 Matthew J. Ryan1,2,3
Matthew J. Ryan1,2,3 Rohit Bose2,3,4,5,6*
Rohit Bose2,3,4,5,6*- 1Medical Scientist Training Program, University of California, San Francisco, San Francisco, CA, United States
- 2Biomedical Sciences Graduate Program, University of California, San Francisco, San Francisco, CA, United States
- 3Department of Anatomy, University of California, San Francisco, San Francisco, CA, United States
- 4Departments of Medicine and Urology, University of California, San Francisco, San Francisco, CA, United States
- 5UCSF Helen Diller Family Comprehensive Cancer Center, San Francisco, CA, United States
- 6UCSF Benioff Initiative for Prostate Cancer Research, San Francisco, CA, United States
The increasing number of patients with sequenced prostate cancer genomes enables us to study not only individual oncogenic mutations, but also capture the global burden of genomic alterations. Here we review the extent of tumor genome mutations and chromosomal structural variants in various clinical states of prostate cancer, and the related prognostic information. Next, we discuss the underlying mutational processes that give rise to these various alterations, and their relationship to the various molecular subtypes of prostate cancer. Finally, we examine the relationships between the tumor mutation burden of castration-resistant prostate cancer, DNA repair defects, and response to immune checkpoint inhibitor therapy.
Introduction
Prostate cancer is the second-most common cancer in men worldwide (1), and advanced forms of the disease cause debilitating bone pain, pathologic fractures, and severe anemia. The era of profiling patients' tumors using next generation sequencing (NGS) has yielded both scientific and clinical advances including: the comprehensive detection of BRCA mutations for PARP inhibitor therapy; the identification of poorer prognosis RB1 mutations; and the full extent of AR genomic alterations associated with androgen receptor signaling inhibitor (ARSI) resistance (2–6). Equally important, NGS profiling has expanded our insight beyond a handful of known loci to capture our first snapshots of the prostate tumor genome in its entirety. This raises the question: does the global burden of mutations and chromosomal structural variants reveal information beyond individual driver mutation analysis?
Here we examine the genomic alteration burden in various states of prostate cancer, a disease with a heterogenous clinical course. Next, we delve into the various mutational processes underlying those alterations and highlight associations with molecular subtypes. Finally, we evaluate how a tumor's mutation burden may help predict response to certain therapies. There are several caveats: factors beyond the tumor genome, such as the transcriptome, epigenome, and the microenvironment are undoubtedly relevant, but beyond the scope of this mini review. Secondly, the analyzed cohorts are predominantly comprised of patients of European ancestry. Finally, this review of global genomic alterations is simply designed to augment, not supersede, the relevance of individual mutations and traditional clinical parameters.
Burden of Genomic Alterations in Different Clinical States
