 Fernando Vieira Pericole1
Fernando Vieira Pericole1 Mariana Lazarini1,2
Mariana Lazarini1,2 Luciana Bueno de Paiva1
Luciana Bueno de Paiva1 Adriana da Silva Santos Duarte1Karla Priscila Vieira Ferro1
Adriana da Silva Santos Duarte1Karla Priscila Vieira Ferro1 Fernanda Soares Niemann1
Fernanda Soares Niemann1 Fernanda Marconi Roversi1,3
Fernanda Marconi Roversi1,3 Sara Teresinha Olalla Saad1*
Sara Teresinha Olalla Saad1*- 1Hematology and Transfusion Medicine Center, Instituto Nacional de Ciência e Tecnologia do Sangue, University of Campinas, Hemocentro-Unicamp, São Paulo, Brazil
- 2Department of Pharmaceutical Sciences, Federal University of São Paulo, São Paulo, Brazil
- 3Universidade São Francisco (USF), Bragança Paulista, São Paulo, Brazil
Myelodysplastic syndromes (MDS) are clonal hematopoietic stem cell-based disorders characterized by ineffective hematopoiesis, increased genomic instability and a tendency to progress toward acute myeloid leukemia (AML). MDS and AML cells present genetic and epigenetic abnormalities and, due to the heterogeneity of these molecular alterations, the current treatment options remain unsatisfactory. Hypomethylating agents (HMA), especially azacitidine, are the mainstay of treatment for high-risk MDS patients and HMA are used in treating elderly AML. The aim of this study was to investigate the potential role of the epigenetic reader bromodomain-containing protein-4 (BRD4) in MDS and AML patients. We identified the upregulation of the short variant BRD4 in MDS and AML patients, which was associated with a worse outcome of MDS. Furthermore, the inhibition of BRD4 in vitro with JQ1 or shRNA induced leukemia cell apoptosis, especially when combined to azacitidine, and triggered the activation of the DNA damage response pathway. JQ1 and AZD6738 (a specific ATR inhibitor) also synergized to induce apoptosis in leukemia cells. Our results indicate that the BRD4-dependent transcriptional program is a defective pathway in MDS and AML pathogenesis and its inhibition induces apoptosis of leukemia cells, which is enhanced in combination with HMA or an ATR inhibitor.
Introduction
Myelodysplastic syndromes (MDS) encompass a heterogeneous group of disorders characterized by molecular alterations in hematopoietic stem cells, leading to ineffective hematopoiesis and risk of acute leukemia. Progression to acute myeloid leukemia (AML) occurs in one third of the patients, and it is believed to be the result of increased genomic instability (1). The disease predominantly affects older patients, with a median age of around 70 years and annual incidence in the range of 4–8 cases/100,000 individuals (2). Elderly AML (>65 years) presents a median survival of just 5–6 months (3). MDS and AML cells exhibit aberrant methylation in promoter regions of tumor-suppressor genes, which has been tracked to the most immature cells (4, 5) and is associated to gene mutations involving epigenetic modifiers (6, 7). Increased DNA damage and alterations in the DNA damage response pathway (DDR) are critical features of gene instability that are implicated in the pathogenesis of MDS and AML (8).
Hypomethylating agents (HMA), especially azacitidine, are the current mainstay for the treatment of advanced MDS and elderly AML, inducing hematological improvement, partial and complete responses, resulting in long-term survival (3, 9). However, hematological improvement occurs in only 30% of patients on HMA therapy (10). Therefore, drug combinations aiming to increase HMA efficiency are needed. Bromodomain containing proteins (BET proteins) are the most prominent group of epigenetic reader proteins, recognizing and binding to acetylated lysine residues within histone tails. BET proteins influence gene expression, cell-cycle regulation and maintain an association with chromatin throughout mitosis, facilitating “gene bookmarking.” As a member of the BET family, BRD4 bromodomain containing 4 (BRD4) acts by inducing the expression of growth-promoting genes and has been described as a therapeutic target for AML (11). BRD4 generates two major transcriptional variants: long and short. The C-terminal domain of the long isoform of BRD4 has been described as crucial for maintaining normal chromatin structure (12). Conversely, BRD4 short variant function is less known. It has been shown as an endogenous inhibitor of DDR signaling, recruiting the condensing II chromatin remodeling complex to acetylated histones (13).
Herein, we describe an increased expression of the short variant of BRD4 in MDS and AML patients and establish BRD4 short variant overexpression as a new independent MDS prognostic factor. The combination of a BRD4 inhibitor (JQ1) and the HMA, azacitidine, was more effective than azacitidine or JQ1 alone for inducing cell apoptosis. JQ1 and AZD6738 (a specific ATR inhibitor) also synergized to induce apoptosis, suggesting a role for the combination of BET inhibitors with HMA or DDR inhibitors in MDS and AML treatment.
Materials and Methods
Patient Samples
Bone marrow samples were collected from patients with MDS (n = 58), AML with MDS-related changes AML (AML-MRC) (n = 16), de novo AML (n = 34), and healthy donors (n = 24). All patients included in the study were untreated at the time of sample collection. MDS patients were classified according to 2016 World Health Organization (WHO) classification (14) and according to revised international prognostic staging system (R-IPSS) (15). The cytogenetic risk for MDS and AML was defined according to R-IPSS (15) and to the Medical Research Council cytogenetic classifications (16), respectively. Healthy donors' and patients' characteristics are described in Table 1. All healthy donors and patients signed informed consent forms under a local research protocol. This study was approved by the Institutional Ethical Review Board in accordance to the Helsinki Declaration.

Table 1. Characteristics of healthy donors and patients.
CD34+ Cell Separation
Bone marrow mononuclear cells were isolated by Ficoll-Paque (GE, Uppsala, Sweden) from diagnostic samples of AML patients and cord blood units (CBU) samples from full-term deliveries. After that, primary human CD34+ hematopoietic stem and progenitor cells (CD34+ cells) were selected using immunomagnetic activated cell sorting columns (Miltenyi Biotech, Auburn, CA, USA), obtaining purity of at least 90%.
Cell Culture
A panel of human myeloid leukemia cell lines (HEL, HL60, K562, and U937) was cultured in Roswell Park Memorial Institute medium-1640 (RPMI) (Sigma) containing 10% FBS, glutamine (2 mM), penicillin (100 μg/mL), streptomycin (100 μg/mL), and amphotericin B (0.25 μg/mL). The cells were maintained in a humidified atmosphere at 37°C and 5% CO2 and the experiments were performed when they reached exponential growth.
