 Javier Oliver1,2,3†
Javier Oliver1,2,3† Rosalía Quezada Urban3,4†
Rosalía Quezada Urban3,4† Claudia Alejandra Franco Cortés2
Claudia Alejandra Franco Cortés2 Clara Estela Díaz Velásquez3Ana Lorena Montealegre Paez5
Clara Estela Díaz Velásquez3Ana Lorena Montealegre Paez5 Rafael Adrián Pacheco-Orozco5
Rafael Adrián Pacheco-Orozco5 Carlos Castro Rojas5Reggie García-Robles5
Carlos Castro Rojas5Reggie García-Robles5 Juan Javier López Rivera6
Juan Javier López Rivera6 Sandra Gaitán Chaparro7Ana Milena Gómez8
Sandra Gaitán Chaparro7Ana Milena Gómez8 Fernando Suarez Obando8,9
Fernando Suarez Obando8,9 Gustavo Giraldo10Maria Isabel Maya10Paula Hurtado-Villa11,12Ana Isabel Sanchez12,13Norma Serrano14Ana Isabel Orduz Galvis14Sandra Aruachan15Johanna Nuñez Castillo15
Gustavo Giraldo10Maria Isabel Maya10Paula Hurtado-Villa11,12Ana Isabel Sanchez12,13Norma Serrano14Ana Isabel Orduz Galvis14Sandra Aruachan15Johanna Nuñez Castillo15 Cecilia Frecha16Cecilia Riggi17Federico Jauk2Eva María Gómez García18
Cecilia Frecha16Cecilia Riggi17Federico Jauk2Eva María Gómez García18 Claudia Lorena Carranza19
Claudia Lorena Carranza19 Vanessa Zamora19Gabriela Torres Mejía20Isabelle Romieu20,21
Vanessa Zamora19Gabriela Torres Mejía20Isabelle Romieu20,21 Carlos Arturo Castañeda22
Carlos Arturo Castañeda22 Miluska Castillo23Rina Gitler24Adriana Antoniano24Ernesto Rojas Jiménez3,4
Miluska Castillo23Rina Gitler24Adriana Antoniano24Ernesto Rojas Jiménez3,4 Luis Enrique Romero Cruz3,4Fernando Vallejo Lecuona3,4Iván Delgado Enciso25
Luis Enrique Romero Cruz3,4Fernando Vallejo Lecuona3,4Iván Delgado Enciso25 Abril Bernardette Martínez Rizo26Alejandro Flores Carranza27Verónica Benites Godinez27Claudia Fabiola Méndez Catalá3Luis Alonso Herrera28
Abril Bernardette Martínez Rizo26Alejandro Flores Carranza27Verónica Benites Godinez27Claudia Fabiola Méndez Catalá3Luis Alonso Herrera28 Yolanda Irasema Chirino4Luis Ignacio Terrazas3,4
Yolanda Irasema Chirino4Luis Ignacio Terrazas3,4 Sandra Perdomo5,29*and
Sandra Perdomo5,29*and  Felipe Vaca Paniagua3,4,30* on behalf of the LACAM Study
Felipe Vaca Paniagua3,4,30* on behalf of the LACAM Study- 1Medical Oncology Service, Hospitales Universitarios Regional y Virgen de la Victoria, Institute of Biomedical Research in Malaga, CIMES, University of Málaga, Málaga, Spain
- 2Laboratorio de Secuenciación, Instituto de Medicina Traslacional e Ingeniería Biomédica, Hospital Italiano de Buenos Aires, Buenos Aires, Argentina
- 3Laboratorio Nacional en Salud, Diagnóstico Molecular y Efecto Ambiental en Enfermedades Crónico-Degenerativas, Facultad de Estudios Superiores Iztacala, Tlalnepantla de Baz, Mexico
- 4Unidad de Biomedicina, Facultad de Estudios Superiores Iztacala, UNAM, Tlalnepantla de Baz, Mexico
- 5Instituto de Nutrición, Genética y Metabolismo, Facultad de Medicina, Universidad El Bosque, Bogota, Colombia
- 6Grupo INPAC, Organización Keralty, Departamento de Genética, Clínica Universitaria Colombia, Bogotá, Colombia
- 7Grupo INPAC, Organización Keralty, Facultad de Medicina, Fundación Universitaria Sanitas, Bogotá, Colombia
- 8Servicio de Genética, Hospital Universitario San Ignacio, Bogotá, Colombia
- 9Instituto de Genética Humana, Facultad de Medicina, Pontificia Universidad Javeriana, Bogotá, Colombia
- 10Clínica Universitaria Bolivariana, Pontificia Universidad Bolivariana, Medellín, Colombia
- 11Departamento Ciencias Básicas de Salud, Facultad de Ciencias de la Salud, Pontificia Universidad Javeriana Cali, Cali, Colombia
- 12Centro Médico Imbanaco, Cali, Colombia
- 13Departamento Materno Infantil, Facultad de Ciencias de la Salud, Pontificia Universidad Javeriana Cali, Cali, Colombia
- 14Fundación Cardiovascular de Colombia, Centro de Investigaciones, Floridablanca, Colombia
- 15Departamento de Investigación y Estudios Clínicos, IMAT - Oncomédica S.A., Montería, Colombia
- 16Instituto de Medicina Traslacional e Ingeniería Biomédica, CONICET-Instituto Universitario del Hospital Italiano-Hospital Italiano de Buenos Aires, Buenos Aires, Argentina
- 17Servicio de Ginecología, Hospital Italiano de Buenos Aires, Buenos Aires, Argentina
- 18Centro Oncológico Estatal ISSEMyM, Toluca de Lerdo, Mexico
- 19INVEGEM, Guatemala, Guatemala
- 20Instituto Nacional de Salud Pública, Cuernavaca, Mexico
- 21Hubert Department of Global Health, Emory University, Atlanta, GA, United States
- 22Departamento de Oncología Médica, Instituto Nacional de Enfermedades Neoplásicas, Lima, Peru
- 23Departamento de Investigación, Instituto Nacional de Enfermedades Neoplásicas, Lima, Peru
- 24Fundación Alma, Ciudad de México, Mexico
- 25Instituto Estatal de Cancerología de Colima, Colima, Mexico
- 26Laboratorio de Investigación Biomédica, Unidad Académica de Medicina, Universidad Autónoma de Nayarit, Tepic, Mexico
- 27Instituto Mexicano del Seguro Social, Ciudad de México, Mexico
- 28Unidad de Investigación Biomédica en Cáncer, Instituto de Investigaciones Biomédicas-Instituto Nacional de Cancerología, Ciudad de México, Mexico
- 29Departamento de Patología, Hospital Universitario Fundación Santa Fe de Bogotá, Bogota, Colombia
- 30Instituto Nacional de Cancerología, Ciudad de México, Mexico
Purpose: Hereditary Breast and Ovarian Cancer (HBOC) syndrome is responsible for ~5–10% of all diagnosed breast and ovarian cancers. Breast cancer is the most common malignancy and the leading cause of cancer-related mortality among women in Latin America (LA). The main objective of this study was to develop a comprehensive understanding of the genomic epidemiology of HBOC throughout the establishment of The Latin American consortium for HBOC-LACAM, consisting of specialists from 5 countries in LA and the description of the genomic results from the first phase of the study.
