Kuss AW, Knodel M, Berberich-Siebelt F, Lindemann D, Schimpl A, Berberich I. A1 expression is stimulated by CD40 in B cells and rescues WEHI 231 cells from anti-IgM-induced cell death. Eur J Immunol. (1999) 29:3077–88. doi: 10.1002/(SICI)1521-4141(199910)29:10<3077::AID-IMMU3077>3.0.CO;2-R
139. Kucharczak JF, Simmons MJ, Duckett CS, Gelinas C. Constitutive proteasome-mediated turnover of Bfl-1/A1 and its processing in response to TNF receptor activation in FL5.12 pro-B cells convert it into a prodeath factor. Cell Death Differ. (2005) 12:1225–39. doi: 10.1038/sj.cdd.4401684
140. Herold MJ, Zeitz J, Pelzer C, Kraus C, Peters A, Wohlleben G, et al. The stability and anti-apoptotic function of A1 are controlled by its C terminus. J Biol Chem. (2006) 281:13663–71. doi: 10.1074/jbc.M600266200
141. Brien G, Trescol-Biemont MC, Bonnefoy-Berard N. Downregulation of Bfl-1 protein expression sensitizes malignant B cells to apoptosis. Oncogene (2007) 26:5828–32. doi: 10.1038/sj.onc.1210363
142. Zong WX, Edelstein LC, Chen C, Bash J, Gelinas C. The prosurvival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-kappaB that blocks TNFalpha-induced apoptosis. Genes Dev. (1999) 13:382–7.
143. Gibson L, Holmgreen SP, Huang DC, Bernard O, Copeland NG, Jenkins NA, et al. bcl-w, a novel member of the bcl-2 family, promotes cell survival. Oncogene (1996) 13:665–75.
144. Merino D, Khaw SL, Glaser SP, Anderson DJ, Belmont LD, Wong C, et al. Bcl-2, Bcl-x(L), and Bcl-w are not equivalent targets of ABT-737 and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood (2012) 119:5807–16. doi: 10.1182/blood-2011-12-400929
145. Ott G, Rosenwald A. Molecular pathogenesis of follicular lymphoma. Haematologica (2008) 93:1773–6. doi: 10.3324/haematol.2008.001495
146. Lapham A, Adams JE, Paterson A, Lee M, Brimmell M, Packham G. The Bcl-w promoter is activated by beta-catenin/TCF4 in human colorectal carcinoma cells. Gene (2009) 432:112–7. doi: 10.1016/j.gene.2008.12.002
147. Xu Y, Zhao F, Wang Z, Song Y, Luo Y, Zhang X, et al. MicroRNA-335 acts as a metastasis suppressor in gastric cancer by targeting Bcl-w and specificity protein 1. Oncogene (2012) 31:1398–407. doi: 10.1038/onc.2011.340
148. Cao J, Cai J, Huang D, Han Q, Yang Q, Li T, et al. miR-335 represents an invasion suppressor gene in ovarian cancer by targeting Bcl-w. Oncol Rep. (2013) 30:701–6. doi: 10.3892/or.2013.2482
149. Eischen CM, Packham G, Nip J, Fee BE, Hiebert SW, Zambetti GP, et al. Bcl-2 is an apoptotic target suppressed by both c-Myc and E2F-1. Oncogene (2001) 20:6983–93. doi: 10.1038/sj.onc.1204892
150. Eischen CM, Woo D, Roussel MF, Cleveland JL. Apoptosis triggered by Myc-induced suppression of Bcl-X(L) or Bcl-2 is bypassed during lymphomagenesis. Mol Cell Biol. (2001) 21:5063–70. doi: 10.1128/MCB.21.15.5063-5070.2001
151. Advani PP, Paulus A, Masood A, Sher T, Chanan-Khan A. Pharmacokinetic evaluation of oblimersen sodium for the treatment of chronic lymphocytic leukemia. Expert Opin Drug Metab Toxicol. (2011) 7:765–74. doi: 10.1517/17425255.2011.579105
152. Galatin PS, Advani RH, Fisher GA, Francisco B, Julian T, Losa R, et al. Phase I trial of oblimersen (Genasense(R)) and gemcitabine in refractory and advanced malignancies. Invest New Drugs (2011) 29:971–7. doi: 10.1007/s10637-010-9416-4