Chemical Reagents
A BRD4 specific inhibitor (JQ1) was kindly provided by Dr. James Bradner (Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA). The compound was diluted in dimethylsulfoxide (DMSO) to a 10 mM stock solution and was added to the cells at final concentrations of 0.24 μM to 100 μM for 48 h. Azacitidine (AZA) was also diluted in DMSO to a 100 mM stock concentration and added to cells at final concentrations of 1–3 μM for 48 h. The AZA concentration range was chosen based on the established AZA treatment schedule for MDS patients (75 mg/m2/day), which achieved plasma levels in the range of 3–11 μM (17, 18). The dose of 1 μM was used for both drugs when JQ1 and azacitidine were combined. AZD6738 was obtained from Selleck Chemicals (Houston, TX) diluted in DMSO and added to the cells at final concentrations of 1–20 μM. GI30 (drug inhibition of 30%) was determined after 48 h of exposure and used in combination with 1 μM of JQ1 or azacitidine.
Gene Expression Analysis
RNA was extracted using the RNeasy Plus Micro Kit (Qiagen, Valencia, CA, USA) and reversely transcribed into cDNA with the RevertAid H Minus First Strand cDNA Synthesis Kit (MBI Fermentas, St. Leon-Rot, Germany). The quantitative RT-PCR (qRT-PCR) reaction was run with SYBR Green Master Mix PCR (Fermentas) using the ABI 7500 Sequence Detection System (Applied-Biosystem, Foster City, CA, USA). The values of the relative quantification of gene expression was calculated through the equation 2−ΔΔCT (19). A negative no “template control” was included for each primer pair and the amplification specificity was verified using a dissociation curve at the end of each run. Three replicas were run on the same plate for each sample. Sense and antisense primers were designed to be complementary to the sequences contained in different exons. The following primers were used: BRD4 long variant (BRD4L), 5′- AAAGGACCTGAAAATCAAGAACATG-3′ and 5′-GAAGCTGTCGCTGGATGACTT-3′; BRD4 short variant (BRD4S) 5′-CTGACAGCGAAGACTCCGAAA-3′ and 5′-GCTATAGCTTGCTGGGAAGGAA-3′, hypoxanthine phosphoribosyltransferase (HPRT), 5′-GAACGTCTTGCTCGAGATGTGA-3′ and 5′-TCCAGCAGGTCAGCAAAGAAT-3′.
Western Blotting
Equal amounts of protein were submitted to electrophoresis on SDS polyacrylamide gels under reducing conditions and the nitrocellulose membrane was blotted with specific antibodies. Polyclonal antibodies against cleaved PARP-1 (sc-56196), CDK6 (sc-56282), GAPDH (sc-32233), and ACTIN (sc-1616) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-phospho-histone H2AX (Ser139) (pH2AX) (9718), P53 (2524), and anti-caspase 3 (9665) were purchased from Cell signaling (Danvers, MA, USA) and P21 were obtained from Abcam (Cambridge, MA, USA). The target proteins were analyzed by chemiluminescence using an ECL Plus Kit (GE-Healthcare, Buckinghamshire, England). Quantitative analyses of the optical intensities of protein bands were determined with UN-SCAN-IT graph digitalizing software (Silk scientific, UT, USA) and normalized by actin or GAPDH for protein expression.
Assessment of Cell Growth
Cell growth was measured by methylthiazoletetrazolium (MTT) assay. Briefly, 2.5 × 104 cells per well were plated in 96–well plates in RPMI/10% FBS for 48 h. MTT solution (5 mg/mL) was added to each well and incubated at 37°C for 4 h. The reaction was stopped by 0.1 N HCl in anhydrous isopropanol. Cell growth was evaluated by measuring the absorbance at 570 nm, using an automated plate reader (Multiscan MS, Labsystems).
Assessment of Apoptosis
Cell death was measured by annexin-V and PI assay. Briefly, the cells were seeded in 24-well plates and treated or not with different concentrations of JQ1 and/or azacitidine for 48 h. After this period, the cells were collected and incubated with 1 μg/mL propidium iodide (PI) and 1 μg/mL APC-Annexin-V for 15 min at room temperature in the dark. All specimens were analyzed on a FACSCalibur (BD Biosciences, CA, USA) and 10,000 events were acquired for each sample.
Transduction With Lentivirus
U937 cells were transduced with lentivirus-mediated shRNA non-specific control (sc-108080) or lentivirus-mediated shRNA targeting BRD4 (sc-43639-V) from Santa Cruz Biotechnology (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and named shControl and shBRD4 cells, respectively. Pooling of multiple BRD4 shRNAs (three different sequences) was used to reduce possible off-target effects of the shRNAs. BA/F3 cells were transduced with lentivirus particles produced using pCDH-MSV-MCS-EF1α empty vector (control) or containing the full lengths of BRD4S or BRD4L. Briefly, 2 × 105 cells were transduced with lentivirus by spinoculation at multiplicity of infection (MOI) equal to 1 and selected with specific antibiotics for at least 7 days before using in the experiments. BA/F3 cells were also sorted using a FACsAria Fusion (Becton–Dickinson Biosciences) in addition to antibiotic selection.
Cell Cycle Analysis
Cells were seeded on a 24-well plate and treated or not with different concentrations of JQ1 for 48 h. Next, the cells were collected and fixed overnight in 70% ethanol. DNA was stained with Pipes buffer containing PI (20 μg/mL) and RNase A (10 μg/mL). Cell fluorescence was detected with a FACSCalibur. The proportions of cells in the cell cycle phases were analyzed by Modifit (Verify Software House Inc., Topsham, ME, USA), according to DNA distributions.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism5 software (GraphPad Software, Inc., San Diego, CA, USA) or SAS System for Windons 9.2 (SAS Institute, Inc., Cary, NC, USA). Data were expressed as the median [minimum-maximum]. For comparisons, an appropriate Mann–Whitney test, Student's t-test or ANOVA was used. Comparison between patient groups was performed by analysis of t-test and covariance (ANCOVA) controlled for age, followed by post-hoc comparisons using the Tukey test. All experiments were repeated at least four times. Cox regression model was used to estimate overall survival (OS) and event-free survival (EFS) for MDS patients. The stepwise process of selection was used for multivariate analysis. OS was defined as the time (in months) between the date of sampling and the date of death (for deceased patients) or last follow-up (for censored patients). EFS was defined as the time (in months) between the date of sampling and the first event (death or MDS progression or leukemic transformation) or last follow-up (for censored patients). All tests were two-tailed. P ≤ 0.05 were considered statistically significant.
Results
BRD4 Short Variant Expression Is Increased in Total Bone Marrow Cells From MDS and AML Patients and Associates With Worse Outcomes in MDS
The first step of this study comprised the evaluation of mRNA levels of both BRD4 variants in total bone marrow cells from healthy donors (n = 24), MDS (n = 58), and AML (n = 50) patients. In order to exclude confounders, we carried out an ANCOVA analysis, which showed that age and gender did not interfere in our results.
