 Xiaohong Han1,2†Qiaoyun Tan1†Sheng Yang1Junling Li1Jianping Xu1Xuezhi Hao1Xingsheng Hu1Puyuan Xing1Yutao Liu1Lin Lin1Lin Gui1Yan Qin1
Xiaohong Han1,2†Qiaoyun Tan1†Sheng Yang1Junling Li1Jianping Xu1Xuezhi Hao1Xingsheng Hu1Puyuan Xing1Yutao Liu1Lin Lin1Lin Gui1Yan Qin1 Jianliang Yang1Peng Liu1Xingyuan Wang1
Jianliang Yang1Peng Liu1Xingyuan Wang1 Wumin Dai1Dongmei Lin3Hua Lin4
Wumin Dai1Dongmei Lin3Hua Lin4 Yuankai Shi1*
Yuankai Shi1*- 1Department of Medical Oncology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing Key Laboratory of Clinical Study on Anticancer Molecular Targeted Drugs, Beijing, China
- 2Department of Clinical Laboratory, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
- 3Department of Pathology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
- 4Department of Medical Record, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
Background: Lung adenocarcinoma (LUAD) possesses a poor prognosis with a low 5-year survival rate even for stages I-III resected patients, it is thus critical to understand the determinants that affect the survival and discover new potentially prognostic biomarkers. Somatic copy number alterations (CNAs) are major source of genomic variations driving tumor evolution, CNAs screening may identify prognostic biomarkers.
Methods: Oncoscan MIP array was used to analyze the patterns of CNAs on formalin fixed paraffin embedded(FFPE) tumor specimens from 163 consecutive stage I-III resected LUAD patients, 145 out of which received platinum-based adjuvant chemotherapy.
Results: Of the 163 patients, 91(55.8%) were recurred within 3 years after surgery. The most common aberrations in our cohort were 1q, 5p, 5q, 7p, 8q, 14p, 16p, 17q, 20q for copy number gains and 8p, 9p, 13p, 16q, 18q for losses. The GISTIC2 analysis produced 45 amplification peaks and 40 deletion peaks, involving some reported genes TERT, EGFR, MYC, CCND1, CDK4, MDM2, ERBB2, NKX2-1, CCNE1, and CDKN2A, most of which were consistent with TCGA database. The amplifications of 12p12.1 (CMAS, GOLT1B, YS2, LDHB, RECQL, ETNK1, IAPP, PYROXD1, KRAS) and KDM5A were correlated with worse prognosis in our cohort, this result was further validated in 506 LUAD patients from TCGA. In addition, 163 patients could be well-classified into five groups, and the clinical outcomes were significantly different based on threshold copy number at reoccurring alteration peaks. Among the 145 patients who received adjuvant chemotherapy, focal amplification of ERBB2 and deletion of 4q34.3 were found to be specific in relapsed patients, this result was validated in an independent group of Imielinski et al., demonstrating these two CNAs may contribute to resected LUAD recurrence after adjuvant chemotherapy.
Conclusion: This study suggests that CNAs profiling may be a potential prognostic classifier in resected LAUD patients. Amplifications of 12p12.1 and KDM5A might be prognostic biomarkers for LUAD, and amplification of ERBB2 and deletion of 4q34.3 predicted early relapse after adjuvant chemotherapy. These novel findings may provide implication for better implementation of precision therapy for lung cancer patients.
Introduction
About 85 % of lung cancers are non-small cell lung cancer (NSCLC), the majority of which are lung adenocarcinoma (LUAD) and squamous cell carcinoma subtypes (1). Patients diagnosed with NSCLC usually present with advanced stages and experience a dismal survival rate. Even for patients with surgically resectable stage I to III diseases, relapse is unfortunately common (2). Currently, the clinical principle prognosis prediction for NSCLC is tumor extension, characterized by tumor node metastasis (TNM) staging (3). However, the clinical outcome varies among individuals sharing the same clinical features, suggesting the existence of unknown factors that influence the disease outcomes, this necessitates the discovery of new potentially prognostic biomarkers which can identify patients with higher risk of relapse after surgical resection.
As a major source of genomic variations driving tumor evolution, somatic copy number alterations (CNAs) may provide potential prognostic information. Several studies have addressed the prognosis value of CNA patterns in various cancers. Yu et al. evaluated CNAs in 81 esophageal squamous cell cancer patients, and found four genes were significantly associated with the patients‘ prognosis (4). In breast cancer, gene CNA signature patterns were proven to be correlated with clinical features and survival (5). In NSCLC, a genotype classifier based on CNA differences between LUAD and squamous cell carcinoma has been reported and gained our understanding of carcinogenesis of lung cancer (6, 7). As a fact that LUAD composed of the majority of lung cancer, and the CNA spectrum difference do exist between LUAD and squamous cell carcinoma, it is thus important to explore the prognostic predictive value of CNA in LUAD subtype. On the other hand, most of the samples in clinical settings are formalin-fixed and paraffin-embedded (FFPE), and DNA-based tests are more robust when applied to FFPE tissues, thus, identifying prognostic markers based on CNA from FFPE samples may be of potential clinical significance.
The goal of this study was to evaluate the genetic CNA profiles in LUAD and to assess the predictive value of CNA for survival benefits, subsequently we investigated potential CNA associated with adjuvant chemotherapy in a group of patients who received platinum-based adjuvant chemotherapy treatment. To this end, we extensively analyzed the CNA data obtained by Oncoscan MIP array using FFPE samples from 163 LUAD patients and correlated these results with the clinical survival outcomes.
Materials and Methods
Sample Collection and DNA Extraction
A total of 190 patients were consecutively recruited in National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College between 2010 and 2014. All patients were pathologically confirmed with LUAD and treated with operation, clinical information was obtained from hospital electronic medical database. This was a retrospective study in nature, the study was conducted in accordance with the ethical guidelines of the National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Peking Union Medical College and Chinese Academy of Medical Sciences (NCC2017GKZ-01), and BGI Genomics (BGI-IRB18004). Tumor specimens were collected and paraffin blocks was prepared following standard process at the Department of Pathology, about 3–5 slices of 10 μm of thickness were collected from the paraffin block and made into an H&E-stained slide for DNA extraction. The tumoral area was manually dissected with scalpel following identification by superimposition of a H&E stained glass, and digested at 56°C in ATL buffer over-night with proteinase K (Qiagen) in. DNA extraction was then continually performed with QIAamp DNA micro kit (Qiagen) following manufacturer's instructions. Spectrophotometric (Nanodrop) were used to quantify DNA concentration.
