 Thibault Kervarrec1,2,3Mahtab Samimi2,4Serge Guyétant1,2
Thibault Kervarrec1,2,3Mahtab Samimi2,4Serge Guyétant1,2 Bhavishya Sarma3Jérémy Chéret5
Bhavishya Sarma3Jérémy Chéret5 Emmanuelle Blanchard1,6Patricia Berthon2
Emmanuelle Blanchard1,6Patricia Berthon2 David Schrama3Roland Houben3
David Schrama3Roland Houben3 Antoine Touzé2*
Antoine Touzé2*- 1Department of Pathology, Centre Hospitalier Universitaire de Tours, Tours, France
- 2ISP “Biologie des infections à polyomavirus” team, UMR INRA 1282, University of Tours, Tours, France
- 3Department of Dermatology, Venereology and Allergology, University Hospital Würzburg, Würzburg, Germany
- 4Departement of Dermatology, Centre Hospitalier Universitaire de Tours, Tours, France
- 5Monasterium Laboratory, Skin and Hair Research Solutions GmbH, Münster, Germany
- 6Plateforme IBiSA de Microscopie Electronique, INSERM 1259, Université de Tours, Tours, France
Merkel cell carcinoma (MCC) is a primary neuroendocrine carcinoma of the skin. This neoplasia features aggressive behavior, resulting in a 5-year overall survival rate of 40%. In 2008, Feng et al. identified Merkel cell polyomavirus (MCPyV) integration into the host genome as the main event leading to MCC oncogenesis. However, despite identification of this crucial viral oncogenic trigger, the nature of the cell in which MCC oncogenesis occurs is actually unknown. In fact, several hypotheses have been proposed. Despite the large similarity in phenotype features between MCC tumor cells and physiological Merkel cells (MCs), a specialized subpopulation of the epidermis acting as mechanoreceptor of the skin, several points argue against the hypothesis that MCC derives directly from MCs. Alternatively, MCPyV integration could occur in another cell type and induce acquisition of an MC-like phenotype. Accordingly, an epithelial as well as a fibroblastic or B-cell origin of MCC has been proposed mainly based on phenotype similarities shared by MCC and these potential ancestries. The aim of this present review is to provide a comprehensive review of the current knowledge of the histogenesis of MCC.
Introduction
Merkel cell carcinoma (MCC) is an aggressive neoplasm defined as a primary neuroendocrine carcinoma of the skin. The incidence is still low, with for example 0.7 cases per 100,000 person-years in the United States in 2013, but has increased by 95% from 2000 to 2013, and a further increase in incidence has been predicted (1). MCC occurs essentially in older people, with known risk factors being sun exposure (2) and immunosuppression (3, 4). MCC is characterized by aggressive behavior resulting in a 5-year overall survival rate of 40% (5). Combined radiotherapy and surgery is considered the mainstay of treatment for patients with localized disease, but until recently, those with advanced, inoperable disease received various regimens of cytotoxic chemotherapy, without a significant effect on survival (6). Recently, restoration of T-cell responses by inhibitors targeting programmed cell death 1 (PD-1) and its ligand (PD-L1) checkpoints has been identified as an effective approach in such patients (7). Indeed, after failure of first-line chemotherapy, treatment with avelumab resulted in objective tumoral responses in 32% of MCC patients with advanced disease (7), and avelumab has been approved for advanced MCC both in the United States and European Union (7, 8). Avelumab is being investigated as first-line therapy in this setting, with objective responses in approximately 60% of patients in preliminary reports (9).
MCC is diagnosed on the basis of histological examination, which reveals infiltration of the dermis or hypodermis by proliferating tumor cells harboring high-grade neuroendocrine carcinoma features (10) (Figure 1). Blastic lymphomas as well as other small round blue cell tumors must be considered in the differential diagnosis. Immunohistochemical investigation of MCC cases (Figure 1) reveals the expression of both epithelial (pancytokeratin AE1/AE3) and neuroendocrine markers such as chromogranin A (11), synaptophysin (11), CD56 (10) and INSM1 (insulinoma-associated 1) (12). In addition, the combination of cytokeratin 20 (CK20) positivity with thyroid transcription factor-1 negativity (13) is currently used to distinguish MCC from other metastatic neuroendocrine carcinomas. Neurofilament and special AT-rich sequence-binding protein 2 (SATB2) have been proposed as additional markers providing high diagnostic accuracy (14, 15).
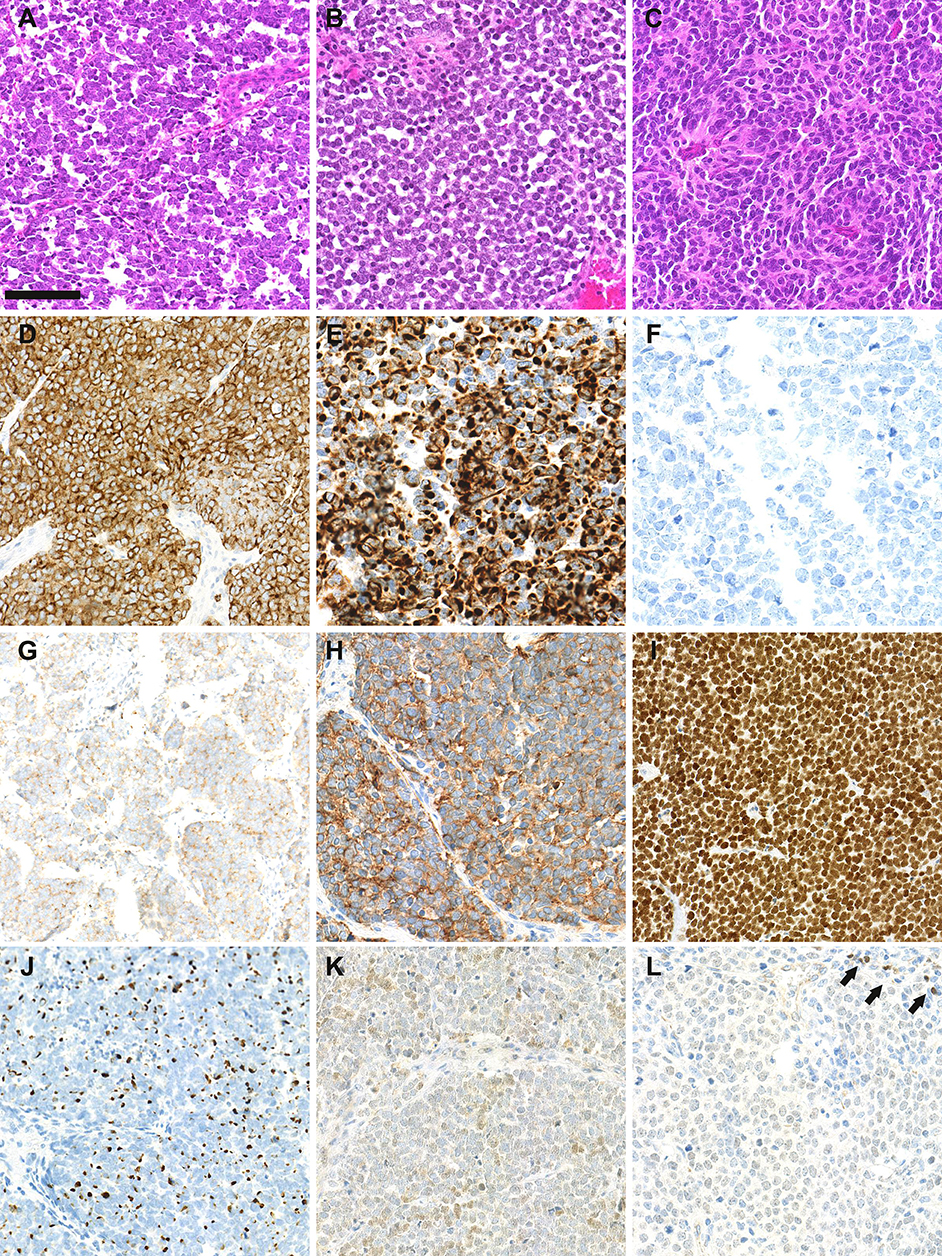
Figure 1. Morphological and immunohistochemical features of Merkel cell carcinoma: (A–C): hematein-phloxin-saffron staining revealed sheet of tumor cells with high mitotic activity (bar = 100 μm). Whereas, MCPyV-positive MCC (A,B) harbor scant cytoplasm, round nucleus and dusty chromatin, MCPyV negative tumor cells have more abondant clear cytoplasm and irregular nucleus (C). (D) chromogranin A cytoplasmic positivity, (E) cytokeratin 20 expression with paranuclear dot-pattern; (F) thyroid transcription factor-1 negativity; (G) membranous synaptophysin expression; (H) membranous CD56 expression; (I) special AT-rich sequence-binding protein 2 (SATB2) nuclear expression; (J) neurofilament expression with a dot-pattern; (K) terminal deoxy nucleotidyl transferase weak/moderate expression, (L) paired box 5 weak expression in tumor cells in comparison with intratumor lymphocytes (arrows).
Significant progress in understanding the MCC pathogenesis occurred in 2008, when Feng et al. reported a yet undescribed virus, the Merkel cell polyomavirus (MCPyV), whose genome was integrated in 80% of MCC tumors (16). MCPyV was further found to be an ubiquitous virus responsible for an asymptomatic life-long infection, because the episomal genome of MCPyV can be detected in the skin flora of most healthy people (17) and antibodies directed against the viral capsid are highly prevalent in the general population (18, 19).
Despite the high population prevalence of MCPyV, viral integration probably occurs very rarely, which accounts for the rarity of MCC tumors, and constitutes the main oncogenetic event leading to MCC oncogenesis. MCPyV integration together with mutations of the viral sequence (20) result in loss of replicative abilities of the virus before MCC development. As a consequence, MCPyV-positive MCC tumors do not produce MCPyV virions but are characterized by permanent nuclear expression of the viral T-antigen proteins (small T [sT] and large T [LT] antigen in a truncated form). Both sT and LT antigens bear oncogenic properties, by targeting various host cell proteins involved in cell cycle control and proliferation, and are now considered as the key actors of oncogenesis in MCPyV-positive MCC (21). By contrast, MCPyV-negative MCC, which accounts for approximately 20% of MCC cases, have a high mutational burdens, with a prominent UV signature, which affects various oncogenes. Among these, mutations of the tumor suppressor genes RB1 and TP53 appear to be critical oncogenic events (22).