Methods: We have recruited 403 individuals that fulfilled the criteria for HBOC from 11 health institutions of Argentina, Colombia, Guatemala, Mexico and Peru. A pilot cohort of 222 individuals was analyzed by NGS gene panels. One hundred forty-three genes were selected on the basis of their putative role in susceptibility to different hereditary cancers. Libraries were sequenced in MiSeq (Illumina, Inc.) and PGM (Ion Torrent-Thermo Fisher Scientific) platforms.
Results: The overall prevalence of pathogenic variants was 17% (38/222); the distribution spanned 14 genes and varied by country. The highest relative prevalence of pathogenic variants was found in patients from Argentina (25%, 14/57), followed by Mexico (18%, 12/68), Guatemala (16%, 3/19), and Colombia (13%, 10/78). Pathogenic variants were found in BRCA1 (20%) and BRCA2 (29%) genes. Pathogenic variants were found in other 12 genes, including high and moderate risk genes such as MSH2, MSH6, MUTYH, and PALB2. Additional pathogenic variants were found in HBOC unrelated genes such as DCLRE1C, WRN, PDE11A, and PDGFB.
Conclusion: In this first phase of the project, we recruited 403 individuals and evaluated the germline genetic alterations in an initial cohort of 222 patients among 4 countries. Our data show for the first time in LA the distribution of pathogenic variants in a broad set of cancer susceptibility genes in HBOC. Even though we used extended gene panels, there was still a high proportion of patients without any detectable pathogenic variant, which emphasizes the larger, unexplored genetic nature of the disease in these populations.
Introduction
Breast and ovarian cancers have been recognized as heterogeneous diseases comprising several molecular subtypes, each one with an associated risk profile and specific treatment recommendations (1–3). The majority of the cases are sporadic; however, 5–10% of breast and ovarian cancers occur in patients with germline predisposition variants (4). Patients predisposed to breast and ovarian cancer have the phenotype that is defined as the Hereditary Breast and Ovarian Cancer (HBOC) syndrome.
Latin America (LA) is a region encompassing 33 countries, with diverse culture, geography, and ethnicity. Their resources, health care systems, and socioeconomic status are highly diverse among countries. Genetic studies have shown a unique history of migration to this region, combined with a high diversity of native population per country and important population bottlenecks (5, 6). Thus, LA is considered the most genetically admixed population in the world, and its specific combinations of genetic ancestries may impact on the genomic determinants of diseases (7). In Latin America, breast cancer is the most common malignancy and the leading cause of cancer-related mortality among women (8). By country, the highest standardized incidence rates of breast cancer by age are present in Argentina, Brazil and Uruguay (between 67.7 and 71.9) and the lowest in Bolivia (12.7) and El Salvador (7.9). Geographic variation of breast cancer incidence in LA may be explained by reproductive patterns, lifestyle factors, possibility of early detection and access to healthcare (9) together with ethnicity and different ancestral genetic components (7, 10). In terms of mortality, Uruguay (20.5), Argentina (19.4), and Cuba (14.9) show the higher rates whereas Guatemala (3.9) and El Salvador (4.1) show the lowest (9). Moreover, both incidence and mortality proportions for Latin American women aged under 44 years have been shown to be higher when compared with those found in high income countries (20 vs. 12% and 14 vs. 7%, respectively), suggesting an important contribution in LA of prevalent risk factors and genetic background (11).
Germline variation in BRCA1 and BRCA2 (BRCA1/2) genes is responsible for up to half of the heritable pathogenic variants in HBOC. However, germline variants in other recognized HBOC susceptibility genes have been identified including ATM, CDH1, CHEK2, PALB2, PTEN, STK11, and TP53 (12). Recently, germline pathogenic variants in other genes present in the same DNA repair pathway as BRCA1/2 have been identified and are associated with HBOC, including RAD51C, BRIP1, BARD1, and MRE11A (12, 13). In addition, recent reports have shown a potential implication of pathogenic heterozygotic alterations of genes related to other DNA repair mechanisms such as non-homologous end joining (NHEJ), Fanconi anemia pathway (FANCM, FANCI, FANCL, FANCC, FANCB, FANCF, FANCE), base-excision repair (MUTYH), nucleotide excision repair (ERCC2, ERCC6), and mismatch repair (MLH1, MLH3, MSH2, MSH5, MSH6) (12, 14–16). Nevertheless, clinical guidelines for appropriate testing and interventions are not yet available for these genes and pathogenic variants are reported exclusively in the research context.
Currently, there is a limited volume of studies of genetic susceptibility to HBOC in Latin America, and the majority of them are focused on the BRCA1/2 genes. Only a few publications have described the association of pathogenic variants in genes other than BRCA1/2 in selected HBOC cases in patients from Colombia (17), Mexico (18), Brazil (19, 20), and Chile (21). Therefore, a network of collaborators and cancer specialists in LA joined common efforts to create a The Latin American consortium for HBOC-LACAM. The LACAM study aims to recruit a large series of 3,000 cases from multiple centers in LA in 4 years, which will make it the largest study of HBOC in these populations. This population size will ensure accurate and robust findings on the role of germline pathogenic variants in cancer susceptibility genes and modifying risk factors. The purpose of this work is to describe the standard procedures for patient recruitment, data collection, sample preparation and next-generation sequencing (NGS) analysis of cancer susceptibility gene panels for the LACAM consortium. Additionally, we describe the frequency of germline variations in an initial cohort of 222 individuals who fulfilled the criteria for HBOC from Argentina, Colombia, Guatemala and México using two NGS gene panels.