Copy Number Analysis
Genome-wide estimation of CNAs was performed using the OncoScan FFPE Assay Kit (8) following the manufacturer's instructions. Briefly, 80 ng of FFPE DNA at a concentration of 6.6 mL at 12 ng/mL was used for the analysis. The DNA was incubated with biotin-labeled molecular inversion probes 16 h at 58°C. Subsequently, the circularized DNA is subjected to endonuclease cleavage, which generates fragments with common regions on the 30 and 50 ends that are subsequently used to amplify the fragments. Post amplification, the fragments are subjected to HaeIII endonuclease digestion and hybridized to the array overnight, stained, and scanned. Scanning results from both arrays are combined by the OncoScan Console software version 1.2 (Affymetrix) and subjected to further analysis and visualization by ASCAT (9) as implemented in the Nexus Express software. As per the manufacturer's recommendation, a gene copy number of one or less was considered as a copy number loss, copy number of three as gain, and four or more as high gain. Significantly focal copy number gains or losses were evaluated with GISTIC 2.0 (10) using default parameters.
NMF Analysis
Non-negative matrix factorization (NMF) (11) is a dimensionality reduction method which we can factorize a high-dimensional matrix profile to several matrices, thus, making the resulting matrices easier to inspect. Results from NMF vary slightly based on initial conditions. Here, we used different initial conditions with k = 2–10 for thresholded copy number profile of focal alteration peaks, Hence, we obtain a consensus plot (and cophenetic coefficient) for each k = 2 to 10 and seek values of k for which k is close to 1. Our data provided compelling evidence in support of k = 4 clusters (Supplementary Figure 4), suggesting that there was evidence for four consensus clusters. This result was also observed in two validation datasets.
Validation Datasets
Validation datasets included 159 LUADs from the cohort of Imielinski et al. (12), and 506 LUADs from TCGA of which whole genome CNAs (Affymetrix SNP 6.0 SNP array) and mRNA expressions (RNA-seq V2 RSEM) data were downloaded from TCGA portal.
Statistical Analysis
Statistical analysis was performed using R software (version R 3.2.3). The Student's t-test was used to compare the significant differences between two groups. All statistical tests were two-sided. The Fisher's exact test was used to determine association between two categories. Statistical significance was declared if the P-value was <0.05. Survival time distribution in the survival analysis was assessed by the Kaplan–Meier method using log-rank tests. We constructed a multivariate model to compute the HR based on the CNA status of select gene, including gender (male vs. female), age at surgery (≥58 vs. <58 years), family history (yes vs. no), smoking history (yes vs. no), high blood pressure (yes vs. no), diabetes (yes vs. no), tumor location (right vs. left), TNM stage (III vs. I and II), blood vessel invasion (yes vs. no), pleura invasion (yes vs. no), lymphnode metastasis (yes vs. no), and chemotherapy (yes vs. no). Interactions were assessed by including the cross-product of the CNA status and another variable of interest in a Cox model.
Results
Patient Clinical Characteristics
Of 190 LUAD patients consecutively recruited between 2010 and 2014, 27 failed the CNA genotyping might because of degradation and insufficient quantity of DNA obtained from FFPE samples, leaving 163 patients eligible for CNA and survival analyses. The clinical features of these 163 patients are shown in Supplementary Table 1 and Figure 1. The proportion of male (53%) and female (47%) were similar, and more than half (53%) were non-smokers. The median age in diagnosis was 58 (25–78 years) and the median follow-up was 5.62 years (0.58–7.19 years). The cohort included 1 stage I, 104 stage II, 58 stage III cases. All 163 patients received operation and 145 (88.9%) out of which received adjuvant chemotherapy treatment. All tumors were chemotherapy-naïve, primary resection specimens. Sample acquisition and processing details are provided in the Experimental Procedures.
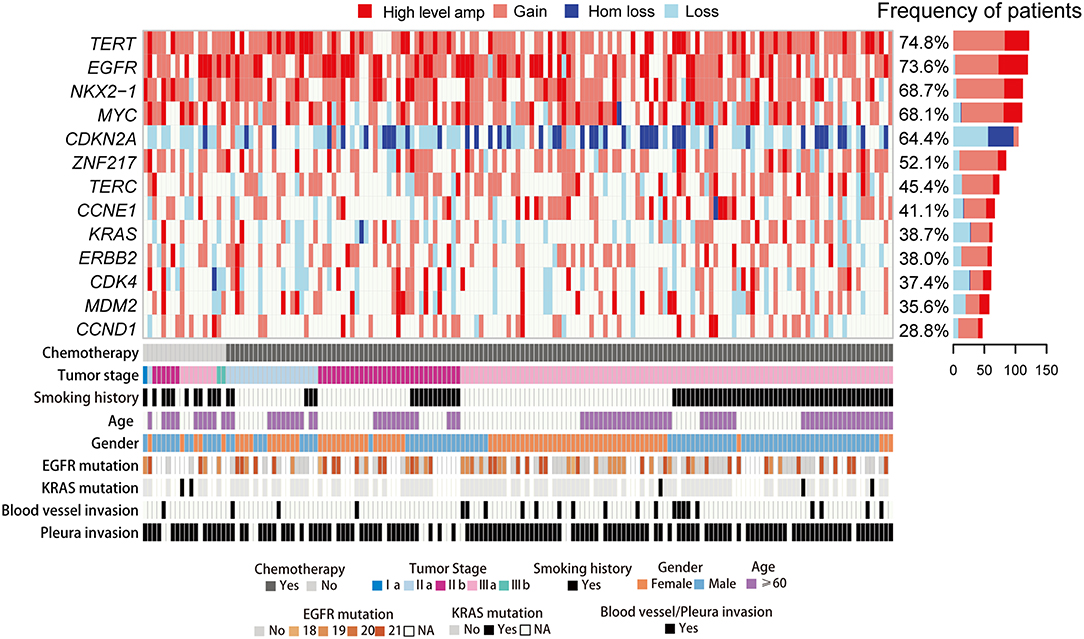
Figure 1. CNA spectrum and clinical characteristics of 163 LUAD cases. Heat map shows somatic CNAs with different status. Bars in the right represent the proportion of the CNAs identified in LUAD. The panel in the bottom shows key clinicopathological characteristics.
CNA Detection and Validation
Eighty nanogram FFPE gDNA has been extracted from 163 FFPE samples and purified as input to the OncoScan CNA Plus Assay to explore the whole genome CNAs and capture the alleles of nearly 220,000 SNPs at carefully selected genomic locations. Microarray data was transformed into genome-wide segmentation and allele imbalance data by using ASCAT (9). Our results showed the very similar landscape of somatic CNAs with TCGA cohort. The most frequent arm-level aberrations identified were 1q, 5p, 5q, 7p, 8q, 14p, 16p, 17q, 20q for copy number gains and 8p, 9p, 13p, 16q, 18q for losses (Figure 2A). GISTIC2 analysis generated 45 recurrent amplification peaks and 40 recurrent deletion peaks including some previously reported genes TERT, EGFR, MYC, CCND1, CDK4, MDM2, ERBB2, NKX2-1, CCNE1, and CDKN2A (Figure 2B), most of which were consistent with previous results in TCGA analyses (13). We next investigated CNAs of these genes across 163 LUADs, and found copy number gain was prominent in TERT (74.8%), EGFR (73.6%), NKX2-1 (68.7%), MYC (68.1%), whereas loss was predominant in CDKN2A (64.4%) (Figures 1, 2B).
