 Zhi-Wen Pan1†Xiao-Jia Wang2†
Zhi-Wen Pan1†Xiao-Jia Wang2† Tianhui Chen3Xiao-Wen Ding4
Tianhui Chen3Xiao-Wen Ding4 Xiyi Jiang3Yun Gao5Wen-Ju Mo4Yuan Huang2Cai-Jin Lou2
Xiyi Jiang3Yun Gao5Wen-Ju Mo4Yuan Huang2Cai-Jin Lou2 Wen-Ming Cao2*
Wen-Ming Cao2*- 1Department of Clinical Laboratory, Zhejiang Cancer Hospital, Hangzhou, China
- 2Department of Breast Medical Oncology, Zhejiang Cancer Hospital, Hangzhou, China
- 3Group of Molecular Epidemiology & Cancer Precision Prevention (GMECPP), Zhejiang Academy of Medical Sciences (ZJAMS), Hangzhou, China
- 4Department of Breast Cancer Surgery, Zhejiang Cancer Hospital, Hangzhou, China
- 5Institute of Cancer Research, Zhejiang Cancer Hospital, Hangzhou, China
Introduction: FANCC is reported as a novel susceptibility gene for breast cancer, however, its mutation remains unclear in Chinese population. We aimed to identify the germline mutations of FANCC in high-risk breast cancer patients in China.
Methods: 255 BRCA1/2-negative Chinese familial breast and/or ovarian cancer (FBOC) patients were recruited for FANCC germline mutations screen. For whom 90 patients were detected by PCR-sequencing assay, and another 165 patients were detected by a 98-gene panel sequencing assay. The 98-gene panel sequencing assay was also used to screen other possible gene mutations for the patients with FANCC mutations detected by PCR-sequencing assay. Two hundred and fifty sporadic breast cancer (SBC) patients and 248 female non-cancer controls (FNCCs) were recruited for the genotyping analysis. Immunohistochemistry (IHC) analysis was used to evaluate the FANCC expression in patients with FANCC mutation.
Results: We found one rare FANCC deleterious mutation (c.339G>A, p.W113X, 0.4%) and two novel non-synonymous variants (c.51G>C, p.Q17H, 0.4% and c.758C>A, p.A253E, 0.4%) in FBOC patients, whereas none of above mutations was identified in SBC patients or FNCCs. We also found that one novel synonymous variant (c.903A>G, p.A301A) existed in one FBOC patient. Additionally, two non-synonymous SNPs rs201407189 (c.973G>A, p.A325T) and rs1800367 (c.1345G>A, p.V449M), and two synonymous SNPs rs55719336 (c.816C>T, p.I272I) and rs79722116 (c.1407G>A, p.T469T) were identified in FBOC patients.
Conclusion: FANCC deleterious mutations exist in Chinese FBOC patients and investigations on the penetrance and spectrum of FANCC mutations need to be further conducted.
Introduction
Breast cancer is the most common malignancy in Chinese women, which accounts for 11.2% newly diagnosed cases and 9.2% deaths from breast cancer worldwide (1). The onset age is ~ 10 years younger in Chinese women compared to women in western countries (2). The study from our group (3), summarizing the characteristics of germline mutations in breast cancer susceptibility genes in Chinese women with high-risk breast cancer, found that BRCA1/2 mutations accounted for the majority of hereditary breast cancer, while other genes such as TP53, BRIP1, PALB2, CHEK2, RAD50, NBS1, and RAD51C were only responsible for a smaller fraction (3). However, the genetic etiology for more than 80% Chinese women with high-risk breast cancer still remains unknown.
Increasing efforts have been invested to identify novel susceptibility genes that predispose individuals to breast cancer. Several rare moderate-penetrance susceptibility genes including XRCC2, (4) BLM, (5) FANCC, (5) RECQL, (6) MCPH1, (7), and (8) are identified by the next-generation sequencing assay. Thompson et al. (5) found four found four deleterious mutations in DNA repair gene FANCC in 1,410 breast cancer families. Among these mutations, two truncating mutations were found in 15 BRCA1/2-negative high-risk breast cancer families by whole-exome sequencing, and another two were found in 438 validating BRCA1/2-negative breast cancer families screened over the entire coding region by Sanger sequencing. In addition, one mutation in additional 957 BRCA1/2 uninformative breast cancer families was found through the mutation hotspot screening. However, none of these mutations were identified either in the healthy controls or in the 1,000 Genomes Project database. These results suggested that deleterious mutations in FANCC gene play a role in breast cancer predisposition.
Investigations on the germline mutation of FANCC in high-risk breast cancer patients are sparse. Therefore, we aimed at, for the first time in mainland China, assessing the germline mutation of FANCC in high-risk breast cancer patients in women from eastern China, by screening the complete coding regions and exon-intron boundaries of FANCC.
