 Tanja Eisemann
Tanja Eisemann Barbara Costa
Barbara Costa Heike Peterziel1,3,4†
Heike Peterziel1,3,4† Peter Angel
Peter Angel- 1Division of Signal Transduction and Growth Control, DKFZ/ZMBH Alliance, Heidelberg, Germany
- 2Faculty of Biosciences, University Heidelberg, Heidelberg, Germany
- 3Translational Program, Hopp Children's Cancer Center at NCT Heidelberg (KiTZ), University Hospital and DKFZ Heidelberg, Heidelberg, Germany
- 4Clinical Cooperation Unit Pediatric Oncology, DKFZ, German Consortium for Translational Cancer Research (DKTK), Heidelberg, Germany
The dynamic and interactive tumor microenvironment is conceived as a considerable parameter in tumor development and therapy response. Implementing this knowledge in the development of future cancer treatments could provide novel options in the combat of highly aggressive and difficult-to-treat tumors such as gliomas. One compartment of the tumor microenvironment that has gained growing interest is the immune system. As endogenous defense machinery the immune system has the capacity to fight against cancer cells. This, however, is frequently circumvented by tumor cells engaging immune-regulatory mechanisms that disable tumor-directed immune responses. Thus, in order to unlock the immune system against cancer cells, it is crucial to characterize in great detail individual tumor-associated immune cell subpopulations and dissect whether and how they influence immune evasion. In this study we investigated the function of a tumor-associated myeloid cell subpopulation characterized by podoplanin expression on the development of high-grade glioma tumors. Here, we show that the deletion of podoplanin in myeloid cells results in increased (CD8+) T-cell infiltrates and significantly prolonged survival in an orthotopic transplantation model. In vitro co-cultivation experiments indicate a podoplanin-dependent transcriptional regulation of arginase-1, a well-known player in myeloid cell-mediated immune suppression. These findings identify podoplanin positive myeloid cells as one novel mediator of the glioma-induced immune suppression. Thus, the targeted ablation of podoplanin positive myeloid cells could be included in combinatorial cancer therapies to enhance immune-mediated tumor elimination.
Introduction
Emerging evidence supports the concept that the tumor microenvironment fundamentally impacts on tumor progression and treatment response [reviewed by (1)]. The microenvironment of most solid tumors is comprised of various non-neoplastic cell types, including fibroblasts, pericytes, endothelial, and immune cells. Reprogrammed by tumor cells, each of these stromal cell types has been shown to secrete growth factors, chemokines, anti-apoptotic, and pro-angiogenic factors or extracellular matrix components changing the local environment into a tumor-promoting milieu [as reviewed by (2–5)]. This knowledge has raised the intriguing possibility to inhibit tumor progression by targeting its supportive microenvironment. There is great hope that the development of adjuvant therapies against the tumor microenvironment will provide additional new treatment options for highly resistant and difficult to treat tumors such as gliomas.
High-grade gliomas, the most common primary brain tumors in adults, belong to the tumors with worst prognosis and highest mortality (6). Although the brain has been considered for decades as “immune privileged” with restricted access for immune cells, the immune system has more and more been recognized as a critical player in brain tumor biology (7, 8). In brain tumors, resident microglia, and infiltrated macrophages represent the most prevalent immune cells. Dependent on the genetic make-up and grade of the tumor, microglia and infiltrating macrophages can account for 30–50% of the entire tumor mass (9). Previously, it has been shown that tumor cells “educate” these myeloid cells to promote tumor cell proliferation (10), invasion (11), angiogenesis (12), and suppression of apoptosis (9). The “re-education” or depletion of tumor-associated microglia and macrophages by colony-stimulating factor 1 receptor (CSF1R) inhibition significantly reduces glioma progression (9, 13). However, there is compelling evidence that subsets of glioma-associated macrophages do not only act directly on tumor cells but furthermore create a local or even systemic immune suppression, helping tumor cells to evade rejection by the adaptive immune system (14–17). Importantly, it has been shown that microglia and macrophages do not only differ in their origin (18) but also in their response to glioma growth (17, 19), and are consequently suspected to influence tumor progression and anti-cancer therapy in different ways (20). Thus, in order to inhibit the pro-tumoral interaction of innate immune cells and tumor cells most efficiently, it is essential to comprehensively characterize individual tumor-associated immune cell subpopulations and decipher how they contribute to tumor progression.
Previously, the glioma-associated myeloid compartment has been reported to express the transmembrane protein podoplanin (PDPN) (21, 22). PDPN is a highly glycosylated protein that is constitutively expressed by various cell types of the human and rodent body. Despite its abundant expression, the function of PDPN has remained elusive for many cell types [for review see (23)]. Similarly, the effect of increased Pdpn expression in many pathologies has not been clarified yet. Here, Pdpn is expressed in neoplastic cells and cancer-associated fibroblasts of various cancer entities (24–27), in the endothelial vessel wall during venous thrombosis (28), in fibroblastic reticular cells during lymph node expansion (29) and in multiple immune cell populations (25, 30), including macrophages during inflammation (31–33). Interestingly, although PDPN on inflammatory macrophages has been reported as a critical player in the inflammation control during sepsis and acute respiratory distress syndrome (34, 35), the function of PDPN positive (PDPN+) macrophages in cancer has remained unexplored. Thus, in this study we examined tumor-associated PDPN+ myeloid cells and their effect on glioma development and immune cell infiltration. Here we show that the deletion of Pdpn in myeloid cells results in increased T-cell infiltrates and significantly prolonged survival, identifying the PDPN+ myeloid cell population as one mediator of the glioma-induced immune suppression.
Materials and Methods
Tumor Cell Cultivation and Transduction
Tlx-CreERT2; Ptenflox/flox mice (27) crossed with Tp53flox/flox animals (The Jackson Laboratory) spontaneously developed high grade glioma tumors, from which primary murine tumor cells DKO11804 were isolated. Tumor tissue was minced and digested in Leibovitz medium supplemented with 12 U/ml papain, 100 U/ml DNase and 0.5 mM EDTA for 15 min at 37°C. After filtration (70 μm) and lysis of erythrocytes tumor cells were cultured as spheroids in DMEM/F12 medium (life technologies) containing N2 supplement (life technologies), 20 ng/ml of each EGF and FGFb (promokine), 2 mM L-glutamine and 100 U/ml penicillin/streptomycin at 37°C and 5% CO2. Lentiviral transduction with a construct encoding mCherry was performed in order to label the murine cells for subsequent transplantation assays. For virus production we transfected one 10 cm dish HEK293T cells with 8 μg target vector; 4 μg psPAX2; 2 μg pVSVg and 42 μg polyethylenimine (Alfa Aesar). HEK293T cells were cultivated in N2-supplemented serum-free medium. Virus-containing medium was transferred from HEK293T cells to the target cells and replaced by cultivation medium after 24 h. Upon recovery from infection recipient cells were sorted for mCherry expression by fluorescence activated cell sorting (FACS).
