 Jingyuan Li1
Jingyuan Li1 H. Rosie Xing
H. Rosie Xing- 1Laboratory of Translational Cancer Stem Cell Research, Institute of Life Sciences, Chongqing Medical University, Chongqing, China
- 2State Key Laboratory of Ultrasound Engineering in Medicine, Chongqing Medical University and the Ministry of Science and Technology, Chongqing, China
Lung cancer management remains a challenge due to its asymptomatic and late presentation when it is metastatic. The clinical response to the first-line platinum-based chemotherapy in patients with advanced lung cancer is disappointing due to the development of chemoresistance. Chemoresistance is a complex phenomenon. Mechanistic research using experimental models has yielded limited clinical results to help increase understanding for overcoming resistance. While the role of lung CSCs in conferring multidrug resistance has been postulated, experimental evidence remains associative and lacks in depth mechanistic inquisition. In the present study, using mouse and human lung adenocarcinoma cell lines and their respective paired CSC derivative cell lines that we generated, we identified cancer stem cell component of lung adenocarcinoma as the source that confers multidrug resistance phenotype. Mechanistically, Gstp1 confers cisplatin resistance in mouse and human lung CSC models, both in vitro and in vivo. Further, transcriptional activation of Gstp1 expression by MEK/ERK signaling underlies cisplatin resistance in lung CSC cells. Moreover, we show that GSTP1 expression is a poor diagnostic and prognostic marker for human lung adenocarcinoma, thus is of high clinical relevance. Taken together, we have provided mechanistic understanding of the lung CSC in mediating chemoresistance.
Highlights
• The cancer stem cell component of lung adenocarcinoma is the source that confers multidrug resistance phenotype.
• Gstp1 expression is elevated in lung CSC cells which can be further increased upon treatment with a panel of chemotherapy drugs.
• Gstp1 confers cisplatin resistance in mouse and human lung CSC models, both in vitro and in vivo.
• Providing the first experimental evidence that transcriptional activation of Gstp1 expression by MEK/ERK signaling underlies cisplatin resistance in lung CSC cells.
• GSTP1 expression is a poor diagnostic and prognostic marker for human lung adenocarcinoma thus is of high clinical relevance.
Introduction
Lung cancer is the most common cause of cancer-related deaths in the world (1). The high mortality rate (51–99%) of lung adenocarcinoma is due to it being asymptomatic, it having late presentation when it is metastatic and becoming resistant to anti-cancer therapies (2). In spite of the development of new therapeutic strategies, the outcome of patients with metastatic lung cancer has barely improved over the past few decades, and the overall 5-year survival rate remains very low (10–15%) (3, 4). Lung adenocarcinoma is the most common histological type of lung cancer, comprising ~60% of non-small cell lung cancers (NSCLC) (5). Although platinum-based chemotherapy represents the standard first-line treatment for patients with advanced NSCLC, therapeutic outcome is disappointing due to the development of chemo-resistance, relapse, and distant metastases (6, 7). Mechanistic understanding of the involvement of commonly studied multidrug resistant genes using human lung adenocarcinoma cell lines has yielded limited clinical success in overcoming chemo-resistance thus far.
According to the CSCs theory, tumorigenesis, and cancer progression are due to a subset of phenotypically distinct cells characterized by unlimited self-renewal and enhanced clonogenic potential (8–10). Lung CSCs are shown to be associated with higher recurrence rates (11, 12). In agreement with this hypothesis, lung cancers that manifest stem cell signatures are associated with multidrug resistance (including cisplatin) and with disease relapse (12–14). However, in depth characterization and mechanistic investigation of multidrug resistance in lung CSCs were lacking, partially due to the lack of stable cellular models of lung CSC.
Glutathione S-transferases (GSTs) are phase II detoxifying enzymes involved in the maintenance of cell integrity, oxidative stress and protection against DNA damage by catalyzing the conjugation of glutathione to a wide variety of electrophilic substrates (15–17). Gstp1, a member of the GST family, is directly involved in the detoxification of cisplatin via the formation of cisplatin-glutathione adducts, which indicates that Gstp1 may play a role in the acquisition of resistance to this platinum compound (18, 19). Even though a growing number of studies have demonstrated that Gstp1 plays a key role in the development and maintenance of malignancy in several tumor types (20–22), mechanistic understanding of Gstp1 in mediating chemoresistance in lung cancer is sketchy. Its role in mediating chemoresistance in CSCs is unknown.
The MAPK pathway, including MEK/ERK, JNK, and p38 kinase, plays a pivotal role in cell survival, proliferation and migration of tumor cells (23–25). While several studies reported activation of the MEK/ERK cascade in response to cisplatin treatment in several forms of cancer, the consequence of such activation on cell survival remains controversial (26–32). Few studies reported the activation of GST gene expression by MEK/ERK signaling in breast cancer (33–35). Up until the present study, regulation of Gstp1 expression in lung CSCs has not been examined.
In the present study, we employed the lung CSCs derived from mouse parental Lewis lung carcinoma cell line (LLC-Parental) and human cancer cell lines H1299, which were named LLC-SD and H1299-SD, respectively. The stem cell properties of LLC-SD in vitro and in vivo had been characterized (36–38). Using the stable mouse and human lung CSC models that we generated and characterized, we clearly revealed lung CSCs as the cellular component that confer multidrug chemoresistance both in vitro and in vivo. We also demonstrate that the underlying mechanism of cisplatin chemo-resistance is the transcriptional activation of Gstp1 by MEK/ERK signaling. We also show the clinical relevance of these findings and establish GSTP1 as a poor diagnostic and prognostic marker, as well as a therapeutic target for human lung adenocarcinoma. Since the expression changes of Gstp1 in response to chemo-therapeutic agents were specific to the lung CSCs, not the non-stem-like parental cells, Gstp1 may also serve a lung CSC marker for evaluating the CSC content in clinical lung samples.
Materials and Methods
Cell Culture and Cell Lines
LLC-Parental and H1299-Parental cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM high glucose supplemented (Hyclone) containing 10% FBS (Gibco). LLC-SD and H1299-SD cells were stem counterpart cells, which were maintained in DMEM/F12-based normal stem cell media (Hyclone), supplemented with 20 ng/ml EGF (BD), 20ng/ml FGF (BD) and 2% B27 (Gibco). All cell lines were cultured at 37°C, in a 5% CO2 atmosphere with 95% humidity.
Reverse Transcription Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR)
Total RNA was isolated using TRIZOL (Takara) according to the manufacturer's instructions. cDNA was reverse transcribed from 2 μg of total RNA. 39 cycles of PCR amplification were performed using 95°C for 30s, 95°C for 5s, and 60°C for 30s for each cycle. TBP was used as a loading control. The sequences of PCR primers are listed in Table 1.
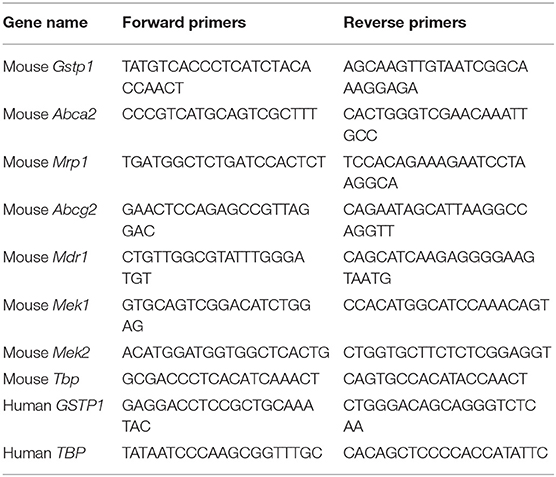
Table 1. PCR primer sequence.
Flow Cytometric Apoptosis Assay
At 24 h following transfection or inhibitor treatment, the indicated cells were treated with cisplatin at a final concentration of 10 μM. After 24 h of cisplatin treatment, the floating and attached cells were collected separately and then combined. 0.5–1 million cells of each single-cell suspension was stained with the Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI), and analyzed with a BD LSR. Unstained cells were included as negative controls for each FACS analysis. The data were analyzed using BD FACS software.
Cck8 Assay and Drug Treatment
Cck8 colorimetric assay was used to detect the sensitivity of cells to anticancer drugs, cell viability and proliferation in response to drug treatments in vitro. They were also used to determine the concentration of the drug that inhibited cell growth by 50% (IC50) after 24 h of treatment. Cells were re-suspended in a final concentration of 2 × 103 cells/well, seeded into 96 well plates and subjected to different concentrations of chemotherapeutic drugs after pre-incubation for 48 h. For drug combination treatment experiments, to investigate the reversal effects of cisplatin, different concentrations of cisplatin were added after pre-incubation with Ezatiostat or PD98059 for 48 h. After 24 h of incubation with cisplatin, Cck8 reagent (10 μl/well) was added and the plates were further incubated for 2 h. Subsequently, absorbance was determined at 450 nm by NanoQuant microplate reader (M200 PRO, Switzerland). The IC50 values (concentration required to inhibit the growth by 50%) were calculated from the survival curves using modified Bliss method. Resistance fold (Rf) was calculated by dividing the IC50 for the resistant cells with or without an inhibitor by that of the parental cells without an inhibitor. The concentrations of Ezatiostat or PD98059 used in this study were in the range of 5~150 μM.