Tumor mutation burden (TMB) (7) is measured differently among various prostate cancer cohorts. Sometimes, it is reported as the load of non-synonymous mutations (NS) with a minimum allele frequency of 0.5–10%. Other times, it is reported as the load of any single nucleotide variants (SNVs). Some studies additionally report the rate of indels (8, 9). The TMB of unselected and usually treatment-naïve locoregional prostate adenocarcinoma cohorts typically falls between 0.94 and 1.74 NS per megabase (Mb) (Table 1). Average TMB appears to correlate with the patient's age at diagnosis (~0.5 NS/Mb for those diagnosed in their 40s vs. ~0.9 NS/Mb in their 60s) (12). Primary tumor grade is a major clinical feature and described by the Gleason score (currently being updated to the Grade Group system) (33). The SNV burden has been reported as 1.5 × higher in intermediate pattern Gleason 7 tumors vs. well-differentiated pattern Gleason 6 tumors (p = 1.05 × 10−3) (16), consistent with other reports (12). Interestingly, a small cohort of South African patients of African ancestry with high-risk locoregional disease were found to have a roughly 4-fold increase of TMB (3.0–4.7 SNVs plus indels/Mb) (Table 1) compared with a control cohort of European ancestry (23). On the other hand, a study of African-American men with primary prostate cancer had a rate of 0.83 SNVs/Mb, in line with cohorts of predominantly European-Americans (17).
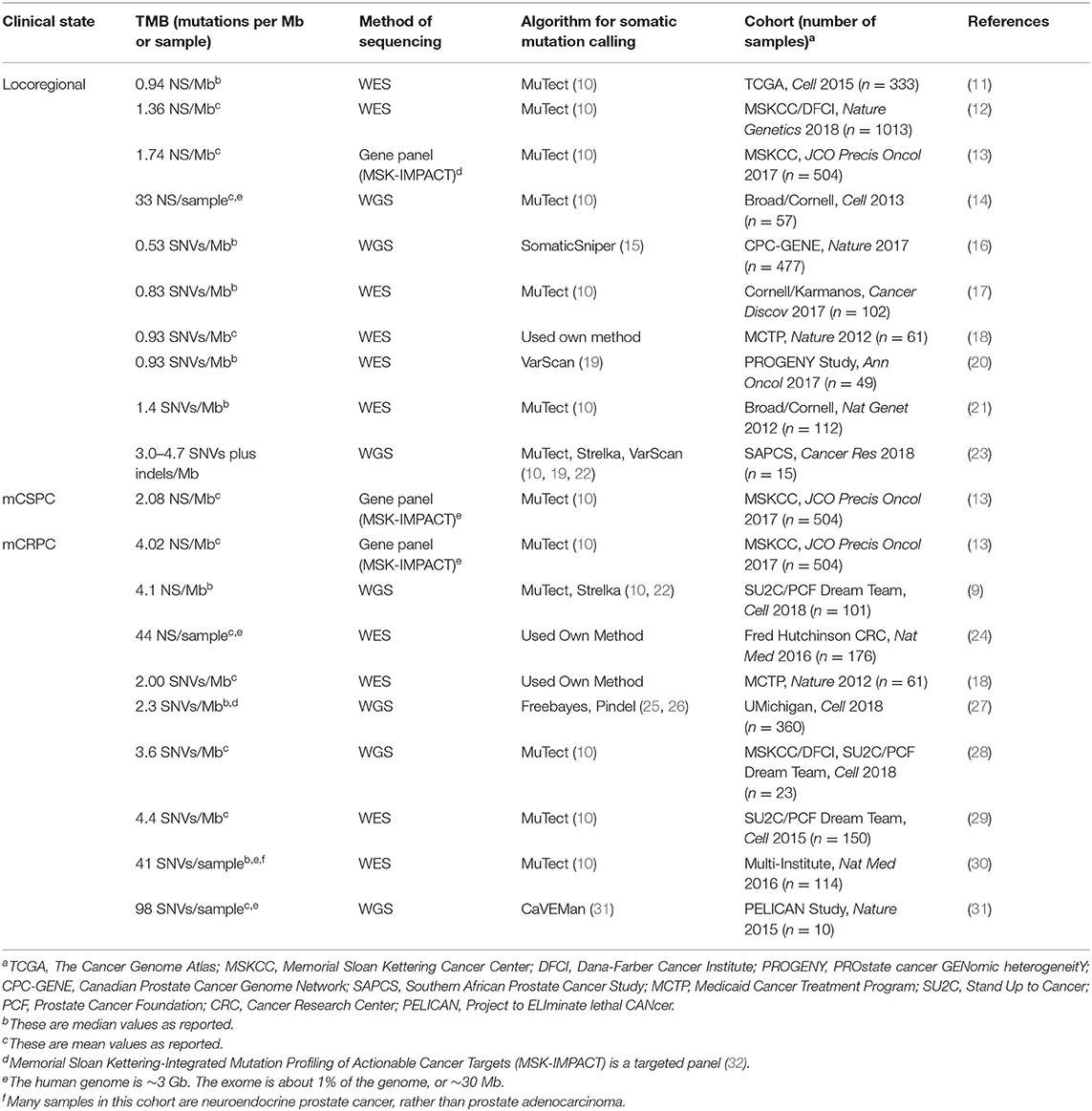
Table 1. Tumor mutation burden (TMB) in locoregional, metastatic castration-sensitive (mCSPC), and metastatic castration-resistant (mCRPC) prostate cancer samples.
Prostate cancer that presents as de novo metastases, or reappears as macro-metastases following definitive prostatectomy/radiotherapy, is termed metastatic castration-sensitive prostate cancer (mCSPC) (34–36). Just as the pattern of individual mutations is similar between locoregional disease and mCSPC, so is the mean TMB (1.74 vs. 2.08 NS/Mb) (13). Likewise, a separate study showed that patients presenting with markedly elevated PSAs (≥15) and a biopsied MRI-positive primary lesion had no significant TMB difference compared to those found to have mCSPC disease (20). However, as the disease advances beyond mCSPC, so too does the TMB. Metastatic castration-resistant prostate cancer (mCRPC) can no longer be controlled with androgen ablation and is the most morbid and lethal clinical state. Several groups have noted that the TMB of mCRPC is accordingly increased (4.02 vs. 2.08 NS/Mb in mCSPC in one study) (Table 1) (13, 18, 27, 29, 31).
However, analyzing prostate tumor genomes solely via TMB misses many alterations, since the disease has a higher burden than many other cancers of chromosomal structural variants including insertions, deletions, inversions, translocations, gene-fusions, and tandem duplications (14, 37, 38). Locoregional prostate cancer cohorts have a highly variable structural variant burden, with a median of 19 structural variants per genome (range between 0 and 499, Table 2) (16). Like TMB, the structural variant burden correlates with Gleason score (17 in Gleason 6 disease compared to 22 in Gleason 7, p < 0.001) (Table 2) (16). The mCRPC cohorts have a much higher structural variant burden than in locoregional disease; median lies between 230 and 337 per study (Table 2), keeping in mind structural variant measurement is not standardized (9, 28, 29).
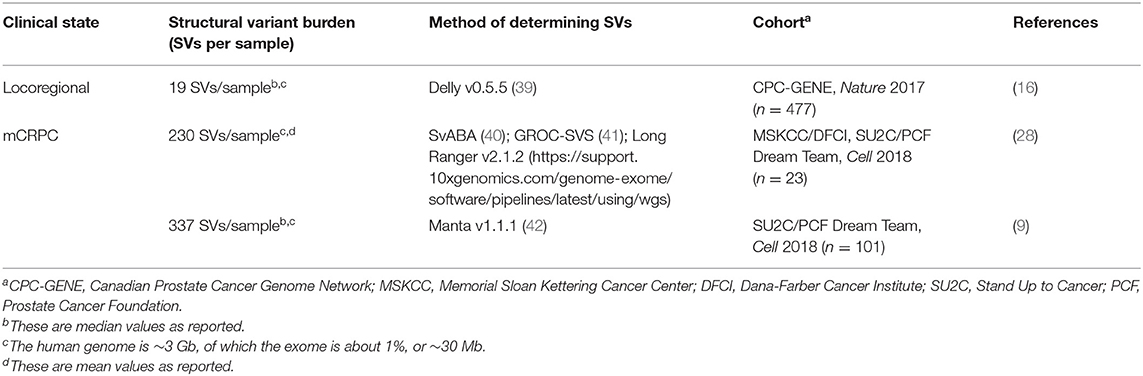
Table 2. Structural variant burden (SVB) in locoregional and mCRPC samples.
