 Niaz Mahmood
Niaz Mahmood Shafaat A. Rabbani
Shafaat A. Rabbani- Department of Medicine, McGill University Health Centre, Montréal, QC, Canada
DNA methylation is a major epigenetic process that regulates chromatin structure which causes transcriptional activation or repression of genes in a context-dependent manner. In general, DNA methylation takes place when methyl groups are added to the appropriate bases on the genome by the action of “writer” molecules known as DNA methyltransferases. How these methylation marks are read and interpreted into different functionalities represents one of the main mechanisms through which the genes are switched “ON” or “OFF” and typically involves different types of “reader” proteins that can recognize and bind to the methylated regions. A tightly balanced regulation exists between the “writers” and “readers” in order to mediate normal cellular functions. However, alterations in normal methylation pattern is a typical hallmark of cancer which alters the way methylation marks are written, read and interpreted in different disease states. This unique characteristic of DNA methylation “readers” has identified them as attractive therapeutic targets. In this review, we describe the current state of knowledge on the different classes of DNA methylation “readers” identified thus far along with their normal biological functions, describe how they are dysregulated in cancer, and discuss the various anti-cancer therapies that are currently being developed and evaluated for targeting these proteins.
Dna Methylation And Cancer: An Introduction
The identity, behavior, and functionality of cells are dictated by the set of genes that are expressed at a given time. These genes are under tight regulation by different regulatory factors which makes sure that cells behave and function normally. However, in cancer, the cells lose their ability to behave normally which can be seen and measured by the qualitative and quantitative changes in the expression pattern of the active genes (1, 2). Historically, much attention has been given to the qualitative changes of gene expression which mainly focuses on the gene sequence-based alteration in the tumor cells (1). Therefore, cancer has been classically known as a predominantly genetic disease (3). However, the realization that quantitative changes in gene expression, due to different types of epigenetic modification, also plays an essential role in the development and progression of cancer has been one of the most significant breakthroughs in cancer biology over the last two decades (4).
Among the various types of epigenetic modifications of the genome, DNA methylation is the first one to be identified where the 5th carbon of the cytosine residue at CpG dinucleotides is methylated by the action of writer molecules known as DNA methyltransferases (5). Recent studies have revealed that methylation can take place beyond the CpG context. For example, in the brain and embryonic stem cells methylation can take place in non-CpG sites (CpH where H = A/T/C) (6, 7). However, methylation at non-CpG sites is not as frequent as CpG methylation. The distribution of methylation across the genome is cell type-specific and dynamic (8). Recent studies have demonstrated that DNA methylation-mediated transcriptional regulation is context dependent where methylation at the promoter region is typically related to repression of gene expression and methylation at the gene bodies is associated with activation of gene expression (9).
DNA methylation at the regulatory region of the gene can either directly hinder transcription factor binding and thereby cause transcriptional repression (10), or recruit “reader” molecules known as methyl-binding proteins (MBPs) at the methylated site which can then attract different members of the chromatin remodeling complex to cause transcriptional activation/repression depending on the cellular context (9, 11). However, the mechanisms by which the DNA methylation signatures are “read” and interpreted in different genomic context is not fully elucidated and may provide valuable information on how the gene expression programs are controlled in normal biological processes as well as in pathological conditions like cancer. The role of methyl-CpG-binding domain (MBD) proteins, that belongs to one of the three main families of MBPs (classification given in the next section), in the pathophysiology of different diseases including cancer has most recently been reviewed by Du et al. (12). On the other hand, the role of all the three known families of MBPs in relation to cancer was last reviewed by Parry and Clarke (13). However, since then, the field has gained newer insights into the functions and therapeutic targeting of different MBPs in cancer. In addition, several new proteins have been discovered to have methyl binding activities which warranted an update in the classification of MBPs. This review focuses on the known families of DNA methylation readers or MBPs and provides an updated summary of their roles in the context of cancer.
Classification Of Mbps
Even though the existence of DNA methylation was first described in the late 1940s (14), it was not until the next 30 years when the importance of the process started to get appreciated due to the findings that revealed its role in the regulation of gene expression and cellular differentiation (15, 16). Several years after that two proteins with methyl binding activities were reported in mammals which were termed as methyl-CpG binding protein 1 (MeCP1) and MeCP2 (17, 18). However, later studies have demonstrated that MeCP1 is, in fact, a complex containing multiple proteins involved in chromatin remodeling (19–21). Therefore, MeCP2 is regarded as the first ever single MBP to be identified (17).
At the structural level, MeCP2 contains a MBD domain comprising 70–85 amino acids that can recognize and bind to methylated CpGs (22). The MBD domain was later used to identify other proteins with methyl-binding potentials (23). At present, there are 11 known proteins with MBD domains which are classified as the family of “MBD-containing proteins.” More than a decade after the discovery of MeCP2, a second family of MBPs were identified that recognizes the methylated DNA using the Zinc finger motifs. Hence, they are called the “Methyl-CpG binding zinc fingers” (24). This particular family has seen the most rapid expansion over the last few years and, at present, there are at least 8 members in this family (25). The third family of MBPs was identified based on their ability to bind methylated DNA using the Set and RING-associated (SRA) domain and hence called the “SRA domain-containing proteins”. A schematic classification of the three main families of MBPs is shown in Figure 1, and the general and pathophysiological functions (in cancer) of each of the proteins is summarized in Table 1.
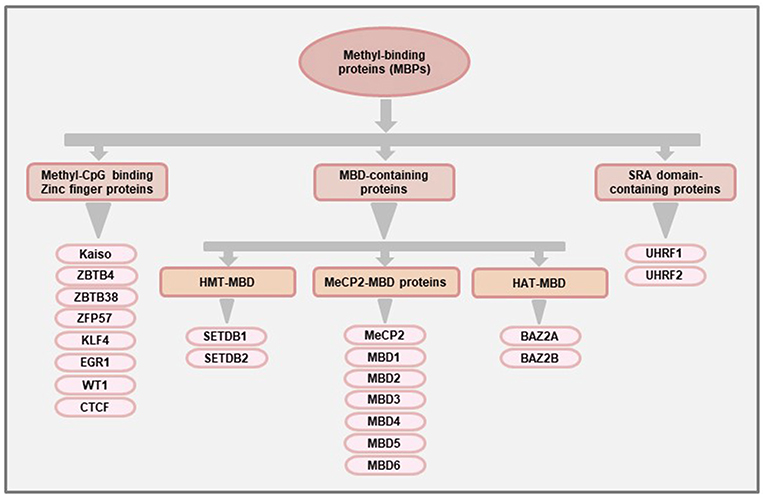
Figure 1. Classification of methyl-binding proteins (MBPs). The proteins with methyl-CpG binding abilities are broadly classified into three families based on the functional domains used for binding to methylated DNA. The “MBD-containing proteins” were the first group of MBPs to be identified and are further classified into three subfamilies (MeCP2-MBD, HMT-MBD, and HAT-MBD) based on the presence of functional domains other than MBD. The members of the HMT-MBD and HAT-MBD subfamilies have protein methyltransferase and acetylase activities respectively. The “Methyl-CpG binding Zinc finger proteins” have at least 8 members (Kaiso, ZBTB4, ZBTB38, ZFP57, KLF4, EGR1, WT1, CTCF) that can bind to methylated region using the Zinc finger motifs while the third family of MBPs consisting of UHRF1 and UHRF2 proteins uses their Set and RING-associated (SRA) domain to bind methylated DNA.
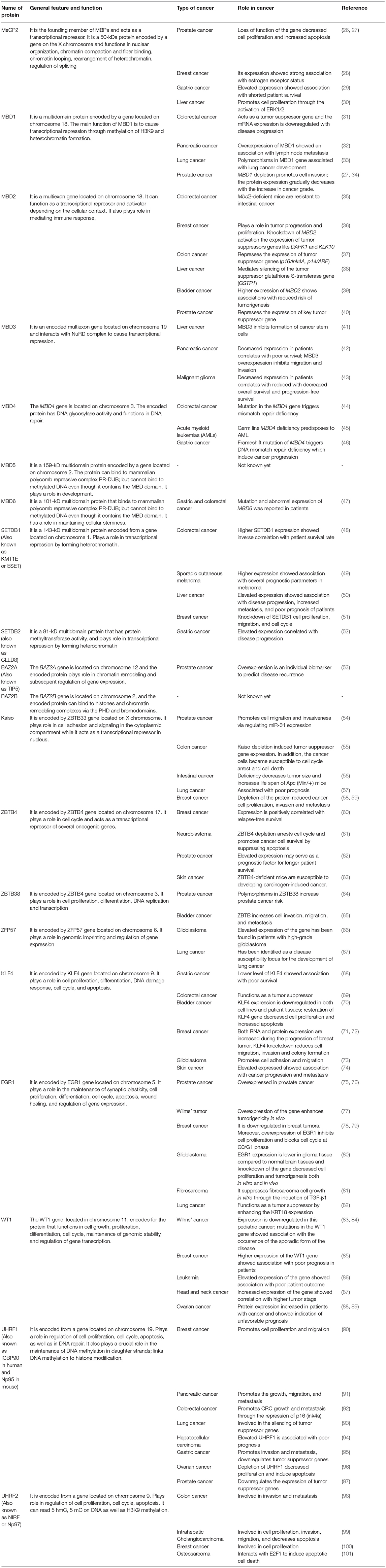
Table 1. Summarized features and functions of different MBPs.
MBD-Containing Proteins
This is the first family of MBPs to be identified. All members of this family have the conserved MBD domain (NCBI Conserved Domain Database ID: cl00110 and cd00122). However, they also have some additional domains which provide them with specific features (Figure 2) and functions (Table 1). Based on the presence of domains other than MBD, the members of this family are further classified into three categories: (1) MeCP2-MBD, (2) HMT-MBD, and (3) HAT-MBD. The three subfamilies of MBD-containing proteins are phylogenetically distinct from each other (102). In addition, not all the proteins can bind to methylated DNA but are historically classified together based on their structural features rather than methyl-binding abilities.
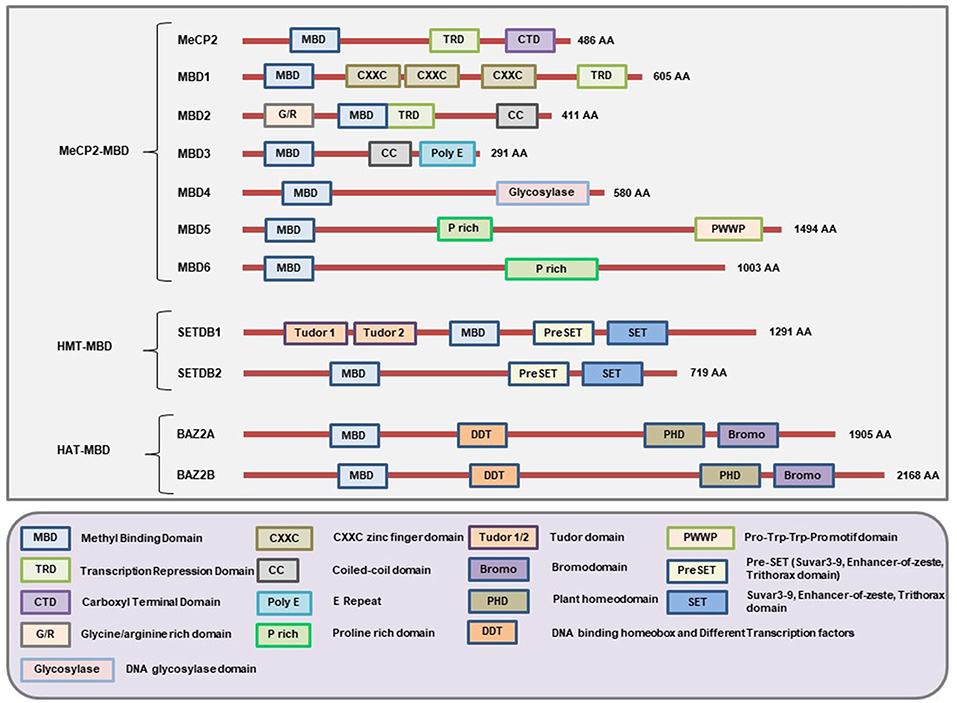
Figure 2. Schematic representation of the major domains present in different MBD-containing proteins. All members of this family of proteins have the MBD domain. However, not all 11 members under this family can bind methylated DNA but were classically grouped under the same family because of having the MBD domain. Each member of this family has domains other than MBD that gives them unique characteristics to carry out different cellular functionalities. The MeCP2, MBD1, and MBD2 contain a TRD domain that helps them to recruit chromatin remodeling corepressors and thereby cause transcriptional silencing. In addition, the MBD1 protein may have two or three CXXC-domains due to splicing of its gene. The first two CXXC domains of MBD1 bind to methylated DNA while the third CXXC domain binds to unmethylated DNA. MBD2 has unique G/R rich domain that allows protein-protein interaction as well as post-translational modification. Due to a mutation in the region of the gene encoding the MBD domain of MBD3, it cannot bind to methylated DNA. However, MBD3 can interact with other chromatin remodeling complex and play a role in the regulation of gene expression. The glycosylase domain at the C-terminal end of MBD4 provides it with the unique function of having DNA glycosylase activity while the N-terminal MBD helps it to bind methylated DNA. MBD5 and MBD6 cannot bind to methylated DNA but can interact with the mammalian polycomb deubiquitinase complex PR-DUB. In addition, the PWWP (Pro-Try-Try-Pro) motif of MBD5 helps it bind to the methylated histones. The SET domain provides SETDB1 and SETDB2 the ability to act as protein methyltransferase and the Tudor domains help SETDB1 to bind to methylated histones. In addition, these proteins also have PreSET domain located N-terminus to the SET domain that functions in stabilizing the SET domain. Both BAZ2A and BAZ2B contains a PHD domain to bind unmodified histone while bromodomain and DDT domains allow them to recognize the acetylated histones and DNA binding abilities respectively. (Figure not drawn to the exact scale of the proteins).
MeCP2
The MeCP2 is a 50-kD multidomain transcriptional repressor protein encoded from an X chromosome gene containing four exons that give rise to two isoforms MECP2_e1 and MeCP2_e2 depending on the presence or absence of the second exon (103). These isoforms provide different functionalities to each of these proteins.
The MBD domain of MeCP2 can bind to a single methylated CpG. However, the binding of MBD requires an A/T-rich sequence near the methylated CpG regions (104). Apart from the MBD domain, MeCP2 also has the transcription repression domain (TRD) involved in mediating gene silencing through the recruitment of chromatin remodeling complex comprising the Sin3A co-repressor and histone deacetylases (HDAC1 and HDAC2) (11) (Figure 2). Some of the other functions of the MeCP2 protein include nuclear organization, chromatin compaction and fiber binding, chromatin looping, rearrangement of heterochromatin, regulation of splicing, and DNA methylation (105). Mutations of the MECP2 gene may cause a severe neurodegenerative disease known as the Rett syndrome that mainly affects females due to the presence of the gene on the X chromosome (106).
MeCP2 is also involved in cancer through it's binding to the hypermethylated regions of promoters of tumor suppressor genes and thereby cause their subsequent repression in breast cancer (107), prostate cancer (108), lung cancer (109), liver cancer (110), and colorectal cancer (111). Elevated expression of the MeCP2 gene has been reported in different cancers (28, 29, 112). In the case of liver cancer cells, MeCP2 promotes proliferation via the activation of ERK1/2 with simultaneous inhibition of p38 activity (30). Increased MeCP2 expression has shown an association with shorter overall survival and disease-free survival of gastric cancer patients (29). Loss of function of MeCP2 has been reported to inhibit cell proliferation and increased apoptosis of prostate cancer cells in vitro (26, 27). In addition, treatment with several natural compounds has shown to downregulate the elevated MeCP2 expression in prostate and breast cancer cells in vitro (113, 114).
MBD1
MBD1 is a multidomain protein consisting of an N-terminal MBD domain that can bind methylated DNA, a C-terminal TRD domain that can mediate protein-protein interaction, and two or three CXXC-type Zinc fingers in between MBD and TRD domains (103). It is encoded from a multiexon gene located on chromosome 18. MBD1 has 13 isoforms due to alternative splicing of the gene (13). The major distinction between the encoded proteins from the differentially spliced isoforms is the retention of two or three CXXC-type Zinc finger motifs (115). The isoforms having the first two CXXC-type Zinc finger motifs can bind to methylated CpG to cause transcriptional repression, whereas the isoforms having the third CXXC-type Zinc finger motif has the ability to bind unmethylated DNA (115, 116). This implies that the isoforms having all three CXXC-type Zinc finger motifs can cause transcriptional repression regardless of the DNA methylation status.
MBD1 interacts with MBD1-containing chromatin-associated factor 1 (MCAF1) and histone methyltransferase SETDB1 to form MBD1:SETDB1:MCAF1 complex that methylates lysine residue 9 of histone H3 to promote heterochromatin formation (117–119). MBD1 can also interact with another histone methyltransferase known as Suv39h and cause H3K9 methylation. Mbd1 knockout mice (Mbd1−/−) are viable and healthy but show some defects during neural stem cell differentiation along with increased chromosome instability (120).
