 Nawale Hajjaji
Nawale Hajjaji Yves Marie Robin2,3
Yves Marie Robin2,3- 1Breast Cancer Department, Oscar Lambret Cancer Center, Lille, France
- 2Prism Laboratory, Inserm, University of Lille, Lille, France
- 3Pathology Department, Oscar Lambret Cancer Center, Lille, France
- 4School of Medicine, University of Lille, Lille, France
Background: Multigene signatures refine the risk of recurrence and guide adjuvant chemotherapy decision in luminal breast cancers. The decision to perform the assay is highly variable among oncologists. In order to guide the appropriate clinical group in whom to perform a genomic signature, our study analyzed in a homogeneous cohort which clinical risk groups triggered the use of the PAM50-based signature and their concordance with the genomic risk.
Methods: A real life cohort of 222 early breast cancer patients with hormone receptor positive and HER2 negative disease had a commercial PAM50-based assay (Prosigna®) performed at our institution. The assay provided the risk group, the 10-year risk of distant recurrence and the intrinsic molecular subtype of breast cancer.
Results: Based on nodal involvement, Ki67, tumor grade, mitotic index, and tumor size, no clinical pattern could identify a specific genomic risk group. The discordance with the genomic risk was high in patients with clinical low risk tumors, both in node negative and node positive patients. Up to 60% of them had a 10% or more risk of distant recurrence. Moreover, we identified a subgroup of luminal A tumors with a high genomic risk of recurrence. Genomic risk and intrinsic subtype were strong determinants of chemotherapy decision.
Conclusions: Clinical profiles could not reliably identify genomic risk groups and guide the decision to use a multigene signature. Significant discordance with the genomic risk was observed within low clinical risk and luminal A tumors.
Introduction
Luminal breast cancer has a wide range of outcomes not fully predicted by clinical or pathological features. Multigene signatures are used to refine the risk of distant recurrence, and guide adjuvant chemotherapy decision (1–3). These signatures are intended as a prognostic tool for postmenopausal women with early-stage breast cancer, after surgery, when tumors are hormone receptor-positive and HER2 negative.
There is no specific guidance regarding when to use a multigene assay. It is usually recommended in equivocal clinical situations in both node negative or node positive patients. However, this covers a wide and heterogeneous spectrum of clinical cases. The decision to use or not a multigene assay depends on the level of risk perceived by the oncologist. This estimation is based on clinical and pathological factors, mainly age, tumor size, nodal involvement, grade, Ki67, mitotic index, and hormone receptors (3, 4). However, the integration and interpretation of these factors in the decision process is highly variable among oncologists, which could impact patients' access to these assays. There is a need to rationalize the indications to use or not a multigene signature, especially given their financial burden.
The study aims were to analyze in a homogeneous real life cohort (i) which clinical risk groups triggered the use of the PAM50-based Prosigna® assay, and (ii) their concordance with the genomic risk. This genomic signature is among the commercialized multiparameter prognostic tests. It provides an estimate of the 10-year risk of relapse and the breast cancer intrinsic molecular subtype (5–8).
Patients and Methods
Patient Population
A population of early breast cancer patients with hormone receptor positive and HER2 negative tumors, all treated at our institution, whose risk of relapse was estimated using the commercial PAM50-based assay Prosigna® was identified through our clinical database. Two cohorts of patients tested with Prosigna® were available and pooled totalizing 222 cases: a cohort of 159 breast cancer cases (cohort 1) tested prospectively since the assay was commercialized in France, from June 2016 to August 2018, and a retrospective cohort of 63 cases with a clinical intermediate risk of relapse, tested for research purposes (9). Their characteristics are detailed in Supplementary Table 1. Additionally, the records of all the patients with HR+ HER2 negative tumors and less than 4 positive nodes not tested with Prosigna® and available in our database (initiated in 2016) during the same period as cohort 1 were analyzed. Treatment decision was collected and analyzed in cohort 1 (tested prospectively with Prosigna®). The study was approved by our institutional review committee and all procedures performed in this study were in accordance with the ethical standards of our institutional research committee. Informed consent was obtained from all individual participants.
PAM50-Based Prosigna® Assay
Formalin-fixed, paraffin embedded blocks from the original tumors were collected to delineate an area with >70% of cancer cells for RNA extraction. Analyses were performed using the nCounter automated platform (nanoString® Technologies, Seattle, WA) following the manufacturers' instructions. The ROR (risk of recurrence) score was calculated for each sample using the expression profile plus tumor size to classify the tumor within a risk category: genomic low, intermediate or high risk. The test also provided an estimate of the 10-year risk of distant recurrence (in %). Each tumor sample was assigned to an intrinsic molecular subtype of breast cancer (luminal A, luminal B, HER2 enriched, or basal like) based on PAM50 subtype prediction.
Clinical and Histological Data
Clinical and histopathological parameters were collected from patients' records. Immunohistochemical (IHC) staining was performed for estrogen receptor (ER) and progesterone receptor (PR) expression, HER2, and Ki67. The Allred score was used to evaluate ER and PR expression (10). Hormone receptor expression was defined as positive if ≥ 10% of the cells were stained positive. HER2 scoring followed ASCO/ACP guidelines (11). Scores of 0 and 1 were considered negative for HER2 expression and a score of 3 positive. FISH assay was performed for HER2 scores of 2. Histological grading was performed according to the Nottingham protocol (12). The guidelines of van Diest et al. were applied for the correct identification of mitotic figures (13). Ki67 assessment by IHC followed international recommendations (14). The Ki67 score was expressed as the percentage of positively stained cells among the total number of invasive cells in the area scored. Ki67 cut offs in our practice were 14% and 20% based on the 2013 St. Gallen recommendations (15, 16).
