 Fangyu Chen
Fangyu Chen Liuwei Chen2†
Liuwei Chen2† Xinchen Sun
Xinchen Sun- 1Department of Radiation Oncology, The First Affiliated Hospital of Nanjing Medical University, Nanjing, China
- 2The First School of Clinical Medicine, Nanjing Medical University, Nanjing, China
Salt-inducible kinase (SIK), which belongs to the sucrose non-fermenting 1/AMP-activated protein kinase family, was first discovered in the adrenal cortex of a rat on a high-salt diet. As an isoform of the SIK family, SIK2 modulates various biological functions and acts as a signal transmitter in various pathways. Compared with that in adjacent normal tissues, the expression of SIK2 is significantly higher in multiple types of tumors, which indicates its pivotal effect in oncogenesis. Studies on SIK2 have recently underlined its role in several signaling pathways, including the PI3K-Akt-mTOR pathway, the Hippo-YAP pathway, the LKB1-HDAC axis, and the cAMP-PKA axis. Moreover, a few small-molecule SIK2 inhibitors have been found to be able to rescue the oncogenicity of SIK2 during tumor development and reverse its abnormal activation of downstream pathways. In this mini-review, we discuss the results of in vivo and in vitro studies regarding the SIK2 mechanism in different signaling pathways, particularly their regulation of cancer cells. This work may provide new ideas for targeting SIK2 as a novel therapeutic strategy in tumor therapy.
Introduction
Plasma ion balances regulate a wide range of cellular processes from cell proliferation to mitochondrial functions. The plasma concentrations of Na+ and K+ have been proven to play a vital role in the biosynthesis of aldosterone in the adrenal cortex. Studies have shown that changes in plasma ion concentration can target biomembrane ion channels, such as Na+-K+-ATPase to regulate extra- and intracellular ion balances (1, 2). As a major part of this ion modulation network, salt-inducible kinase (SIK) was first discovered in 1999 by Okamoto et al. in the adrenal cortex of a rat on a high-salt diet. SIK is a serine/threonine protein kinase that belongs to the sucrose non-fermenting 1/AMP-activated protein kinase (SNF1/AMPK) family. The SIK family comprises three isoforms, namely, SIK1, SIK2, and SIK3, all of which may act as metabolic transmitters. The SIK2 gene is located on chromosome 11 and encodes for the SIK2 protein, which has 926 amino acids and three domains (3, 4). The C-terminal domain of the SIK protein contains numerous unique sites that can be phosphorylated by different protein kinases and transmit various stimulation signals involved in different biological processes, including cell growth and apoptosis (4–8). In many malignant tumors, such as breast cancer, lung cancer, melanoma, primary liver cancer, and ovarian cancer, SIK expression is significantly different from that in adjacent tissues (9–14).
Growing evidence has proven that the expression and action of SIK2 are tissue-specific. The cellular and subcellular distributions of SIK should be considered to determine its mechanism. Earlier investigations demonstrate that SIK2 maintains cell homeostasis via modulation of cAMP response element binding protein (CREB)-mediated gene transcription during starvation, which may be a possible mechanism for cancer cell survival under stress, such as chemoradiotherapy (15). SIK2 reduces glucose uptake in muscle cells and white adipocytes and downregulates lipogenesis and ketogenesis by phosphorylating the glucose-activated histone acetyltransferase coactivator p300 (16). SIK2 modulates several subtle cellular signaling pathways, and its abundant expression in melanoma and ovarian tumors is suggestive of its pivotal function in tumor development (13, 17). Thus, in this mini-review, we discuss the specific role and related signaling pathways of SIK2 in tumorigenesis. Our findings indicate the potential application of SIK2 as a therapeutic target for cancers.