MBD1 plays a role in tumorigenesis by repressing tumor suppressor genes like CDH1, RASSF1A, TIMP3, P14ARF, and Rb (121). In human pancreatic carcinomas, elevated expression of MBD1 showed association with lymph node metastasis (32). Furthermore, knockdown of MBD1 inhibited cell proliferation, invasion, and increased apoptosis of pancreatic cancer cells (122, 123). However, the oncogenic role of MBD1 is not typical for all cancers. For example, MBD1 may also act as a tumor suppressor in colorectal cancer (31). A hypermethylation-mediated downregulation of the MBD1 gene is seen along with the progression of metastatic colorectal cancer (31). Moreover, polymorphisms in the MBD1 gene have shown an association with the increased risk of developing lung cancer (33). In prostate cancer cells, depletion of MBD1 increased cell invasion with no change in apoptosis compared to control cells where MBD1 expression was intact (27). Moreover, analysis of patient biopsies revealed that the MBD1 protein expression gradually decreased with the increase of prostate cancer grade (34). Taken together, these observations indicate a dual role of MBD1 both as a tumor suppressor and oncogene depending on the type of cancer.
MBD2
The MBD2 is a multiexon gene located on chromosome 18 of both human and mouse genomes. The encoded protein (MBD2) from this locus shows more than 70% amino acid sequence similarity with another protein called MBD3 (23). In addition, a high level of gene-sequence homology exists between human and mouse MBD2 and MBD3 genes which is suggestive a gene duplication event during the course of evolution (102). Initial studies proposed that MBD2 functions as a transcriptional repressor by recruiting co-repressors like the NuRD (Nucleosome Remodeling Deacetylase) complex to the methylated sites (19).
MBD2 protein has three main isoforms due to the use of alternative translational start site and alternative splicing: MBD2a, MBD2b, and MBD2c (also called MBD2t) (23, 124). The presence of different domains which give the different MBD2 isoforms the ability to interact with different binding partners and thereby carry out different functions (Figure 3). However, all three isoforms have the MBD domain to bind to methylated CpG. MBD2a is the canonical isoform and contains an N-terminal glycine-arginine (GR) repeat that can undergo post-translational modification, followed by the MBD domain, the TRD domain, and lastly the coiled-coil (CC) domain at the c-terminal region that has the ability to mediate protein-protein interactions (12, 125). The MBD2b uses an alternative start site during translation, and the only difference from MBD2a is the absence of the N-terminal GR repeat. The presence of the MBD and TRD domain in both of these isoforms helps them to bind different corepressor complexes to mediate transcriptional repression (126). The third isoform MBD2c is devoid of the TRD and CC domains due to the inclusion of an alternative exon 3 that produces a truncated protein (23). The MBD2c may function differently than the other isoforms. For example, in human pluripotent stem cells (hPSC), MBD2a interacts with NuRD to promote cell differentiation while MBD2c mediates the reprogramming to pluripotency (127).
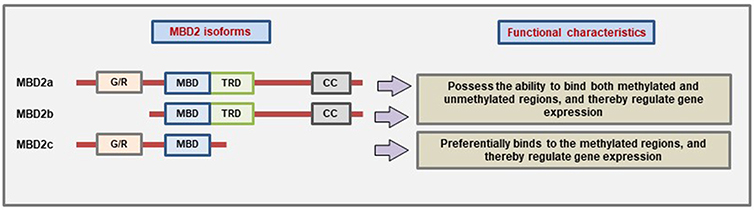
Figure 3. Characteristic domain architecture and function of different MBD2 isoforms. MBD2a is the canonical isoform containing four domains while MBD2b lacks the N-terminal G/R-repeat due to the use of alternative start site during translation. On the other hand, the MBD2c isoform is formed due to the inclusion of an alternative third exon which produces a premature stop codon, and as a result, the MBD2c lacks the TRD and CC domains (Figure not drawn to the exact scale of the proteins).
It has been shown that MBD2 can also bind to unmethylated DNA to cause changes in gene expression (128). However, the TRD-domain deficient MBD2c isoform cannot bind to unmethylated DNA which suggests that MBD2 binding to the unmethylated regions of the DNA is dependent on the interaction between TRD domain and NuRD complex (128). More recent evidence suggests that the MBD2 protein can also mediate the activation of gene expression (129–131). MBD2 has been proposed to function as a demethylase enzyme that can remove or “erase” the DNA methylation marks (132). However, this finding has been contested by several others (126, 133). The Mbd2 knockout mice (Mbd2−/−) are viable and do not show any abnormalities during embryonic development even though the female Mbd2−/− mice have been reported to show some abnormalities related to maternal behavior (134).
MBD2 plays an important role in cancer by silencing key tumor suppressor genes in prostate cancer (40), colon cancer (37), and liver cancer (38). On the other hand, in several cancer-types, MBD2 has been shown to mediate transcriptional repression of human telomerase reverse transcriptase (hTERT) which is suggestive of a tumor suppressive function of MBD2 (135). In breast cancer, Müller et al. could not detect any discernable difference in MBD2 expression (28), while Billard et al. detected a statistically significant upregulation of MBD2 in the mammary tumor (136).
Stable knockdown of the MBD2 gene suppressed the proliferation of several breast cancer cell lines in vitro and decreased tumor volume in vivo (36). Furthermore, it was demonstrated that tumor suppressor genes like DAPK1 and KLK10 are de-repressed upon depletion of MBD2 in breast cancer cells. Interestingly, MBD2 has been also shown to function in the maintenance and spread of DNA methylation at specific regulatory regions of prostate cancer cells (137). Moreover, knockout of Mbd2 gene mice protected against tumorigenesis when crossed with ApcMin/+ mice (a rodent model for colorectal cancer) (35). The loss of MBD2 function causes downregulation of the Wnt signaling pathway which plays a major role in the development of colorectal cancer (35, 138). However, later studies have found that depletion of several other epigenetic and chromatin binding factors like Kaiso, DNMTs, and Brg1 also downregulated the Wnt signaling pathway and thereby protected from tumorigenesis (56, 139, 140). This suggests that the downregulation of Wnt signaling is not MBD2-specific, and it is instead a result of a general perturbation of chromatin remodeling complex (8).
Emerging evidence supports that MBD2 plays a role in immunity partly because of its tissue localization pattern. Among the various members of the MeCP2-MBD family, MBD2 shows the highest expression in spleen which is a major site for both adaptive and innate immune responses (8, 141). Wang et al. demonstrated that MBD2 regulates the expression of Foxp3 which is the master regulator of regulatory T cells (Tregs) (129). They have shown that MBD2 binds to the Treg-specific demethylation region (TSDR) located upstream of the Foxp3 gene and thereby facilitates TET2-mediated demethylation to induce Foxp3 expression. Furthermore, knockout of Mbd2 gene reduced the number of Tregs and impaired the immunosuppressive function meditated by the Tregs. Interestingly, the Mbd2−/− mice did not develop autoimmunity which makes it an attractive target in pathological conditions like cancer where the Tregs have been suggested as potential targets for immunotherapy (142).
Even though MBD2 is an attractive anti-cancer target, drugs that can specifically target MBD2 have not been identified to date. Our group has previously shown that the treatment of cancer cells with the universal methyl group donor S-adenosylmethionine (SAM) downregulates MBD2 expression and shows anti-proliferative and anti-metastatic effects both in vitro and in vivo (143, 144). Moreover, antisense oligonucleotides against MBD2 gene also showed promising anti-cancer effects in xenograft models (145). However, caution must be taken while targeting MBD2 to avoid any potential negative ramification (13).
MBD3
MBD3 is encoded from a multiexon gene located on chromosome 19 that produces three isoforms: MBD3a, MBD3b, and MBD3c (23, 146). As mentioned before, MBD3 has a high degree of protein sequence similarity with MBD2 but lacks the GR-rich domain found in the full-length canonical isoform of MBD2a. Due to the presence of a point mutation in the region coding for the MBD domain, the selective binding affinity for methylated CpG is abolished in case of MBD3 protein (8, 147). However, MBD3 has been shown to preferentially bind to 5-hydroxymethylated (5 hmC) CpGs in vitro (148). Furthermore, MBD3 colocalizes with the DNA demethylase TET1 in vivo and is required maintenance of 5 hmC (148). MBD3 is a component of NuRD repressive complex and causes a transcriptional repression of genes (149). The association of MBD3 and MBD2 with NuRD complex is mutually exclusive which is indicative of different functional properties (149). The MBD3-NuRD complex plays a role in pluripotency and differentiation of embryonic stem cells (146, 148). Moreover, knockout of Mbd3 gene is embryonic lethal (134).
MBD3 has been shown to function as a tumor suppressor. In clinical specimens obtained from pancreatic cancer patients, decreased MBD3 gene expression showed an association with poor patient survival (42). A similar observation was noted in malignant glioma where patient samples with reduced MBD3 expression showed a correlation with decreased overall survival and progression-free survival (43). Moreover, knockdown of MBD3 gene promoted cancer cell migration and invasion while overexpression inhibited those effects (42). In liver cancer, the MBD3-NuRD complex inhibited the induction of cancer stem cells (CSC) whereas knockdown of MBD3 upregulated the expression of CSC-related genes (41). In addition, knockdown of MBD3 also increased the c-Jun protein expression (41). Other studies have shown that N-terminal phosphorylation of c-Jun inhibits the recruitment of MBD3-NuRD complex and thereby causes the upregulation of tumorigenesis promoting genes (150). While anti-cancer targeting of c-Jun may serve as a viable and attractive strategy to induce MBD3 expression, further research is needed in this regard.
MBD4
MBD4 is encoded from a gene located on chromosome 3 and functions in DNA repair mechanism. The N-terminal MBD domain allows it to bind to the methylated DNA. However, the unique feature of the protein lies at the C-terminal glycosylase domain that makes it the only MBP to have DNA glycosylase activity to repair mismatches of hypermutable CpG (151). Spontaneous deamination of 5-methylcytosine (5 mC) is one of the primary sources of somatic mutation that the body accumulates with time (45), and MBD4 protects from such mutations by interacting with mutL homolog 1 (MLH1) to trigger the DNA repair mechanism (152).
The homozygous Mbd4 gene knockout mice are viable and do not show any defective phenotype (153). When these knockout mice were crossed with the rodent model of colorectal cancer (ApcMin/+ mice), there was an increase in tumorigenesis (153). Mutations in the MBD4 gene has also been observed in human colorectal and gastric cancer patients that trigger mismatch repair deficiency (44, 46). A recent study has shown that germline mutation of MBD4 increases the susceptibility to develop acute myeloid leukemia (AML) (45). Taken together these studies support a tumor suppressive function of MBD4 in cancer even though more research is needed to elucidate whether this effect is common for all cancer or specific for certain types of cancer.
MBD5 and MBD6
The MBD5 and MBD6 proteins are encoded from their respective genes located on the chromosomes 2 and 6. They were initially identified through protein homology search and classified as MBD proteins due to the presence of the MBD domain (154). They still remain the least functionally characterized members of the MeCP2-MBD subfamily of proteins. It has been shown that both MBD5 and MBD6 localize to the heterochromatin in vitro and cannot bind to the methylated DNA (155). Both these proteins contain a proline-rich domain that can mediate protein-protein interactions during intracellular signaling. In addition, MBD5 has a C-terminal PWWP (Pro-Try-Try-Pro) motif that may bind to the methylated histones.
MBD5 and MBD6 can both interact with the mammalian polycomb repressive complex PR-DUB that can cause deubiquitination of different cellular targets (156). They are highly expressed in the brain and testis, which is suggestive of their possible role in development (155, 157, 158). Indeed, abnormalities in the MBD5 gene has been reported in patients with mental retardation (159, 160). Moreover, the Mbd5 knockout mice showed deficiencies in postnatal growth and aberrations in glucose homeostasis (161). The MBD6 protein has been reported to be involved in cellular stemness by regulating the activity of Oct4 (octamer-binding transcription factor 4) (162). Whether these proteins have any role in cancer, is still not clear. Mutation and abnormal expression of the MBD6 gene have been reported in gastric and colorectal cancers (47). Further studies are needed to know whether these genes are implicated in cancer.
HMT-MBD
This subgroup of MBD-containing proteins consists of two members: SET domain bifurcated 1, SETDB1 (also known as KMT1E or ESET) and SETDB2 (also known as CLLD8). Apart from having the MBD domain, these proteins also have the SET (SuVar3-9, enhancer of Zeste, Trithorax) domain for interacting with other proteins (Figure 2). Both SETDB1 and SETDB2 have the protein lysine methyltransferase activity which enables them to cause transcriptional repression (163, 164). The exact molecular mechanism on how SETDB2 mediates gene repression is still not clear (52). However, the mechanism of SETDB1 mediated transcriptional repression is known. Briefly, the SETDB1 protein interacts with MBD1 which directs it to the methylated CpG region. Then SETDB1, through its methyltransferase activity, mediates trimethylation of histone H3 on lysine 9 (H3K9me3) to cause heterochromatin formation and thereby repress transcription. Both SETDB1 and SETDB2 are involved in embryonic development (165, 166).
Emerging evidence supports that SETDB1 functions as an oncogene in many cancers that include sporadic cutaneous melanoma (49), liver cancer (50), and colorectal cancer (48). It has been shown that SETDB1 can directly interact with the de novo DNA methyltransferase DNMT3A in order to attach at the promoter region and mediate the transcriptional silencing of well-known tumor suppressor genes RASSF1A (in breast cancer cells) and P53BP2 (in cervical cancer cells) (167). Moreover, knockdown of SETDB1 led to the suppression of breast cancer proliferation and migration in vitro, and also inhibited tumorigenesis in vivo (51).
The gene encoding for the SETDB2 protein was initially identified as a potential candidate for leukemogenesis (168). Elevated expression of SETDB2 has shown association with gastric cancer progression (52). Further studies are warranted to know whether the oncogenic characteristics of SETDB1 and SETDB2 are universal for all cancers or just specific to particular cancer type.
HAT-MBD
This subgroup also consists of two members: BAZ2A (bromodomain adjacent to zinc finger domain 2A, also known as TIP5) and BAZ2B. Apart from the MBD domain, they also contain the DDT (DNA binding homeobox and Different Transcription factors), PHD (Plant homeodomain), and bromodomains (Figure 2). The PHD domain helps to bind to the unmodified histone, and the bromodomain helps to recognize the acetylated lysine residues of a protein (169–171). These proteins have histone acetyltransferase activities and are involved in chromatin remodeling (13). The MBD domain of these proteins cannot bind to the oligonucleotides that are methylated (13). However, the MBD domain of BAZ2A binds to unmethylated DNA in vitro (172).
Not much is known about the involvement of these proteins in cancer. The BAZ2A protein showed an association with the epigenetic alteration in prostate cancer patients, and its overexpression has been suggested to be an individual biomarker to predict disease recurrence (53). Further studies are needed to know the exact roles of these proteins in cancer.
Methyl-CpG Binding Zinc Finger Proteins
The members of this family of MBPs have Zinc finger motifs at the C-terminal region which allow them to bind both methylated and unmethylated DNA. The initially identified member of the family is Kaiso whose methylated DNA-binding ability was demonstrated by the labs of Egor Prokhortchouk and Adrian Bird in 2001 (24). Later on, two homologs of the Kaiso proteins, Zinc-finger and BTB domain containing 4 (ZBTB4) and ZBTB38 were discovered (173). Like Kaiso, ZBTB4 and ZBTB38 both have the N-terminal broad-complex, tramtrack, and bric-a-brac/poxvirus and zinc finger (BTB/POZ) domain (Figure 4). For several years, this branch of MBPs only comprised of these three known members. More recently, several other members (ZFP57, KLF4, WT1, EGR1, and CTCF) of this branch have been identified (25). These proteins have the Zinc finger motifs in the C-terminal region for binding to the methylated DNA. However, the N-terminal domain is not similar to Kaiso, ZBTB4, and ZBTB38 (Figure 4). So, according to the current literature, there are at least 8 members in this family of MBPs (25).
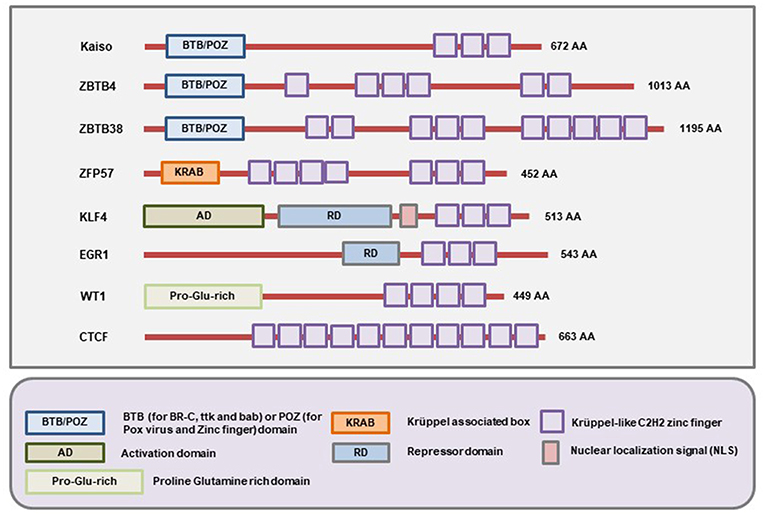
Figure 4. Schematic representation of the domains present in different Methyl-CpG Binding Zinc finger proteins. All members of this family contain C-terminal Zinc finger motifs that allow them to bind to both methylated and unmethylated DNA. In addition, Kaiso, ZBTB4, and ZBTB38 contain the BTB/POZ domain while ZFP57, WT1, EGR1 contain the Krüppel associated box (KRAB), Proline-Glutamine rich (Pro-Glu-rich), and repressor domains (RD) respectively. On the other hand, the KLF4 protein contains an activation domain (AD), repressor domain (RD), and an NLS (nuclear localization signal/sequence) apart from the Zinc fingers (Figure not drawn to the exact scale of the proteins).