Statistical Analysis
Statistical analyses were performed with the Graphpad Prism 8 software (GraphPad Software, USA). A Mann Whitney or Kruskal Wallis test was used to analyze the association between the ROR score or the risk of recurrence and tumor grade, mitotic index, Ki67, tumor size and nodal status. A Spearman test was used to explore the correlation between continuous variables and the PAM50 ROR score or risk of recurrence. A Chi2 test was used to compare categorical variables. P < 0.05 was considered significant.
Results
Cohort Characteristics
The genomic signature was performed in 222 cases. Patients and tumor characteristics were described in Table 1. Median patient age was 59 years; tumor size was T1 (≤2 cm) in 46% of the cases. Patients were node negative in 51% of the cases, and 84% were grade 2. Ki67 was <14% in 31% of the cases, and >20% in 34% of the cases. Mitotic index was classified as 1 in 61% of the cases. Among the 222 cases, the genomic signature classified 42 cases (19%) in the low risk group, 99 cases (45%) as intermediate risk and 81 cases (36%) as high risk. In genomic low risk tumors, 85% were luminal A and 15% were basal. In genomic intermediate risk tumors, 76% were luminal A and 20% luminal B. In genomic high-risk tumors, 31% were luminal A and 69% were luminal B.
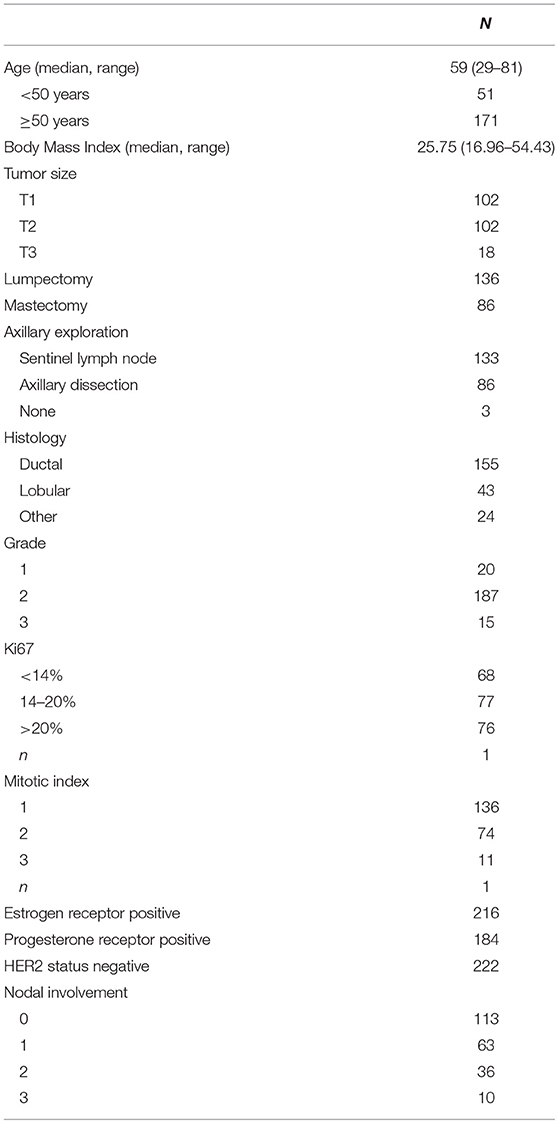
Table 1. Patients and tumor characteristics.
Clinical Profiles Triggering the Use of the Genomic Signature
The main prognostic factors used at our institution to discuss whether a genomic test should be performed were: nodal involvement (0 to 3 positive nodes), three proliferative markers (Ki67, tumor grade and mitotic index), and tumor size. These factors were integrated into clinical profiles and a clinical risk estimate was attributed to each profile as per our practice. The number of Prosigna assay performed according to each clinical risk profile was detailed in node negative and in 1 to 3 node positive cases (Figure 1A). Among the tumors tested, 58% had a clinical intermediate risk, but 24% were clinical low and 18% clinical high-risk tumors.
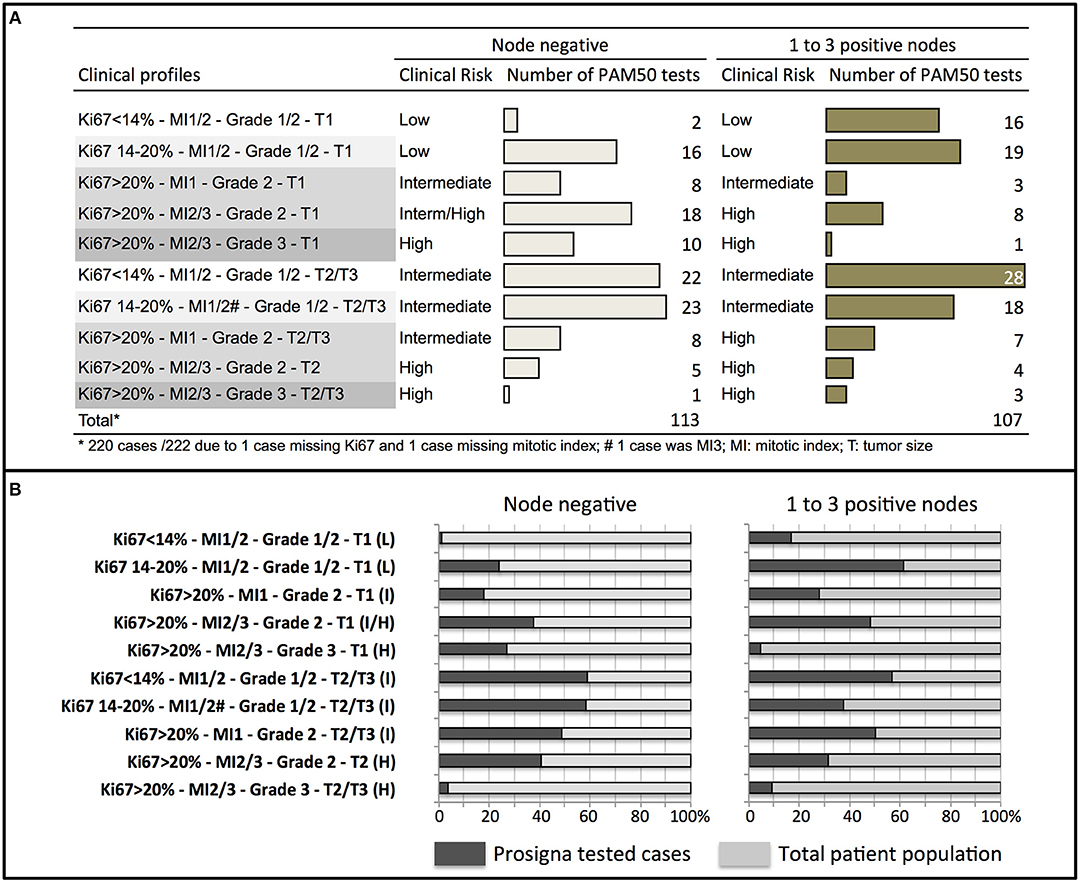
Figure 1. Distribution of clinical profiles and clinical risk groups in patients tested with Prosigna® in node negative or node positive (1–3 nodes) patients (A), and proportion (in%) of cases tested with Prosigna® in the total patient population treated during the same period of time than cohort 1 (B). H, high clinical risk; I, intermediate clinical risk; L, low clinical risk.