Despite identification of both viral and UV-induced oncogenetic triggers in MCC, the nature of the cell where MCC oncogenesis occurs remains unknown (23). Actually, several hypotheses have been advanced. The aim of this article is to provide a comprehensive review of current knowledge of the histogenesis of MCC.
The Merkel Cell: the Historical Candidate
According to Boyd et al. rare cancer types identified before the molecular biology era were “either tumors presumed to originate from or resemble a cell type that infrequently gave rise to cancer, or histologically defined subsets within a more common type of cancer” (24). MCC, a perfect illustration of the first group, was classified according to its similarities with skin physiological Merkel cells (MCs). MCs are highly specialized epithelial cells located in the basal layer of the epidermis and in the external part of the hair follicle (Figure 2). They have been shown to act as mechanoreceptors by transforming tactile stimuli into Ca2+-action potentials (25) and serotonin release (26) and pass these signals on to Aβ-afferent nerve endings. The protein allowing transformation of mechanic into electric signals is the ion channel Piezo2 (25), which is also highly expressed by MCC cells [(27), unpublished data]. Expression of this MC-characteristic molecule is only one of many features shared by MCs and MCC cells. Originally described as “trabecular carcinomas of the skin” by Toker (28), ultrastructural studies of such cases revealed numerous neuroendocrine dense cores neuroendocrine granules, which are hallmarks of MCs (28, 29) (Figure 2). Hence, these “trabecular carcinomas” were suggested to derive from MCs, leading to their reclassification as MCC (29). Further immunohistochemical studies corroborated these initial findings by revealing a shared expression of many common markers in MCs and MCC (10, 30) but only a limited number of markers distinguishing them from each other (Table 1; Figures 1, 2). Indeed, both MCs and MCC express cytokeratin 20 (CK20) (13, 15, 31), neuroendocrine markers chromogranin A and synaptophysin (11, 37) and neuropeptides (30, 47). In contrast, the expression of vasoactive intestinal peptide and metenkephalin (44) are specific to MCs, whereas CD117 and CD171 are detected in only MCC cells (49, 61).
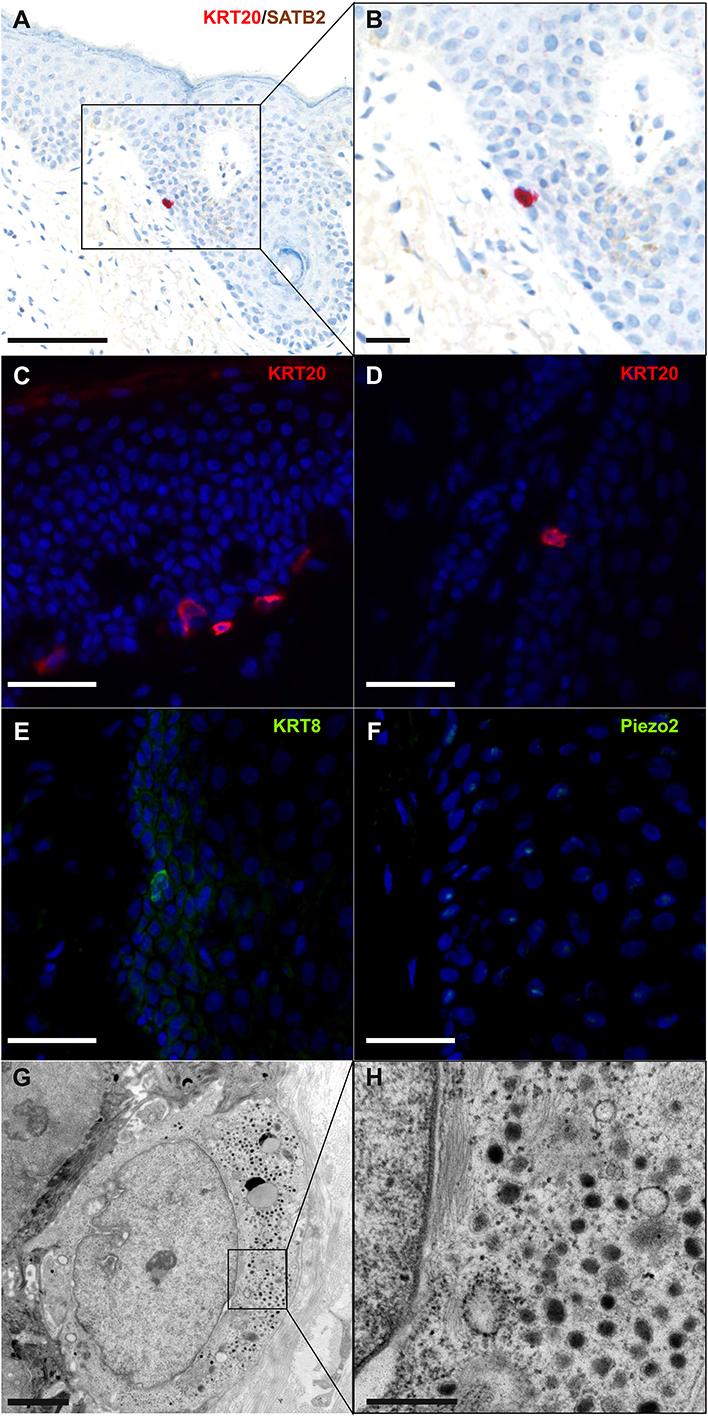
Figure 2. Immunohistochemical and ultrastructural features of physiological Merkel cells: immunohistochemical staining of normal skin (A,B) revealed one Merkel cell located in the infundibulum of a hair follicle and coexpressing cytokeratin 20 (cytoplasmic expression in red) and SATB2 (nuclear expression in brown) (bar = 100 and 50 μm for A,B). Immunofluorescence staining of healthy skin revealed some Merkel cells expressing cytokeratin 20 (C,D), cytokeratin 8 (E) and Piezo2 (F) in the epidermis (C) and in hair follicles (D–F) (bar = 40 μm for C–F). Electron microscopy of a Merkel cell (G,H) revealed numerous dense-core granules (bars = 2 and 0.5 μm for G,H, respectively). A cropped region is shown in the inset (H).
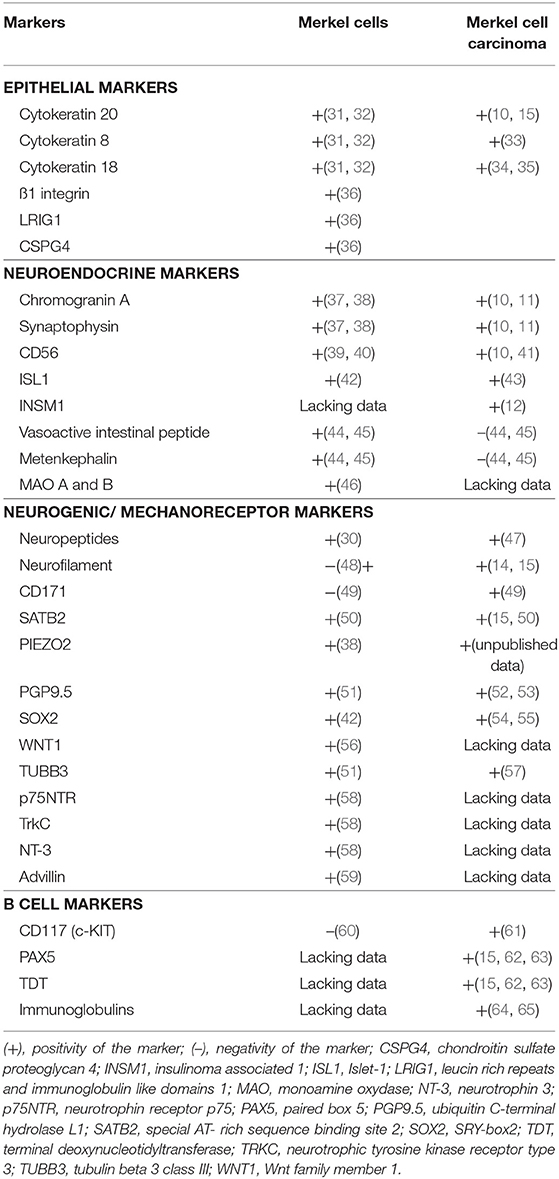
Table 1. Markers expressed by physiological Merkel cells and Merkel cell carcinoma.
Despite the large similarity in phenotypic features, several points argue against MCC deriving directly from MCs. First, in other organs such as lung, strong data suggest that neuroendocrine carcinoma derives more from epithelial progenitors rather than an neuroendocrine cell (66, 67). Second, MCs are mainly post-mitotic cells (31) and thus have low sensitivity to oncogenic stimuli. Accordingly, ectopic expression of sT antigen in MCs failed to induce cell proliferation or transformation in a transgenic mouse model (68). Of note, hyperplasia of MCs as well as mitotic activity in keratin 20-positive cells has been reported in pathologic conditions (69, 70); however, whether these observations are due to proliferation of already differentiated MCs or MC precursor cells is still unclear. Third, MCs are most frequently present in the palm and sole in humans (71, 72), whereas MCC occurs mainly in sun-exposed areas [head and neck, legs (2, 73)]. Moreover, no infection of MCs by MCPyV has been reported (74). Finally, in an in vitro model, MCPyV pseudovirions could barely infect CK20-positive cells obtained from the fetal scalp (0.8%) (75), which argues against an efficient MCPyV infection triggering MCC oncogenesis in an already differentiated MC.
Putative Mechanism of a “Non-MC” Origin for MCC
The tumor classification system is based on tumor differentiation and should not be considered a direct indicator of tumor histogenesis (76). Indeed, several phenotypic changes occurring during the oncogenic process contribute to the final differentiation profile of tumor cells, which consequently differ from the primary cell in which the first oncogenic event took place (76). Accordingly, acquisition of an MC-like phenotype including neuroendocrine differentiation (77) during MCC oncogenesis could explain the similarities between MCs and MCC (23). In MCC, both UV and virus-induced oncogenic triggers are thought to act on shared molecular pathways, accounting for the similar phenotype between MCPyV-positive and -negative tumors (78). In this respect, disruption of pRB function occurs by somatic mutations and repression of protein expression in virus-negative tumors (22), whereas sequestration by MCPyV LT antigen inactivates pRB1 in virus-positive MCC cells (79). Interestingly, disruption of this pathway has been identified as a main contributor driving acquisition of a neuroendocrine phenotype in tumors of other organs (80–82).