Materials and Methods
Patient/Sample Ascertainment
The eligibility criteria for this study were breast cancer patients who had at least one first- or second-degree relative affected with breast cancer and/or ovarian cancer, regardless of the diagnosis age. A total of 335 unrelated breast cancer patients fulfilled this criterion. All of the participants were ascertained between the years 2008 and 2018 in the Zhejiang Cancer Hospital in Zhejiang, an eastern province of China. Small mutations in BRCA1/2 were analyzed in 133 patients using polymerase chain reaction (PCR)-sequencing assay and also in another 202 patients using 98-gene panel sequencing assay. Large genomic rearrangements (LGRs) in BRCA1/2 were analyzed in all 335 patients using a multiplex ligation-dependent probe amplification (MLPA) assay. Among them, we found 73 breast cancer patients carrying BRCA1/2 small mutations and seven patients carrying BRCA1 LGRs (9, 10). Finally, blood samples from 255 unrelated BRCA1/2-negative breast cancer patients were enrolled for DNA sequencing in this study. Additionally, 250 sporadic breast cancer (SBC) patients and 248 female non-cancer controls (FNCCs) were selected for genotyping analyses in the Zhejiang Cancer Hospital. The use of tissue samples in this study was approved by the Research and Ethical Committee of the Zhejiang Cancer Hospital. All experiments were performed in accordance with the approved guidelines. Written informed consent was obtained from all participating patients prior to clinical data and peripheral blood collection. Peripheral blood samples were collected from each proband and from as many affected relatives as possible. All of the blood samples were collected in EDTA tubes and stored at −80°C.
FANCC Mutation Analysis
Genomic DNA was prepared from peripheral blood leukocytes from the proband of each family using the QIAamp DNA Blood Mini kit (Qiagen, Hilden, Germany). The complete coding regions and exon-intron boundaries of FANCC [NM_000136.2] were screened in the first phase for 90 patients using a PCR-sequencing assay, and another 165 patients were also screened for mutations of FANCC using a 98-gene panel sequencing assay. Furthermore, the patients found with FANCC deleterious mutations in the first phase were further screened for mutations in other 97 genes by the 98-gene panel sequencing assay.
The primers for PCR-sequencing assay were designed using Primer Premier 5.0 (Premier, CA). Overall 14 pairs of primers for amplifying the whole coding sequences and their flanking sequences in introns were synthesized by Invitrogen (Invitrogen, Carlsbad, CA, USA). Information on the primers was listed in Table 1. The reaction conditions were as follows: an initial denaturation at 94°C for 5 min, followed by 30 cycles of denaturing at 94°C for 30 s, annealing at 60°C for 30 s, and the extension at 72°C for 60 s; finally, the reaction was elongated at 72°C for 5 min. All fragments were sequenced using a BigDye Mix and an ABI 3730xl Genetic Analyzer (Applied Biosystems, Foster City, CA), and the data were analyzed by Mutation Surveyor (Softgenetics Inc., USA). Each mutation was confirmed by duplicate independent PCR-sequencing assays. All variants were named according to the Human Genome Variation Society (HGVS) sequence systematic nomenclature (http://www.hgvs.org/mutnomen/). Mutalyzer Name Checker (http://mutalyzer.nl) was used to check variant descriptions.

Table 1. Primers for entire coding exons and intron-exon boundaries of FANCC.
A 98-gene panel was designed using the NEBNext direct sequencing technology conducted by New England Biolabs (Ipswich, MA). The panel contains 98 genes including 24 known and candidate breast cancer susceptibility genes (ATM, BARD1, BLM, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, FANCC, FANCM, FANCR/RAD51, FANCU/XRCC2, MCPH1, MRE11A, NBN, NF1, PLAB2, PETN, RAD50, RAD51C, RAD51D, RECQL, STK11, and TP53), 48 other cancer susceptibility genes (ALK, APC, AXIN2, BAP1, BMPR1A, CASR, CDC73, CDK4, CDKN2A, CEBPA, DICER1, EPCAM, FLCN, GATA2, GPC3, HRAS, KIT, MEN1, MLH1, MSH2, MSH6, MUTYH, MET, NF2, PAX5, PGDFRA, PHOX2B, PMS1, PMS2, POLD1, POLE, PRF1, PRKAR1A, PTCH1, RB1, RET, RUNX1, SDHA, SDHB, SDHC, SDHD, SMAD4, SMARCE1, SUFU, TERT, VHL, WRN, WT1) and other 26 genes (ABRAXAS1, AIP, CDKN1B, CDKN1C, DIS3L2, FANCA, FANCB, FANCD2, FANCE, FANCF, FANCG/XRCC9, FANCI, FANCL, FANCP/SLX4, FANCQ/ERCC4, FANCT/UBE2T, FH, MAX, RECQL4, SDHAF2, SMARCA4, SMARCB1, TERC, TMEM127, TSC1 and TSC2). The 98-gene panel sequencing assay was performed on 165 BRCA1/2-negative unrelated patients and on the patients with FANCC deleterious mutations by Hiseq X sequencing (Illumina, CA, USA). A minimum 500x mean coverage was achieved for the aforementioned genes. Variants were called from raw FASTQ data by running through the in-house bioinformatics pipeline. Variants were strictly interpreted according to the Standards and Guideline from American College of Medical Genetics (ACMG), Genomics, and the Association for Molecular Pathology (11).