Western Blot
Western blot was performed according to a standard method. The total protein was extracted with RIPA buffer (Beyotime), according to the manufacturer's instructions. The concentration was determined by the BCA method. Twenty-five micrograms of protein was used to run on a 10% polyacrylamide (Beyotime) gel and transferred to a PVDF membrane underneath. The membrane was blocked in 5% non-fat milk (Bio-Rad). Primary antibodies and secondary antibodies (Proteintech) were used according to manufacturer's protocol. ECL method was used and the blot was developed under the gel electrophoresis imager (Bio-Rad). The following primary antibodies were used: anti–Gstp1 (Abcam), anti–Mek1 (upstate), anti–p-Mek1/2 (Cell Signaling Technology), and anti–GAPDH (Proteintech).
siRNA Transient Interference Assay
To knockdown Gstp1 or Mek1/2 expression, LLC-Parental and LLC-SD cells were transfected with 5 nM control siRNA or siRNA against Gstp1 (CCUCAUCUACACCAACUAUTT) or Mek1/2 (AGUCGGACAUCUGGAGCAUTT) (GenePharma, China) for 72h using Lipofectamine 2000 transfect reagent (Invitrogen, USA).
Lentivirus Packaging and Infection
Stable knockdown or overexpression of Gstp1 in LLC-SD cells was achieved with infection of LLC-SD cells with either pLL3.7-shN.C.(mouse) or pLL3.7-shGstp1(mouse) and LV4-Vector(mouse) or LV4-OEGstp1(mouse), respectively, in the presence of 8 μg/ml polybrene (Genepharma, China) for 12 h, and the medium was refreshed. Seventy-two hours after the infection, the efficiency of infection was measured under a fluorescent microscope and by RT-qPCR.
Clinical Samples
We collected 43 tumor samples from The Affiliated Hospital of Southwest Medical University after informed consent approved by Southwest Medical University of Institutional Review Boards. Specimens were prepared from paraffin embedded (FFPE) tissue. Genomic Total RNA from FFPE tissues was extracted using the Tiangen RNAprep Pure FFPE Kit (Tiangen, China). RNA yield was evaluated using a Nanodrop 2000 spectrometer (NanoDrop Technologies Inc., DE) and Qubit 3.0 Fluorometer (Thermo Fisher Scientific, MA). All of the formalin fixed paraffin embedded was obtained from the department of pathology in the affiliated hospital of southwest medical university, which was used to identify the cancer progress as the stage of lung cancer and received the approval of the institutional ethics committee of southwest medical university medical center. Moreover, this study obtained written informed consent from the participants.
Cisplatin Resistance Assay
Single cell suspension (1 × 103) mixed with 50 μL Matrigel Matrix (Corning) at a 1:1 ratio was injected subcutaneously into both insides of the hind legs of each 6–8 weeks old female BALB/c nude mice (Beijing Huafukang bioscience company, Beijing, China). Tumor growth was measured every 2 days, and tumor volume was calculated as V = (length × width2)/2. Treatment with cisplatin started when the tumor volume of the subcutaneous xenograft tumors reached about 100 mm3. 16 mg/kg cisplatin was administrated every other day intraperitoneally (qilu-pharmaceutical). After 9 days of treatment, when mouse body weights were decreased by 20%, mice were killed in a humane manner. Experimental mice were raised in an accredited specific pathogen-free Animal Facility at Chongqing medical University. All protocols were approved by the Institutional Animal Care and Use Committee of Chongqing medical University and conducted with humane animal care.
Bioinformatics Analyses
The lung cancer dataset, which included 535 lung adenocarcinoma tissues and 59 normal lung tissues, was obtained from The Cancer Genome Atlas project (TCGA, https://portal.gdc.cancer.gov/). Student's t-test was then used to compare GSTP1 expression levels between lung cancer and normal lung tissues. Gene set enrichment analysis (GSEA) was performed to identify pathways associated with GSTP1 mRNA expression levels in the TCGA lung cancer dataset. GSEA software was obtained from the Broad Institute (http://www.broad.mit.edu/gsea).The expression levels of GSTP1 genes in the selected cancers were analyzed using Oncomine. For this, we compared clinical specimens of cancer vs. normal patient datasets. In order to reduce our false discovery rate, we selected p < 0.01 and gene rank, with the top 10% as a threshold. We analyzed the results for their p-values, fold change, and cancer subtype. In many instances we found several significant correlations in different tumor types, but we showed only lung adenocarcinoma except for sub-forms. TCGA survival analysis used the website http://www.oncolnc.org/. Additionally, Kaplan-Meier Plotter database was used to draw the overall survival (OS), progression-free survival (FS) and post-progression survival (PPS) curves.
Statistical Analysis
Data were analyzed by one-way analysis of variance and Student's independent t-test using GraphPad Prism software and was presented as mean ± SD or SEM indicated in figure legends. Differences were considered statistically significant when P < 0.05.
Results
The Multi-drug Resistant Phenotypes of Lung Cancer Stem Cells Are Manifested by Their Resistance to a Wide Range of Structurally and Functionally Unrelated Chemotherapy Drugs
The lung CSCs LLC-SD cells were derived from the LLC-Parental cell line and demonstrated high capability of self-renewal and enhanced tumorigenicity as we characterized and reported (36–38). LLC-SD cells exhibited spheroid growth in vitro and can be stably maintained in culture (Figure 1A). The multi-drug resistant phenotype of LLC-SD was manifested by their resistance to a wide range of structurally and functionally unrelated chemotherapy drugs. LLC-SD cells exhibited more than 3-folds more resistance to cisplatin, paclitaxel and 5-fluorouracil compared with that in LLC-Parental cells (Figure 1B). LLC-SD cells were more resistant to undergo apoptosis upon all drug treatments than the paired LLC-Parental cells (Figure 1B). LLC-SD exhibited 12.8-, 30.0-, and 50.6-fold resistance to cisplatin, paclitaxel and 5-fluorouracil, respectively, as determined by IC50 (Table 2). To discern the potential genes that mediate multidrug resistance in the LLC-SD cells, we examined the expression of known multidrug resistance genes, including Gstp1, Abca2, Mrp1, Abcg2 and Mdr1, by RT-qPCR. Gstp1, Abca2, Mrp1 expression was robustly upregulated in LLC-SD, compared with that in LLC-Parental (Figure 1C).
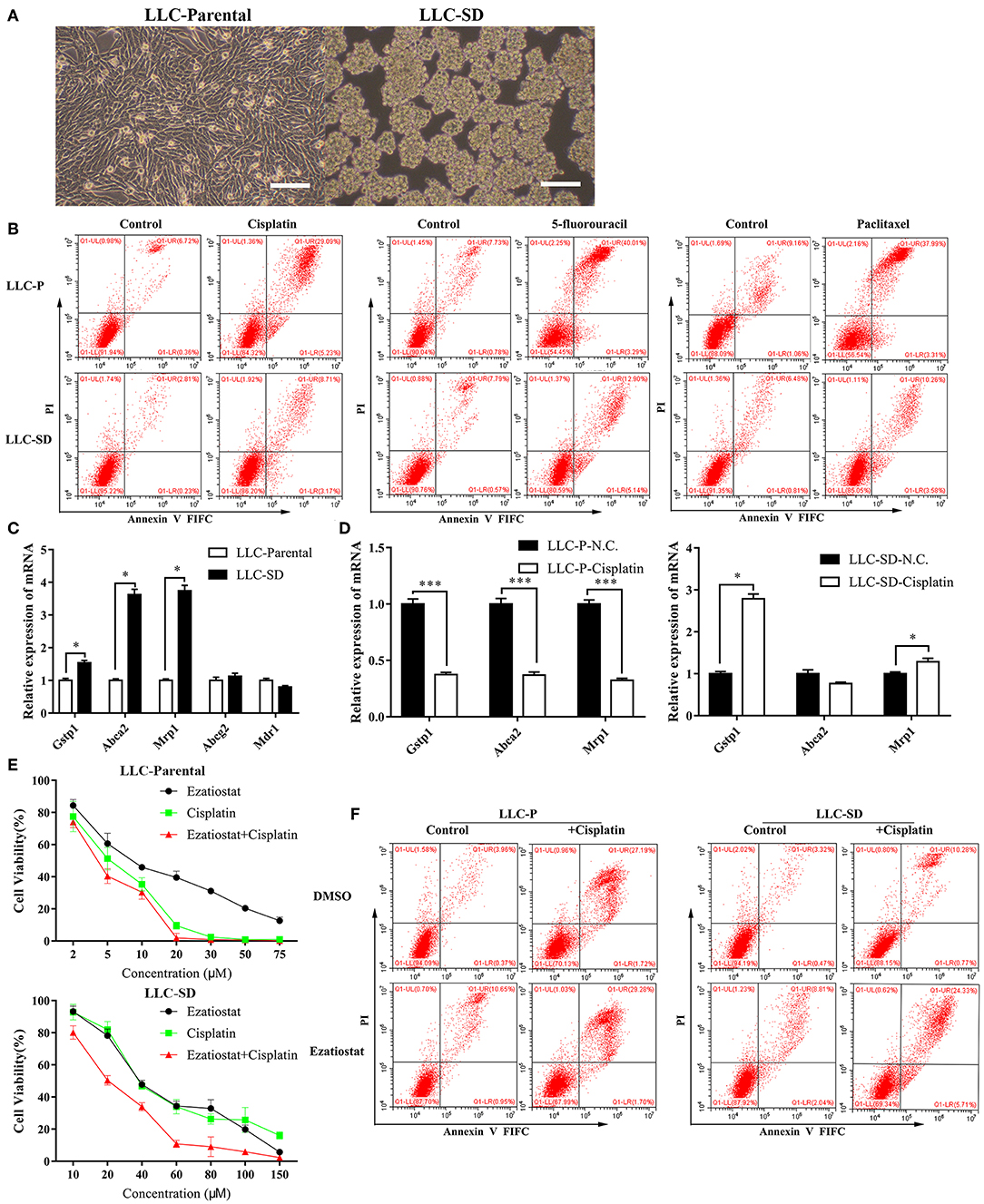
Figure 1. The multi-drug resistant phenotype of Lung cancer stem cells is manifested by their resistance to a wide range of structurally and functionally unrelated drugs. (A) Morphological differences between the spindle attached LLC-Parental cell line and its derivative CSC spheroid forming floating LLC-SD cell line, bar = 120 μm. (B) Annexin V-FITC/PI staining of apoptotic LLC-Parental and LLC-SD cells under treatment of cisplatin(10 μM), 5-fluorouracil(10 μM) and paclitaxel(10 μM) for 24 h. (C) The expression of resistance-related genes (Gstp1, Abca2, Mrp1, Abcg2, and Mdr1) in LLC-SD cell lines and LLC-Parental by RT-qPCR, the tata-Box binding protein gene (Tbp) was used as a reference gene. (D) The expression of resistance-related genes (Gstp1, Abca2, and Mrp1) after cisplatin treatment in LLC-SD cell lines by RT-qPCR, the tata-Box binding protein gene (Tbp) was reference gene. (E) Single-cell suspensions LLC-Parental and LLC-SD cell lines were seeded at 2,000/well in sphere medium and treated with increasing concentrations of cisplatin or Gstp1 inhibitor Ezatiostat alone or in combination. After 24 h of treatment, Cell growth curves (%) was derived and compared to the DMSO control. (F) Annexin V-FITC/PI staining of apoptotic LLC-Parental and LLC-SD cells under treatment of cisplatin(10 μM) and/or Ezatiostat(5 μM) for 24 h. Results are statistically significant if *p < 0.05, ***p < 0.001(Student's t-test).