BRD4S expression was significantly increased in both MDS (4.21 [0.01–56.17]) and AML (4.01 [0.33–26.58]) patients, when compared to healthy donors (2.11 [0.04–10.32]; all P < 0.01) (Figure 1A). No difference in BRD4L expression was observed between healthy donors, MDS and AML patients (Figure 1B). There were no differences when MDS patients were stratified according BM blasts or when AML patients were grouped into de novo AML or AML with myelodysplasia related changes (AML-MRC).
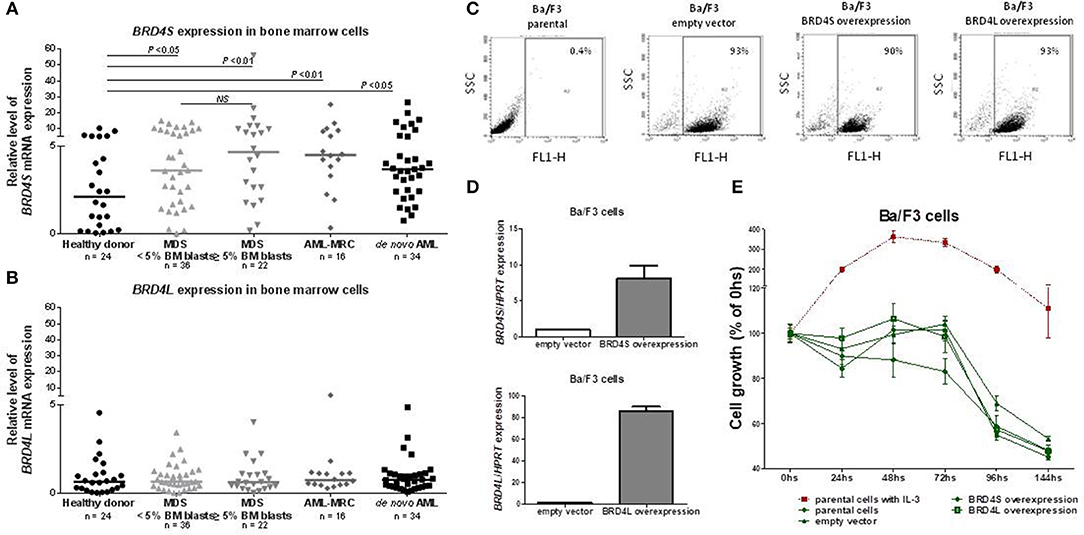
Figure 1. BRD4 short variant gene is overexpressed in MDS and AML patients. BRD4S mRNA expression in total bone marrow cells from healthy donors, MDS <5% BM blasts, >5% BM blasts, AML-MRC and de novo AML patients (A); BRD4L mRNA expression in total bone marrow cells from healthy donors, MDS <5% BM blasts, >5% BM blasts, AML-MRC, and de novo AML patients (B); Efficiency of GFP-positive BA/F3 cells transduced with empty vector, BRD4S and BRD4L, in that order, measured by flow cytometry (C) and by quantitative PCR (D); (E) Comparative growth (normalized by the initial number of cells) of BA/F3 parental cells (with and without IL-3), control (empty vector) or transduced with full-length BRD4S or BRD4L. The number of subjects and significant P-values (Mann–Whitney test) are indicated in the graph. MDS, myelodysplastic syndromes; AML, acute myeloid leukemia; AML-MRC, acute myeloid leukemia with myelodysplastic related changes.
With a median follow-up time of 34.4 months, BRD4S expression appeared as one of the variables with significant impact in event-free survival (EFS) and overall survival (OS) of MDS patients in univariate analysis. The bone marrow blast percentages (absolute values) and R-IPSS (stratified into very low/low, intermediate and high/very high) also significantly altered EFS and OS. Multivariate analysis demonstrated that higher BRD4S expression, along with intermediate and high/very high R-IPSS risk MDS, were the only independent factors related to worse outcomes (Table 2).
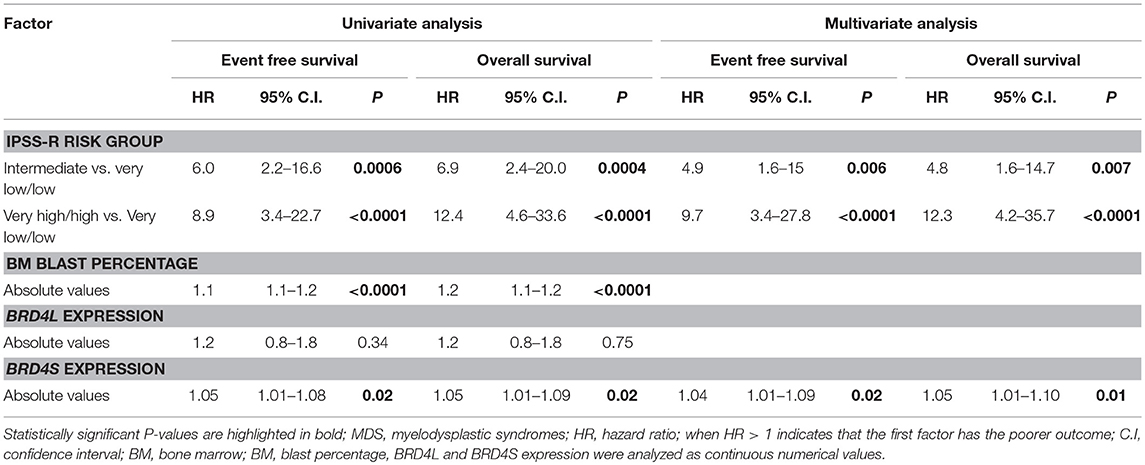
Table 2. Univariate and multivariate analysis for event-free survival and overall survival of MDS patients.
Aiming to explore a possible function of BRD4 as an oncogene, we overexpressed BRD4L or BRD4S using GFP tagged in normal hematopoietic murine cells (BA/F3), which are dependent on IL-3, and accessed daily the cell growth for 144 h in the presence or absence of 20 ηg/mL IL-3. The overexpression of both variants was effective (Figures 1C,D), but overexpression of BRD4L or BRD4S did not lead to IL-3 independency (Figure 1E). In the presence of IL-3, the growth of BRD4S or BRD4L overexpressed cells were also similar to the control (data not shown). These results suggest that BRD4 does not act as an oncogene alone.