Materials and Methods
LACAM Participating Countries and Institutions
We identified countries in the Latin American region with few or absent genomic studies for HBOC. Aiming to cover the diversity of populations of this region in terms of ancestry and sociocultural factors, we included representative countries from North America: Mexico; Central America: Guatemala; and South America: Colombia, Peru; and the Southern cone, including Argentina.
Participants are being recruited at 11 centers, including 2 centers from Estado de Mexico and Mexico City in Mexico (Centro Oncológico Estatal de Toluca, Instituto Mexicano del Seguro Social Siglo XXI), 6 centers from different regions in Colombia (Clínica Universitaria Colombia, Hospital Universitario San Ignacio, UPB Clínica Universitaria Bolivariana, IMAT-Oncomédica S.A., Centro Médico Imbanaco, Fundación cardiovascular de Colombia), one center in Peru (Instituto Nacional de Enfermedades Neoplásicas), Argentina (Hospital Italiano de Buenos Aires) and Guatemala (Instituto para la Investigación Científica y la Educación Acerca de las Enfermedades Genéticas y Metabólicas Humanas). The LACAM protocol was approved by the Ethics Committee of each center (INEN-18-06, FM-CIE-0409-17, HI-2730, COE/UEI/PT/02/2018, INSP-CI:1065, INSP-341, CE/INVEGEM 1-2017, UEB.471-2018, ONC-CEI-801-2018, UPB-2018, ISEM 28-09-2015, CEICANCL290515-05GENCMAHER) and it is conducted in accordance with the Declaration of Helsinki.
LACAM Recruitment Protocol and Standard Operating Procedures (SOPs)
All individuals were enrolled based on the criteria established in the Genetic/Familial High-Risk Assessment: Breast and Ovarian of the National Comprehensive Cancer Network (NCCN) guidelines, version 2.2018 (22). Participating individuals were identified in the Clinical genetics, Oncology or Gynecology consultation of each institution (incident cases) or through the review of clinical records (prevalent cases). Prevalent cases were also interviewed to confirm that they met the inclusion criteria. Each study participant received a unique identification number based on the center number and a 4-digit patient number. This code is allocated upon patient's agreement to participate in the study. All patients were provided with written informed consent for the participation in this study.
Individuals underwent an interview and completion of a questionnaire about their lifestyle and basic demographic information was requested. The lifestyle questionnaire is structured to collect the following information: (i) Demographic details (age, sex, ethnic origin, city and place of residence and educational status); (ii) History of tobacco use; (iii) History of alcohol consumption, including types of alcoholic beverages; and (iv) Reproductive factors including parity, contraceptive use, age of menarche, and age of menopause.
In addition, we recorded anthropometric measurements: weight and height at time of recruitment, abdominal perimeter, weight up to 2 years before the interview, as well as weight at the age of 20 years. For all patients diagnosed with cancer, there is an additional clinical questionnaire which gathers all the available information on clinical and histopathological diagnosis and treatment. Study data are collected and managed using REDCap electronic data capture tools hosted at Hospital Universitario San Ignacio. REDCap (Research Electronic Data Capture) is a secure, web-based application designed to support data captured from research studies, therefore providing (1) an intuitive interface for validated data entry; (2) audit trails for tracking data manipulation and export procedures; (3) automated export procedures for seamless data downloads to common statistical packages; and (4) procedures for importing data from external sources (23). In all participating centers, clinical geneticists or health care personnel trained in genetic counseling were involved during recruitment in order to guarantee pre- and post-test communication and guidance.
Standardization of Sequencing Protocol and Analysis
After completion of the first phase of the study (first year of recruitment), we standardized methods for sample preparation, sequencing, and variant analysis using the first 222 individuals included in this study.
Sample Preparation
Blood extraction was performed by trained personnel at the time of the interview unless a more convenient time was available. 10-mL samples of blood are collected with EDTA in order to obtain: whole blood, buffy coat (leukocytes/platelets), red blood cells, and plasma. All samples are stored at each center at −80°C. In any case the amount of time between extraction and freezing does not exceed 12 h. When available, H&E slides and FFPE tumor tissue are collected.
DNA Extraction
Peripheral blood DNA was extracted with the DNeasy Blood & Tissue Kit (Qiagen) and the Magna Pure System (Roche) following the manufacturer's instructions. The DNA concentration was quantified with the Qubit dsDNA HS Assay Kit (Invitrogen) and the integrity and purity of the material was verified by agarose gel electrophoresis and spectrophotometry.
Preliminary Information on Sequencing Variants
To evaluate the prevalence of germline pathogenic variants in individuals with criteria for hereditary disease from Latin America, we performed NGS of peripheral blood DNA of 68 individuals from Mexico, 57 from Argentina and 19 from Guatemala and analyzed a panel of 143 cancer susceptibility genes. The resulting pathogenic variants were polled with those from 78 Colombian patients sequenced with the Trusight kit (94 cancer susceptibility genes). All pathogenic variants correspond to genes shared by both panels, with the exception those found in DCLRE1C, MLH3, PDE11A, PDGFB.
Library Preparation and Massive Parallel Sequencing
Library preparation and sequencing of the Mexican, Argentinian and Guatemalan samples was performed with the GeneRead Cancer Predisposition V2 Kit (Qiagen), as previously described (18). Briefly, the kit targets 143 genes associated with inherited cancers. The genes have been selected on the basis of their putative role in the susceptibility of different hereditary cancers. Most, but not all, are associated with hereditary breast and ovarian cancer and with an increased risk to 88 oncological syndromes such as colorectal, ovarian, endometrial, prostate, gastric, and pancreatic cancers among others; and almost all these genes have evidence supporting a >2-fold increase in risk. The libraries were sequenced in a MiSeq instrument (Illumina, Inc.; 2X150 cycles) to reach an average theoretical mean coverage of 80X for each sample (Supplementary Figures 1, 2). The DNA samples from Argentina were prepared with Generead Library L kit (Qiagen) and sequenced in a PGM (Ion Torrent-Thermo fisher- Scientific). The libraries from Colombia were prepared using the Trusight cancer panel (Illumina, Inc.), which includes a panel of 96 genes (40 genes overlap with the 143 gene panel) and sequenced in a MiSeq instrument (Illumina, Inc.; 2X150 cycles).
