 Shiv K. Gupta1*
Shiv K. Gupta1* Emily J. Smith1
Emily J. Smith1 Ann C. Mladek1
Ann C. Mladek1 Shulan Tian2Paul A. Decker2
Shulan Tian2Paul A. Decker2 Sani H. Kizilbash3
Sani H. Kizilbash3 Gaspar J. Kitange1
Gaspar J. Kitange1 Jann N. Sarkaria1*
Jann N. Sarkaria1*- 1Departments of Radiation Oncology, Mayo Clinic, Rochester, MN, United States
- 2Division of Biomedical Statistics and Informatics, Department of Health Sciences Research, Mayo Clinic, Rochester, MN, United States
- 3Departments of Oncology, Mayo Clinic, Rochester, MN, United States
Prognosis of patients with glioblastoma (GBM) remains dismal despite maximal surgical resection followed by aggressive chemo-radiation therapy. Almost every GBM, regardless of genotype, relapses as aggressive recurrent disease. Sensitization of GBM cells to chemo-radiation is expected to extend survival of patients with GBM by enhancing treatment efficacy. The PARP family of enzymes has a pleiotropic role in DNA repair and metabolism and has emerged as an attractive target for sensitization of cancer cells to genotoxic therapies. However, despite promising results from a number of preclinical studies, progress of clinical trials involving PARP inhibitors (PARPI) has been slower in GBM as compared to other malignancies. Preclinical in vivo studies have uncovered limitations of PARPI-mediated targeting of base excision repair, considered to be the likely mechanism of sensitization for temozolomide (TMZ)-resistant GBM. Nevertheless, PARPI remain a promising sensitizing approach for at least a subset of GBM tumors that are inherently sensitive to TMZ. Our PDX preclinical trial has helped delineate MGMT promoter hyper-methylation as a biomarker of the PARPI veliparib-mediated sensitization. In clinical trials, MGMT promoter hyper-methylation now is being studied as a potential predictive biomarker not only for response to TMZ therapy alone, but also PARPI-mediated sensitization of TMZ therapy. Besides the combination approach being investigated, IDH1/2 mutant gliomas associated with 2-hydroxygluterate (2HG)-mediated homologous recombination (HR) defect may potentially benefit from PARPI monotherapy. In this article, we discuss existing results and provide additional data in support of potential alternative mechanisms of sensitization that would help identify potential biomarkers for PARPI-based therapeutic approaches to GBM.
Background
Glioblastoma (GBM) is a fatal disease with less than 2% of patients surviving 5 years after initial diagnosis and treatment (1, 2). GBM therapy, which includes aggressive surgical resection, high dose external beam radiation therapy (RT) and temozolomide (TMZ) chemotherapy, is associated with a median time to progression of approximately 6 months and a median overall survival of 15 months (3). Sensitizing strategies to enhance efficacy of radiation and chemotherapy may prolong patient survival. TMZ, used as standard of care for newly diagnosed GBM, is a mono-alkylating agent that induces cytotoxic lesions including N7-methylguanine (N7MeG), N3-methyladenine (N3MeA) and O6-methylguanine (O6MeG) (4, 5). N7MeG and N3MeA are repaired by base-excision repair (BER) and contribute minimally to overall cytotoxicity of TMZ, while O6MeG is repaired by O6-methylguanine-DNA-methyl transferase (MGMT), found suppressed by promoter methylation in ~40% of GBM tumors. Lack of MGMT expression results in persistent O6MeG lesions that trigger replicative stress and cytotoxicity via futile cycles of mismatch repair (MMR) (5, 6). The poly (ADP ribose) polymerase (PARP) family of enzymes coordinates the DNA damage response. Binding of PARP1 to nicked DNA provides the necessary scaffold that recruits BER components (7, 8). Therefore, PARP inhibitors (PARPI) were thought to potentiate TMZ by disrupting BER (9). Indeed, PARPI potentiate TMZ efficacy in numerous pre-clinical models (4, 9), providing a rationale for clinical development of PARPI to potentiate TMZ therapy in GBM. In addition to the established role of PARP in BER, destabilization of stalled replication forks by allosteric trapping of PARP also contributes toward mechanisms of TMZ sensitization by PARPI (10).
However, like other novel drugs for GBM, several promising PARPI agents have limited distribution across the blood-brain barrier (BBB) or demonstrate heterogeneous in vivo response (11). For example, talazoparib and rucaparib are potent PARPI that are substrates for the efflux transporters P-glycoprotein (PgP) and/or breast cancer resistance protein (BCRP) that are active in brain endothelial cells (12, 13). In keeping with poor brain penetration, these drugs have limited distribution and no appreciable TMZ sensitization in orthotopically implanted GBM patient-derived xenografts (PDXs). In contrast, the PARPI veliparib is brain penetrant and an effective TMZ-sensitizer in a subset of GBM PDX models (4, 14, 15). Based on previously published data and additional experimental results, the focus of this article is to explore potential biomarkers critical to a PARPI-based sensitization approach to GBM therapy.
Discordance Between in vitro Versus in vivo Preclinical Data
Numerous preclinical studies have investigated the combination of PARPI with RT, TMZ or RT/TMZ and other chemotherapy agents in glioma models (14, 16, 17). Models including established glioma cell lines (16, 18–20), zebrafish embryos (21), genetically engineered mouse models (GEMM) (22) and PDXs (14) have been used. While each of these models has helped to characterize PARPI combinations, discordance between in vitro vs. in vivo data needs to be considered when developing therapies based on preclinical studies. Specifically, the in vitro sensitizing effects of the PARPI veliparib were pronounced in TMZ-resistant models, while these models did not benefit from the combination in vivo. In contrast, in vivo sensitization by veliparib was pronounced in TMZ-sensitive models, although the in vitro sensitization was limited (4). This discordance is due to in vivo drug achievability, which was lower than concentrations required for DNA damage induction in resistant tumors (4). These results highlight the importance of using clinically relevant concentrations of both TMZ and PARPI for in vitro assays and raise the possibility that molecular mechanisms defined by using supratherapeutic drug concentrations may not be applicable to in vivo sensitization.
PDX models are translationally relevant because they preserve the genetic characteristics of the tumor, and orthotopically implanted PDXs represent tumor microenvironment and vascular structures found in human GBM (23–25). Furthermore, pharmacokinetic profiles of PARPI in murine models mimic drug exposures reported in human clinical trials (12, 18). GEMMs are ideal to study gliomagenesis; however, GEMMs cannot recapitulate genetic heterogeneity or epigenetic features, such as MGMT promoter methylation found in human GBM. Use of large panels of PDXs for drug evaluation may accurately model tumor heterogeneity and the variability in response. As reported previously, veliparib-mediated in vivo sensitization is associated with inherent TMZ sensitivity (4, 14). This concept was further tested in a preclinical PDX trial using orthotopic therapy models of 28 different GBM PDX lines with or without MGMT promoter methylation, a marker of TMZ sensitivity (15). In this study, profound survival extension with TMZ/veliparib over TMZ alone was observed in ~45% of PDX models with MGMT hyper-methylation, while MGMT unmethylated models had no meaningful survival benefit (15). This result helped delineate MGMT promoter methylation as a predictive biomarker for veliparib-mediated sensitization (15).