Table 2. Chemosensitivity of LLC-Parental/LLC-SD cells to different chemotherapy drugs.
Cisplatin is one of a number of platinum (Pt)-containing compounds used for the treatment of a variety of malignancies, including ovarian and testicular cancer, and remains the first-line of chemotherapy for patients with advanced NSCLC (39). Hence, we further determined the expression of Gstp1, Abca2, Mrp1 before and after cisplatin treatment in LLC-SD and LLC-Parental cells. While the expression of all three genes in LLC-Parental cells was inhibited upon cisplatin treatment, Gstp1 and Mrp1 expression was elevated in LLC-SD cells after cisplatin treatment (Figure 1D) and the most pronounced increase was seen in Gstp1 (Supplementary Figure 3). Thereafter, we chose to focus on the Gstp1 gene and cisplatin for mechanistic investigation as GSTs are known to form adducts with platinum compound (18). Treatment of LLC-SD cells with an Gstp1 inhibitor, Ezatiostat, reversed the resistance to cisplatin at all doses evaluated, while Ezatiostat had less of an effect on LLC-Parental cells determined by Cck-8 assay (Figure 1E). Flow cytometric analysis confirmed these findings (Figure 1F). Taken together, these observations demonstrate that it is the CSC component of the LLC-Parental cells, i.e., the LLC-SD cells, that confer resistance to cisplatin, which appears to be mediated by Gstp1.
Blocking Gstp1 Reversed Cisplatin Resistance Both in vitro and in vivo
To confirm the involvement of Gstp1 in mediating chemoresistance of lung CSCs, we deregulated Gstp1 expression by generating LLC-SD cell lines: (i) in which Gstp1 was constitutively inhibited by lentiviral-mediated Gstp1 shRNA (shGstp1) silencing, and (ii) in which Gstp1 was up-regulated by lentiviral-mediated Gstp1 (OEGstp1) over-expression (Figures 2A,B). Chemosensitivity of LLC-SD to cisplatin varied with changes in Gstp1 expression. Inhibition of Gstp1 by shGstp1 resulted in a left-shifted cell survival curve indicating sensitization (Figure 2C, left). In contrast, Gstp1 overexpression led to enhanced resistance to cisplatin, as evident by the right-shifted cell survival curve (Figure 2C, right). The Cck-8 assay results were confirmed by the apoptosis assay (Figure 2D).
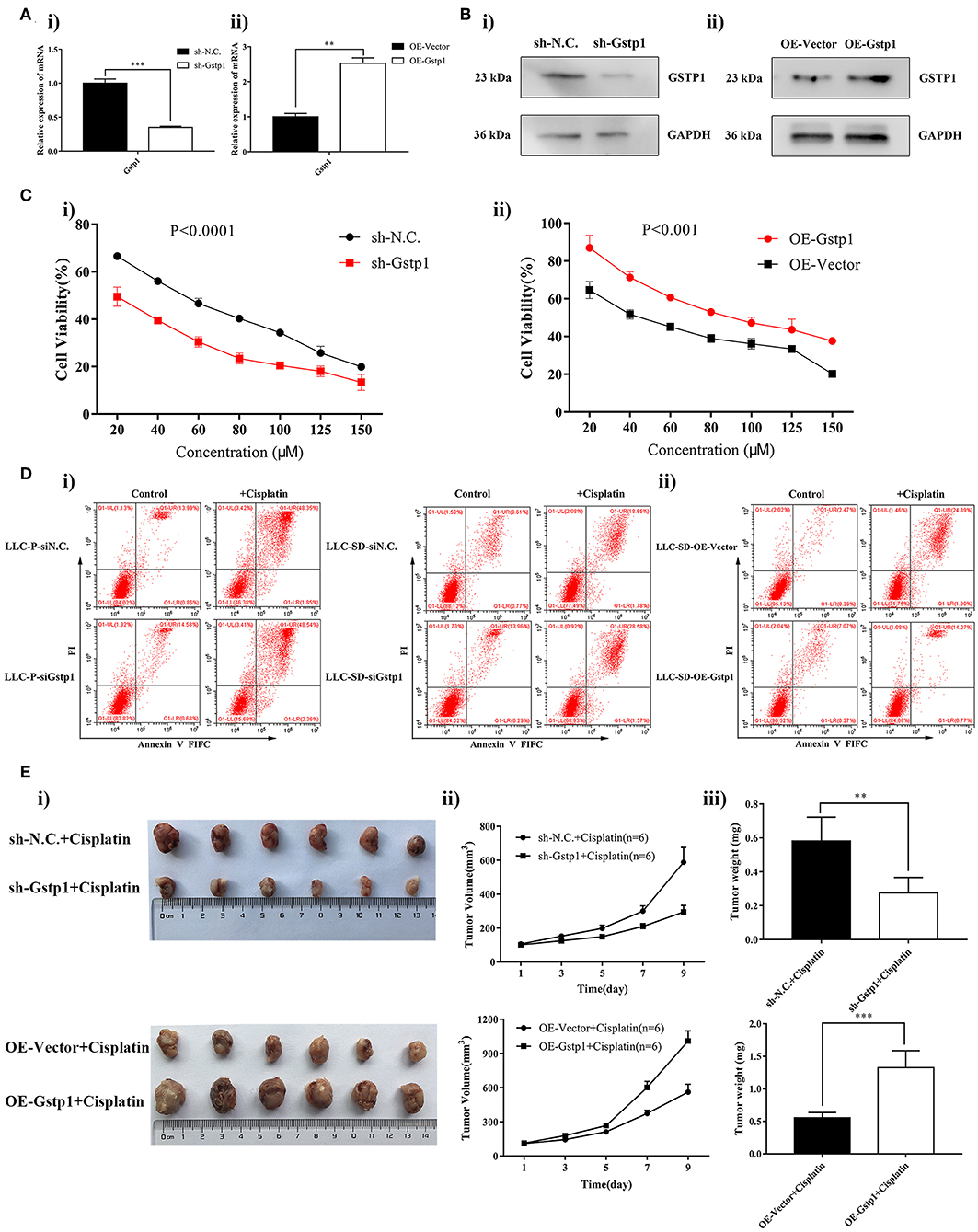
Figure 2. Gstp1 regulates cisplatin sensitivity in LLC-SD cells. (Ai,ii) Expression of Gstp1 in LLC-SD cells infected with control shRNA (shN.C.), shRNA to Gstp1 (shGstp1), control Vector (Vector), and overexpression of Gstp1 (OEGstp1) by RT-qPCR, with tata-Box binding protein gene (Tbp) as the reference gene, **p < 0.01, ***p < 0.001. (Bi,ii) Expression of Gstp1 in shN.C., shGstp1, Vector, and OEGstp1 Group by western blotting, with GAPDH as the reference protein. (Ci,ii) Single-cell suspensions of shN.C. and shGstp1/Vector and OEGstp1 groups were seeded at 2,000/well in sphere medium and treated with increasing concentrations of cisplatin. Growth curves (%) was derived and compared to the DMSO control. (Di,ii) Annexin V-FITC/PI staining of apoptotic siN.C. and siGstp1/Vector and OEGstp1 groups under treatment of cisplatin(10 μM) for 24 h. (E) Gstp1 expression regulates LLC-SD tumor sensitivity to cisplatin in vivo. Subcutaneous tumors harvested after cisplatin treatment in BALB/c nude mice on Day 9. (i) tumor volume; (ii) tumor growth curve; (iii) tumor weigh, **p < 0.01, ***p < 0.001.