JQ1 Treatment Reduces Cell Viability, Along With the Induction of Apoptosis and G0/G1 Cell Cycle Arrest in Myeloid Leukemia Cell Lines, but Not in Normal CD34+ Cells
In order to characterize the role of BRD4 in the disease pathogenesis, a panel of myeloid leukemia cell lines was treated with JQ1, an inhibitor of both BRD4 isoforms and other BET protein members (20, 21). Treatment with JQ1 induced a dose-dependent reduction in cell viability, in association with a similar induction of apoptosis. U937 and K562 cells were more resistant to the treatment, and presented higher GI50 (growth inhibitory concentration, with 50% reduction in cell viability) levels compared with HL60 and HEL cells (Figure 2A). Normal CD34+ cells from four samples of cord blood units exhibited greater tolerability to JQ1, with sustained cell viability and lower apoptosis rate compared to neoplastic cells (Figure 2B). JQ1 treatment induced an increase in G0/G1 cell cycle in cell lines, indicating cell cycle arrest (Figure 2C). We also observed an increase in p-H2AX in both HEL (more sensitive) and U937 (more resistant) cell lines, along with decreasing levels of CDK6 and with higher P53 and P21 protein concentrations (Figure 2D). This result suggests that BRD4 pharmacological inhibition causes DDR pathway activation and cell cycle arrest, even when the cells were exposed to a lower dose of JQ1.
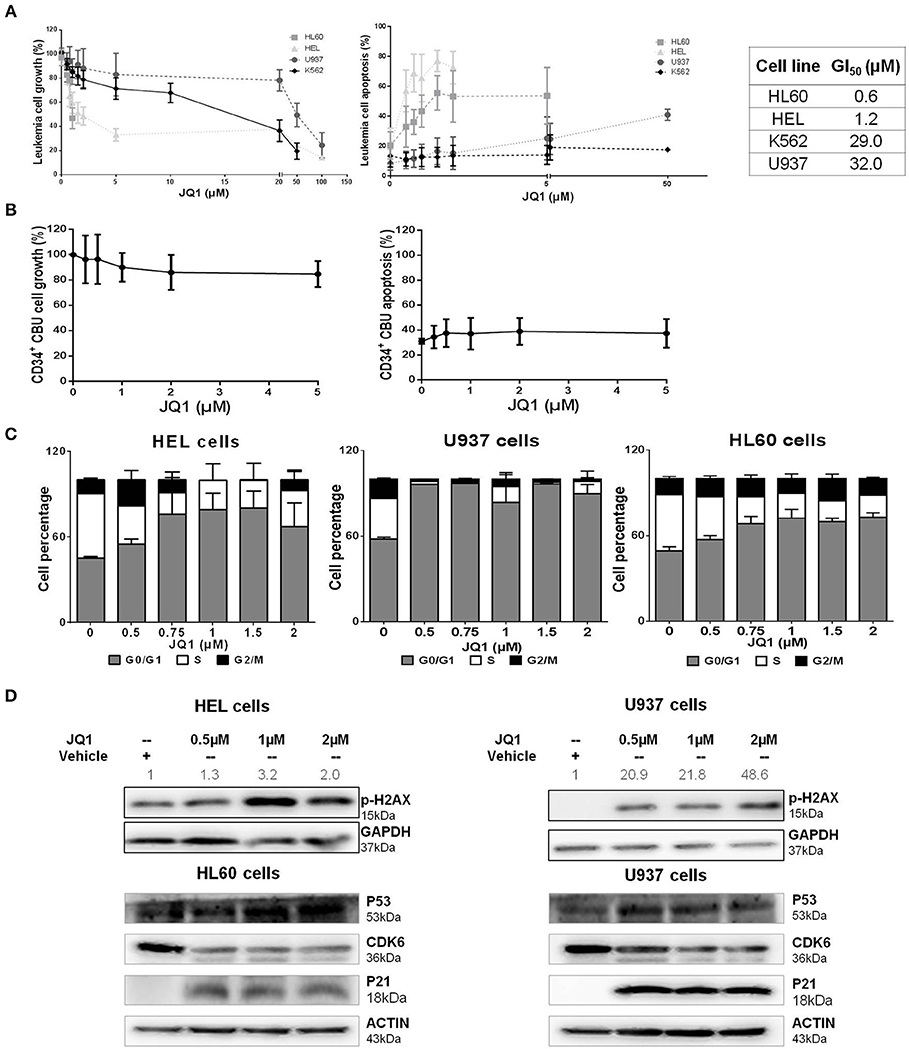
Figure 2. JQ1, a BRD4 inhibitor, reduces cell viability, induces apoptosis and cell cycle arrest of leukemia cell lines, with variable drug sensitivity. (A) Cell viability and apoptosis were determined by MTT assays and flow cytometry after 48 h of JQ1 treatment and growth inhibition (GI50) of four leukemia cell lines (HL60, HEL, K562, and U937). GI50 values determined from at least five independent experiments, by non-linear curve fitting to a sigmoid dose-response model; (B) Treatment of CD34+ cells from cord blood units (CBU) with increasing doses of JQ1 showed no significant cell viability reduction or apoptosis induction in that order; (C) Cell cycle phases were detected by flow cytometry. The cell lines tested are indicated in the graph. Results are shown as the percentage of total cells and bars represent increasing doses of JQ1. Graph bars represent a minimum of 6 independent experiments. (D) Western blotting analysis of total cell extracts from HEL, HL60, and U937 cells after 48 h of treatment with JQ1. The membrane was blotted anti-pH2AX (15 kDa), P53 (53 kDa), CDK6 (36 kDa), P21 (18 kDa), and for GAPDH (37 kDa) or ACTIN (43 kDa) as a control for equal sample loading, and developed with the ECL Western Blotting Analysis System. Densitometry was performed and the ratio of p-H2AX vs. GAPDH compared with the normalized value of control is shown. Western blot figures are representative of all experiments performed. All bar graphs represent mean ± SD of at least five independent experiments.
JQ1 Exhibits an Additive Effect Together With Azacitidine on the Apoptosis of Leukemia Cell Lines and CD34+ AML Primary Cells
With the aim of further characterizing the potential combinatory effects of BRD4 inhibition and a standard treatment for MDS, HEL, and U937 cells were treated with JQ1 and azacitidine for 48 h. JQ1 + AZA treatment increased the apoptotic rate of both cell lines, particularly in U937 cells (Figure 3A). JQ1 treatment increased p-H2AX and cleaved PARP-1, suggesting that activation of DDR is an important mechanism for apoptotic induction after JQ1 treatment. Although annexin V positive cells were increased with the co-treatment, we did not observe the same synergism in pH2AX and cleaved PARP-1 expression, possibly because high levels of these proteins were already detected when JQ1 was used alone (Figure 3B). CD34+ cells were isolated from diagnostic samples of 5 AML patients and treated with JQ1, AZA or both. In all samples, the combination of JQ1 and AZA induced a higher apoptosis rate than JQ1 or AZA alone (Figure 3C).