Sequencing Normalization and Pathogenic Variant Detection
The variant reduction to identify pathogenic variants was done as reported previously (18). Briefly, FastQC files were aligned to the human genome reference hg19 with BWA-MEM; indels were realigned and the bases were recalibrated. Adaptors were soft-clipped and reads with <20 bp were eliminated. Variant calling was done with HaplotypeCaller (Broad Institute) and annotation with ANNOVAR and InterVar (24, 25). Pathogenic variant description was done following Human Genome Variation Society (HGVS) nomenclature (http://www.hgvs.org/) and variant classification followed the five-tier criteria of the American College of Medical Genetics and Genomics (ACMG) (26) and was manually curated. Synonymous variants and those with depth <5X or with mutant allele fraction <20% were excluded. Splicing and null variants (stop-gain/loss, frameshift indels) and missense variants previously defined as pathogenic in ClinVar were considered unequivocally pathogenic (https://www.ncbi.nlm.nih.gov/clinvar). Null variants present at the 3′ end of the gene that were reported as conflicting in ClinVar were classified as “unknown clinical significance” (VUS). Minor allelic frequency <0.001 in the gnomeAD (http://gnomad.broadinstitute.org/) database was used to capture rare, potentially pathogenic, null and missense variants. Low frequency (<0,001) missense variants predicted as deleterious by SIFT or PolyPhen-2 but with no further evidence of in vitro/vivo or clinical pathogenicity were classified as VUS. All filtered variants were manually curated by inspection of the BAM files with the IGV software (Broad Institute). All pathogenic and likely pathogenic variants were confirmed experimentally by two independent Sanger sequencing assays. Variants in BRCA1 and BRCA2 were further assessed in the Huntsman Cancer Institute Breast Cancer Genes Prior Probabilities site (http://priors.hci.utah.edu/PRIORS/index.php) to evaluate their potential impact. Variants in MLH1, MSH2, MSH6, PMS2 were also investigated in the Leiden Open Variation Database (http://hci-lovd.hci.utah.edu/home.php). Both pathogenic and likely pathogenic variant categories were grouped as “pathogenic” to simplify presentation and discussion of our results.
Detection of Exon 9-12 Deletion in BRCA1
The founder mutation consisting in a deletion in exons 9–12 was detected by PCR amplification of the mutant and wild-type alleles, using specific primers based the method published in Weitzel et al. (18, 27). The PCR products were resolved in 1.5% agarose gels to identify the amplification of the truncated allele and sequenced.
Large Rearrangement (LR) Analysis
Patients from Colombia without BRCA1 or BRCA2 pathogenic variants were tested for MLPA (Multiplex Ligation-dependent Probe Amplification), using SALSA® MLPA® probemix P002 BRCA1 and P090 BRCA2 kits.
Statistical Analysis
Characteristics of all recruited individuals and cases with confirmed diagnosis of cancer were summarized with descriptive statistics. The association between demographic and clinical characteristics on the presence of pathogenic variants was assessed using univariate analyses (unadjusted logistic regression model). Age at recruitment, age at diagnosis for cancer cases and body mass index (BMI) were included as continuous variables, whereas the rest of the factors were considered as categorical variables. The logistic regression model utilized all available data (complete and missing). p < 0.05 was considered to indicate statistical significance. All the analyses were conducted using STATA 13.0.
Results
Initial LACAM Recruitment in 5 Countries
Four hundred and three individuals have been recruited in the first phase of the LACAM study from October 2017 to March 2019 (Supplementary Table 2). Epidemiological characteristics with complete information were obtained for more than 80% of individuals and are listed in Table 1. Fifty-seven percent of individuals had a family history of cancer, 72% reported at least one pregnancy and the average parity was three children (SD: 1.7), 37% never used oral contraceptives and 65% reported not being current or former alcohol consumers or smokers. Importantly, 48% percent of all individuals were overweight or obese at time of recruitment.
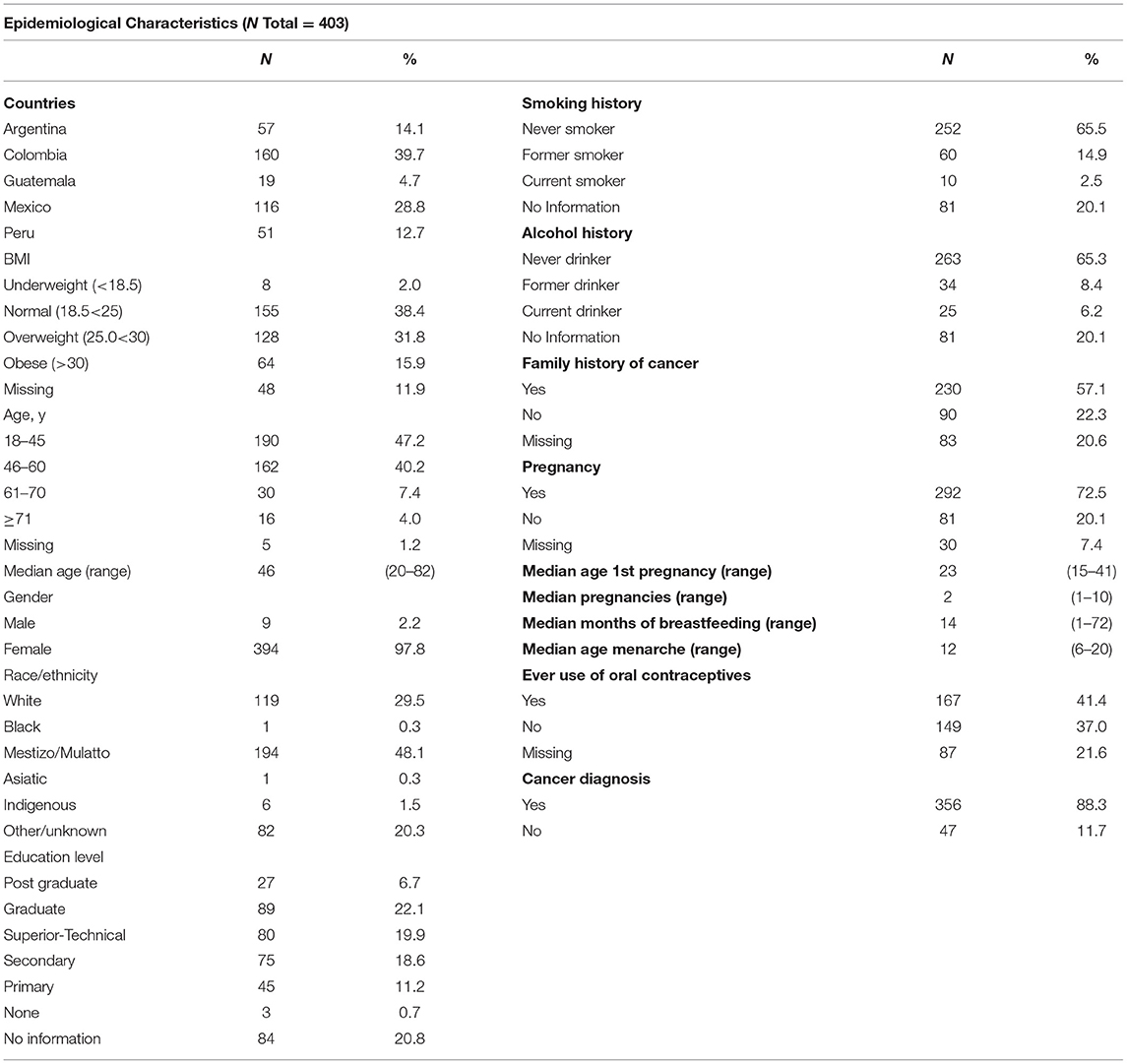
Table 1. Epidemiological characteristics of the 403 individuals recruited in the first phase of the LACAM study.