Established cell lines LN308; LN319; GL261 and SMA-560 were cultivated as adherent monolayers in DMEM supplemented with 10% FBS, 2 mM L-glutamine and 100 U/ml penicillin/streptomycin at 37°C and 5% CO2. GL261 and SMA-560 were provided by Dr. Michael Platten (DKFZ/University Hospital Heidelberg). Human glioma cell lines LN308 and LN319 were provided by Dr. Wolfgang Wick (DKFZ/University Hospital Heidelberg) and authenticated in April 2018 using Multiplex Cell Authentication by Multiplexion (Heidelberg, Germany) as described recently (36). The SNP profiles matched known profiles.
Intracranial Injections
For orthotopic injections of DKO11804 glioma cells we used a motorized stereotaxic instrument (Neurostar). 5 × 105 tumor cells were injected in 2 μl PBS 2 mm lateral (right) and 3 mm ventral to the bregma with a speed of 0.2 μl/min. Eight to ten weeks old control [Pdpnflox/flox, (37)] or conditional knockout animals (cKO, Csf1r-Cre (38); Pdpnflox/flox) were used as recipients. Mice were sacrificed when exhibiting termination criteria such as loss of > 20% body weight or poor general condition. Length of animal survival was measured by means of Kaplan–Meier estimate. All animal experiments were approved by the responsible authority for animal experiments (Regierungspräsidium Karlsruhe, Germany) and performed in conformity with the German Law for Animal Protection.
Immunohistochemistry and Immunofluorescence
When animals had to be sacrificed, brains were fixed in 4% PFA and embedded in paraffin for histological examination. 6 μm histological sections were stained according to standard immunohistochemistry protocols and counterstained with haematoxylin. The antibodies used were specific for mCherry (abcam, ab167453), CD3 (Biorad, MCA1477T), Pdpn (The Developmental Studies Hybridoma Bank), and Iba1 (Wako, 01919741). The analysis of CD3+ cell numbers was performed using an ImageJ macro programmed by Dr. Damir Krunic (DKFZ core facility for light microscopy).
Preparation and Flow Cytometry of Blood, Lymph Nodes, and Brain
Blood was collected in an EDTA-coated vessel and erythrocytes lysed by ACK lysing buffer. After washing in 1% FBS PBS, cells were counted.
Right mandibular lymph nodes were removed and gathered in 1% FBS PBS. Subsequently, tissue was minced between two frosted object slides which were finally rinsed with 1% FBS PBS. After filtering through a 70 μm strainer cells were once washed and counted.
Both hemispheres of unchallenged or the tumor-bearing hemisphere of treated mice were isolated and chopped using a scalpel. As the proteins of interest CD4, CD8, and PDPN were sensitive to papain treatment, we used 1.5 ml accumax supplemented with 0.75 mM EDTA and 200 U DNase I for tissue dissociation. After 15–20 min incubation at RT and thorough resuspension in 10 ml washing medium (DMEM, Sigma), cell suspension was filtered through a 70 μm cell strainer and centrifuged for 4 min at 1,500 rpm. After erythrocyte lysis and another washing step, the pellet was resuspended in 10 ml 50% isotonic Percoll/washing medium and centrifuged at 764 g at 4°C for 20 min. Half of the supernatant was removed and fresh washing medium added. After another centrifugation step of 10 min cells were counted.
Cells were stained for flow cytometry using following antibodies: CD3 (Biolegend, 100306), CD4 (eBioscience, 17-0042-81), CD8 (Biolegend, 100707), CD11b (Biolegend, 101207), CD45 (BD Biosciences, 553080), Pdpn (BioLegend, 127409), Syrian hamster isotype control (Biolegend, 402012). CD16/CD32 block (eBioscience, 14-0161-82) was performed prior to staining. Live/dead discrimination was based on 7AAD staining.
Isolation of Microglia, Bone Marrow-Derived Macrophages, and Spleen Macrophages
Microglia cells were isolated from newborn pups (P0-P5). Brains were isolated and meninges removed. Cortices were mechanically dissociated using a glass pestle and a 70 μm cell strainer. After washing with plain DMEM medium, cell suspension of one brain was transferred in DMEM (10% FBS, 2 mM L-glutamine, 100 U/ml penicillin/streptomycin) to one poly-L-lysine coated 3 cm dish. Debris was washed off the next day and medium refreshed. By this protocol, cells are kept in co-cultivation with astrocytes (mixed glia culture).
Bone marrow-derived macrophages were generated by flushing femur and tibia of adult mice. After erythrocyte lysis and a washing step with plain DMEM, cells obtained from one leg were seeded on one non-coated 10 cm petri dish. For cultivation we used RMPI 1640 GlutaMAX (thermofisher) supplemented with 10% FBS, 100 U/ml penicillin/streptomycin, 50 mM β-mercaptoethanol and 30% L929-MCSF-conditioned medium (39).
Spleen macrophages were isolated by mechanical dissociation of adult spleens using the piston of a 5 ml syringe and a 70 μm cell strainer. After erythrocyte lysis and a washing step, cells were plated on non-coated plates. Non-adherent cells were washed off after 24 h. Spleen macrophage medium contained 10% FBS, 100 U/ml penicillin/streptomycin, 50 mM β-mercaptoethanol and 10% L929-MCSF-conditioned medium in RMPI 1640 GlutaMAX (thermofisher).
Co-cultivation Experiments
To assess the effect of tumor cells on Pdpn expression of myeloid cells, 2 × 105 BMDM or spleen macrophages were co-cultivated with 0.5 × 105 LN308 tumor cells for 48 h in coated 6 wells. In case of microglia, LN308 were added to confluent mixed glia cultures. After 48 h, co-cultures of tumor cells and BMDM or spleen macrophages were detached by 5 min incubation with accutase and gentle usage of a cell lifter. For tumor cell and microglia co-cultures, a mild trypsinization protocol (0.05% trypsin in serum-free medium) (40) was followed in order to reduce the number of astrocytes and tumor cells in flow cytometry analysis. Subsequently, microglia cells were detached as described for the other myeloid cell types. Prior to staining of CD11b and PDPN, a CD16/CD32 block was performed.
Transwell Permeable Supports (Falcon, 353090) were utilized for indirect co-cultivation of distinct cell types to examine the effect of tumor cells on the transcription of M1/M2 markers in macrophages. 2 × 105 BMDM were seeded per 6 well in above described macrophage medium. 1 × 105 LN308, LN319, GL261, or SMA-560 tumor cells were seeded on top of the transwell membrane. After 72 h, the transwell and medium was removed and macrophages lysed for RNA isolation using RNeasy kit (Qiagen). Five hundred nanograms of RNA was transcribed in cDNA using RevertAid M-MuLV reverse transcriptase (Thermo Fisher Scientific).