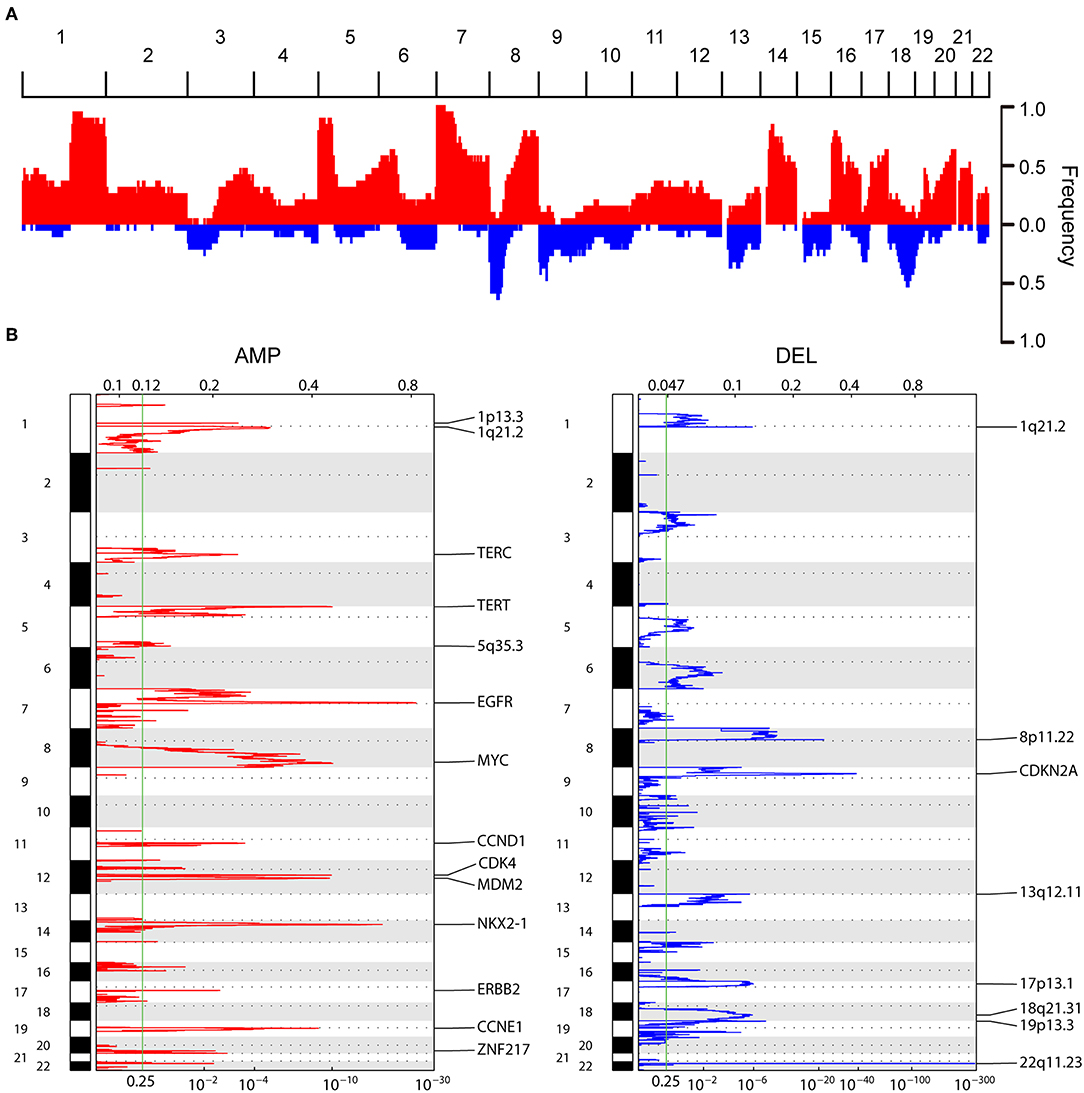
Figure 2. Composite copy number profiles for LUAD tumors. (A) Frequency plot s of CNAs distribution in 163 LUAD patients, red and blue indicate gain and loss, respectively (B) GISTIC2.0 focal amplifications (red) and deletions (blue) for 163 LUADs. Peaks with an FDR < 0.25 are annotated with candidate oncogenes, tumor suppressors or cytobands.
Association Analysis With Prognosis
The multivariate proportional hazards analysis was applied to clinical variables, TNM stage and chemotherapy were found to be independent prognostic factors in our data (Supplementary Figure 1). Compared with patients received only operation, adjuvant chemotherapy significantly improved the overall survival (OS) (p < 0.001). This significance which remained in the multiple analysis confirmed the conclusion as a result of several large clinical trials. There were no additional parameters other than tumor stage robustly associated with OS in the multivariate analysis (including all factors with p-values <0.05 from the univariate analysis). Stage III patients had a 1.82-fold increased risk of death compared with stage I and II LUAD patients.
To explore the potential prognostic role of CNAs changes, we applied the univariate proportional hazards analysis to CNAs of all genes, and discovered 16 significantly amplified CNAs that derived increased hazards of death. We further determined whether the association of these CNAs on cancer-specific survival was modified by any of the clinical pathologic variables evaluated. Among 16 genes of interest, 11 genes from 2 locations (12p12.1 and 12p13.33) remained associated with OS in multiple analyses (Table 1). Kaplan–Meier analysis indicated amplifications of 12p12.1 (CMAS, GOLT1B, GYS2, LDHB, RECQL, ETNK1, IAPP, PYROXD1, and KRAS) and 12p13.33 (KDM5A) were associated with worse prognosis in our cohort (Figure 3).
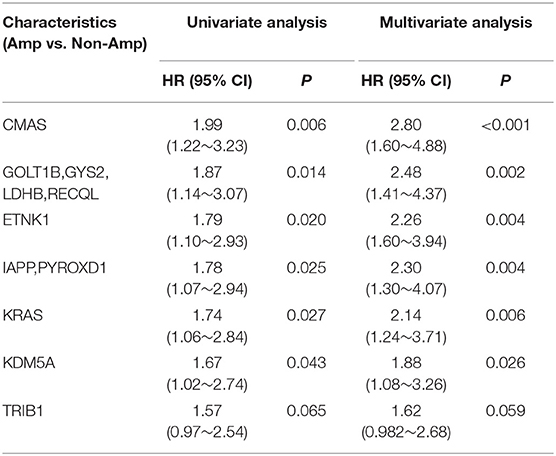
Table 1. Cox regression analyses for CNAs associated with survival.