Kaiso
Kaiso is a transcription factor that was initially identified as the cytoplasmic binding partner of p120-catenin (174). It is encoded by ZBTB33 gene located on the X chromosome of the human genome (175). Its ability to bind a pair of methylated CpG dinucleotides was demonstrated experimentally (24). Kaiso can also bind to unmethylated DNA at Kaiso binding sites (KBS) with a consensus sequence of TCCTGCNA, where N is any nucleotide (175). The Kaiso binding affinity at KBS is higher than that of methylated CpG dinucleotides. Qin et al. have further demonstrated that Kaiso cannot bind the hydroxymethylated CpG dinucleotides (176).
Kaiso plays roles both at the cell surface and the nucleus. At the cell surface, it plays roles in cell adhesion as well as signal transmission by regulating the stability of the cadherins (177). On the other hand, in the nucleus, it directs the Nuclear Receptor Corepressor Complex (N-CoR) to both methylated and unmethylated regions on the DNA and thereby promotes a repressive chromatin state (178). In general, Kaiso is considered as a transcriptional repressor even though it can also bind to the unmethylated regions of actively expressed genes (179). Kaiso also localizes to the centrosomes and mitotic spindles during cell cycle progression (180).
Kaiso expression and localization has shown association with different types of cancer that include lung (57), prostate (54), breast (58, 59), and colon (55). Kaiso causes transcriptional repression of several well-known tumor suppressor genes like Retinoblastoma (Rb), Hypermethylated In Cancer 1 (HIC1) and Cyclin-Dependent Kinase Inhibitor 1A (CDKN1A) (55, 181). Depletion of Kaiso inhibited breast cell proliferation and survival by increasing apoptotic cell death (58). Moreover, Kaiso-deficient mice crossed with Apc(Min/+) mice have shown lower susceptibility of developing intestinal tumors compared to the control mice where the gene encoding for the Kaiso protein was not knocked out (56).
Since Kaiso binds to both methylated and unmethylated DNA, it is difficult to understand how crucial a role its methylated CpG binding property plays in cancer. For example, Kaiso can cause transcriptional repression of unmethylated Matrix Metallopeptidase 7 (MMP7) and Cyclin D1 (CCND1) genes as well as methylated Metastasis Associated 1 family member 2 (MTA2) gene in lung cancer (182). In this case, all three genes have roles in cancer progression and, therefore, further research is needed to pinpoint the precise role of Kaiso's methylated CpG binding property in cancer.
Even though Kaiso's role as a potent repressor is now well-established and Kaiso-deficient mice are viable, therapeutic agents to inhibit Kaiso have not been developed so far. Through computer-aided drug designing methods, it has been shown that a natural compound (chem. ID 28127) can strongly inhibit Kaiso (183). However, detailed experimental evidence is needed in this regard.
ZBTB4
ZTBT4 can bind to a single methylated CpG which is different from Kaiso that needs two methylated CpG dinucleotides. Moreover, ZBTB4 can also bind to the unmethylated KBS (173). This makes ZBTB4 a bimodal protein that can bind to both methylated and unmethylated DNA. However, the ZBTB4 binding affinity for methylated CpG is higher than that of KBS. ZBTB4 can function as a transcriptional repressor by recruiting Sin3/histone deacetylases and thereby silence the expression of the target gene (61). Furthermore, ZBTB4 has been shown to localize at the highly methylated centromeric and pericentromeric repeats (173, 184). This localization is abolished in the cells that lack methylation of DNA.
Downregulation of ZBTB4 has been found in several common malignancies that include neuroblastoma (61), breast (60) and prostate cancer (62). Decreased levels of ZBTB4 in the tumor correlated with higher genomic instability (63). In breast cancer patients, ZBTB4 protein and mRNA are both downregulated, and its expression showed an association with relapse-free survival (60). Elevated levels of ZBTB4 also correlated with longer survival of prostate cancer patients (62). ZBTB4 is involved in the crosstalk between DNA methylation and histone modification. It has been shown that ZBTB4 causes transcriptional silencing of specificity protein (Sp) transcription factors (Sp1, Sp3, and Sp4) that are required for histone methyltransferase EZH2 expression (185). EZH2 is currently considered a target for anti-cancer therapy since it has a role in cell proliferation and cell division. ZBTB4 also causes repression of key angiogenesis factors, vascular endothelial growth factor (VEGF) and its receptor (VEGFR) (60). The Zbtb4-deficient mice showed more susceptibility to carcinogen-induced skin cancer as compared to the wild-type counterpart (63). Taken together, these studies indicate a tumor-suppressive role of ZBTB4 in cancer. However, more studies are needed as enough literature is not available on this protein.
ZBTB38
ZBTB38 can also bind to a single methylated CpG. It is also a bimodal protein that can bind to both methylated and unmethylated DNA. The ZBTB38 gene that encodes the protein is located on chromosome 3. ZBTB38 plays roles in various cellular processes that include cell proliferation (186) and differentiation (187), control of DNA replication and genomic stability (188) as well as regulation of gene expression (173).
Results from a genome-wide association study (GWAS) done on prostate cancer patients revealed that polymorphisms in ZBTB38 gene increase the risk of developing prostate cancer in men (64). In bladder cancer cells, ZBTB38 increased cell migration, invasion, and metastasis via regulating genes belonging to the Wnt/β-catenin signaling pathway but decreased bladder cancer cell proliferation (65). Depletion of ZBTB38 increases the cytotoxic ability of DNMT inhibitors in different solid and hematological cancer cell lines by increasing the expression of Cyclin-Dependent Kinase Inhibitor 1C (CDKN1C) which opens up a new avenue to increase the therapeutic response to DNMT inhibitors (189).
ZFP57
This protein is encoded from the ZFP57 gene located on chromosome 6. In addition to the Zinc finger domain, this protein also has an N-terminal Krueppel-associated box (KRAB) domain. ZFP57 preferentially binds to methylated CpGs within the TGCCGC hexanucleotide (190), and its primary functions include genome imprinting (191), regulation of gene expression, and cell signaling (192). Since it functions during early embryogenesis and is downregulated upon differentiation (193), it is also categorized as a stem cell transcription factor.
Ectopic expression of ZFP57 enhanced the anchorage-independent growth properties of human fibrosarcoma HT1080 cells via the regulation of insulin-like growth factor 2 (IGF2) expression (194). In addition, knockdown of ZFP57 gene caused suppression of HT1080 tumor formation while overexpression of the gene enhanced tumor formation in immunocompromised mice (194). The ZFP57 gene expression has been shown to be elevated in patients with high-grade glioblastoma (66). Moreover, a recent GWAS revealed that ZFP57 is a potential disease susceptibility gene for lung cancer development (67). Previous studies have shown that elevated IGF2 level is associated with a reduced survival rate in lung cancer (195). Since ZFP57 regulates IGF2 expression, this axis may well be a potential target for lung cancer treatment.
KLF4
The Krüppel-like factor 4 (KLF4) is a 55 kD protein that has been demonstrated to function as one of the four factors needed for the generation of iPS (induced pluripotent stem) cells (196). The other functions of the protein include regulation of cell cycle (197), cell proliferation (198), DNA damage response (199), genomic stability (200), and apoptosis (201). KLF4 can recognize methylated CpG as well as unmethylated CpG or TpG within a specific DNA sequence (202, 203). It has been demonstrated that KLF4 binding to certain methylated CpGs attracts the recruitment of chromatin remodeling complex and thereby cause transcriptional activation (73), which is a paradigm shift from the common notion that DNA methylation readers mainly function as transcriptional repressors.
Both tumor suppressive and oncogenic function of KLF4 has been reported (69, 71, 72). KLF4 expression is downregulated in several malignancies like gastric cancer (68), colorectal cancer (69), and bladder cancer (70) where it mainly functions as a tumor suppressor gene. It has been shown that transduction of the KLF4 gene using adenoviral vectors decreased proliferation and induced apoptosis of bladder cancer cells (70). As mentioned above, KLF4 also functions as an oncogene in some cancers. For example, both mRNA and protein levels of KLF4 are elevated during breast cancer progression (71). Knockdown of KLF4 gene suppressed breast cancer cell migration, invasion, and colony formation in vitro and inhibited tumorigenesis in immunocompromised mice (72). KLF4 promotes cell adhesion and migration in glioblastoma cells (73). In addition, increased KLF4 has shown association with the progression and metastasis of human skin squamous cell carcinoma (74). In oral squamous cell carcinoma, KLF4 showed dual functionality as a tumor suppressor as well as an oncogene (204). Taken together, these observations indicate a context-dependent role of KLF4 in cancer.
EGR1
The Early growth response protein 1, EGR1 (also known as ZIF268 or NGFI-A or KROX24) is a transcription factor that is induced following exposure to different cellular and external stimuli or stress signals (205, 206). The EGR1 gene, located on chromosome 5, belongs to the category of immediate early gene and the encoded protein from this locus function in a plethora of cellular processes that include maintenance of synaptic plasticity (207), wound healing (208), inflammation (209), and differentiation (210). It recognizes and binds to the GCC(T/G)GGGCG consensus sequence near the target gene promoter regardless of the CpG methylation status (211, 212). In addition, the EGR1 binding affinity for cytosine (C) or 5 mC is much higher than that of 5-hydroxymethylcytosine (5 hmC) or 5-formylcytosine (5fC) (212). This implies that EGR1 can differentiate between oxidized and unoxidized C but not between methylated and unmethylated C moiety.
EGR1 expression is altered in several cancers. It functions both as a tumor suppressor and an oncogene depending on the type of cancer. The tumor-promoting effect of EGR1 has been demonstrated in Wilms' tumor (77) and prostate cancer (75). In contrast, it acts as a tumor suppressor in glioblastoma (80), fibrosarcoma (81), breast (78, 79), and lung cancer (82). Moreover, both male and female homozygous knockout (Egr1−/−) mouse cannot reproduce (213, 214). So careful considerations should be taken before pharmacologically targeting EGR1.
WT1
This protein is encoded by the Wilms' tumor 1 (WT1) gene located on chromosome 11. It was initially identified as a tumor suppressor gene that was inhibited in the most common form of pediatric kidney cancer called the Wilms' tumor 1 (83). Hence, it was named after that specific tumor even though later studies have demonstrated its presence in several other tissues and malignancies. It plays roles in the regulation of cell growth (215), differentiation (216), cell cycle (217), cell division and maintenance of genome stability (218). Like EGR1, WT1 also recognizes the GCC(T/G)GGGCG consensus sequence near the promoter of target genes (212). However, EGR1 and WT1 have differential binding sensitivity to the oxidative derivatives of 5mC (212).
WT1 may function either as a tumor suppressor or an oncogene depending on the type of cancer. Higher expression of WT1 has shown association with the poor prognosis of breast cancer (85), ovarian cancer (88, 89), leukemia (86), and head and neck cancer (87). In contrast, knockdown of WT1 gene increased apoptosis of different cancer cell lines (219, 220). Moreover, WT1 has been ranked at the top position in the National Cancer Institute's (NCI) list of cancer antigens with the highest prioritization for vaccine development (221). The use of peptide vaccines against the WT1 antigen showed beneficial therapeutic outcomes in several clinical trials (222, 223).
CTCF
The CCCTC-binding factor (CTCF) is a well-known transcription factor that contains 11 highly conserved Zinc finger domains which allow it to bind at different locations on the genome (224) (Figure 4). One of the main functions of the CTCF protein is to act as a barrier/insulator to inhibit the interaction between the promoter and enhancer region (225). Other functions of CTCF include cellular context-dependent regulation of chromatin architecture, RNA splicing, and gene expression (225, 226). The DNA binding consensus sequence of CTCF contains CpG (227). It can bind to methylated DNA even though the binding preference for unmethylated DNA is much higher (228). For example, CTCF provides epigenetic stability to the retinoblastoma (RB) gene by binding at a specific region of the promoter sequence and thereby protecting the site from undergoing epigenetic silencing (181). Interestingly, CpG methylation at this particular region of the RB gene promoter leads to the loss of CTCF binding and promotes the binding of another methylation reader Kaiso (181). In this context, CTCF can be defined as a reader protein that can read methylated CpG but not necessarily binds to it.
CTCF can act as both tumor suppressor and oncogene depending on the type of cancer (229, 230). In ovarian cancer, CTCF expression is upregulated and shows association with poor prognosis (231). Higher expression CTCF in breast cancer cells has been shown to provide a survival advantage by inhibiting apoptosis (232). Depletion of the CTCF gene decreased proliferation and induced apoptosis of breast cancer cells (229). However, given that it plays a crucial role in the maintenance of chromatin architecture, targeting the downstream effectors of CTCF may serve as a more feasible option than targeting the protein itself.
SRA Domain-Containing Proteins
The SRA family of methylation readers is comprised of two members: Ubiquitin-like with PHD and RING Finger domains 1 (UHRF1) and UHRF2. Both of these proteins contain at least five distinct functional domains that include the Ubiquitin-like Domain (UBL) (also known as the NIRF_N domain according to the NCBI Conserved Domain Database), Tandem Tudor Domain (TTD), Plant Homeodomain (PHD), SRA domain, and Really Interesting New Gene (RING) domain all of which confer tremendous functional complexities to these proteins (Figure 5). The N-terminal UBL domain and C-terminal RING domains are involved in the ubiquitin-proteasome system, the TTD and PHD domains can bind to the methylated histone proteins, and the SRA domain binds to the methylated CpG (233). The SRA domain is distinct from MBD/MeCP2 in their ability to recognize and localize to methylated sites on DNA since they possess a higher binding affinity toward the hemimethylated regions on the DNA (234, 235) while the MBD/MeCP2 domains tend to bind at the symmetrically methylated DNA (236).
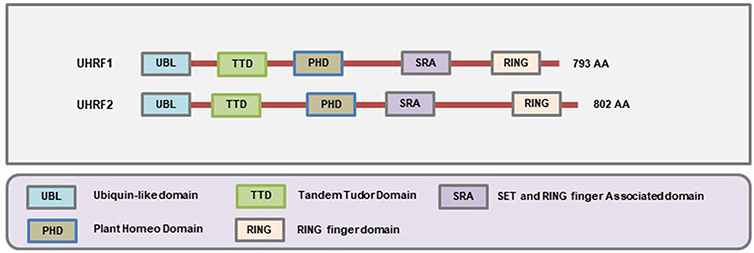
Figure 5. Schematic representation of the domains present in different SRA-domain containing proteins. The SRA domain allows them to bind to hemi-methylated DNA, the Ubl and RING domains are involved in ubiquitination, and the TTD and PHD domains allow to interact with the histones (Figure not drawn to the exact scale of the proteins).
UHRF1
The UHRF1 [also known as ICBP90 (human) or Np95 (mouse)] protein recognizes the hemimethylated DNA and subsequently recruits DNA methyltransferase 1 (DNMT1) to ensure that the sequence becomes faithfully methylated following replication (237, 238). It is a 793 amino acid containing protein encoded by the UHRF1 gene located on chromosome 19 of the human genome. It was initially identified during the screening of proteins that can bind to the inverted CCAAT box (ICB2) region of topoisomerase Iiα promoter (239). During the earlier studies, the human version of the protein was called ICBP90, and the mouse homolog was called Np95. Hence, there was some confusion in the nomenclature of the protein (240). At present, it is most commonly referred to as UHRF1.
The ability of the SRA domain of the UHRF1 protein to bind the methylated DNA was first reported by Unoki et al. in human breast cancer cells where an elevated expression of the protein showed association with hypermethylation-mediated downregulation of early growth response 2 (EGR2) gene (241). More recent evidence showed that UHRF1 acts as a linker or adaptor between DNA methylation and histone modification (242). The interaction between UHFR1 and histone marks plays a crucial role in recruiting DNMT1 to the daughter stands. The other functions of the protein include regulation of cell cycle (243), cell proliferation (244), DNA damage repair (245, 246), apoptosis (as an anti-apoptotic protein) (247) as well as the progression of tumors (248, 249). Aberrant expression of the UHRF1 gene has been found in different types of cancer like breast cancer (241), gastric cancer (95), hepatocellular carcinoma (250), renal cell carcinoma (251), squamous cell carcinoma (252), and osteosarcoma (253). Moreover, elevated levels of UHRF1 in the blood of breast and gastric cancer patients has been shown to provide diagnostic and prognostic value as an independent biomarker (254, 255). Since UHRF1 is overexpressed in almost all major types of cancer, its role as a universal biomarker for cancer has also been proposed (256).