Four hundred eighty-three cases of hormone receptor positive HER2 negative early breast cancer with less than 4 positive nodes were treated during the same period of time than cohort 1 at our institution (including cases tested and not tested with Prosigna). The proportion of clinical profiles tested with Prosigna was analyzed according to the total patient population (Figure 1B). In node negative patients, the clinical profiles that triggered the genomic signature were mostly large tumors (T2/T3) with low or discordant proliferative markers. In node positive patients, the same clinical profiles were observed and additionally small tumors (T1) with intermediate or discordant proliferative markers.
Relationship Between Clinical Factors and Genomic Risk
In our cohort, a significant association was observed between the five prognostic factors used to define clinical profiles and the ROR score (with the exception of tumor size) or the genomic risk of recurrence (Figure 2). No correlation was observed between the ROR score or the genomic risk and the expression of estrogen or progesterone receptors (Supplementary Figure 1).
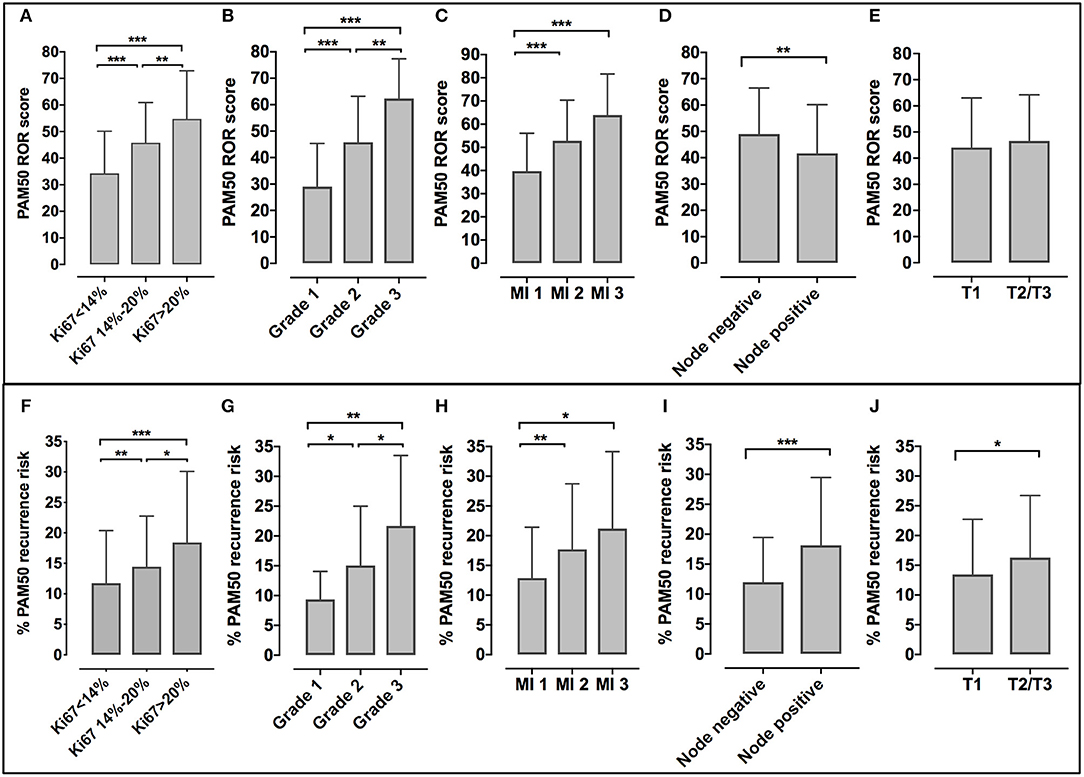
Figure 2. Relationship between the ROR score or the PAM50 risk of recurrence and Ki67 (A,F), tumor grade (B,G), mitotic index (MI) (C,H), nodal status (D,I), and tumor size (T1 or T2/T3) (E,J). Bars represent mean and standard deviation. *p < 0.05, **p < 0.01, ***p < 0.0001.
Distribution of Intrinsic Breast Cancer Subtypes and Genomic Risk Groups Among the Clinical Profiles
The luminal A subtype was observed across all the clinical risk groups in node negative patients. In node positive tumors, it was more frequent in tumors with Ki67 ≤ 20% (Figure 3). The luminal B subtype was less frequent in small low proliferative tumors (Ki67 < 14%).