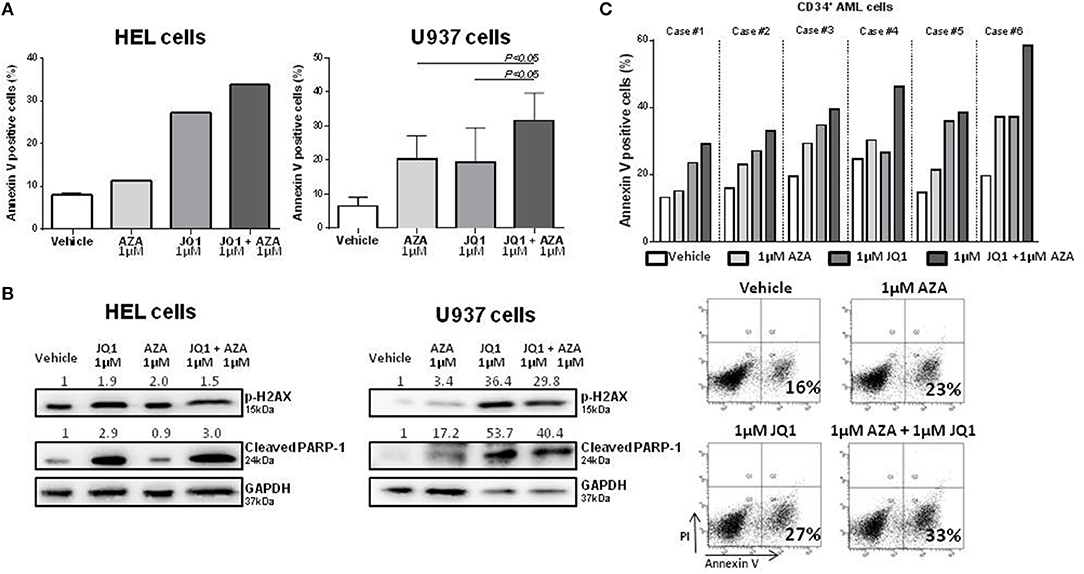
Figure 3. Combined treatment of JQ1 and azacitidine induces an additive effect on apoptosis of HEL, U937, and primary AML cells.(A) Apoptosis rate of HEL and U937 cell lines treated with JQ1, AZA, or JQ1+AZA for 48 h: Bar graphs show mean ± SD of at least four independent experiments. (B) Western blotting of HEL and U937 cell lines extract showing increased p-H2AX and cleaved PARP expression after JQ1+AZA treatment. Densitometry was performed and the ratio of target proteins vs. GAPDH compared with the normalized value of control is shown. (C) Apoptosis rate of 5 primary CD34+ AML cells treated with JQ1, AZA, and JQ1+AZA and a representative plot showing increasing apoptosis under JQ1+AZA combination.
BRD4 Silencing Decreases Proliferation and Apoptosis and Potentiates the Proapoptotic Effect of Azacitidine
We next sought to investigate whether BRD4 gene silencing would produce similar results to that of JQ1 treatment, in order to exclude possible therapy-related off target effects. For this purpose, U937 cells were stably transduced with lentivirus-mediated shRNA targeting BRD4 (shBRD4) or an appropriate control (shControl) and the efficacy of the transduction was confirmed by qRT-PCR (Figure 4A).
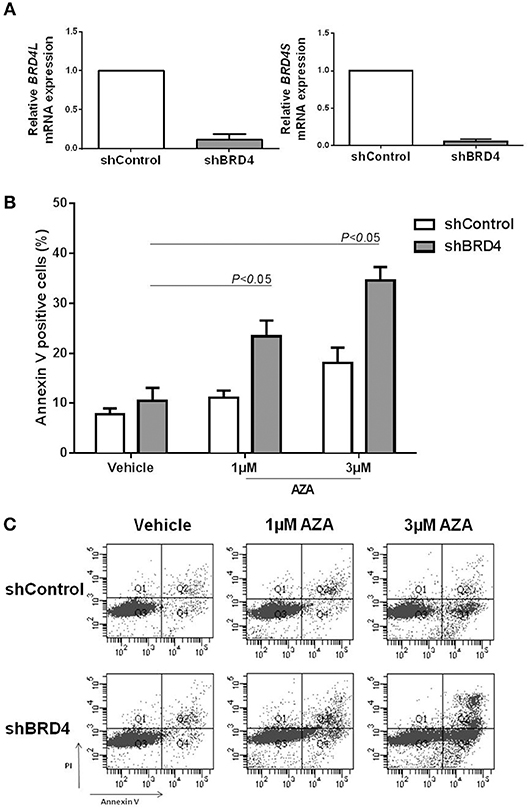
Figure 4. BRD4 silencing increases apoptosis rate of U937 cells under azacitidine treatment. (A) Efficacy of BRD4 silencing in U937 cells transduced with lentivirus-mediated control (shControl) or BRD4 (shBRD4) measured by RT-PCR. (B) Silenced and control cells were treated for 48 h with vehicle (DMSO) or azacitidine (1 or 3 μM): and apoptosis was detected by flow cytometry. (C) Representative plot of increasing apoptosis of shBRD4 cells under AZA treatment. Significant P-values (one-way Anova test) are indicated in the graph. All bar graphs represent mean ± SD of at least four independent experiments.
Apoptosis was evaluated in silenced cells treated or not with 1 or 3 μM AZA for 48 h. Following AZA treatment, shBRD4 cells showed a significant increase in apoptotic rate (P < 0.05) (Figures 4B,C), similarly to that observed for the drug combination.
JQ1 Potentiates the Effects of the ATR Inhibitor (AZD6738) on the Apoptosis of Leukemia Cells
Ataxia telangiectasia and Rad3 related (ATR) inhibitors have been reported to display synergism with JQ1 in the induction of apoptosis in lymphoma and melanoma cells (22, 23). Therefore, we aimed to test and compare the effects of JQ1 combined with a specific ATR inhibitor (AZD6738) or with azacitidine in leukemia cells. The GI30 of AZD6738 was firstly calculated in HEL, U937, and HL60 cell lines (Figure S1). Subsequently, these cell lines were exposed to 1 μM JQ1, 1 μM azacitidine or the GI30 dose of AZD6738 monotherapy or double combined treatment courses (JQ1 + azacitidine, JQ1 + AZD8768, azacitidine + AZD6738) for 48 h. In addition to U937 and HEL cells, a synergic effect of JQ1 and azacitidine was observed in HL60 cells (Figures 5A–F). In HL60 cells, the effects of JQ1 + AZD6738 were stronger than JQ1 + azacitidine (Figure 5A), whereas JQ1 + azacitidine was more potent in U937 cells (Figure 5C) and both combinations produced similar effects in HEL cells (Figure 5E). AZA + AZD6738 was also a promising combination, especially in U937 cells (Figure 5B). pH2AX was induced by all treatment schemes (monotherapy or combinations) in the three cell lines (Figures 5G–I). Cleaved caspase-3 expression was consistent with annexin V and cleaved PARP-1 results (Figures 5G–I).