Mechanism of PARPI-Mediated Sensitization:
Understanding mechanisms of sensitization is important to delineate biomarkers and new therapeutic targets. Synthetic lethality of PARPI with HR is the hallmark of single-agent PARPI therapy in breast and ovarian cancers (26, 27). PARPI also potentiate efficacy of genotoxic agents, including DNA alkylating agents and RT (28). Mechanistically, enzymatic activation of PARP consumes NAD+ and generates poly-ADP-ribose (PAR) moieties to modify interacting proteins and itself via a phenomenon known as PARylation (29). PARP auto-PARylation at DNA lesions initiates recruitment of repair proteins, while also keeping PARP-DNA interactions unstable allowing repair machinery access to the lesion (7, 30). PARPI blocks auto-PARylation and prevents dissociation of PARP-DNA interactions, thereby trapping PARP at the damage site, leading to replicative stress and replication-associated double strand DNA breaks (10, 31).
Prior studies suggest that PARPI-mediated in vivo sensitization of TMZ depends on replicative stress caused by persistent O6MeG (4, 14, 15). The significance of PARP trapping to O6MeG-mediated replicative stress is unclear as PARP is known to engage at N7MeG and N3MeA lesions. PARPI-mediated BER inhibition and PARP trapping contribute more robustly at supratherapeutic drug concentrations used in vitro; whether PARPI concentrations achievable in vivo induce detectable PARP trapping remains to be seen (30, 32, 33). Furthermore, PARPI with high trapping capacity are not well tolerated in combination with TMZ (30), and dose-reduced regimens tested have not shown greater sensitization than veliparib, a weak trapping agent (12–15). In a head-to-head comparison of PARPI agents, the trapping capacity was found to be inversely correlated with in vivo efficacy (30), suggesting that the trapping ability of PARPI may not be fully exploited for TMZ sensitization. However, this can be important to PARPI monotherapy or combinations where higher doses of trapping agents can be safely administered. Robust in vitro radio-sensitizing effects of PARPI talazoparib used at clinically relevant concentrations have been reported (34). However, evaluation of radio-sensitizing effects of talazoparib in in vivo orthotopic GBM models will be important as talazoparib concentrations in intracranial tumors may not reach clinically relevant concentrations based on plasma level (12).
Veliparib-mediated in vivo sensitization is limited to a subset of tumors that are inherently sensitive to TMZ (4, 14), suggesting that N7MeG or N3MeA lesions may have little effect on in vivo sensitization (4, 12). Consistent with this idea, here we demonstrate that depletion of XRCC1 or MPG, the essential proteins in the BER pathway, had no further increase in sensitization at clinically relevant veliparib concentrations in U251TMZ cells (Figures 1A,B). However, knockdown (KD) of BRCA1 or RAD51 in U251TMZ cells increased sensitivity to veliparib or TMZ alone, but also led to robust TMZ sensitization (Figures 1A,B). Surprisingly, BRCA2 KD had no increase in sensitivity toward veliparib or TMZ; additionally, veliparib-mediated sensitization was modest in BRCA2 KD cells compared to that in BRCA1 or RAD51 KD cells (Figures 1A,B). The differential response among BRCA2 vs. BRCA1 or RAD51 KD cells was intriguing as HR efficiency was equally suppressed in BRCA1, BRCA2 or RAD51 KD cells (Figure 1C). BRCA1, BRCA2 and RAD51 have also been reported to regulate replication fork stability, a function considered unrelated to HR (35, 36). Thus, compromised fork protection by BRCA1 or RAD51 depletion can be a new mechanism of PARPI-mediated TMZ sensitization.
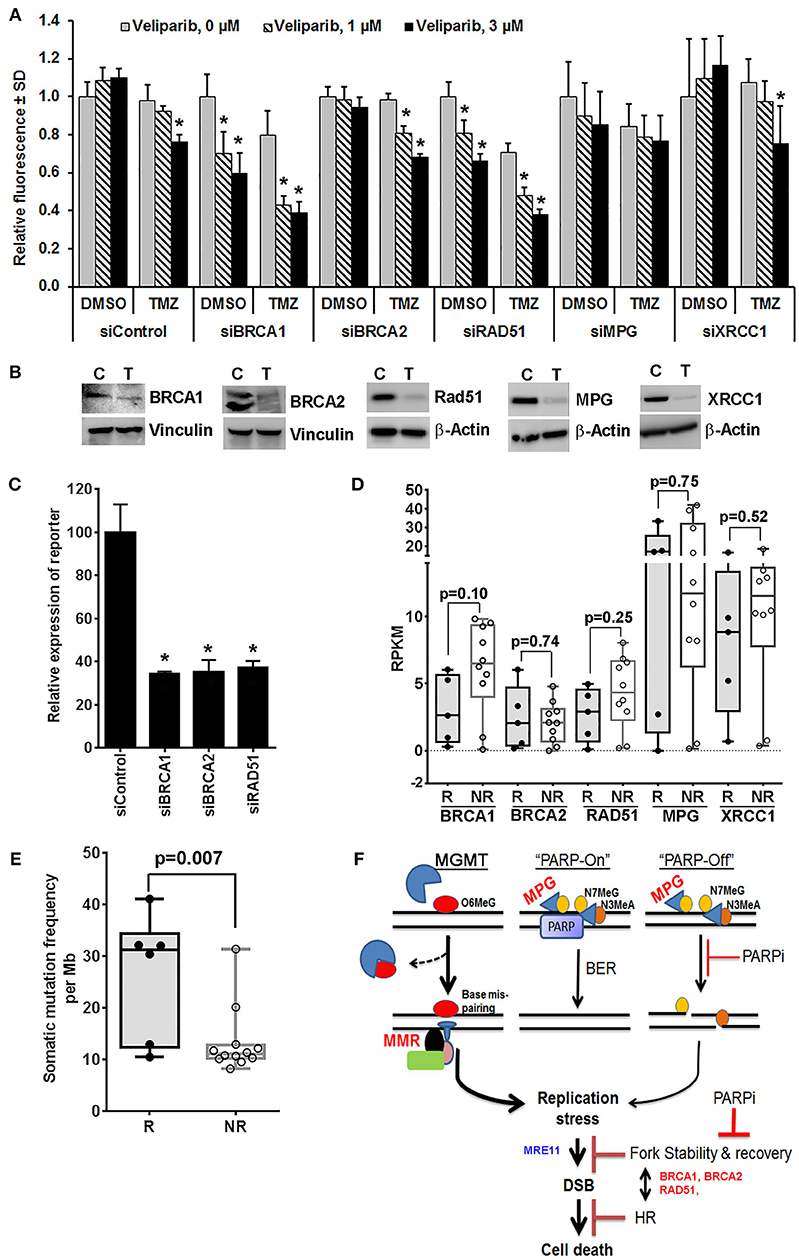
Figure 1. Mechanistic insights into veliparib-mediated sensitization of TMZ therapy in GBM cells (see Supplementary Material for Materials and Methods). (A) Effect of homologous recombination (HR) vs. base excision repair (BER) pathway disruption on veliparib-mediated sensitization in U251TMZ cells. Cells transfected with specified siRNA were seeded in 96 well plates (500 cells per well), treated with the vehicle or 30 μM TMZ ± 1 or 3 μM veliparib for 5 days and cell growth measured by CyQuant assay. Bar graphs demonstrate change in average fluorescence intensity relative to control, error bars represent standard deviation calculated from 3 replicates in a representative experiment, and *p < 0.05 compared to corresponding control. (B) Western blot analysis to determine level of knockdown for cells used in (A), lanes marked with T represent cells transfected with targeted siRNA and C represent cells transfected with control siRNA. (C) Bar graphs showing effects of BRCA1, BRCA2 or RAD51 knockdown on HR efficiency. U251TMZ-DRGFP cells were transfected with specific siRNA along with plasmid pCBASceI encoding I-SceI restriction enzyme and incubated for 72 h followed by quantification of GFP expressing population by FACS analysis. *p < 0.05 as compared to control. (D) Box plots showing expression levels for specified genes, RKPM values were extrapolated from RNA-Seq data of PDX lines differentially sensitized by veliparib in preclinical PDX trial. Data shown are for 5 of 6 responders (R) lines, which had significantly improved survival vs. 10 of 16 non-responder (NR) lines, which had no significant survival improvement with veliparib/TMZ therapy over TMZ alone in preclinical PDX trial reported previously. two tailed p-values reported were calculated by unpaired t-test. (E) Box plots showing mutation burden based on whole exome seq data available for 21 MGMT methylated PDX lines used in PDX pre-clinical trial and plotted grouped as TMZ/veliparib Responsive (R) vs. Non-responsive (NR) models. SNVs and INDELs across 346 genes involved in DNA damage recognition or repair were analyzed for mutation burden, two tailed p-values reported were calculated by unpaired t-test. (F) Hypothetical model of potential mechanism of the sensitizing effect of PARP inhibition on TMZ therapy in vivo. O6MeG, O6-methylguanine; N7MeG, N7-methylguanine; N3MeA, N3-methyladenine; MPG, methyl purine glycosylase; PARP, poly-ADP-ribose polymerase; PARPi, poly-ADP-ribose polymerase inhibitor; MMR, mismatch repair; DSB, double strand DNA breaks; MRE11, Meiotic Recombination 11 Homolog; BRCA1 and BRCA2, BReast CAncer genes 1 and 2; and HR, homologous recombination.