SIK Family and Their Functions
The structures of the SIK isoforms are shown in Figure 1. The three isoforms are similar to one another, particularly in three domains: a kinase domain near the N-terminal, a central SNF1 protein kinase homology (SNH) domain, and a phosphorylation domain near the C-terminal (3). SIK1 is a 776-amino acid protein with a kinase domain in the region of residues 27–278, an SNH domain in the region of residues 301–354, and a domain enriched with PKA-dependent phosphorylation sites in the region of residues 567–613. Similarly, SIK2 is a 931-amino acid protein with a kinase domain in the region of residues 20–271, an SNH domain in the region of residues 293–346, and a phosphorylation domain in the region of residues 577–623. Finally, SIK3 is a 1,263-amino acid protein with a kinase domain in the region of residues 8–259, an SNH domain in the region of residues 283–336, and a phosphorylation domain in the region of residues 486–518. Initial studies have found that SIK1 is most abundant in the adrenal cortex and an important regulator in the early phase of hormonal stimulation of the adrenal cortex (4, 18), adipose tissue (6), and neural tissue (19). It may overexpress in several non-adipose tissues, such as in the ovaries and lungs, and act as an oncogenic signal transmitter during the occurrence and progression of tumors in the aforementioned organs (18–20). Unlike SIK1, SIK2 modulates several subtle cellular signaling pathways, and the increased expression of SIK2 in adipose and neuronal tissues indicates its pivotal role in lipid metabolism and neural physiology. SIK2 promotes insulin resistance and diabetes by reducing glucose uptake in muscles and white adipose tissues and inhibiting gluconeogenesis (7). SIK2 is overexpressed in several cancer cell lines and boosts cancer cell tolerance to different stresses, such as deprivation of nutrients and taxol chemotherapy (21). It plays a proinflammatory role by repressing IL-10 secretion of regulatory macrophages (22). However, little is known about why the structural similarity of the SIK family leads to different biological functions.
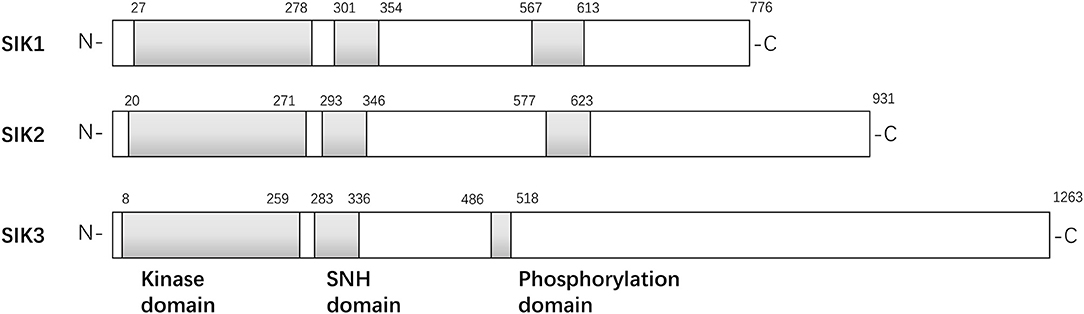
Figure 1. Structures of isoforms in SIK family.
SIK2 and the PI3K-Akt-mTOR Pathway
The expression level of SIK2 in cancers is significantly higher than that in adjacent and surrounding normal tissues, which suggests that SIK2 is critical in tumorigenesis and tumor development. Miranda et al. found that the loss of SIK2 reduces G1/S transition, delays mitotic progression, and decreases Akt phosphorylation levels (17). They also confirmed that SIK2 is overexpressed in adipocyte-rich metastatic deposits compared with ovarian primary lesions and that adipocytes activate SIK2 in ovarian cancer cells in a calcium-dependent manner. Following adipocyte-induced stimulation, the activated SIK2 alters metabolic effects in ovarian cancer cells by inhibiting acetyl-CoA carboxylase and promoting fatty acid oxidation. p85α, the regulatory subunit of the PI3K complex, was previously identified as a putative SIK2 substrate during chemical genetic screening. The identified p85α phosphorylation site (S154) resides in the known SIK2 phosphorylation consensus sequence L-x-[HKR]-[ST]-x-S-X(3)-L at L149–L158 (LYRTQSSSNL). Incubation of recombinant full-length SIK2 or its kinase domain with a peptide corresponding to L149–L158 of p85α confirmed that SIK2 catalyzes the phosphorylation of this sequence. More importantly, full-length SIK2, but not the kinase-inactive mutant, phosphorylated p85α was confirmed in isotopic labeling assay. Phosphopeptide mapping of p85α following incubation with SIK2 (kinase domain or full-length) revealed that the former was phosphorylated at S154 in the BH domain. The BH domain is thought to bind to proteins that modulate PI3K activity. Downstream S154 phosphorylation also appears to increase in an SIK2-dose-dependent manner. siRNA-mediated depletion or chemical inhibition confirms that SIK2 is required for p85α S154 phosphorylation. Moreover, p85α phosphorylation and concomitant Akt phosphorylation can be triggered by calcium-mediated SIK2 activation. Consistent with these observations, incubation of the PI3K complex with recombinant SIK2 leads to a profound increase in PI3K activity in vitro (up to 13.8-fold), while chemical inhibition of SIK2 induces a dose-dependent reduction in PI3K activity to its basal level. These data confirm that p85α is a direct catalytic substrate of SIK2 and that SIK2 S154 phosphorylation significantly increases the activity of the PI3K-Akt pathway in ovarian cancer cells.