153. Ebrahim AS, Kandouz M, Liddane A, Sabbagh H, Hou Y, Li C, et al. PNT2258, a novel deoxyribonucleic acid inhibitor, induces cell cycle arrest and apoptosis via a distinct mechanism of action: a new class of drug for non-Hodgkin's lymphoma. Oncotarget (2016) 7:42374–84. doi: 10.18632/oncotarget.9872
154. Ebrahim AS, Sabbagh H, Liddane A, Raufi A, Kandouz M, Al-Katib A. Hematologic malignancies: newer strategies to counter the BCL-2 protein. J Cancer Res Clin Oncol. (2016) 142:2013–22. doi: 10.1007/s00432-016-2144-1
155. Durig J, Duhrsen U, Klein-Hitpass L, Worm J, Hansen JB, Orum H, et al. The novel antisense Bcl-2 inhibitor SPC2996 causes rapid leukemic cell clearance and immune activation in chronic lymphocytic leukemia. Leukemia (2011) 25:638–47. doi: 10.1038/leu.2010.322
156. van de Donk NW, Kamphuis MM, van Dijk M, Borst HP, Bloem AC, Lokhorst HM. Chemosensitization of myeloma plasma cells by an antisense-mediated downregulation of Bcl-2 protein. Leukemia (2003) 17:211–9. doi: 10.1038/sj.leu.2402768
157. van de donk N, Kamphuis M, van Dijk M. Evaluation of Bcl-2 antisense oligonucleotide drugs in multiple myeloma. Blood (2000) 96:757a. doi: 10.1182/blood.V96.757a
158. Marcucci G, Stock W, Dai G, Klisovic RB, Liu S, Klisovic MI, et al. Phase I study of oblimersen sodium, an antisense to Bcl-2, in untreated older patients with acute myeloid leukemia: pharmacokinetics, pharmacodynamics, and clinical activity. J Clin Oncol. (2005) 23:3404–11. doi: 10.1200/JCO.2005.09.118
159. Pro B, Leber B, Smith M, Fayad L, Romaguera J, Hagemeister F, et al. Phase II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in combination with rituximab in patients with recurrent B-cell non-Hodgkin lymphoma. Br J Haematol. (2008) 143:355–60. doi: 10.1111/j.1365-2141.2008.07353.x
160. Grant S. Rational combination strategies to enhance venetoclax activity and overcome resistance in hematologic malignancies. Leuk Lymphoma (2018) 59:1292–9. doi: 10.1080/10428194.2017.1366999
161. Anderson MA, Huang D, Roberts A. Targeting BCL2 for the treatment of lymphoid malignancies. Semin Hematol. (2014) 51:219–27. doi: 10.1053/j.seminhematol.2014.05.008
162. Paoluzzi L, Gonen M, Bhagat G, Furman RR, Gardner JR, Scotto L, et al. The BH3-only mimetic ABT-737 synergizes the antineoplastic activity of proteasome inhibitors in lymphoid malignancies. Blood (2008) 112:2906–16. doi: 10.1182/blood-2007-12-130781
163. Vogler M, Dinsdale D, Sun XM, Young KW, Butterworth M, Nicotera P, et al. A novel paradigm for rapid ABT-737-induced apoptosis involving outer mitochondrial membrane rupture in primary leukemia and lymphoma cells. Cell Death Differ. (2008) 15:820–30. doi: 10.1038/cdd.2008.25
164. Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. (2007) 117:112–21. doi: 10.1172/JCI28281
165. Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. (2008) 68:3421–8. doi: 10.1158/0008-5472.CAN-07-5836
166. Inoue-Yamauchi A, Jeng PS, Kim K, Chen HC, Han S, Ganesan YT, et al. Targeting the differential addiction to anti-apoptotic BCL-2 family for cancer therapy. Nat Commun. (2017) 8:16078. doi: 10.1038/ncomms16078
167. Wilson WH, O'Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. (2010) 11:1149–59. doi: 10.1016/S1470-2045(10)70261-8
168. Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. (2011) 29:909–16. doi: 10.1200/JCO.2010.31.6208
169. Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. (2012) 30:488–96. doi: 10.1200/JCO.2011.34.7898
170. Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, et al. Programmed anuclear cell death delimits platelet life span. Cell (2007) 128:1173–86. doi: 10.1016/j.cell.2007.01.037
171. Park CM, Bruncko M, Adickes J, Bauch J, Ding H, Kunzer A, et al. Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins. J Med Chem. (2008) 51:6902–15. doi: 10.1021/jm800669s
172. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. (2013) 19:202–8. doi: 10.1038/nm.3048
173. Itchaki G, Brown JR. The potential of venetoclax (ABT-199) in chronic lymphocytic leukemia. Ther Adv Hematol. (2016) 7:270–87. doi: 10.1177/2040620716655350
174. Vogler M, Dinsdale D, Dyer MJ, Cohen GM. ABT-199 selectively inhibits BCL2 but not BCL2L1 and efficiently induces apoptosis of chronic lymphocytic leukaemic cells but not platelets. Br J Haematol. (2013) 163:139–42. doi: 10.1111/bjh.12457
175. Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. (2016) 374:311–22. doi: 10.1056/NEJMoa1513257
176. Stilgenbauer S, Eichhorst B, Schetelig J, Hillmen P, Seymour JF, Coutre S, et al. Venetoclax for patients with chronic lymphocytic leukemia with 17p deletion: results from the full population of a phase II pivotal trial. J Clin Oncol. (2018) 36:1973–80. doi: 10.1200/JCO.2017.76.6840
177. Jones JA, Mato AR, Wierda WG, Davids MS, Choi M, Cheson BD, et al. Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: an interim analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. (2018) 19:65–75. doi: 10.1016/S1470-2045(17)30909-9
178. Coutre S, Choi M, Furman RR, Eradat H, Heffner L, Jones JA, et al. Venetoclax for patients with chronic lymphocytic leukemia who progressed during or after idelalisib therapy. Blood (2018) 131:1704–11. doi: 10.1182/blood-2017-06-788133
179. Wierda W, Davids MS, Furman RR, Cheson BD, Barr PM, Eradat H, et al. Venetoclax (VEN) is active in chronic lymphocytic leukemia (CLL) relapsed or refractory to more than one B-cell receptor pathway inhibitor (BCRi). Blood (2017) 130:3025. doi: 10.1182/blood.v130.suppl1.3025
180. Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med. (2015) 7:279ra40. doi: 10.1126/scitranslmed.aaa4642
181. Seymour JF, Kipps TJ, Eichhorst B, Hillmen P, D'Rozario J, Assouline S, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. (2018) 378:1107–20. doi: 10.1056/NEJMoa1713976
182. Seymour JF, Ma S, Brander DM, Choi MY, Barrientos J, Davids MS, et al. Venetoclax plus rituximab in relapsed or refractory chronic lymphocytic leukaemia: a phase 1b study. Lancet Oncol. (2017) 18:230–40. doi: 10.1016/S1470-2045(17)30012-8
183. Seymour JF, Kipps TJ, Eichhorst B, Hillmen P, D'Rozario JM, Assouline S, et al. Venetoclax plus rituximab is superior to bendamustine plus rituximab in patients with relapsed/refractory chronic lymphocytic leukemia–results from pre-planned interim analysis of the randomized phase 3 murano study. Blood (2017) 130:LBA-2. doi: 10.1182/blood.v130.suppl1.LBA-2