In the skin, MC differentiation occurs in specific epithelial precursors upon expression of one main transcription factor, atonal homolog 1 (ATOH1) (31). Under physiologic conditions, ATOH1 expression in the skin is restricted to MCs (31). Because ATOH1 is also observed in MCC, its expression could explain the shared phenotype between MCs and MCC (83). Moreover, genetic ablation of Rb1 and the related Rb-family protein p130 in the intestinal epithelium in a mouse model led to increased expression of Atoh1 (84), which suggests that Atoh1 induction could occur during an oncogenic process associated with Rb inactivation.
Considering these findings, a non-MC could also be candidate for the ancestry of MCC, and an epithelial non-MC as well as a fibroblastic and B-cell origin has been proposed (Figure 3; Table 2).
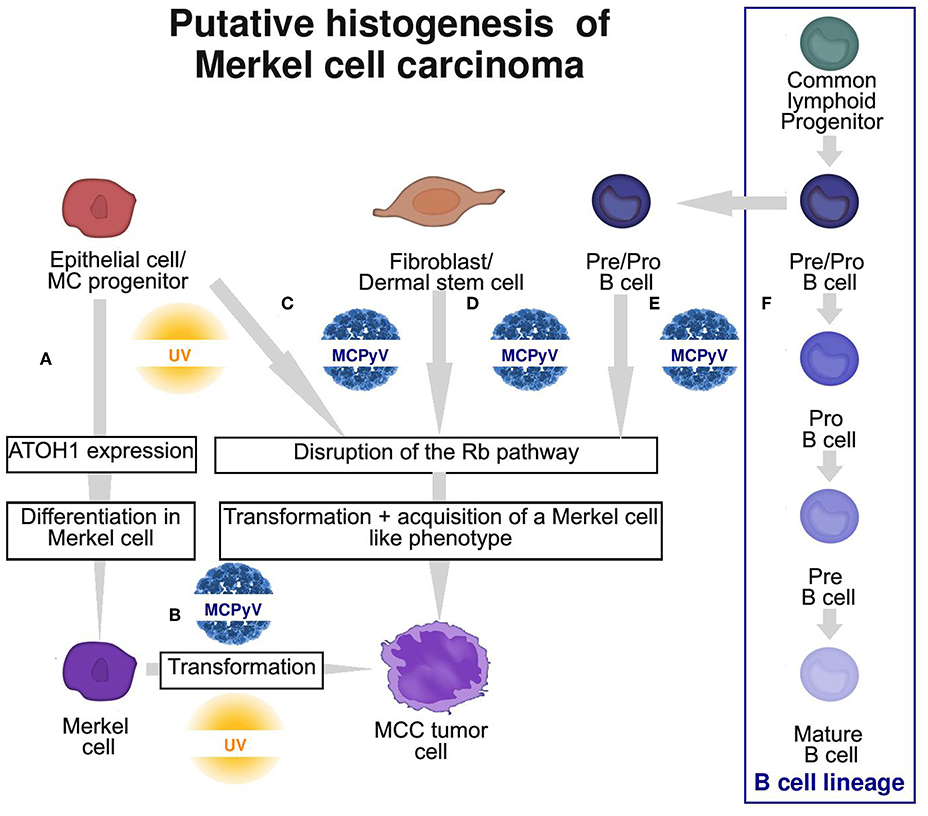
Figure 3. Graphic summary of the 4 putative cells of origin of Merkel cell carcinoma (MCC). (A) Physiological MC differentiation (B) First hypothesis: physiological MC as the cell of origin of MCC, suggesting that T antigens can induce transformation in this cell type. (C–E) Second hypothesis: oncogenic events occur in a non-MC and induce transformation and acquisition of an MC-like phenotype. Potential ancestries are epithelial progenitors (C), fibroblast/dermal stem cells (D) or pre/pro B cells (E) from the B cell lineage (F). MC, Merkel cell; MCPyV, Merkel cell polyomavirus.
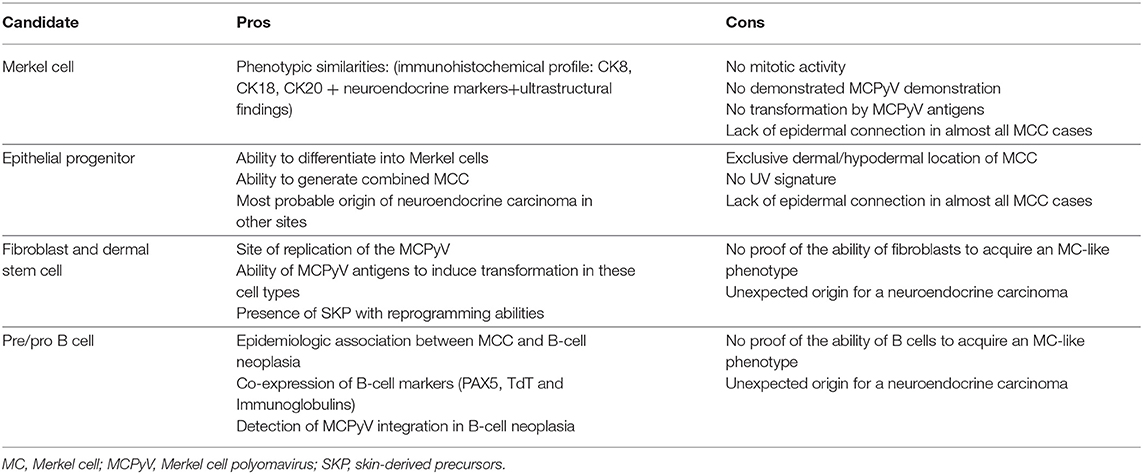
Table 2. Pros and cons of current hypotheses for the potential cell of origin of Merkel cell carcinoma (MCC).
A Non-MC Epithelial Origin
For quite some time it has been a matter to debate whether MCs derive from the neural crest or epidermal lineage. Of note both neural crest and epidermal lineages derived from the same embryologic structure and this common ectodermal origin might explain the mixed phenotype observed in Merkel cell Indeed, ultrastructural studies of MC revealed on the one hand intracytoplasmic neuroendocrine granules suggesting a neural crest origin (85) and on the other hand frequent desmosomes and cytokeratins, two hallmarks of the epithelial subset (86). Accordingly, also immunohistochemistry demonstrated both expression of “neural crest” as well as epithelial markers (Table 1). Although the neural crest origin hypothesis was additionally supported by chimeric chicken/quail models (87, 88), xenograft of human fetal skin free of neural crest progenitors in immunocompromised mice led to the development of human Merkel cell suggesting an epidermal origin of this population (89).
An epithelial origin of Merkel cells in mammals was finally demonstrated in 2009 by two consecutive transgenic mouse studies (31, 90). In both studies it was shown that deletion of Atoh1 in epidermal progenitors resulted in a complete absence of MCs. Additionally, Morrison and colleagues demonstrated that Atoh1 deletion in the neural crest lineage did not affect the MC population (90).
Additional studies in mice models revealed that MC phenotype acquisition upon Atoh1 expression seems to be restricted to a specific subpopulation of keratinocyte progenitors characterized by an activated Sonic Hedgehog pathway (91, 92). Indeed, Atoh1 expression failed to induce MC differentiation in other keratinocyte populations (31) and gave rise to distinct differentiation in other cell types (93–95).
A thorough characterization of the MC progenitor population in humans is still missing (96). Therefore, our current knowledge of this cellular subset is mainly based on findings in mice, in which cells bearing MC differentiation potential are mainly located in the outer root sheet and bulge region of the hair follicle (97, 98) but are also present in the interfollicular epidermis in specialized structures called touch domes (92). Interestingly, these hair follicle- and touch- dome–derived stem cells have been found as preferentially the origin of basal cell carcinomas (99). Therefore, their ability to acquire an MC phenotype and to proliferate, as well as their high sensitivity to oncogenic stimuli, should promote their transformation into MCC, rendering them likely candidates as cells of origin. Of note, MCC developing within follicular cysts (100) as well as preferential MCPyV infection of the dermal cells around hair follicles (75) support MCPyV(+) MCC as being derived from hair follicles.
A hair-follicle origin of MCC would also weaken one argument frequently used against an epithelial origin of MCC. Because MCC cells are mostly found in the dermis and subcutis lacking a connection to the epidermis, an epidermal origin is unlikely (62). However, some appendage tumors such as trichoblastoma and spiradenoma (101, 102) are well known to lack an epidermal connection (10).
The observation of so-called combined MCC or MCC with divergent differentiation further supports an epithelial origin of MCC. Combined MCC represents 5 to 10% of cases and is characterized by the association of an MCC component with a tumor of another differentiation lineage (103–105). Although several divergent additional components have been described (sarcomatous, adnexal) (104, 106), MCC is most frequently found associated with squamous/eccrine carcinoma (105, 107) (Figure 4). For individual cases, the same genetic alterations have been reported for both components, which implies a common progenitor (108), whereas other cases gave proof of a collision tumor (109). Furthermore, similar aberrant p53 expression is frequently observed in both components of combined MCC (105). In some combined MCC cases, intra-epidermal neoplasia such as actinic keratosis or Bowen's disease (107) was detected close to the squamous cell carcinoma component. Bowen's disease originates from the epidermis, and invasive squamous cell carcinoma can derive from Bowen's disease; hence, the clonality between squamous cell carcinoma and the MCC component (108) favors an epidermal origin of MCC (97). Of note, the hyperplasia of MCs in the squamous cell carcinoma component of combined tumors (70) might suggest that such components contain precursors with the ability to acquire an MC phenotype.