Available RNA-Seq data from the PDX lines, used in the previously reported preclinical trial (15), showed that among analyzed HR and BER pathway genes, the expression of BRCA1 was trending lower in all five TMZ/veliparib responsive lines as compared to 10 non-responsive GBM lines that were analyzed (p = 0.10, Figure 1D). Interestingly, the BRCA1 expression, found upregulated in GBM, appears to be a prognostic factor in a Rembrandt GBM patient data set (Supplementary Figure 1). Our limited RNA Seq results suggest that low BRCA1 expression could be useful to identify tumors likely to respond to PARPI-mediated sensitization. The mechanism of BRCA1 downregulation in responder PDXs is not yet clear, although the promoter hyper-methylation, microRNA or the epigenetic modifier RBBP4, have been previously reported to influence BRCA1 expression (37–40). Similarly, analysis of available whole exome-seq data for PDX lines used in our preclinical trial showed a significantly higher average mutation burden in responder lines than non-responder lines (Figure 1E), suggesting that the GBM tumors with genomic instability are likely to respond to PARPI-mediated sensitization. This idea that BRCA1 downregulation in responder PDXs correlates with increased mutation frequency will need further validation.
TMZ induces replicative stress via futile attempts of MMR at O6MeG:T mismatches, while PARPI may further enhance the stress by compromising stability of stalled replication forks (41, 42). Association of BRCA1 levels with TMZ/veliparib response in the PDX trial indicates that BRCA1 synthetic lethality with PARPI can be important to fork protection (43) in the context of TMZ/veliparib treatment. Understanding the relationship between PARP and other proteins involved in fork protection may reveal key determinants of PARPI-mediated sensitization. Figure 1F shows an overview of potential mechanisms of PARPI-mediated sensitization.
Efflux Liability and Delivery Across BBB: A key Determinant of in vivo Sensitizing Effects
Drug exclusion from the brain by the BBB undermines the efficacy of many CNS-directed pharmaceutical agents including PARPI (11, 44). The BBB is a complex neurovascular unit comprised of specialized brain capillary endothelium expressing ATP-binding cassette transporters. The distribution of contrast enhancement agents on magnetic resonance imaging (MRI) is commonly used to assess BBB integrity in gliomas. However, infiltrating GBM cells invade brain tissues beyond margins of contrast enhancement (45, 46). The invasive front of GBM tumors is not accessible to cyto-reductive surgery or chemotherapies that do not adequately penetrate the brain. BBB breakdown in GBM is regional and heterogeneous (44), and therefore drug distribution can be significantly lower at infiltrating edges as compared to the necrotic tumor core (44). Thus, delivery to infiltrating glioma cells is limited for many chemotherapy drugs in GBM (13, 47–49). Considerable effort has been made to understand the brain pharmacokinetics of PARPI, and several PARPI, especially the trapping agents, talazoparib and rucaparib, have efflux liabilities at the BBB and therefore lack sensitizing activity in orthotopic tumor models despite their excellent activity in heterotopic tumor models (13, 30). These findings are consistent with the notion that the delivery of targeted drugs into normal brain or orthotopically implanted tumors can model their efficacy in GBM (11, 49).
We have previously reported that the talazoparib concentration in a normal mouse brain (0.5 ng/g, or 1.3 nmol/L) after drug administration was lower than required for effective PARP inhibition in vitro. Comparing the pharmacokinetics of talazoparib to other PARPI in healthy rodents, the brain-to-plasma concentration ratio for talazoparib (0.02) was lower than that of rucaparib (0.11), which also lacks efficacy in orthotopic glioma models (13). Olaparib is another PARP trapping agent known to have efflux liability and restricted delivery across the BBB (50, 51). Although a phase I clinical trial in patients with recurrent GBM has shown that olaparib can reach the core and the margins of GBM tumors (50), this data has to be interpreted cautiously because GBM cells invade tissues beyond the margins defined by the MRI. Veliparib, on the other hand, has a much higher brain-to-plasma concentration ratio (0.47) than either talazoparib or rucaparib despite the efflux liability of veliparib to MDR1 and BCRP (15, 52). Furthermore, unlike talazoparib and rucaparib, veliparib sensitized orthotopic GBM models despite being significantly less potent in terms of PARP trapping (15). A comparison of the properties of drugs from the same class provides insight on the relative significance of variables such as drug potency, BBB penetrability, and efflux liability for efficacy in orthotopic glioma models. These considerations emphasize the importance of brain pharmacokinetics, drug tolerability, and efficacy evaluation in animal models for the successful design of novel therapies for GBM.