To determine whether Gstp1 mediates cisplatin resistance in vivo, four cell lines that differ in Gstp1 expression were injected subcutaneously into the nude mice (Methods, 1000 cells/side). After 11 days of injection, when subcutaneous xenograft tumors growth reached ~100 mm3, cisplatin treatment was administrated (Methods). The experiment was terminated on Day 9 since the start of the treatment after 4 treatments wereadministered. Consistent with our in vitro finding, Gstp1 regulates LLC-SD tumor sensitivity to cisplatin, measured by visual examination of tumor size (Figure 2Ei), tumor growth rate (Figure 2Eii) and tumor weight (Figure 2Eiii).
Taken together, Gstp1 regulates LLC-SD sensitivity to cisplatin both in vitro and in vivo (Figures 1, 2).
MEK/ERK Signaling Pathway Mediates Cisplatin Resistance in LLC-SD Cells
It is reported that activation of the mitogen activated protein kinase (MEK/ERK) pathway may mediate resistance to cisplatin in ovarian cancer cells by regulating GST-π expression (40). Gene set enrichment analysis (GSEA) was performed to evaluate signaling pathways that were associated with Gstp1 expression in the TCGA lung cancer samples. The results revealed that Gstp1 expression was positively correlated with the MEK/ERK signaling pathway (Supplementary Figure 1A), which implied that Gstp1 may be affected by MEK/ERK signaling pathway in lung cancer. To investigate the relationship between MEK/ERK signaling pathway and Gstp1, we first evaluated whether Gstp1 expression is subjected to MEK/ERK signaling pathway regulation. Treatment of LLC-SD cells with MEK/ERK inhibitor PD98059, at the same time of receiving cisplatin treatment, prevented the up-regulation of Gstp1 expression (Figure 3A). In LLC-SD cells that exhibit resistance to cisplatin both in vitro and in vivo (Figures 1, 2), LLC-SD proliferative activity was sensitive to the inhibition of the MEK/ERK signaling pathway by PD98059 (Figure 3Bii). Further, PD98059 treatment significantly reduced LLC-SD resistance to cisplatin (Figure 3Bii). In contrast, PD98059 treatment did not alter cisplatin sensitivity in LLC-P cells (Figure 3Bi) that were sensitive to cisplatin's growth inhibition (Figure 1B). In agreement with the Cck8 analyses, PD98059 augmented cisplatin-induced apoptosis in LLC-SD cells (Figure 3Cii), and it restored the ability of cisplatin to induce apoptosis in LLC-SD to a level that is comparable to cisplatin-induced apoptosis in LLC-P cells (Figure 3Ci). Based on this set of observations, we hypothesized that cisplatin-induced increase in Gstp1 expression (Figure 1D), that was subjected to PD98059 inhibition (Figure 3A), might be a result of cisplatin-induced activation of the MEK/ERK signaling pathway leading to transcriptional activation of Gstp1 expression. Consistent with this hypothesis, enhancement of Mek1 phosphorylation was only seen in cisplatin-resistant LLC-SD cells and not in the LLC-Parental cells that were sensitive to cisplatin (Figure 3D).
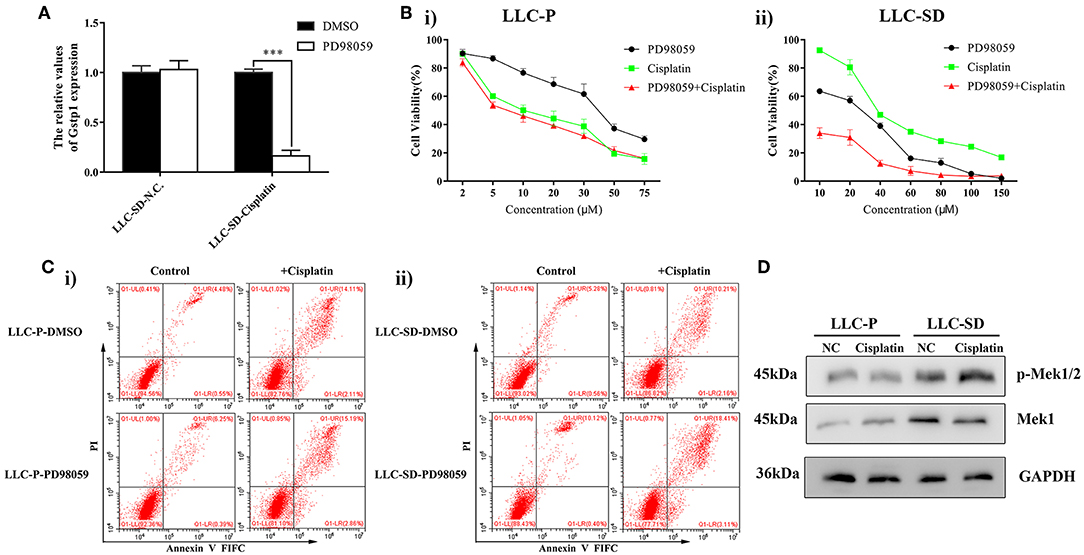
Figure 3. MEK/ERK inhibitor treatment sensitize LLC-SD to cisplatin. (A) Expression of Gstp1 in LLC-SD cells under treatment of cisplatin or PD98059 alone, or in combination by RT-qPCR, with tata-Box binding protein gene (Tbp) as the reference gene, ***p < 0.001. (B) Single-cell suspensions LLC-Parental and LLC-SD cell lines were seeded at 2,000/well in sphere medium and treated with increasing concentrations of cisplatin or PD98059 alone or in simultaneous combination. After 24 h of treatment, growth curves (%) were constructed and compared. (i) LLC- Parental; (ii) LLC-SD. (C) Annexin V-FITC/PI staining of apoptotic LLC-Parental and LLC-SD cells under treatment of cisplatin (10 μM) and/or PD98059 (5 μM) for 24 h. Results are statistically significant if ***p < 0.001(Student's t-test). (i) LLC-Parental; (ii) LLC-SD. (D) Expression of p-Mek1/2 and Mek1 in LLC-Parental or LLC-SD cell line after treatment with cisplatin by western blotting, with GAPDH as the reference protein.
To further investigate the role of the MEK/ERK signaling pathway in mediating cisplatin resistance in LLC-SD cells, we generated LLC-SD derivative cell lines in which Mek1/2 expression was stably inhibited by RNA interference (shMek1/2) (Figure 4A). Mek1/2 silencing prevented cisplatin-induced increase in Gspt1 expression in LLC-SD cells (Figure 4B). Consequently, cisplatin resistance was reduced, as determined by the Cck8 assay (Figure 4C) and apoptosis assay (Figure 4D) in vitro and by tumor growth and tumor weight in vivo (Figures 4Ei–iii), upon inactivation of the MEK/ERK pathway by Mek1/2-knockdown. This set of observations are consistent with PD98059 treatment, confirming the role of MEK/ERK signaling in mediating resistance to cisplatin in LLC-SD lung CSC cells.
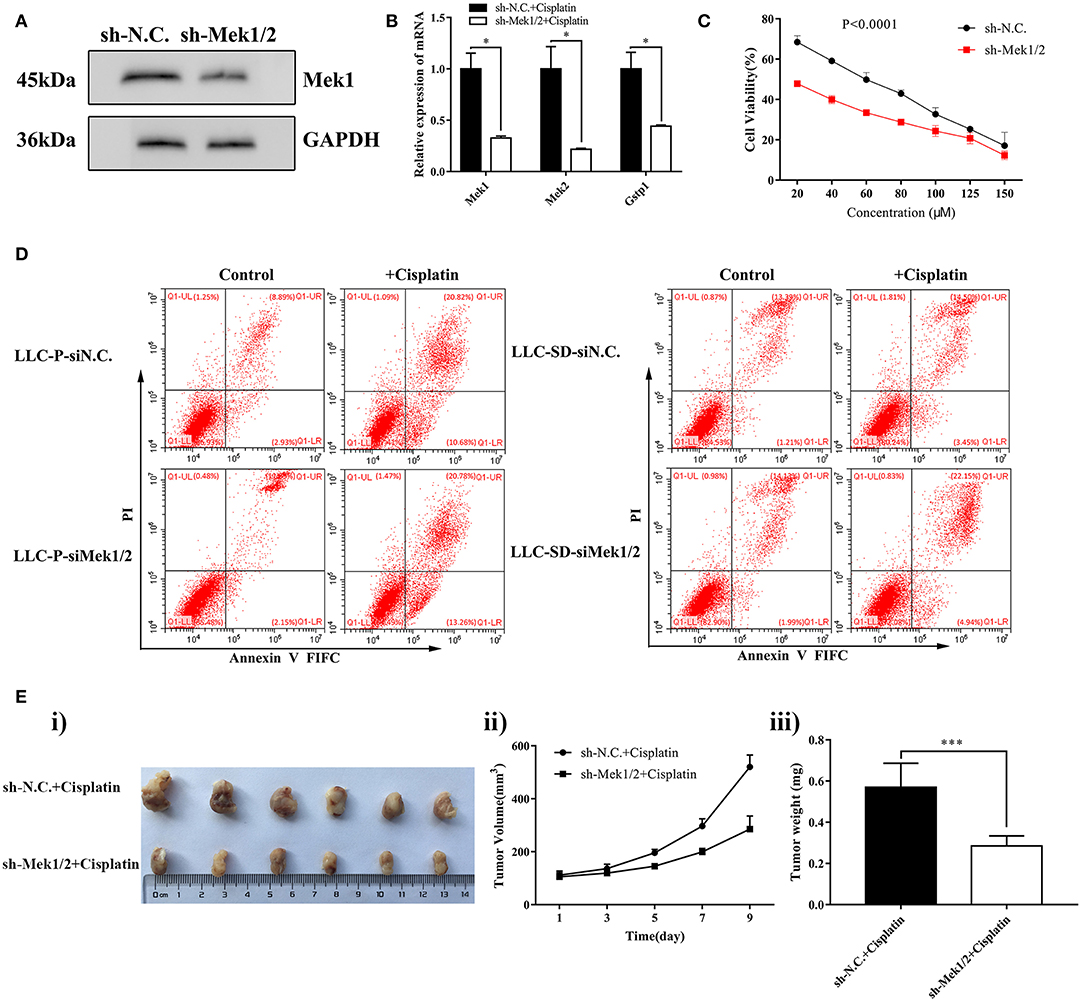
Figure 4. Stable inhibition of MEK/ERK signaling via shMek1/2 restored chemosensitivity to cisplatin in LLC-SD cells. (A) Expression of Gstp1 in LLC-SD-shN.C. and shMek1/2cells by western blotting, with GAPDH as a reference. (B) Expression of Mek1, Mek2, and Gstp1 in shN.C. and shMek1/2 groups after treatment with cisplatin by RT-qPCR, with tata-Box binding protein gene (Tbp) as the reference gene, *p < 0.05. (C) Single-cell suspensions of LLC-SD-shN.C. and LLC-SD-shMek1/2 cells were seeded at 2,000/well in sphere medium and treated with increasing concentrations of cisplatin. After 24 h of treatment, growth curves (%) were constructed and compared. (D) Annexin V-FITC/PI staining of apoptotic LLC-SD-siN.C. and siMek1/2 cells under treatment of cisplatin (10 μM) for 24 h. Results are statistically significant if *p < 0.05, ***p < 0.001(Student's t-test). (E) Mek1/2 silencing restored LLC-SD tumor sensitivity to cisplatin in vivo. Subcutaneous tumors harvested after cisplatin treatment in BALB/c nude mice. (i) tumor volume; (ii) tumor growth curve; (iii) tumor weigh, ***p < 0.001.