At the molecular level, UHRF1 acts as a transcriptional repressor since it binds to and recruits HDAC1 to the methylated CpG sites near the promoters of tumor suppressor genes (257). For example, UHRF1 binding at the promoters of CDKN2A, RASSF1 (93), KiSS1 (258), and PAX1 (259) genes causes transcriptional repression of these genes. Loss of function studies showed that UHRF1 depletion decreased cancer cell proliferation and increased apoptotic cell death (260, 261). Depletion of UHRF1 also enhanced radio sensitivity of highly aggressive, triple negative MDA-MB-231 human breast cancer cells (262). Moreover, inoculation of UHRF1 depleted MKN45 gastric cancer cells into immunocompromised mice showed a marked reduction in tumor volume and weight compared to the control tumors where UHRF1 gene was not depleted (95). Polyphenols have shown promising effects in downregulating the expression of UHRF1 and thereby reduce tumor cell proliferation (263–265).
The UHRF1 protein also has some beneficial functions in cancer. It has been shown that UHRF1 directly binds the promoter region of the MDR1 gene (also known as ABCB1) and thereby represses its expression (266). The MDR1 gene encodes for P-glycoprotein (P-gp) which provides resistance to the cancer cells against various cytotoxic agents. It has been suggested that the UHRF1 mediated transcriptional repression of MDR1 has the potential to overcome multidrug resistance during the treatment of breast cancer (266). Therefore, caution should be maintained while targeting UHRF1 for cancer treatment.
UHRF2
The UHRF2 [also known as NIRF or Np97] was initially identified as a protein implicated during the regulation of cell cycle (267). It is an 802 amino acid containing protein encoded by UHRF2 gene located on chromosome 9 of the human genome. Even though it has a high degree of sequence similarity with UHRF1 at the amino acid level, it cannot functionally substitute UHRF1 in the maintenance of DNA methylation (268). In addition, structural studies have revealed that the SRA domain of UHRF2 protein but not UHRF1 preferentially binds to 5-hydroxymethylated DNA (5 hmc) (269). Therefore, UHRF2 has the unique ability to read and interpret at least three types of epigenetic marks that includes 5 hmC, 5 mC, and H3K9 methylation. It has been shown that UHRF2 can also enhance the enzymatic activity of Ten-eleven translocation methylcytosine dioxygenase 1 (TET1) (270). The expression of UHRF2 is higher in proliferating cells where PEST-containing nuclear protein (PCNP) acts as its substrate for ubiquitination (267, 271). Elevated expression of UHRF2 has been found in intrahepatic cholangiocarcinoma (99), colon cancer (98), breast cancer (100). Loss of function of UHRF2 decreased breast cancer cell proliferation while forced overexpression of UHRF2 gene into non-tumorigenic MCF10A breast cells induced cell proliferation (100). On the contrary, several groups have also reported on the tumor suppressor role of UHRF2 (101, 272). So, at present, it seems that UHRF2 plays roles as both oncogene and tumor suppressor depending on the cancer cell type. More detailed studies are warranted to establish its role in cancer and develop appropriate therapeutic strategies to attain clinically favorable outcomes.
EPI-Therapies Targeting The MBPs In Cancer
Epigenetic therapies (“Epi-therapies”) targeting the components of the epigenome are emerging as potential therapeutic modalities for many pathological conditions. In cancer, several “Epi-therapies” are already being approved by the Food and Drug Administration (FDA) for the treatment of several hematological cancers. These drugs are mainly the inhibitors of DNA methylation and histone deacetylation. In the case of DNA methylation inhibitors, the two approved drugs are Vidaza and Dacogen (Decitabine) (273). While these two inhibitors can reverse the hypermethylated state at the promoters of tumor suppressor genes, they also induce the activation of several prometastatic genes (274). Therefore, targeting the DNA methylation readers/MBPs may serve a suitable alternative for the next generations of precision Epi-therapies.
Even though the field is still at its infancy, several studies have shown promising effects in terms of developing anti-cancer therapeutic strategies against the MBPs. A summary of the currently described anti-cancer strategies targeting different known MBPs is shown in Figure 6.
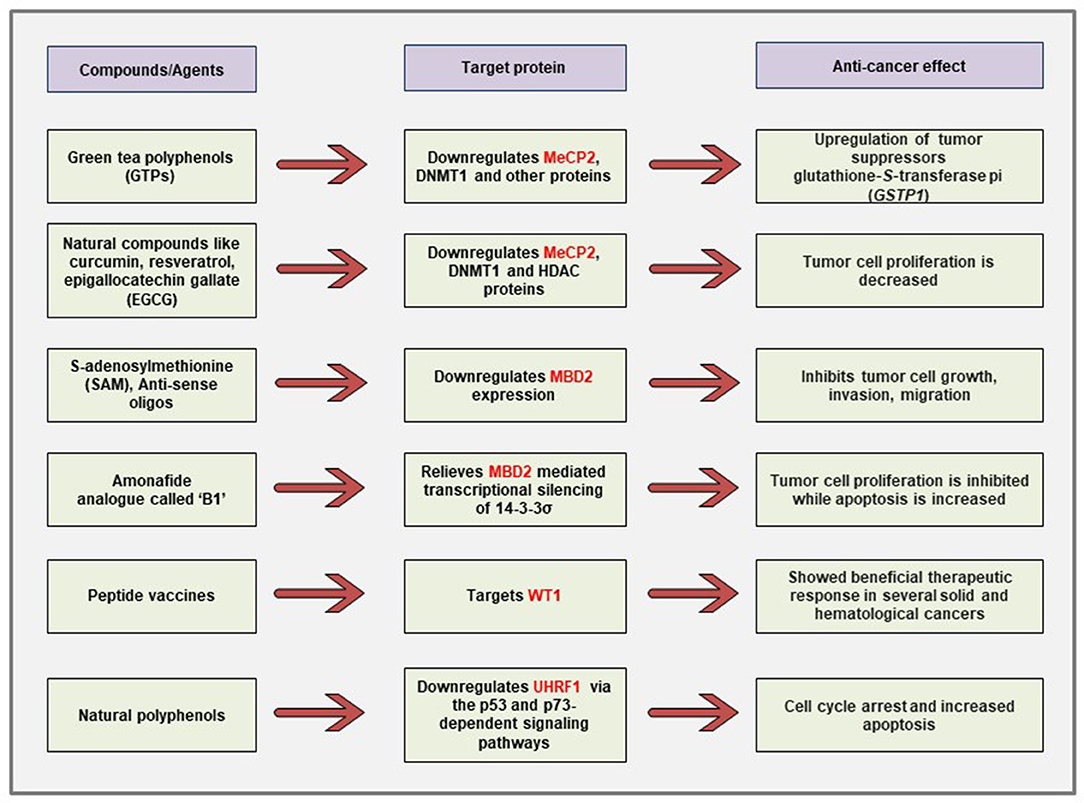
Figure 6. Schematic representation of the currently described anti-cancer strategies against different MBPs. Polyphenols obtained from natural compounds can downregulate the aberrantly expressed MeCP2, UHRF1 in cancer cells via differential regulation of cancer-related signaling pathways. The naturally occurring physiologic compound S-adenosylmethionine as well as anti-sense oligonucleotides can downregulate the elevated expression of MBD2 gene and cause inhibition of tumor growth, invasion, and metastasis. Immunotherapy against WT1 antigen has shown promising effects in clinical trials for several malignancies.
It has been demonstrated that treatment of prostate cancer cells with green tea polyphenols (GTPs) reversed the DNA hypermethylation-mediated silencing of the known tumor suppressor gene glutathione-S-transferase pi (GSTP1) through the downregulation of DNMT1, MeCP2, and several other MBD proteins (113). The authors have shown that GTP treatment causes demethylation at the promoter of GSTP1. Furthermore, chromatin immunoprecipitation (ChIP) assays revealed that GTP treatment also reduced the association of the transcriptional repressor MBD2 with Sp1 binding site that leads to the increased transcriptional activation of the GSTP1 gene. In other studies, it has been shown that natural compounds like curcumin, resveratrol, guggulsterone, EGCG, withaferin A, and genistein can also cause the reversal of epigenetic state in cancer cells through the reduction of DNMT1, HDAC1, and MeCP2 protein expression (114).
Interestingly, polyphenols obtained from natural products have also been shown to decrease cancer cell proliferation through the downregulation of UHRF1 predominantly via the p53 and p73-dependent signaling pathways (263–265). Limoniastrum guyonianum aqueous gall extract (G extract), as well as luteolin, independently inhibited proliferation of cervical cancer HeLa cells by arresting the cells in G2/M phase and induced apoptosis through the inhibition of UHRF1 along with the upregulation of p16 tumor suppressor (275). In a mouse model of colon cancer, red wine polyphenols (RWPs) inhibited tumor growth, metastasis, angiogenesis, and increased apoptosis through the downregulation of UHRF1 and other proliferation markers like ki67, cyclin D1 (276). The UHRF1 expression has also been shown to be downregulated in mechanisms independent of the p53 and p73 signaling pathways. For example, in chronic lymphocytic leukemia patients, polyphenols from Bilberry extract (Antho 50) decreased UHRF1 expression and increased apoptosis via targeting the Bcl-2/Bad pathway (277).
The expression of MBD2 gene was downregulated when the cancer cells were treated with the naturally occurring methyl group donor SAM that shows anti-proliferative and anti-metastatic effects (143, 144). This approach is particularly attractive because SAM is non-toxic to cancer cells and has been shown to cause downregulation of several other oncogenes and prometastatic genes without changing the expression of the known tumor suppressor genes in vitro and in vivo (278). Moreover, antisense oligonucleotides against MBD2 gene decreased tumorigenesis in human lung and colorectal cancer cells both in vitro and in vivo (145). In human promyelocytic leukemia cells, an amonafide analog named B1 [chemical name: N-(2-(dimethylamino)ethyl)-2-aminothiazonaphthalimide] has been demonstrated to cause relief from the MBD2-mediated repression of 14-3-3σ tumor suppressor gene (279). Moreover, KCC07, a brain-permeable small molecule inhibitor of the MBD2 pathway, have been demonstrated to suppress medulloblastoma in vivo through the activation of BAI1/p53 axis (280).
Cancer immunotherapies have shown great promise as therapeutic strategies in patients. There are several forms of immunotherapies that include the use of checkpoint inhibitors, monoclonal antibody therapies, and vaccine immunotherapies against the tumor-associated antigens (TAAs) (281). With the advancements of cancer immunology, several TAAs have been identified, and one of them is a WT1 product (282, 283). Indeed, immunotherapies against the WT1 antigen showed promising outcomes in clinical trials on patients with several solid and hematological cancers (222, 223, 281, 284).
Conclusion
This review summarized the roles of different MBPs based on the current literature with an aim to highlight their potential use as therapeutic targets and disease biomarkers. With the advances in the genome-wide binding studies, the specific functions of many MBPs are now clear even though more research is needed as some MBPs are still relatively less explored. In addition, the human proteome contains a wide array of Zinc finger motif-containing proteins, and recent evidence supports that many of these proteins may potentially bind to methylated DNA (5). However, in-depth experimental evidence is needed before classifying them as MBPs.
For the known family of MBPs, loss-of-function studies have aided to decipher their roles in different types of cancer. Since many of MBPs are cancer-type dependent, it opens a new avenue to develop targeted therapies against those subtypes. This will overcome the non-specificity issues related to currently approved epigenetic drugs like Decitabine and Vidaza, and thereby allow for a more specific approach to reduce cancer-associated morbidity and mortality. However, caution is warranted before the use of such targeted agents to avoid any potential negative ramification.
Author Contributions
NM wrote the initial draft of the manuscript. SR revised it to its final version. Both authors have made a substantial intellectual contribution and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the Canadian Institutes for Health Research (MOP 130410 and PJT-156225) to SR. NM is the recipient of a Ruth and Alex Dworkin Fellowship from the Faculty of Medicine, McGill University.
References
1. Jones PA, Laird PW. Cancer-epigenetics comes of age. Nat Genetics. (1999) 21:163. doi: 10.1038/5947
2. Mahmood N, Rabbani SA. DNA methylation and breast cancer: mechanistic and therapeutic applications. Trends Cancer Res. (2017) 12:1–18.
3. Prokhortchouk E, Hendrich B. Methyl-CpG binding proteins and cancer: are MeCpGs more important than MBDs? Oncogene. (2002) 21:5394. doi: 10.1038/sj.onc.1205631
4. Ballestar E, Esteller M. Methyl-CpG-binding proteins in cancer: blaming the DNA methylation messenger. Biochem Cell Biol. (2005) 83:374–84. doi: 10.1139/o05-035
5. Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet. (2016) 17:551–65. doi: 10.1038/nrg.2016.83
6. Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B, et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci. (2014) 17:215–22. doi: 10.1038/nn.3607
7. Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. (2009) 462:315–22. doi: 10.1038/nature08514
8. Wood KH, Zhou Z. Emerging molecular and biological functions of MBD2, a reader of DNA methylation. Front Genet. (2016) 7:93. doi: 10.3389/fgene.2016.00093
9. Schübeler D. Function and information content of DNA methylation. Nature. (2015) 517:321–26. doi: 10.1038/nature14192
10. Tate PH, Bird AP. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr Opin Genet Dev. (1993) 3:226–31. doi: 10.1016/0959-437X(93)90027-M
11. Nan X, Ng H-H, Johnson CA, Laherty CD, Turner BM, Eisenman RN, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. (1998) 393:386–9. doi: 10.1038/30764
12. Du Q, Luu P-L, Stirzaker C, Clark SJ. Methyl-CpG-binding domain proteins: readers of the epigenome. Epigenomics. (2015) 7:1051–73. doi: 10.2217/epi.15.39
13. Parry L, Clarke AR. The roles of the methyl-CpG binding proteins in cancer. Genes Cancer. (2011) 2:618–30. doi: 10.1177/1947601911418499
14. Hotchkiss RD. The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J Biol Chem. (1948) 175:315–32.
15. Holliday R, Pugh J. DNA modification mechanisms and gene activity during development. Science. (1975) 187:226–32. doi: 10.1126/science.1111098
16. Compere SJ, Palmiter RD. DNA methylation controls the inducibility of the mouse metallothionein-I gene in lymphoid cells. Cell. (1981) 25:233–40. doi: 10.1016/0092-8674(81)90248-8
17. Meehan RR, Lewis JD, McKay S, Kleiner EL, Bird AP. Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell. (1989) 58:499–507. doi: 10.1016/0092-8674(89)90430-3
18. Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, et al. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. (1992) 69:905–14. doi: 10.1016/0092-8674(92)90610-O
19. Ng H-H, Zhang Y, Hendrich B, Johnson CA, Turner BM, Erdjument-Bromage H, et al. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat Genetics. (1999) 23:58–61. doi: 10.1038/12659
20. Feng Q, Cao R, Xia L, Erdjument-Bromage H, Tempst P, Zhang Y. Identification and functional characterization of the p66/p68 components of the MeCP1 complex. Mol Cell Biol. (2002) 22:536–46. doi: 10.1128/MCB.22.2.536-546.2002
21. Feng Q, Zhang Y. The MeCP1 complex represses transcription through preferential binding, remodeling, and deacetylating methylated nucleosomes. Genes Dev. (2001) 15:827–32. doi: 10.1101/gad.876201
22. Nan X, Meehan RR, Bird A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. (1993) 21:4886–92. doi: 10.1093/nar/21.21.4886
23. Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl CpG-binding proteins. Genet Res. (1998) 72:59–72. doi: 10.1017/S0016672398533307
24. Prokhortchouk A, Hendrich B, Jørgensen H, Ruzov A, Wilm M, Georgiev G, et al. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. (2001) 15:1613–8. doi: 10.1101/gad.198501
25. Hudson NO, Buck-Koehntop BA. Zinc finger readers of methylated DNA. Molecules. (2018) 23:2555. doi: 10.3390/molecules23102555
26. Bernard D, Gil J, Dumont P, Rizzo S, Monte D, Quatannens B, et al. The methyl-CpG-binding protein MECP2 is required for prostate cancer cell growth. Oncogene. (2006) 25:1358–66. doi: 10.1038/sj.onc.1209179
27. Yaqinuddin A, Abbas F, Naqvi SZ, Bashir MU, Qazi R, Qureshi SA. Silencing of MBD1 and MeCP2 in prostate-cancer-derived PC3 cells produces differential gene expression profiles and cellular phenotypes. Biosci Rep. (2008) 28:319–26. doi: 10.1042/BSR20080032
28. Müller H, Fiegl H, Goebel G, Hubalek M, Widschwendter A, Müller-Holzner E, et al. MeCP2 and MBD2 expression in human neoplastic and non-neoplastic breast tissue and its association with oestrogen receptor status. Br J Cancer. (2003) 89:1934–9. doi: 10.1038/sj.bjc.6601392
29. Zhang J, Zhao J, Gao N, Wang Y, Chen Y, Han J. MECP2 expression in gastric cancer and its correlation with clinical pathological parameters. Medicine. (2017) 96:7691. doi: 10.1097/MD.0000000000007691
30. Trappe R, Laccone F, Cobilanschi J, Meins M, Huppke P, Hanefeld F, et al. MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am J Hum Genet. (2001) 68:1093–101. doi: 10.1086/320109
31. Qi L, Ding Y. Screening of Tumor Suppressor Genes in Metastatic Colorectal Cancer. BioMed Res Int. (2017) 2017:2769140. doi: 10.1155/2017/2769140
32. Di Y, Long J, Fu D, Yu X, Jin C, Ni Q, et al. The elevation of methyl-CpG binding domain protein 1 in pancreatic carcinoma and its significance. J Surg Concepts Pract. (2006) 11:35–8.