While most reports suggest that SIK2 is an oncogenic marker, one study in Turkey claimed that SIK2 is a potential tumor suppressor in breast cancer (23); SIK2 expression was reportedly reduced in tumor tissues and breast cancer cell lines compared with that in normal counterparts. The researchers also found SIK2-mediated attenuation of proliferation and survival of breast cancer cells with parallel inhibition of the Ras-Erk and PI3K-Akt pathways. However, the mechanisms underlying the reduction of SIK2 levels in cancer tissues were not discussed. Thus, research into the mechanism of SIK2 loss will help future scholars better understand tumor transformation in breast tissue and design new treatment strategies.
SIK2 and the Hippo-YAP Pathway
The Hippo pathway is a highly conserved growth regulatory signaling pathway that was first discovered in Drosophila. It can block the downstream pro-growth transcriptional co-activator Yorkie (Yki), which is homologous to mammalian Yes-associated protein (YAP), and exert its regulatory effects on organ size, cell proliferation, and apoptosis during organ development (24, 25). YAP has been shown to be highly expressed in various human tumors, such as endometrial carcinoma, primary liver cancer, and oral squamous cell carcinoma. Activation of YAP can remove tumor cell contact inhibition, leading to tumor metastasis (25–27). Tsujiura et al. immunohistochemically analyzed YAP in endometrial carcinoma tissue samples and found that the high expression of YAP in the nucleus is closely associated with higher tumor grading and staging, lymphatic/blood vessel invasion, increased recurrence, and metastasis. They then confirmed these results at the cellular level in knockdown and overexpression assays. Recent studies have demonstrated that YAP restricts the activity of the cell cycle checkpoints ATM and ChK2 to enable cancer cells to enter the cell cycle and mitosis after chemoradiotherapy despite unrepaired DNA damage, resulting in tumor growth, chemoradiotherapy resistance, and ongoing proliferation (28).
Wehr et al. characterized Drosophila salt-inducible kinase (sik2) as an upstream inhibitor of the Hippo pathway (29). sik2 has been identified as the ortholog of human SIK2. Activated sik2 phosphorylates Ser413 of the scaffold protein Salvador (Sav), a major part of the core kinase complex of the Hippo pathway, and subsequently abolishes the inhibition of the proto-oncogene Yki. In addition, sik2 directly induces the expression of Yki and facilitates Yki-dependent tissue overgrowth. Coincidentally, both SIK2 and YAP have been proven to be oncogenes in ovarian cancer. Research has confirmed a close interaction between the PI3K-Akt-mTOR and Hippo-YAP pathways via SIK2 (Figure 2). On the one hand, YAP directly activates PI3K-Akt-mTOR and alters cellular biological functions (30, 31). YAP also increases pAkt-S473 levels and suppresses apoptosis by induction of insulin-like growth factor 2 expression (28). On the other hand, mTOR complex 2 enhances the oncogenicity of YAP through phosphorylation of the Hippo pathway component AMOTL2 (32). These observations reveal that mutual activation between the PI3K-Akt-mTOR and Hippo-YAP pathways caused by SIK2 may be crucial in tumorigenesis. However, the precise role of SIK2 in these intersecting pathways is not well-understood, and future studies are still desperately needed to elucidate the related detailed mechanisms.
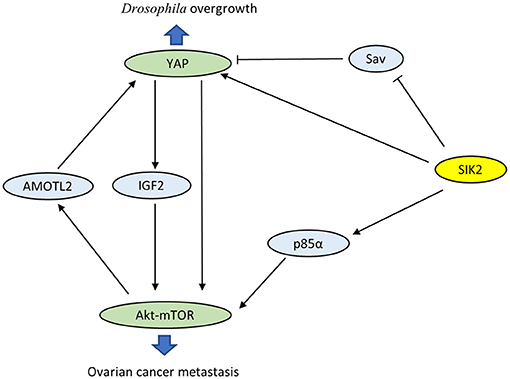
Figure 2. Crosstalk between the PI3K-Akt-mTOR pathway and the Hippo-Yap pathway via SIK2.