Gstp1 Confers MEK/ERK Mediation of Cisplatin Chemoresistance in LLC-SD Cells
To order the molecular relationship between activation of MEK/ERK signaling and transcriptional activation of Gstp1 in mediating cisplatin chemoresistance, Gstp1 expression was restored in Mek1/2 inhibited LLC-SD-shMek1/2 cells by lentiviral-mediated overexpression of Gstp1 (Figures 5A,B). Restoration of Gstp1 expression in LLC-SD-shMek1/2 cells reversed the cisplatin sensitive phenotype seen in LLC-SD-shMek1/2 cells to cisplatin resistance seen in shMek1/2+OEGstp1 cells, measured by Cck-8 proliferation assay (Figure 5C) and the apoptosis assay (Figure 5D) in vitro, and the tumor transplantation assay in vivo (Figure 5E). These results indicate that Gstp1 acts downstream of MEK/ERK signaling pathway. Further, MEK/ERK signaling mediates cisplatin resistance in lung CSC LLC-SD cells by transcriptional activation of Gstp1.
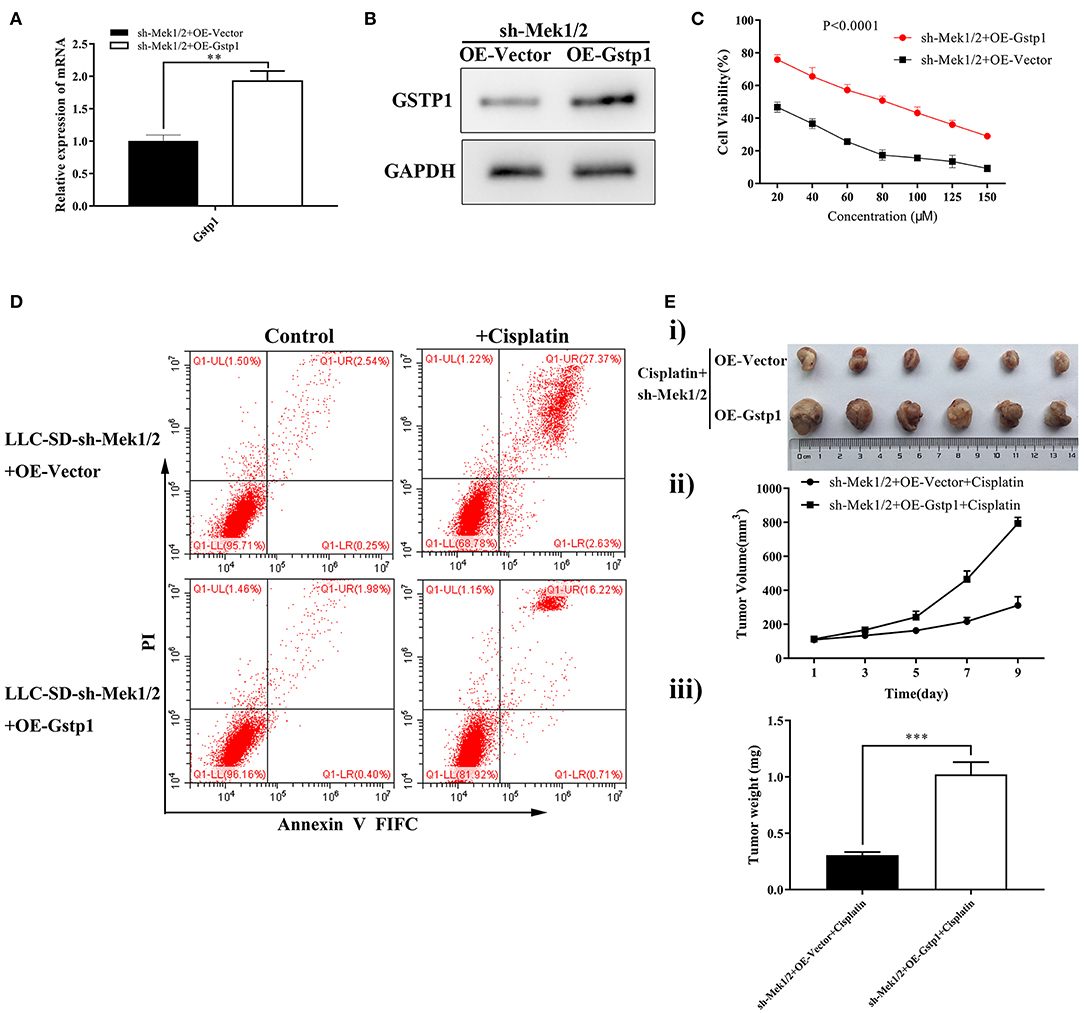
Figure 5. Overexpression of Gstp1 in LLC-SD-shMek1/2 cells restored resistance to cisplatin. (A) Expression of Gstp1 in LLC-SD-shMek1/2+Vector and LLC-SD-shMek1/2+OEGstp1 cells by RT-qPCR, with tata-Box binding protein gene (Tbp) as the reference gene. (B) Expression of Gstp1 in LLC-SD-shMek1/2+Vector and LLC-SD-shMek1/2+OEGstp1 cells by western blotting, with GAPDH as the reference protein. (C) Single-cell suspensions of LLC-SD-shMek1/2+Vector and LLC-SD-shMek1/2+OEGstp1 cells were seeded at 2,000/well in sphere medium and treated with increasing concentrations of cisplatin. After 24 h of treatment growth curves (%) were constructed and compared. (D) Annexin V-FITC/PI staining of apoptotic LLC-SD-shMek1/2+Vector and LLC-SD- shMek1/2+OEGstp1 cells under treatment of cisplatin (10 μM) for 24 h. (E) Overexpression of Gstp1 in LLC-SD-shMek1/2 cells restored resistance to cisplatin in vivo. Subcutaneous tumors harvested after cisplatin treatment in BALB/c nude mice. (i) tumor volume; (ii) tumor growth curve; (iii) tumor weigh, **p < 0.01, ***p < 0.001.
High Gstp1 mRNA Levels Are Associated With Clinical Accelerated Disease Progression and Poorer Survival in Lung Adenocarcinoma
To verify whether the findings we made in LLC-SD mouse model of lung adenocarcinoma are applicable to lung CSC models derived from human adenocarcinoma, the human lung cancer cell line H1299 was used to generate H1299-SD cells (Figure 6Ai) using the same approach that we developed (36–38). Cisplatin resistance phenotypes observed in LLC-SD cells were similarly manifested in H1299-SD cells (Figures 6Aii,iii). GSTP1 expression was also increased in H1299-SD compared with that in H1299 cells (Figure 6Aiv). More importantly, inhibition of GSTP1 restored cisplatin sensitivity in H1299-SD cells in vitro (Figures 6Bi–iii). Further, the promoting effect of GSTP1 on cisplatin resistance is regulated by MEK/ERK signaling (Figure 6C). This set of observations confirmed our main findings in the LLC-SD model and both collectively demonstrate that “lung CSC component is the cause of chemoresistance to cisplatin, which is conferred by MEK/ERK transcriptional activation of GSTP1.”