184. Hillmen P, Munir T, Rawstron A, Brock K, Vicente SM, Yates F, et al. Initial results of ibrutinib plus venetoclax in relapsed, refractory CLL (Bloodwise TAP CLARITY Study): high rates of overall response, complete remission and mrd eradication after 6 months of combination therapy. Blood (2017) 130:428. doi: 10.1182/blood.v130.suppl1.428
185. Jain N, Thompson PA, Ferrajoli A, Burger JA, Borthakur G, Takahashi K, et al. Combined venetoclax and ibrutinib for patients with previously untreated high-risk CLL, and relapsed/refractory CLL: a phase II trial. Blood (2017) 130:429. doi: 10.1182/blood.v130.suppl1.429
186. Fischer K, Al-Sawaf O, Fink AM, Dixon M, Bahlo J, Warburton S, et al. Venetoclax and obinutuzumab in chronic lymphocytic leukemia. Blood (2017) 129:2702–5. doi: 10.1182/blood-2017-01-761973
187. Stilgenbauer S, Morschhauser F, Wendtner CM, Cartron G, Hallek M, Eichhorst B, et al. Phase Ib study (GO28440) of venetoclax with bendamustine/rituximab or bendamustine/obinutuzumab in patients with relapsed/refractory or previously untreated chronic lymphocytic leukemia. Blood (2016) 128:4393. doi: 10.1182/blood.V128.22.4393
188. Flinn IW, Gribben JG, Dyer MJ, Wierda W, Maris MB, Furman RR, et al. Safety, efficacy and MRD negativity of a combination of venetoclax and obinutuzumab in patients with previously untreated chronic lymphocytic leukemia–results from a phase 1b study (GP28331). Blood (2017) 130:430. doi: 10.1182/blood.v130.suppl1.430
189. Flinn IW, Brunvand M, Choi MY, Dyer MJ, Gribben JG, Hillmen P, et al. Safety and efficacy of a combination of venetoclax (GDC-0199/ABT-199) and obinutuzumab in patients with relapsed/refractory or previously untreated chronic lymphocytic leukemia–results from a phase 1b study (GP28331). Blood (2015) 126:494. doi: 10.1182/blood.v126.23.494
190. Rogers KA, Huang Y, Ruppert AS, Awan FT, Heerema NA, Hoffman C, et al. Phase 1b study of obinutuzumab, ibrutinib, and venetoclax in relapsed and refractory chronic lymphocytic leukemia. Blood (2018) 132:1568–72. doi: 10.1182/blood-2018-05-853564
191. Rogers KA, Huang Y, Stark A, Awan FT, Maddocks KJ, Woyach J, et al. Initial results of the phase 2 treatment naive cohort in a phase 1b/2 study of obinutuzumab, ibrutinib, and venetoclax in chronic lymphocytic leukemia. Blood (2017) 130:431. doi: 10.1182/blood.v130.suppl1.431
192. Cramer P, von Tresckow J, Bahlo J, Robrecht S, Langerbeins P, Al-Sawaf O, et al. Bendamustine followed by obinutuzumab and venetoclax in chronic lymphocytic leukaemia (CLL2-BAG): primary endpoint analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. (2018) 19:1215–28. doi: 10.1016/S1470-2045(18)30414-5
193. Zelenetz AD, Salles GA, Mason KD, Casulo C, Le Gouill S, Sehn LH, et al. Results of a phase Ib study of venetoclax plus R- or G-CHOP in patients with B-Cell non-Hodgkin lymphoma. Blood (2016) 128:3032. doi: 10.1200/JCO.2016.34.15_suppl.7566
194. Zinzani PL, Topp MS, Yuen SLS, Rusconi C, Fleury I, Pro B, et al. Phase 2 study of venetoclax plus rituximab or randomized ven plus bendamustine + rituximab (BR) versus BR in patients with relapsed/refractory follicular lymphoma: interim data. Blood (2016) 128:617. doi: 10.1182/blood.v128.22.617
195. Tam CS, Anderson MA, Pott C, Agarwal R, Handunnetti S, Hicks RJ, et al. Ibrutinib plus venetoclax for the treatment of mantle-cell lymphoma. N Engl J Med. (2018) 378:1211–23. doi: 10.1056/NEJMoa1715519