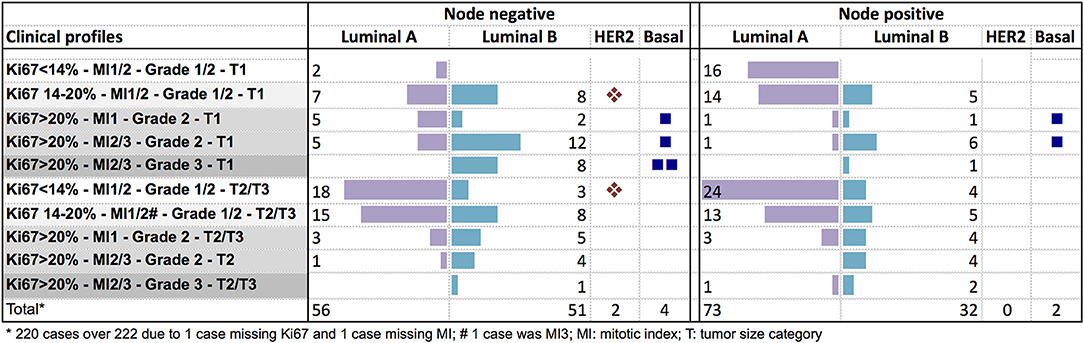
Figure 3. Distribution of PAM50 breast cancer subtypes according to clinical profiles in node negative or node positive (1–3 nodes) patients.
The genomic risk group of each clinical profile was described in Figure 4. In node negative tumors, no specific clinical pattern was observed for the genomic low or intermediate risk groups. The genomic high-risk group was more frequent in T2/T3 tumors, or in small tumors with Ki67 > 20%. Only 9% of node positive T1 tumors were classified as low genomic risk. The high genomic risk group was observed across all the clinical node positive profiles.
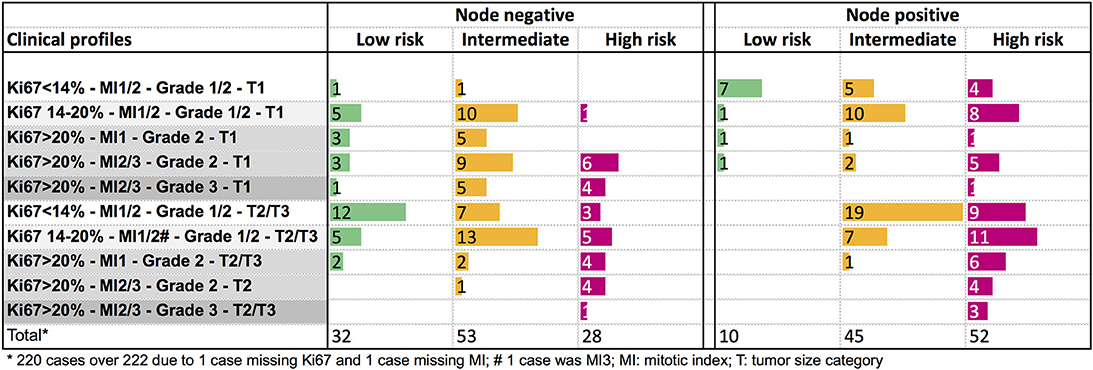
Figure 4. Distribution of the PAM50 risk categories according to clinical profiles in node negative or node positive (1–3 nodes) patients.
Concordance Between Clinical Risk, Risk of Recurrence and Intrinsic Subtypes
In clinical high-risk patients, the genomic risk was concordant in node positive patients (1 to 3 nodes, Ki67 > 20%, grade 2, T2/T3). However, among node negative patients with these tumor characteristics, 15% had a risk of recurrence lower than 10% (Figure 5A).
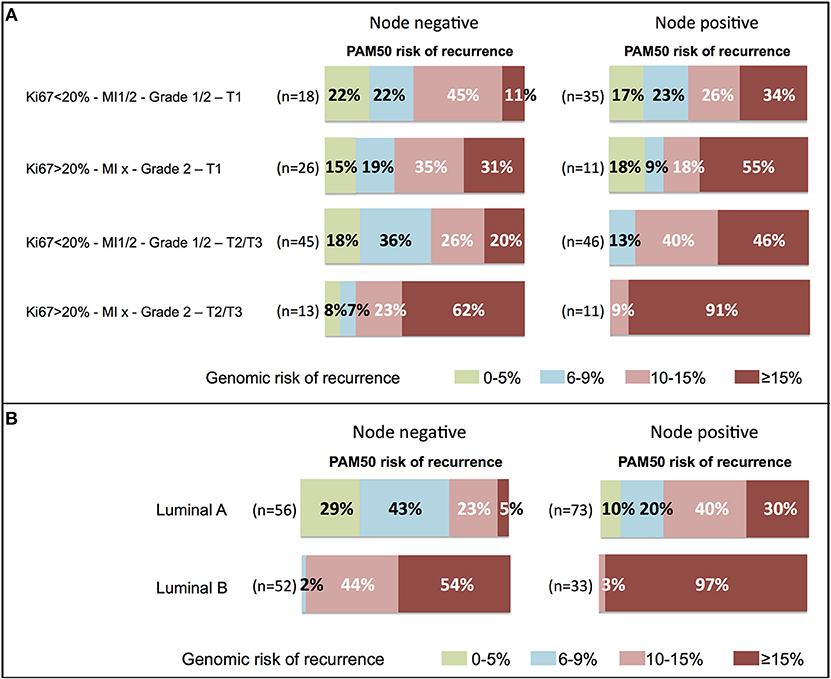
Figure 5. Distribution of the 10-year risk of distant recurrence estimated by PAM50 according to (A) clinical profiles or (B) luminal A or B subtypes, in node negative or node positive (1–3 nodes) patients. The range of risk was color-coded and the proportion of patients (%) indicated within the corresponding area. MI, mitotic index; T, tumor size.