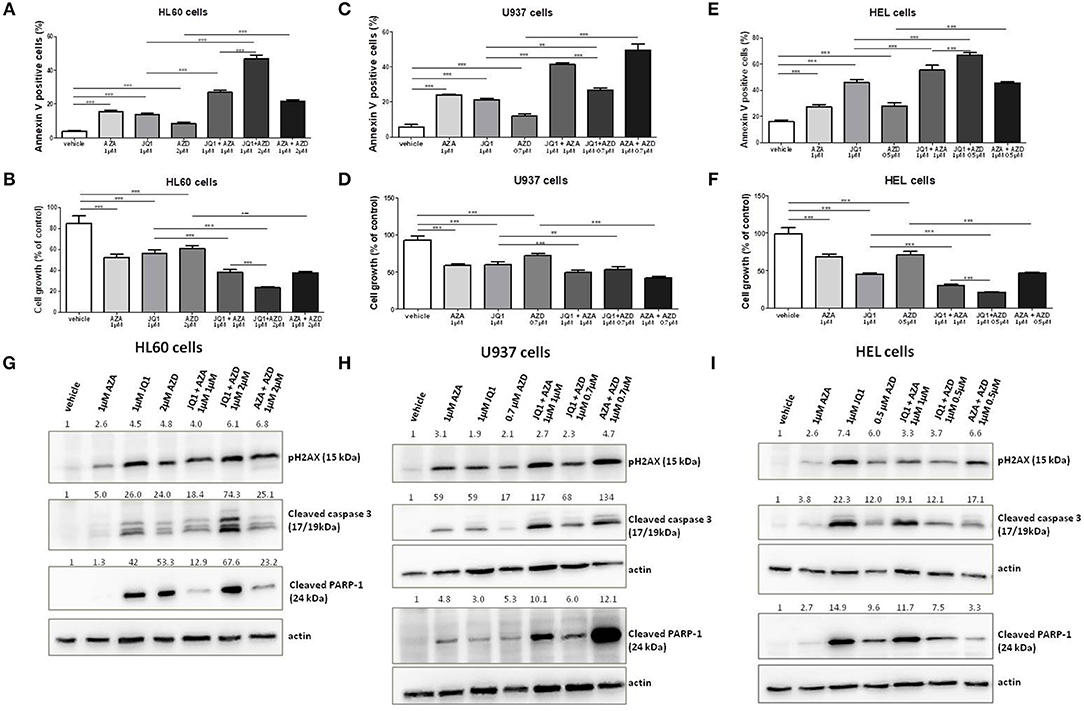
Figure 5. JQ1 effects on apoptosis of leukemia cell lines are increased with the ATR inhibitor AZD6738. Apoptosis rate and cell growth percentage in HL60 (A,B), U937 (C,D), and HEL (E,F) leukemia cell lines treated with AZA, JQ1, or AZD6738 monotherapy or in double combinations in the indicated doses for 48 h. Protein expression levels of pH2AX, cleaved caspase 3, and cleaved PARP-1 in HL60 (G), U937 (H), and HEL (I) leukemia cell lines treated with AZA, JQ1, or AZD6738 monotherapy or in double combinations in the indicated doses for 48 h. Actin (42 kDa) was used as a control for equal sample loading and the membrane was developed with the ECL Western Blotting Analysis System. Densitometry was performed and the ratio of target proteins vs. actin compared with the normalized value of control is shown. All bar graphs represent mean ± SD of at least four independent experiments. **P ≤ 0.01; ***P ≤ 0.001.
Discussion
Our results suggest that the short variant of BRD4 is a biological determinant of MDS phenotype and aggressiveness, since the two MDS subtypes (with lower and higher blast percentages) and AML patients exhibited overexpression of BRD4S, but particularly those with higher-risk disease. Moreover, a higher expression of this gene predicted a worse overall outcome in our cohort, in accordance with similar results for BRD4 overexpression and worse outcomes in solid tumors (24–26).
The overexpression of BRD4 isoforms in BA/F3 cells did not induce IL-3 independence, suggesting that this gene it is not and oncogene by itself. Moreover, in vitro studies showed that BRD4 inhibition, combined with azacitidine or AZD6738, synergistically induced apoptosis of leukemia cells.
Floyd et al. (13) described the BRD4 short isoform as an endogenous inhibitor of DNA damage response since cell lines with forced BRD4 hyper expression exhibited attenuated DNA damage response signaling (13). In this context, we speculate that the BRD4 short isoform could decrease the DNA damage response, favoring the disease toward genetic instability and clonal evolution. Our results using the BA/F3 cell model indicate that BRD4 does not function as an oncogene when overexpressed alone, which is in accordance with previous studies that have demonstrated that BET bromodomain proteins are not oncogenes themselves, but may act as a coactivators or direct modulators of other oncogenes, such as MYC and E2F proteins (27, 28).
In accordance with experimental data from other cancers, our data showed that BRD4 inhibition leads to variable apoptosis induction. However, since BRD4 also causes an arrest in G0/G1, and cells unable to differentiate undergo apoptosis, we cannot affirm that BRD4 acts directly on apoptosis. Importantly, we observed an additive effect of azacitidine combined with either JQ1 treatment or BRD4 silencing. To our knowledge, this is the first evidence for an additive effect between the standard treatment for MDS and elderly AML (the hypomethylating agent azacitidine) and a drug targeting an epigenetic modulator other than histone deacetylase.
Interestingly, JQ1 treatment increased the levels of p-H2AX, indicating a DDR pathway activation, even when using a lower dose. Double strand-breaks (DSB) in damaged DNA cause the phosphorylation of the neighboring histone H2AX at Ser139 via ATM. The functional significance of p-H2AX is a signal that facilitates DSB repair, presumably by causing the chromatin to be more accessible for DNA repair. Once DNA repair fails, the cells undergo death by activating apoptosis. Thus, apoptosis induction following DNA damage is a protective mechanism that prevents carcinogenesis (29). Some recent evidence suggests that DNA methyltransferase 1 (DNMT1) inhibition could also activate DDR, reinforcing the idea that epigenetic and genetic stability are intrinsically linked to one another (30, 31). JQ1 is a Brodomain and Extra-terminal (BET) inhibitor with activity blocking BRD4 (short and long isoforms), but also against other BET protein members, such as BRD2, BRD3, and BRDT. Therefore, this non-selective effect of JQ1 needs to be taken into consideration in the interpretation of the data.