196. Tam C, Roberts A, Anderson MA, Dawson SJ, Hicks R, Pott C, et al. The combination of ibrutinib and venetoclax (ABT-199) to achieve complete remissions in patients with relapsed/refractory mantle cell lymphoma: preliminary results of the phase II AIM study. J Clin Oncol. (2016) 34:7548. doi: 10.1200/JCO.2016.34.15_suppl.7548
197. Montero J, Letai A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. (2018) 25:56–64. doi: 10.1038/cdd.2017.183
198. Lessene G, Czabotar PE, Sleebs BE, Zobel K, Lowes KN, Adams JM, et al. Structure-guided design of a selective BCL-X(L) inhibitor. Nat Chem Biol. (2013) 9:390–7. doi: 10.1038/nchembio.1246
199. Koehler MF, Bergeron P, Choo EF, Lau K, Ndubaku C, Dudley D, et al. Structure-guided rescaffolding of selective antagonists of BCL-XL. ACS Med Chem Lett. (2014) 5:662–7. doi: 10.1021/ml500030p
200. Tao ZF, Hasvold L, Wang L, Wang X, Petros AM, Park CH, et al. Discovery of a potent and selective BCL-XL inhibitor with in vivo activity. ACS Med Chem Lett. (2014) 5:1088–93. doi: 10.1021/ml5001867
201. Muppidi A, Doi K, Edwardraja S, Drake EJ, Gulick AM, Wang HG, et al. Rational design of proteolytically stable, cell-permeable peptide-based selective Mcl-1 inhibitors. J Am Chem Soc. (2012) 134:14734–7. doi: 10.1021/ja306864v
202. Smith BJ, Lee EF, Checco JW, Evangelista M, Gellman SH, Fairlie WD. Structure-guided rational design of alpha/beta-peptide foldamers with high affinity for BCL-2 family prosurvival proteins. Chembiochem (2013) 14:1564–72. doi: 10.1002/cbic.201300351
203. Varadarajan S, Vogler M, Butterworth M, Dinsdale D, Walensky LD, Cohen GM. Evaluation and critical assessment of putative MCL-1 inhibitors. Cell Death Differ. (2013) 20:1475–84. doi: 10.1038/cdd.2013.79
204. Belmar J, Fesik SW. Small molecule Mcl-1 inhibitors for the treatment of cancer. Pharmacol Ther. (2015) 145:76–84. doi: 10.1016/j.pharmthera.2014.08.003
205. Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci USA. (2007) 104:19512–7. doi: 10.1073/pnas.0709443104
206. O'Brien SM, Claxton DF, Crump M, Faderl S, Kipps T, Keating MJ, et al. Phase I study of obatoclax mesylate (GX15-070), a small molecule pan-Bcl-2 family antagonist, in patients with advanced chronic lymphocytic leukemia. Blood (2009) 113:299–305. doi: 10.1182/blood-2008-02-137943
207. Mohammad RM, Goustin AS, Aboukameel A, Chen B, Banerjee S, Wang G, et al. Preclinical studies of TW-37, a new nonpeptidic small-molecule inhibitor of Bcl-2, in diffuse large cell lymphoma xenograft model reveal drug action on both Bcl-2 and Mcl-1. Clin Cancer Res. (2007) 13:2226–35. doi: 10.1158/1078-0432.CCR-06-1574
208. Schelman WR, Mohammed TA, Traynor AM, Kolesar JM, Marnocha RM, Eickhoff J, et al. A phase I study of AT-101 with cisplatin and etoposide in patients with advanced solid tumors with an expanded cohort in extensive-stage small cell lung cancer. Invest New Drugs (2014) 32:295–302. doi: 10.1007/s10637-013-9999-7
209. Bruncko M, Wang L, Sheppard GS, Phillips DC, Tahir SK, Xue J, et al. Structure-guided design of a series of MCL-1 inhibitors with high affinity and selectivity. J Med Chem. (2015) 58:2180–94. doi: 10.1021/jm501258m
210. Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. (2015) 6:e1590. doi: 10.1038/cddis.2014.561