At the chromosomal level, mCRPC genomes frequently demonstrate polyploidy and/or aneuploidy. There are several NGS studies confirming that roughly ≥40% of mCRPC samples are triploid or more (9, 27, 43), a status itself associated with more translocations and SNVs (9). Regarding aneuploidy, about 75% of locoregional prostate cancer genomes have chromosomal arm-level alterations, and 23% possessed ≥5 arm-level alterations (44). As with TMB and structural variants, the degree of arm-level alterations correlates with Gleason score: only ~3% of Gleason 6 tumors have ≥5 arm-level alterations compared to ~40% of the very poorly differentiated Gleason 9–10 tumors. Even after adjusting for Gleason score, the degree of tumor aneuploidy predicted future lethal prostate cancer risk with a median follow-up of 15 years: patients with ≥5 arm-level alterations had a odds ratio for lethality of 5.34 (95% CI 2.18–13.1) compared with those with no aneuploidy (44).
The majority of NGS-based clinical testing involves targeted panels, rather than whole genome sequencing (WGS), making direct detection of some structural variants challenging. However, copy number alterations (CNA) of individual genes and genomic regions can be robustly detected, and they are an indirect measure of unbalanced structural variants and aneuploidy. The tumor CNA burden (TCB) is reported as the fraction of the measured genome with broad CNA. The median TCB of locoregional disease is ~7% of the genome altered (12, 13, 45). TCB differs statistically with age at diagnosis and Gleason score in a similar way to TMB: those diagnosed in their 40s have a median TCB of ~2% genome altered whereas those diagnosed in their 60s have ~9% altered (12). Gleason 6 tumors have median TCBs of ~1% genome altered, whereas Gleason ≥8 tumors have ~13% altered (12), consistent with other reports (46). The TCB of tumors confers considerable prognostic information (43, 45, 47, 48): it is significantly associated with biochemical recurrence (each 1% increase in TCB was associated with a 5–8% decrease in 5-years relapse-free survival) and future metastasis (45). This was independently verified (43), even after adjustment for Gleason score and TMB (47). TCB was also found to be associated with prostate cancer-specific death after adjustment for clinical parameters, such as CAPRA (CAncer of the Prostate Risk Assessment) score (49) or Gleason score (per 5% TCB, HR 1.49; 95% 1.30–1.70) (47). Unlike TMB, median TCB of mCSPC tumors is higher than locoregional disease (20–30% genome altered) and even higher in mCRPC tumors (cohort medians between 23 and ~38%) (9, 12, 13, 20, 24). TCB is negatively associated with overall survival in metastatic tumors in multivariate analysis even after adjustment for TMB (per 5% TCB, HR = 1.08; 95% CI 1.02–1.15) (47).
There is a subset of prostate cancer that emerges clinically in the treated mCRPC state, whereby the dominant metastatic histology is now either small-cell carcinoma or possesses neuroendocrine features. This treatment-emergent small-cell/neuroendocrine prostate cancer (t-SCNC) (30, 50) has both characteristic molecular and aggressive clinical features: there is an enrichment for RB1/TP53 genomic alterations and rapidly progressive visceral metastases. In one analysis, there were no statistically significant differences between TMB, TCB, or ploidy between t-SCNC vs. mCRPC adenocarcinoma (30). This is consistent with the postulation that neuroendocrine transdifferentiation may be driven substantially by epigenetic mechanisms (51).
Longitudinal analysis of prostate tumor genomes reveals further complexity in interpreting TMB and TCB, since mutational processes are dynamic, interrelated, can arise in a multi-focal setting, and evolve with different degrees of clonality (13, 31, 46, 52). One study reports that 40% of primary prostate tumors appear to be monoclonal i.e., one dominant clone is detected (46). The remaining 60% of primary tumors demonstrate subclonal populations (78% biclonal, 20% triclonal) originating from an ancestral clone. These polyclonal tumors have inferior clinical outcomes with respect to biochemical relapse following definitive prostatectomy/radiotherapy (HR 2.64; CI 1.36–5.15), and persisting after adjustment for standard clinical parameters and TCB (46). The polyclonality-related risk appears to be additive to those derived from the combination of TMB and TCB. Interestingly, triclonal tumors have a higher median PSA level at diagnosis (9.7 vs. ~7 for monoclonal/biclonal; p < 0.01), and polyclonal primary tumors are also more likely to develop metastases later on (OR = 4.01; p < 0.05). In polyclonal primary tumors, most of the TMB is truncal (median 87% of total SNVs) whereas the TCB is more evenly distributed between being truncal (55%) vs. branch-specific (45%). Moreover, the individual truncal CNAs are larger (median 11.5 vs. 6.5 Mb for branch-specific CNAs) and biased toward deletions (84% of all deletions are truncal). CNAs are also observed at chromosome ends, and the median telomere length of polyclonal tumors is 500 bp shorter than monoclonal tumors (46).
Altogether, the burden of genomic alterations correlates with key clinical information for prostate cancer patients. The TMB, structural variants, and TCB all tend to increase with advancing clinical state, Gleason score, and age. However, clonality analysis hints that how these mutational processes combine during tumor evolution is quite complex.
Mutational Processes Underlying Prostate Cancer Genomic Alterations
Next, we examine the biologic processes that generate these genomic alterations, starting with SNVs. Comparison of tumor-derived patterns of SNVs within their trinucleotide context to pre-defined signatures (53) can suggest the underlying etiology; for example, cancers with known exogenous risk factors reveal robust signatures associated with tobacco or UV exposure. However, the majority of mCRPC tumors with intact DNA repair pathways reveal a robust signature that is endogenous, age-related and likely results from deamination of 5-methylcytosine to thymine at mCpG dinucleotides (COSMIC signature 1) (12, 54). If not repaired before DNA replication, this results in a permanent C > T transition. This signature is contributory in most cancer types and the frequency of associated SNVs correlates with the age at diagnosis in pan-cancer analysis, although not necessarily meeting statistical significance when analyzing each tissue individually (54). Nevertheless, the rate of prostate cancer SNVs attributed to this age-related signature loosely fits a slope of ~6 SNVs/Gb/year and may contribute to the increased TMB of patients diagnosed at older ages. Clonality analysis reveals that this age-related signature is most dominant early in prostate tumor evolution (46).