Despite not having yet been analyzed specifically in MDS patients, there is increasing evidence supporting a role for BRD4 in AML pathogenesis and its potential therapeutic applicability. Herrmann et al. (32) described the overexpression of BRD4 in AML patient samples, even in highly enriched CD34+/CD38− and CD34+/CD38+ stem and progenitor cells and showed that JQ1 was capable of inducing the apoptosis of these cells. They also demonstrated that JQ1 synergized with cytarabine, reducing the cell viability of AML cells. Chen et al. (33) showed that BRD4 inhibition induced differentiation and death of IDH2 mutated AML, whereas Dawson et al (34) reported a role for NPM1 mutation in the induction of a BRD4 transcriptional program and that BET inhibition restored the NPM1 nuclear localization and abrogated the BRD4-induced oncogenic transcriptional program.
Stewart et al. (35) showed that JQ1 causes caspase 3/7-mediated apoptosis and a DNA damage response in DNMT3a/NPM1-mutated AML, suggesting that JQ1 might sensitize AML cells to p53-mediated cell death. Moreover, recent data suggest a synergistic effect of the histone deacetylase inhibitor, panobinostat, and JQ1 in AML cells (36). DDR activation seems to function as a biomarker of BRD4 inhibition efficacy, as cell lines presenting DDR activation also showed apoptosis induction. Chen et al. (37) recently showed that AZA-resistant MDS/AML cells have significantly increased expressions of BRD4, BRD2, and DNMT1. However, many DNA damage-induced cell death pathways are active in mammalian cells and the involved mechanisms and protein networks are complex and not fully understood (38). Therefore, our study indicates (not confirm) that BRD4 inhibition induces apoptosis through the activation of DDR.
Li et al. (39) recently demonstrated similar results regarding the role of BRD4 in prostate cancer, where higher gene expression was associated with worse outcomes and BRD4 inhibition activated H2AX. The authors showed that BRD4 is essential for the repair of DNA double-strand breaks (39). Muralidharan et al showed a synergistic effect of BRD4 inhibition with ATR inhibitors, inducing DDR in lymphoma cell lines (23), as demonstrated here in leukemia cells.
Larger and different cohorts of patients are necessary to confirm our results, since our cohort had low number of high-risk cytogenetics patients. Our findings however are in accordance with those from other authors supporting the existence of a defective BRD4-dependent transcriptional program in MDS and AML. The in vivo efficacy of JQ1 or other BET-targeting drugs for MDS patients is currently unknown, even though preliminary experimental and clinical data from BET inhibition in AML patients showed that BET-targeting drugs presented low toxicity profile and promising efficacy. Importantly, cord blood CD34 cells tolerated the effects of BRD4, with sustained cell viability and lower apoptosis, suggesting that BRD4 has a different role in normal vs. leukemic progenitor cells and, therefore, leukemic cells could be more BRD4 dependent than normal progenitor cells.
In conclusion, the BRD4 short variant is upregulated in MDS and AML patients and functions as an independent MDS prognostic factor, predicting worse outcomes. Our in vitro results further demonstrated that BRD4 inhibition shows a synergism with azacitidine or AZD6738, activating apoptosis, possibly through DDR pathway activation.
Author Contributions
FP designed and performed the experiments, participated in patient follow-up, collected, analyzed, and interpreted the data, and wrote the manuscript. ML performed experiments, analyzed, and interpreted data and wrote the manuscript. LdP performed experiments. FR contributed to the experiments and analyzed and interpreted data. AD, KV, and FN performed experiments. SO provided the study conception, directed the research, provided financial support, revised, and gave final approval of the manuscript.
Funding
This study was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). The Hematology and Transfusion Medicine Center–UNICAMP forms part of the National Institute of Blood, Brazil (INCT de Sangue–CNPq/MCT). Funding from FAPESP thematic grant of SO number 2011/51959-0.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. James E. Bradner, MD (Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA) who kindly provided the compound JQ1 for this study. We also thank Prof. Nicola Amanda Conran Zorzetto (Hematology and Blood Transfusion Center, University of Campinas, São Paulo, Brazil) for the English revision and Tereza Salles for her valuable technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.00016/full#supplementary-material
Abbreviations
MDS, myelodysplastic syndromes; AML, acute myeloid leukemia; BET, bromodomain containing proteins; HMA, hypomethylating agents; BRD4, Bromodomain-containing protein 4; AZA, azacitidine.
References
1. Zhou T, Hasty P, Walter CA, Bishop AJ, Scott LM, Rebel VI. Myelodysplastic syndrome: an inability to appropriately respond to damaged DNA? Exp Hematol. (2013) 41:665–74. doi: 10.1016/j.exphem.2013.04.008
2. Scott LJ. Azacitidine: a review in myelodysplastic syndromes and acute myeloid leukaemia. Drugs (2016) 76:889–900. doi: 10.1007/s40265-016-0585-0
3. Dombret H, Seymour JF, Butrym A, Wierzbowska A, Selleslag D, Jang JH, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood (2015) 126:291–9. doi: 10.1182/blood-2015-01-621664
4. Aggerholm A, Holm MS, Guldberg P, Olesen LH, Hokland P. Promoter hypermethylation of p15INK4B, HIC1, CDH1, and ER is frequent in myelodysplastic syndrome and predicts poor prognosis in early-stage patients. Eur J Haematol. (2006) 76:23–32. doi: 10.1111/j.1600-0609.2005.00559.x
5. Elias HK, Schinke C, Bhattacharyya S, Will B, Verma A, Steidl U. Stem cell origin of myelodysplastic syndromes. Oncogene (2014) 33:5139–50. doi: 10.1038/onc.2013.520
6. Xu L, Gu ZH, Li Y, Zhang JL, Chang CK, Pan CM, et al. Genomic landscape of CD34+ hematopoietic cells in myelodysplastic syndrome and gene mutation profiles as prognostic markers. Proc Natl Acad Sci USA. (2014) 111:8589–94. doi: 10.1073/pnas.1407688111
7. Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia (2014) 28:241–7. doi: 10.1038/leu.2013.336
8. Popp HD, Naumann N, Brendel S, Henzler T, Weiss C, Hofmann WK, et al. Increase of DNA damage and alteration of the DNA damage response in myelodysplastic syndromes and acute myeloid leukemias. Leuk Res. (2017) 57:112–8. doi: 10.1016/j.leukres.2017.03.011