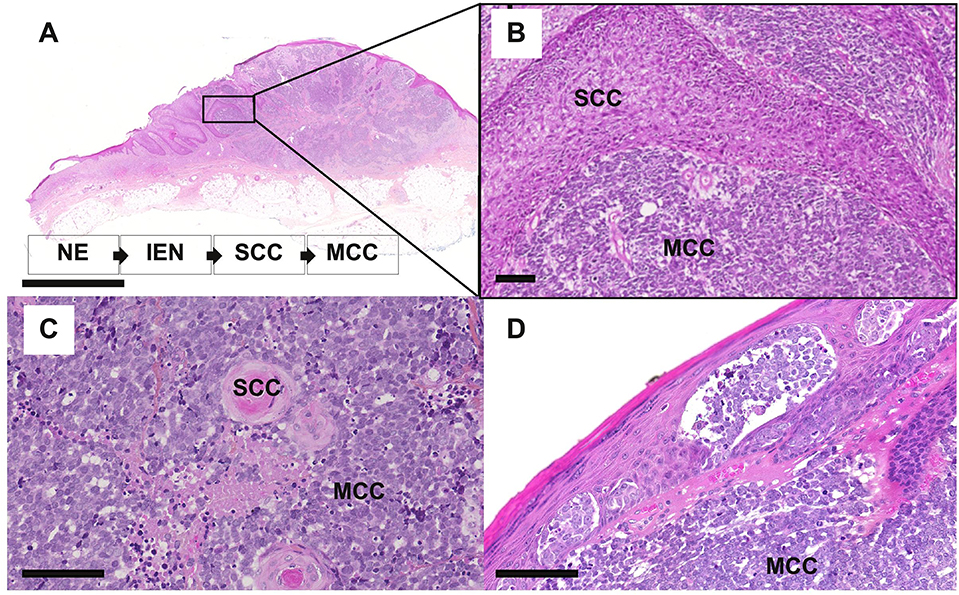
Figure 4. Microscopy features of MCC with divergent differentiation or intra-epidermal involvement [bars = 5 mm and 200 μm (A,B) and 100 μm (C,D). (A–C) combined MCC is characterized by the association of MCC with another differentiation subset, mainly squamous cell carcinoma (SCC). In some specimens, intra-epidermal neoplasia (IEN) such as Bowen disease, deriving from the non-neoplasic epidermis (NE) can be detected in tumor in close contact. (D) MCC harboring an intra-epidermal component.
Importantly, such combined cases have been described to be usually typical UV-induced MCCs, harboring morphologic and immunohistochemical features distinct from MCPyV-positive MCC and high mutational load (104, 106, 108) as depicted in Table 3. Of note low viral load of MCPyV in some cases is probably related to an episomal viral genome present in the skin (105). In our experience [(118), Figure 4], rare cases of MCC with intra-epidermal involvement [2% in our previously reported cohort (73)] are also related to the UV-induced subset. Hence, although combined cases imply that MCPyV-negative cases derive from some epidermal progenitors of the interfollicular epidermis, they provide no information about MCPyV-induced tumors (119).
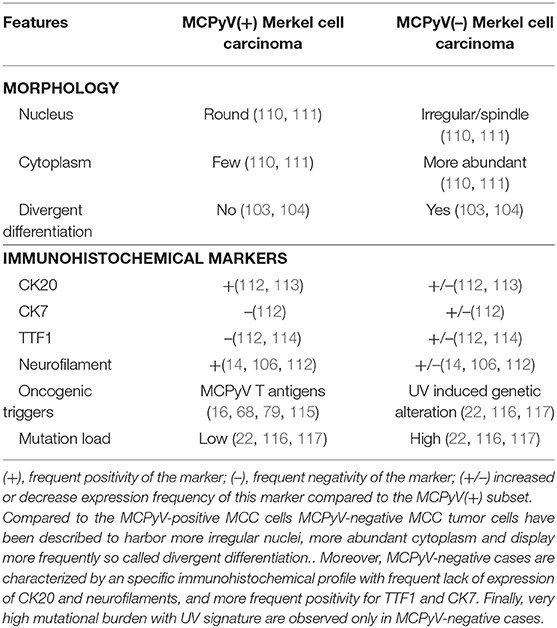
Table 3. Distinct features of MCPyV-positive and -negative MCC cases.
In agreement with this observation, Sunshine et al. hypothesized that there might be two different cells of origin for the two MCC subtypes (119). They provided several arguments for this conclusion. For example while the UV-mutation signature of virus-negative MCC favors an epidermal origin the failure of epidermis targeted TA-expression to produce tumors resembling human MCC in mouse models (68, 120, 121) suggests that other cells in the skin such as dermal fibroblast may serve as origin of MCC (119). Since both UV- and virus-induced MCC occur in sun-exposed areas where frequent UV-induced mutations are observed in keratinocytes (122), but only MCPyV-negative cases are characterized by high mutational load and UV signature (22, 119) Sunshine and colleagues excluded an epithelial and instead proposed a fibroblastic origin of MCPyV(+) MCC (119). However, low mutational burden as well as lack of UV-signature in MCPyV(+) MCC might also be explained by MCPyV integration into a cell from the hair follicle which like dermal fibroblasts is located deeper in the skin then normal epidermal keratinocytes.
In conclusion and acting on the assumption that MCC generally has an epithelial origin, one could speculate that UV-induced MCC derives from a keratinocytic progenitor from the interfollicular epidermis that acquires the ability to differentiate into MCs during the oncogenic process, whereas MCPyV-driven oncogenesis is initiated in a progenitor from a hair follicle.
A Fibroblastic Origin
Another hypothesis is MCC developing from fibroblastic cells. This hypothesis might account for the quasi-exclusive dermal location of MCC, discussed above. Furthermore, the fibroblastic origin of MCCs would be consistent with our knowledge of the MCPyV cycle because fibroblasts of the papillary dermis have been identified as the main site of replicative MCPyV infection (75). Although infectious MCPyV particles can enter several cell types including keratinocytes with various efficiency rates (75, 123), fibroblasts remain the only host cell evidencing early and late viral protein expression. One could argue that replication and transformation can occur in independent cell types, as was previously demonstrated for polyomavirus SV40 (124); however, the ability of fibroblasts to allow replication of the MCPyV genome increases the likelihood of accidental integration of the viral genome. Moreover, the in vitro transforming potential of sT antigen has until now been demonstrated only in fibroblasts (68, 124, 125). Notably, ectopic expression of SV40 T antigens in fibroblastic cells (126) triggered the induction of cytokeratin expression, which suggests that polyomavirus infection can influence a differentiation lineage. In such a setting, acquisition of an MCC phenotype induced by viral protein expression could require a transient pluripotent stage. Indeed, fibroblasts are widely used for reprogramming to pluripotent cells. The resulting induced pluripotent stem cells (127) can be differentiated into epithelial cells in vitro. Furthermore, physiological stem cells of the papillar dermis [i.e., dermal skin precursors or skin-derived precursors (128)] share phenotypic similarities with induced pluripotent stem cells, such as expression of the stem cell factors c-Myc and Sox2 (129), two markers also expressed by MCC (54, 130). These dermal skin precursors are able to differentiate into epithelial or neuronal cells in vitro. Hence, because of the close proximity of these cells to dermal fibroblasts, which can support productive MCPyV infection (75), as well as their expression of pluripotent factors and their differentiation abilities, MCPyV integration in such cells could lead to MCC oncogenesis and acquisition of an MCC phenotype.
A Pre/Pro or Pre–B-Cell Origin
Because of the recurrent association between MCC and B-cell neoplasias (131–134) as well as phenotypic similarities and the occasional integration of MCPyV in hematopoietic cells, a lymphoid pre/pro B-cell origin is also discussed (62, 64).
Indeed, chronic lymphocytic leukemia is the most frequent neoplasia associated with MCC development. Whether this is due to a common transforming event or the first tumor creating an immunological microenvironment facilitating the development of the second tumor or merely due to both tumors appearing in older immunocompromised subject has yet to be determined (131).
Moreover, MCC shares morphological features with other small round blue cell tumors, which explains why B-cell neoplasia must be considered a differential diagnosis of MCC. In addition, the coexpression of terminal deoxy nucleotidyl transferase (TdT), paired box 5 (Pax5) and immunoglobulin chains, all markers expressed during B-cell differentiation, has been observed in MCC tumors (62, 64). Initially, the frequency of TdT and Pax5 positivity was reported to be about 65% (N = 187) and 90% (N = 143) of MCC cases (64); however, recently observed rates were lower, 26% (N = 217) or 23% (N = 213) (15, 63). Of note, expression of immunoglobulin chains was restricted to the MCPyV(+) subset and detected in about 70% of cases (65). In addition, rare observations of MCC cases with monoclonal immunoglobulin rearrangement of heavy chain as well as monoclonal expression of Kappa light chain were reported (62, 65). As already discussed, determination of the histogenesis based on phenotype similarities between terminally differentiated tumor and physiological cells does not account for phenotypic changes during oncogenesis (76). In this regard, induction of immunoglobulin expression during the oncogenic process has been reported for several epithelial and soft-tissue neoplasias (135, 136) and may contribute to tumor aggressiveness (137). Furthermore, immunoglobulin rearrangement due to the expression of essential enzymes required for gene rearrangement and class switch recombination has been described in non-hematopoietic neoplasia (136). Hence, immunoglobulin expression and rearrangement might result from the oncogenic process, and their occurrence in MCC cannot rule out a non-lymphoid cell origin. Induction of immunoglobulin expression in epithelial cells has been reported to result from Epstein-Barr virus infection (138) and was also observed in papillomavirus-induced neoplasia (139). These findings, combined with the exclusive expression of immunoglobulins in MCPyV(+) MCC, led Murakami and colleagues to hypothesize that the immunoglobulin expression in MCC cells is induced by MCPyV oncoproteins (65). In the same manner, the concomitant expression of TdT and Pax5 is restricted to immature B cells and thymocytes under physiological conditions (140) and is also observed in MCC. While co-expression have not yet been described positivity of one of these markers has also been demonstrated in several epithelial neoplasias (141, 142), which indicates that these markers can be acquired during the oncogenic process. Moreover, MCPyV genome integration (143) associated with a deletion leading to a truncated LT antigen (144), the two hallmarks of MCC oncogenesis, have been evidenced in some cases of chronic lymphocytic leukemia and tropism of other tumor viruses for the Pre-Pro B cells has been previously emphasized (145). Although these findings demonstrate that MCPyV integration associated with transformation can occur in B cells, lack of acquisition of an MCC phenotype in these cases argue against a B-cell origin of MCC.