In prostate tumors possessing DNA repair defects, SNVs are associated with different dominant signatures. A recent NGS analysis of one cohort revealed 3% of genomes possess somatic DNA mismatch repair defects (MMRD) (55) caused by loss-of-function mutations in the canonical genes MLH1, MSH2, MSH6, or PMS2, and consistent with other cohorts (56–58). If one of the allelic mutations is germline, the patient has Lynch syndrome and possesses increased lifetime risk for several cancer types including prostate cancer (59, 60). These tumor genomes are 10- to 100-fold less likely to repair base pair substitutions prior to DNA replication, and their TMB is elevated (20–80 SNVs/Mb in mCRPC) although not necessarily as high as other MMRD cancer types (29). Analysis as above reveals dominant SNV signatures associated with MMRD, as expected (9, 27, 55, 61). MMRD tumors also possess high rates of indels (9), leading to higher instability of DNA microsatellite lengths, a way in which such tumors can be detected (55). MMRD tumors have distinct genomes from those that are MMR proficient: they are usually diploid, and have the lowest TCB (27). We further discuss MMRD tumors in the next section.
A third class of SNVs is observed in tumors with homologous recombination deficiency (HRD) from 6 to 20% of patients with either somatic or germline alterations of BRCA1 or BRCA2, frequently biallelic (9, 13, 27, 29, 62). Since DNA homologous recombination coordinates the repair of double stranded DNA breaks, HRD not only results in a high TCB, but also a reliance on alternative error-prone DNA repair pathways (63) and a distinct dominant group of SNV signatures (9, 27). Accordingly, BRCA-mutant tumors in mCRPC possess the highest SNV rate among MMR proficient tumors (7.0 muts/Mb), in addition to higher TCB (9).
There are other SNV signatures observed to varying degrees in prostate tumors, some of which have not yet been associated with an etiology (27, 53). Moreover, SNVs are not evenly distributed throughout a given tumor's genome, but rather dependent on many interrelated factors, including the underlying mutational process, the timing of the locus within DNA replication, as well as whether the locus affects transcription and/or translation. The phenomenon of localized regions of SNV-based hypermutation is called kataegis, and is found in 23% of primary tumors (16); it is coincident with genomic instability, likely altered DNA repair (64), and enriched for deletions of the chromatin remodeler CHD1 (33% of kataegis-positive tumors compared with only 11% of kataegis-null tumors) (16). Kataegis is associated with increasing Gleason score, and present in 40% of Gleason 4 + 3 tumors.
Just as specific processes lead to increased TMB, others lead to increased TCB. For example, BRCA-mutant tumors have markedly higher frequencies of copy number deletions as well as classic genomic “scars” due to their HRD (9). On the other hand, specifically in HR proficient tumors, chromothripsis can occur: evidence of “shattering” of regions in one or a few chromosomes followed by intrachromosomal reassembly in a stochastic manner, resulting in large numbers of both deletions and inversions (9). It is found in 20% of non-indolent primary prostate cancer samples (16) and 23% of mCRPC samples (9). Although the exact mechanism is unknown, there are some clear correlations: chromothripsis positive genomes are enriched for biallelic TP53 loss (83% of chromothripsis positive tumors vs. 35% of chromothripsis null tumors), although this event is not likely sufficient to cause chromothripsis (9, 16). Others have noted a correlation between genomic loss of CHD1 and chromothripsis (14). From a clinical standpoint, chromothripsis is associated with the primary tumor T-stage, but was not found to differ by age or Gleason grade (16).
About 5% of mCRPC cases have a significantly higher number of genomic tandem duplications, and 90% of these genomes have biallelic CDK12 alterations (27, 28). In such cases there is a median of 150 tandem duplications per sample with a median duplicated region size of 1.3 Mb (28). Accordingly, such tumors possess large numbers of focal CNAs, and also have the highest gene-fusion burden (100 per tumor vs. 25 in other tumors), due directly to the genomic duplication phenomenon (27). CDK12-mutant tumors are usually diploid and trend toward mutual exclusivity from HRD biallelic BRCA-mutant tumors (9). Clonality analysis reveals that CDK12 alterations are usually truncal; in these samples, the accompanying SNVs are more likely to occur after tandem duplication than before, and in many cases in branch-specific subclones (28). It is unknown whether CDK12 alterations directly cause the tandem duplications, or are merely associated with it, but there is evidence to support the former (28).
Finally, some mutational processes occur without directly affecting TMB or TCB. The most common gene fusions in prostate cancer occur between androgen-driven upstream elements of genes like TMPRSS2, and oncogenic ETS transcription factors like ERG, and are present in up to 50–60% of men of European descent (65). The underlying chromosomal rearrangements that cause such gene fusions are initially balanced, frequently complex and involve multiple chromosomes in a phenomenon termed chromoplexy (14). Some degree of chromoplexy is present in 50–90% prostate tumors (9, 14). Moreover, in tumors possessing ETS gene fusions, the chromoplexy has more than double the number of interchromosomal rearrangements compared to ETS fusion-null tumors (14). There is evidence of successive rounds of chromoplexy occurring, for example initially leading to ETS fusion formation, and then subsequently to inactivation of tumor suppressor genes. It is not known exactly how chromoplexy occurs; there is no enrichment for TP53 mutations in such tumors, but the process may be related to androgen-related chromatin configuration (9, 14). Notably, the small cohort of South African men possessed lower frequencies of larger genomic rearrangements, such as chromothripsis and chromoplexy, and lower frequencies of ETS gene fusions, than the comparable cohort with European ancestry (23).
In summary, we have just begun to understand the processes that contribute to the burden of prostate cancer genomic alterations. SNVs possess distinct mutational signatures including those associated with aging and DNA repair defects; moreover, many tumor genomes have localized hypermutated regions. Complex chromosomal alterations, such as chromothripsis, tandem duplication, and chromoplexy tend to stratify by specific alterations in TP53, CHD1, CDK12, and BRCA1/2 and underlie many CNAs and fusion events, such as the canonical TMPRSS2-ERG fusion.