Quantitative Real-Time PCR
For quantitative gene expression analysis, 40 cycles of real-time PCR were performed on the StepOnePlus real-time detection system (Applied Biosystems). Every PCR reaction was carried out in duplicates with 2.5 ng of cDNA in a final volume of 12.5 μl Power SYBR® Green PCR Master Mix (Applied Biosystem). StepOneTM Software v2.2 was used for data analysis. Ppia was used as housekeeping gene to normalize target gene expression. We used the following primer sequences for detection of Pdpn transcripts (41): FW 5′-agagaacacgagagtacaacc-3′; RV 5′-caacaatgaagatccctccgac-3′, Arg1 (42): FW 5′-TTGGGTGGATGCTCACACTG-3′; RV 5′-TTGCCCATGCAGATTCCC-3′; Ppia: FW 5′-AATTCATGTGCCAGGGTGGTG-3′; RV 5′-TGCCTTCTTTCACCTTCCCAA-3′. Additional primer sequences are given in Table S1.
Results
Glioma Tumors Harbor a PDPN+ Myeloid Subpopulation
We analyzed the myeloid compartment of high-grade glioma tumors obtained from orthotopic injections of syngeneic C57BL/6 glioma cells. These cells were derived from an endogenous glioma model based on the Tlx-CreERT2-driven deletion of Pten and Tp53 in neuronal stem cells. Immunofluorescence staining (Figure 1A) and flow cytometry (Figure 1B and Figure S1A) of glioma-bearing mouse brains revealed the presence of a PDPN+ myeloid subpopulation. Interestingly, CD11b+ PDPN+ cells were only present in glioma-bearing brains but not in unchallenged brain tissue. This either indicates that brain tumor growth triggers de novo Pdpn expression in the resident microglial compartment or that the CD11b+ PDPN+ population is constituted by infiltrating myeloid cells, in line with the finding of a PDPN+ subpopulation of peripheral myeloid cells (in the mandibular lymph node in tumor-bearing and untreated control mice Figure 1B and Figure S1A). Of note, no PDPN+ myeloid cells were detected in blood (Figure 1B and Figure S1A).
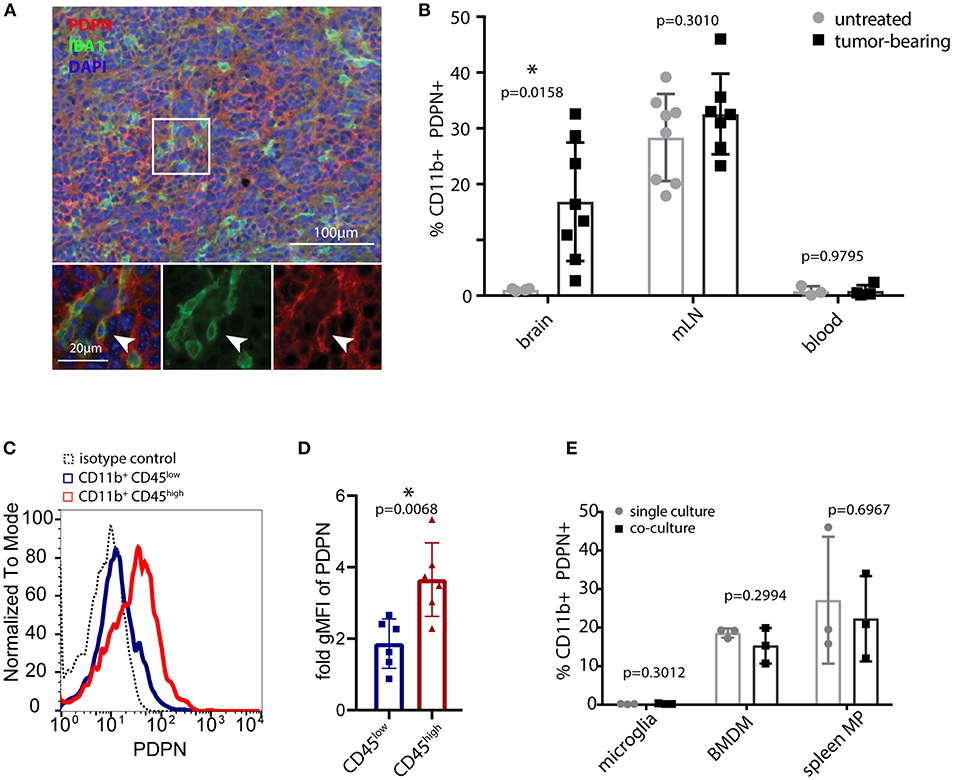
Figure 1. PDPN+ myeloid cells are present in lymphoid tissue and glioma tumors. (A) Immunofluorescence staining of high-grade murine glioma for PDPN (red) and IBA1 (green) shows co-expression in a subset of cells. Nuclei were stained with DAPI and pseudocolored in blue. Arrowhead indicates a cell co-expressing PDPN and IBA1. (B) Flow cytometry of brain, mandibular lymph nodes (mLN) and blood of untreated and tumor-bearing mice. Data represent mean and SD. Statistical analysis: Student's t-test. n = 4 and n = 8 for brain of untreated and tumor-bearing mice respectively. n = 8 and n = 7 mLN of untreated and tumor-bearing mice respectively. n = 3 and n = 4 for blood of untreated and tumor-bearing mice respectively. (C) Flow cytometry of glioma-associated myeloid cells shows PDPN is predominantly expressed by CD11b+ CD45high cells. (D) Quantification of geometric mean fluorescence intensity representative for PDPN levels on CD11b+CD45low and CD11b+CD45high cells, normalized to isotype control staining. Statistical analysis (Student's t-test of logarithmized values) revealed significantly higher expression of PDPN in CD11b+CD45high cells, p = 0.007; n = 6. (E) Flow cytometry of PDPN expression of microglia, bone marrow-derived macrophages (BMDM) and spleen macrophages in single- or co-culture with glioma cell line LN308. Bar graphs indicate percentage of PDPN+ cells of all CD11b+ cells, data represent mean and SD. Statistical analysis: Student's t-test. n = 3 for all samples. *p < 0.05, **p < 0.001.
To determine the PDPN+ myeloid cell type in gliomas we combined the CD11b and PDPN staining with CD45. Our analysis indicated that Pdpn is primarily expressed by CD11b+ CD45high SSChigh cells (Figures 1C,D), a population that has previously been described as infiltrating macrophages (43–45). This is in line with in vitro experiments, where we cultivated murine microglia, bone marrow-derived macrophages (BMDM) and spleen macrophages in the absence and presence of LN308 glioma cells (Figure 1E). In contrast to BMDM and spleen macrophages that both comprise a PDPN+ subpopulation at any condition, we could not detect PDPN protein on microglial cells, suggesting that the PDPN+ myeloid cell type we found in glioma tumors are not activated microglia. Taken together, we show that gliomas harbor a myeloid subpopulation that expresses Pdpn and that is most likely of peripheral origin.