Clinical Trials of PARPI in GBM
PARPI have shown significant promise as a specific RT and/or TMZ-sensitizing strategy. Ever since the rucaparib/TMZ combination was found safe to administer in patients with solid tumors (53), several studies have been launched to assess the safety and efficacy of various PARPI in patients with GBM (Table 1). The majority of early clinical trials involved patients with recurrent GBM. However, recently launched trials have involved not only newly diagnosed patients, but have also stratified patients by MGMT promoter methylation status to enrich the patient population likely to benefit from the therapy (NCT02152982, PARADIGM-2, and NCT03150862). Phase I or phase I/II studies in patients with recurrent GBM are helpful in determining MTD and toxicity. For example, phase I trial NCT00770471 showed that combining veliparib with RT/TMZ is not adequately tolerated (54), and based on this data, later studies planned to evaluate veliparib in combination with RT alone and/or velipaib combined with adjuvant TMZ (NCT03581292, NCT02152982), thus avoiding toxicities reported with the triple combination. Although triple combination of veliparib has been excluded from further development, other PARPI agents in combination with RT/TMZ continue to be tested (NCT03212742, NCT03150862 and PARADIGM-2).
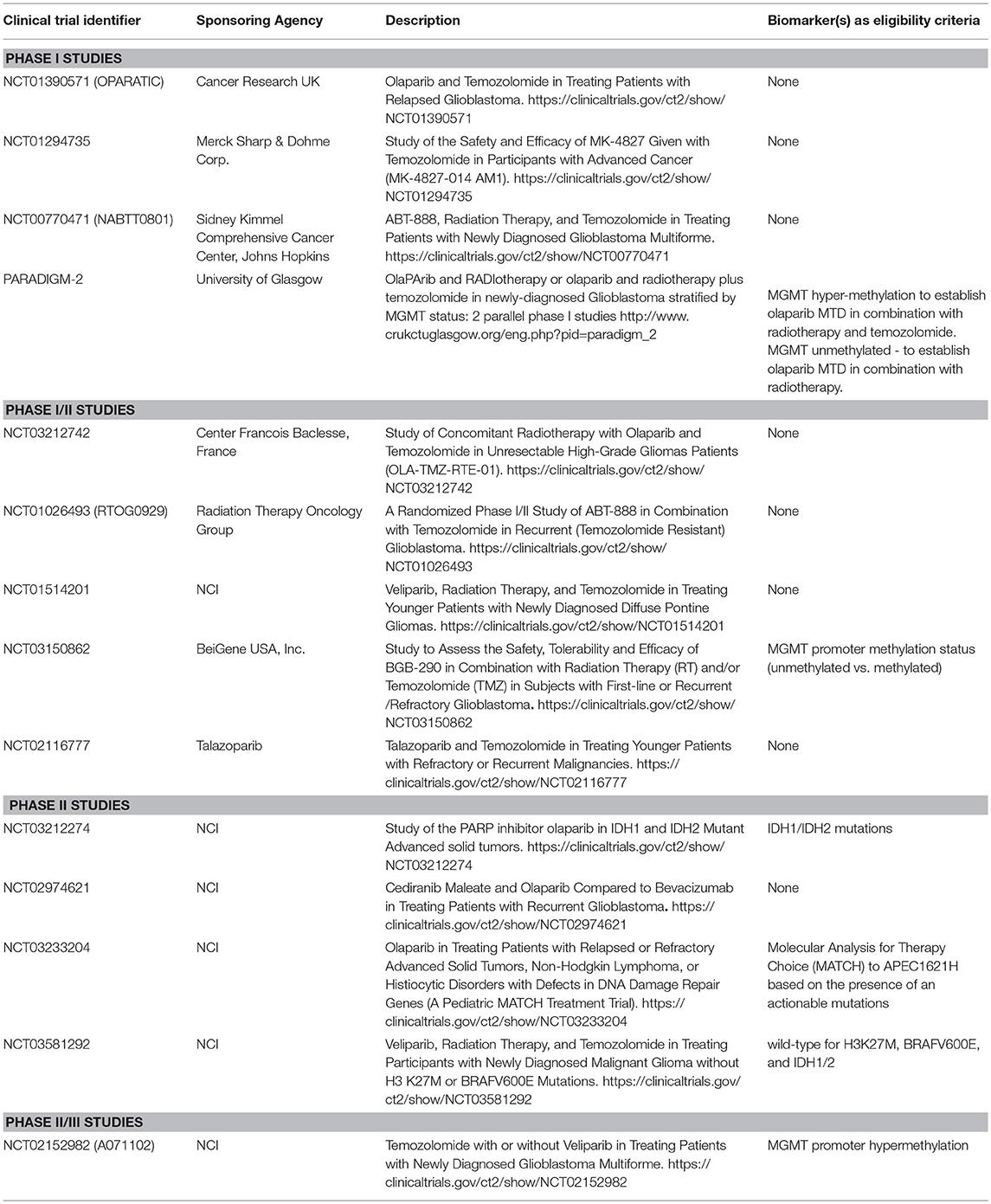
Table 1. Clinical trials of various PARP inhibitors in patients with low grade gliomas and GBM.
Another important phase I trial has been OPARATIC (NCT01390571), demonstrating that olaparib reaches tumor core and the margins in patients with recurrent GBM, and that the olaparib combined with low dose extended TMZ is well tolerated (50). This data has generated enthusiasm for the olaparib combinations in GBM. A second phase I trial (PARADIGM-2) stratifies newly diagnosed GBM based on MGMT hypermethylation to receive olaparib/TMZ/radiation (MGMT methylated) or olaparib/radiation (MGMT unmethylated) (55). Besides these clinical trials evaluating olaparib combinations, phase II studies NCT03233204 and NCT03212274 aim to investigate single-agent activity of olaparib in pediatric patients with mutated or altered DNA damage repair genes (NCT03233204) or in patients with IDH1/2-mutant tumors (NCT03212274). A phase-II study plans to compare the antitumor activity of olaparib combined with cediranib, an inhibitor of VEGF receptor, vs. bevacizumab monotherapy in patients with recurrent GBM (NCT02974621). An ongoing phase I-II study is investigating PARPI talazoparib combined with TMZ (NCT02116777). Children with refractory or recurrent solid tumors on this trial will receive talazoparib orally either once or twice daily on days 1–6 and TMZ on days 2–6, with therapy repeating every 28 days for up to 24 cycles until disease progression or unacceptable toxicity occurs. Due to limited distribution into the CNS in preclinical mouse models for several of these PARPI agents, concerns remain about the effectiveness of these therapies in gliomas that all have at least a partially intact BBB (56).
