 Sara P. Neves1
Sara P. Neves1 Nanashara C. de Carvalho1
Nanashara C. de Carvalho1 Monize M. da Silva2
Monize M. da Silva2 Ana Carolina B. C. Rodrigues1Larissa M. Bomfim1
Ana Carolina B. C. Rodrigues1Larissa M. Bomfim1 Rosane B. Dias1
Rosane B. Dias1 Caroline B. S. Sales3
Caroline B. S. Sales3 Clarissa A. Gurgel Rocha1
Clarissa A. Gurgel Rocha1 Milena B. P. Soares1
Milena B. P. Soares1 Alzir A. Batista2
Alzir A. Batista2 Daniel P. Bezerra1*
Daniel P. Bezerra1*- 1Gonçalo Moniz Institute, Oswaldo Cruz Foundation (IGM-FIOCRUZ/BA), Salvador, Brazil
- 2Department of Chemistry, Federal University of São Carlos, São Carlos, Brazil
- 3Department of Biomorphology, Institute of Health Sciences, Federal University of Bahia, Salvador, Brazil
Herein, ruthenium complexes containing heterocyclic thioamidates [Ru(mmi)(bipy)(dppb)]PF6 (1), [Ru(tzdt)(bipy)(dppb)]PF6 (2), [Ru(dmp)(bipy)(dppb)]PF6 (3) and [Ru(mpca)(bipy)(dppb)]PF6 (4) were investigated for their cellular and molecular effects in cancer cell lines. Complexes 1 and 2 were the most potent of the four compounds against a panel of different cancer cell lines in monolayer cultures and showed potent cytotoxicity in a 3D model of multicellular spheroids that formed from human hepatocellular carcinoma HepG2 cells. In addition, both complexes were able to bind to DNA in a calf thymus DNA model. Compared to the controls, a reduction in cell proliferation, phosphatidylserine externalization, internucleosomal DNA fragmentation, and the loss of the mitochondrial transmembrane potential were observed in HepG2 cells that were treated with these complexes. Additionally, coincubation with a pan-caspase inhibitor (Z-VAD(OMe)-FMK) reduced the levels of apoptosis that were induced by these compounds compared to those in the negative controls, indicating that cell death through apoptosis occurred via a caspase-dependent pathway. Moreover, these complexes also induced the phosphorylation of ERK1/2, and coincubation with an MEK inhibitor (U0126), which is known to inhibit the activation of ERK1/2, but not JNK/SAPK and p38 MAPK inhibitors, reduced the complexes-induced apoptosis compared to that in the negative controls, indicating that the induction of apoptotic cell death occurred through ERK1/2 signaling in HepG2 cells. On the other hand, no increase in oxidative stress was observed in HepG2 cells treated with the complexes, and the complexes-induced apoptosis was not reduced with coincubation with the antioxidant N-acetylcysteine or a p53 inhibitor compared to that in the negative controls, indicating that apoptosis occurred via oxidative stress- and p53-independent pathways. Finally, these complexes also reduced the growth of HepG2 cells that were engrafted in C.B-17 SCID mice compared to that in the negative controls. These results indicated that these complexes are novel anticancer drug candidates for liver cancer treatment.
Introduction
Liver cancer is the sixth most common cancer and the fourth most common cause of cancer lethality worldwide. The estimated incidence of liver cancer in 2018 included approximately 841,080 new cases and 781,631 deaths, which represents an overall ratio of mortality to incidence of 0.93, indicating that the prognosis for liver cancer is very poor (1). Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer, accounting for approximately 75% of cases. For advanced HCC, sorafenib, a tyrosine kinase inhibitor, is a validated systemic therapy, but this treatment prolongs survival by only 3 months (2–4). Therefore, new drugs to treat liver cancer are needed.
In regard to metallodrugs, platinum-based drugs are widely used in the treatment of a variety of cancers, and ruthenium-based drugs have emerged as a novel potential class of chemotherapeutic and antineoplastic drugs (5). Ruthenium-based drugs have been considered an attractive alternative to platinum-based drugs because some ruthenium complexes have been shown to be more selective for cancer cells. This property has been attributed to the ability of ruthenium to mimic iron in binding to several biological molecules, including serum proteins (e.g., transferrin and albumin) (6). Therefore, many ruthenium complexes have been synthesized as novel anticancer agents (7–13). In particular, the ruthenium complexes [ImH]trans-[RuCl4(Im)(dmso-S)] (NAMI-A, where Im = imidazole) and [IndH]trans-[RuCl4(Ind)2] (KP1019, where Ind = indazole) completed phase I/II clinical trials in 2004 and 2008, respectively (14, 15). NAMI-A has been reported to interfere with the regulation of cell cycle and extracellular matrix invasion, thus preventing tumor metastasis, and KP1019 causes cell death through apoptosis via the formation of reactive oxygen species and the activation of the intrinsic mitochondrial pathway (16, 17).
Recently, ruthenium complexes containing heterocyclic thioamidates [Ru(mmi)(bipy)(dppb)]PF6 (1), [Ru(tzdt)(bipy)(dppb)]PF6 (2), [Ru(dmp)(bipy)(dppb)]PF6 (3) and [Ru(mpca)(bipy)(dppb)]PF6 (4) [where mmi = mercapto-1-methyl-imidazole; tzdt = 1,3-thiazolidine-2-thione; dmp = 4,6-diamino-2-mercaptopyrimidine; mpca = 6-mercaptopyridine-3-carboxylic acid; bipy = 2,2′-bipyridine; and dppb = 1,4-bis(diphenylphosphino)butane] (Figure 1A) were synthesized and shown to have potent cytotoxicity in cancer cell lines, to be able to bind to DNA and to inhibit the supercoiled DNA relaxation that is mediated by human topoisomerase IB (18, 19). However, cellular and molecular mechanisms of these compounds in causing cancer cell cytotoxicity have not been extensively explored. In the present work, these ruthenium complexes were investigated for their underlying cellular and molecular mechanisms in cancer cell cytotoxicity.
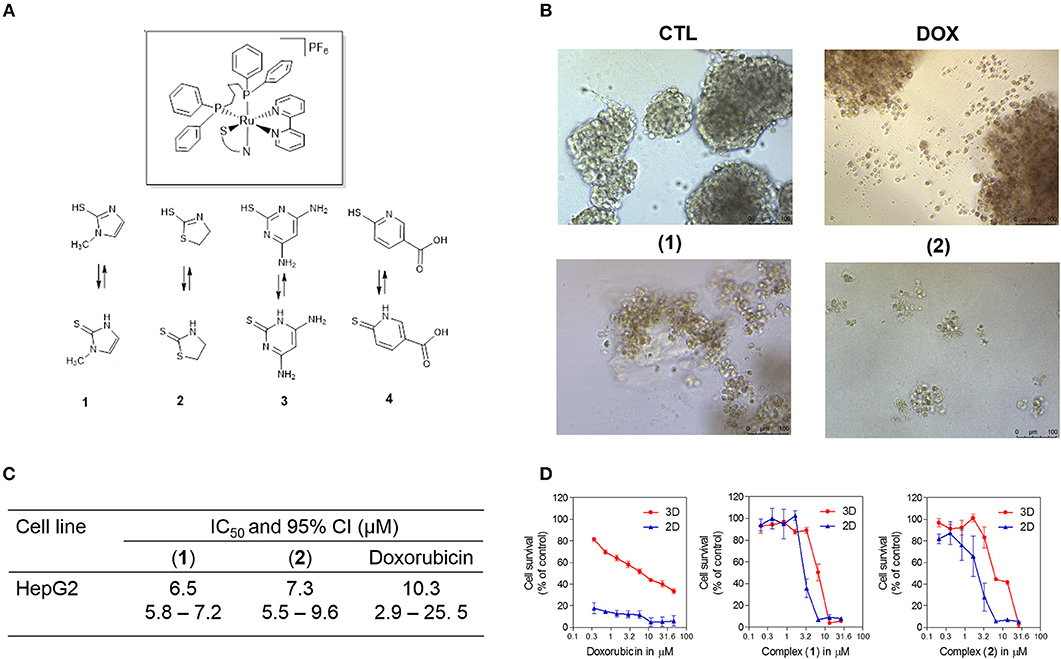
Figure 1. Effect of ruthenium complexes containing heterocyclic thioamidates in a 3D in vitro model of cancer multicellular spheroids formed from HepG2 cells. (A) Chemical structure of the ruthenium complexes containing heterocyclic thioamidates. (B) Cells were examined by light microscopy (bar = 100 μm) at the highest concentration tested after 72 h of incubation. (C) IC50 values and their respective 95% confidence intervals (95% CI) in μM. (D) Cell survival curves of the 3D vs. 2D culture models. The curves were obtained with non-linear regression from at least three independent experiments that were performed in duplicate and were measured with an Alamar blue assay after 72 h of incubation. The negative control (CTL) was treated with the vehicle (0.5% DMSO) that was used to solubilize and dilute the complexes, and doxorubicin (DOX) was used as the positive control.
Materials and Methods
Synthesis of Ruthenium Complexes Containing Heterocyclic Thioamidates
The ruthenium complexes containing heterocyclic thioamidates [Ru(mmi)(bipy)(dppb)]PF6 (1), [Ru(tzdt)(bipy)(dppb)]PF6 (2), [Ru(dmp)(bipy)(dppb)]PF6 (3) and [Ru(mpca)(bipy)(dppb)]PF6 (4) were synthesized as previously reported (18, 19). Briefly, the complexes were prepared by reacting the cis-[RuCl2(dppb)(bipy)] precursor (0.132 mmol) with the ligands (0.150 mmol) and KPF6 (0.132 mmol) in methanol (50 mL) in an Ar atmosphere for 24 h. The final orange solution was concentrated to approximately 2 mL, and diethyl ether was added to obtain an orange precipitate. The solid was filtered, well rinsed with water (5 × 5 mL) and diethyl ether (3 × 5 mL) and dried in vacuo. All manipulations were performed under argon. All reagents were purchased from Sigma-Aldrich (Sigma-Aldrich Co., Saint Louis, MO, USA) and were used as received.
In vitro Assays
Cells
The cell lines HepG2 (human hepatocellular carcinoma), HL-60 (human promyelocytic leukemia), K-562 (human chronic myelogenous leukemia), B16-F10 (mouse melanoma), MCF-7 (human breast carcinoma), HCT116 (human colon carcinoma), HSC-3 (human oral squamous cell carcinoma), SCC-4 (human oral squamous cell carcinoma), MRC-5 (human lung fibroblast), WT SV40 MEF (wild-type immortalized mouse embryonic fibroblast), and BAD KO SV40 MEF (BAD gene knockout immortalized mouse embryonic fibroblasts) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in RPMI 1640 medium (Gibco-BRL, Gaithersburg, MD, USA) with 10% fetal bovine serum (Life, Carlsbad, CA, USA), 2 mM L-glutamine (Vetec Química Fina, Duque de Caxias, RJ, Brazil) and 50 μg/mL gentamycin (Life). For all experiments, adherent cells were collected by treatment with 0.25% trypsin-EDTA solution (Gibco-BRL). All cell lines were cultured in flasks at 37°C in 5% CO2 and were subcultured every 3–4 days to maintain exponential growth. All cell lines were tested for mycoplasma using a mycoplasma staining kit (Sigma-Aldrich Co.) to validate that the cells that were used were free from contamination.
Heparinized blood was collected from 20 to 35-year-old, non-smoking healthy donors who had not taken any drugs for at least 15 days prior to sampling, and peripheral blood mononuclear cells (PBMCs) were isolated using a Ficoll density gradient in GE Ficoll-Paque Plus (GE Healthcare Bio-Sciences AB, Sweden). PBMCs were washed and resuspended at a concentration of 3 × 105 cells/mL in RPMI 1640 medium with 20% fetal bovine serum, 2 mM glutamine and 50 μg/mL gentamycin at 37°C with 5% CO2. Concanavalin A (ConA, Sigma-Aldrich Co.) was used as a mitogen to trigger cell division in T-lymphocytes. ConA (10 μg/mL) was added at the beginning of the culture and the cells were treated with the test compounds after 24 h. Cell viability was examined using a trypan blue exclusion assay for all experiments. The Research Ethics Committee of the Oswaldo Cruz Foundation (Salvador, Bahia, Brazil) approved the experimental protocol (#031019/2013). All participants signed a written informed consent form to participate in the study.