Significant discordance with the genomic risk was observed among clinical low risk patients or with tumors classified as luminal A (Figure 5). 56% of the patients with node negative clinical low risk tumors (Ki67 ≤ 20%, MI 1/2, grade 1/2, T1) had a genomic risk of recurrence of 10% or more. Among patients with node positive clinical low risk tumors, 76% had a luminal A intrinsic subtype, but 60% had a genomic risk of recurrence of 10% or more (Figure 5A).
The relationship between the luminal A intrinsic subtype and the genomic risk of recurrence was analyzed in Figure 5B. Among node negative luminal A tumors, 28% had a 10% or more risk of recurrence. This proportion increased to 70% among node positive luminal A tumors.
Impact of Clinical and Genomic Risks on Adjuvant Chemotherapy Decision
The decision whether to propose adjuvant chemotherapy or not was analyzed according to clinical profiles, genomic risk and intrinsic breast cancer subtype in node negative and node positive patients (Table 2).
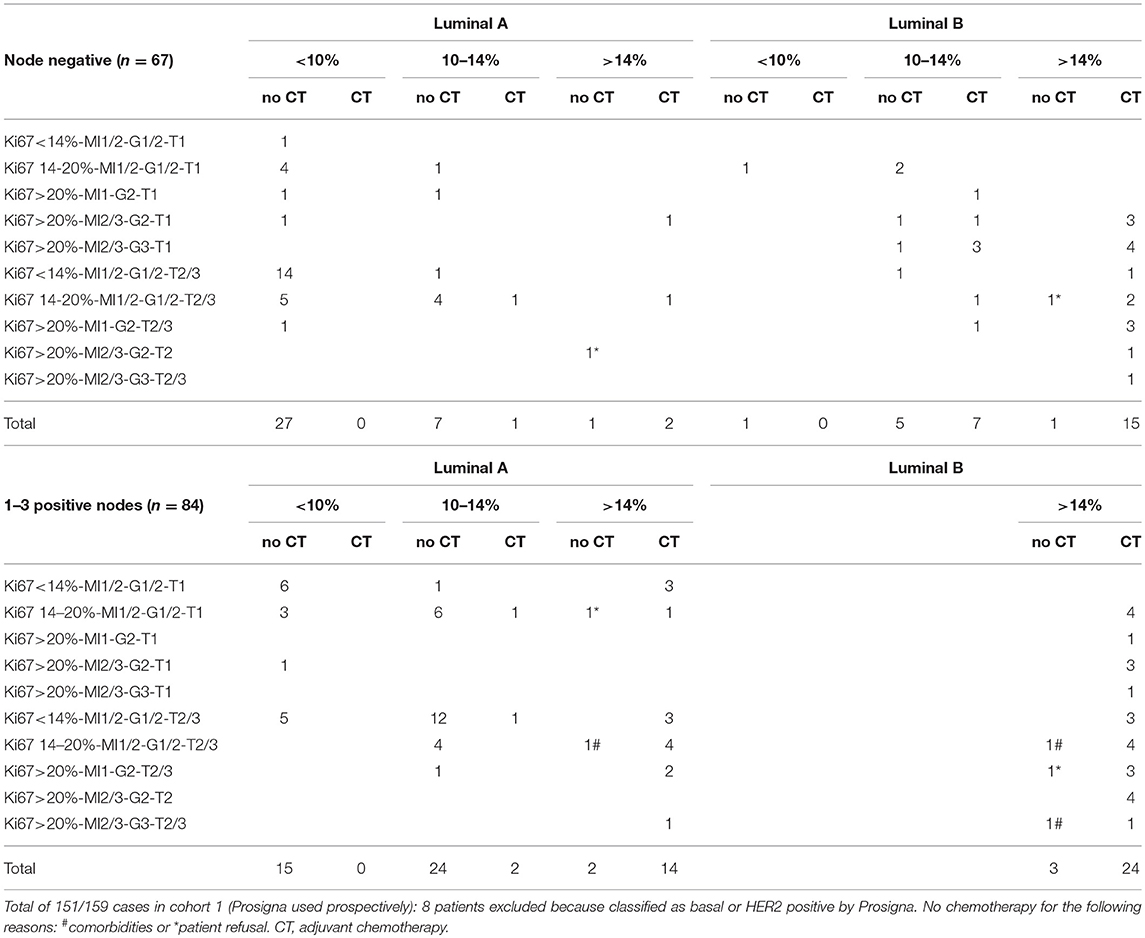
Table 2. Decision regarding adjuvant chemotherapy according to clinical profiles, genomic risk, and intrinsic breast cancer subtype in node negative and node positive patients.
A genomic risk of recurrence < 10% was a main determinant to withhold chemotherapy, whatever the clinical profile. On the other hand, a genomic risk >14% triggered the chemotherapy decision, including in clinical low risk tumors. If the genomic risk was between 10 and 14%, the luminal A subtype was a strong factor to withhold chemotherapy. In luminal B tumors with a genomic risk between 10 and 14%, the decision to propose chemotherapy varied among oncologists.
Discussion
A clinical assessment of the risk of recurrence and the decision with respect to the use of a multigene signature are subjective and highly variable among oncologists. Our study is the first to analyze how a multigene signature is used in a real life cohort, and its concordance with clinical risk estimates. In this cohort, the genomic signature was used mainly in clinical intermediate risk tumors. But the test was also performed in clinical high or low risk patients in case of discordant clinical and/or proliferative factors. It provided additional information about the molecular subtype or confirmed the poor prognosis in debilitated patients. Another reason for using the assay in these cases was the growing pressure from patients to have access to a multigene signature with the hope to better assess their risk of recurrence and therapeutic options.