SIK2 and the LKB1-HDAC Signaling Axis
Epigenetic studies have confirmed that DNA acetylation modification is closely related to tumorigenesis, tumor invasion, and chemoradiotherapy resistance (33–35). The abnormal activation and overexpression of histone deacetylase (HDAC) down-regulates tumor suppressor genes and exhibits tumor-promoting effects. Using kinase domain-focused CRISPR techniques, researchers screened all dependent kinase in acute myeloid leukemia (AML), focusing subsequent experiments on SIK3, which scored strongly in MOLM-13 and MV4-11 AML cells and in a more intermediate fashion in other AML cell lines (36). Liver kinase B1 (LKB1) was also identified to show an AML-biased pattern of dependence. Since SIK3 is homologous to SIK1 and SIK2, further studies were conducted to determine whether a broader requirement exists for SIKs in cancer. By performing dual targeting of each SIK gene combination in 17 AML cell lines, researchers observed a broad AML-specific requirement for SIK2 + SIK3 resembling the pattern of LKB1 dependence with a bias for lines with mixed lineage leukemia fusions. In cDNA rescue assays, LKB1 was found to phosphorylate and activate SIK3 in AML. The SIK3 mutant was unable to maintain the proliferation of MOLM-13 cells, while a phosphomimetic allele of SIK3 rescued the proliferation arrest caused by inactivating LKB1. The reverse of SIK3 dependence for AML proliferation was observed during dual CRISPR targeting of HDAC4. Western blotting revealed reductions in HDAC4 phosphorylation upon genetic targeting of SIK3 or chemical inhibition of SIK. Taken together, these results indicate that the function of SIK3 is critical in AML and that inhibition of HDAC4 is one of the key functions of SIK3 in supporting AML proliferation.
Histone H3 lysine 27 acetylation (H3K27ac) is linked to the relevant downstream activity in the LKB1-SIK pathway, and ChIP-seq has confirmed that LKB1/SIK3-dependent H3K27ac coincides with sites of transcription factor MEF2C occupancy. While LKB1/SIK3 knockout or following SIK inhibitor HG-9-91-01 treatment did not change MEF2C protein expression, HG-9-91-01 exposure led to increased HDAC4 binding to MEF2C-bound sites. Epigenomic analysis suggests that LKB1-SIK signaling is critical in AML to prevent HDAC4 from inactivating the function of MEF2C on chromatin. These genetic experiments suggest that co-inhibition of SIK2 + SIK3 could be the ideal strategy to achieve potent MEF2C inhibition in AML. Since MEF2C is maladjusted in lymphoid malignancies, LKB1-SIK signaling is likely to be important in other hematopoietic cancers (37).
SIK2 and the cAMP-PKA Signaling Axis
The G protein αs (GNAS) gene encodes the Gαs stimulatory subunit of G proteins, which mediate G-protein-coupled receptor signaling, a major mechanism that links multiple environmental stimuli with intracellular responses (38). The primary target is adenylyl cyclase, which generates the second messenger cAMP, which, in turn, activates downstream protein kinase A (PKA). In many tissues, GNAS–cAMP-PKA signaling is required during cell dormancy and cell growth (39–43). However, multiple types of human cancers show gain-of-function variations in this pathway (38). For example, loss of p53 promotes the advent of GNAS R201C mutations and induces malignant transformation in pancreatic benign tumors in the KGC mice model, which can rapidly develop cystic pancreatic tumors (44–47). Mutated GNAS R201C supports pancreatic tumor growth via cAMP-PKA signaling, which subsequently phosphorylates SIKs (SIK1, SIK2, and SIK3) and prevents them from phosphorylating downstream targets (48). Also, small molecule pan-SIK inhibitors (HG-9-91-01 and KIN-112) prevent the growth of KGC organoids after silencing GNAS, and their effects are directly proportional to the degree of SIK inhibition. Compared with wild-type SIK2, the SIK2-S4A mutant, which is resistant to cAMP-PKA activation, strongly inhibits the proliferation of KGC-like organs. In particular, SIKKO rescues both organoid growth in vitro and subcutaneous tumor growth following GNAS R201C silencing, and these findings have been confirmed in human pancreatic ductal adenocarcinomas (PDA). Thus, the cAMP-PKA-SIK2 signaling pathway is a conserved tumorigenic mechanism in pancreatic tumor cells. The mutant GNAS drives downstream PKA-SIK2 axis and promotes lipid hydrolysis in addition to lipid synthesis and remodeling. While SIK2 is known to maintain cell homeostasis and energetic metabolism, particularly glucose and fatty acid oxidation (15), the suppression of SIK2 mediated by GNAS-PKA will inhibit the phosphorylation of its downstream CREB-regulated transcription co-activator (CRTC) and others (Figure 3). Then it will promote lipids absorption and synthesis, and the abundant lipids in tumor cells provide substrates for structural, signaling, and metabolic purposes, which explains why SIK2 act as a tumor suppressor in PDA.