Cytotoxicity Assay
Cytotoxicity was measured colorimetrically using an Alamar blue method that was performed as previously described (20, 21). Cells were seeded in 96-well plates for all experiments (7 × 104 cells/mL for adherent cells or 3 × 105 cells/mL for suspension cells in 100 μL of medium) and incubated overnight at 37°C with 5% CO2 to allow for adherence. The complexes were dissolved in 0.5% dimethyl sulfoxide (DMSO, Vetec Química Fina) at eight different concentrations from 0.19 to 25 μg/mL and were added to each well and incubated for 72 h. The negative control was treated with the vehicle (0.5% DMSO) that was used to solubilize and dilute the complexes. Doxorubicin (purity ≥ 95%, doxorubicin hydrochloride, Laboratory IMA S.A.I.C., Buenos Aires, Argentina) and oxaliplatin (Sigma-Aldrich Co.) were used as the positive controls. Four (for cell lines) or 24 h (for PBMCs) before the end of incubation, 20 μL of a stock solution (0.312 mg/mL) of resazurin (Sigma-Aldrich Co.) was added to each well. Absorbance was measured at 570 and 600 nm using a SpectraMax 190 Microplate Reader (Molecular Devices, Sunnyvale, CA, USA).
3D Multicellular Spheroid Culture
HepG2 cells were cultivated as 3D multicellular spheroids in a 96-well plate with a cell-repellent surface. Briefly, 100 μL of a suspension of cells (5 × 105 cells/mL) was seeded into a 96-well plate with a cell-repellent surface (Greiner Bio-One, Kremsmünster, Austria) and cultured in RPMI 1640 medium with 10% fetal bovine serum, 2 mM glutamine plus 3% Matrigel (BD Biosciences, San Jose, CA, EUA) and 50 μg/mL gentamycin at 37°C with 5% CO2. Spheroids with stable structures formed after 3 days. Then, the complexes were dissolved in 0.5% DMSO at eight different concentrations from 0.19 to 25 μg/mL, were added to each well and incubated for 72 h. The negative control was treated with the vehicle (0.5% DMSO) that was used to solubilize and dilute the complexes, and doxorubicin was used as the positive control. At the end of the experiment, morphological changes were examined by light microscopy (Olympus BX41, Tokyo, Japan) using Image-Pro software (Media Cybernetics, Inc. Silver Spring, USA), and cytotoxicity was quantified with the Alamar blue method as described above.
DNA Interaction Assay
DNA interaction was measured in a cell-free system as previously described (22). In this assay, the ability of the complexes to translocate ethidium bromide, a DNA intercalator, from calf thymus DNA (ctDNA, Sigma-Aldrich Co.) was examined. The assay was conducted in 96-well plates, and the reaction mixture contained 15 μg/mL ctDNA, 1.5 μM ethidium bromide (Sigma-Aldrich Co.) and 10 or 20 μM of complexes in 100 μL of saline solution. The negative control was treated with the vehicle (0.2% DMSO) that was used to solubilize and dilute the complexes, and doxorubicin was used as the positive control. After a 15 min incubation at room temperature, fluorescence was measured using the excitation and emission wavelengths of 320 and 600 nm, respectively, using a Spectramax Microplate Reader (Molecular Devices).
Trypan Blue Exclusion Assay
For all subsequent in vitro experiments, 2 mL of a suspension of HepG2 cells were seeded (7 × 104 cells/mL) and incubated in a 24-well plate overnight to allow the cells to adhere to the plate surface. Then, the cells were incubated with 5 and/or 10 μM of complex 1 and 2 and/or 4 μM of complex 2 for 24 and/or 48 h (or until an indicated time point). The negative control was treated with the vehicle (0.2% DMSO) that was used to solubilize and dilute the complexes, and doxorubicin (2 μM) was used as the positive control.
In the trypan blue exclusion assay, the number of viable cells and non-viable cells (stained with trypan blue) were counted. Briefly, 90 μL was removed from the cell suspension, and 10 μL of trypan blue (0.4%) was added. Cell counting was performed using a light microscope with a hemocytometer filled with an aliquot of the homogenized cell suspension.
May-Grunwald-Giemsa Staining
To evaluate alterations in the morphology, cells were cultured on a coverslip and stained with May-Grunwald-Giemsa. Morphological changes were examined by light microscopy (Olympus BX41) using Image-Pro software (Media Cybernetics).
Flow Cytometric Assays
Internucleosomal DNA fragmentation and cell cycle distribution were measured by the quantification of the DNA content (23). In this assay, cells were harvested in a permeabilization solution containing 0.1% Triton X-100, 2 μg/mL propidium iodide, 0.1% sodium citrate and 100 μg/mL RNAse (all from Sigma-Aldrich Co.) and incubated in the dark for 15 min at room temperature.
A FITC Annexin V Apoptosis Detection Kit I (BD Biosciences) was used for apoptosis quantification, and the analysis was performed according to the manufacturer's instructions. Cell fluorescence and light scattering features were determined by flow cytometry. The percentages of viable, early apoptotic, late apoptotic and necrotic cells were determined. Protection assays using a pan-caspase inhibitor (Z-VAD(Ome)-FMK, Cayman Chemical; Ann Arbor, MI, USA), the antioxidant N-acetylcysteine (NAC, Sigma-Aldrich Co.), a Jun kinase (JNK/SAPK) inhibitor (SP600125; Cayman Chemical), a p38 MAPK inhibitor (PD169316; Cayman Chemical), a MEK (mitogen-activated protein kinase kinase) inhibitor (U0126; Cayman Chemical), and a p53 inhibitor (cyclic pifithrin-α; Cayman Chemical) were also performed. In these assays, the cells were preincubated for 2 h with 50 μM Z-VAD(Ome)-FMK, 5 μM SP600125, 5 μM PD169316, 5 μM U0126 or 10 μM cyclic pifithrin-α, followed by incubation with the complexes at an established concentration (10 μM for complex 1 and 4 μM for complex 2) for 48 h.
The mitochondrial transmembrane potential was evaluated by the retention of the rhodamine 123 dye (24). Cells were incubated with rhodamine 123 (5 μg/mL, Sigma-Aldrich Co.) at 37°C for 15 min in the dark and washed with saline. The cells were then incubated again in saline at 37°C for 30 min in the dark, and the cell fluorescence was determined by flow cytometry.
The reactive oxygen species (ROS) levels were measured using 2′,7′-dichlorofluorescin diacetate (DCF-DA) (Sigma-Aldrich Co.) (25). The cells were treated with the complexes for 1 or 3 h. Then, the cells were collected, washed with saline and resuspended in tubes with saline containing 5 μM DCF-DA for 30 min. Finally, the cells were washed with saline, and cell fluorescence was determined by flow cytometry.
Phosphorylated extracellular signal-regulated kinase (ERK), JNK/SAPK and p38 MAPK were quantified by flow cytometry as previously described (26). The cells were collected and resuspended in 0.5–1 mL of 4% formaldehyde and fixed for 10 min at 37°C. Then, the tubes were chilled on ice for 1 min. The cells were permeabilized by slowly adding ice-cold 100% methanol to the cooled cells while gently vortexing to a final concentration of 90% methanol and incubating for 30 min on ice. After washing with incubation buffer (0.5% bovine serum albumin in PBS), PE mouse anti-JNK/SAPK (pT183/pY185) (ID 562480), PE mouse anti-p38 MAPK (pT180/pY182) (ID 612565), PE mouse anti-ERK1/2 (pT202/pY204) (ID 612566) or PE mouse IgG1,κ isotype control (ID 555749) antibodies, all from BD Biosciences, were added and incubated for 1 h at room temperature. Finally, the cells were washed with PBS, and the cell fluorescence was measured by flow cytometry.
For all flow cytometry analyses, at least 104 events were recorded per sample using a BD LSRFortessa cytometer, BD FACSDiva Software (BD Biosciences) and FlowJo Software 10 (FlowJo Lcc; Ashland, OR, USA). Cellular debris was omitted from the analysis.
In vivo Assays
Animals
A total of 64 C.B-17 severe-combined immunodeficiency (SCID) mice (females, 25–30 g) were obtained and maintained at the animal facilities from Gonçalo Moniz Institute-FIOCRUZ (Salvador, Bahia, Brazil). Animals were housed in cages with free access to food and water. All animals were kept under a 12:12-h light-dark cycle (lights on at 6:00 a.m.). The animals were treated according to the ethical principles for animal experimentation of SBCAL (Brazilian Association of Laboratory Animal Science), Brazil. The Animal Ethics Committee of Gonçalo Moniz Institute–FIOCRUZ (Salvador, Bahia, Brazil) approved the experimental protocol (number 06/2015). Animal welfare was monitored throughout the study, and the pain and suffering were minimized.
Human Hepatocellular Carcinoma Xenograft Model
A human hepatocellular carcinoma xenograft model was generated by subcutaneously implanting HepG2 cells (1 × 107 cells per 500 μL) into the left front armpit of C.B-17 SCID mice. At the beginning of the experiment, the mice were randomly divided into six groups: group 1 animals received injections of vehicle with 5% DMSO solution (n = 10); group 2 animals received injections of doxorubicin (0.3 mg/kg/day, n = 8); group 3 animals received injections of complex 1 (0.5 mg/kg/day, n = 10); group 4 animals received injections of complex 1 (1 mg/kg/day, n = 10); group 5 animals received injections of complex 2 (0.5 mg/kg/day, n = 10); and group 6 animals received injections of complex 2 (1 mg/kg/day, n = 10). The treatments were initiated one day after the cancer cell injection. The animals were treated intraperitoneally (200 μL per animal) once a day for 21 consecutive days. On the 22nd day, the animals were anesthetized, and peripheral blood samples were collected from the brachial artery. Animals were euthanized by an anesthetic overdose, and tumors were excised and weighed.
Toxicological Evaluation
To evaluate the toxicological aspects, mice were weighed at the beginning and at the end of the experiment. Animals were observed for signs of abnormalities throughout the study. Hematological analysis was performed using an Advia 60 hematology system (Bayer, Leverkusen, Germany). Before fixation in 4% formaldehyde, tumors, livers, kidneys, hearts, and lungs were examined for size, color, and hemorrhaging. Histological analyses were performed under optical microscopy using hematoxylin/eosin and periodic acid-schiff (liver) staining by an experienced pathologist.
Statistical Analysis
Data are presented as the means ± S.E.M. or as the half-maximal inhibitory concentration (IC50) values with 95% confidence intervals, which were obtained by non-linear regression. Differences among experimental groups were compared using analysis of variance (ANOVA) followed by the Student–Newman–Keuls test (P < 0.05). All statistical analyses were performed using GraphPad Prism (Intuitive Software for Science; San Diego, CA, USA).
Results
Ruthenium Complexes Containing Heterocyclic Thioamidates Exhibit Potent Cytotoxicity to Different Cancer Cells
The cytotoxic potential of ruthenium complexes containing heterocyclic thioamidates was evaluated in eight different cancer cell lines (HepG2, HL-60, K-562, B16-F10, MCF-7, HCT116, HSC-3, and SCC-4) and in two non-cancerous cell lines (PBMCs and MRC-5) with the Alamar blue method after 72 h of incubation. Table 1 shows the results that were obtained. Complexes 1 and 2 were the most potent cytotoxic agents. Complex 1 had IC50 values that ranged from 1.4 to 2.6 μM for the cancer cell lines K-562 and HepG2, respectively; complex 2 had IC50 values that ranged from 0.9 to 7.3 μM for the cancer cell lines K-562 and MCF-7, respectively; complex 3 had IC50 values that ranged from 3.9 to 8.7 μM for the cancer cell lines B16-F10 and MCF-7, respectively; and complex 4 had IC50 values that ranged from 8.7 to 17.7 μM for the cancer cell lines HL-60 and B16-F10, respectively. Doxorubicin had IC50 values that ranged from 0.2 to 4 μM for the cancer cell lines HL-60/B16-F10 and SCC-4, respectively, while oxaliplatin had IC50 values that ranged from 0.6 to 7.7 μM for the cancer cell lines HL-60 and SCC-4, respectively. The selectivity index (SI) of each complex was calculated using the following formula: SI = IC50 [noncancerous cells]/IC50 [cancer cells]. Table 2 shows the calculated selectivity index. For most cancer cell lines, the complexes exhibited a selectivity index similar to that of the positive controls doxorubicin and oxaliplatin. Since complexes 1 and 2 were the most potent cytotoxic agents, these complexes were selected for further experiments.