Delineation of Predictive Biomarkers to PARPI-Mediated Sensitization
HR deficiency (also known as BRCAness) and PARP expression are predictive biomarkers for PARPI efficacy (57–59). However, unlike breast and ovarian cancers, BRCAness is uncommon in GBM. Although homozygous PTEN deletion, mutant STAG2, or IDH-mutations found in GBM have been reported to disrupt the HR pathway, these studies were performed in established cell lines (60–62). In a PDX preclinical trial, PTEN alterations had no correlation with the TMZ-sensitizing effects of veliparib (15). Similarly, veliparib had neither single agent activity nor any significant sensitization in two different IDH1-mutant GBM PDX models (data not shown). These results suggest that HR deficiency, a conventional marker of PARPI sensitivity, may not be a robust biomarker for veliparib-mediated in vivo sensitization in GBM.
As reported previously, PARPI-mediated in vivo sensitization is associated with inherent TMZ sensitivity (4, 14), whereas MGMT hypermethylation is a marker of TMZ sensitivity (5, 63). We assessed the utility of MGMT methylation status as a biomarker of veliparib-mediated sensitization in a PDX preclinical trial involving 28 GBM PDX models (15). In this preclinical trial, PDX lines with unmethylated MGMT had no survival benefit with TMZ/veliparib over TMZ alone, while profound survival extension with TMZ/veliparib was observed in ~45% of PDX lines with MGMT promoter hyper-methylation (15). Based on this result, the A071102 clinical trial uses MGMT promoter methylation as selection criterion for a randomized clinical trial of adjuvant TMZ combined with veliparib or placebo (NCT02152982). MGMT promoter methylation status has been integrated in clinical trial designs for at least two other studies testing TMZ/PARPI in GBM (PARADIGM-2, NCT03150862). However, as only a fraction of patients with MGMT hyper-methylation expected to benefit from TMZ/PARPI therapy, refinement of predictive biomarkers is necessary to guide optimal use of PARPI in GBM.
Lack of Schlafen Family Member 11 (SLFN11) is known to confer resistance to DNA damaging agents (64, 65). Mechanistically, SLFN11 interacts with replication protein A (RPA), destabilizes RPA-ssDNA complexes and inhibits HR (66). Like MGMT, SLFN11 expression is epigenetically suppressed through promoter hypermethylation in nearly 50% of solid tumors (67). In a recent study, SLFN11 expression correlated with in vivo tumor response to talazoparib in patient-derived xenograft (PDX) models of small cell lung cancer (SCLC) (68). Interestingly, in this study, response to TMZ/Talazoparib had no clear association with SLFN11. However, a phase II clinical trial testing TMZ plus veliparib (or placebo) in patients with SCLC showed that SLFN11-positive tumors, as defined by immunohistochemistry (n = 12) had improved progression-free and overall survival relative to patients with SLFN11-negative tumors (69). Based on this promising data in SCLC, SLFN11 is a potential biomarker to be examined in GBM.
IDH1 mutations are oncogenic mutations found in 74% of low-grade gliomas and 9% of GBM (70). Mechanistically, 2-HG produced by the neomorphic mutant-IDH1 enzyme inhibits α-ketoglutarate (αKG)-dependent ALKBH2-3 enzymes and prevents repair of endogenous DNA damage, rendering vulnerability to alkylation therapies (71). A recent study by Salkowski et al. suggests that 2-HG can disrupt HR activity and sensitize cells to PARPI (62). This finding was further confirmed in GBM cell lines modified to express mutant IDH1 constructs (72). However, exogenously expressed mutant IDH1 may not recapitulate all the genetic and phenotypic changes that occur in IDH1-mutant gliomas. NAD+ deficiency is one of the striking features of IDH1-mutant glioma cells, which are highly vulnerable to NAD+ depletion via TMZ treatment or NAMPT inhibition (73). Since NAD+ is consumed by PARP activation during genotoxic therapy, PARPI can be counterproductive. This hypothesis was proven by Tateishi et al. whereby TMZ/olaparib had lesser cytotoxicity than TMZ alone in glioma cells in vitro (74, 75). Comprehensive analysis of metabolic vulnerability is necessary to understand conflicting results of PARPI sensitivity in IDH1 mutant gliomas.
Conclusions and Future Directions
Increased DNA repair compromises therapeutic efficacy of anti-cancer genotoxic therapies (76). Based on the pleotropic role of PARP in DNA repair, there is immense interest in clinical development of PARPI as cancer monotherapy (for HR defective tumors) and as a chemo-radiation sensitizer (76). Preclinical studies using orthotopic GBM models suggest that the efficacy of PARPI in GBM may be limited due to restricted delivery across the BBB and heterogeneous tumor response (4, 12, 13, 15). Pronounced TMZ sensitization by the brain penetrant PARPI veliparib was observed in a subset of tumors inherently sensitive to TMZ, while TMZ-resistant tumors lacked in vivo sensitization, suggesting that potentiation of replication stress rather than BER inhibition or PARP trapping is a key mechanism involved in in vivo sensitization (4, 14, 15). Based on these findings, MGMT promoter methylation was delineated as a predictive biomarker and is being increasingly used in PARPI clinical trials in GBM. However, as only a fraction of MGMT methylated tumors responded in preclinical trial, discovery of precise biomarkers is necessary.
One particular area of interest is to dissect the role of PARP in replication stress resolution. Whereas TMZ induces replicative stress via repetitive MMR at O6MeG:T sites, PARPI may potentially compromise stability of stalled replication forks (5, 7, 77). However, compromised fork protection is a complex biological process, where PARPI may act more robustly in context of vulnerabilities such as loss of BRCA1 or other factors involved in fork protection. Identification of critical regulators of fork protection in context the of TMZ/PARPI combinations will help identify new biomarkers. Endogenous replicative stress in cells with compromised fork protection may result in genomic instability and higher mutation burden. Analysis of mutation burden in the context of TMZ/PARPI therapy can be another crucial marker of PARPI-mediated sensitization. Ongoing PARPI trials are poised to generate data and biospecimens that will allow correlative analysis of putative biomarkers identified through preclinical studies in GBM models.
Author Contributions
SG and JS conception and design. SG, ES, AM, ST, and PD acquisition of data. SG, JS, ST, and GK analysis and interpretation of data. SG, JS, SK, and GK writing, review, and/or revision of the manuscript. SG and JS study supervision.