33. Liu H, Jin G, Wang H, Wu W, Liu Y, Qian J, et al. Methyl-CpG binding domain 1 gene polymorphisms and lung cancer risk in a Chinese population. Biomarkers. (2008) 13:607–17. doi: 10.1080/13547500802168031
34. Patra SK, Patra A, Zhao H, Carroll P, Dahiya R. Methyl-CpG–DNA binding proteins in human prostate cancer: expression of CXXC sequence containing MBD1 and repression of MBD2 and MeCP2. Biochem Biophys Res Commun. (2003) 302:759–66. doi: 10.1016/S0006-291X(03)00253-5
35. Sansom OJ, Berger J, Bishop SM, Hendrich B, Bird A, Clarke AR. Deficiency of Mbd2 suppresses intestinal tumorigenesis. Nat Gen. (2003) 34:145–7. doi: 10.1038/ng1155
36. Mian OY, Wang SZ, Zu Zhu S, Gnanapragasam MN, Graham L, Bear HD, et al. Methyl-binding domain protein 2–dependent proliferation and survival of breast cancer cells. Mol Cancer Res. (2011) 9:1152–62. doi: 10.1158/1541-7786.MCR-11-0252
37. Magdinier F, Wolffe AP. Selective association of the methyl-CpG binding protein MBD2 with the silent < em>p14/p16 < /em> locus in human neoplasia. Proc Natl Acad Sci USA. (2001) 98:4990–5. doi: 10.1073/pnas.101617298
38. Bakker J, Lin X, Nelson WG. Methyl-CpG binding domain protein 2 represses transcription from hypermethylated π-class glutathione S-transferase gene promoters in hepatocellular carcinoma cells. J Biol Chem. (2002) 277:22573–80. doi: 10.1074/jbc.M203009200
39. Zhu Y, Spitz MR, Zhang H, Grossman HB, Frazier ML, Wu X. Methyl-CpG-binding domain 2: a protective role in bladder carcinoma. Cancer Interdisc Int J Am Cancer Soc. (2004) 100:1853–8. doi: 10.1002/cncr.20199
40. Pulukuri S, Rao JS. CpG island promoter methylation and silencing of 14–3-3σ gene expression in LNCaP and Tramp-C1 prostate cancer cell lines is associated with methyl-CpG-binding protein MBD2. Oncogene. (2006) 25:4559–72. doi: 10.1038/sj.onc.1209462
41. Li R, He Q, Han S, Zhang M, Liu J, Su M, et al. MBD3 inhibits formation of liver cancer stem cells. Oncotarget. (2016) 8:6067–78. doi: 10.18632/oncotarget.13496
42. Xu M, He J, Li J, Feng W, Zhou H, Wei H, et al. Methyl-CpG-binding domain 3 inhibits epithelial–mesenchymal transition in pancreatic cancer cells via TGF-β/Smad signalling. Br J Cancer. (2016) 116:91–99. doi: 10.1038/bjc.2016.397
43. Cui Y, Li J, Weng L, Wirbisky SE, Freeman JL, Liu J, et al. Regulatory landscape and clinical implication of MBD3 in human malignant glioma. Oncotarget. (2016) 7:81698–714. doi: 10.18632/oncotarget.13173
44. Bader S, Walker M, Hendrich B, Bird A, Bird C, Hooper M, et al. Somatic frameshift mutations in the MBD4 gene of sporadic colon cancers with mismatch repair deficiency. Oncogene. (2000) 18:8044–7. doi: 10.1038/sj.onc.1203229
45. Sanders MA, Chew E, Flensburg C, Zeilemaker A, Miller SE, al Hinai AS, et al. MBD4 guards against methylation damage and germ line deficiency predisposes to clonal hematopoiesis and early-onset AML. Blood. (2018) 132:1526–34. doi: 10.1182/blood-2018-05-852566
46. Yamada T, Koyama T, Ohwada S, Tago K-i, Sakamoto I, Yoshimura S, et al. Frameshift mutations in the MBD4/MED1 gene in primary gastric cancer with high-frequency microsatellite instability. Cancer Lett. (2002) 181:115–20. doi: 10.1016/S0304-3835(02)00043-5
47. Choi YJ, Yoo NJ, Lee SH. Mutation and expression of a methyl-binding protein 6 (MBD6) in gastric and colorectal cancers. Pathol Oncol Res. (2015) 21:857–8. doi: 10.1007/s12253-015-9904-0
48. Chen K, Zhang F, Ding J, Liang Y, Zhan Z, Zhan Y, et al. Histone methyltransferase SETDB1 promotes the progression of colorectal cancer by inhibiting the expression of TP53. J Cancer. (2017) 8:3318–30. doi: 10.7150/jca.20482
49. Kostaki M, Manona AD, Stavraka I, Korkolopoulou P, Levidou G, Trigka EA, et al. High-frequency p16(INK) (4A) promoter methylation is associated with histone methyltransferase SETDB1 expression in sporadic cutaneous melanoma. Exp Dermatol. (2014) 23:332–8. doi: 10.1111/exd.12398
50. Wong CM, Wei L, Law CT, Ho DW, Tsang FH, Au SL, et al. Up-regulation of histone methyltransferase SETDB1 by multiple mechanisms in hepatocellular carcinoma promotes cancer metastasis. Hepatology. (2016) 63:474–87. doi: 10.1002/hep.28304
51. Wu M, Fan B, Guo Q, Li Y, Chen R, Lv N, et al. Knockdown of SETDB1 inhibits breast cancer progression by miR-381–3p-related regulation. Biol Res. (2018) 51:39. doi: 10.1186/s40659-018-0189-0
52. Nishikawaji T, Akiyama Y, Shimada S, Kojima K, Kawano T, Eishi Y, et al. Oncogenic roles of the SETDB2 histone methyltransferase in gastric cancer. Oncotarget. (2016) 7:67251–65. doi: 10.18632/oncotarget.11625
53. Gu L, Frommel SC, Oakes CC, Simon R, Grupp K, Gerig CY, et al. BAZ2A (TIP5) is involved in epigenetic alterations in prostate cancer and its overexpression predicts disease recurrence. Nat Genet. (2015) 47:22–30. doi: 10.1038/ng.3165
54. Jones J, Wang H, Zhou J, Hardy S, Turner T, Austin D, et al. Nuclear kaiso indicates aggressive prostate cancers and promotes migration and invasiveness of prostate cancer cells. Am J Pathol. (2012) 181:1836–46. doi: 10.1016/j.ajpath.2012.08.008
55. Lopes EC, Valls E, Figueroa ME, Mazur A, Meng F-G, Chiosis G, et al. Kaiso contributes to DNA methylation-dependent silencing of tumor suppressor genes in colon cancer cell lines. Cancer Res. (2008) 68:7258–63. doi: 10.1158/0008-5472.CAN-08-0344
56. Prokhortchouk A, Sansom O, Selfridge J, Caballero IM, Salozhin S, Aithozhina D, et al. Kaiso-deficient mice show resistance to intestinal cancer. Mol Cell Biol. (2006) 26:199–208. doi: 10.1128/MCB.26.1.199-208.2006
57. Dai S-D, Wang Y, Miao Y, Zhao Y, Zhang Y, Jiang G-Y, et al. Cytoplasmic Kaiso is associated with poor prognosis in non-small cell lung cancer. BMC Cancer. (2009) 9:178. doi: 10.1186/1471-2407-9-178
58. Bassey-Archibong BI, Rayner LGA, Hercules SM, Aarts CW, Dvorkin-Gheva A, Bramson JL, et al. Kaiso depletion attenuates the growth and survival of triple negative breast cancer cells. Cell Death Dis. (2017) 8:e2689. doi: 10.1038/cddis.2017.92
59. Kwiecien JM, Bassey-Archibong BI, Dabrowski W, Rayner LG, Lucas AR, Daniel JM. Loss of Kaiso expression in breast cancer cells prevents intra-vascular invasion in the lung and secondary metastasis. PLoS ONE. (2017) 12:e0183883. doi: 10.1371/journal.pone.0183883
60. Kim K, Chadalapaka G, Lee SO, Yamada D, Sastre-Garau X, Defossez PA, et al. Identification of oncogenic microRNA-17–92/ZBTB4/specificity protein axis in breast cancer. Oncogene. (2012) 31:1034–44. doi: 10.1038/onc.2011.296
61. Weber A, Marquardt J, Elzi D, Forster N, Starke S, Glaum A, et al. Zbtb4 represses transcription of P21CIP1 and controls the cellular response to p53 activation. Embo J. (2008) 27:1563–74. doi: 10.1038/emboj.2008.85
62. Kim K, Chadalapaka G, Pathi SS, Jin UH, Lee JS, Park YY, et al. Induction of the transcriptional repressor ZBTB4 in prostate cancer cells by drug-induced targeting of microRNA-17–92/106b-25 clusters. Mol Cancer Ther. (2012) 11:1852–62. doi: 10.1158/1535-7163.MCT-12-0181
63. Roussel-Gervais A, Naciri I, Kirsh O, Kasprzyk L, Velasco G, Grillo G, et al. Loss of the Methyl-CpG-binding protein ZBTB4 Alters mitotic checkpoint, increases aneuploidy, and promotes tumorigenesis. Cancer Res. (2017) 77:62–73. doi: 10.1158/0008-5472.CAN-16-1181
64. Kote-Jarai Z, Olama AA, Giles GG, Severi G, Schleutker J, Weischer M, et al. Seven prostate cancer susceptibility loci identified by a multi-stage genome-wide association study. Nat Genet. (2011) 43:785–91. doi: 10.1038/ng.882
65. Jing J, Liu J, Wang Y, Zhang M, Yang L, Shi F, et al. The role of ZBTB38 in promoting migration and invasive growth of bladder cancer cells. Oncol Rep. (2018). doi: 10.3892/or.2018.6937
66. Di Salle A, Petillo O, Melone MAB, Grimaldi G, Bellotti A, Torelli G, et al. High grade glioblastoma is associated with aberrant expression of ZFP57, a protein involved in gene imprinting, and of CPT1A and CPT1C that regulate fatty acid metabolism AU - Cirillo, Alessandra. Cancer Biol Ther. (2014) 15:735–41. doi: 10.4161/cbt.28408
67. Lamontagne M, Joubert P, Timens W, Postma DS, Hao K, Nickle D, et al. Susceptibility genes for lung diseases in the major histocompatibility complex revealed by lung expression quantitative trait loci analysis. Eur Respiratory J. (2016) 48:573–6. doi: 10.1183/13993003.00114-2016
68. Wei D, Gong W, Kanai M, Schlunk C, Wang L, Yao JC, et al. Drastic down-regulation of Kruppel-like factor 4 expression is critical in human gastric cancer development and progression. Cancer Res. (2005) 65:2746–54. doi: 10.1158/0008-5472.CAN-04-3619
69. Zhao W, Hisamuddin IM, Nandan MO, Babbin BA, Lamb NE, Yang VW. Identification of Kruppel-like factor 4 as a potential tumor suppressor gene in colorectal cancer. Oncogene. (2004) 23:395–402. doi: 10.1038/sj.onc.1207067
70. Ohnishi S, Ohnami S, Laub F, Aoki K, Suzuki K, Kanai Y, et al. Downregulation and growth inhibitory effect of epithelial-type Kruppel-like transcription factor KLF4, but not KLF5, in bladder cancer. Biochem Biophys Res Commun. (2003) 308:251–6. doi: 10.1016/S0006-291X(03)01356-1
71. Foster KW, Frost AR, McKie-Bell P, Lin CY, Engler JA, Grizzle WE, et al. Increase of GKLF messenger RNA and protein expression during progression of breast cancer. Cancer Res. (2000) 60:6488–95.
72. Yu F, Li J, Chen H, Fu J, Ray S, Huang S, et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. (2011) 30:2161–72. doi: 10.1038/onc.2010.591
73. Wan J, Su Y, Song Q, Tung B, Oyinlade O, Liu S, et al. Methylated cis-regulatory elements mediate KLF4-dependent gene transactivation and cell migration. Elife. (2017) 6:e20068. doi: 10.7554/eLife.20068
74. Chen YJ, Wu CY, Chang CC, Ma CJ, Li MC, Chen CM. Nuclear Kruppel-like factor 4 expression is associated with human skin squamous cell carcinoma progression and metastasis. Cancer Biol Ther. (2008) 7:777–82. doi: 10.4161/cbt.7.5.5768
75. Baron V, Adamson ED, Calogero A, Ragona G, Mercola D. The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGFbeta1, PTEN, p53, and fibronectin. Cancer Gene Ther. (2006) 13:115–24. doi: 10.1038/sj.cgt.7700896
76. Thigpen AE, Cala KM, Guileyardo JM, Molberg KH, McConnell JD, Russell DW. Increased expression of early growth response-1 messenger ribonucleic acid in prostatic adenocarcinoma. J Urol. (1996) 155:975–81. doi: 10.1016/S0022-5347(01)66361-4
77. Scharnhorst V, Menke AL, Attema J, Haneveld JK, Riteco N, van Steenbrugge GJ, et al. EGR-1 enhances tumor growth and modulates the effect of the Wilms' tumor 1 gene products on tumorigenicity. Oncogene. (2000) 19:791–800. doi: 10.1038/sj.onc.1203390
78. Ronski K, Sanders M, Burleson JA, Moyo V, Benn P, Fang M. Early growth response gene 1 (EGR1) is deleted in estrogen receptor-negative human breast carcinoma. Cancer. (2005) 104:925–30. doi: 10.1002/cncr.21262
79. Huang RP, Fan Y, de Belle I, Niemeyer C, Gottardis MM, Mercola D, et al. Decreased Egr-1 expression in human, mouse and rat mammary cells and tissues correlates with tumor formation. Int J Cancer. (1997) 72:102–9. doi: 10.1002/(SICI)1097-0215(19970703)72:1<102::AID-IJC15>3.0.CO;2-L
80. Chen D-g, Zhu B, Lv S-q, Zhu H, Tang J, Huang C, et al. Inhibition of EGR1 inhibits glioma proliferation by targeting CCND1 promoter. J Exp Clin Cancer Res. (2017) 36:186. doi: 10.1186/s13046-017-0656-4
81. Liu C, Adamson E, Mercola D. Transcription factor EGR-1 suppresses the growth and transformation of human HT-1080 fibrosarcoma cells by induction of transforming growth factor beta 1. Proc Natl Acad Sci USA. (1996) 93:11831–6. doi: 10.1073/pnas.93.21.11831
82. Zhang H, Chen X, Wang J, Guang W, Han W, Zhang H, et al. EGR1 decreases the malignancy of human non-small cell lung carcinoma by regulating KRT18 expression. Sci Rep. (2014) 4:5416. doi: 10.1038/srep05416
83. Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms' tumor locus. Cell. (1990) 60:509–20. doi: 10.1016/0092-8674(90)90601-A
84. Little M, Wells C. A clinical overview of WT1 gene mutations. Hum Mutat. (1997) 9:209–25. doi: 10.1002/(SICI)1098-1004(1997)9:3<209::AID-HUMU2>3.0.CO;2-2
85. Miyoshi Y, Ando A, Egawa C, Taguchi T, Tamaki Y, Tamaki H, et al. High expression of Wilms' tumor suppressor gene predicts poor prognosis in breast cancer patients. Clin Cancer Res. (2002) 8:1167–71.
86. Inoue K, Sugiyama H, Ogawa H, Nakagawa M, Yamagami T, Miwa H, et al. WT1 as a new prognostic factor and a new marker for the detection of minimal residual disease in acute leukemia. Blood. (1994) 84:3071–9.