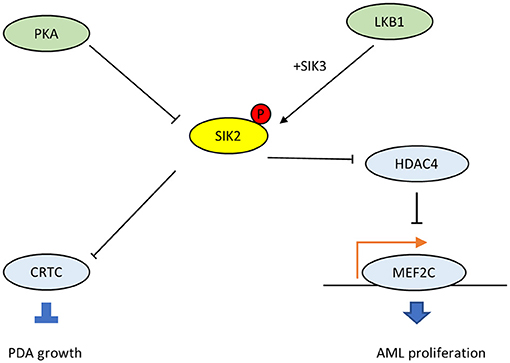
Figure 3. The dichotomous oncogenic roles of SIK2 in the LKB1-HDAC axis and the cAMP-PKA axis.
While SIK2 is deemed to be a tumor promoter in most cases, in the context of GNAS mutated PDA, it is supposed to be a tumor suppressor, mainly because SIK2 plays different roles in different tissue and cells, similar to cAMP/PKA signaling. Given the context-dependent tumor-promoting and -suppressing roles of SIK2, administration of SIK2 inhibitors in GPCR-mutated or other overactive cAMP-PKA cancer types should be attempted with extremely caution to avoid potential pro-tumor effects. More investigations are necessary to clarify these issues and promote the use of SIK2 inhibitors in tumor therapy.
SIK2 in Cancer Therapy
Previous studies on SIK2 have reported its regulation of energetic metabolism, mostly based on its signaling pathways and the downstream role of LKB1 in adipocytes. Studies on SIK2 have recently underlined its role in several signaling pathways related to tumorigenesis. Clinical and pathological data indicate that SIK2 is a potential oncogenic marker in ovarian (17, 49), prostate (50), osteosarcoma (51), and colorectal (52) cancers by controlling different cellular mechanisms. Intriguingly, two studies report that SIK2 may act as a tumor suppressor in breast cancer and PDA. Since SIK2 plays a distinct role in different tissues and divergent pathways, its dysregulation may lead to conflicting phenotypes. Initial studies on SIK2 maily focused on its role in energetic metabolism, particularly in glucose, and lipids oxidation during starvation. The functions of SIK2 may be unique in cells that are involved in glycolipid metabolism, such as hepatocyte and pancreatic cells. As a consequence, SIK2 may act as both tumor promoter and suppressor due to the diversity of cancer cell types or different genetic background. The SIK2 inhibitors HG-9-91-01, ARN-3236, and KIN-112 have succeeded in cancer therapy approaches, validated in cultured cells and in vivo animal models (17, 36, 48), although additional optimization of these small molecules is required for therapeutic investigation. Further evaluation of these small molecules is necessary to achieve potent SIK2 inhibition in the uncontrolled signaling pathways of tumor cells while preserving the homeostatic and tumor-protective functions of SIK2 in other cell types.
Conclusion
In this mini-review, we discussed the role of the newly identified protein kinase, SIK2, in tumorigenesis, specifically focusing on different signaling pathways involving SIK2. SIKs present significant physiological functions, including novel roles in tumorigenesis and tumor progression. While most studies reveal SIK2 to be a tumor promoter, some claims indicate that SIK2 provides protection from cancer. Thus, the dichotomous function and mechanism between SIK2 and cancer must be further elucidated. As described earlier, SIK2 targeting may be applied as a novel strategy for treating multiple cancer types. Future studies to investigate the molecular mechanisms underlying the precise role of SIK2 in intersecting signaling pathways, as well as the therapeutic effects of SIK2 in preclinical and clinical trials, are recommended.
Author Contributions
FC, LC, and QQ contributed to conception and manuscript writing. XS participated in its coordination and modification. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81472809, No. 81502653, No. 81672983, No.81874217), A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) (JX10231801), Young Medical Key Talents of Jiangsu Province (grant number QNRC2016572) and the Six Major Talent Peak Project of Jiangsu Province (No.2013-WSN-040).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Muller J. Regulation of aldosterone biosynthesis. Physiological and clinical aspects. Monogr Endocrinol. (1987) 29:1–364.