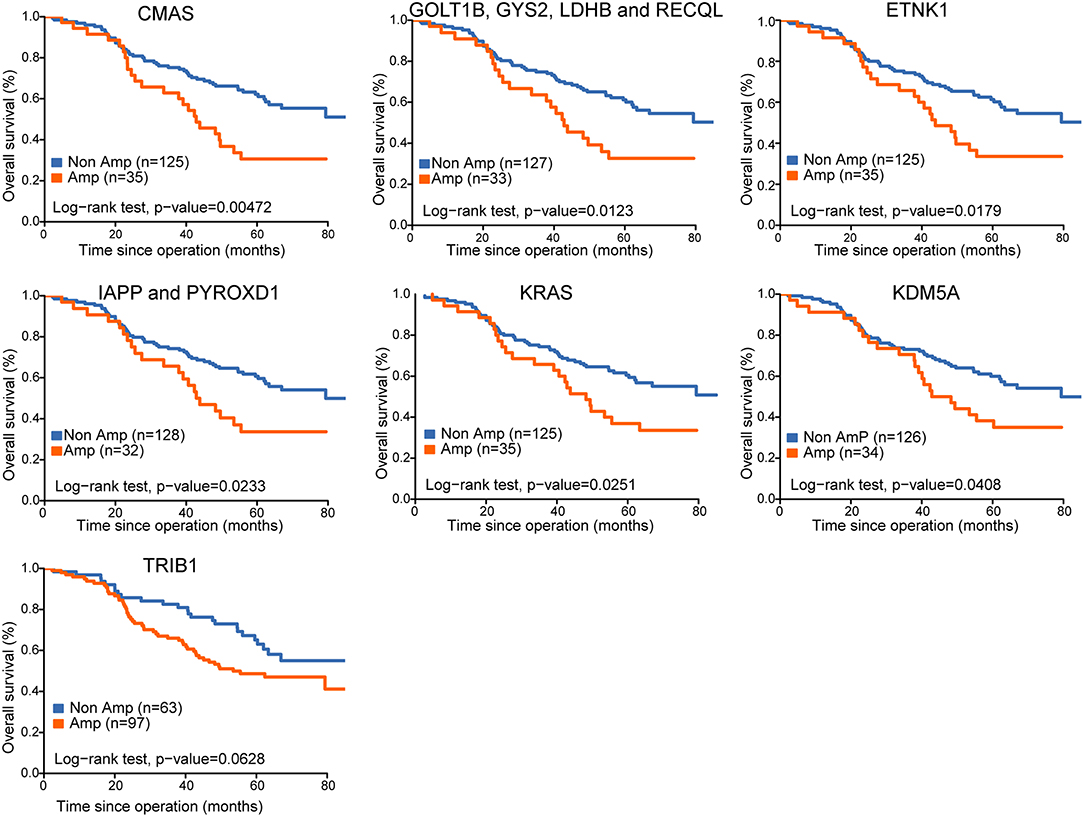
Figure 3. Kaplan-Meier survival curves for survival of lung adenocarcinoma with CNA of select genes.
Whole genome CNAs and gene expression data of 506 lung adenocarcinoma were downloaded from TCGA database, as shown in Supplementary Figure 2. In comparison to the unamplified genes, amplifications of 12p12.1 (CMAS, GOLT1B, GYS2, IAPP, KRAS, LDHB, PYROXD1, RECQL, and ETNK1) predicted poorer OS (p < 0.05), and the expression of these genes increased significantly accompanying the increased copy number (Supplementary Figure 3). There was a trend toward an OS benefit from TRIB1 non-amplification vs. amplification group in our cohort (p = 0.062), which was further validated in TCGA with a marginally significant association (p = 0.049). The prognostic impact of KDM5A amplification was seen on both univariable and multivariable analysis, with a significant difference in gene expression (p < 0.001). Of note, this is the first time to find KDM5A CNA was significantly associated with prognosis in resected LUAD.
Classification of LUAD Based on CNA
To explore the intratumor heterogeneity of LUAD, we performed molecular classification analysis by using NMF to cluster copy number profiles of 163 LUAD tumors. These tumors were clustered based on thresholded copy number of focal alteration peaks which was identified by GISTIC 2.0 analysis. This analysis defined four clusters with highest cophenetic coefficient (Supplementary Figure 4). We found that these clusters exhibit exclusive focal amplification of well-known oncogenes in lung cancer, finally, the 163 lung adenocarcinomas were reliably divided into five groups: LUAD1 (n = 33, 20.2%), LUAD2 (n = 54, 33.1%), LUAD3 (n = 28, 17.2%), LUAD4 (n = 38, 23.3%), LUAD5 (n = 10, 6.2%) (Figure 4A). The first group of tumors was significantly enriched for amplification of 8q on which a driver gene MYC is located. MYC is associated with genomic instability by promoting cell cycle progression and chromosome aneuploidy (14). The second group was enriched for a widely used clinical target gene EGFR (LUAD2). Focal amplification of MDM2 which play a role in tumorigenesis through inhibition of P53 protein was concentrated in LUAD3 group. The remaining group 4 was enriched for overexpression of TERC and ERBB2 (HER2), ERBB2 is a member of the human epidermal growth factor receptor family involved in receptor tyrosine kinases which regulate the modulation of cell proliferation, apoptosis, and cell mortality (15). Subsequently, we manually ascertained several tumors without any amplification of genes mentioned above, and assigned them a new group (LUAD5). The classifier was also validated in two additional independent sets [Imielinski et al. (12) and TCGA (13)], all subtypes were identified with comparable proportions of samples (Supplementary Figures 4, 5).