87. Oji Y, Inohara H, Nakazawa M, Nakano Y, Akahani S, Nakatsuka S, et al. Overexpression of the Wilms' tumor gene WT1 in head and neck squamous cell carcinoma. Cancer Sci. (2003) 94:523–9. doi: 10.1111/j.1349-7006.2003.tb01477.x
88. Hylander B, Repasky E, Shrikant P, Intengan M, Beck A, Driscoll D, et al. Expression of Wilms tumor gene (WT1) in epithelial ovarian cancer. Gynecol Oncol. (2006) 101:12–7. doi: 10.1016/j.ygyno.2005.09.052
89. Netinatsunthorn W, Hanprasertpong J, Dechsukhum C, Leetanaporn R, Geater A. WT1 gene expression as a prognostic marker in advanced serous epithelial ovarian carcinoma: an immunohistochemical study. BMC Cancer. (2006) 6:90. doi: 10.1186/1471-2407-6-90
90. Li XL, Xu JH, Nie JH, Fan SJ. Exogenous expression of UHRF1 promotes proliferation and metastasis of breast cancer cells. Oncol Rep. (2012) 28:375–83. doi: 10.3892/or.2012.1792
91. Cui L, Chen J, Zhang Q, Wang X, Qu J, Zhang J, et al. Up-regulation of UHRF1 by oncogenic Ras promoted the growth, migration, and metastasis of pancreatic cancer cells. Mol Cell Biochem. (2015) 400(1–2):223–32. doi: 10.1007/s11010-014-2279-9
92. Wang F, Yang YZ, Shi CZ, Zhang P, Moyer MP, Zhang HZ, et al. UHRF1 promotes cell growth and metastasis through repression of p16(ink(4)a) in colorectal cancer. Ann Surg Oncol. (2012) 19:2753–62. doi: 10.1245/s10434-011-2194-1
93. Daskalos A, Oleksiewicz U, Filia A, Nikolaidis G, Xinarianos G, Gosney JR, et al. UHRF1-mediated tumor suppressor gene inactivation in nonsmall cell lung cancer. Cancer. (2011) 117:1027–37. doi: 10.1002/cncr.25531
94. Liu X, Ou H, Xiang L, Li X, Huang Y, Yang D. Elevated UHRF1 expression contributes to poor prognosis by promoting cell proliferation and metastasis in hepatocellular carcinoma. Oncotarget. (2017) 8:10510–22. doi: 10.18632/oncotarget.14446
95. Zhou L, Shang Y, Jin Za, Zhang W, Lv C, Zhao X, et al. UHRF1 promotes proliferation of gastric cancer via mediating tumor suppressor gene hypermethylation. Cancer Biol Ther. (2015) 16:1241–51. doi: 10.1080/15384047.2015.1056411
96. Yan F, Wang X, Shao L, Ge M, Hu X. Analysis of UHRF1 expression in human ovarian cancer tissues and its regulation in cancer cell growth. Tumour Biol. (2015) 36:8887–93. doi: 10.1007/s13277-015-3638-1
97. Babbio F, Pistore C, Curti L, Castiglioni I, Kunderfranco P, Brino L, et al. The SRA protein UHRF1 promotes epigenetic crosstalks and is involved in prostate cancer progression. Oncogene. (2012) 31:4878–87. doi: 10.1038/onc.2011.641
98. Lu S, Yan D, Wu Z, Jiang T, Chen J, Yuan L, et al. Ubiquitin-like with PHD and ring finger domains 2 is a predictor of survival and a potential therapeutic target in colon cancer. Oncol Rep. (2014) 31:1802–10. doi: 10.3892/or.2014.3035
99. Peng R, Huang X, Zhang C, Yang X, Xu Y, Bai D. Overexpression of UHRF2 in intrahepatic cholangiocarcinoma and its clinical significance. OncoTargets Ther. (2017) 10:5863–5872. doi: 10.2147/OTT.S149361
100. Wu J, Liu S, Liu G, Dombkowski A, Abrams J, Martin-Trevino R, et al. Identification and functional analysis of 9p24 amplified genes in human breast cancer. Oncogene. (2012) 31:333–41. doi: 10.1038/onc.2011.227
101. Lu H, Hallstrom TC. The nuclear protein UHRF2 is a direct target of the transcription factor E2F1 in the induction of apoptosis. J Biol Chem. (2013) 288:23833–43. doi: 10.1074/jbc.M112.447276
102. Hendrich B, Tweedie S. The methyl-CpG binding domain and the evolving role of DNA methylation in animals. TRENDS Genet. (2003) 19:269–77. doi: 10.1016/S0168-9525(03)00080-5
103. Lopez-Serra L, Esteller M. Proteins that bind methylated DNA and human cancer: reading the wrong words. Br J Cancer. (2008) 98:1881–85. doi: 10.1038/sj.bjc.6604374
104. Klose RJ, Sarraf SA, Schmiedeberg L, McDermott SM, Stancheva I, Bird AP. DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol Cell. (2005) 19:667–78. doi: 10.1016/j.molcel.2005.07.021
105. Hite KC, Adams VH, Hansen JC. Recent advances in MeCP2 structure and function. Biochem Cell Biol. (2009) 87:219–27. doi: 10.1139/O08-115
106. Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Gen. (1999) 23:185–8. doi: 10.1038/13810
107. Magdinier F, Billard L-M, Wittmann G, Frappart L, BenchaÏb M, Lenoir GM, et al. Regional methylation of the 5′ end CpG island of BRCA1 is associated with reduced gene expression in human somatic cells. FASEB J. (2000) 14:1585–94. doi: 10.1096/fj.99-0817com
108. Pulukuri S, Patibandla S, Patel J, Estes N, Rao JS. Epigenetic inactivation of the tissue inhibitor of metalloproteinase-2 (TIMP-2) gene in human prostate tumors. Oncogene. (2007) 26:5229–37. doi: 10.1038/sj.onc.1210329
109. Lin R-K, Hsu H-S, Chang J-W, Chen C-Y, Chen J-T, Wang Y-C. Alteration of DNA methyltransferases contributes to 5′ CpG methylation and poor prognosis in lung cancer. Lung Cancer. (2007) 55:205–13. doi: 10.1016/j.lungcan.2006.10.022
110. Sohn BH, Park IY, Lee JJ, Yang SJ, Jang YJ, Park KC, et al. Functional switching of TGF-β1 signaling in liver cancer via epigenetic modulation of a single CpG site in TTP promoter. Gastroenterology. (2010) 138:1898–908. e12. doi: 10.1053/j.gastro.2009.12.044
111. Pancione M, Sabatino L, Fucci A, Carafa V, Nebbioso A, Forte N, et al. Epigenetic silencing of peroxisome proliferator-activated receptor γ is a biomarker for colorectal cancer progression and adverse patients' outcome. PLoS ONE. (2010) 5:e14229. doi: 10.1371/journal.pone.0014229
112. Neupane M, Clark AP, Landini S, Birkbak NJ, Eklund AC, Lim E, et al. MECP2 is a frequently amplified oncogene with a novel epigenetic mechanism that mimics the role of activated RAS in malignancy. Cancer Disc. (2016) 6:45–58. doi: 10.1158/2159-8290.CD-15-0341
113. Pandey M, Shukla S, Gupta S. Promoter demethylation and chromatin remodeling by green tea polyphenols leads to re-expression of GSTP1 in human prostate cancer cells. Int J Cancer. (2010) 126:2520–33. doi: 10.1002/ijc.24988
114. Mirza S, Sharma G, Parshad R, Gupta SD, Pandya P, Ralhan R. Expression of DNA methyltransferases in breast cancer patients and to analyze the effect of natural compounds on DNA methyltransferases and associated proteins. J Breast Cancer. (2013) 16:23–31. doi: 10.4048/jbc.2013.16.1.23
115. Jørgensen HF, Ben-Porath I, Bird AP. Mbd1 is recruited to both methylated and nonmethylated CpGs via distinct DNA binding domains. Mol Cell Biol. (2004) 24:3387–95. doi: 10.1128/MCB.24.8.3387-3395.2004
116. Nakao M, Matsui S-i, Yamamoto S, Okumura K, Shirakawa M, Fujita N. Regulation of transcription and chromatin by methyl-CpG binding protein MBD1. Brain Dev. (2001) 23:S174–S6. doi: 10.1016/S0387-7604(01)00348-5
117. Ichimura T, Watanabe S, Sakamoto Y, Aoto T, Fujita N, Nakao M. Transcriptional repression and heterochromatin formation by MBD1 and MCAF/AM family proteins. J Biol Chem. (2005) 280:13928–35. doi: 10.1074/jbc.M413654200
118. Sarraf SA, Stancheva I. Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol Cell. (2004) 15:595–605. doi: 10.1016/j.molcel.2004.06.043
119. Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. (2006) 31:89–97. doi: 10.1016/j.tibs.2005.12.008
120. Zhao X, Ueba T, Christie BR, Barkho B, McConnell MJ, Nakashima K, et al. Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proc Natl Acad Sci USA. (2003) 100:6777–82. doi: 10.1073/pnas.1131928100
121. Xu J, Liu C, Yu X, Jin C, Fu D, Ni Q. Activation of multiple tumor suppressor genes by MBD1 siRNA in pancreatic cancer cell line BxPC-3. Zhonghua yi xue za zhi. (2008) 88:1948–51. doi: 10.3321/j.issn:0376-2491.2008.28.002
122. Xu J, Zhu W, Xu W, Yao W, Zhang B, Xu Y, et al. Up-regulation of MBD1 promotes pancreatic cancer cell epithelial-mesenchymal transition and invasion by epigenetic down-regulation of E-cadherin. Curr Mol Med. (2013) 13:387–400. doi: 10.2174/156652413805076740
123. Luo G, Jin C, Long J, Fu D, Yang F, Xu J, et al. RNA interference of MBD1 in BxPC-3 human pancreatic cancer cells delivered by PLGA-poloxamer nanoparticles. Cancer Biol Ther. (2009) 8:594–8. doi: 10.4161/cbt.8.7.7790
124. Hendrich B, Abbott C, McQueen H, Chambers D, Cross S, Bird A. Genomic structure and chromosomal mapping of the murine and human Mbd1, Mbd2, Mbd3, and Mbd4 genes. Mammalian Genome. (1999) 10:906–12. doi: 10.1007/s003359901112
125. Gnanapragasam MN, Scarsdale JN, Amaya ML, Webb HD, Desai MA, Walavalkar NM, et al. p66α-MBD2 coiled-coil interaction and recruitment of Mi-2 are critical for globin gene silencing by the MBD2–NuRD complex. Proc Natl Acad Sci USA. (2011) 108:7487–92. doi: 10.1073/pnas.1015341108
126. Boeke J, Ammerpohl O, Kegel S, Moehren U, Renkawitz R. The minimal repression domain of MBD2b overlaps with the methyl-CpG-binding domain and binds directly to Sin3A. J Biol Chem. (2000) 275:34963–7. doi: 10.1074/jbc.M005929200
127. Lu Y, Loh Y-H, Li H, Cesana M, Ficarro Scott B, Parikh Jignesh R, et al. Alternative splicing of MBD2 supports self-renewal in human pluripotent stem cells. Cell Stem Cell. (2014) 15:92–101. doi: 10.1016/j.stem.2014.04.002
128. Baubec T, Ivánek R, Lienert F, Schübeler D. Methylation-dependent and-independent genomic targeting principles of the MBD protein family. Cell. (2013) 153:480–92. doi: 10.1016/j.cell.2013.03.011
129. Wang L, Liu Y, Han R, Beier UH, Thomas R, Wells AD, et al. Mbd2 promotes Foxp3 demethylation and T-regulatory cell function. Mol Cell Biol. (2013) 33:4106–15. doi: 10.1128/MCB.00144-13
130. Weaver IC, Hellstrom IC, Brown SE, Andrews SD, Dymov S, Diorio J, et al. The methylated-DNA binding protein MBD2 enhances NGFI-A (egr-1)-mediated transcriptional activation of the glucocorticoid receptor. Phil Trans R Soc B. (2014) 369:20130513. doi: 10.1098/rstb.2013.0513
131. Angrisano T, Lembo F, Pero R, Natale F, Fusco A, Avvedimento VE, et al. TACC3 mediates the association of MBD2 with histone acetyltransferases and relieves transcriptional repression of methylated promoters. Nucleic Acids Res. (2006) 34:364–72. doi: 10.1093/nar/gkj400
132. Bhattacharya SK, Ramchandani S, Cervoni N, Szyf M. A mammalian protein with specific demethylase activity for mCpG DNA. Nature. (1999) 397:579–83. doi: 10.1038/17533
133. Zhang Y, Ng H-H, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. (1999) 13:1924–35. doi: 10.1101/gad.13.15.1924
134. Hendrich B, Guy J, Ramsahoye B, Wilson VA, Bird A. Closely related proteins MBD2 and MBD3 play distinctive but interacting roles in mouse development. Genes Dev. (2001) 15:710–23. doi: 10.1101/gad.194101
135. Chatagnon A, Bougel S, Perriaud L, Lachuer J, Benhattar J, Dante R. Specific association between the methyl-CpG-binding domain protein 2 and the hypermethylated region of the human telomerase reverse transcriptase promoter in cancer cells. Carcinogenesis. (2008) 30:28–34. doi: 10.1093/carcin/bgn240
136. Billard L-M, Magdinier F, Lenoir GM, Frappart L, Dante R. MeCP2 and MBD2 expression during normal and pathological growth of the human mammary gland. Oncogene. (2002) 21:2704–12. doi: 10.1038/sj/onc/1205357
137. Stirzaker C, Song J, Ng W, Du Q, Armstrong N, Locke W, et al. Methyl-CpG-binding protein MBD2 plays a key role in maintenance and spread of DNA methylation at CpG islands and shores in cancer. Oncogene. (2017) 36:1328–38. doi: 10.1038/onc.2016.297
138. Phesse TJ, Parry L, Reed KR, Ewan KB, Dale TC, Sansom OJ, et al. Deficiency of Mbd2 attenuates Wnt signaling. Mol Cell Biol. (2008) 28:6094–103. doi: 10.1128/MCB.00539-08
139. Cai Y, Geutjes E-J, De Lint K, Roepman P, Bruurs L, Yu L, et al. The NuRD complex cooperates with DNMTs to maintain silencing of key colorectal tumor suppressor genes. Oncogene. (2014) 33:2157–68. doi: 10.1038/onc.2013.178
140. Holik AZ, Young M, Krzystyniak J, Williams GT, Metzger D, Shorning BY, et al. Brg1 loss attenuates aberrant wnt-signalling and prevents wnt-dependent tumourigenesis in the murine small intestine. PLoS Genet. (2014) 10:e1004453. doi: 10.1371/journal.pgen.1004453
141. Wood KH, Johnson BS, Welsh SA, Lee JY, Cui Y, Krizman E, et al. Tagging methyl-CpG-binding domain proteins reveals different spatiotemporal expression and supports distinct functions. Epigenomics. (2016) 8:455–73. doi: 10.2217/epi-2015-0004
142. Shitara K, Nishikawa H. Regulatory T cells: a potential target in cancer immunotherapy. Ann N Y Acad Sci. (2018) 1417:104–15. doi: 10.1111/nyas.13625
143. Shukeir N, Pakneshan P, Chen G, Szyf M, Rabbani SA. Alteration of the methylation status of tumor-promoting genes decreases prostate cancer cell invasiveness and tumorigenesis in vitro and in vivo. Cancer Res. (2006) 66:9202–10. doi: 10.1158/0008-5472.CAN-06-1954
144. Pakneshan P, Szyf M, Farias-Eisner R, Rabbani SA. Reversal of the hypomethylation status of urokinase (uPA) promoter blocks breast cancer growth and metastasis. J Biol Chem. (2004) 279:31735–44. doi: 10.1074/jbc.M401669200
145. Campbell PM, Bovenzi V, Szyf M. Methylated DNA-binding protein 2 antisense inhibitors suppress tumourigenesis of human cancer cell lines in vitro and in vivo. Carcinogenesis. (2004) 25:499–507. doi: 10.1093/carcin/bgh045
146. Kaji K, Caballero IM, MacLeod R, Nichols J, Wilson VA, Hendrich B. The NuRD component Mbd3 is required for pluripotency of embryonic stem cells. Nat Cell Biol. (2006) 8:285–92. doi: 10.1038/ncb1372
147. Spruijt CG, Vermeulen M. DNA methylation: old dog, new tricks? Nat Struc Mol Biol. (2014) 21:949–54. doi: 10.1038/nsmb.2910
148. Yildirim O, Li R, Hung J-H, Chen PB, Dong X, Ee L-S, et al. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell. (2011) 147:1498–510. doi: 10.1016/j.cell.2011.11.054
149. Le Guezennec X, Vermeulen M, Brinkman AB, Hoeijmakers WA, Cohen A, Lasonder E, et al. MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol Cell Biol. (2006) 26:843–51. doi: 10.1128/MCB.26.3.843-851.2006
150. Aguilera C, Nakagawa K, Sancho R, Chakraborty A, Hendrich B, Behrens A. c-Jun N-terminal phosphorylation antagonises recruitment of the Mbd3/NuRD repressor complex. Nature. (2011) 469:231. doi: 10.1038/nature09607
151. Hashimoto H, Liu Y, Upadhyay AK, Chang Y, Howerton SB, Vertino PM, et al. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. (2012) 40:4841–9. doi: 10.1093/nar/gks155
152. Bellacosa A, Cicchillitti L, Schepis F, Riccio A, Yeung AT, Matsumoto Y, et al. MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc Natl Acad Sci USA. (1999) 96:3969–74. doi: 10.1073/pnas.96.7.3969
153. Wong E, Yang K, Kuraguchi M, Werling U, Avdievich E, Fan K, et al. Mbd4 inactivation increases C → T transition mutations and promotes gastrointestinal tumor formation. Proc Natl Acad Sci USA. (2002) 99:14937–42. doi: 10.1073/pnas.232579299
154. Roloff TC, Ropers HH, Nuber UA. Comparative study of methyl-CpG-binding domain proteins. BMC Genomics. (2003) 4:1. doi: 10.1186/1471-2164-4-1
155. Laget S, Joulie M, Le Masson F, Sasai N, Christians E, Pradhan S, et al. The human proteins MBD5 and MBD6 associate with heterochromatin but they do not bind methylated DNA. PLOS ONE. (2010) 5:e11982. doi: 10.1371/journal.pone.0011982
156. Baymaz HI, Fournier A, Laget S, Ji Z, Jansen PW, Smits AH, et al. MBD5 and MBD6 interact with the human PR-DUB complex through their methyl-CpG-binding domain. Proteomics. (2014) 14:2179–89. doi: 10.1002/pmic.201400013
157. Williams SR, Mullegama SV, Rosenfeld JA, Dagli AI, Hatchwell E, Allen WP, et al. Haploinsufficiency of MBD5 associated with a syndrome involving microcephaly, intellectual disabilities, severe speech impairment, and seizures. Eur J Hum Genet. (2010) 18:436–41. doi: 10.1038/ejhg.2009.199
158. Lim J, Hao T, Shaw C, Patel AJ, Szabó G, Rual J-F, et al. A protein–protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. (2006) 125:801–14. doi: 10.1016/j.cell.2006.03.032
159. Grosso S, Pucci L, Curatolo P, Coppola G, Bartalini G, Di Bartolo R, et al. Epilepsy and electroencephalographic anomalies in chromosome 2 aberrations: a review. Epilepsy Res. (2008) 79:63–70. doi: 10.1016/j.eplepsyres.2007.12.011
160. Talkowski ME, Mullegama SV, Rosenfeld JA, van Bon BW, Shen Y, Repnikova EA, et al. Assessment of 2q23. 1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet. (2011) 89:551–63. doi: 10.1016/j.ajhg.2011.09.011
161. Du Y, Liu B, Guo F, Xu G, Ding Y, Liu Y, et al. The essential role of Mbd5 in the regulation of somatic growth and glucose homeostasis in mice. PLoS ONE. (2012) 7:e47358. doi: 10.1371/journal.pone.0047358
162. Jung JS, Jee MK, Cho HT, Choi JI, Im YB, Kwon OH, et al. MBD6 is a direct target of Oct4 and controls the stemness and differentiation of adipose tissue-derived stem cells. Cell Mol Life Sci. (2013) 70:711–28. doi: 10.1007/s00018-012-1157-4
163. Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. (2002) 16:919–32. doi: 10.1101/gad.973302
164. Falandry C, Fourel G, Galy V, Ristriani T, Horard B, Bensimon E, et al. CLLD8/KMT1F is a lysine methyltrasferase that is important for chromosome segregation. J Biol Chem. (2010) 285:20234–41. doi: 10.1074/jbc.M109.052399
165. Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H, et al. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature. (2010) 464:927–31. doi: 10.1038/nature08858
166. Du TT, Xu PF, Dong ZW, Fan HB, Jin Y, Dong M, et al. Setdb2 controls convergence and extension movements during zebrafish gastrulation by transcriptional regulation of dvr1. Dev Biol. (2014) 392:233–44. doi: 10.1016/j.ydbio.2014.05.022
167. Li H, Rauch T, Chen Z-X, Szabó PE, Riggs AD, Pfeifer GP. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J Biol Chem. (2006) 281:19489–500.