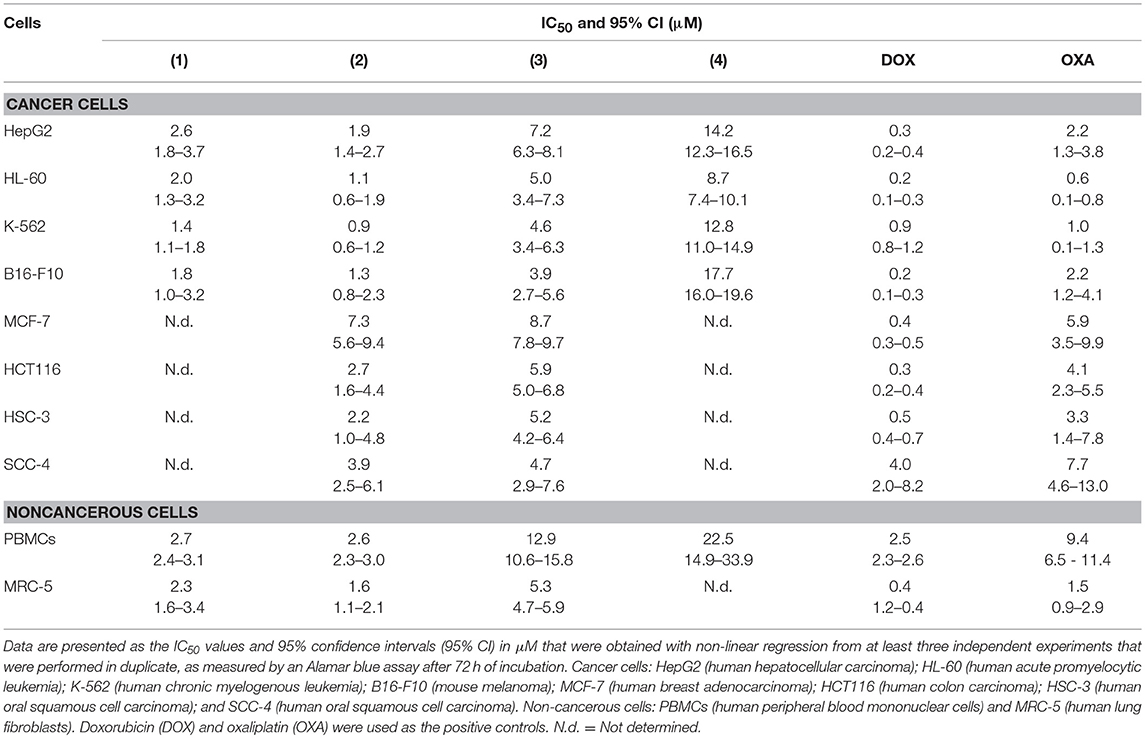
Table 1. Cytotoxic activity of ruthenium complexes containing heterocyclic thioamidates.
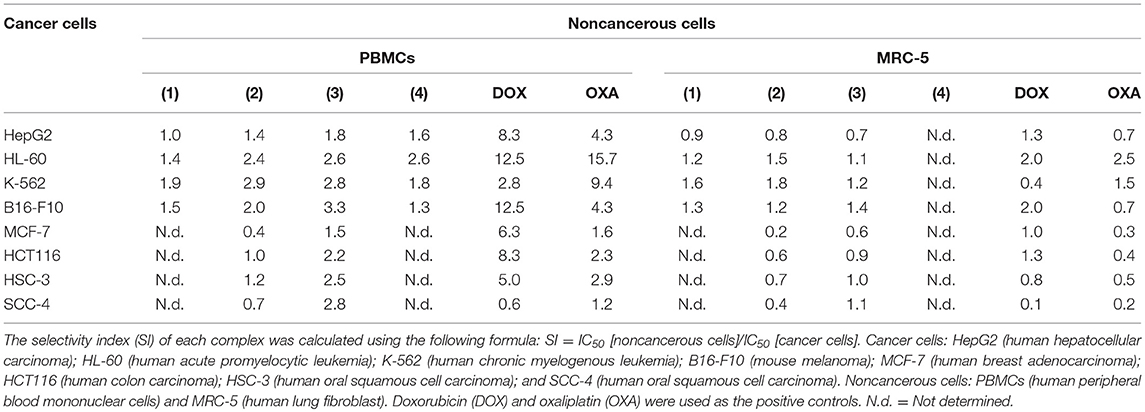
Table 2. Selectivity index of ruthenium complexes containing heterocyclic thioamidates.
Next, the cytotoxicity of complexes 1 and 2 were evaluated in an in vitro 3D model with cancer multicellular spheroids that formed from HepG2 cells, by using the Alamar blue method after 72 h of incubation. This cell line was chosen as a cellular model because it was among the most sensitive cell lines to the complexes. After treatment with both complexes, the cells in the spheroids were dissociated, indicating that drug penetrance and cytotoxicity had occurred in the 3D model (Figure 1B). The IC50 values were 6.5 and 7.3 μM for complexes 1 and 2, respectively (Figure 1C). Doxorubicin had an IC50 value of 10.3 μM. Figure 1D shows the cell survival curves of 3D vs. 2D culture models for these complexes.
To complement the Alamar blue method, the cell viability was evaluated after 24 and 48 h of treatment with complexes 1 and 2 with the trypan blue exclusion method in HepG2 cells. Both complexes significantly reduced (p < 0.05) the number of viable cells (Figure 2) compared to that of the negative controls. At concentrations of 5 and 10 μM, complex 1 reduced the number of viable cells by 40.1 and 78.9% after 24 h and 80.9 and 93.9% after 48 h, respectively, compared to that of the negative controls. At concentrations of 2 and 4 μM, complex 2 reduced the number of viable cells by 42.1 and 58.6% after 24 h and 67.3 and 93.1% after 48 h, respectively, compared to that of the negative controls. None of the complexes induced a significant (p > 0.05) increase in the number of non-viable cells compared to that in the negative controls. Doxorubicin also reduced the number of viable cells by 53.1 and 74.7% after 24 and 48 h of incubation, respectively, compared to that of the negative controls.
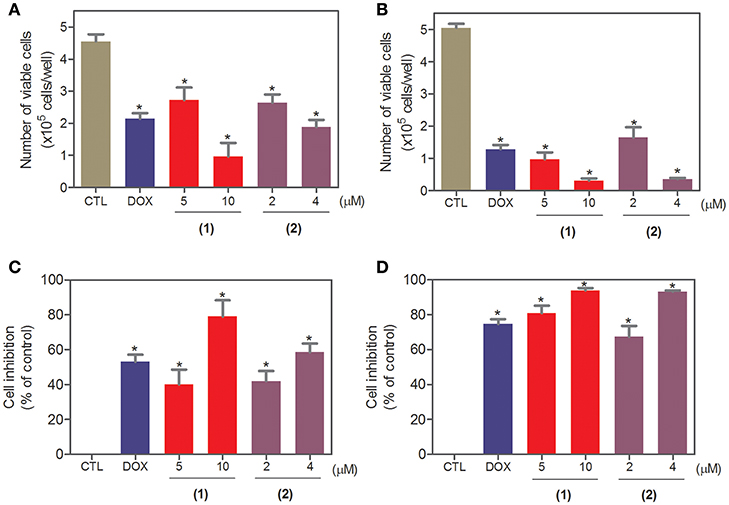
Figure 2. Effect of ruthenium complexes containing heterocyclic thioamidates on the cell viability of HepG2 cells, as determined by the trypan blue exclusion method after 24 (A,C) and 48 (B,D) h of incubation. The negative control (CTL) was treated with the vehicle (0.2% DMSO) that was used to solubilize and dilute the complexes, and doxorubicin (DOX, 2 μM) was used as the positive control. Data are presented as the mean ± S.E.M. of at least three independent experiments that were performed in duplicate. *P < 0.05 compared with the negative control, as determined with ANOVA followed by the Student-Newman-Keuls test.
Ruthenium Complexes Containing Heterocyclic Thioamidates Interact With DNA
The DNA-binding ability of ruthenium complexes containing heterocyclic thioamidates was assessed in a cell-free system using a ctDNA model. In this assay, the ability of the drugs to intercalate with the DNA is quantified by an ethidium bromide replacement method, where the drugs displace intercalated ethidium bromide from ctDNA and subsequently reduce the fluorescence of the ethidium bromide-DNA complex by releasing ethidium bromide in the solution. Both complexes significantly reduced (p < 0.05) the fluorescence intensity compared to that of the negative controls, indicating that both complexes are DNA intercalators (Figure 3). Compared to the negative controls, at concentrations of 5, 10, and 20 μM, complex 1 reduced the fluorescence intensity by 41.6, 34.0, and 30.4%, respectively, while complex 2 reduced the fluorescence intensity by 36.9, 42.9, and 45.4%, respectively, at the same concentrations. Doxorubicin, a known DNA intercalator, was used at 20 μM as the positive control, and reduced the fluorescence intensity by 89.0% compared to that of the negative controls.
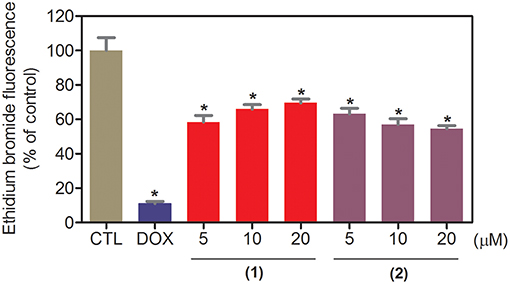
Figure 3. DNA interaction of ruthenium complexes containing heterocyclic thioamidates, as evaluated by the ability of the complexes to displace ethidium bromide from calf thymus DNA. The negative control (CTL) was treated with the vehicle (0.2% DMSO) that was used to solubilize and dilute the complexes, and doxorubicin (DOX, 20 μM) was used as the positive control. Data are presented as the mean ± S.E.M. of three independent experiments that were performed in duplicate. *P < 0.05 compared with the negative control, as determined by ANOVA, followed by the Student-Newman-Keuls test.
Ruthenium Complexes Containing Heterocyclic Thioamidates Trigger Caspase-Mediated Apoptosis in HepG2 Cells
Next, the DNA content was quantified by flow cytometry to measure the internucleosomal DNA fragmentation and cell cycle distribution in HepG2 cells treated with ruthenium complexes containing heterocyclic thioamidates after 24 and 48 h of incubation, and the results are shown in Figure 4. All DNA that was subdiploid (sub-G0/G1) was considered fragmented. Both complexes caused significant DNA fragmentation (p < 0.05) compared to that in the negative controls. Doxorubicin caused cell cycle arrest at the G2/M phase, followed by DNA fragmentation.