PDPN+ Myeloid Cells Have a Tumor-Promoting Function
In order to investigate the effect of PDPN+ myeloid cells on glioma development, we generated a mouse line that carries a myeloid-specific deletion of Pdpn by crossing a Pdpnflox/flox with a Csf1r-Cre transgenic line (Figure 2A). Although the colony-stimulating factor-1 receptor (CSF1R) is largely restricted to mononuclear phagocytes, the Csf1r-Cre line has also been reported to delete loxP-flanked alleles in lymphocytes (38). As Pdpn deletion in T-cells has recently been shown to significantly delay melanoma growth due to PDPN's newly discovered role as co-inhibitory receptor (46, 47), we tested whether the application of the Csfr1-Cre line resulted in the undesired deletion of Pdpn in T-lymphocytes. Flow cytometry of tumor-infiltrated CD3+ T-cells revealed that only a subset of ~10% expressed Pdpn and that this is not changed in Csf1r-Cre; Pdpnflox/flox conditional knockout (cKO) mice (Figure S2A). In contrast, we found significantly fewer PDPN+ CD11b+ myeloid cells in mandibular lymph nodes of cKO compared to control animals (Figure 2B), albeit we could not achieve deletion of the gene in the entire population. We continued with orthotopic injections of syngeneic C57BL/6 glioma cells. These cells formed tumors with hallmarks of high-grade gliomas including extensive invasion in both control and cKO animals (Figure 2C). However, we observed a significantly prolonged survival of cKO animals (Figure 2D, p = 0.016). In summary, our observation of the prolonged survival of glioma-bearing cKO animals indicates that the PDPN+ myeloid subpopulation promotes glioma progression.
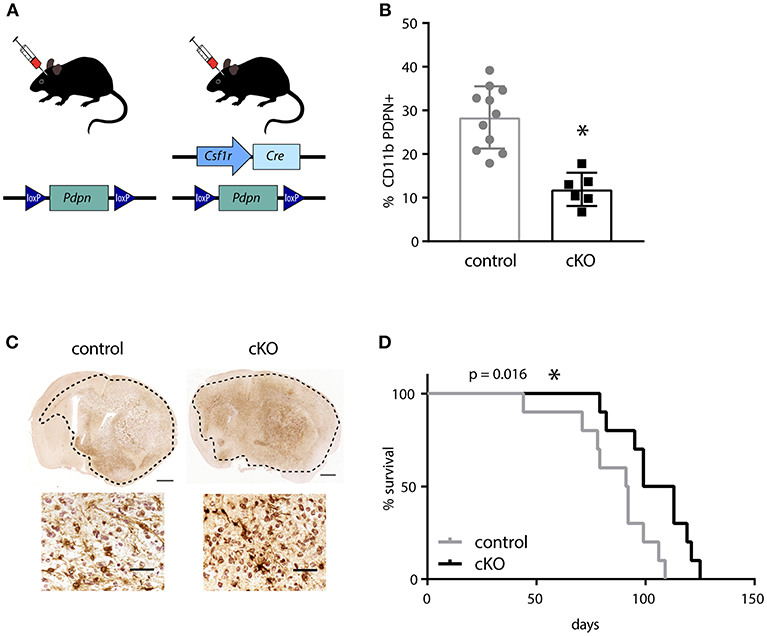
Figure 2. The deletion of Pdpn in myeloid cells improves survival of glioma bearing mice. (A) Schematic representation of genotypes applied for syngeneic transplantations of high-grade glioma cells. (B) Percentage of PDPN+ cells (of all CD11b+ cells) in mandibular lymph nodes of control and conditional knockout (cKO) animals, assessed by flow cytometry, data represent mean and SD, p = 0.0001; Student's t-test. (C) Immunohistochemical staining of mCherry positive glioma cells implanted in control and cKO brains, scale bar 1 mm and 50 μm for magnifications. (D) Kaplan-Meier survival analysis showed significantly prolonged survival of cKO animals, p = 0.016 (log-rank test); n = 10. *p < 0.05, **p < 0.001.
The Deletion of Pdpn in Myeloid Cells Increases T-Cell Infiltrates
We next examined how the beneficial effect of the myeloid cell-specific Pdpn knockout is mediated. Myeloid cells, and especially macrophages, have been shown to acquire a tumor-promoting phenotype in cancer patients and mouse models of cancer (4, 20). Tumor-associated macrophages support tumor growth by diverse mechanisms of immune suppression (15, 48–50). Although there seems to be no consensus about the significance of immune infiltrates in brain tumors due to local immune suppression, there is increasing evidence that associates a beneficial outcome with increased CD3+/CD8+ infiltrates in glioblastoma patients (51, 52). Thus, we investigated T-cell numbers in control and cKO animals.
First, we monitored CD3+, CD4+, and CD8+ cells in the blood during tumor development by flow cytometry. T-cell percentages and the balance between CD4+ and CD8+ T-cells up to day 54 post tumor cell injection were comparable to unchallenged mice (data not shown). However, in subsequent analyses we found slightly decreasing T-cell numbers during tumor development in all animals, especially of the CD4+ compartment (Figure 3B). This is in line with peripheral lymphocyte deficiencies and CD4 lymphopenia observed in glioblastoma patients (53), indicating a systemic immune suppression. Interestingly, cKO mice showed in general a slightly higher CD3+ T-cell number compared to control animals (Figures 3A,B). This becomes more apparent during later stages of tumor development (p = 0.04 for d82 and p = 0.055 for d96 post tumor cell injection). Furthermore, we analyzed T-cell populations in the mandibular lymph node which has been shown to be one of the draining lymph nodes of the brain (54). When animals had to be sacrificed, the mandibular lymph nodes were removed and cells isolated for analysis by flow cytometry. Lymph nodes of the majority of analyzed cKO animals appeared to be enlarged compared to control mice (Figure S2B), indicating a response to tumor growth. However, we did not find a significant difference in the percentage of CD3+ T-cells or the ratio of CD4+/CD8+ cells in the lymph node when analyzed by flow cytometry (Figure 3C). Taken together, brain tumor growth seemed to impact on peripheral T-cells and PDPN+ myeloid cells. Control animals appeared slightly more susceptible to glioma-induced systemic immune suppression than mice carrying a myeloid-specific deletion of Pdpn.