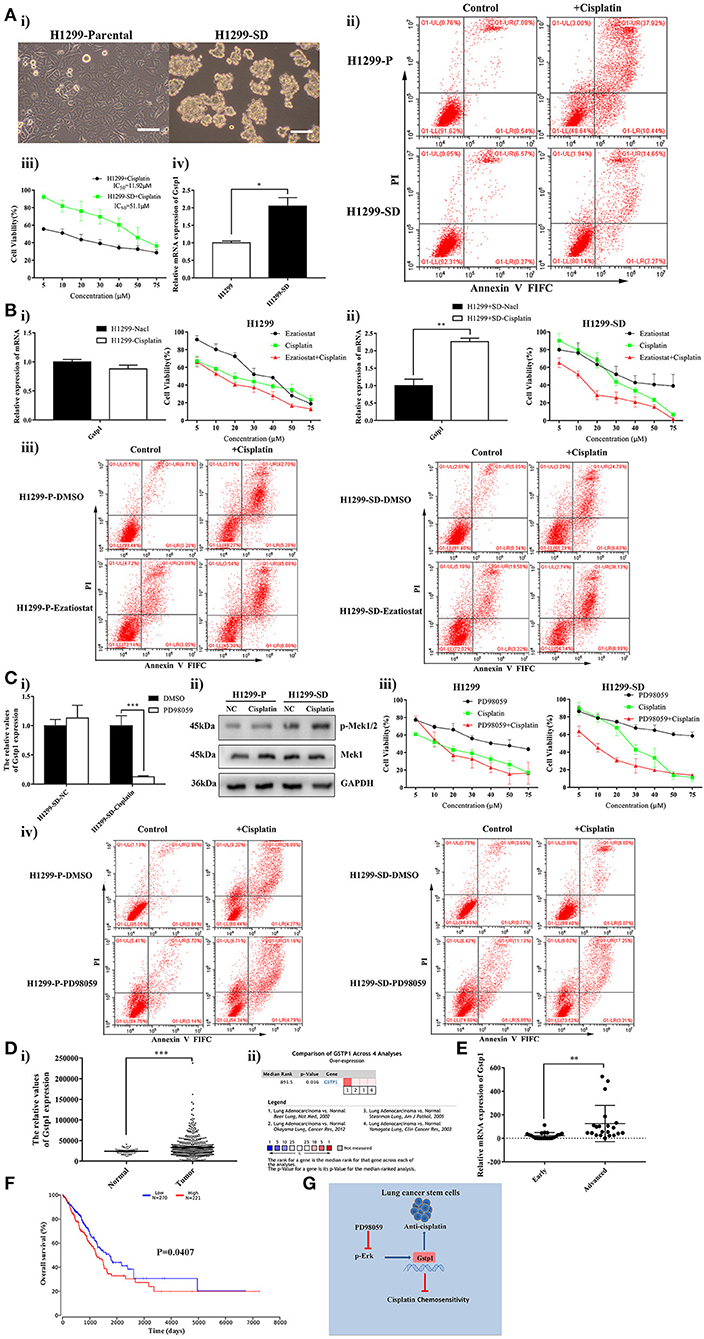
Figure 6. High GSTP1 mRNA levels are linked with disease progression and poorer survival in lung adenocarcinoma. (Ai) Morphological differences between the spindle attached human lung adenocarcinoma H1299-Parental cell line and its derivative CSC spheroid forming floating H1299-SD cell line, bar = 120 μm. (ii) Annexin V-FITC/PI staining of apoptotic H1299-Parental and H1299-SD cells under treatment of cisplatin (15 μM) for 24 h. (iii) The IC50 assay of H1299-parental and H1299-SD. (iv) Expression of GSTP1 in H1299-Parental and H1299-SD cell lines by RT-qPCR. (Bi) Expression of GSTP1 after cisplatin treatment in H1299-Parental and H1299-SD cell lines, with tata-Box binding protein gene (TBP) as the reference gene, *p < 0.05. (ii) Single-cell suspensions H1299-Parental and H1299-SD cell lines were seeded at 2,000/well in sphere medium and treated with increasing concentrations of cisplatin or Ezatiostat alone or in simultaneous combination. After 24 h of treatment growth curves (%) were constructed and compared. (iii) Annexin V-FITC/PI staining of apoptotic H1299-Parental and H1299-SD cells under treatment of cisplatin (15 μM) and/or GSTP1 inhibitor Ezatiostat (10 μM) for 24 h. (Ci) Expression of GSTP1 in H1299-SD cells under treatment of cisplatin or PD98059 alone or in simultaneous combination by RT-qPCR, with tata-Box binding protein gene (TBP) as the reference gene, ***p < 0.001. (ii) Expression of p-Mek1/2 and Mek1 in H1299-parental or H1299-SD cell line after treatment with cisplatin by western blotting, with GAPDH as the reference protein. (iii) Single-cell suspensions H1299-Parental and H1299-SD cell lines were seeded at 2,000/well in sphere medium and treated with increasing concentrations of cisplatin or PD98059 alone or in combination. After 24 h of treatment, the growth curves (%) were constructed and compared. (iv) Annexin V-FITC/PI staining of apoptotic H1299-Parental and H1299-SD cells under treatment of cisplatin (15 μM) and/or PD98059 (10 μM) for 24 h. (Di) GSTP1 expression level in normal lung tissues and LUAD tissues in the mRNA sequencing dataset of LUAD from the TCGA (Normal, n = 59; Lung adenocarcinoma, n = 535). (ii) Meta-analysis of the 4 datasets from 4 studies (Beer Lung, Stearman Lung, Hou Lung, and Okayama Lung) on GSTP1 mRNA levels in LUAD vs. normal lung tissue searched by Oncomine database. (E) A total of 43 lung adenocarcinoma patient samples were collected. For each sample, the expression levels of GSTP1 were determined by RT-qPCR and normalized to TBP as an internal control (early = 22, advanced = 21), **p < 0.01. (F) Kaplan–Meier analysis of overall curves of patients with lung adenocarcinoma in high GSTP1 expression (n = 221) and low GSTP1 expression (n = 270). P < 0.05, log-rank test. (G) Hypothetical model illustrating cisplatin resistance mechanism in lung CSC cells. Lung CSC component is the cause of chemoresistance to cisplatin which is conferred by MEK/ERK transcriptional activation of GSTP1.
To assess the clinical relevance of GSTP1 in lung adenocarcinoma, lung adenocarcinoma mRNA sequencing datasets from The Cancer Genome Atlas (TCGA) and ONCOMINE database were analyzed. The differential GSTP1 expression between the cancer tissue and the normal lung was drastic and the elevation in lung adenocarcinoma tissues was profound (Figures 6Di,ii, Supplementary Figure 1B). Moreover, we collected 43 clinical paraffin samples of lung adenocarcinoma, including 22 cases of early adenocarcinoma (T1 and T2) and 21 cases of advanced adenocarcinoma (TIII and TIV). We analyzed GSTP1 mRNA expression in this clinical cohort and found higher GSTP1 expression in advanced lung adenocarcinoma compared to that in early stage lung adenocarcinoma (Figure 6E). Thus, GSTP1 expression appears to correlate with disease staging. The low GSTP1 expression in the non-cancerous lung tissue and staging-wise elevation could be explored for lung cancer diagnosis and merits further investigation.
We next analyzed the prognostic value of GSTP1 expression by examining the relationship between GSTP1 expression and lung adenocarcinoma progression using the publicly accessible TCGA (analyzed in Oncolnc website) and Kaplan-Meier Plotter (analyzed in Kaplan-Meier Plotter website) databases. High GSTP1 mRNA levels are correlated with faster disease progression and a lower rate of survival in lung adenocarcinoma (Figure 6F, Supplementary Figure 1C). Thus, high GSTP1 expression predicts poor prognosis.
Taken together, the bioinformatics analyses indicate that the findings we made using the mouse and human lung carcinoma CSC cellular models, regarding GSTP1 mediation of cisplatin resistance, are of clinical relevance and importance.
Discussion
Lung cancer management remains a challenge due to it being asymptomatic, having late presentation when it is metastatic and becoming resistant to anti-cancer therapies. Although platinum-based chemotherapy represents the standard first-line treatment for patients with advanced NSCLC, therapeutic outcome is compromised by the development of chemoresistance, relapse and disease recurrence (6, 7). Chemoresistance is a complex phenomenon, which may involve but is not limited to the reduction of drug accumulation, enhancement of DNA repair, impediment to apoptosis and alterations in cell cycle. Mechanistic research of the involvement of commonly studied multidrug resistant genes using human lung adenocarcinoma cell lines has yielded limited clinical results to help increase the understanding for overcoming resistance. While the role of lung CSCs in conferring multidrug resistance has been postulated, experimental evidence remains associative and lacks in depth mechanistic inquisition (12–14), partially due to the lack of stable cellular models of lung CSC.
The present study has made the following novel findings that contribute to an improved mechanistic understanding of clinically observed chemoresistance to platinum-based chemotherapies in advanced lung cancer:
First, lung CSCs are the source of chemoresistance. We developed and characterized the paired mouse Lewis lung adenocarcinoma cell line, LLC-Parental, and its CSC derivative cell line, LLC-SD (36–38), as well as paired human lung adenocarcinoma cell line, H1299-Parental, and its CSC derivative cell line, H1299-SD, that we generated using the same approach for this study (Figure 6) to overcome the obstacle of lacking stable lung CSC cellular models for mechanistic investigation. The sensitivity of both the LLC-Parental and H1299-Parental to chemotherapies, in deer contrast of the multidrug resistant phenotypes observed in the LLC-SD and H1299-SD CSCs, helped us to clearly identify the CSC component of both the mouse and human lung adenocarcinoma cell lines as the cellular component to confer multidrug resistance phenotype, in particular to cisplatin (Figures 1, 6). Thus, we have provided concrete experimental evidence demonstrating the involvement of lung CSCs in chemoresistance and have overcome the obstacle of the lack of stable cellular models of CSCs, including lung CSCs.