3. Katoh Y, Takemori H, Horike N, Doi J, Muraoka M, Min L, et al. Salt-inducible kinase (SIK) isoforms: their involvement in steroidogenesis and adipogenesis. Mol Cell Endocrinol. (2004) 217:109–12. doi: 10.1016/j.mce.2003.10.016
4. Wang Z, Takemori H, Halder SK, Nonaka Y, Okamoto M. Cloning of a novel kinase (SIK) of the SNF1/AMPK family from high salt diet-treated rat adrenal. FEBS Lett. (1999) 453:135–9. doi: 10.1016/S0014-5793(99)00708-5
5. Dentin R, Liu Y, Koo SH, Hedrick S, Vargas T, Heredia J, et al. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature (2007) 449:366–9. doi: 10.1038/nature06128
6. Horike N, Takemori H, Katoh Y, Doi J, Min L, Asano T, et al. Adipose-specific expression, phosphorylation of Ser794 in insulin receptor substrate-1, and activation in diabetic animals of salt-inducible kinase-2. J Biol Chem. (2003) 278:18440–7. doi: 10.1074/jbc.M211770200
7. Muraoka M, Fukushima A, Viengchareun S, Lombes M, Kishi F, Miyauchi A, et al. Involvement of SIK2/TORC2 signaling cascade in the regulation of insulin-induced PGC-1alpha and UCP-1 gene expression in brown adipocytes. Am J Physiol Endocrinol Metab. (2009) 296:E1430–9. doi: 10.1152/ajpendo.00024.2009
8. Wang Y, Li G, Goode J, Paz JC, Ouyang K, Screaton R, et al. Inositol-1,4,5-trisphosphate receptor regulates hepatic gluconeogenesis in fasting and diabetes. Nature (2012) 485:128–32. doi: 10.1038/nature10988
9. Ahmed AA, Lu Z, Jennings NB, Etemadmoghadam D, Capalbo L, Jacamo RO, et al. SIK2 is a centrosome kinase required for bipolar mitotic spindle formation that provides a potential target for therapy in ovarian cancer. Cancer Cell (2010) 18:109–21. doi: 10.1016/j.ccr.2010.06.018
10. Bricambert J, Miranda J, Benhamed F, Girard J, Postic C, Dentin R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J Clin Invest. (2010) 120:4316–31. doi: 10.1172/JCI41624
11. Charoenfuprasert S, Yang YY, Lee YC, Chao KC, Chu PY, Lai CR, et al. Identification of salt-inducible kinase 3 as a novel tumor antigen associated with tumorigenesis of ovarian cancer. Oncogene (2011) 30:3570–84. doi: 10.1038/onc.2011.77
12. Cheng H, Liu P, Wang ZC, Zou L, Santiago S, Garbitt V, et al. SIK1 couples LKB1 to p53-dependent anoikis and suppresses metastasis. Sci Signal. (2009) 2:ra35. doi: 10.1126/scisignal.2000369
13. Horike N, Kumagai A, Shimono Y, Onishi T, Itoh Y, Sasaki T, et al. Downregulation of SIK2 expression promotes the melanogenic program in mice. Pigment Cell Melanoma Res. (2010) 23:809–19. doi: 10.1111/j.1755-148X.2010.00760.x
14. Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell (2012) 150:1107–20. doi: 10.1016/j.cell.2012.08.029
15. Du J, Chen Q, Takemori H, Xu H. SIK2 can be activated by deprivation of nutrition and it inhibits expression of lipogenic genes in adipocytes. Obesity (2008) 16:531–8. doi: 10.1038/oby.2007.98
16. Zhang ZN, Gong L, Lv S, Li J, Tai X, Cao W, et al. SIK2 regulates fasting-induced PPAR alpha activity and ketogenesis through p300. Sci Rep. (2016) 6:23317. doi: 10.1038/srep23317
17. Miranda F, Mannion D, Liu S, Zheng Y, Mangala LS, Redondo C, et al. Salt-inducible kinase 2 couples ovarian cancer cell metabolism with survival at the adipocyte-rich metastatic niche. Cancer Cell (2016) 30:273–89. doi: 10.1016/j.ccell.2016.06.020
18. Lin X, Takemori H, Katoh Y, Doi J, Horike N, Makino A, et al. Salt-inducible kinase is involved in the ACTH/cAMP-dependent protein kinase signaling in Y1 mouse adrenocortical tumor cells. Mol Endocrinol. (2001) 15:1264–76. doi: 10.1210/mend.15.8.0675
19. Feldman JD, Vician L, Crispino M, Hoe W, Baudry M, Herschman HR. The salt-inducible kinase, SIK, is induced by depolarization in brain. J Neurochem. (2000) 74:2227–38. doi: 10.1046/j.1471-4159.2000.0742227.x