Whether the sequence variants were previously reported was checked in public databases, including the 1,000 Genomes Browser (http://browser.1000genomes.org/), the Genome Aggregation database (gnomAD, http://gnomad.broadinstitute.org/), the NCBI SNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/), the Leiden Open Variation Database 3.0 (LOVD 3.0, http://databases.lovd.nl/shared/genes/FANCC), the Exome Variant Server (http://evs.gs.washington.edu/EVS/), Online Mendelian Inheritance in Man (OMIM) (http://www.omim.org/), and ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/).
In silico Prediction
To identify the non-synonymous variants (regardless of whether or not FANCC function was disrupted), we conducted in silico prediction using four comparative evolutionary bioinformatics programs: SIFT (http://http://sift.jcvi.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph/), PROVEAN (http://provean.jcvi.org/index.php), and PANTHER (http://www.pantherdb.org/tools/csnpScoreForm.jsp). MutationTaster (http://www.mutationtaster.org/) analyses were also conducted for functional prediction.
Genotyping in Breast Cancer Cases and Non-cancer Controls
Associations between the selected variants and breast cancer were evaluated in further studies. Three variants were genotyped in a set of 250 SBC patients and 248FNCCs. Genotyping of the variants c.339G>A and c.973G>A was performed with the MassARRAY platform (Sequenom, San Diego, CA, USA) using the iPLEX Gold Assay. The amplification and its extended primers were designed by MassARRAY Designer of Sequenom. The information on the primers was listed in Table 2. The amplification reaction conditions were as follows: an initial denaturation at 94°C for 15 min, followed by 45 cycles of denaturing at 94°C for 20 s, annealing at 56°C for 30 s, and the extension at 72°C for 60 s; finally, the reaction was elongated at 72°C for 3 min. Reaction parameters of single-base extension were an initial incubation at 94°C for 30 s, followed by 40 cycles at 94°C for 5 s with 5 nested cycles of 52°C for 5 s and 80°C for 5 s, respectively. Finally, singe-base extension was completed at 72°C for 3 min. Experimental data were analyzed by Typer software version 4.0 (Sequenom, San Diego, CA, USA). Genotyping of the variants c.51G>C and c.758C>A was done by PCR-sequencing assay. The primers and reaction conditions were the same as those used in the mutation screening for FANCC gene exon1 and exon7.
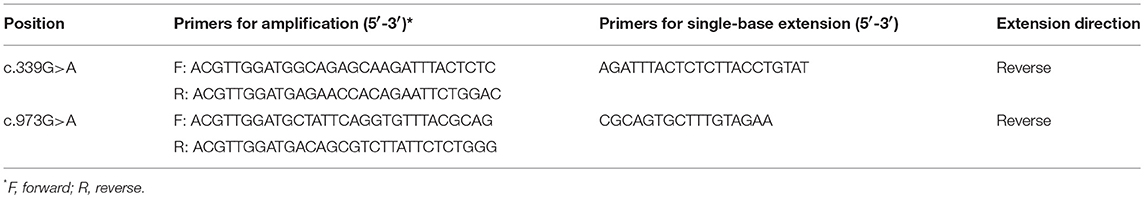
Table 2. Primers for genotyping analysis by MassARRAY platform.
Immunohistochemistry Analysis
The expression of estrogen receptor (ER), progesterone receptor (PR), human epidermal growth factor receptor 2 (HER2), and FANCC was analyzed by immunohistochemistry (IHC) assay in breast cancer tissues. A panel of antibodies was used in this study: anti-ER (clone: SP1 from Roche, #05278406001), anti-PR (clone: 1E2 from Roche, #05277990001), anti-HER-2 (clone: 4B5 from Roche, #05999570001), and anti-FANCC (GeneTex #GTX100400, dilution 1:400). ER and PR positive were defined as ≥1% of tumor cells showing positive nuclear staining. HER2 testing was performed according to the guideline of the American Society of Clinical Oncology/College of American Pathologists for HER2 testing in breast cancer (12). HER2 positive was defined as membrane staining with a score of 3+; when the score ranged 2–3, a fluorescence in situ hybridization (FISH) assay was performed to confirm the HER2 status. FANCC displayed cytoplasmic staining. The scores of FANCC expression were classified as below: 0 for no staining; 1 for weak staining; 2 for moderate staining; and 3 for strong staining; staining score ≥1 was considered positive. All immunostains were assessed independently by three pathologists from Zhejiang Cancer Hospital, Hangzhou, China.
Statistical Analysis
Data were analyzed using the SPSS 17.0 statistical package (SPSS, Chicago, IL, USA). The genotype frequencies of variants in SBC cases and FNCCs were compared using a χ2 test. Two-side P < 0.05 was considered statistically significant.