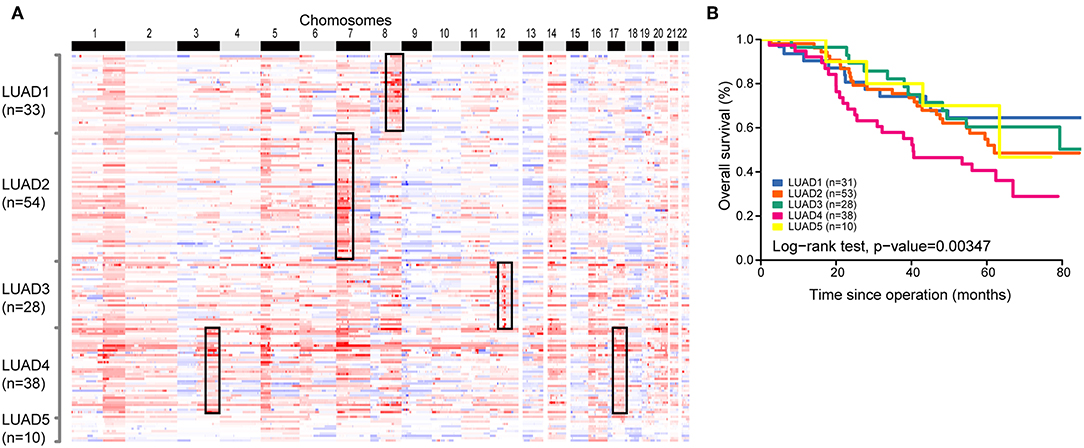
Figure 4. Copy number subtypes of 163 LUAD genomes. (A) In the heat map, SCNAs of each tumor which was assigned to five subgroups (vertical axis) are plotted by chromosomal position (horizontal axis). (B) The Kaplan-Meier plot depicting overall survival (OS) within 163 LUADs stratified by the classification.
Evaluation of the clinical characteristics of these subtypes suggested no significant correlation between them. Moreover, Kaplan-Meier survival analysis revealed significant differences in OS of the five subgroups and confirmed a poor prognosis for patients with LUAD4 subgroup (Figure 4B). Similar prognosis result was also observed in validation dataset (Supplementary Figure 5). Cumulatively, our classification system might provide useful information for risk stratification and treatment.
ERBB2 Amplification Associated With Relapse After Adjuvant Chemotherapy
The prognosis of LUAD has improved due to the standardization of adjuvant chemotherapy and the adoption of target therapy, however, the survival states of patients with recurrence after surgical resection are not well-studied. We speculated that CNA may become molecular biomarkers for helping inform clinical therapeutic strategies for post-surgical recurrence. Here, we included 145 patients who have received adjuvant chemotherapy and separated them into two groups based on whether LUAD was recurred within 3 years (91 relapse and 54 relapse-free). GISTIC 2.0 analyses were applied to these two groups to identify mutually exclusive foal amp/del makers. We saw a recurrent amplification at 17q12 at the locus containing ERBB2 only in relapse tumors, which was further validated in cohort of Imielinski et al. (Figure 5). Amplification or over expression of this gene has occurred in ~2–5% of LUAD (16). They have been identified as oncogenic drivers and associated with a poor prognosis in lung cancer (17). Previous studies have also suggested that ERBB2 overexpression is strongly associated with increased chemotherapy resistance leading to increase disease recurrence in breast cancers (18). Our result indicated that ERBB2 amplification may also have a role in regulating recurrence in LUAD. There were many other specific CNAs, such as deletion of 4q34.3 for relapse tumors, amplifications of 16p12.3 and 16q12.1 and deletion of 21q22.3 for relapse-free tumors (Supplementary Figures 6, 7). More detailed integrated genomic and transcriptomic sequencing analyses will be needed to elaborate the underlying mechanisms in the future.
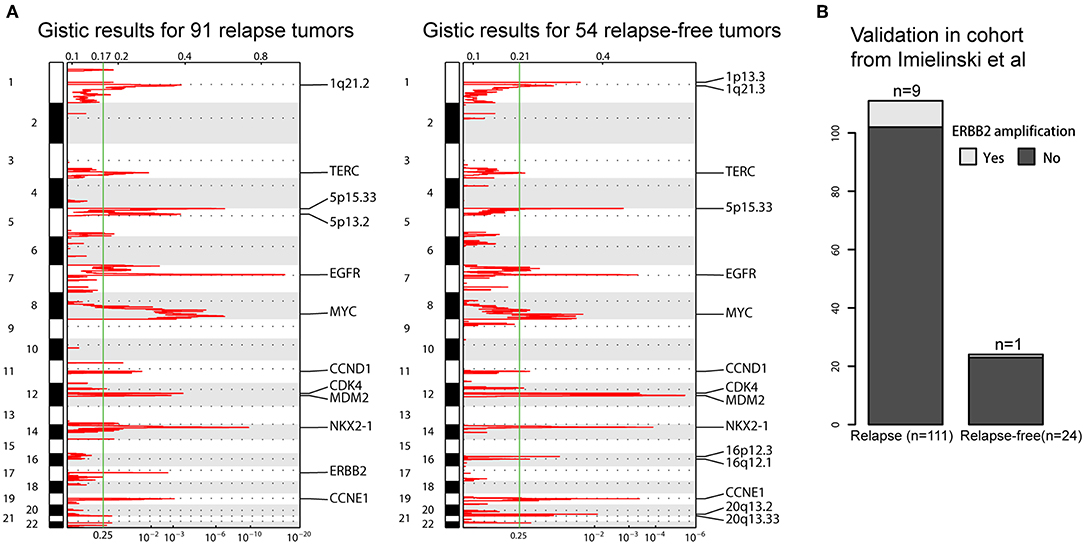
Figure 5. Focal copy number peaks specified for post-operative relapse LUADs. (A) GISTIC2.0 focal amplifications (red) for post-operative relapse and relapse-free LUADs. Peaks with an FDR < 0.25 are annotated with candidate oncogenes or cytobands. (B) Barplot shows the comparison on the ERBB2 amplification in validation sets.
Discussion
In this study, we sought to explore the CNA spectrum of lung adenocarcinoma patients and determine whether the CNA profiling could provide more insight into the different prognosis in patients receiving adjuvant chemotherapy.
Here, we tested the CNA in 163 LUAD patients. Our results show the greatest defections involved gains in 1q, 5p, 5q, 7p, 8q, 14p, 16p, 17q, 20q and losses in 8p, 9p, 13p, 16q, 18q, which shared similar CNAs landscape with TCGA database. Consistent with previous data, LUAD frequently displays gains in driver genes (19), and we identified frequent amplifications in known genes: TERT, EGFR, MYC, CCND1, CDK4, MDM2, ERBB2, NKX2-1, CCNE1, and this was further validated in TCGA database. In line with previous report (6), telomerase reverse transcriptase (TERT), located in 5p15, is the most frequently amplified gene in our result. Gains of oncogenes including EGFR (7p12), ERBB2 (17q12), KRAS in the EGFR pathway were characteristic for LUAD and contribute to cell proliferation and tumor development, suggesting this is a core requirement for LUAD pathogenesis. NKX2-1 was firstly known to contribute to normal lung cells differentiation (20, 21), later it was found to be a specific tissue marker in LUAD as patients carry increased NKX2-1 gene amplification and protein expression compared to normal cells (22), which was not identified in other cancers types of breast, prostate, colon and pancreas (23–25), indicating NKX2-1 was an oncogene specific for lung cancer, and now it's commonly used for differential diagnosis in clinical practice (26). However, the oncogene role and function mechanism remain incompletely clarified. Recently, Deborah et al. reported that NKX2-1 targeted on Selenium binding protein 1 (Selenbp1) led to LUAD growth inhibition (27). In our study, we showed that NKX2-1 amplification is commonly occurred in LUAD, which permitted the possibility of further function investigation for LUAD. Apart from gene gains, CDKN2A, which is frequently mutated or deleted in a wide variety of tumors, was significantly deleted in our cohort. CDKN2A is known to be a tumor suppressor gene (28, 29). For malignant mesothelioma, CDKN2A deletion was reported to be potential diagnostic and prognostic marker (30). Though different mechanisms of CDKN2A deletion exist in various human cancers (31), the CDKN2A tumor-suppression functions can be attenuated through point mutation, promoter hypermethylation, or deletion, and this could be one reason why this locus bears a high frequency of homozygous deletion.