20. Ruiz JC, Conlon FL, Robertson EJ. Identification of novel protein kinases expressed in the myocardium of the developing mouse heart. Mech Dev. (1994) 48:153–64. doi: 10.1016/0925-4773(94)90056-6
21. Du WQ, Zheng JN, Pei DS. The diverse oncogenic and tumor suppressor roles of salt-inducible kinase (SIK) in cancer. Expert Opin Ther Targets (2016) 20:477–85. doi: 10.1517/14728222.2016.1101452
22. MacKenzie KF, Clark K, Naqvi S, McGuire VA, Noehren G, Kristariyanto Y, et al. PGE(2) induces macrophage IL-10 production and a regulatory-like phenotype via a protein kinase A-SIK-CRTC3 pathway. J Immunol. (2013) 190:565–77. doi: 10.4049/jimmunol.1202462
23. Zohrap N, Saatci O, Ozes B, Coban I, Atay HM, Battaloglu E, et al. SIK2 attenuates proliferation and survival of breast cancer cells with simultaneous perturbation of MAPK and PI3K/Akt pathways. Oncotarget (2018) 9:21876–92. doi: 10.18632/oncotarget.25082
24. Gumbiner BM, Kim NG. The Hippo-YAP signaling pathway and contact inhibition of growth. J Cell Sci. (2014) 127 (Pt. 4):709–17. doi: 10.1242/jcs.140103
25. Zeng Q, Hong W. The emerging role of the hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer Cell (2008) 13:188–92. doi: 10.1016/j.ccr.2008.02.011
26. Fernandez LA, Kenney AM. The Hippo in the room: a new look at a key pathway in cell growth and transformation. Cell Cycle (2010) 9:2292–9. doi: 10.4161/cc.9.12.11919
27. Zhao B, Ye X, Yu J, Li L, Li W, Li S, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. (2008) 22:1962–71. doi: 10.1101/gad.1664408
28. Fernandez LA, Squatrito M, Northcott P, Awan A, Holland EC, Taylor MD, et al. Oncogenic YAP promotes radioresistance and genomic instability in medulloblastoma through IGF2-mediated AKT activation. Oncogene (2012) 31:1923–37. doi: 10.1038/onc.2011.379
29. Wehr MC, Holder MV, Gailite I, Saunders RE, Maile TM, Ciirdaeva E, et al. Salt-inducible kinases regulate growth through the Hippo signalling pathway in Drosophila. Nat Cell Biol. (2013) 15:61–71. doi: 10.1038/ncb2658
30. Jiang J, Chang W, Fu Y, Gao Y, Zhao C, Zhang X, et al. SAV1 represses the development of human colorectal cancer by regulating the Akt-mTOR pathway in a YAP-dependent manner. Cell Prolif. (2017) 50:e12351. doi: 10.1111/cpr.12351
31. Zhang Y, Yuan J, Zhang X, Yan F, Huang M, Wang T, et al. Angiomotin promotes the malignant potential of colon cancer cells by activating the YAP-ERK/PI3K-AKT signaling pathway. Oncol Rep. (2016) 36:3619–26. doi: 10.3892/or.2016.5194
32. Artinian N, Cloninger C, Holmes B, Benavides-Serrato A, Bashir T, Gera J. Phosphorylation of the hippo pathway component AMOTL2 by the mTORC2 kinase promotes YAP signaling, resulting in enhanced glioblastoma growth and invasiveness. J Biol Chem. (2015) 290:19387–401. doi: 10.1074/jbc.M115.656587
33. Frame FM, Pellacani D, Collins AT, Simms MS, Mann VM, Jones GD, et al. HDAC inhibitor confers radiosensitivity to prostate stem-like cells. Br J Cancer (2013) 109:3023–33. doi: 10.1038/bjc.2013.691
34. Marampon F, Megiorni F, Camero S, Crescioli C, McDowell HP, Sferra R, et al. HDAC4 and HDAC6 sustain DNA double strand break repair and stem-like phenotype by promoting radioresistance in glioblastoma cells. Cancer Lett. (2017) 397:1–11. doi: 10.1016/j.canlet.2017.03.028
35. Roos WP, Krumm A. The multifaceted influence of histone deacetylases on DNA damage signalling and DNA repair. Nucleic Acids Res. (2016) 44:10017–30. doi: 10.1093/nar/gkw922
36. Tarumoto Y, Lu B, Somerville TDD, Huang YH, Milazzo JP, Wu XS, et al. LKB1, Salt-inducible kinases, and MEF2C are linked dependencies in acute myeloid leukemia. Mol Cell (2018) 69:1017–27 e6. doi: 10.1016/j.molcel.2018.02.011