168. Mabuchi H, Fujii H, Calin G, Alder H, Negrini M, Rassenti L, et al. Cloning and characterization of CLLD6, CLLD7, and CLLD8, novel candidate genes for leukemogenesis at chromosome 13q14, a region commonly deleted in B-cell chronic lymphocytic leukemia. Cancer Res. (2001) 61:2870–7.
169. Tallant C, Valentini E, Fedorov O, Overvoorde L, Ferguson FM, Filippakopoulos P, et al. Molecular basis of histone tail recognition by human TIP5 PHD finger and bromodomain of the chromatin remodeling complex NoRC. Structure. (2015) 23:80–92. doi: 10.1016/j.str.2014.10.017
170. Bortoluzzi A, Amato A, Lucas X, Blank M, Ciulli A. Structural basis of molecular recognition of helical histone H3 tail by PHD finger domains. Biochem J. (2017) 474:1633–51. doi: 10.1042/BCJ20161053
171. Jones MH, Hamana N, Nezu J-i, Shimane M. A novel family of bromodomain genes. Genomics. (2000) 63:40–5. doi: 10.1006/geno.1999.6071
172. Strohner R, Nemeth A, Jansa P, Hofmann-Rohrer U, Santoro R, Längst G, et al. NoRC—a novel member of mammalian ISWI-containing chromatin remodeling machines. EMBO J. (2001) 20:4892–900. doi: 10.1093/emboj/20.17.4892
173. Filion GJP, Zhenilo S, Salozhin S, Yamada D, Prokhortchouk E, Defossez P-A. A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol Cell Biol. (2006) 26:169–81. doi: 10.1128/MCB.26.1.169-181.2006
174. Daniel JM, Reynolds AB. The catenin p120 ctn interacts with Kaiso, a novel BTB/POZ domain zinc finger transcription factor. Mol Cell Biol. (1999) 19:3614–23. doi: 10.1128/MCB.19.5.3614
175. Daniel JM, Spring CM, Crawford HC, Reynolds AB, Baig A. The p120 ctn-binding partner Kaiso is a bi-modal DNA-binding protein that recognizes both a sequence-specific consensus and methylated CpG dinucleotides. Nucleic Acids Res. (2002) 30:2911–9. doi: 10.1093/nar/gkf398
176. Qin S, Zhang B, Tian W, Gu L, Lu Z, Deng D. Kaiso mainly locates in the nucleus in vivo and binds to methylated, but not hydroxymethylated DNA. Chinese J Cancer Res. (2015) 27:148–55. doi: 10.3978/j.issn.1000-9604.2015.04.03
177. Ireton RC, Davis MA, Van Hengel J, Mariner DJ, Barnes K, Thoreson MA, et al. A novel role for p120 catenin in E-cadherin function. J Cell Biol. (2002) 159:465–76. doi: 10.1083/jcb.200205115
178. Yoon H-G, Chan DW, Reynolds AB, Qin J, Wong J. N-CoR mediates DNA methylation-dependent repression through a methyl CpG binding protein Kaiso. Mol Cell. (2003) 12:723–34. doi: 10.1016/j.molcel.2003.08.008
179. Blattler A, Yao L, Wang Y, Ye Z, Jin VX, Farnham PJ. ZBTB33 binds unmethylated regions of the genome associated with actively expressed genes. Epigenetics Chromatin. (2013) 6:13. doi: 10.1186/1756-8935-6-13
180. Soubry A, Staes K, Parthoens E, Noppen S, Stove C, Bogaert P, et al. The transcriptional repressor kaiso localizes at the mitotic spindle and is a constituent of the pericentriolar material. PLoS ONE. (2010) 5:e9203. doi: 10.1371/journal.pone.0009203
181. De La Rosa-Velazquez IA, Rincon-Arano H, Benitez-Bribiesca L, Recillas-Targa F. Epigenetic regulation of the human retinoblastoma tumor suppressor gene promoter by CTCF. Cancer Res. (2007) 67:2577–85. doi: 10.1158/0008-5472.CAN-06-2024
182. Dai SD, Wang Y, Zhang JY, Zhang D, Zhang PX, Jiang GY, et al. Upregulation of delta-catenin is associated with poor prognosis and enhances transcriptional activity through Kaiso in non-small-cell lung cancer. Cancer Sci. (2011) 102:95–103. doi: 10.1111/j.1349-7006.2010.01766.x
183. Chikan NA, Vipperla B. KAISO inhibition: an atomic insight. J Biomol Struc Dyn. (2015) 33:1794–804. doi: 10.1080/07391102.2014.974072
184. Almouzni G, Probst AV. Heterochromatin maintenance and establishment: lessons from the mouse pericentromere. Nucleus. (2011) 2:332–8. doi: 10.4161/nucl.2.5.17707
185. Yang WS, Chadalapaka G, Cho SG, Lee SO, Jin UH, Jutooru I, et al. The transcriptional repressor ZBTB4 regulates EZH2 through a MicroRNA-ZBTB4-specificity protein signaling axis. Neoplasia. (2014) 16:1059–69. doi: 10.1016/j.neo.2014.09.011
186. Nishii T, Oikawa Y, Ishida Y, Kawaichi M, Matsuda E. CtBP-interacting BTB zinc finger protein (CIBZ) promotes proliferation and G1/S transition in embryonic stem cells via Nanog. J Biol Chem. (2012) 287:12417–24. doi: 10.1074/jbc.M111.333856
187. Oikawa Y, Omori R, Nishii T, Ishida Y, Kawaichi M, Matsuda E. The methyl-CpG-binding protein CIBZ suppresses myogenic differentiation by directly inhibiting myogenin expression. Cell Res. (2011) 21:1578–90. doi: 10.1038/cr.2011.90
188. Miotto B, Chibi M, Xie P, Koundrioukoff S, Moolman-Smook H, Pugh D, et al. The RBBP6/ZBTB38/MCM10 axis regulates DNA replication and common fragile site stability. Cell Rep. (2014) 7:575–87. doi: 10.1016/j.celrep.2014.03.030
189. Marchal C, de Dieuleveult M, Saint-Ruf C, Guinot N, Ferry L, Olalla Saad ST, et al. Depletion of ZBTB38 potentiates the effects of DNA demethylating agents in cancer cells via CDKN1C mRNA up-regulation. Oncogenesis. (2018) 7:82. doi: 10.1038/s41389-018-0092-0
190. Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell. (2011) 44:361–72. doi: 10.1016/j.molcel.2011.08.032
191. Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, et al. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell. (2008) 15:547–57. doi: 10.1016/j.devcel.2008.08.014
192. Shamis Y, Cullen DE, Liu L, Yang G, Ng S-F, Xiao L, et al. Maternal and zygotic < em>Zfp57 < /em> modulate NOTCH signaling in cardiac development. Proc Natl Acad Sci USA. (2015) 112:E2020–E9. doi: 10.1073/pnas.1415541112
193. Loh YH, Zhang W, Chen X, George J, Ng HH. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. (2007) 21:2545–57. doi: 10.1101/gad.1588207
194. Tada Y, Yamaguchi Y, Kinjo T, Song X, Akagi T, Takamura H, et al. The stem cell transcription factor ZFP57 induces IGF2 expression to promote anchorage-independent growth in cancer cells. Oncogene. (2014) 34:752–60. doi: 10.1038/onc.2013.599
195. Takanami I, Imamuma T, Hashizume T, Kikuchi K, Yamamoto Y, Yamamoto T, et al. Insulin-like growth factor-II as a prognostic factor in pulmonary adenocarcinoma. J Surg Oncol. (1996) 61:205–8. doi: 10.1002/(SICI)1096-9098(199603)61:3<205::AID-JSO8>3.0.CO;2-F
196. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
197. Chen X, Johns DC, Geiman DE, Marban E, Dang DT, Hamlin G, et al. Kruppel-like factor 4 (gut-enriched Kruppel-like factor) inhibits cell proliferation by blocking G1/S progression of the cell cycle. J Biol Chem. (2001). doi: 10.1074/jbc.M101194200
198. Zhang W, Geiman DE, Shields JM, Dang DT, Mahatan CS, Kaestner KH, et al. The gut-enriched kruppel-like factor (Kruppel-Like Factor 4) mediates the transactivating effecto of p53 on the p21WAF1/Cip1 promoter. J Biol Chem. (2000) 275:18391–8. doi: 10.1074/jbc.C000062200
199. Yoon HS, Ghaleb AM, Nandan MO, Hisamuddin IM, Dalton WB, Yang VW. Krüppel-like factor 4 prevents centrosome amplification following γ-irradiation-induced DNA damage. Oncogene. (2005) 24:4017–25. doi: 10.1038/sj.onc.1208576
200. Hagos E, Ghaleb A, Dalton W, Bialkowska A, Yang V. Mouse embryonic fibroblasts null for the Krüppel-like factor 4 gene are genetically unstable. Oncogene. (2009) 28:1197–1205. doi: 10.1038/onc.2008.465
201. Yang Y, Goldstein BG, Chao H-H, Katz J. KLF4 and KLF5 regulate proliferation, apoptosis and invasion in esophageal cancer cells. Cancer Biol Ther. (2005) 4:1216–21. doi: 10.4161/cbt.4.11.2090
202. Hashimoto H, Wang D, Steves AN, Jin P, Blumenthal RM, Zhang X, et al. Distinctive Klf4 mutants determine preference for DNA methylation status. Nucleic Acids Res. (2016) 44:10177–85. doi: 10.1093/nar/gkw774
203. Liu Y, Olanrewaju YO, Zheng Y, Hashimoto H, Blumenthal RM, Zhang X, et al. Structural basis for Klf4 recognition of methylated DNA. Nucleic Acids Res. (2014) 42:4859–67. doi: 10.1093/nar/gku134
204. Li W, Liu M, Su Y, Zhou X, Liu Y, Zhang X. The Janus-faced roles of Kruppel-like factor 4 in oral squamous cell carcinoma cells. Oncotarget. (2015) 6:44480–94. doi: 10.18632/oncotarget.6256
205. Pagel J-I, Deindl E. Early growth response 1—a transcription factor in the crossfire of signal transduction cascades. Indian J Biochem Biophys. (2011) 48:226–35.
206. Yan SF, Fujita T, Lu J, Okada K, Shan Zou Y, Mackman N, et al. Egr-1, a master switch coordinating upregulation of divergent gene families underlying ischemic stress. Nat Med. (2000) 6:1355–61. doi: 10.1038/82168
207. Bozon B, Davis S, Laroche S. A requirement for the immediate early gene zif268 in reconsolidation of recognition memory after retrieval. Neuron. (2003) 40:695–701. doi: 10.1016/S0896-6273(03)00674-3
208. Khachigian LM, Lindner V, Williams AJ, Collins T. Egr-1-induced endothelial gene expression: a common theme in vascular injury. Science. (1996) 271:1427–31. doi: 10.1126/science.271.5254.1427
209. Pawlinski R, Pedersen B, Kehrle B, Aird WC, Frank RD, Guha M, et al. Regulation of tissue factor and inflammatory mediators by Egr-1 in a mouse endotoxemia model. Blood. (2003) 101:3940–7. doi: 10.1182/blood-2002-07-2303
210. Zhang W, Tong H, Zhang Z, Shao S, Liu D, Li S, et al. Transcription factor EGR1 promotes differentiation of bovine skeletal muscle satellite cells by regulating MyoG gene expression. J Cell Physiol. (2018) 233:350–62. doi: 10.1002/jcp.25883
211. Zandarashvili L, White MA, Esadze A, Iwahara J. Structural impact of complete CpG methylation within target DNA on specific complex formation of the inducible transcription factor Egr-1. FEBS Lett. (2015) 589:1748–53. doi: 10.1016/j.febslet.2015.05.022
212. Hashimoto H, Olanrewaju YO, Zheng Y, Wilson GG, Zhang X, Cheng X. Wilms tumor protein recognizes 5-carboxylcytosine within a specific DNA sequence. Genes Dev. (2014) 28:2304–13. doi: 10.1101/gad.250746.114
213. Lee SL, Sadovsky Y, Swirnoff AH, Polish JA, Goda P, Gavrilina G, et al. Luteinizing hormone deficiency and female infertility in mice lacking the transcription factor NGFI-A (Egr-1). Science. (1996) 273:1219–21. doi: 10.1126/science.273.5279.1219
214. Topilko P, Schneider-Maunoury S, Levi G, Trembleau A, Gourdji D, Driancourt MA, et al. Multiple pituitary and ovarian defects in Krox-24 (NGFI-A, Egr-1)-targeted mice. Mol Endocrinol. (1998) 12:107–22. doi: 10.1210/mend.12.1.0049
215. Kim HS, Kim MS, Hancock AL, Harper JC, Park JY, Poy G, et al. Identification of novel Wilms' tumor suppressor gene target genes implicated in kidney development. J Biol Chem. (2007) 282:16278–87. doi: 10.1074/jbc.M700215200
216. Rae FK, Martinez G, Gillinder KR, Smith A, Shooter G, Forrest AR, et al. Anlaysis of complementary expression profiles following WT1 induction versus repression reveals the cholesterol/fatty acid synthetic pathways as a possible major target of WT1. Oncogene. (2004) 23:3067–79. doi: 10.1038/sj.onc.1207360
217. Kerst G, Bergold N, Viebahn S, Gieseke F, Kalinova M, Trka J, et al. WT1 protein expression in slowly proliferating myeloid leukemic cell lines is scarce throughout the cell cycle with a minimum in G0/G1 phase. Leukemia Res. (2008) 32:1393–9. doi: 10.1016/j.leukres.2008.03.006
218. Shandilya J, Roberts SG. A role of WT1 in cell division and genomic stability. Cell Cycle. (2015) 14:1358–64. doi: 10.1080/15384101.2015.1021525
219. Tatsumi N, Oji Y, Tsuji N, Tsuda A, Higashio M, Aoyagi S, et al. Wilms' tumor gene WT1-shRNA as a potent apoptosis-inducing agent for solid tumors. Int J Oncol. (2008) 32:701–11. doi: 10.3892/ijo.32.3.701
220. Ito K, Oji Y, Tatsumi N, Shimizu S, Kanai Y, Nakazawa T, et al. Antiapoptotic function of 17AA(+)WT1 (Wilms' tumor gene) isoforms on the intrinsic apoptosis pathway. Oncogene. (2006) 25:4217–29. doi: 10.1038/sj.onc.1209455
221. Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. (2009) 15:5323–37. doi: 10.1158/1078-0432.CCR-09-0737
222. Ohno S, Okuyama R, Aruga A, Sugiyama H, Yamamoto M. Phase I trial of Wilms' Tumor 1 (WT1) peptide vaccine with GM-CSF or CpG in patients with solid malignancy. Anticancer Res. (2012) 32:2263–9.