When we analyzed the association of CNAs with long time survival, our analysis revealed 11 genes from two locations associated with worse prognosis, including amplifications of 12p12.1 (CMAS, GOLT1B, YS2, LDHB, RECQL, ETNK1, IAPP, PYROXD1, KRAS) and 12p13.33 (KDM5A). Former studies detected amplification of 12p12.1 in testicular germ cell tumors (32, 33). In lung adenocarcinoma, 18 genes (SLCO1A2, PYROXD1, RECQL, LDHB, CMAS, KIAA0528, ETNK1, ASUN, FGFR1OP2, TM7SF3, MED21, MRPS35, KLHDC5, CCDC91, FGD4, DNM1L, YASR2, and KRAS) in 12p12.1 were found to have increased copy number and coamplified with KRAS, 88.9% (8/9) genes in our study were included. Among them, LDHB amplification was found to be associated with RAS pathway activation. The elevated LDHB level in serum was validated to correlate with clinical stage and poor outcome of lung cancer (34, 35), indicating its potential to predict prognosis in lung cancer patients. CMAS is a protein-coding gene and participates in protein metabolism, it was reported to significantly associated with metastasis which decreases breast cancer survival (36). KDM5A was identified as a histone demethylase which represses transcription of its targeted genes (37, 38). It was reported that KDM5A is required to develop a metastable chromatin state that enables drug resistance in breast cancer (39) and lung cancer treated with EGFR-TKI (40). As far as we know, this is the first time to find KDM5A correlated with clinical outcome in LUAD patients receiving adjuvant chemotherapy in our study. Together, the critical role of KDM5A played in chemotherapy and targeted therapy drug tolerance yield an opportunity to develop pharmacologic inhibitors of KDM5A which potentially prevent drug resistance and improve treatment efficacy.
Moreover, we have built a molecular classifier for LUAD based on CNA profile, and identified five distinct subtypes (LUAD1, LUAD2, LUAD3, LUAD4, and LUAD5). These five distinct subtypes were validated in independent primary data sets. Our findings revealed that common CNA alterations are helpful for subtype classification. Each subgroup CNA with previously published focal amplifications helped improving the biological relevance of the stratification. Indeed, this analysis suggested that different types of LUAD may exhibit distinct focal amplifications, including MYC, ERBB2, EGFR, and MDM2. The ERBB2-amplificated subtype was well-identified with poor prognosis. This LUAD4 subtype is also observed with amplification of TERC, a gene located at chromosome 3 which serves as a template for the telomere repeat, its overexpression may be involved in oncogenesis (41). The further detection and investigation of the LUAD4 tumors will potentially give rise to optimized therapy for LUAD patients. In addition, there was no significant relationship between our classifier and clinical pathological stage, indicating that tumor subgroup might be established at the initial stages.
In the present study, we also attempted to explore the association between copy number profile and post-operative chemotherapy recurrence. ERBB2 amplification and 4q34.3 deletion were specially identified in LUAD patients who had tumor recurrence occurring within 3 years after therapy. Previous studies are mainly concentrated on ERBB2-mutated metastatic NSCLC (42), to our knowledge, our study is the first to show ERBB2 amplification is associated with the resistance of adjuvant chemtherapy and recurrence in LUAD patients, monoclonal antibody trastuzumab and pertuzumab which target ERBB2 may help guide therapeutic strategies for post-operative relapse for LUAD. This result was also validated in an independent data set. Collectively, our findings suggested that ERBB2 amplification plays a key role in mediating resistance to chemotherapy in LUAD, and further studies involving large LUAD cohorts with completely clinical data and other omics, may facilitate guiding therapeutic strategies.
In conclusion, we have shown that CNAs might be a potential prognostic classifier in resected LUAD patients. In addition, we found that amplifications of 12p12.1 and KDM5A were associated with worse clinical outcome for resected LUAD, and amplification of ERBB2 and deletion of 4q34.3 were associated with adjuvant chemotherapy resistance. These novel findings support a prognostic potential of CNAs and may provide certain implication for better clinical decision.
Data Availability
All data included in this study are available upon request by contact with the corresponding author.
Ethics Statement
This was a retrospective study in nature, and was conducted in accordance with the Declaration of Helsinki and all participants were above the age of 18. The study was approved by the Independent Review Board (IRB) of Cancer Hospital, Chinese Academy of Medical Sciences and BGI Genomics. The data will be available on request.
Author Contributions
YS and XHan contributed to the concept and design of this study. YS, XHan, QT, SY, JL, JX, XHao, XHu, PX, YL, LL, LG, YQ, JY, PL, XW, WD, DL, and HL performed acquisition of data (collected samples of patients, provided patients clinical information etc.). QT, XHan, and YS wrote, reviewed and revised the manuscript. QT, SY, and HL performed administrative, technical, or material support (i.e., reporting or organizing data, constructing databases). YS and XHan supervised the study.
Funding
This study was funded by National Key Technology Support Program (2014BAI09B01), and Chinese National Major Project for New Drug Innovation (2017ZX09304015, 2019ZX09201002), CAMS Innovation Fund for Medical Sciences CIFMS (2016-I2M-1-001), Beijing Municipal Science and Technology Commission Major Project (D141100000214005).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Teng Xiong, Bhaskar Roy, and Yong Zhou of BGI Genomics (BGI-Shenzhen) for sequencing and bioinformatics analysis. Part of this study was displayed in abstract for 2018 American Society of Clinical Oncology annual meeting (ASCO2018, June 1–5, Chicago, No. e24278) (43) and as a poster at the World Conference on Lung Cancer (WCLC 2018, September 23–26, Toronto, No. 13314) (44).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.00556/full#supplementary-material
Supplementary Figure 1. Multivariate analysis of clinical information with prognosis. (A) The panel shows hazard ratios (HR) of clinicopathological characteristics for cancer-specific survival. (B) The Kaplan-Meier graphs depicting overall survival (OS) associated with chemotherapy and TNM stage.