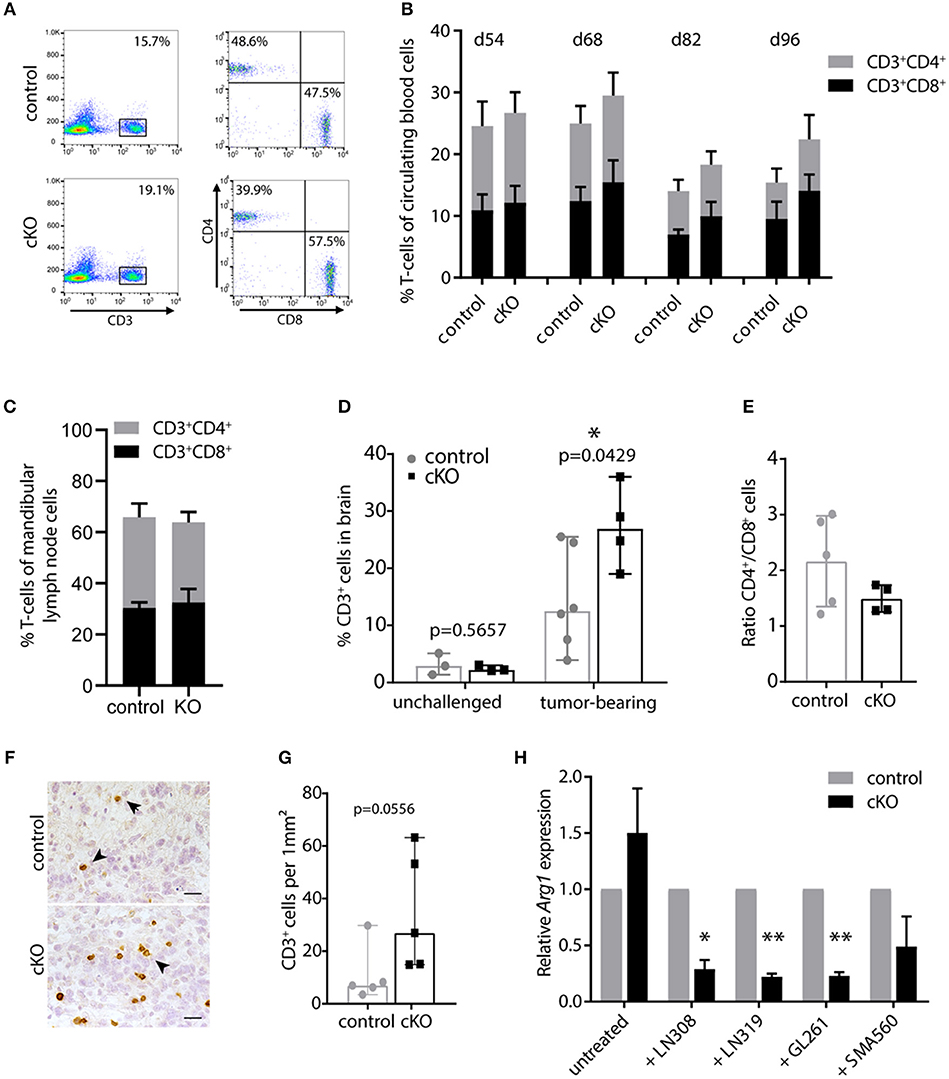
Figure 3. PDPN+ myeloid cells execute an immune inhibitory function. (A) Flow cytometry plots of one control and one cKO animal 82 days after tumor cell transplantation. CD3+ T-cells were pre-gated for lack of 7AAD and analyzed for CD4 and CD8 expression. (B) Time course of flow cytometric analysis of circulating CD3+CD4+ and CD3+CD8+ T-cells of tumor-bearing control and cKO animals. Days post tumor cell injection are indicated on top. Statistical comparison (Student's t-test) of T-cell percentages between control and cKO animals showed following p-values: 0.05; 0.25; 0.047 and 0.056 at d54; d68; d82 and d96, respectively, n = 8. (C) Percentage of CD3+CD4+ and CD3+CD8+ T-cells in mandibular lymph nodes is not altered between glioma-bearing control and cKO mice. Statistical comparison (Student's t-test) of T-cell percentages between control and cKO animals showed following p-value 0.6647; n = 5 (control) and n = 4 (cKO). Data (B,C) represent mean and SD. (D) Percentages of brain-infiltrating CD3+ T-cells are increased by tumor growth and are significantly elevated in tumors of cKO mice. Statistical analysis: Student's t-test, n = 3 (each unchallenged control and cKO); n = 6 (tumor-bearing control); n = 4 (tumor-bearing cKO). (E) CD4+/CD8+ ratio of CD3+ cells detected in the brain by flow cytometry. Statistical analysis: Student's t-test. p = 0.1609; n = 5 (control) and n = 4 (cKO). (F) Representative immunohistochemical staining of CD3, scale bar 25 μm (G) Corresponding quantification of CD3+ cells in tumors of control and cKO animals shows a clear tendency to increased T-cell infiltration in cKO mice (p = 0.0556, Mann-Whitney test, n = 5). Data (D,E,G) represent median and 95% CI. (H) Relative Arg1 expression in BMDM isolated from control or cKO animals analyzed by quantitative Real-Time PCR, performed with biological triplicates. Values normalized to house-keeping gene Ppia. Arg1 expression of cKO macrophages was normalized to expression of control macrophages for each condition. Arg1 expression is significantly attenuated in Pdpn knockout macrophages when co-cultivated with LN308 (p = 0.0173), LN319 (p = 0.002), or GL261 (p = 0.0029), p-value for SMA-560-co-cultivation = 0.111, paired Student's t-test of logarithmized values, n = 3. *p < 0.05, **p < 0.001.
We then continued analyzing the T-cell infiltration of brain tumors. Brain tissue of untreated mice contained only few CD3+ T-cells (~3% of isolated cells, Figure 3D). These cell numbers were elevated in the tumor setting, however, probably due to the high immune suppressive potential of glioma tumors, this increase was rather mild in tumors of control mice with a median of about 13% of all isolated cells. Remarkably, cKO animals showed significantly elevated T-cell infiltrates with about 27% CD3+ cells (Figure 3D). This increase in CD3+ immune cell infiltrates in tumors of cKO compared to control animals was also shown by immunohistochemistry staining (Figures 3F,G). Moreover, we observed that most tumors of cKO animals harbored higher numbers of infiltrated CD8+ T-cells, also in relation to CD4+ cells (lower CD4+/CD8+ ratio), than control tumors (Figure 3E). The overall increase in infiltrated T-cells, together with elevated cytotoxic CD8+ cell infiltrates and an altered CD4+/CD8+ ratio potentially accounts for the prolonged survival of cKO animals.