Summary
To conclude, reviewing the current knowledge of MCC histogenesis allows for also underlining the basis of the current tumor classification system. Indeed, tumors are mostly classified according to their differentiation status and their level of similarities with physiological cells at the same location (24). However, we should keep in mind that the final phenotype of a given tumor cell may result from strong differentiation changes occurring during oncogenesis and thus does not necessarily directly reflect the cell ancestry (76). Accordingly, despite strong similarities, MCC likely does not derive from already differentiated MCs, which suggests that acquisition of an MC-like phenotype occurs during the oncogenic process (Figure 3). From the observations of combined MCC tumors, high somatic pathologic variant loads and detection of an UV signature in this subset, UV-induced MCC cases probably derive from a progenitor cell of the epidermis. By contrast, the nature of the cell in which MCPyV integration occurs remains to be clarified. The lack of connection between tumor cells and the epidermis as well as lack of a UV signature could favor a non-epithelial origin but alternatively could be explained by integration of MCPyV in cutaneous appendage enriched with MC precursors. Use of experimental models in addition to phenotypic characterization of MCC to monitor phenotype changes induced by MCPyV in several cell types are needed to fully address this question.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the Foundation ARC and the Ligue nationale contre le cancer, Comités 16, 18, 28.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Paulson KG, Park SY, Vandeven NA, Lachance K, Thomas H, Chapuis AG, et al. Merkel cell carcinoma: current US incidence and projected increases based on changing demographics. J Am Acad Dermatol. (2018) 78:457–63 e2. doi: 10.1016/j.jaad.2017.10.028
2. Miller RW, Rabkin CS. Merkel cell carcinoma and melanoma: etiological similarities and differences. Cancer Epidemiol Biomark Prev. (1999) 8:153–8.
3. Asgari MM, Sokil MM, Warton EM, Iyer J, Paulson KG, Nghiem P. Effect of host, tumor, diagnostic, and treatment variables on outcomes in a large cohort with Merkel cell carcinoma. JAMA Dermatol. (2014) 150:716–23. doi: 10.1001/jamadermatol.2013.8116
4. Cook M, Baker K, Redman M, Lachance K, Nguyen MH, Parvathaneni U, et al. Differential outcomes among immunosuppressed patients with Merkel cell carcinoma: impact of immunosuppression type on cancer-specific and overall survival. Am J Clin Oncol. (2019) 42:82–8. doi: 10.1097/COC.0000000000000482
5. Lemos BD, Storer BE, Iyer JG, Phillips JL, Bichakjian CK, Fang LC, et al. Pathologic nodal evaluation improves prognostic accuracy in Merkel cell carcinoma: analysis of 5823 cases as the basis of the first consensus staging system. J Am Acad Dermatol. (2010) 63:751–61. doi: 10.1016/j.jaad.2010.02.056
6. Nghiem P, Kaufman HL, Bharmal M, Mahnke L, Phatak H, Becker JC. Systematic literature review of efficacy, safety and tolerability outcomes of chemotherapy regimens in patients with metastatic Merkel cell carcinoma. Future Oncol Lond Engl. (2017) 13:1263–79. doi: 10.2217/fon-2017-0072
7. Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D'Angelo SP, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. (2016) 17:1374–85. doi: 10.1016/S1470-2045(16)30364-3
8. Colunga A, Pulliam T, Nghiem P. Merkel cell carcinoma in the age of immunotherapy: facts and hopes. Clin Cancer Res. (2018) 24:2035–43. doi: 10.1158/1078-0432.CCR-17-0439
9. D'Angelo SP, Russell J, Lebbé C, Chmielowski B, Gambichler T, Grob J-J, et al. Efficacy and safety of first-line avelumab treatment in patients with stage IV metastatic Merkel cell carcinoma: a preplanned interim analysis of a clinical trial. JAMA Oncol. (2018) 4:e180077. doi: 10.1001/jamaoncol.2018.0077
11. Koljonen V, Haglund C, Tukiainen E, Böhling T. Neuroendocrine differentiation in primary Merkel cell carcinoma–possible prognostic significance. Anticancer Res. (2005) 25:853–8.
12. Lilo MT, Chen Y, LeBlanc RE. INSM1 is more sensitive and interpretable than conventional immunohistochemical stains used to diagnose Merkel cell carcinoma. Am J Surg Pathol. (2018) 42:1541–48. doi: 10.1097/PAS.0000000000001136
13. Cheuk W, Kwan MY, Suster S, Chan JK. Immunostaining for thyroid transcription factor 1 and cytokeratin 20 aids the distinction of small cell carcinoma from Merkel cell carcinoma, but not pulmonary from extrapulmonary small cell carcinomas. Arch Pathol Lab Med. (2001) 125:228–31. doi: 10.1043/0003-9985(2001)125<0228:IFTTFA>2.0.CO;2
14. Stanoszek LM, Chan MP, Palanisamy N, Carskadon S, Siddiqui J, Patel RM, et al. Neurofilament is superior to cytokeratin 20 in supporting cutaneous origin for neuroendocrine carcinoma. Histopathology. (2019) 74:504–3. doi: 10.1111/his.13758
15. Kervarrec T, Tallet A, Miquelestorena-Standley E, Houben R, Schrama D, Gambichler T, et al. Diagnostic accuracy of a panel of immunohistochemical and molecular markers to distinguish Merkel cell carcinoma from other neuroendocrine carcinomas. Mod Pathol. (2019) 32:499–510. doi: 10.1038/s41379-018-0155-y
16. Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. (2008) 319:1096–100. doi: 10.1126/science.1152586
17. Foulongne V, Kluger N, Dereure O, Mercier G, Molès JP, Guillot B, et al. Merkel cell polyomavirus in cutaneous swabs. Emerg Infect Dis. (2010) 16:685–7. doi: 10.3201/eid1604.091278
18. Martel-Jantin C, Pedergnana V, Nicol JTJ, Leblond V, Trégouët D-A, Tortevoye P, et al. Merkel cell polyomavirus infection occurs during early childhood and is transmitted between siblings. J Clin Virol. (2013) 58:288–91. doi: 10.1016/j.jcv.2013.06.004
19. Tolstov YL, Knauer A, Chen JG, Kensler TW, Kingsley LA, Moore PS, et al. Asymptomatic primary Merkel cell polyomavirus infection among adults. Emerg Infect Dis. (2011) 17:1371–80. doi: 10.3201/eid1708.110079
20. Shuda M, Feng H, Kwun HJ, Rosen ST, Gjoerup O, Moore PS, et al. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci USA. (2008) 105:16272–7. doi: 10.1073/pnas.0806526105
21. Houben R, Angermeyer S, Haferkamp S, Aue A, Goebeler M, Schrama D, et al. Characterization of functional domains in the Merkel cell polyoma virus Large T antigen. Int J Cancer. (2015) 136:E290–300. doi: 10.1002/ijc.29200
22. Harms PW, Vats P, Verhaegen ME, Robinson DR, Wu Y-M, Dhanasekaran SM, et al. The distinctive mutational spectra of polyomavirus-negative Merkel cell carcinoma. Cancer Res. (2015) 75:3720–7. doi: 10.1158/0008-5472.CAN-15-0702
23. Harms PW, Harms KL, Moore PS, DeCaprio JA, Nghiem P, Wong MKK, et al. The biology and treatment of Merkel cell carcinoma: current understanding and research priorities. Nat Rev Clin Oncol. (2018) 15:763–76. doi: 10.1038/s41571-018-0103-2
24. Boyd N, Dancey JE, Gilks CB, Huntsman DG. Rare cancers: a sea of opportunity. Lancet Oncol. (2016) 17:e52–61. doi: 10.1016/S1470-2045(15)00386-1
25. Ikeda R, Cha M, Ling J, Jia Z, Coyle D, Gu JG. Merkel cells transduce and encode tactile stimuli to drive Aβ-afferent impulses. Cell. (2014) 157:664–75. doi: 10.1016/j.cell.2014.02.026
26. Chang W, Kanda H, Ikeda R, Ling J, DeBerry JJ, Gu JG. Merkel disc is a serotonergic synapse in the epidermis for transmitting tactile signals in mammals. Proc Natl Acad Sci USA. (2016) 113:E5491–500. doi: 10.1073/pnas.1610176113
27. Harms PW, Patel RM, Verhaegen ME, Giordano TJ, Nash KT, Johnson CN, et al. Distinct gene expression profiles of viral- and nonviral-associated merkel cell carcinoma revealed by transcriptome analysis. J Invest Dermatol. (2013) 133:936–45. doi: 10.1038/jid.2012.445
28. Toker C. Trabecular carcinoma of the skin. Arch Dermatol. (1972) 105:107–10. doi: 10.1001/archderm.1972.01620040075020
29. Tang CK, Toker C. Trabecular carcinoma of the skin: an ultrastructural study. Cancer. (1978) 42:2311–21. doi: 10.1002/1097-0142(197811)42:5<2311::AID-CNCR2820420531>3.0.CO;2-L
30. Godlewski J, Kowalczyk A, Kozielec Z, Pidsudko Z, Kmieć A, Siedlecka-Kroplewska K. Plasticity of neuropeptidergic neoplasm cells in the primary and metastatic Merkel cell carcinoma. Folia Histochem Cytobiol. (2013) 51:168–73. doi: 10.5603/FHC.2013.0015
31. Van Keymeulen A, Mascre G, Youseff KK, Harel I, Michaux C, De Geest N, et al. Epidermal progenitors give rise to Merkel cells during embryonic development and adult homeostasis. J Cell Biol. (2009) 187:91–100. doi: 10.1083/jcb.200907080
32. Perdigoto CN, Dauber KL, Bar C, Tsai P-C, Valdes VJ, Cohen I, et al. Polycomb-mediated repression and sonic hedgehog signaling interact to regulate Merkel cell specification during skin development. PLoS Genet. (2016) 12:e1006151. doi: 10.1371/journal.pgen.1006151
33. Baudoin C, Meneguzzi G, Portier MM, Demarchez M, Bernerd F, Pisani A, et al. Peripherin, a neuronal intermediate protein, is stably expressed by neuroendocrine carcinomas of the skin, their xenograft on nude mice, and the corresponding primary cultures. Cancer Res. (1993) 53:1175–81.