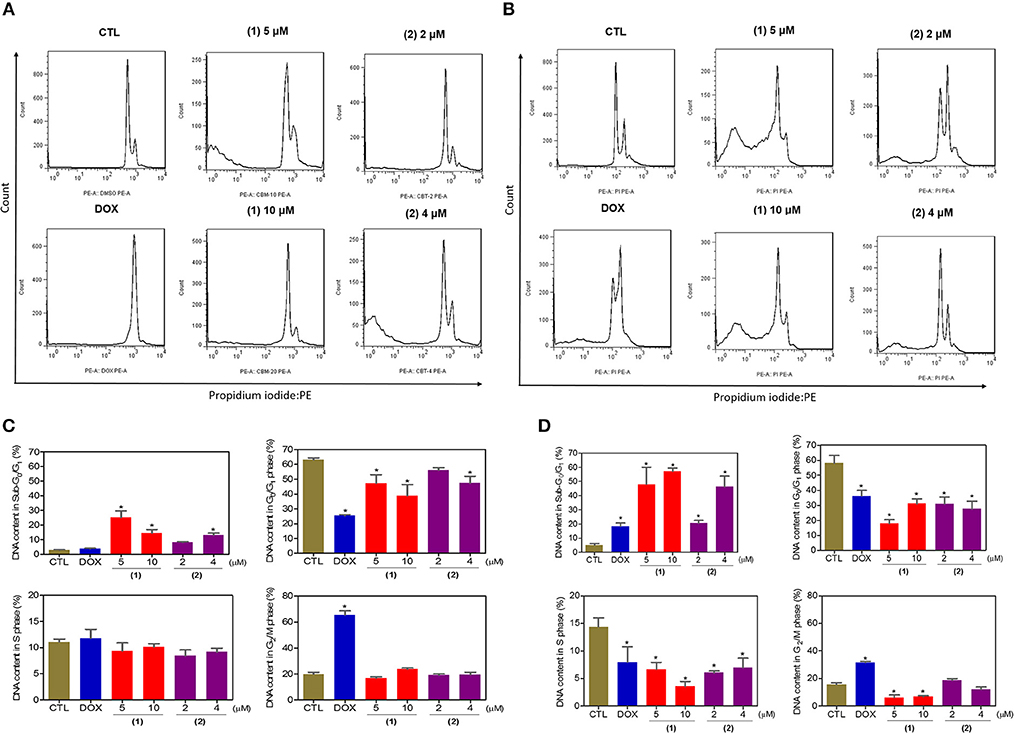
Figure 4. Effect of ruthenium complexes containing heterocyclic thioamidates on the cell cycle distribution of HepG2 cells after 24 (A,C) and 48 (B,D) h of incubation. The negative control (CTL) was treated with the vehicle (0.2% DMSO) that was used to solubilize and dilute the complexes, and doxorubicin (DOX, 2 μM) was used as the positive control. Data are presented as the mean ± S.E.M. of three independent experiments that were performed in duplicate. *P < 0.05 compared with the negative control, as determined by ANOVA, followed by the Student-Newman-Keuls test. Ten thousand events were evaluated per experiment, and cellular debris was omitted from the analysis.
Cell morphology was assessed by light microscopy using May-Grunwald-Giemsa staining after 24 and 48 h of incubation (Figure 5). Compared to the negative controls, both complexes induced a reduction in the cell volume, chromatin condensation and fragmentation of the nuclei, which are morphological aspects that are associated with cell death via apoptosis. To complement the light microscopy analysis, light-scattering features were assessed by flow cytometry and compared to the negative controls, both complexes caused cell shrinkage, as observed by the decrease in forward-light scatter, and nuclear condensation, as observed by the increase in side scatter (Figure 6). Doxorubicin also caused apoptosis-related morphological alterations.
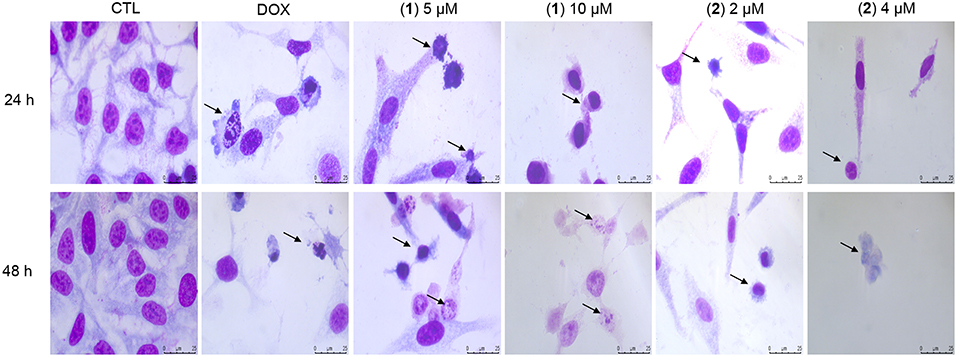
Figure 5. Effect of ruthenium complexes containing heterocyclic thioamidates in the morphological analysis of HepG2 cells after 24 and 48 h of incubation. The cells were stained with May-Grunwald-Giemsa and examined by light microscopy (bar = 20 μm). The negative control (CTL) was treated with the vehicle (0.2% DMSO) that was used to solubilize and dilute the complexes, and doxorubicin (DOX, 2 μM) was used as the positive control. Arrows indicate cell shrinkage or cells with fragmented DNA.
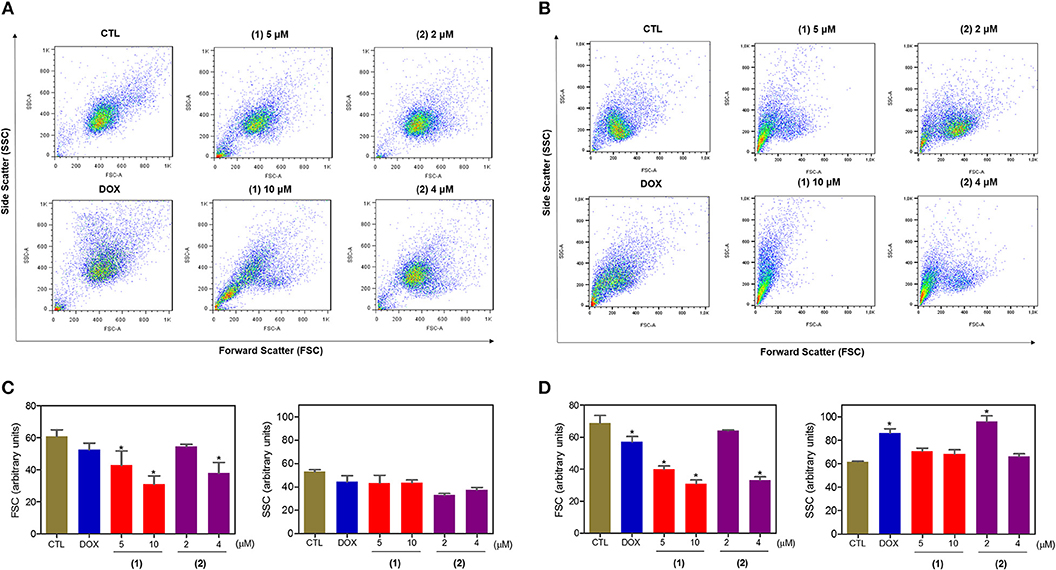
Figure 6. Effect of ruthenium complexes containing heterocyclic thioamidates on the morphology of HepG2 cells, as determined by light-scattering features detected by flow cytometry after 24 (A,C) and 48 (B,D) h of incubation. The negative control (CTL) was treated with the vehicle (0.2% DMSO) that was used to solubilize and dilute the complexes, and doxorubicin (DOX, 2 μM) was used as the positive control. Data are presented as the mean ± S.E.M. of three independent experiments that were performed in duplicate. *P < 0.05 compared with the negative control, as determined by ANOVA followed by the Student-Newman-Keuls test. Ten thousand events were evaluated per experiment, and cellular debris was omitted from the analysis.
Apoptosis quantification was performed with annexin-V/PI double staining using flow cytometry after 24 and 48 h of incubation (Figure 7). Annexin-V positive cells were used as indicators of apoptosis. Compared to the negative controls, both complexes significantly increased the percentage of apoptotic cells at both incubation times (P < 0.05). No significant increase in the number of necrotic cells was observed in HepG2 cells treated with the complexes compared to that in the negative controls (P > 0.05). Furthermore, compared to the negative controls, coincubation with Z-VAD(OMe)-FMK, a pan-caspase inhibitor, partly reduced the apoptosis induced by both complexes (Figure 8), and the mitochondrial transmembrane potential was reduced after the treatment of HepG2 cells with these complexes (Figure 9), indicating that ruthenium complexes containing heterocyclic thioamidates induce cell death by apoptosis via caspase-dependent pathways. On the other hand, since coincubation with Z-VAD(OMe)-FMK did not fully block the complexes-induced apoptotic cell death, other cell death pathways, e.g., necroptosis and autophagy, may also be involved in the cell death that was caused by these complexes.
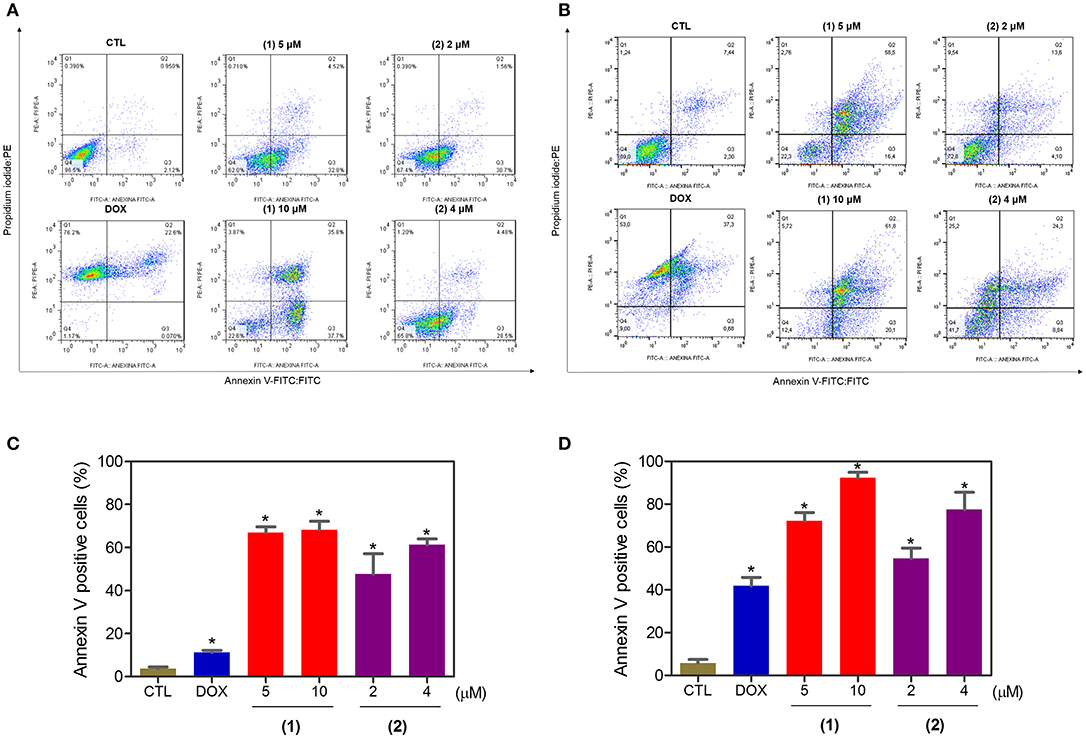
Figure 7. Effect of ruthenium complexes containing heterocyclic thioamidates on the induction of apoptosis in HepG2 cells, as determined by flow cytometry using annexin V-FITC/PI staining after 24 (A,C) and 48 (B,D) h of incubation. The negative control (CTL) was treated with the vehicle (0.2% DMSO) that was used to solubilize and dilute the complexes, and doxorubicin (DOX, 2 μM) was used as the positive control. Data are presented as the mean ± S.E.M. of three independent experiments that were performed in duplicate. *P < 0.05 compared with the negative control, as determined by ANOVA followed by the Student-Newman-Keuls test. Ten thousand events were evaluated per experiment, and cellular debris was omitted from the analysis.