Our study could not identify specific clinical patterns associated with genomic risk groups. However, in clinical high-risk patients, the genomic risk estimate was mostly concordant, especially in node positive patients, and did not add useful information. On the contrary, the discordance with the genomic risk was surprisingly high in node negative or node positive clinical low risk tumors (Ki67 ≤ 20%, MI 1/2, grade 1/2, T1). A significant proportion of clinical low risk patients had a ≥10% genomic risk of recurrence. These results were troublesome, particularly in node negative patients because the signature should have been omitted in these patients based on the excellent prognosis reported in the TAILORx and MINDACT trials (17, 18). In the TAILORx trial, node negative patients selected with the 21-gene signature (11–25 RS score) were mostly clinical low risk. Their 9-year distant risk of relapse was 5% and no additional benefit of chemotherapy was observed in patients over 50 years (17). The TAILORx study excluded genomic high-risk patients. In the MINDACT trial, 592 node negative patients had a low clinical risk/high genomic risk as assessed by Adjuvant Online and the 70-gene signature, respectively. In the patient population, 65% of the patients had a T1 tumor, 70% were grade 2 and 16% were grade 1; Ki67 and mitotic index were not displayed. Their distant metastasis free survival at 5 years was 4–6% (18). This prognosis and the lack of numerical difference in outcomes between the groups treated with or without chemotherapy suggest that a multigene assay in this population could be safely omitted (18, 19).
Nevertheless, in an era of personalized medicine where patients are asking for individualized care, how confidently can we extrapolate the 5-year results from MINDACT to node negative clinical low but genomic high risk patients tested with another signature? How should a clinical low risk patient with a genomic risk of 10% or more at 10 years be managed? In fact, the multigene signatures use different technical approaches and measure different genes. The PAM50-based assay used in the present study includes a higher proportion of genes involved in proliferation compared to other signatures (20), and classifies more patients in the high-risk group than the 21-gene signature (21). The comparison of several multigene signatures showed significant discrepancies both in disease subtyping and risk prediction (21, 22), highlighting their low level of concordance, which is quite problematic at an individual patient level. For this reason and until a long term follow up of MINDACT is available, extrapolations between genomic signatures should be cautious.
How confident can we be about the lack of benefit of chemotherapy in clinical low risk/ genomic high-risk node negative patients? Recently, a PAM50-based chemo-endocrine score was developed and validated to predict tumor sensitivity to treatment in the neoadjuvant setting (23). Tumors classified high-risk by the PAM50-based score or tumors with a luminal B intrinsic subtype were associated with sensitivity to chemotherapy. Based on these data, some benefit from chemotherapy cannot be excluded in clinical low risk but PAM50 high-risk patients. This patient subgroup represented almost 10% of our node negative population. This proportion may have been underestimated given that the use of the genomic signature was not systematic in clinical low risk node negative patients.
In node positive, the RxPONDER trial is ongoing (NCT01272037) to explore the benefit of adjuvant chemotherapy in 1 to 3 node positive patients selected according to their genomic risk of relapse with the 21-gene signature. Until more data is available, clinical low-risk patients may benefit from a multigene assay to adequately discuss their risk and therapeutic options.
Contrary to the study by Wallden et al. (8), our real life cohort showed that a significant proportion of tumors with an intrinsic luminal A subtype had a risk of recurrence of 10% or more. Luminal A tumors are considered to have a favorable prognosis as suggested by retrospective analyses of adjuvant trials. These studies also suggested a lack of additional benefit of chemotherapy compared to endocrine therapy (24–29). In these reports, luminal A tumors were identified based on criterion contributing to the surrogate luminal A definition (30). The ESMO 2015 guidelines recommended endocrine therapy alone for luminal A tumors, except in cases of high tumor burden (16). The St. Gallen guidelines recommended omission of adjuvant chemotherapy in stage 1 or 2 luminal A-like cancers (31). However, molecular studies characterizing the genomic and transcriptomic landscape of luminal A tumors (defined using PAM50 or genomic databases) revealed a molecular heterogeneity within this subtype, which also translated to variability in survival (32, 33). A significantly worse outcome was observed in luminal A tumors with specific molecular characteristics such as increased genomic instability, p53 alteration, amplification or overexpression of genes involved in mitosis regulation (32, 33). Whether chemotherapy could be beneficial in these high-risk luminal A tumors has not been prospectively analyzed and warrants further exploration.
Beside patients' pressure, handling of multigene signatures has proven difficult for several reasons. There is no specific recommendation to guide the decision whether these costly assays should be used. Our results showed that clinical profiles used in routine care could not reliably provide guidance. These signatures are prognostic but not predictive tools. Finally, as observed in our study, genomic risk assessment may overshadow clinical factors and become the strongest determinant for treatment decision while no randomized trial has demonstrated the superiority of a genomic driven strategy over clinical factors on patients' outcome.
Our study has some limitations. Assessment of clinical risk relied partly on proliferative markers, which have a poor reproducibility (13, 34–37). However, routine scoring of Ki67, tumor grade and mitotic index followed validated guidelines and reflected real life conditions. Moreover, these proliferative markers were highly correlated to the ROR score and the PAM50 risk of recurrence in our cohort.
In conclusion, our results showed that clinical profiles could not reliably identify genomic risk groups. The underuse of a multigene signature in clinical low risk groups may lead to inadequate risk assessment and patients' under treatment. Our study supports the use of a multigene assay in node negative or positive T1 tumors. It also highlighted a subgroup of luminal A tumors at high risk of relapse, in both node negative and node positive patients. Further work is needed to define the optimal treatment strategy in these subgroups.
Ethics Statement
This retrospective study was carried out in accordance with the recommendations of Center Oscar Lambret Clinical Research Committee with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. This retrospective study was approved by Center Oscar Lambret Clinical Research Committee.
Author Contributions
NH generated the idea and study design. NH and YR performed data collection. Data analysis and writing of the manuscript was performed by NH and JB. All authors approved the final version of the manuscript.