Eighty-eight percent of the individuals had a cancer diagnosis from which 75% corresponded to breast cancer cases (Table 2). Complete staging (pathological or clinical) information was available for 42% of cases.
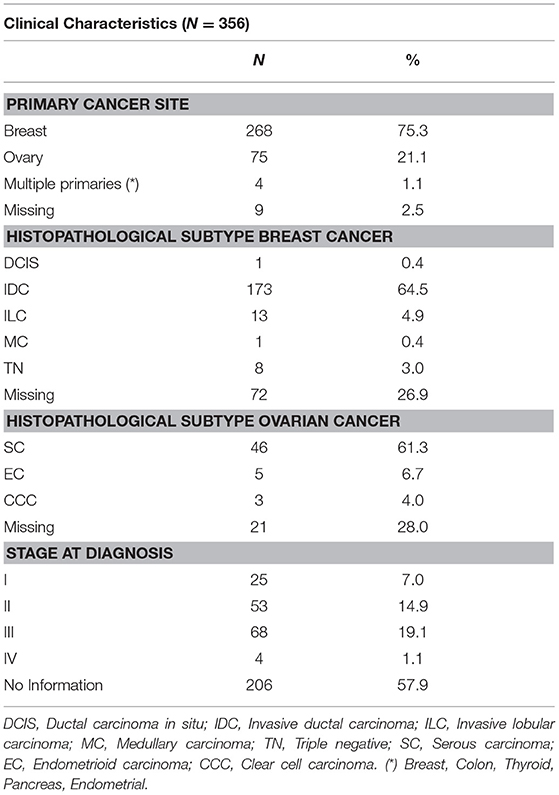
Table 2. Clinical characteristics of 356 cancer patients recruited in the first phase of the LACAM study.
Preliminary Sequencing Results
Initial sequencing results on 222 individuals showed that the overall prevalence of individuals with pathogenic variants was 17% (38/222) and the distribution spanned 14 genes and varied by country (Figure 1, Table 3). The highest relative prevalence of pathogenic variants was found in patients from Argentina (25%, 14/57), followed by Mexico (18%, 12/68), Guatemala (16%, 3/19), and Colombia (13%, 10/78). Twenty-five percent (1/4) of the pathogenic variants found in BRCA1 in the Mexican patients corresponded to the founder mutation ex9-12del. All pathogenic variants were heterozygous. The overall prevalence of patients with a negative result (without any detectable pathogenic variants or VUS) was 48% (106/222). By country the prevalence of patients with a negative result was Guatemala 53% (10/19), México 52% (36/68), Colombia 47% (37/78), Argentina 40% (23/57). No additional large rearrangements (LR) were found in the 33 individuals from Colombia.
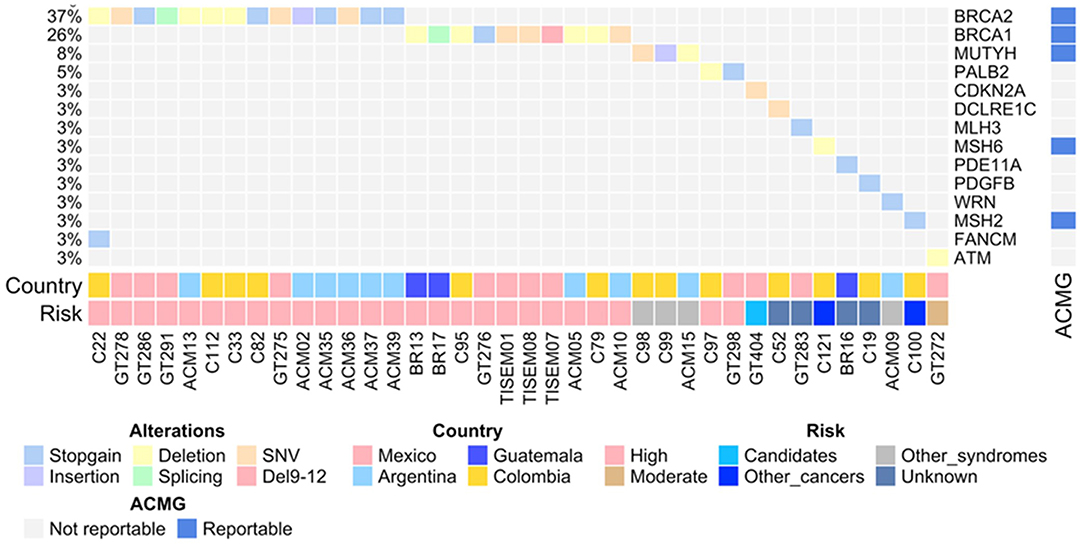
Figure 1. The OncoPrint shows the allelic distribution of the pathogenic variants in patients with cancer. The grid panel depicts the pathogenic mutations found in each patient color-coded for each type. Right panel: gene reportable by the suggestion of the ACMG (blue: yes, gray: no). Bottom axis: patient ID. Left axis: relative frequency of mutations per gene. Right axis: mutated gene. Right bar plot: absolute frequency and type of pathogenic mutation per gene. Bottom panel indicates: Country; risk associated with the variant.