Supplementary Figure 2. Kaplan-Meier survival curves for survival of lung adenocarcinoma with CNA of select genes in TCGA dataset.
Supplementary Figure 3. Analyses of select genes expression levels by corresponding CNA status in 506 LUAD patients' tumors from the TCGA Project.
Supplementary Figure 4. NMF consensus clustering analysis and cophenetic coefficient for cluster k = 2 to k = 10. (A) our cohort. (B) The cohort from Imielinski et al. (C) TCGA cohort.
Supplementary Figure 5. Classification of validation datasets. (A) The cohort from Imielinski et al. (B) TCGA cohort.
Supplementary Figure 6. GISTIC2.0 focal deletions (red) for post-operative relapse and relapse-free LUADs. Peaks with an FDR < 0.25 are annotated with candidate tumor suppressors or cytobands.
Supplementary Figure 7. Barplot shows the comparison on the LOC285501 deletion in validation sets.
Supplementary Table 1. Patient characteristics of 163 patients.
References
1. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. (2018) 553:446–54. doi: 10.1038/nature25183
2. Winton T, Livingston R, Johnson D, Rigas J, Johnston M, Butts C, et al. Vinorelbine plus cisplatin vs. observation in resected non-small-cell lung cancer. N Engl J Med. (2005) 352:2589–97. doi: 10.1056/NEJMoa043623
3. Vansteenkiste J, Crino L, Dooms C, Douillard JY, Faivre-Finn C, Lim E, et al. 2nd ESMO Consensus Conference on Lung Cancer: early-stage non-small-cell lung cancer consensus on diagnosis, treatment and follow-up. Ann Oncol. (2014) 25:1462–74. doi: 10.1093/annonc/mdu089
4. Yu Y, Cao J, Wu W, Zhu Q, Tang Y, Zhu C, et al. Genome-wide copy number variation analysis identified ANO1 as a novel oncogene and prognostic biomarker in esophageal squamous cell cancer. Carcinogenesis. (2019). doi: 10.1093/carcin/bgz077. [Epub ahead of print].
5. Rennstam K, Ahlstedt-Soini M, Baldetorp B, Bendahl PO, Borg A, Karhu R, et al. Patterns of chromosomal imbalances defines subgroups of breast cancer with distinct clinical features and prognosis. A study of 305 tumors by comparative genomic hybridization. Cancer Res. (2003) 63:8861–8. Available online at: http://cancerres.aacrjournals.org/content/63/24/8861.full-text.pdf
6. Qiu ZW, Bi JH, Gazdar AF, Song K. Genome-wide copy number variation pattern analysis and a classification signature for non-small cell lung cancer. Genes Chromosomes Cancer. (2017) 56:559–69. doi: 10.1002/gcc.22460
7. Wellcome Trust Case Control C, Craddock N, Hurles ME, Cardin N, Pearson RD, Plagnol V, et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. (2010) 464:713–20. doi: 10.1038/nature08979
8. Foster JM, Oumie A, Togneri FS, Vasques FR, Hau D, Taylor M, et al. Cross-laboratory validation of the OncoScan(R) FFPE Assay, a multiplex tool for whole genome tumour profiling. BMC Med Genomics. (2015) 8:5. doi: 10.1186/s12920-015-0079-z
9. Van Loo P, Nordgard SH, Lingjaerde OC, Russnes HG, Rye IH, Sun W, et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci USA. (2010) 107:16910–5. doi: 10.1073/pnas.1009843107
10. Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. (2011) 12:R41. doi: 10.1186/gb-2011-12-4-r41
11. Brunet JP, Tamayo P, Golub TR, Mesirov JP. Metagenes and molecular pattern discovery using matrix factorization. Proc Natl Acad Sci USA. (2004) 101:4164–9. doi: 10.1073/pnas.0308531101
12. Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. (2012) 150:1107–20. doi: 10.1016/j.cell.2012.08.029
13. Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. (2014) 511:543–50. doi: 10.1038/nature13385
14. Prochownik EV. c-Myc: linking transformation and genomic instability. Curr Mol Med. (2008) 8:446–58. doi: 10.2174/156652408785747988
15. Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. (2006) 366:2–16. doi: 10.1016/j.gene.2005.10.018
16. Heinmoller P, Gross C, Beyser K, Schmidtgen C, Maass G, Pedrocchi M, et al. HER2 status in non-small cell lung cancer: results from patient screening for enrollment to a phase II study of herceptin. Clin Cancer Res. (2003) 9:5238–43. Available online at: http://clincancerres.aacrjournals.org/content/9/14/5238.short
17. Kim EK, Kim KA, Lee CY, Shim HS. The frequency and clinical impact of HER2 alterations in lung adenocarcinoma. PLoS ONE. (2017) 12:e0171280. doi: 10.1371/journal.pone.0171280
18. Yu MTD. Molecular Mechanisms of ErbB2-Mediated Breast Cancer Chemoresistance Breast Cancer Chemosensitivity. New York, NY: Springer (2007). p. 119–29. doi: 10.1007/978-0-387-74039-3_9
19. Pfarr N, Penzel R, Klauschen F, Heim D, Brandt R, Kazdal D, et al. Copy number changes of clinically actionable genes in melanoma, non-small cell lung cancer and colorectal cancer-A survey across 822 routine diagnostic cases. Genes Chromosomes Cancer. (2016) 55:821–33. doi: 10.1002/gcc.22378
20. Kimura S, Hara Y, Pineau T, Fernandez-Salguero P, Fox CH, Ward JM, et al. The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. (1996) 10:60–9. doi: 10.1101/gad.10.1.60
21. Minoo P, Su G, Drum H, Bringas P, Kimura S. Defects in tracheoesophageal and lung morphogenesis in Nkx2.1(-/-) mouse embryos. Dev Biol. (1999) 209:60–71. doi: 10.1006/dbio.1999.9234
22. Kwei KA, Kim YH, Girard L, Kao J, Pacyna-Gengelbach M, Salari K, et al. Genomic profiling identifies TITF1 as a lineage-specific oncogene amplified in lung cancer. Oncogene. (2008) 27:3635–40. doi: 10.1038/sj.onc.1211012
23. Bashyam MD, Bair R, Kim YH, Wang P, Hernandez-Boussard T, Karikari CA, et al. Array-based comparative genomic hybridization identifies localized DNA amplifications and homozygous deletions in pancreatic cancer. Neoplasia. (2005) 7:556–62. doi: 10.1593/neo.04586