Funding
The study was supported by the NIH grants R03 CA201612 to SG, R01 CA176830 to JS, and P50 CA108961. SG is partly supported through Eagles Cancer Funds, Mayo Clinic Cancer Center.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2018.00670/full#supplementary-material
References
1. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. (2009) 10:459–66. doi: 10.1016/S1470-2045(09)70025-7
2. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. (2005) 352:987–96. doi: 10.1056/NEJMoa043330
3. Gilbert MR. Improving patient outcomes in glioblastoma through integration of recent data: introduction. Semin Oncol. (2011) 38:A1–6, S1–34. doi: 10.1053/j.seminoncol.2011.09.006
4. Gupta SK, Mladek AC, Carlson BL, Boakye-Agyeman F, Bakken KK, Kizilbash SH, et al. Discordant in vitro and in vivo chemopotentiating effects of the PARP inhibitor veliparib in temozolomide-sensitive versus -resistant glioblastoma multiforme xenografts. Clin Cancer Res. (2014) 20:3730–41. doi: 10.1158/1078-0432.CCR-13-3446
5. Sarkaria JN, Kitange GJ, James CD, Plummer R, Calvert H, Weller M, et al. Mechanisms of chemoresistance to alkylating agents in malignant glioma. Clin Cancer Res. (2008) 14:2900–8. doi: 10.1158/1078-0432.CCR-07-1719
6. D'Atri S, Tentori L, Lacal PM, Graziani G, Pagani E, Benincasa E, et al. Involvement of the mismatch repair system in temozolomide-induced apoptosis. Mol Pharmacol. (1998) 54:334–41. doi: 10.1124/mol.54.2.334
7. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. (2008) 8:193–204. doi: 10.1038/nrc2342
8. Liu X, Shi Y, Guan R, Donawho C, Luo Y, Palma J, et al. Potentiation of temozolomide cytotoxicity by poly(ADP)ribose polymerase inhibitor ABT-888 requires a conversion of single-stranded DNA damages to double-stranded DNA breaks. Mol Cancer Res. (2008) 6:1621–9. doi: 10.1158/1541-7786.MCR-08-0240
9. Calabrese CR, Almassy R, Barton S, Batey MA, Calvert AH, Canan-Koch S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst. (2004) 96:56–67. doi: 10.1093/jnci/djh005
10. Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. (2012) 72:5588–99. doi: 10.1158/0008-5472.CAN-12-2753
11. Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, et al. Is the blood-brain barrier really disrupted in all glioblastomas? a critical assessment of existing clinical data. Neuro Oncol. (2018) 20:184–91. doi: 10.1093/neuonc/nox175
12. Kizilbash SH, Gupta SK, Chang K, Kawashima R, Parrish KE, Carlson BL, et al. Restricted delivery of talazoparib across the blood-brain barrier limits the sensitizing effects of PARP inhibition on temozolomide therapy in glioblastoma. Mol Cancer Ther. (2017) 16:2735–46. doi: 10.1158/1535-7163.MCT-17-0365
13. Parrish KE, Cen L, Murray J, Calligaris D, Kizilbash S, Mittapalli RK, et al. Efficacy of PARP inhibitor rucaparib in orthotopic glioblastoma xenografts is limited by ineffective drug penetration into the central nervous system. Mol cancer ther. (2015) 14:2735–43. doi: 10.1158/1535-7163.MCT-15-0553
14. Clarke MJ, Mulligan EA, Grogan PT, Mladek AC, Carlson BL, Schroeder MA, et al. Effective sensitization of temozolomide by ABT-888 is lost with development of temozolomide resistance in glioblastoma xenograft lines. Mol Cancer Ther. (2009) 8:407–14. doi: 10.1158/1535-7163.MCT-08-0854
15. Gupta SK, Kizilbash SH, Carlson BL, Mladek AC, Boakye-Agyeman F, Bakken KK, et al. Delineation of MGMT hypermethylation as a biomarker for veliparib-mediated temozolomide-sensitizing therapy of glioblastoma. J Natl Cancer Inst. (2016) 108:1–10. doi: 10.1093/jnci/djv369
16. Barazzuol L, Jena R, Burnet NG, Meira LB, Jeynes JC, Kirkby KJ, et al. Evaluation of poly (ADP-ribose) polymerase inhibitor ABT-888 combined with radiotherapy and temozolomide in glioblastoma. Radiat Oncol. (2013) 8:65. doi: 10.1186/1748-717X-8-65
17. Tentori L, Ricci-Vitiani L, Muzi A, Ciccarone F, Pelacchi F, Calabrese R, et al. Pharmacological inhibition of poly(ADP-ribose) polymerase-1 modulates resistance of human glioblastoma stem cells to temozolomide. BMC Cancer (2014) 14:151. doi: 10.1186/1471-2407-14-151
18. Donawho CK, Luo Y, Penning TD, Bauch JL, Bouska JJ, Bontcheva-Diaz VD, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. (2007) 13:2728–37. doi: 10.1158/1078-0432.CCR-06-3039
19. Palma JP, Rodriguez LE, Bontcheva-Diaz VD, Bouska JJ, Bukofzer G, Colon-Lopez M, et al. The PARP inhibitor, ABT-888 potentiates temozolomide: correlation with drug levels and reduction in PARP activity in vivo. Anticancer Res. (2008) 28:2625–35.
20. Palma JP, Wang YC, Rodriguez LE, Montgomery D, Ellis PA, Bukofzer G, et al. ABT-888 confers broad in vivo activity in combination with temozolomide in diverse tumors. Clin Cancer Res. (2009) 15:7277–90. doi: 10.1158/1078-0432.CCR-09-1245
21. Erice O, Smith MP, White R, Goicoechea I, Barriuso J, Jones C, et al. MGMT Expression predicts PARP-mediated resistance to temozolomide. Mol Cancer Ther. (2015) 14:1236–46. doi: 10.1158/1535-7163.MCT-14-0810
22. Lin F, de Gooijer MC, Roig EM, Buil LC, Christner SM, Beumer JH, et al. ABCB1, ABCG2, and PTEN determine the response of glioblastoma to temozolomide and ABT-888 therapy. Clin Cancer Res. (2014) 20:2703–13. doi: 10.1158/1078-0432.CCR-14-0084
23. Carlson BL, Pokorny JL, Schroeder MA, Sarkaria JN. Establishment, maintenance and in vitro and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery. Curr Protoc Pharmacol. (2011) 52:14.16.1–23. doi: 10.1002/0471141755.ph1416s52.