Prostate Tumor Mutation Burden, DNA Repair Defects, and Therapeutic Response
The initial trials of immune checkpoint inhibitors in unselected prostate cancer patients (66–68) demonstrated no global clinical benefit in prostate cancer. Nevertheless, interest in such therapies remained strong, given case reports of impressive and durable responses among individual prostate cancer patients (69, 70). Experiences from other tumor types illuminated patient subtypes that may derive clinical benefit from existing therapies. Efforts to identify a specific predictive biomarker, such as PD-L1 expression have been challenging (71, 72); however, the association with global genomic processes has been clear. Patients with non-small cell lung cancers (73), bladder cancers (74), and melanoma (75–77) that have a high TMB derive increased clinical benefit to immune checkpoint inhibitors compared to those with low TMB. There are markedly different numerical thresholds of what constitutes a high TMB, with the highest quintile within a given histology usually being associated with longer overall survival when treated with immune checkpoint inhibitors (78). Increased non-synonymous mutations and indel frameshifts lead to increased neoepitopes within MHC Class I-loaded peptides and it is hypothesized these serve as neoantigens in the context of immunotherapy (79–84). Ongoing prostate cancer immunotherapy trials are now beginning to incorporate TMB analyses (85).
As described above, the prostate tumors with the highest TMB are those with MMRD. A series of trials treating patients with MMRD tumors with pembrolizumab, regardless of histology, reported a 53% objective radiographic response rate, and a 21% complete response rate (86). This led to the FDA approval of pembrolizumab for any MMRD metastatic/unresectable solid tumors and reinforces the importance of testing such prostate cancer patients for MMRD. In a recent study of mCRPC patients with MMRD tumors and treated with immune checkpoint inhibitors, 55% achieved a PSA response >50%, and 45% of patients had durable clinical benefit (55). A smaller study revealed that three out of four patients with MMRD tumors achieved soft tissue tumor responses upon treatment with immune checkpoint inhibitors (56). In a separate large analysis of mCRPC samples, MMRD tumors were predicted to have median neoantigen burdens of ~10,000 vs. 1,000 in MMR proficient tumors (27). Approximately 10% of these are further predicted to be “strong binders” of MHC Class I. MMRD prostate tumors were also found to have high degrees of immune infiltration (20, 27), the highest number of T-cell clonotypes, and the highest percent of expanded T-cell clones (27). Other studies have showed a complex relationship between predicted neoantigen load with immune infiltration (20), as well as considerable heterogeneity of tumor T-cell infiltration in MMRD cases (87). The exact mechanism of how predicted neoantigens stimulate a clinically-relevant immunologic response, and how this might inform the next generation of immunotherapies remains an active area of study (77).
Prostate tumors with MMRD may have other unique molecular and biological features, compared to MMR proficient disease. One case series reported an enrichment of MMRD among ductal adenocarcinoma of the prostate, a rare aggressive subtype of prostate adenocarcinoma (about 3% incidence, compared to the common acinar adenocarcinoma) with poor prognosis (56, 57, 88). Ultimately, it is important to understand the natural history of prostate cancer patients with MMRD tumors, particularly prior to any potential treatment with immune checkpoint inhibitors. One study of patients with recurrent disease reports a longer progression free survival following androgen-deprivation therapy when MMRD is detected (median 66 months compared to only 27 months in MMR proficient cases), as well as longer responses to first-line ARSI agents when used (56). On the other hand, among patients with clinically aggressive tumors (56% having metastatic disease at diagnosis), a different retrospective study of clinically aggressive CRPC noted the median overall survival for the MMRD cases was significantly shorter (3.8 years from androgen ablation) than MMR proficient groups (7.0 years), in both univariate and multivariate analysis (87). The studies above cannot be directly compared, but perhaps the biologic context is key to interpreting the clinical relevance of MMRD.
Beyond MMRD, prostate cancers with other DNA damage repair defects are being explored for their responses to immunotherapies. Because CDK12-mutant tumors have increased rates of gene fusions, they possess higher predicted neoantigen burdens (median ~2,000) than other MMR proficient tumors (27). They may also possess high degrees of T cell infiltration and expanded T cell clones. These findings have led to a Phase II trial evaluating the efficiency of combination nivolumab plus ipilimumab in mCRPC patients with CDK12-mutant tumors (89). There are also several immune checkpoint inhibitor therapy combinations being explored, such as one in which the second agent, a PARP inhibitor, alters how the genome repairs itself (90). Interestingly, a recent phase Ib/2 study showed some interesting clinical responses to the combination of pembrolizumab and olaparib, despite no BRCA mutations being detected in the biopsies tested (91). Whether this clinical response is due undetected HRD, or whether the PARP inhibitor synergizes with the immune checkpoint inhibitor by altering the presented neoepitopes or an unknown mechanism remains to be determined.
While high TMB, particularly in the context of altered DNA repair, is important regarding successful immune checkpoint therapy in prostate cancer, it is certainly not the whole story. Roughly half of MMRD prostate tumors do not exhibit substantial clinical responses to such therapy despite relatively high TMB (55). Moreover, when clinical responses are observed, the TMB is often lower compared to that observed in other MMRD cancer types e.g., in a preplanned interim analysis of a small phase II mCRPC study of combination nivolumab plus ipilimumab, responses were observed in tumors above a modest TMB threshold (85). Identifying other genomic factors that modify response to immune checkpoint inhibitors and determining whether they map to specific genes and/or global processes, remains an active area of investigation. Due to NGS-based analysis of patients' tumors, we are just beginning to obtain a comprehensive snapshot of the prostate tumor genome in differing clinical states. A deeper understanding whether and how global genomic measures, such as TMB, TCB, gene-fusion burden and clonality affect responses to targeted and immuno-therapies will help us shape future prostate cancer investigations.