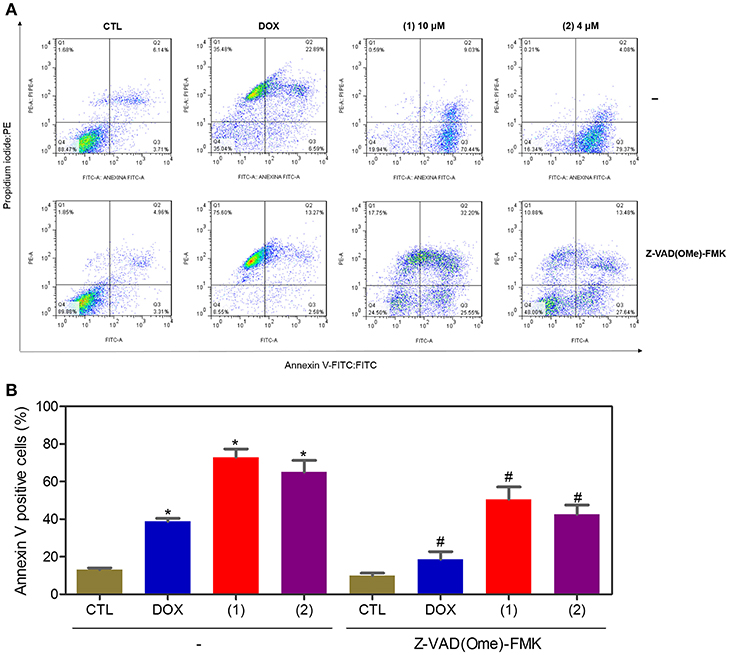
Figure 8. Effect of the pan-caspase inhibitor [Z-VAD(OMe)-FMK] on the apoptosis that was induced by ruthenium complexes containing heterocyclic thioamidates, as determined by flow cytometry using annexin V-FITC/PI staining. (A) Representative flow cytometry dot plots showing the percentage of cells in the viable, early-apoptotic, late-apoptotic, and necrotic stages. (B) Quantification of the apoptotic cells (early- and late-apoptotic cells). For the protection assay, the cells were preincubated for 2 h with 50 μM Z-VAD(Ome)-FMK and were then incubated with 10 μM of complex 1 or 4 μM of complex 2 for 48 h. The negative control (CTL) was treated with the vehicle (0.2% DMSO) that was used to solubilize and dilute the complexes, and doxorubicin (DOX, 2 μM) was used as the positive control. Data are presented as the mean ± S.E.M. of three independent experiments that were performed in duplicate. *P < 0.05 compared with the negative control, as determined by ANOVA followed by the Student-Newman-Keuls test. #P < 0.05 compared with the respective treatment without inhibitor, as determined by ANOVA followed by the Student-Newman-Keuls test. Ten thousand events were evaluated per experiment, and cellular debris was omitted from the analysis.
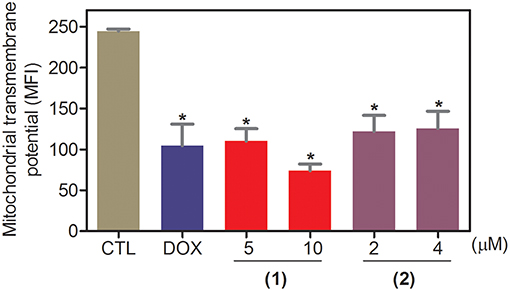
Figure 9. Effect of ruthenium complexes containing heterocyclic thioamidates on the mitochondrial membrane potential of HepG2 cells, as determined by flow cytometry using rhodamine 123 staining after 24 h of incubation. The negative control (CTL) was treated with the vehicle (0.2% DMSO) that was used to solubilize and dilute the complexes, and doxorubicin (DOX, 2 μM) was used as the positive control. Data are presented as the mean ± S.E.M. of three independent experiments that were performed in duplicate. *P < 0.05 compared with the negative control, as determined by ANOVA followed by the Student-Newman-Keuls test. Ten thousand events were evaluated per experiment, and cellular debris was omitted from the analysis. MFI: mean fluorescence intensity.
In complement to the effect of these complexes in cancer cells, the cytotoxicity of the complexes was also determined in the BAD KO SV40 MEF (BAD gene knockout immortalized mouse embryonic fibroblast) cell line and in its parental WT SV40 MEF (wild-type immortalized mouse embryonic fibroblast) cell line with the Alamar blue method after 72 h of incubation (Table S1). The IC50 values for complexes 1 and 2 were 0.7 and 2.9 μM for the BAD KO SV40 MEF cell line and 1.1 and 2.0 μM for the WT SV40 MEF cell line, respectively, indicating that the BAD gene is not essential for the cytotoxicity of these complexes in fibroblasts cells. Doxorubicin had IC50 values of 0.4 and 0.04 μM in the BAD KO SV40 MEF and WT SV40 MEF cell lines, respectively, while cisplatin had IC50 values of 47.3 and 36.9 μM in the BAD KO SV40 MEF and WT SV40 MEF cell lines, respectively.
Ruthenium Complexes Containing Heterocyclic Thioamidates Cause ERK1/2-Mediated Apoptosis in HepG2 Cells Through ROS- and p53-Independent Pathways
Since mitogen-activated protein kinase (MAPK) signaling plays a crucial role in apoptosis that is induced by DNA intercalating agents, we decided to assess the role of the three MAPK proteins, ERK1/2, JNK/SAPK and p38 MAPK, in apoptosis that is induced by ruthenium complexes containing heterocyclic thioamidates in HepG2 cells. Initially, we quantified apoptosis induced by the complexes in HepG2 cells that were coincubated with a JNK/SAPK inhibitor (SP600125), p38 MAPK inhibitor (PD169316), and an MEK inhibitor (U-0126, which inhibits the activation of ERK1/2) (Figure 10). Next, we quantified the phosphorylation status of JNK/SAPK (pT183/pY185), p38 MAPK (pT180/pY182) and ERK1/2 (pT202/pY204) expression in HepG2 cells after acute (15 and 30 min) and prolonged (24 h) incubations with the complexes (Figure 11). Compared to the negative controls, complex 1 induced the phosphorylation of ERK1/2 after 15 min of incubation, but no significant increase was observed in the phosphorylation of JNK/SAPK or p38 MAPK. Furthermore, coincubation with an MEK inhibitor reduced the level of apoptosis that was induced by both complexes compared to negative controls. Coincubation with a JNK/SAPK inhibitor or a p38 MAPK inhibitor did not reduce the level of apoptosis induced by these complexes compared to that in the negative controls.
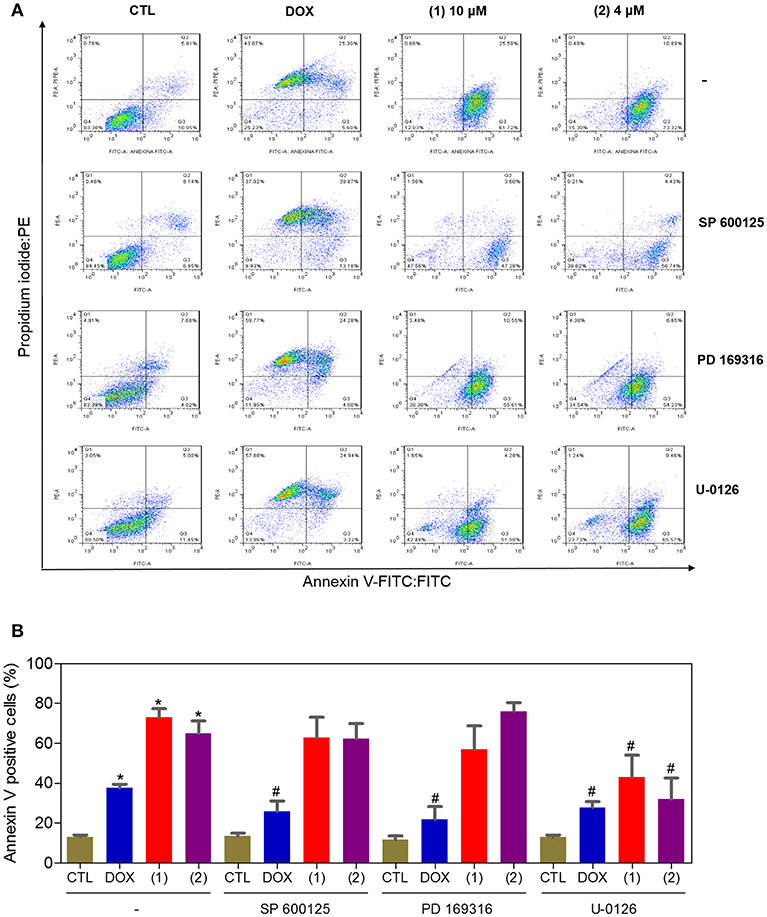
Figure 10. Effect of the JNK/SAPK (SP600125), p38 MAPK (PD169316), and MEK (U0126) inhibitors on the apoptosis that was induced by ruthenium complexes containing heterocyclic thioamidates in HepG2 cells, as determined by flow cytometry using annexin V-FITC/PI staining. (A) Representative flow cytometry dot plots showing the percentage of cells in the viable, early-apoptotic, late-apoptotic and necrotic stages. (B) Quantification of the apoptotic cells (early- and late-apoptotic cells). For the protection assays, the cells were preincubated for 2 h with 5 μM SP600125, 5 μM PD169316 or 5 μM U0126 and were then incubated with 10 μM of complex 1 or 4 μM of complex 2 for 48 h. The negative control (CTL) was treated with the vehicle (0.2% DMSO) that was used to solubilize and dilute the complexes, and doxorubicin (DOX, 2 μM) was used as the positive control. Data are presented as the mean ± S.E.M. of three independent experiments that were performed in duplicate. *P < 0.05 compared with the negative control, as determined by ANOVA followed by the Student-Newman-Keuls test. #P < 0.05 compared with the respective treatment without inhibitor, as determined by ANOVA followed by the Student-Newman-Keuls test. Ten thousand events were evaluated per experiment, and cellular debris was omitted from the analysis.