34. Kontochristopoulos GJ, Stavropoulos PG, Krasagakis K, Goerdt S, Zouboulis CC. Differentiation between merkel cell carcinoma and malignant melanoma: an immunohistochemical study. Dermatol Basel Switz. (2000) 201:123–6. doi: 10.1159/000018454
35. Li Z, Yang J-J, Wu M. Collision tumor of primary merkel cell carcinoma and chronic lymphocytic leukemia/small lymphocytic lymphoma, diagnosed on ultrasound-guided fine-needle aspiration biopsy: a unique case report and review of literature. Diagn Cytopathol. (2015) 43:66–71. doi: 10.1002/dc.23127
36. Tilling T, Wladykowski E, Failla AV, Houdek P, Brandner JM, Moll I. Immunohistochemical analyses point to epidermal origin of human Merkel cells. Histochem Cell Biol. (2014) 141:407–21. doi: 10.1007/s00418-013-1168-8
37. Moll I, Kuhn C, Moll R. Cytokeratin 20 is a general marker of cutaneous Merkel cells while certain neuronal proteins are absent. J Invest Dermatol. (1995) 104:910–5. doi: 10.1111/1523-1747.ep12606183
38. García-Mesa Y, García-Piqueras J, García B, Feito J, Cabo R, Cobo J, et al. Merkel cells and Meissner's corpuscles in human digital skin display Piezo2 immunoreactivity. J Anat. (2017) 231:978–89. doi: 10.1111/joa.12688
39. Mouchet N, Coquart N, Lebonvallet N, Le Gall-Ianotto C, Mogha A, Fautrel A, et al. Comparative transcriptional profiling of human Merkel cells and Merkel cell carcinoma. Exp Dermatol. (2014) 23:928–30. doi: 10.1111/exd.12546
40. Boulais N, Pereira U, Lebonvallet N, Gobin E, Dorange G, Rougier N, et al. Merkel cells as putative regulatory cells in skin disorders: an in vitro study. PLoS ONE. (2009) 4:e6528. doi: 10.1371/journal.pone.0006528
41. Fernández-Figueras M-T, Puig L, Musulén E, Gilaberte M, Lerma E, Serrano S, et al. Expression profiles associated with aggressive behavior in Merkel cell carcinoma. Mod Pathol. (2007) 20:90–101. doi: 10.1038/modpathol.3800717
42. Perdigoto CN, Bardot ES, Valdes VJ, Santoriello FJ, Ezhkova E. Embryonic maturation of epidermal Merkel cells is controlled by a redundant transcription factor network. Dev Camb Engl. (2014) 141:4690–6. doi: 10.1242/dev.112169
43. Agaimy A, Erlenbach-Wünsch K, Konukiewitz B, Schmitt AM, Rieker RJ, Vieth M, et al. ISL1 expression is not restricted to pancreatic well-differentiated neuroendocrine neoplasms, but is also commonly found in well and poorly differentiated neuroendocrine neoplasms of extrapancreatic origin. Mod Pathol. (2013) 26:995–1003. doi: 10.1038/modpathol.2013.40
44. Nguyen BD, McCullough AE. Imaging of Merkel cell carcinoma. Radiogr Rev. (2002) 22:367–76. doi: 10.1148/radiographics.22.2.g02mr14367
45. Hoefler H, Kerl H, Rauch HJ, Denk H. New immunocytochemical observations with diagnostic significance in cutaneous neuroendocrine carcinoma. Am J Dermatopathol. (1984) 6:525–30. doi: 10.1097/00000372-198412000-00002
46. Vitalis T, Alvarez C, Chen K, Shih JC, Gaspar P, Cases O. Developmental expression pattern of monoamine oxidases in sensory organs and neural crest derivatives. J Comp Neurol. (2003) 464:392–403. doi: 10.1002/cne.10804
47. Halata Z, Grim M, Bauman KI. Friedrich Sigmund Merkel and his “Merkel cell”, morphology, development, and physiology: review and new results. Anat Rec A Discov Mol Cell Evol Biol. (2003) 271:225–39. doi: 10.1002/ar.a.10029
48. Narisawa Y, Hashimoto K, Kohda H. Immunohistochemical demonstration of the expression of neurofilament proteins in Merkel cells. Acta Derm Venereol. (1994) 74:441–3. doi: 10.2340/0001555574441443
49. Deichmann M, Kurzen H, Egner U, Altevogt P, Hartschuh W. Adhesion molecules CD171 (L1CAM) and CD24 are expressed by primary neuroendocrine carcinomas of the skin (Merkel cell carcinomas). J Cutan Pathol. (2003) 30:363–8. doi: 10.1034/j.1600-0560.2003.00073.x
50. Fukuhara M, Agnarsdóttir M, Edqvist P-H, Coter A, Ponten F. SATB2 is expressed in Merkel cell carcinoma. Arch Dermatol Res. (2016) 308:449–54. doi: 10.1007/s00403-016-1655-6
51. Sebastian A, Volk SW, Halai P, Colthurst J, Paus R, Bayat A. Enhanced neurogenic biomarker expression and reinnervation in human acute skin wounds treated by electrical stimulation. J Invest Dermatol. (2017) 137:737–47. doi: 10.1016/j.jid.2016.09.038
52. McCluggage WG, Kennedy K, Busam KJ. An immunohistochemical study of cervical neuroendocrine carcinomas: neoplasms that are commonly TTF1 positive and which may express CK20 and P63. Am J Surg Pathol. (2010) 34:525–32. doi: 10.1097/PAS.0b013e3181d1d457
53. Inoue T, Shimono M, Takano N, Saito C, Tanaka Y. Merkel cell carcinoma of palatal mucosa in a young adult: immunohistochemical and ultrastructural features. Oral Oncol. (1997) 33:226–9. doi: 10.1016/S0964-1955(96)00078-4
54. Azmahani A, Nakamura Y, Ishida H, McNamara KM, Fujimura T, Haga T, et al. Estrogen receptor β in Merkel cell carcinoma: its possible roles in pathogenesis. Hum Pathol. (2016) 56:128–33. doi: 10.1016/j.humpath.2016.06.005
55. Lasithiotaki I, Tsitoura E, Koutsopoulos A, Lagoudaki E, Koutoulaki C, Pitsidianakis G, et al. Aberrant expression of miR-21, miR-376c and miR-145 and their target host genes in Merkel cell polyomavirus-positive non-small cell lung cancer. Oncotarget. (2017) 8:112371–83. doi: 10.18632/oncotarget.11222
56. Szeder V, Grim M, Halata Z, Sieber-Blum M. Neural crest origin of mammalian Merkel cells. Dev Biol. (2003) 253:258–63. doi: 10.1016/S0012-1606(02)00015-5
57. Jirásek T, Matej R, Pock L, Knotková I, Mandys V. Merkel cell carcinoma–immunohistochemical study in a group of 11 patients. Cesk Patol. (2009) 45:9–13.
58. Sieber-Blum M, Szeder V, Grim M. The role of NT-3 signaling in Merkel cell development. Prog Brain Res. (2004) 146:63–72. doi: 10.1016/S0079-6123(03)46004-4
59. Hunter DV, Smaila BD, Lopes DM, Takatoh J, Denk F, Ramer MS. Advillin is expressed in all adult neural crest-derived neurons. eNeuro. (2018) 5:e0077-18. doi: 10.1523/ENEURO.0077-18.2018
60. Grichnik JM, Ali WN, Burch JA, Byers JD, Garcia CA, Clark RE, et al. KIT expression reveals a population of precursor melanocytes in human skin. J Invest Dermatol. (1996) 106:967–71. doi: 10.1111/1523-1747.ep12338471
61. Su LD, Fullen DR, Lowe L, Uherova P, Schnitzer B, Valdez R. CD117 (KIT receptor) expression in Merkel cell carcinoma. Am J Dermatopathol. (2002) 24:289–93. doi: 10.1097/00000372-200208000-00001
62. Zur Hausen A, Rennspiess D, Winnepenninckx V, Speel E-J, Kurz AK. Early B-cell differentiation in Merkel cell carcinomas: clues to cellular ancestry. Cancer Res. (2013) 73:4982–7. doi: 10.1158/0008-5472.CAN-13-0616
63. Johansson B, Sahi H, Koljonen V, Böhling T. The expression of terminal deoxynucleotidyl transferase and paired box gene 5 in Merkel cell carcinomas and its relation to the presence of Merkel cell polyomavirus DNA. J Cutan Pathol. (2018). doi: 10.1111/cup.13372
64. Sauer CM, Haugg AM, Chteinberg E, Rennspiess D, Winnepenninckx V, Speel E-J, et al. Reviewing the current evidence supporting early B-cells as the cellular origin of Merkel cell carcinoma. Crit Rev Oncol Hematol. (2017) 116:99–105. doi: 10.1016/j.critrevonc.2017.05.009
65. Murakami I, Takata K, Matsushita M, Nonaka D, Iwasaki T, Kuwamoto S, et al. Immunoglobulin expressions are only associated with MCPyV-positive Merkel cell carcinomas but not with MCPyV-negative ones: comparison of prognosis. Am J Surg Pathol. (2014) 38:1627–35. doi: 10.1097/PAS.0000000000000279
66. Yoshimoto T, Motoi N, Yamamoto N, Nagano H, Ushijima M, Matsuura M, et al. Pulmonary carcinoids and low-grade gastrointestinal neuroendocrine tumors show common MicroRNA expression profiles, different from adenocarcinomas and small cell carcinomas. Neuroendocrinology. (2018) 106:47–57. doi: 10.1159/000461582
67. Yazawa T. Recent advances in histogenesis research of lung neuroendocrine cancers: evidence obtained from functional analyses of primitive neural/neuroendocrine cell-specific transcription factors. Pathol Int. (2015) 65:277–85. doi: 10.1111/pin.12267
68. Shuda M, Guastafierro A, Geng X, Shuda Y, Ostrowski SM, Lukianov S, et al. Merkel cell polyomavirus small T antigen induces cancer and embryonic merkel cell proliferation in a transgenic mouse model. PLoS ONE. (2015) 10:e0142329. doi: 10.1371/journal.pone.0142329
69. Kanitakis J, Bourchany D, Faure M, Claudy A. Merkel cells in hyperplastic and neoplastic lesions of the skin. An immunohistochemical study using an antibody to keratin 20. Dermatol Basel Switz. (1998) 196:208–12. doi: 10.1159/000017900
70. Narisawa Y, Inoue T, Nagase K. Evidence of proliferative activity in human Merkel cells: implications in the histogenesis of Merkel cell carcinoma. Arch Dermatol Res. (2019) 311:37–43. doi: 10.1007/s00403-018-1877-x
71. Vela-Romera A, Carriel V, Martín-Piedra MA, Aneiros-Fernández J, Campos F, Chato-Astrain J, et al. Characterization of the human ridged and non-ridged skin: a comprehensive histological, histochemical and immunohistochemical analysis. Histochem Cell Biol. (2019) 151:57–73. doi: 10.1007/s00418-018-1701-x
72. Fradette J, Godbout MJ, Michel M, Germain L. Localization of Merkel cells at hairless and hairy human skin sites using keratin 18. Biochem Cell Biol. (1995) 73:635–9. doi: 10.1139/o95-070
73. Kervarrec T, Gaboriaud P, Berthon P, Zaragoza J, Schrama D, Houben R, et al. Merkel cell carcinomas infiltrated with CD33+ myeloid cells and CD8+ T cells are associated with improved outcome. J Am Acad Dermatol. (2018) 78:973–82. doi: 10.1016/j.jaad.2017.12.029
74. Tilling T, Moll I. Which are the cells of origin in merkel cell carcinoma? J Skin Cancer. (2012) 2012:680410. doi: 10.1155/2012/680410
75. Liu W, Yang R, Payne AS, Schowalter RM, Spurgeon ME, Lambert PF, et al. Identifying the target cells and mechanisms of Merkel cell polyomavirus infection. Cell Host Microbe. (2016) 19:775–87. doi: 10.1016/j.chom.2016.04.024