Results
Among 255 familial breast and/or ovarian cancer (FBOC) patients without BRCA1/2 mutation, we found one (0.4%) patient carrying a non-sense mutation (c.339G>A, W113X), which was also observed in an individual with hereditary cancer-predisposing syndrome, classified as pathogenic/likely pathogenic in ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/). This mutation was not reported in other public databases, including PubMed (https://www.ncbi.nlm.nih.gov/pubmed/), 1,000 Genomes Browser, NCBI SNP database, LOVD 3.0, and Exome Variant Server. Moreover, this mutation was found in neither 250 SBC cases nor 248 FNCCs in our cohort (Table 3). The pedigree of the mutation carrier and electropherogram for the proband are presented in Figure 1. This patient with FANCC c.339G>A mutation was diagnosed with invasive ductal breast cancer at the age of 44 years. IHC analysis showed positive results for the patient's ER, PR, and HER2 while showing a negative result for FANCC (Figure 2). The patient's mother also had breast cancer whereas blood samples from her mother were not available for the mutation analyses. The 98-gene panel sequencing assay identified 195 germline variants including FANCC c.339G>A (Supplementary Table 1). None of these variants except FANCC c.339G>A could be classified as pathogenic or likely pathogenic through the combination of ClinVar database, in silico prediction and gnomAD.
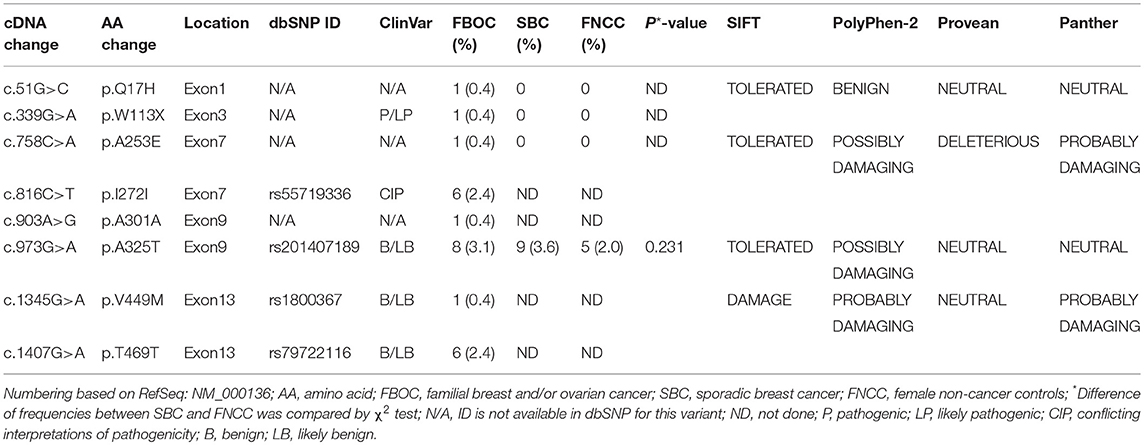
Table 3. The FANCC variants among familial breast and/or ovarian cancer patients (n = 255), sporadic breast cancer patients (n = 250) and female non-cancer controls (n = 248).
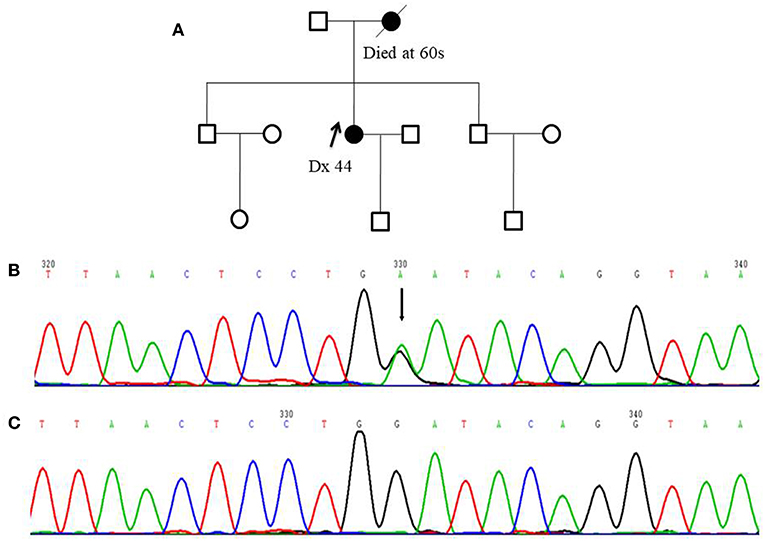
Figure 1. Pedigree of one FANCC deleterious mutation case and an electropherogram for the proband with the identified mutation: (A) The proband of the family carrying FANCC c.339G>A mutation (indicated by black arrow) (B) The electropherogram of FANCC c.339G>A mutation (affected base is indicated by black arrow) (C) Sequence matching wildtype.