24. Joo KM, Kim J, Jin J, Kim M, Seol HJ, Muradov J, et al. Patient-specific orthotopic glioblastoma xenograft models recapitulate the histopathology and biology of human glioblastomas in situ. Cell Rep. (2013) 3:260–73. doi: 10.1016/j.celrep.2012.12.013
25. Xu Z, Kader M, Sen R, Placantonakis DG. Orthotopic patient-derived glioblastoma xenografts in mice. methods Mol Biol. (2018) 1741:183–90. doi: 10.1007/978-1-4939-7659-1_14
26. Boukerroucha M, Josse C, Segers K, El-Guendi S, Freres P, Jerusalem G, et al. BRCA1 germline mutation and glioblastoma development: report of cases. BMC Cancer (2015) 15:181. doi: 10.1186/s12885-015-1205-1
27. Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science (2017) 355:1152–8. doi: 10.1126/science.aam7344
28. Javle M, Curtin NJ. The role of PARP in DNA repair and its therapeutic exploitation. Br J Cancer (2011) 105:1114–22. doi: 10.1038/bjc.2011.382
29. Kim MY, Zhang T, Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: 'PAR-laying' NAD+ into a nuclear signal. Genes Dev. (2005) 19:1951–67. doi: 10.1101/gad.1331805
30. Hopkins TA, Shi Y, Rodriguez LE, Solomon LR, Donawho CK, Digiammarino EL, et al. Mechanistic dissection of PARP1 trapping and the impact on in vivo tolerability and efficacy of PARP inhibitors. Mol Cancer Res. (2015) 13:1465–77. doi: 10.1158/1541-7786.MCR-15-0191-T
31. Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol cancer ther. (2014) 13:433–43. doi: 10.1158/1535-7163.MCT-13-0803
32. Gill SJ, Travers J, Pshenichnaya I, Kogera FA, Barthorpe S, Mironenko T, et al. Combinations of PARP inhibitors with temozolomide drive PARP1 trapping and apoptosis in ewing's sarcoma. PLoS ONE (2015) 10:e0140988. doi: 10.1371/journal.pone.0140988
33. Murai J, Zhang Y, Morris J, Ji J, Takeda S, Doroshow JH, et al. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J Pharmacol Exp Ther. (2014) 349:408–16. doi: 10.1124/jpet.113.210146
34. Lesueur P, Chevalier F, El-Habr EA, Junier MP, Chneiweiss H, Castera L, et al. Radiosensitization effect of talazoparib, a parp inhibitor, on glioblastoma stem cells exposed to low and high linear energy transfer radiation. Sci Rep. (2018) 8:3664. doi: 10.1038/s41598-018-22022-4
35. Schlacher K. PARPi focus the spotlight on replication fork protection in cancer. Nat Cell Biol. (2017) 19:1309–10. doi: 10.1038/ncb3638
36. Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell (2012) 22:106–16. doi: 10.1016/j.ccr.2012.05.015
37. Chang S, Sharan SK. BRCA1 and microRNAs: emerging networks and potential therapeutic targets. Mol Cells (2012) 34:425–32. doi: 10.1007/s10059-012-0118-y
38. Daniels SL, Burghel GJ, Chambers P, Al-Baba S, Connley DD, Brock IW, et al. Levels of DNA methylation vary at CpG sites across the BRCA1 promoter, and differ according to triple negative and “BRCA-Like” status, in both blood and tumour DNA. PLoS ONE. (2016) 11:e0160174. doi: 10.1371/journal.pone.0160174
39. Kitange GJ, Mladek AC, Schroeder MA, Pokorny JC, Carlson BL, Zhang Y, et al. Retinoblastoma binding protein 4 modulates temozolomide sensitivity in glioblastoma by regulating DNA repair proteins. Cell Rep. (2016) 14:2587–98. doi: 10.1016/j.celrep.2016.02.045
40. Weisenberger DJ. Characterizing DNA methylation alterations from the cancer genome atlas. J Clin Invest. (2014) 124:17–23. doi: 10.1172/JCI69740
41. Quinet A, Lemacon D, Vindigni A. Replication fork reversal: players and guardians. Mol Cell (2017) 68:830–3. doi: 10.1016/j.molcel.2017.11.022
42. Ying S, Hamdy FC, Helleday T. Mre11-dependent degradation of stalled dna replication forks is prevented by BRCA2 and PARP1. Cancer Res. (2012) 72:2814–21. doi: 10.1158/0008-5472.CAN-11-3417
43. Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, et al. Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol Cell (2017) 68:414–30 e8. doi: 10.1016/j.molcel.2017.09.036
44. Agarwal S, Mittapalli RK, Zellmer DM, Gallardo JL, Donelson R, Seiler C, et al. Active efflux of dasatinib from the brain limits efficacy against murine glioblastoma: broad implications for the clinical use of molecularly targeted agents. Mol cancer ther. (2012) 11:2183–92. doi: 10.1158/1535-7163.MCT-12-0552
45. Earnest Ft, Kelly PJ, Scheithauer BW, Kall BA, Cascino TL, Ehman RL, et al. Cerebral astrocytomas: histopathologic correlation of MR and CT contrast enhancement with stereotactic biopsy. Radiology (1988) 166:823–7. doi: 10.1148/radiology.166.3.2829270
46. Pafundi DH, Laack NN, Youland RS, Parney IF, Lowe VJ, Giannini C, et al. Biopsy validation of 18F-DOPA PET and biodistribution in gliomas for neurosurgical planning and radiotherapy target delineation: results of a prospective pilot study. Neuro Oncol. (2013) 15:1058–67. doi: 10.1093/neuonc/not002
47. Becker CM, Oberoi RK, McFarren SJ, Muldoon DM, Pafundi DH, Pokorny JL, et al. Decreased affinity for efflux transporters increases brain penetrance and molecular targeting of a PI3K/mTOR inhibitor in a mouse model of glioblastoma. Neuro Oncol. (2015) 17:1210–9. doi: 10.1093/neuonc/nov081
48. Parrish KE, Pokorny J, Mittapalli RK, Bakken K, Sarkaria JN, Elmquist WF. Efflux transporters at the blood-brain barrier limit delivery and efficacy of cyclin-dependent kinase 4/6 inhibitor palbociclib (PD-0332991) in an orthotopic brain tumor model. J Pharmacol Exp Ther. (2015) 355:264–71. doi: 10.1124/jpet.115.228213
49. Pokorny JL, Calligaris D, Gupta SK, Iyekegbe DO Jr, Mueller D, Bakken KK, et al. the efficacy of the wee1 inhibitor MK-1775 combined with temozolomide is limited by heterogeneous distribution across the blood-brain barrier in glioblastoma. Clin cancer res. (2015) 21:1916–24. doi: 10.1158/1078-0432.CCR-14-2588
50. Halford SER, Cruickshank G, Dunn L, Erridge S, Godfrey L, Herbert C, et al. Results of the OPARATIC trial: a phase i dose escalation study of olaparib in combination with temozolomide (TMZ) in patients with relapsed glioblastoma (GBM). J Clin Oncol. (2017) 35:2022. doi: 10.1200/JCO.2017.35.15_suppl.2022
51. Vaidyanathan A, Sawers L, Gannon A-L, Chakravarty P, Scott AL, Bray SE, et al. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br J Cancer (2016) 115:431. doi: 10.1038/bjc.2016.203