76. Fletcher CDM. The evolving classification of soft tissue tumours: an update based on the new WHO classification. Histopathology. (2006) 48:3–12. doi: 10.1111/j.1365-2559.2005.02284.x
77. Chteinberg E, Sauer CM, Rennspiess D, Beumers L, Schiffelers L, Eben J, et al. Neuroendocrine key regulator gene expression in Merkel cell carcinoma. Neoplasia. (2018) 20:1227–35. doi: 10.1016/j.neo.2018.10.003
78. González-Vela MDC, Curiel-Olmo S, Derdak S, Beltran S, Santibañez M, Martínez N, et al. Shared oncogenic pathways implicated in both virus-positive and uv-induced Merkel cell carcinomas. J Invest Dermatol. (2017) 137:197–206. doi: 10.1016/j.jid.2016.08.015
79. Houben R, Adam C, Baeurle A, Hesbacher S, Grimm J, Angermeyer S, et al. An intact retinoblastoma protein-binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. Int J Cancer. (2012) 130:847–56. doi: 10.1002/ijc.26076
80. Shamir ER, Devine WP, Pekmezci M, Umetsu SE, Krings G, Federman S, et al. Identification of high-risk human papillomavirus and Rb/E2F pathway genomic alterations in mutually exclusive subsets of colorectal neuroendocrine carcinoma. Mod Pathol. (2019) 32:290–305. doi: 10.1038/s41379-018-0131-6
81. Meder L, König K, Ozretić L, Schultheis AM, Ueckeroth F, Ade CP, et al. NOTCH, ASCL1, p53 and RB alterations define an alternative pathway driving neuroendocrine and small cell lung carcinomas. Int J Cancer. (2016) 138:927-38. doi: 10.1002/ijc.29835
82. Syder AJ, Karam SM, Mills JC, Ippolito JE, Ansari HR, Farook V, et al. A transgenic mouse model of metastatic carcinoma involving transdifferentiation of a gastric epithelial lineage progenitor to a neuroendocrine phenotype. Proc Natl Acad Sci USA. (2004) 101:4471–6. doi: 10.1073/pnas.0307983101
83. Gambichler T, Mohtezebsade S, Wieland U, Silling S, Höh A-K, Dreißigacker M, et al. Prognostic relevance of high atonal homolog-1 expression in Merkel cell carcinoma. J Cancer Res Clin Oncol. (2017) 143:43–9. doi: 10.1007/s00432-016-2257-6
84. Haigis K, Sage J, Glickman J, Shafer S, Jacks T. The related retinoblastoma (pRb) and p130 proteins cooperate to regulate homeostasis in the intestinal epithelium. J Biol Chem. (2006) 281:638–47. doi: 10.1074/jbc.M509053200
85. Winkelmann RK. The Merkel cell system and a comparison between it and the neurosecretory or APUD cell system. J Invest Dermatol. (1977) 69:41–6. doi: 10.1111/1523-1747.ep12497864
86. Lucarz A, Brand G. Current considerations about Merkel cells. Eur J Cell Biol. (2007) 86:243–51. doi: 10.1016/j.ejcb.2007.02.001
87. Halata Z, Grim M, Christ B. Origin of spinal cord meninges, sheaths of peripheral nerves, and cutaneous receptors including Merkel cells. An experimental and ultrastructural study with avian chimeras. Anat Embryol (Berl). (1990) 182:529–37. doi: 10.1007/BF00186459
88. Grim M, Halata Z. Developmental origin of avian Merkel cells. Anat Embryol (Berl). (2000) 202:401–10. doi: 10.1007/s004290000121
89. Moll I, Lane AT, Franke WW, Moll R. Intraepidermal formation of Merkel cells in xenografts of human fetal skin. J Invest Dermatol. (1990) 94:359–64. doi: 10.1111/1523-1747.ep12874488
90. Morrison KM, Miesegaes GR, Lumpkin EA, Maricich SM. Mammalian Merkel cells are descended from the epidermal lineage. Dev Biol. (2009) 336:76–83. doi: 10.1016/j.ydbio.2009.09.032
91. Xiao Y, Thoresen DT, Williams JS, Wang C, Perna J, Petrova R, et al. Neural Hedgehog signaling maintains stem cell renewal in the sensory touch dome epithelium. Proc Natl Acad Sci USA. (2015) 112:7195–200. doi: 10.1073/pnas.1504177112
92. Xiao Y, Williams JS, Brownell I. Merkel cells and touch domes: more than mechanosensory functions? Exp Dermatol. (2014) 23:692–5. doi: 10.1111/exd.12456
93. Mulvaney J, Dabdoub A. Atoh1, an essential transcription factor in neurogenesis and intestinal and inner ear development: function, regulation, and context dependency. J Assoc Res Otolaryngol. (2012) 13:281–93. doi: 10.1007/s10162-012-0317-4
94. Shi X, Zhang Z, Zhan X, Cao M, Satoh T, Akira S, et al. An epigenetic switch induced by Shh signalling regulates gene activation during development and medulloblastoma growth. Nat Commun. (2014) 5:5425. doi: 10.1038/ncomms6425
95. Aragaki M, Tsuchiya K, Okamoto R, Yoshioka S, Nakamura T, Sakamoto N, et al. Proteasomal degradation of Atoh1 by aberrant Wnt signaling maintains the undifferentiated state of colon cancer. Biochem Biophys Res Commun. (2008) 368:923–9. doi: 10.1016/j.bbrc.2008.02.011
96. Moll I, Troyanovsky SM, Moll R. Special program of differentiation expressed in keratinocytes of human haarscheiben: an analysis of individual cytokeratin polypeptides. J Invest Dermatol. (1993) 100:69–76. doi: 10.1111/1523-1747.ep12354535
97. Narisawa Y, Koba S, Inoue T, Nagase K. Histogenesis of pure and combined Merkel cell carcinomas: an immunohistochemical study of 14 cases. J Dermatol. (2015) 42:445–52. doi: 10.1111/1346-8138.12808
98. Akiyama M, Dale BA, Sun TT, Holbrook KA. Characterization of hair follicle bulge in human fetal skin: the human fetal bulge is a pool of undifferentiated keratinocytes. J Invest Dermatol. (1995) 105:844–50. doi: 10.1111/1523-1747.ep12326649
99. Peterson SC, Eberl M, Vagnozzi AN, Belkadi A, Veniaminova NA, Verhaegen ME, et al. Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem Cell. (2015) 16:400–12. doi: 10.1016/j.stem.2015.02.006
100. Requena L, Jaqueti G, Rütten A, Mentzel T, Kutzner H. Merkel cell carcinoma within follicular cysts: report of two cases. J Cutan Pathol. (2008) 35:1127–33. doi: 10.1111/j.1600-0560.2007.00919.x
101. Tellechea O, Cardoso JC, Reis JP, Ramos L, Gameiro AR, Coutinho I, et al. Benign follicular tumors. An Bras Dermatol. (2015) 90:780-96; quiz 797-8. doi: 10.1590/abd1806-4841.20154114
102. Zheng Y, Tian Q, Wang J, Dong X, Jing H, Wang X, et al. Differential diagnosis of eccrine spiradenoma: a case report. Exp Ther Med. (2014) 8:1097–101. doi: 10.3892/etm.2014.1906
103. Busam KJ, Jungbluth AA, Rekthman N, Coit D, Pulitzer M, Bini J, et al. Merkel cell polyomavirus expression in merkel cell carcinomas and its absence in combined tumors and pulmonary neuroendocrine carcinomas. Am J Surg Pathol. (2009) 33:1378–85. doi: 10.1097/PAS.0b013e3181aa30a5
104. Martin B, Poblet E, Rios JJ, Kazakov D, Kutzner H, Brenn T, et al. Merkel cell carcinoma with divergent differentiation: histopathological and immunohistochemical study of 15 cases with PCR analysis for Merkel cell polyomavirus. Histopathology. (2013) 62:711–22. doi: 10.1111/his.12091
105. Kervarrec T, Samimi M, Gaboriaud P, Gheit T, Beby-Defaux A, Houben R, et al. Detection of the Merkel cell polyomavirus in the neuroendocrine component of combined Merkel cell carcinoma. Virchows Arch Int J Pathol. (2018) 472:825–37. doi: 10.1007/s00428-018-2342-0
106. Pulitzer MP, Brannon AR, Berger MF, Louis P, Scott SN, Jungbluth AA, et al. Cutaneous squamous and neuroendocrine carcinoma: genetically and immunohistochemically different from Merkel cell carcinoma. Mod Pathol. (2015) 28:1023–32. doi: 10.1038/modpathol.2015.60
107. Walsh NM. Primary neuroendocrine (Merkel cell) carcinoma of the skin: morphologic diversity and implications thereof. Hum Pathol. (2001) 32:680–9. doi: 10.1053/hupa.2001.25904
108. Carter MD, Gaston D, Huang W-Y, Greer WL, Pasternak S, Ly TY, et al. Genetic profiles of different subsets of Merkel cell carcinoma show links between combined and pure MCPyV-negative tumors. Hum Pathol. (2017). doi: 10.1016/j.humpath.2017.10.014
109. Falto Aizpurua LA, Wang M, Ruiz HA, Sánchez JL, Chan MP, Andea AA, et al. A case of combined Merkel cell carcinoma and squamous cell carcinoma: molecular insights and diagnostic pitfalls. JAAD Case Rep. (2018) 4:996–9. doi: 10.1016/j.jdcr.2018.08.004
110. Kuwamoto S, Higaki H, Kanai K, Iwasaki T, Sano H, Nagata K, et al. Association of Merkel cell polyomavirus infection with morphologic differences in Merkel cell carcinoma. Hum Pathol. (2011) 42:632–40. doi: 10.1016/j.humpath.2010.09.011
111. Iwasaki T, Matsushita M, Kuwamoto S, Kato M, Murakami I, Higaki-Mori H, et al. Usefulness of significant morphologic characteristics in distinguishing between Merkel cell polyomavirus-positive and Merkel cell polyomavirus-negative Merkel cell carcinomas. Hum Pathol. (2013) 44:1912–7. doi: 10.1016/j.humpath.2013.01.026
112. Pasternak S, Carter MD, Ly TY, Doucette S, Walsh NM. Immunohistochemical profiles of different subsets of Merkel cell carcinoma. Hum Pathol. (2018) 82:232–8. doi: 10.1016/j.humpath.2018.07.022
113. Harms PW, Collie AMB, Hovelson DH, Cani AK, Verhaegen ME, Patel RM, et al. Next generation sequencing of Cytokeratin 20-negative Merkel cell carcinoma reveals ultraviolet-signature mutations and recurrent TP53 and RB1 inactivation. Mod Pathol. (2016) 29:240–8. doi: 10.1038/modpathol.2015.154
114. Czapiewski P, Majewska H, Kutzner H, Kazakov D, Renkielska A, Biernat W. TTF-1 and PAX5 are frequently expressed in combined Merkel cell carcinoma. Am J Dermatopathol. (2016) 38:513–6. doi: 10.1097/DAD.0000000000000464
115. Shuda M, Kwun HJ, Feng H, Chang Y, Moore PS. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J Clin Invest. (2011) 121:3623–34. doi: 10.1172/JCI46323
116. Goh G, Walradt T, Markarov V, Blom A, Riaz N, Doumani R, et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget. (2016) 7:3403–15. doi: 10.18632/oncotarget.6494
117. Wong SQ, Waldeck K, Vergara IA, Schröder J, Madore J, Wilmott JS, et al. UV-associated mutations underlie the etiology of MCV-negative Merkel cell carcinomas. Cancer Res. (2015) 75:5228–34. doi: 10.1158/0008-5472.CAN-15-1877
118. Thibault K, Anne T, Elodie M-S, Roland H, David S, Thilo G, et al. Morphologic and immunophenotypical features distinguishing Merkel cell polyomavirus-positive and -negative Merkel cell carcinoma. Modern Pathol. (accepted).