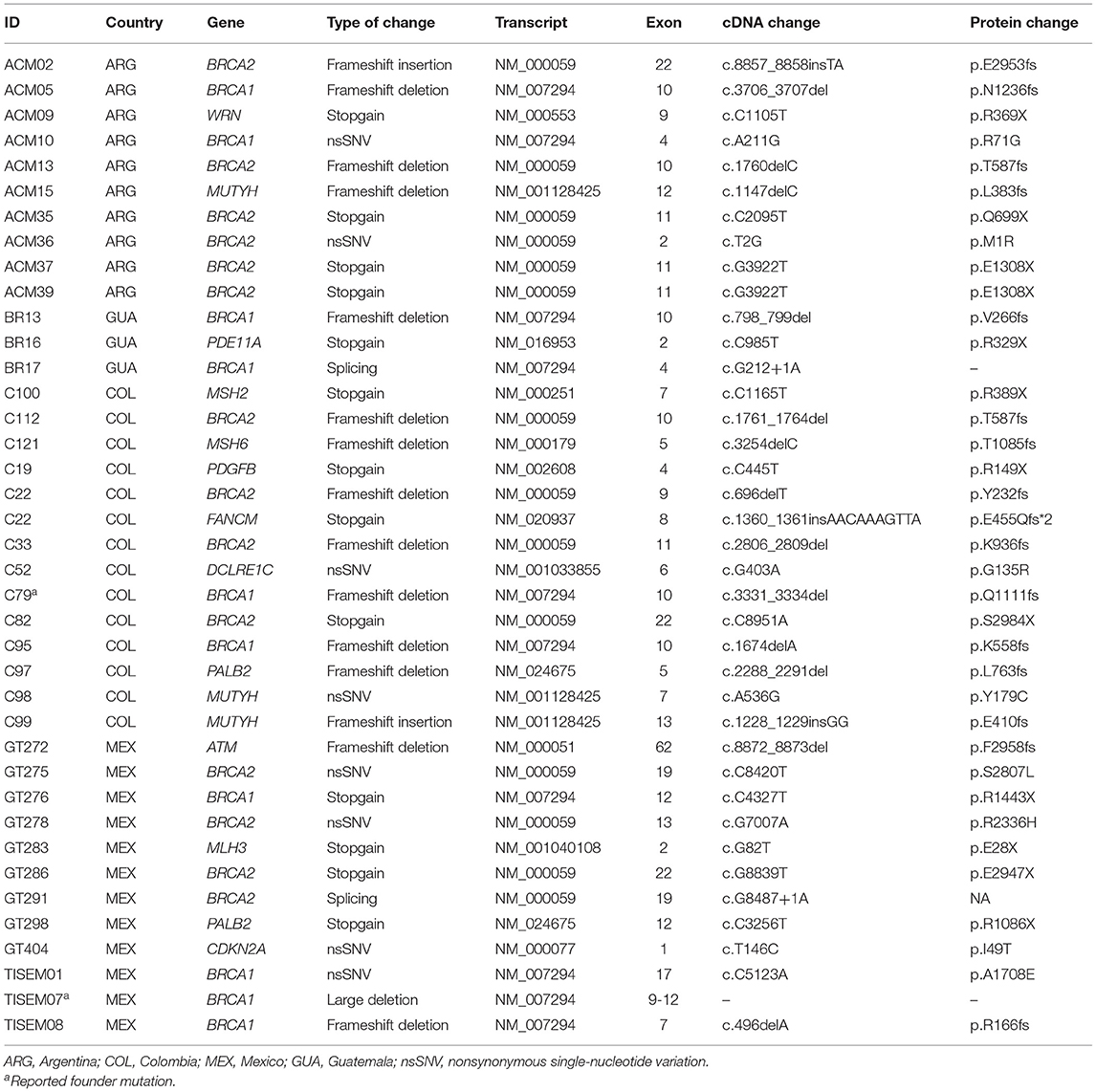
Table 3. Pathogenic variants detected in 222 HBOC individuals from 4 countries.
Forty eight percent (18/38) of pathogenic variants were found in patients diagnosed with breast cancer, 24% (9/38) in ovarian cancer and 21% (8/38) in individuals without cancer diagnosis but with family history of cancer under HBOC criteria.
Pathogenic Variants in BRCA1 and BRCA2 Genes
Twenty percent of the total number of pathogenic variants were found in BRCA1 and 29% in BRCA2 genes in the four countries. Noteworthy, patients from Guatemala and Argentina had the highest country proportion of BRCA1 (40%) and BRCA2 (71%) pathogenic variants, respectively (Figure 1). Only two previously reported founder mutations were found in two patients from Mexico and Colombia. Finally, all of the patients had one monoallelic pathogenic variant, excluding patient C22 from Colombia, who had two monoallelic variants in BRCA2 and in FANCM (Figure 1, Table 3).
Pathogenic Variants in Non-BRCA Genes
Pathogenic variants in non-BRCA genes were found in 12 genes, including those in other high and moderate HBOC risk genes such as MSH2, MSH6, MUTYH, and PALB2. The hereditary melanoma and pancreatic cancer gene CDKN2A was found mutated in one patient from Mexico. Only 3 of these genes, however, are currently suggested as reportable by the ACMG (26) (Table 4). Among the countries included in this study, Colombian individuals were the most affected with pathogenic variants in non-BRCA genes presenting 8 out of 14 participants with these alterations (Table 4). Moreover, pathogenic variants were found in the DNA repair genes DCLRE1C (NHEJ) and WRN (homologous recombination), along with the cyclic-AMP regulator PDE11A in a patient from Guatemala and the growth-promoting gene PDGFB in a Colombian patient. Pathogenic variants in non-BRCA genes were more frequent in patients diagnosed with Breast cancer (21%) compared to those with ovarian cancer (11%).