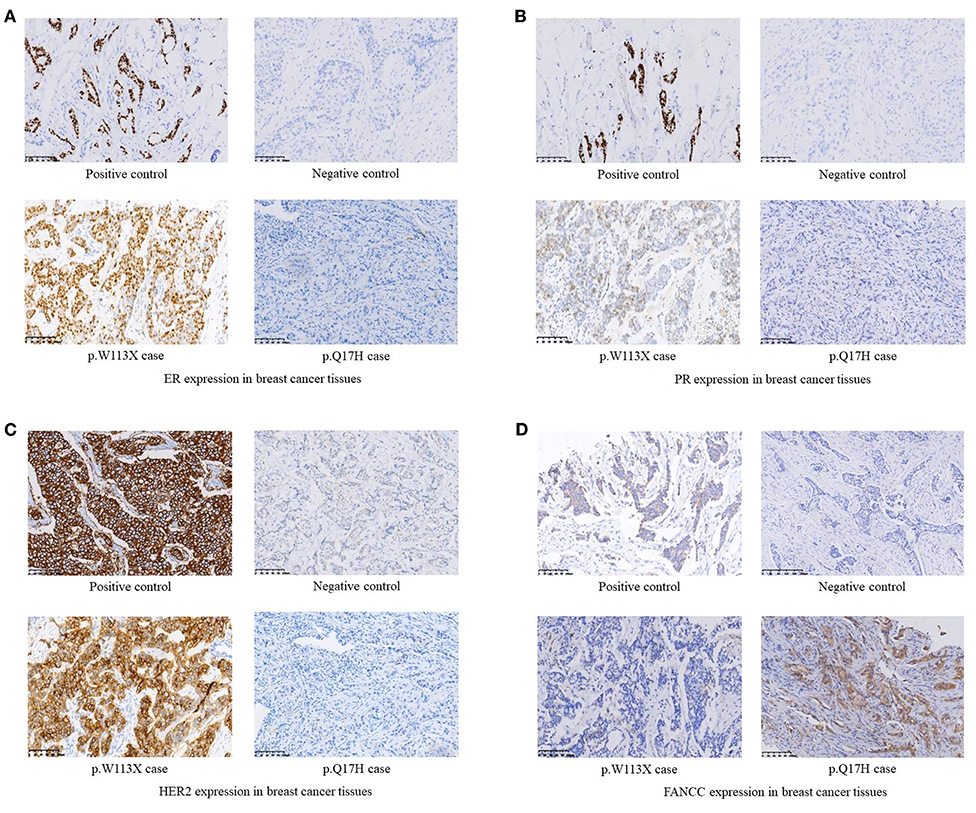
Figure 2. (A) ER expression in breast cancer tissues. (B) PR expression in breast cancer tissues. (C) HER2 expression in breast cancer tissues. (D) FANCC expression in breast cancer tissues.
Two novel non-synonymous variants (c.51G>C, p.Q17H and c.758C>A, p.A253E) and one novel synonymous variant (c.903A>G, p.A301A) were identified in three unrelated patients, which were not reported in any public databases (Table 3). The patient with FANCC c.51G>C mutation was diagnosed with triple-negative (ER-, PR- and HER2-) breast cancer at the age of 41 years (Figure 2). She suffered from bone metastasis 3 years later after a modified radical mastectomy and died 6 years later. Familial clustering of tumors was also found, including ovarian cancer in her maternal grandmother, breast cancer in one of her maternal aunts, and liver cancer in another maternal aunt. In our case-control cohort, c.51G>C (p.Q17H) was not found in neither 250 SBC patients nor 248 FNCCs. In silico analysis suggested that this variant tended to be benign. Moreover, IHC analysis showed that FANCC was positive in this patient (Figure 2). The other non-synonymous variant c.758C>A (p.A253E) was found in another breast cancer patient who was diagnosed at 42 years old, with ER and PR positive, and HER2 negative. Her sister and paternal sister were diagnosed with breast cancer at 54 and 52 years old, respectively. Unfortunately, breast cancer tissues from this patient and blood samples from her sisters were not available for the further analysis. In our case-control cohort, c.758C>A was found in neither 250 SBC patients nor 248 FNCCs. As for in silico analysis, three algorithms suggested that this variant tended to be deleterious while the SIFT algorithm predicted it to be benign.
Among 255 FBOC patients, four SNPs (rs55719336, rs201407189, rs1800367, and rs79722116) were found with variant frequencies (Table 3). Two SNPs (rs201407189 and rs1800367) were non-synonymous (c.973G>A, p.A325T, and c.1345G>A, p.V449M) and the other two (rs55719336 and rs79722116) were synonymous (c.816C>T, p.I272I and c.1407G>A, p.T469T). The frequency of SNP rs201407189 (corresponding to FANCC c.973G>A) was similar in the aforementioned 250 SBC patients and 248 FNCCs (3.6 vs. 2.0%, respectively, P = 0.231). Although some algorithms predicted that p.A325T (rs201407189) and p.V449M (rs1800367) were likely associated with the protein function damage, these two SNPs (rs201407189, rs1800367) and rs79722116 were classified as benign or likely benign in ClinVar database. However, the synonymous variant (c.816C>T, rs55719336) was classified as conflicting interpretations of pathogenicity in ClinVar database. Actually, most of the submitters classified it as benign or likely benign, except one submitter who classified it as uncertain significance.
Discussion
In this study, we identified one rare FANCC deleterious mutation (c.339G>A, W113X) in one patient (0.4%) by screening the germline mutations of FANCC gene in 255 BRCA1/2-negative Chinese women with FBOC. To our knowledge, this is the first report of a FANCC deleterious mutation in Chinese population which might have a distinct genetic landscape of breast cancer compared to the Caucasian population. We also identified two novel non-synonymous variants (c.51G>C, p.Q17H and c.758C>A, p.A253E), one novel synonymous variant (c.903A>G, p.A301A), two non-synonymous SNPs rs201407189 (c.973G>A, p.A325T) and rs1800367 (c.1345G>A, p.V449M), and two synonymous SNPs rs55719336 (c.816C>T, p.I272I) and rs79722116 (c.1407G>A, p.T469T).