Second, expression of Gstp1 is elevated in lung CSC cells, which can be further increased upon treatment with a panel of chemotherapy drugs. Using the two sets of cellular models for the screening of drug resistant genes that mediate multidrug resistant phenotype in the mouse and human lung CSC cells, we identified Gstp1 among other candidates that was consistently expressed at higher levels in LLC-SD and H1299-SD cells and could be further increased upon treatment with a panel of chemotherapy drugs, in particular with cisplatin (Figures 1, 6).
Third, Gstp1 confers cisplatin resistance in mouse and human lung CSC models, both in vitro and in vivo. Using lentiviral-mediated gene transfer, we generated LLC-SD (Figure 2) and H1299-SD (Figure 6) derivative cell lines, in which Gstp1 expression was either inhibited or up-regulated. Through the bi-directional modulation of Gstp1 expression and in vitro (both LLC-SD and H1299-SD) (Figures 2, 6) and in vivo (LLC-SD) (Figure 2) characterization of the sensitivity to cisplatin, we can make a rather convincing conclusion that high Gstp1 expression confers cisplatin resistance in experimental lung CSCs.
In the present study, we found elevated levels of Gstp1 in response to multiple-drug treatment (Figure 1, Supplementary Figure 2), although Gstp1 function is mostly studied under the context of platinum category of chemotherapeutic agents, due to its ability to remove the agents from the cells via the formation of Pt-glutathione. However, our findings are consistent with the literature reports that Gstp1 expression could be regulated by transcriptional factors, including activating protein-1 (AP-1) and nuclear factor erythroid-2-related factor 2 (Nrf2), in response to multiple anticancer drugs, including chlorambucil, cyclophosphamide, cisplatin, DOX and mitoxantrone (41–43). Thus, Gstp1-mediated chemoresistance may not be limited to cisplatin and related anticancer agents, thus merits further studies.
Fourth, we have provided the first experimental evidence that transcriptional activation of Gstp1 expression by MEK/ERK signaling underlies cisplatin resistance in lung CSC cells. Prior to our study, it was reported that in addition to MEK/ERK, both JNK/SAPK and p38 pathways were also affected by cisplatin treatment (44). In addition, Lin et al. reported that Gstp1 expression can be upregulated by phosphorylation of Erk2 and by activating Nrf2 in response to methionine restriction (45). In the present study, we focused on examining MEK/ERK transcriptional regulation of Gstp1 expression in response to cisplatin treatment in lung CSCs. Through lentiviral mediated siRNA interference of Mek1/2 expression, we confirmed the requirement of MEK/ERK activation to confer cisplatin resistance both in vitro and in vivo (Figure 3). We didn't stop here and took one step further which was to order the molecular relationship between activation of MEK/ERK signaling and transcriptional activation of Gstp1 in mediating cisplatin chemoresistance. It was achieved by restoration of Gstp1 expression in Mek1/2 silenced LLC-SD-shMek1/2 cells, which manifested concomitant Gstp1 down-regulation, by overexpressing of Gstp1 (Figures 5A,B). Restoration of Gstp1 expression in LLC-SD-shMek1/2 cells reversed the cisplatin sensitive phenotype seen in LLC-SD-shMek1/2 cells to cisplatin resistance seen in shMek1/2+OEGstp1 cells, both in vitro and in vivo (Figure 5). These results confirm that transcriptional activation of Gstp1 by MEK/ERK signaling pathway mediates cisplatin resistance in experimental lung CSCs.
Fifth, GSTP1 expression is a poor diagnostic and prognostic marker for human lung adenocarcinoma, thus is of high clinical relevance. Prior to this study, the clinical relevance of GSTP1 has not been well-explored. We conducted multi-scale analyses including the analysis of publicly available gene expression databases of human adenocarcinoma of lung, gene expression analysis of clinical paraffin blocks of lung adenocarcinoma as well as mouse and human lung adenocarcinoma CSC models. We found that GSTP1 expression appears to correlate with disease staging (Figure 6). The low GSTP1 expression in the non-cancerous lung tissue and staging-wise elevation could be explored for lung cancer diagnosis and merits further investigation. We also found that high GSTP1 mRNA levels are correlated with faster disease progression and a lower rate of survival in lung adenocarcinoma (Figure 6F, Supplementary Figure 1C). Thus, high GSTP1 expression predicts poor prognosis. Further, since the expression changes of GSTP1 in response to chemo-therapeutic agents were specific to the lung CSCs, not the non-stem-like parental cells, GSTP1 may also serve a lung CSC marker for evaluating the CSC content in clinical lung samples.
Taken together, the bioinformatics analyses indicate that the findings we made using the mouse and human lung carcinoma CSC cellular models, regarding Gstp1 mediation of cisplatin resistance, are of clinical relevance and importance.
Ethics Statement
This study was carried out in accordance with the recommendations of Institutional Animal Care and Use Committee of Chongqing medical University. The protocol was approved by Institutional Animal Care and Use Committee of Chongqing medical University. Experimental mice were raised in an accredited specific pathogen-free Animal Facility at Chongqing medical University. All protocols were approved by the Institutional Animal Care and Use Committee of Chongqing medical University and conducted with humane animal care. All of the formalin fixed paraffin embedded were obtained from department of pathology in the affiliated hospital of southwest medical university and received the approval of the institutional ethics committee of southwest medical university medical center. Moreover, this study obtained written informed consent from the participants.
Author Contributions
JL and TY substantially contributed to conception and design, acquisition of data, analysis and interpretation of data, and drafting the article. YL, LK, ZS, and DL acquired part of the data. JW and HX contributed to conception and design, revising it critically for important intellectual content, and final approval of the version to be published.
Funding
This work was supported by the National Natural Science Fund (Grant No. 81672908) and the National Natural Science Youth Fund (Grant No. 81602596).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the members in the Laboratory of Translational Cancer Stem Cell Research who are not listed in the authors.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.00476/full#supplementary-material
Supplementary Figure 1. (A) GSEA analysis showed that GSTP1 was positively associated with MEK/ERK signaling pathway in the TCGA lung cancer samples. (B) The Oncomine database mining analysis of GSTP1 mRNA levels in Beer Lung, Stearman Lung, Hou Lung, and Okayama Lung grouped by lung cancer and normal specimens. (C) Kaplan-Meier analysis of overall survival (OS), progression-free survival (FS) and post-progression survival (PPS) curves of lung adenocarcinoma (LUAD) patients based on GSTP1 mRNA expression (low vs. high).
Supplementary Figure 2. (A,B) The expression of resistance-related genes (Gstp1, Abca2, Mrp1) after 5-FU or paclitaxel treatment in LLC-SD cell lines by RT-qPCR, the tata-Box binding protein gene (Tbp) was reference gene. *p < 0.05, **p < 0.01.
Supplementary Figure 3. The expression of Gstp1 after cisplatin treatment in LLC-Parental and LLC-SD cells by western blotting, with GAPDH as a reference.
Abbreviations
CSCs, cancer stem cells; LCSCs, lung cancer stem cells, NSCLC, non-small cell lung cancers; LUAD, lung adenocarcinoma.
References
1. Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. (2016) 66:271–89. doi: 10.3322/caac.21349
2. Chalela R, Curull V, Enriquez C, Pijuan L, Bellosillo B, Gea J. Lung adenocarcinoma: from molecular basis to genome-guided therapy and immunotherapy. J Thorac Dis. (2017) 9:2142–58. doi: 10.21037/jtd.2017.06.20
3. Borczuk AC, Gorenstein L, Walter KL, Assaad AA, Wang L, Powell CA. Non-small-cell lung cancer molecular signatures recapitulate lung developmental pathways. Am J Pathol. (2003) 163:1949–60. doi: 10.1016/S0002-9440(10)63553-5
4. Yankelevitz DF, Reeves AP, Kostis WJ, Zhao B, Henschke CI. Small pulmonary nodules: volumetrically determined growth rates based on CT evaluation. Radiology. (2000) 217:251–6. doi: 10.1148/radiology.217.1.r00oc33251
5. Ar Jazieh on Behalf of Saudi Lung Cancer Guidelines Association SLCGM, Jazieh AR, Al Kattan K, Bamousa A, Al Olayan A, Abdelwarith A, et al. Saudi lung cancer management guidelines 2017. Ann Thorac Med. (2017) 12:221–46. doi: 10.4103/atm.ATM_92_17
6. Ou SH, Zell JA, Ziogas A, Anton-Culver H. Prognostic factors for survival of stage I nonsmall cell lung cancer patients: a population-based analysis of 19,702 stage I patients in the California Cancer Registry from 1989 to 2003. Cancer. (2007) 110:1532–41. doi: 10.1002/cncr.22938
7. Asamura H, Goya T, Koshiishi Y, Sohara Y, Eguchi K, Mori M, et al. A Japanese Lung Cancer Registry study: prognosis of 13,010 resected lung cancers. J Thorac Oncol. (2008) 3:46–52. doi: 10.1097/JTO.0b013e31815e8577
8. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. (2006) 66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126
9. Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annu Rev Cell Dev Biol. (2007) 23:675–99. doi: 10.1146/annurev.cellbio.22.010305.104154
10. Rapp UR, Ceteci F, Schreck R. Oncogene-induced plasticity and cancer stem cells. Cell Cycle. (2008) 7:45–51. doi: 10.4161/cc.7.1.5203
11. Miyata T, Oyama T, Yoshimatsu T, Higa H, Kawano D, Sekimura A, et al. The clinical significance of cancer stem cell markers ALDH1A1 and CD133 in lung adenocarcinoma. Anticancer Res. (2017) 37:2541–7. doi: 10.21873/anticanres.11597
12. Pasini A, Paganelli G, Tesei A, Zoli W, Giordano E, Calistri D. Specific biomarkers are associated with docetaxeland gemcitabine-resistant NSCLC cell lines. Transl Oncol. (2012) 5:461–8. doi: 10.1593/tlo.12256
13. Liu YP, Yang CJ, Huang MS, Yeh CT, Wu AT, Lee YC, et al. Cisplatin selects for multidrug-resistant CD133+ cells in lung adenocarcinoma by activating Notch signaling. Cancer Res. (2013) 73:406–16. doi: 10.1158/0008-5472.CAN-12-1733
14. Barr MP, Gray SG, Hoffmann AC, Hilger RA, Thomale J, O'Flaherty JD, et al. Generation and characterisation of cisplatin-resistant non-small cell lung cancer cell lines displaying a stem-like signature. PLoS ONE. (2013) 8:e54193. doi: 10.1371/journal.pone.0054193
15. Chatterjee A, Gupta S. The multifaceted role of glutathione S-transferases in cancer. Cancer Lett. (2018) 433:33–42. doi: 10.1016/j.canlet.2018.06.028
16. Booth J, Boyland E, Sims P. An enzyme from rat liver catalysing conjugations with glutathione. Biochem J. (1961) 79:516–24. doi: 10.1042/bj0790516
17. Tew KD, Manevich Y, Grek C, Xiong Y, Uys J, Townsend DM. The role of glutathione S-transferase P in signaling pathways and S-glutathionylation in cancer. Free Radic Biol Med. (2011) 51:299–313. doi: 10.1016/j.freeradbiomed.2011.04.013
18. Peklak-Scott C, Smitherman PK, Townsend AJ, Morrow CS. Role of glutathione S-transferase P1–1 in the cellular detoxification of cisplatin. Mol Cancer Ther. (2008) 7:3247–55. doi: 10.1158/1535-7163.MCT-08-0250
19. Dong SC, Sha HH, Xu XY, Hu TM, Lou R, Li H, et al. Glutathione S-transferase pi: a potential role in antitumor therapy. Drug Des Devel Ther. (2018) 12:3535–47. doi: 10.2147/DDDT.S169833
20. Fang C, Wei XM, Zeng XT, Wang FB, Weng H, Long X. Aberrant GSTP1 promoter methylation is associated with increased risk and advanced stage of breast cancer: a meta-analysis of 19 case-control studies. BMC Cancer. (2015) 15:920. doi: 10.1186/s12885-015-1926-1
21. Hivet M, Maisel A, Horiot A, Conte J. [Diffuse villous carcinoma of Wirsung's duct. Total pancreatectomy]. Med Chir Dig. (1975) 4:159–623.
22. Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. J Clin Oncol. (2006) 24:2137–50. doi: 10.1200/JCO.2005.05.2308
23. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. (2001) 410:37–40. doi: 10.1038/35065000
24. Eisenmann KM, VanBrocklin MW, Staffend NA, Kitchen SM, Koo HM. Mitogen-activated protein kinase pathway-dependent tumor-specific survival signaling in melanoma cells through inactivation of the proapoptotic protein bad. Cancer Res. (2003) 63:8330–7.
25. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. (2002) 298:1911–2. doi: 10.1126/science.1072682
26. Qin X, Liu C, Zhou Y, Wang G. Cisplatin induces programmed death-1-ligand 1(PD-L1) over-expression in hepatoma H22 cells via Erk /MAPK signaling pathway. Cell Mol Biol (Noisy-le-grand). (2010) 56:OL1366–72. doi: 10.1170/156
27. Wang X, Martindale JL, Holbrook NJ. Requirement for ERK activation in cisplatin-induced apoptosis. J Biol Chem. (2000) 275:39435–43. doi: 10.1074/jbc.M004583200
28. Yeh PY, Chuang SE, Yeh KH, Song YC, Ea CK, Cheng AL. Increase of the resistance of human cervical carcinoma cells to cisplatin by inhibition of the MEK to ERK signaling pathway partly via enhancement of anticancer drug-induced NF kappa B activation. Biochem Pharmacol. (2002) 63:1423–30. doi: 10.1016/S0006-2952(02)00908-5
29. Nowak G. Protein kinase C-alpha and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. J Biol Chem. (2002) 277:43377–88. doi: 10.1074/jbc.M206373200
30. Hayakawa J, Ohmichi M, Kurachi H, Ikegami H, Kimura A, Matsuoka T, et al. Inhibition of extracellular signal-regulated protein kinase or c-Jun N-terminal protein kinase cascade, differentially activated by cisplatin, sensitizes human ovarian cancer cell line. J Biol Chem. (1999) 274:31648–54. doi: 10.1074/jbc.274.44.31648
31. Persons DL, Yazlovitskaya EM, Cui W, Pelling JC. Cisplatin-induced activation of mitogen-activated protein kinases in ovarian carcinoma cells: inhibition of extracellular signal-regulated kinase activity increases sensitivity to cisplatin. Clin Cancer Res. (1999) 5:1007–14.
32. Basu A, Tu H. Activation of ERK during DNA damage-induced apoptosis involves protein kinase Cdelta. Biochem Biophys Res Commun. (2005) 334:1068–73. doi: 10.1016/j.bbrc.2005.06.199
33. Lee J, Kim HH, Ro SM, Yang JH. Capecitabine and cisplatin (XP) combination systemic chemotherapy in heavily pre-treated HER2 negative metastatic breast cancer. PLoS ONE. (2017) 12:e0171605. doi: 10.1371/journal.pone.0171605
34. Kano Y, Akutsu M, Suzuki K, Yoshida M. Effects of carboplatin in combination with other anticancer agents on human leukemia cell lines. Leuk Res. (1993) 17:113–9. doi: 10.1016/0145-2126(93)90055-P
35. Perez EA. Carboplatin in combination therapy for metastatic breast cancer. Oncologist. (2004) 9:518–27. doi: 10.1634/theoncologist.9-5-518
36. Wang J, Sun Z, Liu Y, Kong L, Zhou S, Tang J, et al. Comparison of tumor biology of two distinct cell sub-populations in lung cancer stem cells. Oncotarget. (2017) 8:96852–64. doi: 10.18632/oncotarget.18451
37. Wang J, Zhou T, Sun Z, Ye T, Zhou S, Li J, et al. Zeb1 Regulates the symmetric division of mouse lewis lung carcinoma stem cells through numb mediated by miR-31. Int J Biol Sci. (2018) 14:1399–410. doi: 10.7150/ijbs.27446
38. Ye T, Li J, Sun Z, Liu Y, Kong L, Zhou S, et al. Nr5a2 promotes cancer stem cell properties and tumorigenesis in nonsmall cell lung cancer by regulating Nanog. Cancer Med. (2019) 8:1232–45. doi: 10.1002/cam4.1992
39. Noel EE, Perry J, Chaplin T, Mao X, Cazier JB, Joel SP, et al. Identification of genomic changes associated with cisplatin resistance in testicular germ cell tumor cell lines. Genes Chromosomes Cancer. (2008) 47:604–13. doi: 10.1002/gcc.20564
40. Liu HZ, Yu C, Yang Z, He JL, Chen WJ, Yin J, et al. Tubeimoside I sensitizes cisplatin in cisplatin-resistant human ovarian cancer cells (A2780/DDP) through down-regulation of ERK and up-regulation of p38 signaling pathways. Mol Med Rep. (2011) 4:985–92. doi: 10.3892/mmr.2011.513
41. Louie SM, Grossman EA, Crawford LA, Ding L, Camarda R, Huffman TR, et al. GSTP1 is a driver of triple-negative breast cancer cell metabolism and pathogenicity. Cell Chem Biol. (2016) 23:567–78. doi: 10.1016/j.chembiol.2016.03.017
42. Sweeney C, McClure GY, Fares MY, Stone A, Coles BF, Thompson PA, et al. Association between survival after treatment for breast cancer and glutathione S-transferase P1 Ile105Val polymorphism. Cancer Res. (2000) 60:5621–4.
43. Zeniou M, Ding T, Trivier E, Hanauer A. Expression analysis of RSK gene family members: the RSK2 gene, mutated in Coffin-Lowry syndrome, is prominently expressed in brain structures essential for cognitive function and learning. Hum Mol Genet. (2002) 11:2929–40. doi: 10.1093/hmg/11.23.2929
44. Yin Z, Ivanov VN, Habelhah H, Tew K, Ronai Z. Glutathione S-transferase p elicits protection against H2O2-induced cell death via coordinated regulation of stress kinases. Cancer Res. (2000) 60:4053–7.
Keywords: Gstp1, cancer stem cell, chemotherapeutic resistance, MEK/ERK signaling pathway, lung adenocarcinoma
Citation: Li J, Ye T, Liu Y, Kong L, Sun Z, Liu D, Wang J and Xing HR (2019) Transcriptional Activation of Gstp1 by MEK/ERK Signaling Confers Chemo-Resistance to Cisplatin in Lung Cancer Stem Cells. Front. Oncol. 9:476. doi: 10.3389/fonc.2019.00476
Received: 24 November 2018; Accepted: 20 May 2019;
Published: 11 June 2019.
Edited by:
Luisa Lanfrancone, Istituto Europeo di Oncologia s.r.l., ItalyReviewed by:
Kenneth K. W. To, The Chinese University of Hong Kong, ChinaSabarish Ramachandran, Texas Tech University Health Sciences Center, United States
Copyright © 2019 Li, Ye, Liu, Kong, Sun, Liu, Wang and Xing. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianyu Wang, d2p5MjAwMzEyM0AxNjMuY29t; H. Rosie Xing, eGluZ2xhYjMxMEAxNjMuY29t