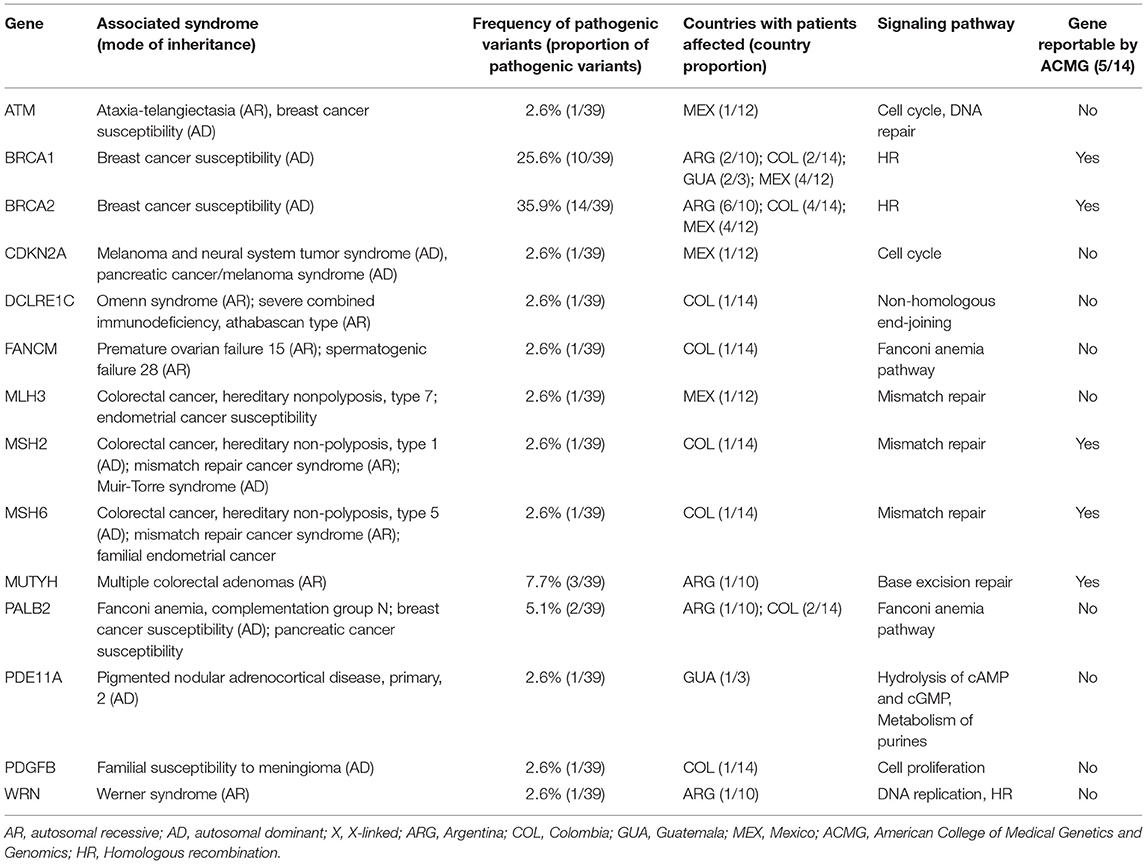
Table 4. Hereditary syndromes associated with the pathogenic variants detected in 222 individuals.
Of note, no association was found between epidemiological or clinical characteristics described in Tables 1, 2 and presence of pathogenic alterations in any of the 143 genes evaluated.
Variants With Unknown Clinical Significance (VUS)
We detected a total number of 132 VUS representing a prevalence of 41% (90/222). These variants were distributed in 76 genes, including high-risk HBOC genes (BRCA1, BRCA2, APC, CDKN2A, CDH1, MSH2, MSH6), moderate risk (ATM, CHEK2, RAD51C, PALB2) and genes with no risk defined to date (Supplementary Table 1). The gene carrying the highest number of VUS was PMS2, probably due to equivocal amplification and alignment of the PMS2 pseudogenes. The following genes with the highest number of VUS were FANCM (6/132) BRCA1 (5/132) APC (4/132) and BRCA2 (4/132). Notably, the proportion of the VUS in non-BRCA genes was 93.2%. Eight percent of individuals (19/222) presented more than one VUS.
Discussion
Breast cancer is a highly prevalent disease in Latin America (LA) and worldwide (28). The epidemiological transition along with the adoption of unhealthy western life-styles together with changing reproductive factors have influenced the increased incidence of breast cancer in LA (9). Several countries of the LA region have acknowledged this as a public health problem and have specific policies to progress in early detection, which is the most effective measure to control this disease.
The LACAM study pursued the primary aim of constructing a network of collaborators to investigate the genetic susceptibility to breast cancer in Latin America focusing mainly on HBOC. We gathered a group of researchers from Argentina, Colombia, Guatemala, Peru and Mexico to recruit individuals from families that fulfill criteria of HBOC. In this first phase of the project we have recruited 403 individuals from five countries, following uniformed procedures for sampling, questionnaire assessment and methods for sequencing analysis.
Fortunately, there has been progress in the region regarding description of HBOC—associated pathogenic variants in the BRCA1 and BRCA2 genes. Relevant works have identified pathogenic recurrent founder mutations in Mexico, Colombia, Argentina, Brazil, Chile, Costa Rica, Cuba and Peru (29). Currently, there are few studies reporting causal pathogenic variants in a more complete set of high and moderate HBOC risk genes in the region (17–21), in comparison with dozens of gene panel studies performed in European and American Caucasian populations. This clearly shows the important disparity in the knowledge of the genetic diversity of this disease as compared with other populations (30). Although the contributions made in LA are of major relevance, much effort still needs to be done for the correct identification of the genetic component for to HBOC susceptibility in this region.
We analyzed the genetic profile using two panels of cancer susceptibility genes by next-generation sequencing and we found that 17% (38/222) of the patients carried pathogenic variants in 14 susceptibility genes. Overall, the prevalence of pathogenic variants in the BRCA1 and BRCA2 genes in Argentina, Mexico, Guatemala and Colombia reached 14.3, 11.8, 10.5, and 8.1%, respectively. Only two previously reported founder mutations in the BRCA1 gene were found (18, 27, 29, 31–33): the ex9-12del founder mutation corresponding to 25% (1/4) of the BRCA1 pathogenic variants found in Mexican patients and the BRCA1 p.Q1111fs mutation found in a Colombian patient.
Although the number of patients with a pathogenic variant was relatively modest (38/222), the total proportion of pathogenic variants in BRCA1/2 as compared with other genes was high, 61.5 vs. 38.5%. Indeed, in Colombia, the proportion of pathogenic variants in non-BRCA genes was the highest, 57.1% (8/14). Moreover, the fact that 39% of the pathogenic variants were found in 12 genes distinct from BRCA1/2 genes, highlights the important locus heterogeneity of this disease that is present in these Latin American populations. Importantly, this is the first report of pathogenic variants in non-BRCA genes in patients with HBOC for Guatemala and Argentina.