FA is an autosomal recessive syndrome characterized as progressive bone marrow failure, congenital abnormalities and susceptibility to cancer. To date, 16 genes have been identified to be associated with FA (13). FA proteins work together with BRCA2/Rad51-mediated homologous recombination in double-stranded DNA repair (14). Biallelic inactivations of BRCA2 and BRCA1 can cause FA complementation group D1 (15) and a new FA subtype (16), respectively. Increasing evidence showed that breast cancer and FA share some susceptibility genes depending upon the monoallelic mutation or biallelic mutation. Recently, the FA susceptibility genes FANCJ/BRIP1 (17), FANCN/PALB2 (18), FANCO/RAD51C (19), FANCP/SLX4 (20), and FANCM (21) have also been identified as breast cancer susceptibility genes.
Homozygous mutations in FANCC are responsible for FA complementation group C. FANCC is an essential substrate for forming a ternary complex together with FANCE and FANCD2, which is responsible for the FA DNA damage response pathway (22). Poor survival in breast cancer patients with alternative FANCC genes suggested that FANCC is a breast cancer suppressor (23). A study investigated the risk of cancer among FA gene heterozygous carriers, which recruited 944 relatives (784 grandparents and 160 other relatives) of FA probands from 312 families. The results showed that breast cancer, but not other cancers, had a significantly higher rate among carriers' grandmothers (SIR: 1.7; 95% CI: 1.1–2.7). Moreover, grandmothers who were FANCC mutation carriers had the highest risk of breast cancer (SIR: 2.4; 95% CI: 1.1–5.2) (24). However, another study which recruited 42 Ashkenazi Jewish women who were FANCC heterozygous carriers showed that the risk of breast cancer was not significantly elevated in the carriers (both 2.2% for the carriers and controls) (25). Additionally, the number of cancer cases among families with FA carriers were significantly fewer than those in the controls (25). These inconsistent results between studies might be related to the small sample size or multiple comparisons and the younger age of FANCC carriers.
The frequency of FANCC mutations in high-risk breast cancer patients has been under-investigated. The first study investigating FANCC germline mutations enrolled 88 BRCA1/2-negative familial breast cancer patients from the United Kingdom, finding no deleterious mutations by conformation sensitive gel electrophoresis followed by a sequencing assay (26). In 2012, Thompson et al. (5) identified the first FANCC deleterious mutations in breast cancer families, with the frequency of FANCC mutations of 0.7% (3/453) in their cohort of BRCA1/2-negative familial breast cancer patients, indicating that FANCC germline mutations in high-risk breast cancer patients would be very rare. Recently, three studies which were conducted in the United States (27), Russia (28), and China (29) used gene panel sequencing to screen the multi-gene germline mutations in high-risk BRCA1/2-negative breast cancer patients, yet none of the FANCC germline mutations were found. In our cohort of 255 BRCA1/2-negative FBOC patients, the frequency of FANCC deleterious mutations was 0.4%, which was comparable to the result reported in the Caucasian population. Since FA is very rare in the Chinese population, the mutation frequency and spectrum of FA genes remain unknown.
FANCC c.339G>A (W113X) was classified as pathogenic/likely pathogenic in ClinVar database while it was not reported in breast cancer patient. This patient had neither small mutations nor LGRs in BRCA1/2 genes. The germline mutations of 98 genes including 24 known and candidate breast cancer susceptibility genes were screened by a gene panel sequencing assay, and no pathogenic or likely pathogenic mutation was found except FANCC c.339G>A. Moreover, IHC analysis showed that FANCC was negative in the breast cancer tissue of this patient. These results suggested that FANCC c.339G>A was a breast cancer susceptibility mutation.
Conclusion
In conclusion, we have identified, for the first time, a FANCC deleterious mutation in one BRCA1/2-negative Chinese FBOC patient. Although this FANCC germline mutation was rare, it might have important clinic implications since FANCC is a breast cancer suppressor and is responsible for FA complementation group C. The number of identified genes responsible for moderate to high-risk susceptibility of breast cancer is increasing, which can account for ~ 1% of the affected families. Further investigations on the penetrance and spectrum of FANCC mutations are highly warranted for the genetic counseling among Chinese FBOC patients.
Clinical Practice Points
1. The genetic etiology in more than 80% of Chinese women with high-risk breast cancer remains unknown. Increasing evidence has shown that breast cancer and Fanconi anemia (FA) share some susceptibility genes depending upon the monoallelic mutation or biallelic mutation.
2. Homozygous mutations in FANCC are responsible for FA complementation group C. FANCC is a breast cancer suppressor (22). Heterozygous FA gene carriers had a significantly higher risk of breast cancer among carriers' grandmothers (SIR: 1.7; 95% CI: 1.1–2.7). Moreover, grandmothers who were FANCC mutation carriers had the highest risk of breast cancer (SIR: 2.4; 95% CI: 1.1–5.2) (23). The study conducted by Thompson et al. (5) showed that FANCC was a novel breast cancer susceptibility gene in the Caucasian population, with the mutation frequency of 0.7% (3/453). Our study provided evidence, for the first time, that the FANCC deleterious mutation exists in Chinese familial breast cancer patients, with a frequency of 0.4% in our cohort.