9. Itzykson R, Fenaux P. Epigenetics of myelodysplastic syndromes. Leukemia (2014) 28:497–506. doi: 10.1038/leu.2013.343
10. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. (2009) 10:223–32. doi: 10.1016/S1470-2045(09)70003-8
11. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature (2011) 478:524–8. doi: 10.1038/nature10334
12. Wang R, Li Q, Helfer CM, Jiao J, You J. Bromodomain protein Brd4 associated with acetylated chromatin is important for maintenance of higher-order chromatin structure. J Biol Chem. (2012) 287:10738–52. doi: 10.1074/jbc.M111.323493
13. Floyd SR, Pacold ME, Huang Q, Clarke SM, Lam FC, Cannell IG, et al. The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature (2013) 498:246–50. doi: 10.1038/nature12147
14. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
15. Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood (2012) 120:2454–65. doi: 10.1182/blood-2012-03-420489
16. Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood (2010) 116:354–65. doi: 10.1182/blood-2009-11-254441
17. Marcucci G, Silverman L, Eller M, Lintz L, Beach CL. Bioavailability of azacitidine subcutaneous versus intravenous in patients with the myelodysplastic syndromes. J Clin Pharmacol. (2005) 45:597–602. doi: 10.1177/0091270004271947
18. Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer (2008) 123:8–13. doi: 10.1002/ijc.23607
19. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (2001) 25:402–8. doi: 10.1006/meth.2001.1262
20. Deeney JT, Belkina AC, Shirihai OS, Corkey BE, Denis GV. BET Bromodomain proteins Brd2, Brd3 and Brd4 selectively regulate metabolic pathways in the pancreatic beta-cell. PLoS ONE (2016) 11:e0151329. doi: 10.1371/journal.pone.0151329
21. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature (2010) 468:1067–73. doi: 10.1038/nature09504
22. Muralidharan SV, Einarsdottir BO, Bhadury J, Lindberg MF, Wu J, Campeau E, et al. BET bromodomain inhibitors synergize with ATR inhibitors in melanoma in melanoma. Cell Death Dis. (2017) 8:e2982. doi: 10.1038/cddis.2017.383
23. Muralidharan SV, Bhadury J, Nilsson LM, Green LC, McLure KG, Nilsson JA. BET bromodomain inhibitors synergize with ATR inhibitors to induce DNA damage, apoptosis, senescence-associated secretory pathway and ER stress in Myc-induced lymphoma cells. Oncogene (2016) 35:4689–97. doi: 10.1038/onc.2015.521
24. Liao YF, Wu YB, Long X, Zhu SQ, Jin C, Xu JJ, et al. High level of BRD4 promotes non-small cell lung cancer progression. Oncotarget (2016) 7:9491–500. doi: 10.18632/oncotarget.7068
25. Zhang P, Dong Z, Cai J, Zhang C, Shen Z, Ke A, et al. BRD4 promotes tumor growth and epithelial-mesenchymal transition in hepatocellular carcinoma. Int J Immunopathol Pharmacol. (2015) 28:36–44. doi: 10.1177/0394632015572070
26. Alsarraj J, Hunter KW. Bromodomain-containing protein 4: a dynamic regulator of breast cancer metastasis through modulation of the extracellular matrix. Int J Breast Cancer (2012) 2012:670632. doi: 10.1155/2012/670632
27. Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. (2013) 24:777–90. doi: 10.1016/j.ccr.2013.11.003
28. Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature (2016) 529:413–7. doi: 10.1038/nature16508
29. Roos WP, Kaina B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. (2013) 332:237–48. doi: 10.1016/j.canlet.2012.01.007
30. Loughery JE, Dunne PD, O'Neill KM, Meehan RR, McDaid JR, Walsh CP. DNMT1 deficiency triggers mismatch repair defects in human cells through depletion of repair protein levels in a process involving the DNA damage response. Hum Mol Genet. (2011) 20:3241–55. doi: 10.1093/hmg/ddr236
31. Ding N, Bonham EM, Hannon BE, Amick TR, Baylin SB, O'Hagan HM. Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage. J Mol Cell Biol. (2015) 8:244–54. doi: 10.1093/jmcb/mjv050
32. Herrmann H, Blatt K, Shi J, Gleixner KV, Cerny-Reiterer S, Mullauer L, et al. Small-molecule inhibition of BRD4 as a new potent approach to eliminate leukemic stem- and progenitor cells in acute myeloid leukemia AML. Oncotarget (2012) 3:1588–99. doi: 10.18632/oncotarget.733
33. Chen C, Liu Y, Lu C, Cross JR, Morris JP, Shroff AS, et al. Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Genes Dev. (2013) 27:1974–85. doi: 10.1101/gad.226613.113
34. Dawson MA, Gudgin EJ, Horton SJ, Giotopoulos G, Meduri E, Robson S, et al. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia (2014) 28:311–20. doi: 10.1038/leu.2013.338
35. Stewart HJ, Horne GA, Bastow S, Chevassut TJ. BRD4 associates with p53 in DNMT3A-mutated leukemia cells and is implicated in apoptosis by the bromodomain inhibitor JQ1. Cancer Med. (2013) 2:826–35. doi: 10.1002/cam4.146
36. Fiskus W, Sharma S, Qi J, Valenta JA, Schaub LJ, Shah B, et al. Highly active combination of BRD4 antagonist and histone deacetylase inhibitor against human acute myelogenous leukemia cells. Mol Cancer Ther. (2014) 13:1142–54. doi: 10.1158/1535-7163.MCT-13-0770
37. Chen L, Li Y, Yue M, Cloe A, Le Beau MM, He C, et al. Bromodomain and extra-terminal motif proteins (BETs) mediate 5-azacitidine resistance in myeloid leukemia through recruitment of an active RNA polymerase II complex. Blood (2016) 128:746. Available online at: http://www.bloodjournal.org/content/128/22/746
38. Matt S, Hofmann TG. The DNA damage-induced cell death response: a roadmap to kill cancer cells. Cell Mol Life Sci. (2016) 73:2829–50. doi: 10.1007/s00018-016-2130-4
Keywords: myelodysplastic syndromes, acute myeloid leukemia, BET member of bromodomain-containing proteins, azacitidine, AZD6738
Citation: Pericole FV, Lazarini M, de Paiva LB, Duarte ASS, Vieira Ferro KP, Niemann FS, Roversi FM and Olalla Saad ST (2019) BRD4 Inhibition Enhances Azacitidine Efficacy in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Front. Oncol. 9:16. doi: 10.3389/fonc.2019.00016
Received: 04 October 2018; Accepted: 04 January 2019;
Published: 29 January 2019.
Edited by:
Cyrus Khandanpour, University Hospital Münster, GermanyReviewed by:
Kavita Raj, Guy's and St Thomas' NHS Foundation Trust, United KingdomGiuseppe Alberto Palumbo, Università degli Studi di Catania, Italy
Copyright © 2019 Pericole, Lazarini, de Paiva, Duarte, Vieira Ferro, Niemann, Roversi and Olalla Saad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sara Teresinha Olalla Saad, c2FyYUB1bmljYW1wLmJy