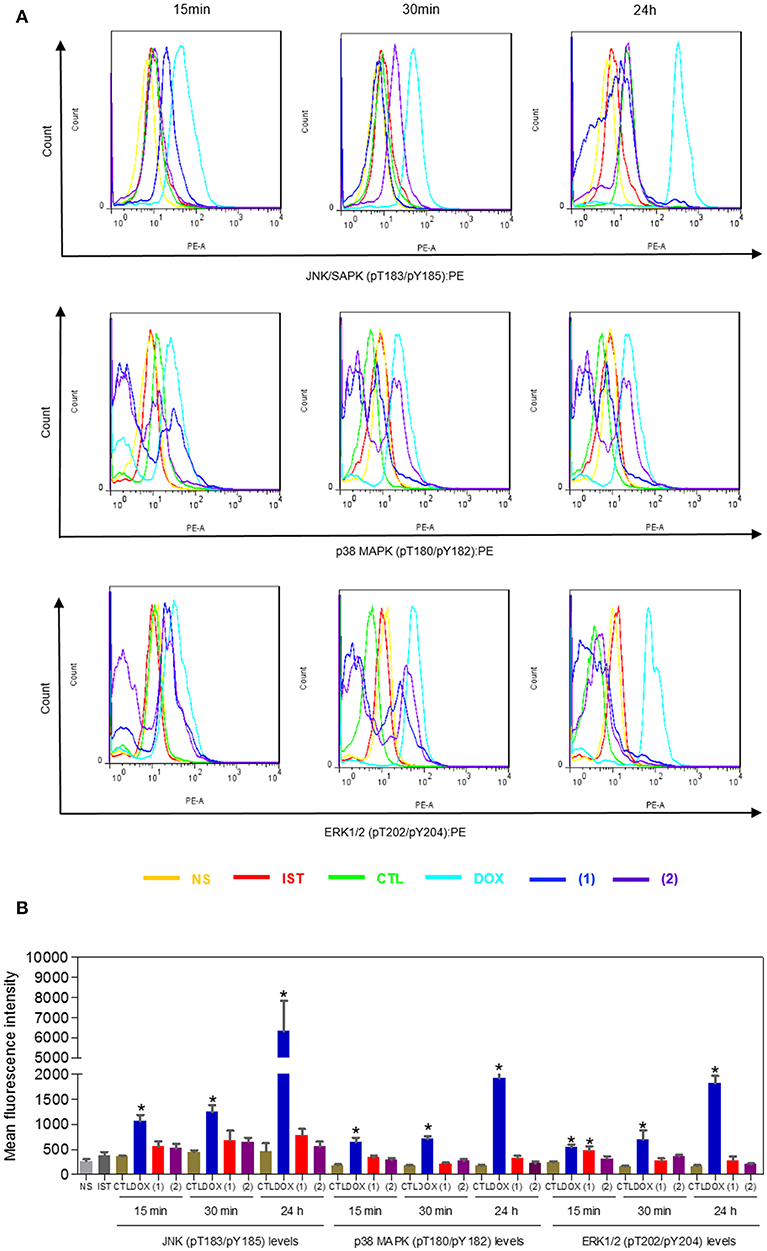
Figure 11. Effect of ruthenium complexes containing heterocyclic thioamidates on the JNK/SAPK (pT183/pY185), p38 MAPK (pT180/pY182), and ERK1/2 (pT202/pY204) levels, as determined by the phosflow analysis of HepG2 cells treated with 10 μM of complex 1 or 4 μM of complex 2 for an acute (15 or 30 min) or prolonged (24 h) incubation. (A) Representative flow cytometry histograms. (B) Quantification of the JNK/SAPK (pT183/pY185), p38 MAPK (pT180/pY182) and ERK1/2 (pT202/pY204) levels. The negative control (CTL) was treated with the vehicle (0.2% DMSO) that was used to solubilize and dilute the complexes, and doxorubicin (DOX, 2 μM) was used as the positive control. Data are presented as the mean ± S.E.M. of three independent experiments that were performed in duplicate. *P < 0.05 compared with the negative control, as determined by ANOVA followed by the Student-Newman-Keuls test. Ten thousand events were evaluated per experiment, and cellular debris was omitted from the analysis. NS, non-stained cells (basal cell fluorescence); IST, isotype control.
Moreover, ERK1/2 activation is associated with the induction of oxidative stress and/or the activation of p53 signaling. Thus, we assessed the role of oxidative stress induction and p53 activation in apoptosis by the complexes in HepG2 cells. First, we quantified ROS levels in HepG2 cells treated with the complexes for 1 and 3 h (Figure S1). Then, we quantified the level of apoptosis induced by the complexes in HepG2 cells that were coincubated with an antioxidant (NAC) (Figure S2) and a p53 inhibitor (cyclic pifithrin-α) (Figure S3). However, compared to the negative controls, the complexes did not induce a significant increase in ROS levels, and coincubation with an antioxidant and a p53 inhibitor did not reduce the apoptosis induced by the complexes, indicating that the complexes induce apoptosis through ROS- and p53-independent pathways in HepG2 cells.
Ruthenium Complexes Containing Heterocyclic Thioamidates Reduce HepG2 Cell Growth in a Xenograft Model
The in vivo anti-liver cancer activity of ruthenium complexes containing heterocyclic thioamidates was assessed in C.B-17 SCID mice engrafted with HepG2 cells. The animals were treated with the complexes at doses of 0.5 and 1 mg/kg/day by intraperitoneal injections for 21 consecutive days. Both complexes were able to inhibit HepG2 cell growth in mice compared to that of the negative controls (Figures 12A,B). At the end of the treatment, the mean tumor mass of the negative control animals was 0.6 ± 0.1 g. In the animals treated with complex 1, the mean tumor masses were 0.4 ± 0.1 g and 0.3 ± 0.1 g at the lower and higher doses, respectively. In the animals treated with complex 2, the mean tumor masses were 0.3 ± 0.1 g and 0.2 ± 0.1 g at the lower and higher doses, respectively. Compared to the negative controls, the tumor mass inhibition rates were 31.5–45.4% and 46.9–67.7% for complexes 1 and 2, respectively. Doxorubicin (at a dose of 0.3 mg/kg/day) reduced the tumor weight by 36.8% compared to that of the negative controls. In the histological analysis, all groups exhibited a solid pattern composed of polygonal and pleomorphic tumor cells, eosinophilic granular cytoplasm, rounded nuclei and prominent nucleoli (Figure 12C). The stroma was fibrous and hypervascular. The group of animals in the negative control group exhibited larger tumors, and the areas of necrosis, dystrophic calcification and inflammation were more evident than those in the other experimental groups. For the doxorubicin group and the groups that were treated with complexes 1 and 2, the tumors were more encapsulated, and the tumor cell islands were smaller, with fewer mitotic figures when compared to those in the negative control group. The most evident fibrosis was observed in the complex 2 group (1 mg/kg/day) compared to that in the other groups.
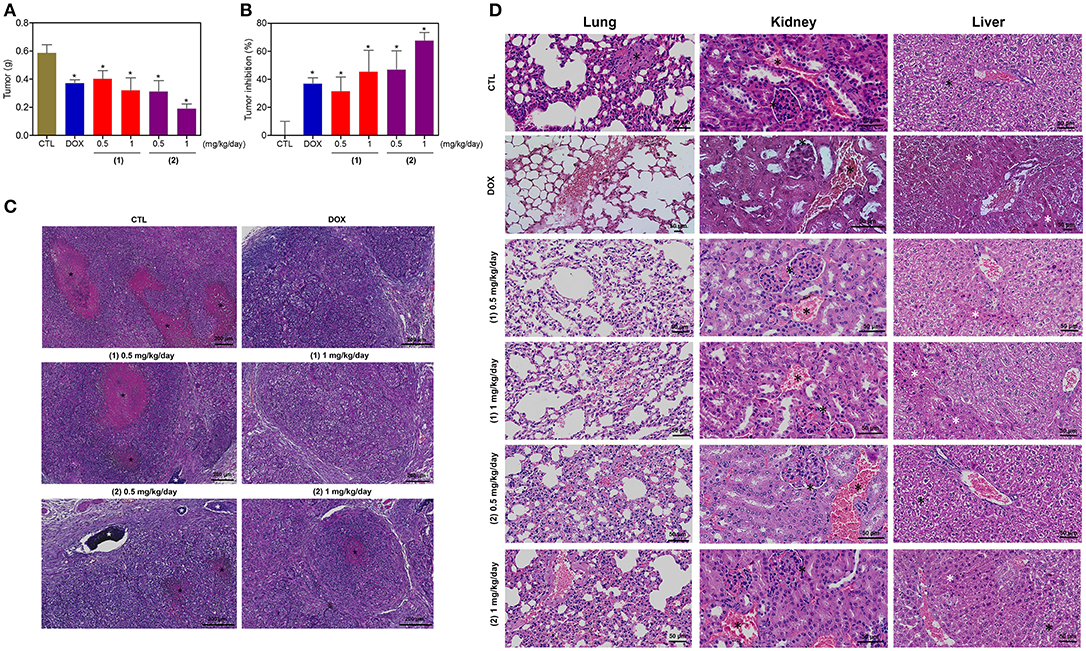
Figure 12. In vivo antitumor activity of ruthenium complexes containing heterocyclic thioamidates in C.B-17 SCID mice with HepG2 cell xenografts. (A) Quantification of the tumor weight (g). (B) Quantification of the tumor inhibition (%). Data are presented as the mean ± S.E.M. of 8–10 animals. *P < 0.05 compared with the negative control, as determined by ANOVA followed by the Student-Newman-Keuls test. (C) Representative histological analysis of the tumors that were stained with hematoxylin and eosin and analyzed by light microscopy. The areas of necrosis and dystrophic calcification are represented by black and white asterisks, respectively. (D) Representative histological analysis of the lungs, kidneys and livers that were stained with hematoxylin and eosin and analyzed by light microscopy. Black asterisks represent metastasis (lung), the thickening of the basal membrane of the renal glomerulus with decreased urinary space or vascular congestion (kidney) and hydropic degeneration (liver). White asterisks represent the coagulation necrosis of hepatic cells. The negative control (CTL) was treated with the vehicle (5% DMSO) that was used to solubilize and dilute the complexes, and doxorubicin (DOX, 0.3 mg/kg/day) was used as the positive control. Beginning 1 day after tumor implantation, the animals were treated via an intraperitoneal route for 21 consecutive days.
Some toxicological aspects were also evaluated in the mice treated with the complexes. There was no significant alteration in the body weight of animals treated with the complexes (P > 0.05) compared to that in the negative controls. No significant alterations were observed in the liver, kidney, lung or heart wet weight of any group (P > 0.05) (Table S2). In addition, the number of white and erythrocyte blood cells in the mice treated with the complexes was also examined (Table S3). The number of leukocytes and erythrocytes remained unchanged after treatment with the complexes compared to that in the controls (P > 0.05).
Morphological analyses of the liver, kidneys, lungs, and heart in all groups were performed (Figure 12D). In the liver, the acinar architecture and centrilobular vein were preserved in all groups. The inflammation observed in the liver portal space was mild in most animals. Other findings, such as congestion and hydropic degeneration, were found in all groups, ranging from mild to moderate. In addition, focal areas of coagulation necrosis were observed in all groups and were more evident in the complex 2 group compared to the other groups, indicating hepatotoxicity. In the kidneys, the tissue architecture was maintained in all experimental groups. Histopathological changes, including vascular congestion and the thickening of the basal membrane of the renal glomerulus with decreased urinary space, were observed in all kidneys. In the lungs, the architecture of the parenchyma was partially maintained in all groups, and a thickening of the alveolar septum with decreased airspace was observed, ranging from mild to moderate. In addition, some animals in the experimental groups showed mild emphysema. Significant inflammation, predominantly of mononuclear cells, edema, congestion and hemorrhage were frequently observed, ranging from mild to severe. Tumor nodules in the lungs were observed only in one animal in the negative control group (DMSO 5%). The heart did not show alterations in any group.
Discussion
The current prospects for the use of ruthenium complexes as potential antineoplastic drugs is both promising, particularly for some specific candidates, as well as disappointing, since none of these molecules has been approved for clinical use in cancer therapy at the present moment (27, 28). The fact that almost 30 years after the first studies with ruthenium complexes were performed, these compounds continue to be viewed as only “promising compounds” is worrisome (27). On the other hand, an impressive number of studies have demonstrated the potential of ruthenium complexes as next-generation anticancer metallotherapeutics that show more selectivity to cancer cells and the ability to overcome the resistance that is observed for platinum-based drugs (28). In this paper, a series of ruthenium complexes containing heterocyclic thioamidates were studied as novel anticancer drug candidates for liver cancer treatment.
Herein, we demonstrated for the first time that the ruthenium complexes containing heterocyclic thioamidates that were studied in this work bind to DNA, inhibit cell proliferation, trigger caspase-mediated apoptosis through ERK1/2 signaling in HepG2 cells, and can reduce the growth of HepG2 cells that are engrafted in C.B-17 SCID mice. The ruthenium complexes containing heterocyclic thioamidates that were tested in this work had potent cytotoxicity against cancer cells with different histological types, with IC50 values below 20 μM, and complexes 1 and 2 were the most potent cytotoxic agents, with IC50 values below 10 μM. These data corroborate the results of Correa et al. (18) and Silva (19), who reported that when these complexes were tested in a small panel of cancer cells (MCF-7, DU-145 [human prostate carcinoma], A549 [human lung carcinoma], and HepG2), they had IC50 values below 10 μM.