Author Contributions
MR and RB drafted, revised, and approved the submitted version.
Funding
RB was supported by grants from the Department of Defense (W81XWH1510277), NCI (1K08CA226348), Prostate Cancer Foundation, and UCSF Benioff Initiative for Prostate Cancer Research.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Rahul Aggarwal, Joshua Armenia, Terence Friedlander, Andrew Hsieh, David Quigley, Sumit Subudhi, and members of the Bose laboratory for their insightful comments.
References
1. Center MM, Jemal A, Lortet-Tieulent J, Ward E, Ferlay J, Brawley O, et al. International variation in prostate cancer incidence and mortality rates. Eur Urol. (2012) 61:1079–92. doi: 10.1016/j.eururo.2012.02.054
2. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. (2015) 373:1697–708. doi: 10.1056/NEJMoa1506859
3. Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci USA. (2019) 116:11428–36. doi: 10.1073/pnas.1902651116
4. Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. (2015) 15:701–11. doi: 10.1038/nrc4016
5. Bose R, Karthaus WR, Armenia J, Abida W, Iaquinta PJ, Zhang Z, et al. ERF mutations reveal a balance of ETS factors controlling prostate oncogenesis. Nature. (2017) 546:671–5. doi: 10.1038/nature22820
6. Adams EJ, Karthaus WR, Hoover E, Liu D, Gruet A, Zhang Z, et al. FOXA1 mutations alter pioneering activity, differentiation and prostate cancer phenotypes. Nature. (2019) 571:408–12. doi: 10.1038/s41586-019-1318-9
7. Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. (2019) 30:44–56. doi: 10.1093/annonc/mdy495
8. Mullaney JM, Mills RE, Stephen Pittard W, Devine SE. Small insertions and deletions (INDELs) in human genomes. Hum Mol Genet. (2010) 19:R131–6. doi: 10.1093/hmg/ddq400
9. Quigley DA, Dang HX, Zhao SG, Lloyd P, Aggarwal R, Alumkal JJ, et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell. (2018) 174:758–69.e9. doi: 10.1016/j.cell.2018.06.039
10. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. (2013) 31:213–9. doi: 10.1038/nbt.2514
11. Abeshouse A, Ahn J, Akbani R, Ally A, Amin S, Andry CD, et al. The molecular taxonomy of primary prostate cancer. Cell. (2015) 163:1011–25. doi: 10.1016/j.cell.2015.10.025
12. Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet. (2018) 50:645–51. doi: 10.1038/s41588-018-0078-z
13. Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, et al. Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol. (2017) 1:1–16. doi: 10.1200/PO.17.00029
14. Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, et al. Punctuated evolution of prostate cancer genomes. Cell. (2013) 153:666–77. doi: 10.1016/j.cell.2013.03.021
15. Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. (2012) 28:311–7. doi: 10.1093/bioinformatics/btr665
16. Fraser M, Sabelnykova VY, Yamaguchi TN, Heisler LE, Livingstone J, Huang V, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature. (2017) 541:359–64. doi: 10.1038/nature20788
17. Huang FW, Mosquera JM, Garofalo A, Oh C, Baco M, Amin-Mansour A, et al. Exome sequencing of African-American prostate cancer reveals loss-of-function ERF mutations. Cancer Discov. (2017) 7:973–83. doi: 10.1158/2159-8290.CD-16-0960
18. Grasso CS, Wu Y-M, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. (2012) 487:239–43. doi: 10.1038/nature11125
19. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. (2012) 22:568–76. doi: 10.1101/gr.129684.111
20. Linch M, Goh G, Hiley C, Shanmugabavan Y, McGranahan N, Rowan A, et al. Intratumoural evolutionary landscape of high-risk prostate cancer: the PROGENY study of genomic and immune parameters. Ann Oncol. (2017) 28:2472–80. doi: 10.1093/annonc/mdx355
21. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. (2012) 44:685–9. doi: 10.1038/ng.2279
22. Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. (2012) 28:1811–7. doi: 10.1093/bioinformatics/bts271
23. Jaratlerdsiri W, Chan EKF, Gong T, Petersen DC, Kalsbeek AMF, Venter PA, et al. Whole-genome sequencing reveals elevated tumor mutational burden and initiating driver mutations in African men with treatment-naïve, high-risk prostate cancer. Cancer Res. (2018) 78:6736–46. doi: 10.1158/0008-5472.CAN-18-0254
24. Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. (2016) 22:369–79. doi: 10.1038/nm.4053
25. Garrison E, Marth G. Haplotype-Based Variant Detection From Short-Read Sequencing. (2012). Available online at: http://arxiv.org/abs/1207.3907 (accessed August 27, 2019).
26. Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. (2009) 25:2865–71. doi: 10.1093/bioinformatics/btp394
27. Wu YM, Cieślik M, Lonigro RJ, Vats P, Reimers MA, Cao X, et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell. (2018) 173:1770–82.e14. doi: 10.1016/j.cell.2018.04.034
28. Viswanathan SR, Ha G, Hoff AM, Wala JA, Carrot-Zhang J, Whelan CW, et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell. (2018) 174:433–47.e19. doi: 10.1016/j.cell.2018.05.036
29. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. (2015) 161:1215–28. doi: 10.1016/j.cell.2015.05.001
30. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. (2016) 22:298–305. doi: 10.1038/nm.4045
31. Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. (2015) 520:353–7. doi: 10.1038/nature14347
32. Cheng DT, Mitchell T, Zehir A, Shah RH, Benayed R, Syed A, et al. MSK-IMPACT: a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. (2015) 17:251–64. doi: 10.1016/j.jmoldx.2014.12.006
33. Epstein JI, Zelefsky MJ, Sjoberg DD, Nelson JB, Egevad L, Magi-Galluzzi C, et al. A contemporary prostate cancer grading system: a validated alternative to the Gleason score. Eur Urol. (2016) 69:428–35. doi: 10.1016/j.eururo.2015.06.046
34. Sweeney CJ, Chen Y-H, Carducci M, Liu G, Jarrard DF, Eisenberger M, et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N Engl J Med. (2015) 373:737–46. doi: 10.1056/NEJMoa1503747
35. Fizazi K, Tran NP, Fein L, Matsubara N, Rodriguez-Antolin A, Alekseev BY, et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N Engl J Med. (2017) 377:352–60. doi: 10.1056/NEJMoa1704174
36. Barata PC, Sartor AO. Metastatic castration-sensitive prostate cancer: abiraterone, docetaxel, or …. Cancer. (2019) 125:1777–88. doi: 10.1002/cncr.32039
37. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. (2010) 18:11–22. doi: 10.1016/j.ccr.2010.05.026
38. Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. (2013) 45:1127–33. doi: 10.1038/ng.2762
39. Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. (2012) 28:i333–9. doi: 10.1093/bioinformatics/bts378
40. Wala JA, Bandopadhayay P, Greenwald NF, O'Rourke R, Sharpe T, Stewart C, et al. SvABA: genome-wide detection of structural variants and indels by local assembly. Genome Res. (2018) 28:581–91. doi: 10.1101/gr.221028.117
41. Spies N, Weng Z, Bishara A, McDaniel J, Catoe D, Zook JM, et al. Genome-wide reconstruction of complex structural variants using read clouds. Nat Methods. (2017) 14:915–20. doi: 10.1038/nmeth.4366
42. Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. (2016) 32:1220–2. doi: 10.1093/bioinformatics/btv710
43. Camacho N, Van Loo P, Edwards S, Kay JD, Matthews L, Haase K, et al. Appraising the relevance of DNA copy number loss and gain in prostate cancer using whole genome DNA sequence data. PLoS Genet. (2017) 13:e1007001. doi: 10.1371/journal.pgen.1007001
44. Stopsack KH, Whittaker CA, Gerke TA, Loda M, Kantoff PW, Mucci LA, et al. Aneuploidy drives lethal progression in prostate cancer. Proc Natl Acad Sci USA. (2019) 116:11390–5. doi: 10.1073/pnas.1902645116
45. Lalonde E, Ishkanian AS, Sykes J, Fraser M, Ross-Adams H, Erho N, et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: a retrospective cohort study. Lancet Oncol. (2014) 15:1521–32. doi: 10.1016/S1470-2045(14)71021-6
46. Espiritu SMG, Liu LY, Rubanova Y, Bhandari V, Holgersen EM, Szyca LM, et al. The evolutionary landscape of localized prostate cancers drives clinical aggression. Cell. (2018) 173:1003–13.e15. doi: 10.1016/j.cell.2018.03.029
47. Hieronymus H, Murali R, Tin A, Yadav K, Abida W, Moller H, et al. Tumor copy number alteration burden is a pan-cancer prognostic factor associated with recurrence and death. Elife. (2018) 7:e37294. doi: 10.7554/eLife.37294
48. Lalonde E, Alkallas R, Chua MLK, Fraser M, Haider S, Meng A, et al. Translating a prognostic DNA genomic classifier into the clinic: retrospective validation in 563 localized prostate tumors. Eur Urol. (2017) 72:22–31. doi: 10.1016/j.eururo.2016.10.013
49. Cooperberg MR, Pasta DJ, Elkin EP, Litwin MS, Latini DM, Duchane J, et al. The University of California, San Francisco cancer of the prostate risk assessment score: a straightforward and reliable preoperative predictor of disease recurrence after radical prostatectomy. J Urol. (2005) 173:1938–42. doi: 10.1097/01.ju.0000158155.33890.e7
50. Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a multi-institutional prospective study. J Clin Oncol. (2018) 36:2492–503. doi: 10.1200/JCO.2017.77.6880
51. Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. (2011) 1:487–95. doi: 10.1158/2159-8290.CD-11-0130
52. Cooper CS, Eeles R, Wedge DC, Van Loo P, Gundem G, Alexandrov LB, et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat Genet. (2015) 47:367–72. doi: 10.1038/ng.3221
53. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. (2013) 500:415–21. doi: 10.1038/nature12477
54. Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, et al. Clock-like mutational processes in human somatic cells. Nat Genet. (2015) 47:1402–7. doi: 10.1038/ng.3441
55. Abida W, Cheng ML, Armenia J, Middha S, Autio KA, Vargas HA, et al. Analysis of the prevalence of microsatellite instability in prostate cancer and response to immune checkpoint blockade. JAMA Oncol. (2019) 5:471–8. doi: 10.1001/jamaoncol.2018.5801
56. Antonarakis ES, Shaukat F, Isaacsson Velho P, Kaur H, Shenderov E, Pardoll DM, et al. Clinical features and therapeutic outcomes in men with advanced prostate cancer and DNA mismatch repair gene mutations. Eur Urol. (2019) 75:378–82. doi: 10.1016/j.eururo.2018.10.009
57. Guedes LB, Antonarakis ES, Schweizer MT, Mirkheshti N, Almutairi F, Park JC, et al. MSH2 loss in primary prostate cancer. Clin Cancer Res. (2017) 23:6863–74. doi: 10.1158/1078-0432.CCR-17-0955
58. Pritchard CC, Morrissey C, Kumar A, Zhang X, Smith C, Coleman I, et al. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat Commun. (2014) 5:5988. doi: 10.1038/ncomms5988