3. Recently, genetic testing using gene panel sequencing assay and subsequent counseling for cancer (such as breast cancer) in high-risk populations has become more and more popular in China. However, researchers and clinicians might use different gene panels. Since FANCC deleterious mutations existed in Chinese high-risk breast cancer patients according to our data, genetic counseling for breast cancer shall also include FANCC deleterious mutations, though further investigations on the penetrance and spectrum of FANCC mutations are highly warranted.
Author Contributions
W-MC is responsible for the study concept and design. W-MC, X-JW, and TC obtained funding. Z-WP and W-MC acquired data. Z-WP, X-WD, XJ, YG and W-MC analyzed and interpreted data. W-JM, YH, and C-JL collected the patients' samples and clinic data. W-MC and X-JW drafted the manuscript, and all authors revised it for important intellectual content. W-MC is the guarantor of this work.
Funding
This work was supported by grants from Key Research-Development Program of Zhejiang Province (grant number: 2017C03013, 2019C04001), the Natural Science Foundation of Zhejiang Province, China (grant number: LY17H160038), the Science and Technology Program offered by the Health Bureau of Zhejiang Province, China (grant numbers: 2014KYA006 and 2017RC014), Joint Key Program of Zhejiang Province- Ministry of Health (grant number: WKJ-ZJ-1714), and Qianjiang Talents Fund of Zhejiang Province (grant number: QJD1602026).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.00169/full#supplementary-material
Supplementary Table 1. Germline variants of 98 genes in the patient with FANCC deleterious mutation (c.339G>A, p.W113X).
Abbreviations
FA, Fanconi anemia; SNPs, single nucleotide polymorphisms; LGRs, large genomic rearrangements; PCR, polymerase chain reaction; MLPA, multiplex ligation-dependent probe amplification; HGVS, Human Genome Variation Society; LOVD, Leiden Open Variation Database 3.0; ACMG, American College of Medical Genetics; OMIM, Online Mendelian Inheritance in Man; AA, Amino Acid; FBOC, familial breast and/or ovarian cancer; SBC, sporadic breast cancer; FNCC, female non-cancer control; IHC, immunohistochemistry; ER, estrogen receptor; PR, progesterone receptor; HER2, human epidermal growth factor receptor 2; FISH, fluorescence in situ hybridization.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. (2015) 136:E359–86. doi: 10.1002/ijc.29210
2. Fan L, Strasser-Weippl K, Li JJ, St Louis J, Finkelstein DM, Yu KD, et al. Breast cancer in China. Lancet Oncol. (2014) 15:e279–89. doi: 10.1016/S1470-2045(13)70567-9
3. Cao W, Wang X, Li JC. Hereditary breast cancer in the Han Chinese population. J Epidemiologic. (2013) 23:75–84. doi: 10.2188/jea.JE20120043
4. Park DJ, Lesueur F, Nguyen-Dumont T, Pertesi M, Odefrey F, Hammet F, et al. Rare mutations in XRCC2 increase the risk of breast cancer. Am J Hum Genet. (2012) 90:734–9. doi: 10.1016/j.ajhg.2012.02.027
5. Thompson ER, Doyle MA, Ryland GL, Rowley SM, Choong DY, Tothill RW, et al. Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet. (2012) 8: e1002894. doi: 10.1371/journal.pgen.1002894
6. Cybulski C, Carrot-Zhang J, Kluzniak W, Rivera B, Kashyap A, Wokołorczyk D, et al. Germline RECQL mutations are associated with breast cancer susceptibility. Nat Genet. (2015) 47:643–6. doi: 10.1038/ng.3284
7. Mantere T, Winqvist R, Kauppila S, Grip M, Jukkola-Vuorinen A, Tervasmäki A, et al. Targeted next-generation sequencing identifies a recurrent mutation in MCPH1 Associating with hereditary breast cancer susceptibility. PLoS Genet. (2016) 12:1–14. doi: 10.1371/journal.pgen.1005816
8. Vijai J, Topka S, Villano D, Ravichandran V, Maxwell KN, Maria A, et al. A recurrent ERCC3 truncating mutation confers moderate risk for breast cancer. Cancer Discov. (2016) 6:1267–75. doi: 10.1158/2159-8290.CD-16-0487
9. Cao WM, Gao Y, Yang HJ, Xie SN, Ding XW, Pan ZW, et al. Novel germline mutations and unclassified variants of BRCA1 and BRCA2 genes in Chinese women with familial breast/ovarian cancer. BMC Cancer. (2016) 16:64. doi: 10.1186/s12885–016-2107–6
10. Cao W, Wang X, Gao Y, Yang H, Li JC. BRCA1 Germ-Line Mutations and Tumor Characteristics in eastern Chinese women with familial breast cancer. Anat Rec. (2013) 296:273–8. doi: 10.1002/ar.22628