Various metal-based complexes have been synthesized containing heterocyclic thioamidates with cytotoxic properties. A silver-chloride complex with the heterocyclic thioamide 5-chloro-2-mercaptobenzothiazole and tri(p-tolyl)phosphine displayed cytotoxicity to leiomyosarcoma cancer cells (LMS) from Wistar rats, with an IC50 value of 13.7 μM (29). On the other hand, different metallocomplexes have been generated using heterocyclic thioamidates that were only cytotoxic at high concentrations. Ruthenium complexes containing the thioamide ligand (2-hydroxy-5-nitrophenyl)(pyrrolidin-1-yl)methanethione were previously synthesized and investigated for their cytotoxic activity, showing IC50 values above 40 μM in human cervical carcinoma (HeLa) and breast adenocarcinoma (MCF-7) cell lines (30). The bismuth complex with the heterocyclic thioamide 2-mercapto-1-methylimidazole had IC50 values above 30 μM for the HeLa and MCF-7 cell lines (31). The antimony bromide complex with the heterocyclic thioamide 2-mercapto-1-methylimidazole had IC50 values above 15 μM in murine leukemia cells (L1210), murine mammary carcinoma cells (FM3A), human T-lymphocyte (Molt4/C8, CEM), and human cervix carcinoma cells (HeLa)(32). In our outcomes, ruthenium-based heterocyclic thioamidates displayed greater cytotoxicity then previous heterocyclic thioamidates metallocomplexes.
As mentioned above, the ruthenium complexes containing heterocyclic thioamidates that were studied in this work were previously reported to bind to DNA and bovine serum albumin and to inhibit DNA supercoiled relaxation that is mediated by human topoisomerase IB (18, 19). Herein, we observed that these ruthenium complexes behave like DNA intercalating agents, which can be caused by drug-DNA interactions at the minor groove or/and electrostatic interactions. These effects can occur with octahedral compounds, such as the ruthenium complexes, that are very bulky and that do not intercalate (33, 34). A ruthenium complex containing guanidine has been reported to bind to DNA and induce DNA damage, cell cycle arrest and to activate the typical apoptosis pathways in MCF-7 cells (35). A ruthenium complex with thymine was reported to bind to DNA and human and bovine serum albumin and to induce caspase-mediated apoptosis in human promyelocytic leukemia HL-60 cells (9, 10). On the other hand, the half-sandwich iridium and ruthenium complexes containing P∧P-chelating ligands cause cell apoptosis in human lung carcinoma A549 cells, and although these types of complexes interact with ctDNA, DNA appears not to be the major target (36). Moreover, the ruthenium complexes with 5-fluorouracil display enhanced cytotoxicity to different cancer cells and are able to induce caspase-mediated apoptosis in HCT116 cells but do not induce DNA intercalation (11). In the present work, we observed that ruthenium complexes containing heterocyclic thioamidates bind to DNA and inhibit cell proliferation, triggering caspase-mediated apoptosis in HepG2 cells.
Additionally, we investigated the role of the MAPK pathway in apoptosis that is induced by ruthenium complexes containing heterocyclic thioamide. MAPKs are serine/threonine protein kinases that have an important function in both cell survival and cell death. The MAPK pathway is currently known to comprise four subpathways: ERK (ERK1 and ERK2), JNK/SAPK (JNK1, JNK2, and JNK3), p38 MAPK (α, β, δ, and γ) and Big MAP kinase 1 (BMK1 or ERK5). The effects of the activation of ERK1/2 include gene expression that promotes cell proliferation, differentiation and cellular survival. The functions of JNK/SAPK and p38 MAPK activation include cell differentiation, apoptosis and survival. However, the function and regulation of BMK1/ERK5 have been less intensively explored, and it has been reported that this subpathway can regulate cell growth, differentiation and survival (37–41).
Herein, we observed that ruthenium complexes containing heterocyclic thioamidates induce apoptosis that is mediated by MAPK ERK1/2 signaling. In fact, although the activation of ERK1/2 has mainly been associated with a prosurvival function, DNA damage-induced ERK1/2 activation causes apoptotic cell death (12, 42–44). The induction of ERK1/2-mediated apoptosis by the ruthenium complexes containing heterocyclic thioamidates that were studied in this work seems to occur via direct DNA damage since these complexes failed to induce ROS production (an indirect mechanism of DNA damage induction) and were able to induce DNA intercalation. Moreover, ERK1/2 activation can induce apoptosis via a p53-dependent or p53-independent pathway (43, 45, 46). The apoptosis induced by the ruthenium complexes containing heterocyclic thioamidates was not reduced by coincubation with a p53 inhibitor, indicating that these complexes cause apoptosis through a p53-independent pathway in HepG2 cells. In support of these data, previous reports have also shown ruthenium-based compounds to induce apoptosis in cancer cells via MAPK pathways that were either ROS- and p53-dependent or independent (12, 47, 48).
In addition, we observed that the ruthenium complexes containing heterocyclic thioamidates reduced the growth of HepG2 cells that were engrafted in C.B-17 SCID mice and had a similar efficacy as the positive control doxorubicin. In relation to toxicity, evidence of hepatotoxicity was observed after treatment with complex 2. On the other hand, although some histological changes were found, these changes may be reversed after treatment. Similarly, ruthenium imidazole complex and association of ruthenium-arene complex and erlotinib were also able to inhibit cancer cell growth in mice engrafted with A549 and A2780 cells, respectively (49, 50).
Overall, this study revealed that ruthenium complexes containing heterocyclic thioamidates bind to DNA and inhibit cell proliferation, triggering caspase-mediated apoptosis through ERK1/2 signaling in HepG2 cells, which occurs via a ROS- and p53-independent pathway (Figure 13). Moreover, these complexes reduce the growth of HepG2 cells that are engrafted in C.B-17 SCID mice. These results indicate that these complexes are novel anticancer drug candidates for liver cancer treatment.
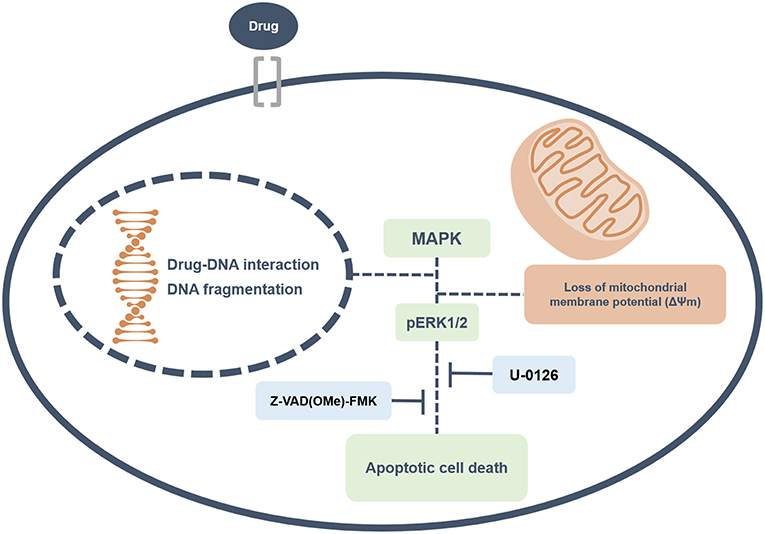
Figure 13. Summary of the molecular mechanisms of ruthenium complexes containing heterocyclic thioamidates in HepG2 cells. Herein, we revealed that these complexes bind to DNA triggering caspase-mediated apoptosis through ERK1/2 signaling in HepG2 cells.
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
PBMC were obtained from peripheral blood of donors voluntary with informed consent (number 031019/2013) approved by the Human Ethics Committee of Gonçalo Moniz Institute from Oswaldo Cruz Foundation (IGM-FIOCRUZ/BA). The animal experiments were conducted in accordance with the experimental protocol (number 06/2015) approved by the Animal Ethics Committee of IGM-FIOCRUZ/BA.
Author Contributions
SN, MdS, MS, CR, AB, and DB conceived and designed the experiments. SN, NdC, AR, and LB performed the in vitro and in vivo experiments. SN, NdC, MdS, AR, LB, RD, CS, CR, and DB analyzed the data. CR, MS, AB, and DB contributed reagents, materials, and analysis tools. DB wrote the paper. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to the flow cytometry and histotechnology cores of FIOCRUZ-Bahia for collecting the flow cytometric data and performing the histological techniques, respectively. This work received financial support and was supported by fellowships from the Brazilian agencies Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação de Amparo à Pesquisa do Estado da Bahia (FAPESB), and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.00562/full#supplementary-material
Abbreviations
ANOVA, analysis of variance; ATCC, American type culture collection; BAD KO SV40 MEF, BAD gene knockout immortalized mouse embryonic fibroblast; bipy, 2,2′-bipyridine; ctDNA, calf thymus DNA; DCF-DA, 2′,7′-dichlorofluorescin diacetate; dmp, 4,6-diamino-2-mercaptopyrimidine; DMSO, dimethyl sulfoxide; dppb, 1,4-bis(diphenylphosphino)butane; ERK, extracellular signal-regulated kinase; HCC, hepatocellular carcinoma; IC50, half-maximal inhibitory concentration; Im, imidazole; Ind, indazole; JNK/SAPK, Jun kinase; KP1019, IndH]trans-[RuCl4(Ind)2]; MAPKs, mitogen-activated protein kinases; mmi, mercapto-1-methyl-imidazole; mpca, 6-mercaptopyridine-3-carboxylic acid; NAC, N-acetylcysteine; NAMI-A, ImH]trans-[RuCl4(Im)(dmso-S); PBMCs, peripheral blood mononuclear cells; ROS, reactive oxygen species; SCID, severe-combined immunodeficiency; SI, selectivity index; TBE, trypan blue exclusion; tzdt, 1,3-thiazolidine-2-thione; WT SV40 MEF, wild-type immortalized mouse embryonic fibroblast.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J-F, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. (2008) 359:378–90. doi: 10.1056/NEJMoa0708857
3. Rosamel L, Blanc JF. Emerging tyrosine kinase inhibitors for the treatment of hepatocellular carcinoma. Expert Opin Emerg Drugs. (2017) 22:175–90. doi: 10.1080/14728214.2017.1336538
4. Meyer T. Treatment of advanced hepatocellular carcinoma: beyond sorafenib. Lancet Gastroenterol Hepatol. (2018) 3:218–20. doi: 10.1016/S2468-1253(17)30255-8
5. Lazarević T, Rilak A, Bugarčić ŽD. Platinum, palladium, gold and ruthenium complexes as anticancer agents: Current clinical uses, cytotoxicity studies and future perspectives. Eur J Med Chem. (2017) 142:8–31. doi: 10.1016/j.ejmech.2017.04.007
6. Sava G, Bergamo A, Dyson PJ. Metal-based antitumour drugs in the post-genomic era: what comes next? Dalton Trans. (2011) 40:9069–75. doi: 10.1039/c1dt10522a
7. D'Sousa Costa CO, Araujo Neto JH, Baliza IRS, Dias RB, Valverde LF, Vidal MTA, et al. Novel piplartine-containing ruthenium complexes: synthesis, cell growth inhibition, apoptosis induction and ROS production on HCT116 cells. Oncotarget. (2017) 8:104367–92. doi: 10.18632/oncotarget.22248
8. Roy S, Das R, Ghosh B, Chakraborty T. Deciphering the biochemical and molecular mechanism underlying the in vitro and in vivo chemotherapeutic efficacy of ruthenium quercetin complex in colon cancer. Mol Carcinog. (2018) 57:700–21. doi: 10.1002/mc.22792
9. Correa RS, Freire V, Barbosa MIF, Bezerra DP, Bomfim LM, Moreira DRM, et al. Ru(II)-thyminate complexes: new metallodrug candidates against tumor cells. New J Chem. (2018) 42:6794–802. doi: 10.1039/C7NJ04368F
10. de Souza Oliveira M, de Santana ÁAD, Correa RS, Soares MBP, Batista AA, Bezerra DP. Ru(II)-thymine complex causes cell growth inhibition and induction of caspase-mediated apoptosis in human promyelocytic leukemia HL-60 cells. Int J Mol Sci. (2018) 19:1609. doi: 10.3390/ijms19061609
11. Silva VR, Corrêa RS, Santos LS, Soares MBP, Batista AA, Bezerra DP. A ruthenium-based 5-fluorouracil complex with enhanced cytotoxicity and apoptosis induction action in HCT116 cells. Sci Rep. (2018) 8:288. doi: 10.1038/s41598-017-18639-6
12. de Carvalho NC, Neves SP, Dias RB, Valverde LF, Sales CBS, Rocha CAG, et al. A novel ruthenium complex with xanthoxylin induces S-phase arrest and causes ERK1/2-mediated apoptosis in HepG2 cells through a p53-independent pathway. Cell Death Dis. (2018) 9:79. doi: 10.1038/s41419-017-0104-6
13. Kljun J, León IE, Peršič Š, Cadavid-Vargas JF, Etcheverry SB, He W, et al. Synthesis and biological characterization of organoruthenium complexes with 8-hydroxyquinolines. J Inorg Biochem. (2018) 186:187–96. doi: 10.1016/j.jinorgbio.2018.05.009
14. Lentz F, Drescher A, Lindauer A, Henke M, Hilger RA, Hartinger CG, et al. Pharmacokinetics of a novel anticancer ruthenium complex (KP1019, FFC14A) in a phase I dose-escalation study. Anticancer Drugs. (2009) 20:97–103. doi: 10.1097/CAD.0b013e328322fbc5
15. Leijen S, Burgers SA, Baas P, Pluim D, Tibben M, van Werkhoven E, et al. Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Invest New Drugs. (2015) 33:201–14. doi: 10.1007/s10637-014-0179-1
16. Hartinger CG, Zorbas-Seifried S, Jakupec MA, Kynast B, Zorbas H, Keppler BK. From bench to bedside–preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. (2006) 100:891–904. doi: 10.1016/j.jinorgbio.2006.02.013
17. Antonarakis ES, Emadi A. Ruthenium-based chemotherapeutics: are they ready for prime time? Cancer Chemother Pharmacol. (2010) 66:1–9. doi: 10.1007/s00280-010-1293-1
18. Correa RS, da Silva MM, Graminha AE, Meira CS, Santos JA, Moreira DR, et al. Ruthenium(II) complexes of 1,3-thiazolidine-2-thione: Cytotoxicity against tumor cells and anti-Trypanosoma cruzi activity enhanced upon combination with benznidazole. J Inorg Biochem. (2016) 156:153–63. doi: 10.1016/j.jinorgbio.2015.12.024
19. Silva MM. Search for Ruthenium II Complexes With Chemotherapeutic Properties: Evaluation of Antitumor Activity, Mutagenicity, and Inhibition of Topoisomerase IB [in Portuguese]. Doctoral Thesis in Chemistry, Federal University of São Carlos, São Carlos, Brazil (2017).