We next tried to understand how PDPN+ myeloid cells inhibit T-cells. Thus, we analyzed the expression of a panel of common M1 (IL-1b, IL-12, iNOS, TNFα) and M2 (TGFβ, IL-10, ARG1) phenotype markers and the immune suppressive PD-L1 (CD274) in control and Pdpn-deleted BMDM co-cultivated with four different glioma cell lines (Figures S2C-K). One examined marker was found to be differentially regulated between control and knockout macrophages; arginase-1 (Figure 3H and Figure S2J). We observed that PDPN-deficient macrophages show a significantly impaired transcriptional upregulation of Arg1 expression in response to three different glioma cell lines. Tumor-associated macrophages and myeloid-derived suppressor cells have been reported to inhibit T-cell activity and viability by the secretion of ARG1 and consequent depletion of arginine (48, 55). Thus, PDPN+ myeloid cells might mediate their immune suppressive function by increased expression of Arg1, which is attenuated upon Pdpn deletion.
Taken together, during tumor development, we observe signs for a systemic immune suppression in both control and cKO animals, which, however, seems to be less pronounced in cKO mice. Strikingly, tumors of mice carrying a myeloid-specific deletion of Pdpn show higher brain tumor infiltrates of CD3+ and CD3+ CD8+ cells than control tumors. Moreover, an altered ratio between CD8+ and CD4+ T-cells was detected between the two groups. These findings indicate an immune suppressive function for PDPN+ myeloid cells. In vitro gene expression analysis suggests that this suppression might be mediated via increased Arg1 expression in PDPN proficient macrophages.
Discussion
The well-established impact of the tumor microenvironment and particularly of cells of the immune system on tumor development and therapy response facilitates the exploration of novel therapeutic approaches calling for the identification and detailed characterization of the involved cell types. Thus, in this study we investigated a myeloid subpopulation, characterized by Pdpn expression, that has previously been reported to critically regulate the inflammatory immune response (34, 35). As PDPN has furthermore been identified as an inhibitory surface protein on T-cells in a tumor setting (47), we questioned whether PDPN might have an immune regulatory function in tumor-associated myeloid cells as well. We performed orthotopic injections of syngeneic high-grade glioma cells, an entity that is characterized by strong local and systemic immune suppression. Here, we detected PDPN+ myeloid cells in glioma tumors, however, not in untreated brains. Furthermore, peripheral lymphoid tissues harbored a PDPN+ myeloid subpopulation in presence but also in absence of glioma growth. Together with the fact that we did not detect PDPN protein on circulating myeloid cells of the blood, we presumed that Pdpn expression is initiated in a subset of peripheral myeloid cells upon tissue infiltration and differentiation. This was further supported by the fact that in brain tumors, Pdpn is predominantly expressed by CD11b+ CD45high SSChigh cells, a population that has been considered as infiltrating macrophages. However, CD45high or CD45low may not clearly discriminate the different myeloid subpopulations as it has been shown that tumor-associated microglia are able to upregulate CD45 expression (56). To assess whether association with tumor cells induces expression of Pdpn, we isolated murine microglia and two other macrophage populations, BMDM and spleen macrophages, and co-cultivated them with the glioma cell line LN308. Our result that, in contrast to macrophages, microglia did not show Pdpn expression at any condition suggests that PDPN+ myeloid cells found in brain tumor tissue are most likely of peripheral origin and recruited by tumor growth.
In our murine glioma model deletion of Pdpn in myeloid cells has a significantly beneficial effect on survival, suggesting a pro-tumoral role for PDPN+ myeloid cells. In fact, the effect on tumor growth in our genetic model may even be underestimated due to the incomplete deletion of Pdpn. Incomplete recombination of Pdpnflox alleles in macrophages has also been reported for the combination with another transgenic Cre-line Vav-Cre; (57), making it questionable whether the employment of another Cre-line would have resulted in more efficient PDPN deletion.
Since GBM-infiltrating macrophages, microglia, and myeloid-derived suppressor cells have been shown to hinder T-cell proliferation, infiltration and activation (14, 16, 58) we addressed the question whether PDPN+ myeloid cells promote tumor development by intervention with the T-cell compartment. Therefore, we analyzed T-cell numbers of the periphery and local brain tumor tissue. In line with published data, we found decreasing T-cell percentages in blood during tumor progression, indicating a systemic immune suppression (53). Of note, most cKO animals showed a less pronounced reduction of circulating T-cells as well as enlarged mandibular lymph nodes, suggesting a general role of PDPN in the regulation of immune suppression. As we exclusively detected a difference in T-cell percentages in (late stage) tumor-bearing and not in unchallenged mice (data not shown), we presume that the PDPN+ myeloid cells are not involved in T-cell development per se, but interfere with T-cell activation/proliferation or survival in a tumor setting.
Consistently, the analysis of brain tumors revealed that CD3+ and CD8+ cells were more abundant in tumors of cKO mice than in controls. Of note, increased CD3+ and CD8+ infiltrates have also been associated with better prognosis of glioma patients (51, 52). Furthermore, we observed a reduced CD4+/CD8+ ratio of tumor infiltrating lymphocytes in the absence of PDPN+ myeloid cells. In line with this, a previous report has indicated that not only the number of T-cells but also their balance determines effective anti-tumor immunity, in particular high CD4+/CD8+ ratios contribute to poor prognosis of glioma patients (59). These findings indicate that PDPN+ myeloid cells contribute to immune suppression and presumably to consequent immune evasion, enhanced tumor growth and poor survival. In vitro characterization of PDPN+ and Pdpn knockout macrophages showed PDPN-dependent enhanced Arg1 expression, an enzyme with known pro-tumorigenic function in the tumor microenvironment (48, 60, 61), in response to the presence of glioma cells. Consistently, Arg1 could be one mediator of the here proposed immune suppressive function of PDPN+ myeloid cells. Additional experiments would be required to validate this cascade in vivo and furthermore, to show that in the absence of PDPN+ myeloid cells, T-cells not only infiltrate the tumors but also execute cytotoxic reactivity against the tumor cells causing the observed prolonged survival. However, as there is increasing evidence for a beneficial outcome in glioblastoma patients with increased CD3+ and CD8+ infiltrates (51, 52), we consider it most likely that the increased infiltration of T-cells in tumors of cKO animals accounts for their prolonged survival. Furthermore, it remains to be determined whether PDPN+ myeloid cells need to be located within the brain to execute their proposed immune inhibitory function or whether they are able to induce a systemic immune suppression sufficient to prevent local T-cell infiltration.
Taken together, we describe a population of immune-inhibitory tumor-associated PDPN+ myeloid cells, which contributes to the poor prognosis in high-grade glioma. Exploiting this knowledge, PDPN+ myeloid cells could be targeted in combinatorial immunotherapies to enhance immune-mediated tumor destruction.