59. Raymond VM, Mukherjee B, Wang F, Huang SC, Stoffel EM, Kastrinos F, et al. Elevated risk of prostate cancer among men with lynch syndrome. J Clin Oncol. (2013) 31:1713–8. doi: 10.1200/JCO.2012.44.1238
60. Haraldsdottir S, Hampel H, Wei L, Wu C, Frankel W, Bekaii-Saab T, et al. Prostate cancer incidence in males with Lynch syndrome. Genet Med. (2014) 16:553–7. doi: 10.1038/gim.2013.193
61. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. (2017) 23:703–13. doi: 10.1038/nm.4333
62. Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. (2016) 375:443–53. doi: 10.1056/NEJMoa1603144
63. Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet. (2017) 49:1476–86. doi: 10.1038/ng.3934
64. Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. (2012) 149:979–93. doi: 10.1016/j.cell.2012.04.024
65. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun X-W, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. (2005) 310:644–8. doi: 10.1126/science.1117679
66. Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, et al. Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol. (2019) 35:40–7. doi: 10.1200/JCO.2016.69.1584
67. Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJM, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. (2014) 15:700–12. doi: 10.1016/S1470-2045(14)70189-5
68. Topalian S, Hodi F. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
69. Cabel L, Loir E, Gravis G, Lavaud P, Massard C, Albiges L, et al. Long-term complete remission with Ipilimumab in metastatic castrate-resistant prostate cancer: case report of two patients. J Immunother Cancer. (2017) 5:1–6. doi: 10.1186/s40425-017-0232-7
70. Basnet A, Khullar G, Mehta R, Chittoria N. A case of locally advanced castration-resistant prostate cancer with remarkable response to nivolumab. Clin Genitourin Cancer. (2017) 15:e881–4. doi: 10.1016/j.clgc.2017.05.005
71. Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-line nivolumab in stage IV or recurrent non–small-cell lung cancer. N Engl J Med. (2017) 376:2415–26. doi: 10.1056/NEJMoa1613493
72. Powles T, Durán I, van der Heijden MS, Loriot Y, Vogelzang NJ, De Giorgi U, et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): a multicentre, open-label, phase 3 randomised controlled trial. Lancet. (2018) 391:748–57. doi: 10.1016/S0140-6736(17)33297-X
73. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. (2015) 348:124–8. doi: 10.1126/science.aaa1348
74. Rosenberg JE, Hoffman-Censits J, Powles T, Van Der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. (2016) 387:1909–20. doi: 10.1016/S0140-6736(16)00561-4
75. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. (2014) 371:2189–99. doi: 10.1056/NEJMoa1406498
76. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. (2016) 165:35–44. doi: 10.1016/j.cell.2017.01.010
77. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. (2015) 350:207–11. doi: 10.1126/science.aad0095
78. Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. (2019) 51:202–6. doi: 10.1038/s41588-018-0312-8
79. Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, et al. Cancer research. Cancer Res. (2008) 20:1561–72. doi: 10.1158/0008-5472.can-07-3095
80. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. (2012) 482:400–4. doi: 10.1038/nature10755
81. van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. (2013) 31:e439–42. doi: 10.1200/JCO.2012.47.7521
82. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. (2015) 348:803–8. doi: 10.1126/science.aaa3828
83. Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. (2014) 515:572–6. doi: 10.1038/nature14001
84. Tran E, Turcotte S, Gros A, Robbins PF, Lu Y-C, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. (2014) 344:641–5. doi: 10.1126/science.1251102
85. Sharma P, Pachynski RK, Narayan V, Flechon A, Gravis G, Galsky MD, et al. Initial results from a phase II study of nivolumab (NIVO) plus ipilimumab (IPI) for the treatment of metastatic castration-resistant prostate cancer (mCRPC; CheckMate 650). J Clin Oncol. (2019) 37:142. doi: 10.1200/JCO.2019.37.7_suppl.142
86. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. (2017) 413:409–13. doi: 10.1126/science.aan6733
87. Rodrigues DN, Rescigno P, Liu D, Yuan W, Carreira S, Lambros MB, et al. Immunogenomic analyses associate immunological alterations with mismatch repair defects in prostate cancer. J Clin Invest. (2018) 128:4441–53. doi: 10.1172/JCI121924
88. Schweizer MT, Cheng HH, Tretiakova MS, Vakar-Lopez F, Klemfuss N, Konnick EQ, et al. Mismatch repair deficiency may be common in ductal adenocarcinoma of the prostate. Oncotarget. (2016) 7:82504–10. doi: 10.18632/oncotarget.12697
89. Reimers MA, Abida W, Chou J, George DJ, Heath EI, McKay RR, et al. IMPACT: immunotherapy in patients with metastatic cancers and CDK12 mutations. J Clin Oncol. (2019) 37: TPS5091. doi: 10.1200/JCO.2019.37.15_suppl.TPS5091
90. Ashworth A, Lord CJ. Synthetic lethal therapies for cancer: what's next after PARP inhibitors? Nat Rev Clin Oncol. (2018) 15:564–76. doi: 10.1038/s41571-018-0055-6
91. Yu EY, Massard C, Retz M, Tafreshi A, Carles Galceran J, Hammerer P, et al. Keynote-365 cohort A: pembrolizumab (pembro) plus olaparib in docetaxel-pretreated patients (pts) with metastatic castrate-resistant prostate cancer (mCRPC). J Clin Oncol. (2019) 37:145. doi: 10.1200/JCO.2019.37.7_suppl.145
Keywords: prostate cancer, tumor mutation burden, copy number alteration, structural variants, aneuploidy, mismatch repair deficiency, immune checkpoint inhibitor
Citation: Ryan MJ and Bose R (2019) Genomic Alteration Burden in Advanced Prostate Cancer and Therapeutic Implications. Front. Oncol. 9:1287. doi: 10.3389/fonc.2019.01287
Received: 05 September 2019; Accepted: 06 November 2019;
Published: 22 November 2019.
Edited by:
Renee de Leeuw, University of Illinois at Chicago, United StatesReviewed by:
Preston C. Sprenkle, Yale University, United StatesKouji Izumi, Kanazawa University, Japan
Francesca Khani, Cornell University, United States
Copyright © 2019 Ryan and Bose. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rohit Bose, cm9oaXQuYm9zZUB1Y3NmLmVkdQ==