11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–424. doi: 10.1038/gim.2015.30
12. Wolff AC, Hammond MEH, Allison KH, Harvey BE, Mangu PB, Bartlett JMS, et al. Human epidermal growth factor receptor 2 testing in breast cancer: American society of clinical oncology/college of American pathologists clinical practice guideline focused update. J Clin Oncol. (2018) 36:2105–22. doi: 10.1200/JCO.2018.77.8738
13. Dong H, Nebert DW, Bruford EA, Thompson DC, Joenje H, Vasiliou V. Update of the human and mouse Fanconi anemia genes. Hum Genomics. (2015) 9:32. doi: 10.1186/s40246-015-0054-y
14. Kitao H, Yamamoto K, Matsushita N, Ohzeki M, Ishiai M, Takata M. Functional interplay between BRCA2/FancD1 and FancC in DNA repair. J Biol Chem. (2006) 281:21312–20. doi: 10.1074/jbc.M603290200
15. Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. (2002) 297:606–9. doi: 10.1126/science.1073834
16. Sawyer SL, Tian L, Kähkönen M, Schwartzentruber J, Kircher M. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. (2015) 5:135–42. doi: 10.1158/2159-8290.CD-14-1156
17. Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. (2006) 38:1239–41. doi: 10.1038/ng1902
18. Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. (2007) 39:165–7. doi: 10.1038/ng1959
19. Meindl A, Hellebrand H, Wiek C, Erven V, Wappenschmidt B, Niederacher D, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. (2010) 42:410–4. doi: 10.1038/ng.569
20. Shah S, Kim Y, Ostrovnaya I, Murali R, Schrader KA, Lach FP, et al. Assessment of SLX4 mutations in hereditary breast cancers. PLoS ONE. (2013) 8:4–8. doi: 10.1371/journal.pone.0066961
21. Kiiski JI, Pelttari LM, Khan S, Freysteinsdottir ES, Reynisdottir I, Hart SN, et al. Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast cancer. Proc Natl Acad Sci USA. (2014) 111:15172–7. doi: 10.1073/pnas.1407909111
22. Gordon SM, Alon N, Buchwald M. FANCC, FANCE, and FANCD2 form a ternary complex essential to the integrity of the fanconi anemia DNA damage response pathway. J Biol Chem. (2005) 280:36118–25. doi: 10.1074/jbc.M507758200
23. Sinha S, Singh RK, Alam N, Roy A, Roychoudhury S, Panda CK. Alterations in candidate genes PHF2, FANCC, PTCH1 and XPA at chromosomal 9q22.3 region: Pathological significance in early- and late-onset breast carcinoma. Mol Cancer. (2008) 7:84. doi: 10.1186/1476–4598-7–84
24. Berwick M, Satagopan JM, Ben-Porat L, Carlson A, Mah K, Henry R, et al. Genetic heterogeneity among fanconi anemia heterozygotes and risk of cancer. Cancer Res. (2007) 67:9591–6. doi: 10.1158/0008-5472.CAN-07-1501
25. Baris HN, Kedar I, Halpern GJ, Shohat T, Magal N, Ludman MD, et al. Prevalence of breast and colorectal cancer in Ashkenazi Jewish carriers of fanconi anemia and bloom syndrome. Isr Med Assoc J. (2007) 9:847–50.
26. Seal S, Barfoot R, Jayatilake H, Smith P, Renwick A, Bascombe L, et al. Evaluation of fanconi anemia genes in familial breast cancer predisposition. Cancer Res. (2003) 63:8596–9.
27. Kurian AW, Hare EE, Mills MA, Kingham KE, McPherson L, Whittemore AS, et al. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol. (2014) 32:2001–9. doi: 10.1200/JCO.2013.53.6607
28. Sokolenko AP, Preobrazhenskaya EV, Aleksakhina SN, Iyevleva AG, Mitiushkina NV, Zaitseva OA, et al. Candidate gene analysis of BRCA1/2 mutation-negative high-risk Russian breast cancer patients. Cancer Lett. (2015) 359:259–61. doi: 10.1016/j.canlet.2015.01.022
Keywords: Chinese, familial breast cancer, familial ovarian cancer, FANCC, deleterious mutation, susceptibility
Citation: Pan Z-W, Wang X-J, Chen T, Ding X-W, Jiang X, Gao Y, Mo W-J, Huang Y, Lou C-J and Cao W-M (2019) Deleterious Mutations in DNA Repair Gene FANCC Exist in BRCA1/2-Negative Chinese Familial Breast and/or Ovarian Cancer Patients. Front. Oncol. 9:169. doi: 10.3389/fonc.2019.00169
Received: 02 July 2018; Accepted: 26 February 2019;
Published: 22 March 2019.
Edited by:
Jianguang Ji, Lund University, SwedenReviewed by:
Zhe Chen, Zhejiang Chinese Medical University, ChinaYueqin Yang, University of Utah Hospital, United States
Copyright © 2019 Pan, Wang, Chen, Ding, Jiang, Gao, Mo, Huang, Lou and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen-Ming Cao, Y2Fvd21AempjYy5vcmcuY24=
†These authors have contributed equally to the work and are first authors