211. Caenepeel SR, Belmontes B, Sun J, Coxon A, Moody G, Hughes PE. Preclinical evaluation of AMG 176, a novel, potent and selective Mcl-1 inhibitor with robust anti-tumor activity in Mcl-1 dependent cancer models. Proc. Am. Assoc. Cancer Res. Annual Meeting 2017 (2017) 77:2027. doi: 10.1158/1538-7445.AM2017-2027
212. Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, et al. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. (2013) 27:1365–77. doi: 10.1101/gad.215871.113
213. Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, et al. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. (2013) 27:1351–64. doi: 10.1101/gad.215855.113
214. Hikita H, Takehara T, Shimizu S, Kodama T, Li W, Miyagi T, et al. Mcl-1 and Bcl-xL cooperatively maintain integrity of hepatocytes in developing and adult murine liver. Hepatology (2009) 50:1217–26. doi: 10.1002/hep.23126
215. Vick B, Weber A, Urbanik T, Maass T, Teufel A, Krammer PH, et al. Knockout of myeloid cell leukemia-1 induces liver damage and increases apoptosis susceptibility of murine hepatocytes. Hepatology (2009) 49:627–36. doi: 10.1002/hep.22664
216. Fresquet V, Rieger M, Carolis C, Garcia-Barchino MJ, Martinez-Climent JA. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood (2014) 123:4111–9. doi: 10.1182/blood-2014-03-560284
217. Song T, Chai G, Liu Y, Yu X, Wang Z, Zhang Z. Bcl-2 phosphorylation confers resistance on chronic lymphocytic leukaemia cells to the BH3 mimetics ABT-737, ABT-263 and ABT-199 by impeding direct binding. Br J Pharmacol. (2016) 173:471–83. doi: 10.1111/bph.13370
218. Tahir SK, Smith ML, Hessler P, Rapp LR, Idler KB, Park CH, et al. Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer (2017) 17:399. doi: 10.1186/s12885-017-3383-5
219. Bodo J, Zhao X, Durkin L, Souers AJ, Phillips DC, Smith MR, et al. Acquired resistance to venetoclax (ABT-199) in t(14;18) positive lymphoma cells. Oncotarget (2016) 7:70000–10. doi: 10.18632/oncotarget.12132
220. Chiron D, Bellanger C, Papin A, Tessoulin B, Dousset C, Maiga S, et al. Rational targeted therapies to overcome microenvironment-dependent expansion of mantle cell lymphoma. Blood (2016) 128:2808–18. doi: 10.1182/blood-2016-06-720490
221. Choudhary GS, Al-Harbi S, Mazumder S, Hill BT, Smith MR, Bodo J, et al. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. (2015) 6:e1593. doi: 10.1038/cddis.2014.525
222. Herling CD, Abedpour N, Weiss J, Schmitt A, Jachimowicz RD, Merkel O, et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat Commun. (2018) 9:727. doi: 10.1038/s41467-018-03170-7
223. Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood (2010) 115:3304–13. doi: 10.1182/blood-2009-07-233304
224. Fernandez-Marrero Y, Spinner S, Kaufmann T, Jost PJ. Survival control of malignant lymphocytes by anti-apoptotic MCL-1. Leukemia (2016) 30:2152–9. doi: 10.1038/leu.2016.213
225. Brennan MS, Chang C, Tai L, Lessene G, Strasser A, Dewson G, et al. Humanized Mcl-1 mice enable accurate pre-clinical evaluation of MCL-1 inhibitors destined for clinical use. Blood (2018) 132:1573–83. doi: 10.1182/blood-2018-06-859405
226. Li Z, He S, Look AT. The MCL1-specific inhibitor S63845 acts synergistically with venetoclax/ABT-199 to induce apoptosis in T-cell acute lymphoblastic leukemia cells. Leukemia (2018). doi: 10.1038/s41375-018-0201-2. [Epub ahead of print].