119. Sunshine JC, Jahchan NS, Sage J, Choi J. Are there multiple cells of origin of Merkel cell carcinoma? Oncogene. (2018) 37:1409–16. doi: 10.1038/s41388-017-0073-3
120. Spurgeon ME, Cheng J, Bronson RT, Lambert PF, DeCaprio JA. Tumorigenic activity of merkel cell polyomavirus T antigens expressed in the stratified epithelium of mice. Cancer Res. (2015) 75:1068–79. doi: 10.1158/0008-5472.CAN-14-2425
121. Verhaegen ME, Mangelberger D, Harms PW, Vozheiko TD, Weick JW, Wilbert DM, et al. Merkel cell polyomavirus small T antigen is oncogenic in transgenic mice. J Invest Dermatol. (2015) 135:1415–24. doi: 10.1038/jid.2014.446
122. Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. (2015) 348:880–6. doi: 10.1126/science.aaa6806
123. Schowalter RM, Reinhold WC, Buck CB. Entry tropism of BK and Merkel cell polyomaviruses in cell culture. PLoS ONE. (2012) 7:e42181. doi: 10.1371/journal.pone.0042181
124. Pipas JM. SV40: Cell transformation and tumorigenesis. Virology. (2009) 384:294–303. doi: 10.1016/j.virol.2008.11.024
125. Angermeyer S, Hesbacher S, Becker JC, Schrama D, Houben R. Merkel cell polyomavirus-positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J Invest Dermatol. (2013) 133:2059–64. doi: 10.1038/jid.2013.82
126. Knapp AC, Franke WW. Spontaneous losses of control of cytokeratin gene expression in transformed, non-epithelial human cells occurring at different levels of regulation. Cell. (1989) 59:67–79. doi: 10.1016/0092-8674(89)90870-2
127. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
128. Naska S, Yuzwa SA, Johnston APW, Paul S, Smith KM, Paris M, et al. Identification of drugs that regulate dermal stem cells and enhance skin repair. Stem Cell Rep. (2016) 6:74–84. doi: 10.1016/j.stemcr.2015.12.002
129. Kwok CK-M, Tam PK-H, Ngan ES-W. Potential use of skin-derived precursors (SKPs) in establishing a cell-based treatment model for Hirschsprung's disease. J Pediatr Surg. (2013) 48:619–28. doi: 10.1016/j.jpedsurg.2012.08.026
130. Kwun HJ, Shuda M, Feng H, Camacho CJ, Moore PS, Chang Y. Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe. (2013) 14:125–35. doi: 10.1016/j.chom.2013.06.008
131. Tadmor T, Aviv A, Polliack A. Merkel cell carcinoma, chronic lymphocytic leukemia and other lymphoproliferative disorders: an old bond with possible new viral ties. Ann Oncol. (2011) 22:250–6. doi: 10.1093/annonc/mdq308
132. Brewer JD, Shanafelt TD, Otley CC, Roenigk RK, Cerhan JR, Kay NE, et al. Chronic lymphocytic leukemia is associated with decreased survival of patients with malignant melanoma and Merkel cell carcinoma in a SEER population-based study. J Clin Oncol. (2012) 30:843–9. doi: 10.1200/JCO.2011.34.9605
133. Howard RA, Dores GM, Curtis RE, Anderson WF, Travis LB. Merkel cell carcinoma and multiple primary cancers. Cancer Epidemiol Biomark Prev. (2006) 15:1545–9. doi: 10.1158/1055-9965.EPI-05-0895
134. Koljonen V, Rantanen M, Sahi H, Mellemkjær L, Hansen BT, Chen T, et al. Joint occurrence of Merkel cell carcinoma and non-Hodgkin lymphomas in four Nordic countries. Leuk Lymphoma. (2015) 56:3315–9. doi: 10.3109/10428194.2015.1040010
135. Chen Z, Gu J. Immunoglobulin G expression in carcinomas and cancer cell lines. FASEB J. (2007) 21:2931–8. doi: 10.1096/fj.07-8073com
136. Chen Z, Li J, Xiao Y, Zhang J, Zhao Y, Liu Y, et al. Immunoglobulin G locus events in soft tissue sarcoma cell lines. PLoS ONE. (2011) 6:e21276. doi: 10.1371/journal.pone.0021276
137. Yang S-B, Chen X, Wu B-Y, Wang M-W, Cai C-H, Cho D-B, et al. Immunoglobulin kappa and immunoglobulin lambda are required for expression of the anti-apoptotic molecule Bcl-xL in human colorectal cancer tissue. Scand J Gastroenterol. (2009) 44:1443–51. doi: 10.3109/00365520903369953
138. Liu H, Zheng H, Li M, Hu D, Tang M, Cao Y. Upregulated expression of kappa light chain by Epstein-Barr virus encoded latent membrane protein 1 in nasopharyngeal carcinoma cells via NF-kappaB and AP-1 pathways. Cell Signal. (2007) 19:419–27. doi: 10.1016/j.cellsig.2006.07.012
139. Li M, Feng D-Y, Ren W, Zheng L, Zheng H, Tang M, et al. Expression of immunoglobulin kappa light chain constant region in abnormal human cervical epithelial cells. Int J Biochem Cell Biol. (2004) 36:2250–7. doi: 10.1016/j.biocel.2004.03.017
140. Buscone S, Garavello W, Pagni F, Gaini RM, Cattoretti G. Nasopharyngeal tonsils (adenoids) contain extrathymic corticothymocytes. PLoS ONE. (2014) 9:e98222. doi: 10.1371/journal.pone.0098222
141. Dong HY, Liu W, Cohen P, Mahle CE, Zhang W. B-cell specific activation protein encoded by the PAX-5 gene is commonly expressed in merkel cell carcinoma and small cell carcinomas. Am J Surg Pathol. (2005) 29:687–92. doi: 10.1097/01.pas.0000155162.33044.4f
142. Mhawech-Fauceglia P, Saxena R, Zhang S, Terracciano L, Sauter G, Chadhuri A, et al. Pax-5 immunoexpression in various types of benign and malignant tumours: a high-throughput tissue microarray analysis. J Clin Pathol. (2007) 60:709–14. doi: 10.1136/jcp.2006.039917
143. Haugg AM, Speel E-JM, Pantulu ND, Pallasch C, Kurz AK, Kvasnicka HM, et al. Fluorescence in situ hybridization confirms the presence of Merkel cell polyomavirus in chronic lymphocytic leukemia cells. Blood. (2011) 117:5776–7. doi: 10.1182/blood-2011-03-339895
144. Pantulu ND, Pallasch CP, Kurz AK, Kassem A, Frenzel L, Sodenkamp S, et al. Detection of a novel truncating Merkel cell polyomavirus large T antigen deletion in chronic lymphocytic leukemia cells. Blood. (2010) 116:5280–4. doi: 10.1182/blood-2010-02-269829
Keywords: merkel cell polyomavirus (MCPyV), epithelial, fibroblast, B cell, Merkel cell carcinoma (MCC), histogenesis, origin
Citation: Kervarrec T, Samimi M, Guyétant S, Sarma B, Chéret J, Blanchard E, Berthon P, Schrama D, Houben R and Touzé A (2019) Histogenesis of Merkel Cell Carcinoma: A Comprehensive Review. Front. Oncol. 9:451. doi: 10.3389/fonc.2019.00451
Received: 07 December 2018; Accepted: 13 May 2019;
Published: 10 June 2019.
Edited by:
Axel zur Hausen, Maastricht University Medical Centre, NetherlandsReviewed by:
Giuseppe Palma, National Cancer Institute G. Pascale Foundation (IRCCS), ItalyAnca Maria Cimpean, Victor Babes University of Medicine and Pharmacy, Romania
Copyright © 2019 Kervarrec, Samimi, Guyétant, Sarma, Chéret, Blanchard, Berthon, Schrama, Houben and Touzé. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antoine Touzé, YW50b2luZS50b3V6ZUB1bml2LXRvdXJzLmZy