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Author Contributions
TE, HP, and PA designed the study. TE performed experiments. BC developed the syngeneic injection model. TE, BC, HP, and PA analyzed and interpreted the data and wrote the manuscript. All authors were involved in revising the manuscript, have read and approved the final version.
Funding
This work was supported by the Helmholtz Alliance Preclinical Comprehensive Cancer Center (to PA).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Christian Clappier, Christine Bauer, Bettina Kast, and Sabrina Lohr for technical assistance, Hai-Kun Liu for providing the animals used to generate the cells of the syngeneic injection model, Susanne Kleber and Ana Martín-Villalba for access to stereotactic devices, Damir Krunic of the DKFZ Light Microscopy Facility for help with image processing and analysis, Juan Rodriguez Vita for providing the primer sequence for Nos2, and Francesca Rampoldi for helpful discussion.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.00187/full#supplementary-material
References
1. Maman S, Witz IP. A history of exploring cancer in context. Nat Rev Cancer. (2018) 18:359–76. doi: 10.1038/s41568-018-0006-7
2. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. (2013) 19:1423–37. doi: 10.1038/nm.3394
3. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. (2016) 16:582–98. doi: 10.1038/nrc.2016.73
4. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
5. De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. (2017) 17:457–74. doi: 10.1038/nrc.2017.51
6. Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol. (2005) 64:479–89. doi: 10.1093/jnen/64.6.479
7. Reardon DA, Freeman G, Wu C, Chiocca EA, Wucherpfennig KW, Wen PY, et al. Immunotherapy advances for glioblastoma. Neuro Oncol. (2014) 16:1441–58. doi: 10.1093/neuonc/nou212
8. Quail DF, Joyce JA. The Microenvironmental Landscape of Brain Tumors. Cancer Cell. (2017) 31:326–41. doi: 10.1016/j.ccell.2017.02.009
9. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. (2013) 19:1264–72. doi: 10.1038/nm.3337
10. Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med. (2012) 18:519–27. doi: 10.2119/molmed.2011.00217
11. Markovic DS, Vinnakota K, Chirasani S, Synowitz M, Raguet H, Stock K, et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci USA. (2009) 106:12530–5. doi: 10.1073/pnas.0804273106
12. De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. (2005) 8:211–26. doi: 10.1016/j.ccr.2005.08.002
13. Stafford JH, Hirai T, Deng L, Chernikova SB, Urata K, West BL, et al. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol. (2016) 18:797–806. doi: 10.1093/neuonc/nov272
14. Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. (2010) 12:1113–25. doi: 10.1093/neuonc/noq082
15. Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res. (2013) 19:3165–75. doi: 10.1158/1078-0432.CCR-12-3314
16. Raychaudhuri B, Rayman P, Huang P, Grabowski M, Hambardzumyan D, Finke JH, et al. Myeloid derived suppressor cell infiltration of murine and human gliomas is associated with reduction of tumor infiltrating lymphocytes. J Neurooncol. (2015) 122:293–301. doi: 10.1007/s11060-015-1720-6
17. Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. (2016) 17:2445–59. doi: 10.1016/j.celrep.2016.10.052
18. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. (2010) 330:841–5. doi: 10.1126/science.1194637
19. Richter N, Wendt S, Georgieva PB, Hambardzumyan D, Nolte C, Kettenmann H. Glioma-associated microglia and macrophages/monocytes display distinct electrophysiological properties and do not communicate via gap junctions. Neurosci Lett. (2014) 583:130–5. doi: 10.1016/j.neulet.2014.09.035
20. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity.(2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
21. Engler JR, Robinson AE, Smirnov I, Hodgson JG, Berger MS, Gupta N, et al. Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PLoS ONE. (2012) 7:e43339. doi: 10.1371/journal.pone.0043339
22. Szulzewsky F, Pelz A, Feng X, Synowitz M, Markovic D, Langmann T, et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS ONE. (2015) 10:e0116644. doi: 10.1371/journal.pone.0116644
23. Ugorski M, Dziegiel P, Suchanski J. Podoplanin - a small glycoprotein with many faces. Am J Cancer Res. (2016) 6:370–86.
24. Martin-Villar E, Scholl FG, Gamallo C, Yurrita MM, Munoz-Guerra M, Cruces J, et al. Characterization of human PA2.26 antigen (T1alpha-2, podoplanin), a small membrane mucin induced in oral squamous cell carcinomas. Int J Cancer. (2005) 113:899–910. doi: 10.1002/ijc.20656
25. Schacht V, Dadras SS, Johnson LA, Jackson DG, Hong YK, Detmar M. Up-regulation of the lymphatic marker podoplanin, a mucin-type transmembrane glycoprotein, in human squamous cell carcinomas and germ cell tumors. Am J Pathol. (2005) 166:913–21. doi: 10.1016/S0002-9440(10)62311-5
26. Hoshino A, Ishii G, Ito T, Aoyagi K, Ohtaki Y, Nagai K, et al. Podoplanin-positive fibroblasts enhance lung adenocarcinoma tumor formation: podoplanin in fibroblast functions for tumor progression. Cancer Res. (2011) 71:4769–79. doi: 10.1158/0008-5472.CAN-10-3228
27. Peterziel H, Muller J, Danner A, Barbus S, Liu HK, Radlwimmer B, et al. Expression of podoplanin in human astrocytic brain tumors is controlled by the PI3K-AKT-AP-1 signaling pathway and promoter methylation. Neuro Oncol. (2012) 14:426–39. doi: 10.1093/neuonc/nos055
28. Payne H, Ponomaryov T, Watson SP, Brill A. Mice with a deficiency in CLEC-2 are protected against deep vein thrombosis. Blood. (2017) 129:2013–20. doi: 10.1182/blood-2016-09-742999
29. Acton S, Farrugia AJ, Astarita JL, Mourão -SáD, Jenkins RP, Nye E, et al. Dendritic cells control fibroblastic reticular network tension and lymph node expansion. Nature. (2014) 514:498–502. doi: 10.1038/nature13814
30. Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, et al. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity. (2011) 35:986–96. doi: 10.1016/j.immuni.2011.10.015
31. Hou TZ, Bystrom J, Sherlock JP, Qureshi O, Parnell SM, Anderson G, et al. A distinct subset of podoplanin (gp38) expressing F4/80+ macrophages mediate phagocytosis and are induced following zymosan peritonitis. FEBS Lett. (2010) 584:3955–61. doi: 10.1016/j.febslet.2010.07.053