52. Li X, Delzer J, Voorman R, de Morais SM, Lao Y. Disposition and drug-drug interaction potential of veliparib (ABT-888), a novel and potent inhibitor of poly(ADP-ribose) polymerase. Drug Metab Dispos. (2011) 39:1161–9. doi: 10.1124/dmd.110.037820
53. Plummer R, Jones C, Middleton M, Wilson R, Evans J, Olsen A, et al. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. (2008) 14:7917–23. doi: 10.1158/1078-0432.CCR-08-1223
54. Kleinberg L, Supko JG, Mikkelsen T, Blakeley JON, Stevens G, Ye X, et al. Phase I adult brain tumor consortium (ABTC) trial of ABT-888 (veliparib), temozolomide (TMZ), and radiotherapy (RT) for newly diagnosed glioblastoma multiforme (GBM) including pharmacokinetic (PK) data. J Clin Oncol. (2013) 31:2065. doi: 10.1200/jco.2013.31.15
55. Fulton B, Short SC, James A, Nowicki S, McBain C, Jefferies S, et al. PARADIGM-2: two parallel phase I studies of olaparib and radiotherapy or olaparib and radiotherapy plus temozolomide in patients with newly diagnosed glioblastoma, with treatment stratified by MGMT status. Clin Transl Radiat Oncol. (2018) 8:12–6. doi: 10.1016/j.ctro.2017.11.003
56. Oberoi RK, Parrish KE, Sio TT, Mittapalli RK, Elmquist WF, Sarkaria JN. Strategies to improve delivery of anticancer drugs across the blood-brain barrier to treat glioblastoma. Neuro Oncol. (2016) 18:27–36. doi: 10.1093/neuonc/nov164
57. Peng G, Chun-Jen Lin C, Mo W, Dai H, Park YY, Kim SM, et al. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat Commun. (2014) 5:3361. doi: 10.1038/ncomms4361
58. Turner NC, Ashworth A. Biomarkers of PARP inhibitor sensitivity. Breast Cancer Res Treat. (2011) 127:283–6. doi: 10.1007/s10549-011-1375-8
59. Watkins JA, Irshad S, Grigoriadis A, Tutt AN. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. (2014) 16:211. doi: 10.1186/bcr3670
60. Bailey ML, O'Neil NJ, van Pel DM, Solomon DA, Waldman T, Hieter P. Glioblastoma cells containing mutations in the cohesin component STAG2 are sensitive to PARP inhibition. Mol Cancer Ther. (2014) 13:724–32. doi: 10.1158/1535-7163.MCT-13-0749
61. McEllin B, Camacho CV, Mukherjee B, Hahm B, Tomimatsu N, Bachoo RM, et al. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res. (2010) 70:5457–64. doi: 10.1158/0008-5472.CAN-09-4295
62. Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med. (2017) 9:eaal2463. doi: 10.1126/scitranslmed.aal2463
63. Kitange GJ, Carlson BL, Schroeder MA, Grogan PT, Lamont JD, Decker PA, et al. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro Oncol. (2009) 11:281–91. doi: 10.1215/15228517-2008-090
64. Tang SW, Bilke S, Cao L, Murai J, Sousa FG, Yamade M, et al. SLFN11 Is a transcriptional target of EWS-FLI1 and a determinant of drug response in ewing sarcoma. Clin Cancer Res. (2015) 21:4184–93. doi: 10.1158/1078-0432.CCR-14-2112
65. Tian L, Song S, Liu X, Wang Y, Xu X, Hu Y, et al. Schlafen-11 sensitizes colorectal carcinoma cells to irinotecan. Anticancer Drugs (2014) 25:1175–81. doi: 10.1097/CAD.0000000000000151
66. Mu Y, Lou J, Srivastava M, Zhao B, Feng XH, Liu T, et al. SLFN11 inhibits checkpoint maintenance and homologous recombination repair. EMBO Rep. (2016) 17:94–109. doi: 10.15252/embr.201540964
67. Nogales V, Reinhold WC, Varma S, Martinez-Cardus A, Moutinho C, Moran S, et al. Epigenetic inactivation of the putative DNA/RNA helicase SLFN11 in human cancer confers resistance to platinum drugs. Oncotarget (2016) 7:3084–97. doi: 10.18632/oncotarget.6413
68. Lok BH, Gardner EE, Schneeberger VE, Ni A, Desmeules P, Rekhtman N, et al. PARP inhibitor activity correlates with SLFN11 expression and demonstrates synergy with temozolomide in small cell lung cancer. Clin Cancer Res. (2017) 23:523–35. doi: 10.1158/1078-0432.CCR-16-1040
69. Pietanza MC, Waqar SN, Krug LM, Dowlati A, Hann CL, Chiappori A, et al. Randomized, double-blind, phase II study of temozolomide in combination with either veliparib or placebo in patients with relapsed-sensitive or refractory small-cell lung cancer. J Clin Oncol. (2018) 36:2386–94. doi: 10.1200/JCO.2018.77.7672
70. Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep. (2013) 13:345. doi: 10.1007/s11910-013-0345-4
71. Johannessen TC, Prestegarden L, Grudic A, Hegi ME, Tysnes BB, Bjerkvig R. The DNA repair protein ALKBH2 mediates temozolomide resistance in human glioblastoma cells. Neuro Oncol. (2013) 15:269–78. doi: 10.1093/neuonc/nos301
72. Lu Y, Kwintkiewicz J, Liu Y, Tech K, Frady LN, Su YT, et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res. (2017) 77:1709–18. doi: 10.1158/0008-5472.CAN-16-2773
73. Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, et al. Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell (2015) 28:773–84. doi: 10.1016/j.ccell.2015.11.006
74. Molenaar RJ, Maciejewski JP, Wilmink JW, van Noorden CJF. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene (2018) 37:1949–60. doi: 10.1038/s41388-017-0077-z
75. Tateishi K, Higuchi F, Miller JJ, Koerner MVA, Lelic N, Shankar GM, et al. The alkylating chemotherapeutic temozolomide induces metabolic stress in IDH1-mutant cancers and potentiates NAD(+) depletion-mediated cytotoxicity. Cancer Res. (2017) 77:4102–15. doi: 10.1158/0008-5472.CAN-16-2263
76. Brown JS, O'Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. (2017) 7:20–37. doi: 10.1158/2159-8290.CD-16-0860
Keywords: PARP (poly(ADP-ribose) polymerase, chemo-radiation sensitivity, DNA Damage, replication stress, DNA repair activity
Citation: Gupta SK, Smith EJ, Mladek AC, Tian S, Decker PA, Kizilbash SH, Kitange GJ and Sarkaria JN (2019) PARP Inhibitors for Sensitization of Alkylation Chemotherapy in Glioblastoma: Impact of Blood-Brain Barrier and Molecular Heterogeneity. Front. Oncol. 8:670. doi: 10.3389/fonc.2018.00670
Received: 20 November 2018; Accepted: 19 December 2018;
Published: 22 January 2019.
Edited by:
Zhi Sheng, Virginia Tech, United StatesReviewed by:
Robin T. Varghese, Edward Via College of Osteopathic Medicine, United StatesThomas DAUBON, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2019 Gupta, Smith, Mladek, Tian, Decker, Kizilbash, Kitange and Sarkaria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shiv K. Gupta, Z3VwdGEuc2hpdkBtYXlvLmVkdQ==
Jann N. Sarkaria, c2Fya2FyaWEuamFubkBtYXlvLmVkdQ==