24. Lapointe J, Li C, Giacomini CP, Salari K, Huang S, Wang P, et al. Genomic profiling reveals alternative genetic pathways of prostate tumorigenesis. Cancer Res. (2007) 67:8504–10. doi: 10.1158/0008-5472.CAN-07-0673
25. Bergamaschi A, Kim YH, Wang P, Sorlie T, Hernandez-Boussard T, Lonning PE, et al. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer. (2006) 45:1033–40. doi: 10.1002/gcc.20366
26. Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y, et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. (2011) 6:244–85. doi: 10.1097/JTO.0b013e318206a221
27. Caswell DR, Chuang CH, Ma RK, Winters IP, Snyder EL, Winslow MM. Tumor Suppressor Activity of Selenbp1, a Direct Nkx2-1 Target, in Lung Adenocarcinoma. Mol Cancer Res. (2018) 16:1737–49. doi: 10.1158/1541-7786.MCR-18-0392
28. Choi M, Kadara H, Zhang J, Parra ER, Rodriguez-Canales J, Gaffney SG, et al. Mutation profiles in early-stage lung squamous cell carcinoma with clinical follow-up and correlation with markers of immune function. Ann Oncol. (2017) 28:83–9. doi: 10.1093/annonc/mdw437
29. Ali SM, Yao M, Yao J, Wang J, Cheng Y, Schrock AB, et al. Comprehensive genomic profiling of different subtypes of nasopharyngeal carcinoma reveals similarities and differences to guide targeted therapy. Cancer. (2017) 123:3628–37. doi: 10.1002/cncr.30781
30. Illei PB, Ladanyi M, Rusch VW, Zakowski MF. The use of CDKN2A deletion as a diagnostic marker for malignant mesothelioma in body cavity effusions. Cancer. (2003) 99:51–6. doi: 10.1002/cncr.10923
31. Raschke S, Balz V, Efferth T, Schulz WA, Florl AR. Homozygous deletions of CDKN2A caused by alternative mechanisms in various human cancer cell lines. Genes Chromosomes Cancer. (2005) 42:58–67. doi: 10.1002/gcc.20119
32. Lopez-Rios F, Chuai S, Flores R, Shimizu S, Ohno T, Wakahara K, et al. Global gene expression profiling of pleural mesotheliomas: overexpression of aurora kinases and P16/CDKN2A deletion as prognostic factors and critical evaluation of microarray-based prognostic prediction. Cancer Res. (2006) 66:2970–9. doi: 10.1158/0008-5472.CAN-05-3907
33. Rodriguez S, Jafer O, Goker H, Summersgill BM, Zafarana G, Gillis AJ, et al. Expression profile of genes from 12p in testicular germ cell tumors of adolescents and adults associated with i(12p) and amplification at 12p11.2-p12.1. Oncogene. (2003) 22:1880–91. doi: 10.1038/sj.onc.1206302
34. McCleland ML, Adler AS, Deming L, Cosino E, Lee L, Blackwood EM, et al. Lactate dehydrogenase B is required for the growth of KRAS-dependent lung adenocarcinomas. Clin Cancer Res. (2013) 19:773–84. doi: 10.1158/1078-0432.CCR-12-2638
35. Chen Y, Zhang H, Xu A, Li N, Liu J, Liu C, et al. Elevation of serum l-lactate dehydrogenase B correlated with the clinical stage of lung cancer. Lung Cancer. (2006) 54:95–102. doi: 10.1016/j.lungcan.2006.06.014
36. Teoh ST, Ogrodzinski MP, Ross C, Hunter KW, Lunt SY. Sialic acid metabolism: a key player in breast cancer metastasis revealed by metabolomics. Front Oncol. (2018) 8:174. doi: 10.3389/fonc.2018.00174
37. Klose RJ, Yan Q, Tothova Z, Yamane K, Erdjument-Bromage H, Tempst P, et al. The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell. (2007) 128:889–900. doi: 10.1016/j.cell.2007.02.013
38. Christensen J, Agger K, Cloos PA, Pasini D, Rose S, Sennels L, et al. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell. (2007) 128:1063–76. doi: 10.1016/j.cell.2007.02.003
39. Hou J, Wu J, Dombkowski A, Zhang K, Holowatyj A, Boerner JL, et al. Genomic amplification and a role in drug-resistance for the KDM5A histone demethylase in breast cancer. Am J Transl Res. (2012) 4:247–56. doi: 10.1158/1538-7445.AM2012-2192
40. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. (2010) 141:69–80. doi: 10.1016/j.cell.2010.02.027
41. Skinner K, Hanning RM, Metatawabin J, Martin ID, Tsuji LJ. Impact of a school snack program on the dietary intake of grade six to ten First Nation students living in a remote community in northern Ontario, Canada. Rural Remote Health. (2012) 12:2122. Available online at: https://www.researchgate.net/profile/Kelly_Skinner/publication/230723709_Impact_of_a_school_snack_program_on_the_dietary_intake_of_grade_six_to_ten_First_Nation_students_living_in_a_remote_community_in_northern_Ontario_Canada/links/00b49518943c959349000000.pdf
42. Chuang JC, Stehr H, Liang Y, Das M, Huang J, Diehn M, et al. ERBB2-mutated metastatic non-small cell lung cancer: response and resistance to targeted therapies. J Thorac Oncol. (2017) 12:833–42. doi: 10.1016/j.jtho.2017.01.023
43. Tan Q, Xiong T, Yang S, Dai W, Lin D, Zhou Y, et al. Comprehensive gene copy number alterations profiling predict efficacy of adjuvant chemotherapy in resected stage II-IIIA lung adenocarcinoma. J Clin Oncol. (2018) 36:e24278. doi: 10.1200/JCO.2018.36.15_suppl.e24278
Keywords: lung adenocarcinoma, copy number alteration, prognosis, predict, adjuvant chemotherapy
Citation: Han X, Tan Q, Yang S, Li J, Xu J, Hao X, Hu X, Xing P, Liu Y, Lin L, Gui L, Qin Y, Yang J, Liu P, Wang X, Dai W, Lin D, Lin H and Shi Y (2019) Comprehensive Profiling of Gene Copy Number Alterations Predicts Patient Prognosis in Resected Stages I–III Lung Adenocarcinoma. Front. Oncol. 9:556. doi: 10.3389/fonc.2019.00556
Received: 27 February 2019; Accepted: 07 June 2019;
Published: 06 August 2019.
Edited by:
Iacopo Petrini, University of Pisa, ItalyReviewed by:
Pinar Ozden Eser, Dana–Farber Cancer Institute, United StatesAlex Friedlaender, Geneva University Hospitals (HUG), Switzerland
Copyright © 2019 Han, Tan, Yang, Li, Xu, Hao, Hu, Xing, Liu, Lin, Gui, Qin, Yang, Liu, Wang, Dai, Lin, Lin and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuankai Shi, c3l1YW5rYWlAY2ljYW1zLmFjLmNu
†These authors have contributed equally to this work