32. Kerrigan AM, Navarro-Nunez L, Pyz E, Finney BA, Willment JA, Watson SP, et al. Podoplanin-expressing inflammatory macrophages activate murine platelets via CLEC-2. J Thromb Haemost. (2012) 10:484–6. doi: 10.1111/j.1538-7836.2011.04614.x
33. Hitchcock JR, Cook CN, Bobat S, Ross EA, Flores-Langarica A, Lowe KL, et al. Inflammation drives thrombosis after Salmonella infection via CLEC-2 on platelets. J Clin Invest. (2015) 125:4429–46. doi: 10.1172/JCI79070
34. Lax S, Rayes J, Wichaiyo S, Haining EJ, Lowe K, Grygielska B, et al. Platelet CLEC-2 protects against lung injury via effects of its ligand podoplanin on inflammatory alveolar macrophages in the mouse. Am J Physiol Lung Cell Mol Physiol. (2017) 313:L1016–L1029. doi: 10.1152/ajplung.00023.2017
35. Rayes J, Lax S, Wichaiyo S, Watson SK, Di Y, Lombard S, et al. The podoplanin-CLEC-2 axis inhibits inflammation in sepsis. Nat Commun. (2017) 8:2239. doi: 10.1038/s41467-017-02402-6
36. Castro F, Dirks WG, Fahnrich S, Hotz-Wagenblatt A, Pawlita M, Schmitt M. High-throughput SNP-based authentication of human cell lines. Int J Cancer. (2013) 132:308–14. doi: 10.1002/ijc.27675
37. Baars S, Bauer C, Szabowski S, Hartenstein B, Angel P. Epithelial deletion of podoplanin is dispensable for re-epithelialization of skin wounds. Exp Dermatol. (2015) 24:785–7. doi: 10.1111/exd.12781
38. Deng L, Zhou JF, Sellers RS, Li JF, Nguyen AV, Wang Y, et al. A novel mouse model of inflammatory bowel disease links mammalian target of rapamycin-dependent hyperproliferation of colonic epithelium to inflammation-associated tumorigenesis. Am J Pathol. (2010) 176:952–67. doi: 10.2353/ajpath.2010.090622
39. Marim FM, Silveira TN, Lima DS Jr, Zamboni DS. A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PLoS ONE. (2010) 5:e15263. doi: 10.1371/journal.pone.0015263
40. Saura J, Tusell JM, Serratosa J. High-yield isolation of murine microglia by mild trypsinization. Glia. (2003) 44:183–9. doi: 10.1002/glia.10274
41. Durchdewald M, Guinea-Viniegra J, Haag D, Riehl A, Lichter P, Hahn M, et al. Podoplanin is a novel fos target gene in skin carcinogenesis. Cancer Res. (2008) 68:6877–83. doi: 10.1158/0008-5472.CAN-08-0299
42. Sharda DR, Yu S, Ray M, Squadrito ML, De Palma M, Wynn TA, et al. Regulation of macrophage arginase expression and tumor growth by the Ron receptor tyrosine kinase. J Immunol. (2011) 187:2181–92. doi: 10.4049/jimmunol.1003460
43. Badie B, Schartner JM. Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery. (2000) 46:957–961; discussion 961–952.
44. Parney IF, Waldron JS, Parsa AT. Flow cytometry and in vitro analysis of human glioma-associated macrophages. Lab Invest J Neurosurg. (2009) 110:572–82. doi: 10.3171/2008.7.JNS08475
45. Bischoff T, Stahl-Hennig C, Matz-Rensing K, Koutsilieri E, Sopper S. Definition of leukocyte subsets in primate central nervous system by polychromatic flow cytometry. Cytometry A. (2011) 79:436–45. doi: 10.1002/cyto.a.21046
46. Peters A, Burkett PR, Sobel RA, Buckley CD, Watson SP, Bettelli E, et al. Podoplanin negatively regulates CD4+ effector T cell responses. J Clin Invest. (2015) 125:129–40. doi: 10.1172/JCI74685
47. Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, et al. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature. (2018) 558:454–9. doi: 10.1038/s41586-018-0206-z
48. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. (2004) 64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465
49. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
50. Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. (2014) 26:623–37. doi: 10.1016/j.ccell.2014.09.006
51. Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin Cancer Res. (2011) 17:4296–308. doi: 10.1158/1078-0432.CCR-10-2557
52. Kmiecik J, Poli A, Brons NH, Waha A, Eide GE, Enger PO, et al. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol. (2013) 264:71–83. doi: 10.1016/j.jneuroim.2013.08.013
53. Gustafson MP, Lin Y, New KC, Bulur PA, O'neill BP, Gastineau DA, et al. Systemic immune suppression in glioblastoma: the interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncol. (2010) 12:631–44. doi: 10.1093/neuonc/noq001
54. Ma Q, Ineichen BV, Detmar M, Proulx ST. Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nat Commun. (2017) 8:1434. doi: 10.1038/s41467-017-01484-6
55. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. (2016) 167:829–42 e813. doi: 10.1016/j.cell.2016.09.031
56. Muller A, Brandenburg S, Turkowski K, Muller S, Vajkoczy P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int J Cancer. (2015) 137:278–88. doi: 10.1002/ijc.29379
57. Lax S, Rayes J, Thickett DR, Watson SP. Effect of anti-podoplanin antibody administration during lipopolysaccharide-induced lung injury in mice. BMJ Open Respir Res. (2017) 4:e000257. doi: 10.1136/bmjresp-2017-000257
58. Razavi SM, Lee KE, Jin BE, Aujla PS, Gholamin S, Li G. Immune Evasion Strategies of Glioblastoma. Front Surg. (2016) 3:11. doi: 10.3389/fsurg.2016.00011
59. Han S, Zhang C, Li Q, Dong J, Liu Y, Huang Y, et al. Tumour-infiltrating CD4(+) and CD8(+) lymphocytes as predictors of clinical outcome in glioma. Br J Cancer. (2014) 110:2560–8. doi: 10.1038/bjc.2014.162
60. Gabrusiewicz K, Ellert-Miklaszewska A, Lipko M, Sielska M, Frankowska M, Kaminska B. Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PLoS ONE. (2011) 6:e23902. doi: 10.1371/journal.pone.0023902
Keywords: tumor immunology, macrophages, immune evasion, tumor-infiltrating lymphocytes, glioblastoma, arginase, tumor microenvironment
Citation: Eisemann T, Costa B, Peterziel H and Angel P (2019) Podoplanin Positive Myeloid Cells Promote Glioma Development by Immune Suppression. Front. Oncol. 9:187. doi: 10.3389/fonc.2019.00187
Received: 21 December 2018; Accepted: 04 March 2019;
Published: 26 March 2019.
Edited by:
Erik P. Sulman, Langone Medical Center, New York University, United StatesReviewed by:
Monica Venere, The Ohio State University, United StatesKrishna Prasad PL Bhat, University of Texas MD Anderson Cancer Center, United States
Copyright © 2019 Eisemann, Costa, Peterziel and Angel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Angel, cC5hbmdlbEBka2Z6LmRl
†These authors have contributed equally to this work