223. Oka Y, Tsuboi A, Nakata J, Nishida S, Hosen N, Kumanogoh A, et al. Wilms' tumor gene 1 (WT1) peptide vaccine therapy for hematological malignancies: from CTL epitope identification to recent progress in clinical studies including a cure-oriented strategy. Oncol Res Treat. (2017) 40:682–90. doi: 10.1159/000481353
224. Filippova GN, Fagerlie S, Klenova EM, Myers C, Dehner Y, Goodwin G, et al. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol Cell Biol. (1996) 16:2802–13. doi: 10.1128/MCB.16.6.2802
225. Kim S, Yu N-K, Kaang B-K. CTCF as a multifunctional protein in genome regulation and gene expression. Exp Mol Med. (2015) 47:e166. doi: 10.1038/emm.2015.33
226. Kornblihtt AR. CTCF: from insulators to alternative splicing regulation. Cell Res. (2012) 22:450–2. doi: 10.1038/cr.2012.22
227. Holwerda SJB, de Laat W. CTCF: the protein, the binding partners, the binding sites and their chromatin loops. Philos Transac R Soc London Series B Biol Sci. 368:20120369. doi: 10.1098/rstb.2012.0369
228. Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. (2011) 480:490–5. doi: 10.1038/nature10716
229. Lee JY, Mustafa M, Kim CY, Kim MH. Depletion of CTCF in breast cancer cells selectively induces cancer cell death via p53. J Cancer. (2017) 8:2124–31. doi: 10.7150/jca.18818
230. Fiorentino FP, Giordano A. The tumor suppressor role of CTCF. J Cell Physiol. (2012) 227:479–92. doi: 10.1002/jcp.22780
231. Zhao L, Yang Y, Yin S, Yang T, Luo J, Xie R, et al. CTCF promotes epithelial ovarian cancer metastasis by broadly controlling the expression of metastasis-associated genes. Oncotarget. (2017) 8:62217–30. doi: 10.18632/oncotarget.19216
232. Docquier F, Farrar D, D'Arcy V, Chernukhin I, Robinson AF, Loukinov D, et al. Heightened expression of CTCF in breast cancer cells is associated with resistance to apoptosis. Cancer Res. (2005) 65:5112–22. doi: 10.1158/0008-5472.CAN-03-3498
233. Sidhu H, Capalash N. UHRF1: The key regulator of epigenetics and molecular target for cancer therapeutics. Tumor Biol. (2017) 39:1010428317692205. doi: 10.1177/1010428317692205
234. Arita K, Ariyoshi M, Tochio H, Nakamura Y, Shirakawa M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. (2008) 455:818–21. doi: 10.1038/nature07249
235. Avvakumov GV, Walker JR, Xue S, Li Y, Duan S, Bronner C, et al. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature. (2008) 455:822–5. doi: 10.1038/nature07273
236. Ho KL, McNae IW, Schmiedeberg L, Klose RJ, Bird AP, Walkinshaw MD. MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol Cell. (2008) 29:525–31. doi: 10.1016/j.molcel.2007.12.028
237. Bostick M, Kim JK, Estève P-O, Clark A, Pradhan S, Jacobsen SE. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. (2007) 317:1760–4. doi: 10.1126/science.1147939
238. Sharif J, Muto M, Takebayashi S-i, Suetake I, Iwamatsu A, Endo TA, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. (2007) 450:908–12. doi: 10.1038/nature06397
239. Hopfner R, Mousli M, Jeltsch J-M, Voulgaris A, Lutz Y, Marin C, et al. ICBP90, a novel human CCAAT binding protein, involved in the regulation of topoisomerase IIα expression. Cancer Res. (2000) 60:121–8.
240. Unoki M, Bronner C, Mousli M. A concern regarding the current confusion with the human homolog of mouse Np95, ICBP90/UHRF1. Radiation Res. (2008) 169:240–4. doi: 10.1667/RR1209.1
241. Unoki M, Nishidate T, Nakamura Y. ICBP90, an E2F-1 target, recruits HDAC1 and binds to methyl-CpG through its SRA domain. Oncogene. (2004) 23:7601–10. doi: 10.1038/sj.onc.1208053
242. Liu X, Gao Q, Li P, Zhao Q, Zhang J, Li J, et al. UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat Commun. (2013) 4:1563. doi: 10.1038/ncomms2562
243. Bonapace IM, Latella L, Papait R, Nicassio F, Sacco A, Muto M, et al. Np95 is regulated by E1A during mitotic reactivation of terminally differentiated cells and is essential for S phase entry. J Cell Biol. (2002) 157:909–14. doi: 10.1083/jcb.200201025
244. Fujimori A, Matsuda Y, Takemoto Y, Hashimoto Y, Kubo E, Araki R, et al. Cloning and mapping of Np95 gene which encodes a novel nuclear protein associated with cell proliferation. Mammal Genome. (1998) 9:1032–5. doi: 10.1007/s003359900920
245. Mistry H, Gibson L, Yun JW, Sarras H, Tamblyn L, McPherson JP. Interplay between Np95 and Eme1 in the DNA damage response. Biochem Biophys Res Commun. (2008) 375:321–5. doi: 10.1016/j.bbrc.2008.07.146
246. Mistry H, Tamblyn L, Butt H, Sisgoreo D, Gracias A, Larin M, et al. UHRF1 is a genome caretaker that facilitates the DNA damage response to γ-irradiation. Genome Integr. (2010) 1:7. doi: 10.1186/2041-9414-1-7
247. ABBADY AQ, Bronner C, TROTZIER MA, Hopfner R, Bathami K, Muller CD, et al. ICBP90 expression is downregulated in apoptosis-induced jurkat cells. Ann N Y Acad Sci. (2003) 1010:300–3. doi: 10.1196/annals.1299.052
248. Sakai A, Kikuchi Y, Muroi M, Masui T, Furihata C, Uchida E, et al. Overexpression of NP95 mRNA by tumor promoters in the promotion phase of a two-stage BALB/3T3 cell transformation assay. Biol Pharmac Bull. (2003) 26:347–51. doi: 10.1248/bpb.26.347
249. Jenkins Y, Markovtsov V, Lang W, Sharma P, Pearsall D, Warner J, et al. Critical role of the ubiquitin ligase activity of UHRF1, a nuclear RING finger protein, in tumor cell growth. Mol Biol Cell. (2005) 16:5621–9. doi: 10.1091/mbc.e05-03-0194
250. Mudbhary R, Hoshida Y, Chernyavskaya Y, Jacob V, Villanueva A, Fiel MI, et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell. (2014) 25:196–209. doi: 10.1016/j.ccr.2014.01.003
251. Ma J, Peng J, Mo R, Ma S, Wang J, Zang L, et al. Ubiquitin E3 ligase UHRF1 regulates p53 ubiquitination and p53-dependent cell apoptosis in clear cell Renal Cell Carcinoma. Biochem Biophys Res Commun. (2015) 464:147–53. doi: 10.1016/j.bbrc.2015.06.104
252. Pi J-T, Lin Y, Quan Q, Chen L-L, Jiang L-Z, Chi W, et al. Overexpression of UHRF1 is significantly associated with poor prognosis in laryngeal squamous cell carcinoma. Med Oncol. (2013) 30:613. doi: 10.1007/s12032-013-0613-9
253. Liu W, Qiao RH, Wang DM, Huang XW, Li B, Wang D. UHRF1 promotes human osteosarcoma cell invasion by downregulating the expression of E-cadherin in an Rb1-dependent manner. Mol Med Rep. (2016) 13:315–20. doi: 10.3892/mmr.2015.4515
254. Geng Y, Gao Y, Ju H, Yan F. Diagnostic and prognostic value of plasma and tissue ubiquitin-like, containing PHD and RING finger domains 1 in breast cancer patients. Cancer Sci. (2013) 104:194–9. doi: 10.1111/cas.12052
255. Ge M, Gui Z, Wang X, Yan F. Analysis of the UHRF1 expression in serum and tissue for gastric cancer detection. Biomarkers. (2015) 20:183–8. doi: 10.3109/1354750X.2015.1061599
256. Ashraf W, Ibrahim A, Alhosin M, Zaayter L, Ouararhni K, Papin C, et al. The epigenetic integrator UHRF1: on the road to become a universal biomarker for cancer. Oncotarget. (2017) 8:51946. doi: 10.18632/oncotarget.17393
257. Jeanblanc M, Mousli M, Hopfner R, Bathami K, Martinet N, Abbady A-Q, et al. The retinoblastoma gene and its product are targeted by ICBP90: a key mechanism in the G1/S transition during the cell cycle. Oncogene. (2005) 24:7337. doi: 10.1038/sj.onc.1208878
258. Zhang Y, Huang Z, Zhu Z, Zheng X, Liu J, Han Z, et al. Upregulated UHRF1 promotes bladder cancer cell invasion by epigenetic silencing of KiSS1. PloS ONE. (2014) 9:e104252. doi: 10.1371/journal.pone.0104252
259. Parashar G, Capalash N. Promoter methylation-independent reactivation of PAX1 by curcumin and resveratrol is mediated by UHRF1. Clin Exp Med. (2016) 16:471–8. doi: 10.1007/s10238-015-0366-1
260. Ge T-T, Yang M, Chen Z, Lou G, Gu T. UHRF1 gene silencing inhibits cell proliferation and promotes cell apoptosis in human cervical squamous cell carcinoma CaSki cells. J Ovar Res. (2016) 9:42. doi: 10.1186/s13048-016-0253-8
261. Yan F, Tan X-Y, Geng Y, Ju H-X, Gao Y-F, Zhu M-C. Inhibition effect of siRNA-downregulated UHRF1 on breast cancer growth. Cancer Biother Radiopharmac. (2011) 26:183–9. doi: 10.1089/cbr.2010.0886
262. Li X, Meng Q, Rosen EM, Fan S. UHRF1 confers radioresistance to human breast cancer cells. Int J Rad Biol. (2011) 87:263–73. doi: 10.3109/09553002.2011.530335
263. Arima Y, Hirota T, Bronner C, Mousli M, Fujiwara T, Niwa Si, et al. Down-regulation of nuclear protein ICBP90 by p53/p21Cip1/WAF1-dependent DNA-damage checkpoint signals contributes to cell cycle arrest at G1/S transition. Genes Cells. (2004) 9:131–42. doi: 10.1111/j.1356-9597.2004.00710.x
264. Sharif T, Alhosin M, Auger C, Minker C, Kim J-H, Etienne-Selloum N, et al. Aronia melanocarpa juice induces a redox-sensitive p73-related caspase 3-dependent apoptosis in human leukemia cells. PLoS ONE. (2012) 7:e32526. doi: 10.1371/journal.pone.0032526
265. Sharif T, Auger C, Alhosin M, Ebel C, Achour M, Étienne-Selloum N, et al. Red wine polyphenols cause growth inhibition and apoptosis in acute lymphoblastic leukaemia cells by inducing a redox-sensitive up-regulation of p73 and down-regulation of UHRF1. Eur J Cancer. (2010) 46:983–94. doi: 10.1016/j.ejca.2009.12.029
266. Jin W, Liu Y, Xu S-g, Yin W-j, Li J-j, Yang J-m, et al. UHRF1 inhibits MDR1 gene transcription and sensitizes breast cancer cells to anticancer drugs. Breast Cancer Res Treat. (2010) 124:39–48. doi: 10.1007/s10549-009-0683-8
267. Mori T, Li Y, Hata H, Ono K, Kochi H. NIRF, a novel RING finger protein, is involved in cell-cycle regulation. Biochem Biophys Res Commun. (2002) 296:530–6. doi: 10.1016/S0006-291X(02)00890-2
268. Zhang J, Gao Q, Li P, Liu X, Jia Y, Wu W, et al. S phase-dependent interaction with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA methylation maintenance. Cell Res. (2011) 21:1723–39. doi: 10.1038/cr.2011.176
269. Zhou T, Xiong J, Wang M, Yang N, Wong J, Zhu B, et al. Structural basis for hydroxymethylcytosine recognition by the SRA domain of UHRF2. Mol Cell. (2014) 54:879–86. doi: 10.1016/j.molcel.2014.04.003
270. Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C, et al. Dynamic readers for 5-(hydroxy) methylcytosine and its oxidized derivatives. Cell. (2013) 152:1146–59. doi: 10.1016/j.cell.2013.02.004
271. Mori T, Li Y, Hata H, Kochi H. NIRF is a ubiquitin ligase that is capable of ubiquitinating PCNP, a PEST-containing nuclear protein. FEBS Lett. (2004) 557(1–3):209–14. doi: 10.1016/S0014-5793(03)01495-9
272. Lu H, Bhoopatiraju S, Wang H, Schmitz NP, Wang X, Freeman MJ, et al. Loss of UHRF2 expression is associated with human neoplasia, promoter hypermethylation, decreased 5-hydroxymethylcytosine, and high proliferative activity. Oncotarget. (2016) 7:76047–61. doi: 10.18632/oncotarget.12583
273. Ahuja N, Sharma AR, Baylin SB. Epigenetic therapeutics: a new weapon in the war against cancer. Ann Rev Med. (2016) 67:73–89. doi: 10.1146/annurev-med-111314-035900
274. Ateeq B, Unterberger A, Szyf M, Rabbani SA. Pharmacological inhibition of DNA methylation induces proinvasive and prometastatic genes in vitro and in vivo. Neoplasia. (2008) 10:266–78. doi: 10.1593/neo.07947
275. Krifa M, Alhosin M, Muller CD, Gies J-P, Chekir-Ghedira L, Ghedira K, et al. Limoniastrum guyonianum aqueous gall extract induces apoptosis in human cervical cancer cells involving p16 INK4A re-expression related to UHRF1 and DNMT1 down-regulation. J Exp Clin Cancer Res. (2013) 32:30. doi: 10.1186/1756-9966-32-30
276. Walter A, Etienne-Selloum N, Brasse D, Khallouf H, Bronner C, Rio M-C, et al. Intake of grape-derived polyphenols reduces C26 tumor growth by inhibiting angiogenesis and inducing apoptosis. he FASEB J. (2010) 24:3360–9. doi: 10.1096/fj.09-149419
277. Alhosin M, León-González AJ, Dandache I, Lelay A, Rashid SK, Kevers C, et al. Bilberry extract (Antho 50) selectively induces redox-sensitive caspase 3-related apoptosis in chronic lymphocytic leukemia cells by targeting the Bcl-2/Bad pathway. Sci Rep. (2015) 5:8996. doi: 10.1038/srep08996
278. Mahmood N, Cheishvili D, Arakelian A, Tanvir I, Ahmed H, Khan A-SP, et al. Methyl donor S-adenosylmethionine (SAM) supplementation attenuates breast cancer growth, invasion, and metastasis in vivo; therapeutic and chemopreventive applications. Oncotarget. (2018) 9:5169–83. doi: 10.18632/oncotarget.23704
279. Liang X, Xu Y, Xu K, Liu J, Qian X. B1, a novel amonafide analogue, overcomes the resistance conferred by Bcl-2 in human promyelocytic leukemia HL60 cells. Mol Cancer Res. (2010) 8:1619–32. doi: 10.1158/1541-7786.MCR-10-0341
280. Zhu D, Osuka S, Zhang Z, Reichert ZR, Yang L, Kanemura Y, et al. BAI1 suppresses medulloblastoma formation by protecting p53 from Mdm2-mediated degradation. Cancer Cell. (2018) 33:1004–16.e5. doi: 10.1016/j.ccell.2018.05.006
281. Tsuboi A, Hashimoto N, Fujiki F, Morimoto S, Kagawa N, Nakajima H, et al. A phase I clinical study of a cocktail vaccine of Wilms' tumor 1 (WT1) HLA class I and II peptides for recurrent malignant glioma. Cancer Immunol Immunother. (2019) 68:331–40. doi: 10.1007/s00262-018-2274-1
282. Gao L, Bellantuono I, Elsasser A, Marley SB, Gordon MY, Goldman JM, et al. Selective elimination of leukemic CD34(+) progenitor cells by cytotoxic T lymphocytes specific for WT1. Blood. (2000) 95:2198–203.
283. Ohminami H, Yasukawa M, Fujita S. HLA class I-restricted lysis of leukemia cells by a CD8(+) cytotoxic T-lymphocyte clone specific for WT1 peptide. Blood. (2000) 95:286–93.
Keywords: methyl-binding proteins, cancer, epigenetics, DNA methylation, MBD
Citation: Mahmood N and Rabbani SA (2019) DNA Methylation Readers and Cancer: Mechanistic and Therapeutic Applications. Front. Oncol. 9:489. doi: 10.3389/fonc.2019.00489
Received: 11 March 2019; Accepted: 23 May 2019;
Published: 11 June 2019.
Edited by:
Zhi Sheng, Virginia Tech, United StatesReviewed by:
Daekyu Sun, University of Arizona, United StatesShiv K. Gupta, Mayo Clinic, United States
Copyright © 2019 Mahmood and Rabbani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shafaat A. Rabbani, c2hhZmFhdC5yYWJiYW5pQG1jZ2lsbC5jYQ==