227. Luedtke DA, Niu X, Pan Y, Zhao J, Liu S, Edwards H, et al. Inhibition of Mcl-1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Signal Transduct Target Ther. (2017) 2:17012. doi: 10.1038/sigtrans.2017.12
228. Choudhary GS, Tat TT, Misra S, Hill BT, Smith MR, Almasan A, et al. Cyclin E/Cdk2-dependent phosphorylation of Mcl-1 determines its stability and cellular sensitivity to BH3 mimetics. Oncotarget (2015) 6:16912–25. doi: 10.18632/oncotarget.4857
229. Aw A, Brown JR. The potential combination of BCL-2 inhibitors and ibrutinib as frontline therapy in chronic lymphocytic leukemia. Leuk Lymphoma (2017) 58:2287–97. doi: 10.1080/10428194.2017.1312387
230. Patel VM, Balakrishnan K, Douglas M, Tibbitts T, Xu EY, Kutok JL, et al. Duvelisib treatment is associated with altered expression of apoptotic regulators that helps in sensitization of chronic lymphocytic leukemia cells to venetoclax (ABT-199). Leukemia (2017) 31:1872–81. doi: 10.1038/leu.2016.382
231. Bojarczuk K, Sasi BK, Gobessi S, Innocenti I, Pozzato G, Laurenti L, et al. BCR signaling inhibitors differ in their ability to overcome Mcl-1-mediated resistance of CLL B cells to ABT-199. Blood (2016) 127:3192–201. doi: 10.1182/blood-2015-10-675009
232. Bui MH, Lin X, Albert DH, Li L, Lam LT, Faivre EJ, et al. Preclinical characterization of BET family bromodomain inhibitor ABBV-075 suggests combination therapeutic strategies. Cancer Res. (2017) 77:2976–89. doi: 10.1158/0008-5472.CAN-16-1793
233. Pham LV, Huang S, Zhang H, Zhang J, Bell T, Zhou S, et al. Strategic therapeutic targeting to overcome venetoclax resistance in aggressive B-cell lymphomas. Clin Cancer Res. (2018) 24:3967–80. doi: 10.1158/1078-0432.CCR-17-3004
234. Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J. (2001) 20:2690–701. doi: 10.1093/emboj/20.11.2690
235. Akl H, Vandecaetsbeek I, Monaco G, Kauskot A, Luyten T, Welkenhuyzen K, et al. HA14-1, but not the BH3 mimetic ABT-737, causes Ca2+ dysregulation in platelets and human cell lines. Haematologica (2013) 98:e49–51. doi: 10.3324/haematol.2012.080598
236. Vervloessem T, Ivanova H, Luyten T, Parys JB, Bultynck G. The selective Bcl-2 inhibitor venetoclax, a BH3 mimetic, does not dysregulate intracellular Ca(2+) signaling. Biochim Biophys Acta Mol Cell Res. (2017) 1864:968–76. doi: 10.1016/j.bbamcr.2016.11.024
237. Szegezdi E, Macdonald DC, Ni Chonghaile T, Gupta S, Samali A. Bcl-2 family on guard at the ER. Am J Physiol Cell Physiol. (2009) 296:C941–53. doi: 10.1152/ajpcell.00612.2008
238. Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Lett. (2010) 584:2981–9. doi: 10.1016/j.febslet.2010.05.061
239. Germain M, Slack RS. MCL-1 regulates the balance between autophagy and apoptosis. Autophagy (2011) 7:549–51. doi: 10.4161/auto.7.5.15098
240. Germain M, Nguyen AP, Le Grand JN, Arbour N, Vanderluit JL, Park DS, et al. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J. (2011) 30:395–407. doi: 10.1038/emboj.2010.327
241. Verfaillie T, Salazar M, Velasco G, Agostinis P. Linking ER stress to autophagy: potential implications for cancer therapy. Int J Cell Biol. (2010) 2010:930509. doi: 10.1155/2010/930509
242. Malik SA, Orhon I, Morselli E, Criollo A, Shen S, Marino G, et al. BH3 mimetics activate multiple pro-autophagic pathways. Oncogene (2011) 30:3918–29. doi: 10.1038/onc.2011.104
243. Malik SA, Shen S, Marino G, BenYounes A, Maiuri MC, Kroemer G. BH3 mimetics reveal the network properties of autophagy-regulatory signaling cascades. Autophagy (2011) 7:914–6. doi: 10.4161/auto.7.8.15785
Keywords: BCL-2, BCL-W, B cell lymphoma, apoptosis, venetoclax, navitoclax, BH-3 mimetic, CLL
Citation: Adams CM, Clark-Garvey S, Porcu P and Eischen CM (2019) Targeting the Bcl-2 Family in B Cell Lymphoma. Front. Oncol. 8:636. doi: 10.3389/fonc.2018.00636
Received: 10 September 2018; Accepted: 05 December 2018;
Published: 08 January 2019.
Edited by:
Silvia Bea, Institut d'Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), SpainReviewed by:
Benjamin Bonavida, University of California, Los Angeles, United StatesAlberto Fabbri, Azienda Ospedaliera Universitaria Senese, Italy
Copyright © 2019 Adams, Clark-Garvey, Porcu and Eischen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christine M. Eischen, Y2hyaXN0aW5lLmVpc2NoZW5AamVmZmVyc29uLmVkdQ==