Conflict of Interest Statement
NH has received a speaker honorarium from NanoString Technologies and has worked as a consultant to Pfizer, Roche and Novartis. YR has received a speaker honorarium from NanoString Technologies. JB has received a speaker honorarium from NanoString Technologies and has worked as a consultant to Pfizer, Lilly, and Novartis.
Acknowledgments
We thank Delphine Bertin for her valuable help in retrieving tumor blocs.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.00454/full#supplementary-material
References
1. Prat A, Ellis MJ, Perou CM. Practical implications of gene-expression-based assays for breast oncologists. Nat Rev Clin Oncol. (2011) 9:48–57. doi: 10.1038/nrclinonc.2011.178
2. Harris LN, Ismaila N, McShane LM, Andre F, Collyar DE, Gonzalez-Angulo AM, et al. Use of biomarkers to guide decisions on adjuvant systemic therapy for women with early-stage invasive Breast Cancer. Am Soc Clin Oncol Clin Prac Guideline. (2016) 34:1134–50. doi: 10.1200/JCO.2015.65.2289
3. Giuliano AE, Connolly JL, Edge SB, Mittendorf EA, Rugo HS, Solin LJ, et al. Breast Cancer-Major changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J Clin. (2017) 67:290–303. doi: 10.3322/caac.21393
4. Isaacs C, Stearns V, Hayes DF. New prognostic factors for breast cancer recurrence. Semin Oncol. (2001) 28:53–67. doi: 10.1053/sonc.2000.20742
5. Gnant M, Sestak I, Filipits M, Dowsett M, Balic M, Lopez-Knowles E, et al. Identifying clinically relevant prognostic subgroups of postmenopausal women with node-positive hormone receptor-positive early-stage breast cancer treated with endocrine therapy: a combined analysis of ABCSG-8 and ATAC using the PAM50 risk of recurrence score and intrinsic subtype. Ann Oncol. (2015) 26:1685–91. doi: 10.1093/annonc/mdv215
6. Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. (2009) 27:1160–7. doi: 10.1200/JCO.2008.18.1370
7. Filipits M, Nielsen TO, Rudas M, Greil R, Stöger H, Jakesz R, et al. The PAM50 risk-of-recurrence score predicts risk for late distant recurrence after endocrine therapy in postmenopausal women with endocrine-responsive early breast cancer. Clin Cancer Res. (2014) 20:1298–305. doi: 10.1158/1078-0432.CCR-13-1845
8. Wallden B, Storhoff J, Nielsen T, Dowidar N, Schaper C, Ferree S, et al. Development and verification of the PAM50–based Prosigna breast cancer gene signature assay. BMC Med Genomics. (2015) 8:54. doi: 10.1186/s12920-015-0129-6
9. Bonneterre J, Prat A, Galván P, Morel P, Giard S. Value of a gene signature assay in patients with early breast cancer and intermediate risk: a single institution retrospective study. Curr Med Res Opin. (2016) 32:835–839. doi: 10.1185/03007995.2016.1146664
10. Allred DC, Harvey JM, Berardo M, Clark GM. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol. (1998) 11:155–68.
11. Wolff AC, Hammond MEH, Allison KH, Harvey BE, Mangu PB, Bartlett JMS, et al. Human epidermal growth factor receptor 2 testing in breast cancer: American society of clinical oncology/college of American pathologists clinical practice guideline focused update. J Clin Oncol. (2018) 36:2105–22. doi: 10.1200/JCO.2018.77.8738
12. Elston CW, Ellis IO. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology. (1991) 19:403–10 doi: 10.1111/j.1365-2559.1991.tb00229.x
13. van Diest PJ, Baak JP, Matze-Cok P, Wisse-Brekelmans EC, van Galen CM, Kurver PH, et al. Reproducibility of mitosis counting in 2,469 breast cancer specimens: results from the Multicenter Morphometric. Mammary Carcinoma Project. Hum Pathol. (1992) 23:603–7. doi: 10.1016/0046-8177(92)90313-R
14. Dowsett M, Nielsen TO, A'Hern R, Bartlett J, Coombes RC, Cuzick J, et al. Assessment of Ki67 in breast cancer: recommendations from the International Ki67 in Breast Cancer working group. J Natl Cancer Inst. (2011) 103:1656–64. doi: 10.1093/jnci/djr393
15. Goldhirsch A, Winer EP, Coates AS, Gelber RD, Piccart-Gebhart M, Thürlimann B, et al. Personalizing the treatment of women with early breast cancer: highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann Oncol. (2013) 24:2206–23. doi: 10.1093/annonc/mdt303
16. Senkus E, Kyriakides S, Ohno S, Penault-Llorca F, Poortmans P, Rutgers E, et al. Primary breast cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2015) 26(Suppl. 5):v8–30. doi: 10.1093/annonc/mdv298
17. Sparano JA, Gray RJ, Makower DF, Pritchard KI, Albain KS, Hayes DF, et al. Adjuvant chemotherapy guided by a 21–gene expression assay in breast cancer. N Engl J Med. (2018) 379:111–121. doi: 10.1056/NEJMoa1804710
18. Cardoso F, van't Veer LJ, Bogaerts J, Slaets L, Viale G, Delaloge S, et al. 70–Gene Signature as an Aid to treatment decisions in early-stage breast cancer. N Engl J Med. (2016) 375:717–29. doi: 10.1056/NEJMoa1602253
19. Krop I, Ismaila N, Andre F, Bast RC, Barlow W, Collyar DE, et al. Use of biomarkers to guide decisions on adjuvant systemic therapy for women with early-stage invasive breast cancer: American society of clinical oncology clinical practice guideline focused update. J Clin Oncol. (2017) 35:2838–47. doi: 10.1200/JCO.2017.74.0472