20. Ahmed SA, Gogal RM, Walsh JE. A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes an alternative to [3H] thymidine incorporation assay. J Immunol Methods. (1994) 170:211–24. doi: 10.1016/0022-1759(94)90396-4
21. Santos LS, Silva VR, Menezes LRA, Soares MBP, Costa EV, Bezerra DP. Xylopine induces oxidative stress and causes G2/M phase arrest, triggering caspase-mediated apoptosis by p53-independent pathway in HCT116 cells. Oxid Med Cell Longev. (2017) 2017:7126872. doi: 10.1155/2017/7126872
22. Glass LS, Bapat A, Kelley MR, Georgiadis MM, Long EC. Semi-automated high-throughput fluorescent intercalator displacement-based discovery of cytotoxic DNA binding agents from a large compound library. Bioorg Med Chem Lett. (2010) 20:1685–8. doi: 10.1016/j.bmcl.2010.01.033
23. Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. (1991) 139:271–9. doi: 10.1016/0022-1759(91)90198-O
24. Sureda FX, Escubedo E, Gabriel C, Comas J, Camarasa J, Camins A. Mitochondrial membrane potential measurement in rat cerebellar neurons by flow cytometry. Cytometry. (1997) 28:74–80. doi: 10.1002/(SICI)1097-0320(19970501)28:1<74::AID-CYTO9>3.0.CO;2-H
25. LeBel CP, Ischiropoulos H, Bondy SC. Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol. (1992) 5:227–31. doi: 10.1021/tx00026a012
26. Chow S, Patel H, Hedley DW. Measurement of MAP kinase activation by flow cytometry using phospho-specific antibodies to MEK and ERK: potential for pharmacodynamic monitoring of signal transduction inhibitors. Cytometry. (2001) 46:72–8. doi: 10.1002/cyto.1067
27. Alessio E. Thirty years of the drug candidate NAMI-A and the myths in the field of ruthenium anticancer compounds: a personal perspective. Eur J Inorg Chem. (2017) 12:1549–60. doi: 10.1002/ejic.201600986
28. Thota S, Rodrigues DA, Crans DC, Barreiro EJ. Ru(II) Compounds: next-generation anticancer metallotherapeutics? J Med Chem. (2018) 61:5805–21. doi: 10.1021/acs.jmedchem.7b01689
29. Kyros L, Kourkoumelis N, Kubicki M, Male L, Hursthouse MB, Verginadis II, et al. Structural properties, cytotoxicity, and anti-inflammatory activity of silver(I) complexes with tris(p-tolyl)phosphine and 5-chloro-2-mercaptobenzothiazole. Bioinorg Chem Appl. (2010) 2010:386860. doi: 10.1155/2010/386860
30. Kumar RR, Ramesh R, Małecki JG. Versatile coordination ability of thioamide ligand in Ru(II) complexes: synthesis, computational studies, in vitro anticancer activity and apoptosis induction. New J Chem. (2017) 41:9130–41. doi: 10.1039/C7NJ01828B
31. Yarar S, Ozturka II, Banti CN, Panagiotou N, Papatriantafyllopoulou C, Manoli M, et al. Synthesis, characterization and cytotoxic properties of bismuth(III) chloride complexes with heterocyclic thioamides. Inorg Chim Acta. (2018) 471:23–33. doi: 10.1016/j.ica.2017.10.026
32. Ozturk II, Hadjikakou SK, Hadjiliadis N, Kourkoumelis N, Kubicki M, Tasiopoulos AJ, et al. New antimony(III) bromide complexes with thioamides: synthesis, characterization, and cytostatic properties. Inorg Chem. (2009) 48:2233–45. doi: 10.1021/ic8019205
33. De Grandis RA, de Camargo MS, da Silva MM, Lopes ÉO, Padilha EC, Resende FA, et al. Human topoisomerase inhibition and DNA/BSA binding of Ru(II)-SCAR complexes as potential anticancer candidates for oral application. Biometals. (2017) 30:321–34. doi: 10.1007/s10534-017-0008-z
34. Barolli JP, Corrêa RS, Miranda FS, Ribeiro JU, Bloch C Jr Ellena J, et al. Polypyridyl ruthenium complexes: novel DNA-intercalating agents against human breast tumor. J Braz Chem Soc. (2017) 28:1879–89. doi: 10.21577/0103-5053.20170019
35. Li C, Ip KW, Man WL, Song D, He ML, Yiu SM, et al. Cytotoxic (salen)ruthenium(III) anticancer complexes exhibit different modes of cell death directed by axial ligands. Chem Sci. (2017) 8:6865–70. doi: 10.1039/C7SC02205K
36. Li J, Tian M, Tian Z, Zhang S, Yan C, Shao C, et al. Half-sandwich iridium(III) and ruthenium(II) complexes containing P∧P-chelating ligands: a new class of potent anticancer agents with unusual redox features. Inorg Chem. (2018) 57:1705–16. doi: 10.1021/acs.inorgchem.7b01959
37. Lu Z, Xu S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life. (2006) 58:621–31. doi: 10.1080/15216540600957438
38. Gough NR. Focus issue: recruiting players for a game of ERK. Sci Signal. (2011) 4:eg9. doi: 10.1126/scisignal.2002601
39. Cossa G, Gatti L, Cassinelli G, Lanzi C, Zaffaroni N, Perego P. Modulation of sensitivity to antitumor agents by targeting the MAPK survival pathway. Curr Pharm Des. (2013) 19:883–94. doi: 10.2174/138161213804547187
40. Burotto M, Chiou VL, Lee JM, Kohn EC. The MAPK pathway across different malignancies: a new perspective. Cancer. (2014) 120:3446–56. doi: 10.1002/cncr.28864
41. Simões AE, Rodrigues CM, Borralho PM. The MEK5/ERK5 signalling pathway in cancer: a promising novel therapeutic target. Drug Discov Today. (2016) 21:1654–63. doi: 10.1016/j.drudis.2016.06.010
42. Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death: is subcellular localization the answer? Cell Cycle. (2009) 8:1168–75. doi: 10.4161/cc.8.8.8147
43. Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death–apoptosis, autophagy and senescence. FEBS J. (2010) 277:2–21. doi: 10.1111/j.1742-4658.2009.07366.x
44. Oliveira MS, Barbosa MIF, de Souza TB, Moreira DRM, Martins FT, Villarreal W, et al. A novel platinum complex containing a piplartine derivative exhibits enhanced cytotoxicity, causes oxidative stress and triggers apoptotic cell death by ERK/p38 pathway in human acute promyelocytic leukemia HL-60 cells. Redox Biol. (2019) 20:182–94. doi: 10.1016/j.redox.2018.10.006
45. Tang D, Wu D, Hirao A, Lahti JM, Liu L, Mazza B, et al. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J Biol Chem. (2002) 277:12710–7. doi: 10.1074/jbc.M111598200
46. Singh S, Upadhyay AK, Ajay AK, Bhat MK. p53 regulates ERK activation in carboplatin induced apoptosis in cervical carcinoma: a novel target of p53 in apoptosis. FEBS Lett. (2007) 581:289–95. doi: 10.1016/j.febslet.2006.12.035
47. Zhao Z, Luo Z, Wu Q, Zheng W, Feng Y, Chen T. Mixed-ligand ruthenium polypyridyl complexes as apoptosis inducers in cancer cells,the cellular translocation and the important role of ROS-mediated signaling. Dalton Trans. (2014) 43:17017–28. doi: 10.1039/C4DT01392A
48. Chen J, Zhang Y, Li G, Peng F, Jie X, She J, et al. Cytotoxicity in vitro, cellular uptake, localization and apoptotic mechanism studies induced by ruthenium(II) complex. J Biol Inorg Chem. (2018) 23:261–75. doi: 10.1007/s00775-017-1528-2
49. Chen L, Li G, Peng F, Jie X, Dongye G, Cai K, et al. The induction of autophagy against mitochondria-mediated apoptosis in lung cancer cells by a ruthenium (II) imidazole complex. Oncotarget. (2016) 7:80716–34. doi: 10.18632/oncotarget.13032
50. Berndsen RH, Weiss A, Abdul UK, Wong TJ, Meraldi P, Griffioen AW, et al. Combination of ruthenium(II)-arene complex [Ru(η6p-cymene)Cl2(pta)] (RAPTA-C) and the epidermal growth factor receptor inhibitor erlotinib results in efficient angiostatic and antitumor activity. Sci Rep. (2017) 7:43005. doi: 10.1038/srep43005
Keywords: ruthenium complexes, heterocyclic thioamidates, apoptosis, ERK1/2, HepG2
Citation: Neves SP, de Carvalho NC, da Silva MM, Rodrigues ACBC, Bomfim LM, Dias RB, Sales CBS, Rocha CAG, Soares MBP, Batista AA and Bezerra DP (2019) Ruthenium Complexes Containing Heterocyclic Thioamidates Trigger Caspase-Mediated Apoptosis Through MAPK Signaling in Human Hepatocellular Carcinoma Cells. Front. Oncol. 9:562. doi: 10.3389/fonc.2019.00562
Received: 28 February 2019; Accepted: 10 June 2019;
Published: 09 July 2019.
Edited by:
Matiullah Khan, AIMST University, MalaysiaReviewed by:
Parames C. Sil, Bose Institute, IndiaAbel Damien Ang, University of Otago, Christchurch, New Zealand
Copyright © 2019 Neves, de Carvalho, da Silva, Rodrigues, Bomfim, Dias, Sales, Rocha, Soares, Batista and Bezerra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel P. Bezerra, ZGFuaWVscGJlemVycmFAZ21haWwuY29t