37. Homminga I, Pieters R, Langerak AW, de Rooi JJ, Stubbs A, Verstegen M, et al. Integrated transcript and genome analyses reveal NKX2–1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell (2011) 19:484–97. doi: 10.1016/j.ccr.2011.02.008
38. O'Hayre M, Vazquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, et al. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev Cancer (2013) 13:412–24. doi: 10.1038/nrc3521
39. Drelon C, Berthon A, Sahut-Barnola I, Mathieu M, Dumontet T, Rodriguez S, et al. PKA inhibits WNT signalling in adrenal cortex zonation and prevents malignant tumour development. Nat Commun. (2016) 7:12751. doi: 10.1038/ncomms12751
40. He X, Zhang L, Chen Y, Remke M, Shih D, Lu F, et al. The G protein alpha subunit Galphas is a tumor suppressor in Sonic hedgehog-driven medulloblastoma. Nat Med. (2014) 20:1035–42. doi: 10.1038/nm.3666
41. Iglesias-Bartolome R, Torres D, Marone R, Feng X, Martin D, Simaan M, et al. Inactivation of a Galpha(s)-PKA tumour suppressor pathway in skin stem cells initiates basal-cell carcinogenesis. Nat Cell Biol. (2015) 17:793–803. doi: 10.1038/ncb3164
42. Pattabiraman DR, Bierie B, Kober KI, Thiru P, Krall JA, Zill C, et al. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science (2016) 351:aad3680. doi: 10.1126/science.aad3680
43. Xing F, Luan Y, Cai J, Wu S, Mai J, Gu J, et al. The anti-warburg effect elicited by the cAMP-PGC1alpha pathway drives differentiation of glioblastoma cells into astrocytes. Cell Rep. (2017) 18:468–81. doi: 10.1016/j.celrep.2016.12.037
44. Amato E, Molin MD, Mafficini A, Yu J, Malleo G, Rusev B, et al. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. J Pathol. (2014) 233:217–27. doi: 10.1002/path.4344
45. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature (2016) 531:47–52. doi: 10.1038/nature16965
46. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. (2017) 23:703–13. doi: 10.1038/nm.4333
47. Cancer Genome Atlas Research Network. Electronic address aadhe, Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell (2017) 32:185–203 e13. doi: 10.1016/j.ccell.2017.07.007
48. Patra KC, Kato Y, Mizukami Y, Widholz S, Boukhali M, Revenco I, et al. Mutant GNAS drives pancreatic tumourigenesis by inducing PKA-mediated SIK suppression and reprogramming lipid metabolism. Nat Cell Biol. (2018) 20:811–22. doi: 10.1038/s41556-018-0122-3
49. Yang FC, Tan BC, Chen WH, Lin YH, Huang JY, Chang HY, et al. Reversible acetylation regulates salt-inducible kinase (SIK2) and its function in autophagy. J Biol Chem. (2013) 288:6227–37. doi: 10.1074/jbc.M112.431239
50. Bon H, Wadhwa K, Schreiner A, Osborne M, Carroll T, Ramos-Montoya A, et al. Salt-inducible kinase 2 regulates mitotic progression and transcription in prostate cancer. Mol Cancer Res. (2015) 13:620–35. doi: 10.1158/1541-7786.MCR-13-0182-T
51. Liu J, Zhu H, Zhong N, Jiang Z, Xu L, Deng Y, et al. Gene silencing of USP1 by lentivirus effectively inhibits proliferation and invasion of human osteosarcoma cells. Int J Oncol. (2016) 49:2549–57. doi: 10.3892/ijo.2016.3752
Keywords: salt-inducible kinase, SIK2, cancer, signaling pathway, target therapy
Citation: Chen F, Chen L, Qin Q and Sun X (2019) Salt-Inducible Kinase 2: An Oncogenic Signal Transmitter and Potential Target for Cancer Therapy. Front. Oncol. 9:18. doi: 10.3389/fonc.2019.00018
Received: 09 October 2018; Accepted: 07 January 2019;
Published: 22 January 2019.
Edited by:
Zhe-Sheng Chen, St. John's University, United StatesReviewed by:
Yun Dai, Virginia Commonwealth University, United StatesLuca Tamagnone, Institute for Cancer Research and Treatment (IRCC), Italy
Xingxiang Pu, Hunan Cancer Hospital, China
Copyright © 2019 Chen, Chen, Qin and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinchen Sun, U3VueGluY2hlbjIwMTJAMTYzLmNvbQ==
†These authors have contributed equally to this work