20. Kwa M, Makris A, Esteva FJ. Clinical utility of gene-expression signatures in early stage breast cancer. Nat Rev Clin Oncol. (2017) 14:595–610. doi: 10.1038/nrclinonc.2017.74
21. Dowsett M, Sestak I, Lopez-Knowles E, Sidhu K, Dunbier AK, Cowens JW, et al. Comparison of PAM50 risk of recurrence score with oncotype DX and IHC4 for predicting risk of distant recurrence after endocrine therapy. J Clin Oncol. (2013) 31:2783–90. doi: 10.1200/JCO.2012.46.1558
22. Bartlett JM, Bayani J, Marshall A, Dunn JA, Campbell A, Cunningham C, et al. Comparing breast cancer multiparameter tests in the OPTIMA prelim trial: no test is more equal than the others. J Natl Cancer Inst. (2016) 108:djw050. doi: 10.1093/jnci/djw050
23. Prat A, Lluch A, Turnbull AK, Dunbier AK, Calvo L, Albanell J, et al. A PAM50–Based Chemoendocrine Score for Hormone Receptor-Positive Breast Cancer with an Intermediate Risk of Relapse. Clin Cancer Res. (2017) 23:3035–44. doi: 10.1158/1078-0432.CCR-16-2092
24. Gao JJ, Swain SM. Luminal a breast cancer and molecular assays: a review. Oncologist. (2018) 23:556–65. doi: 10.1634/theoncologist.2017-0535
25. Cheang MC, Chia SK, Voduc D, Gao D, Leung S, Snider J, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst. (2009) 101:736–50. doi: 10.1093/jnci/djp082
26. Conforti R, Boulet T, Tomasic G, Taranchon E, Arriagada R, Spielmann M, et al. Breast cancer molecular subclassification and estrogen receptor expression to predict efficacy of adjuvant anthracyclines-based chemotherapy: a biomarker study from two randomized trials. Ann Oncol. (2007) 18:1477–1483. doi: 10.1093/annonc/mdm209
27. Hart CD, Sanna G, Siclari O, Biganzoli L, Di Leo A. Defining optimal duration and predicting benefit from chemotherapy in patients with luminal-like subtypes. Breast. (2015) 24:S136–42. doi: 10.1016/j.breast.2015.07.033
28. Coates AS, Colleoni M, Goldhirsch A. Is adjuvant chemotherapy useful for women with luminal a breast cancer? J Clin Oncol. (2012) 30:1260–3. doi: 10.1200/JCO.2011.37.7879
29. Coates AS, Winer EP, Goldhirsch A, Gelber RD, Gnant M, Piccart-Gebhart M., et al. Tailoring therapies – improving the management of early breast cancer: St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer. Ann Oncol. (2015) 26:1533–46. doi: 10.1093/annonc/mdv221
30. Goldhirsch A, Wood WC, Coates AS, Gelber RD, Thürlimann B, Senn HJ. Strategies for subtypes–dealing with the diversity of breast cancer: highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann Oncol. (2011) 22:1736–47. doi: 10.1093/annonc/mdr304
31. Curigliano G, Burstein HJ, Winner EP, Gnant M, Dubsky P, Loibl S, et al. De-escalating and escalating treatments for early-stage breast cancer: the St. Gallen International Expert Consensus Conference on the Primary Therapy of Early Breast Cancer 2017. Ann Oncol. (2017) 28:1700–12. doi: 10.1093/annonc/mdx308
32. Ciriello G, Sinha R, Hoadley KA, Jacobsen AS, Reva B, Perou CM, et al. The molecular diversity of Luminal A breast tumors. Breast Cancer Res Treat. (2013) 141:409–20. doi: 10.1007/s10549-013-2699-3
33. Pérez-Peña J, Alcaraz-Sanabria A, Nieto-Jiménez C, Páez R, Corrales-Sánchez V, Serrano-Oviedo L, et al. Mitotic read-out genes confer poor outcome in luminal A breast cancer tumors. Oncotarget. (2017) 8:21733–40. doi: 10.18632/oncotarget.15562
34. Laenkholm AV, Grabau D, Møller Talman ML, Balslev E, Bak Jylling AM, Tabor TP, et al. An inter-observer Ki67 reproducibility study applying two different assessment methods: on behalf of the Danish Scientific Committee of Pathology, Danish breast cancer cooperative group (DBCG). Acta Oncol. (2018) 57:83–9. doi: 10.1080/0284186X.2017.1404127
35. Penault-Llorca F, Radosevic-Robin N. Ki67 assessment in breast cancer: an update. Pathology. (2017) 49:166–71. doi: 10.1016/j.pathol.2016.11.006
36. Rimm DL, Leung SCY, McShane LM, Bai Y, Bane AL, Bartlett JMS, et al. An international multicenter study to evaluate reproducibility of automated scoring for assessment of Ki67 in breast cancer. Mod Pathol. (2018) 32:59–69. doi: 10.1038/s41379-018-0109-4
37. Skaland I, van Diest PJ, Janssen EA, Gudlaugsson E, Baak JP. Prognostic differences of World Health Organization-assessed mitotic activity index and mitotic impression by quick scanning in invasive ductal breast cancer patients younger than 55 years. Hum Pathol. (2008) 39:584–90. doi: 10.1016/j.humpath.2007.08.016
Keywords: multigene signature, PAM50, luminal breast cancer, risk of recurrence, clinical risk
Citation: Hajjaji N, Robin YM and Bonneterre J (2019) Should a Multigene Signature be Used in all Luminal Early Breast Cancers. Front. Oncol. 9:454. doi: 10.3389/fonc.2019.00454
Received: 13 January 2019; Accepted: 13 May 2019;
Published: 04 June 2019.
Edited by:
José Bines, Brazilian National Cancer Institute (INCA), BrazilReviewed by:
Angel Luis Guerrero-Zotano, Instituto Valenciano de Oncologia, SpainConnie Irene Diakos, Kolling Institute, Australia
Copyright © 2019 Hajjaji, Robin and Bonneterre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nawale Hajjaji, bi1oYWpqYWppQG8tbGFtYnJldC5mcg==