On the other hand, the genes with pathogenic variants associated with other syndromes included the Fanconi Anemia FANCM, Lynch syndrome MLH3, MSH2, MSH6; the colorectal adenoma MUTYH, the pancreatic and melanoma CDKN2A; and the ataxia telangiectasia gene ATM, as previously reported (15, 34–37).
Pathogenic variants were also detected in genes not frequently associated with HBOC, such as DCLRE1C (16), PDGFB, WRN (16), and PDE11A (18). The DCLRE1C gene (cytoband 10p13) codes for the ubiquitously expressed nuclear protein Artemis, which has 5-3′ exonuclease activity and participates in V(D)J recombination and in non-homologous end joining DNA repair, especially in double-strand breaks caused by radiation and oxidative stress (38–41). Germline alterations in this gene have been reported in patients with autosomal recessive conditions such as the Athabascan-type severe combined immunodeficiency (SCIDA) and the Omenn syndrome (42, 43). Artemis is a substrate of phosphorylation by ATM in response to DNA damage and interacts with BRCA1 and the MRN complex composed of MRE11, RAD50, and NBN (44). The pathogenic variant detected in DCLRE1C, p.G135R, is located in the b-lactamase domain, which is the catalytic core for V(D)J recombination (45) and has been classified as a probably pathogenic in ClinVar. The intimate role of this protein in DNA repair is highly suggestive of a cancer susceptibility gene when it is altered in heterozygosity. As for the PDGFB gene (22q13.1), it codes for a potent mitogen protein that participates in the regulation of multiple processes including embryonic development, cell proliferation, cell migration, survival and chemotaxis. The alteration detected in PDGFB (p.R134X) truncates the protein in the N-terminal PDGF/VEGF domain, which is essential for the homo- or heterodimerization with PDGFA and for binding and activation of the PDGFR receptors. Germinal alterations in this gene cause familial susceptibility to meningiomas and brain calcifications, both with an autosomal dominant inheritance pattern (46–48). Particularly, there are no previous reports associating this gene to HBOC susceptibility. The individual carrying this variant (C19) has not been diagnosed with BC, but her family history included a maternal aunt diagnosed with premenopausal breast cancer, a sister with BC at 41 years of age and her mother was diagnosed with lung cancer. It is possible that additional genetic factors or specific exposures could play a role in the aggregation of the disease in this patient and her family.
In a previous study on HBOC we reported WRN and PDE11A alterations in two Mexican patients (18). In the current analysis, pathogenic variants in these genes we also found in cases from Argentina and Guatemala. The helicase WRN participates in the maintenance of genome stability, by interacting with translesion polymerases to prevent collapse of stalled replication forks (49). The WRN protein also interacts with the Ku70/80 helicases to facilitate DNA repair by the NHEJ pathway (50). Alterations in this gene in homozygosity cause Werner syndrome, characterized by premature aging, but monoallelic pathogenic variants in this gene have been previously reported in patients with inherited breast cancer, and its involvement in genome stability is compatible with a potential susceptibility role (16, 18, 51). On the other hand, PDE11A codes for a phosphodiesterase that catalyze the hydrolysis of intracellular concentrations of cyclic AMP and GMP being a negative regulator of growth promoting transduction pathways. Rare germline alterations in PDE11A have been associated with prostate and testicular germ cell tumor susceptibility (52, 53). Of importance, the same pathogenic variant detected in PDE11A, p.R329X, was identified in our previous study in one Mexican patient (18).
Noteworthy, only 5 out of the 14 genes with pathogenic variants described in our study are suggested to be reported as clinically relevant to the patient by the ACMG (26). Therefore, the overwhelming existing evidence shows that the contribution of new susceptibility genes to inherited breast cancer has been underestimated. In this regard, an effort to better establish the role of these non-BRCA genes to HBOC susceptibility in populations from LA is being carried out by the LACAM project and will be discussed in future publications.
Of note, in 48% of individuals there was no evidence of a potential disease causal variant, either VUS or pathogenic. These results show there is a large, unexplored genetic landscape of this disease, known as missing heritability (54). We acknowledge that a fraction of the patients could also be affected by large rearrangements (LR) that were not evaluated in individuals from Argentina and Guatemala. On the contrary, patients from Colombia were negative for LR and patients from Mexico were evaluated for the most common large deletion, the BRCA1 ex9-12del, which comprises almost half of the large rearrangements of this gene in Mexican population. In 2012 Judkins et al. reported that the percentage of LR positivity for Latin American/Caribbean cases in the US who met the criteria for BRCA testing was 6,7% and the ex9-12del of BRCA1 was 45% (44/97) among these cases (31). Using this percentage and considering that a major proportion of the here presented cohort is of Mexican origin, the number of missed LR in our preliminary analysis is not expected to be high. Additional considerations for negative sequencing results in any of the evaluated genes are the presence of variants in non-coding regions, not assessed, and the possibility that common low-risk variation could be acting in some of these families in combinatorial, epistatic ways. The contribution of the low-risk SNPs and the prevalence of LR will be addressed in the following phase of the LACAM project in a larger number of patients.
Unfortunately, the limited number of individuals carrying a pathogenic variant (N = 38) did not allow us to find statistically significant associations with modifying risk factors such as lifestyle and reproductive factors. Therefore, a larger analysis is envisioned for the second phase of our project to better account for the effect of these modifying factors in HBOC as they have never been evaluated in LA populations.
Finally, the ultimate purpose of the LACAM study is to provide solid and detailed scientific evidence on the molecular epidemiology of HBOC in the LA population in order to improve intervention strategies, prevention and risk management for patients affected by this disease. Current national cancer plans among 14 countries in LA include secondary prevention strategies with an emphasis on prevention, early detection, and opportune treatment (55). Also, several countries in the region have national guidelines recommending genetic counseling and testing in high-risk individuals (56). Identifying individuals with a high risk of developing breast and ovarian cancer among other associated HBOC tumors allows for more effective detection of the disease at earlier stages and enables the application of cancer prevention strategies among family members when risk assessment is provided before disease onset (22, 57). In order to efficiently implement these approaches, major efforts should be focused on: (i) Availability of centralized genetic testing to core facilities with certified laboratories to both ensure their cost-effectiveness and increase the technical and bioinformatic expertise in LA; (ii) creating open access databases of HBOC variants and large-scale cancer genomics data sets derived from similar studies in Latin American populations; (iii) strengthening the genetic counseling services in all countries of the region and increasing the number of training programs in clinical genetics; (iv) developing educational awareness programs for the general population; (v) generating formal analyses of the economic impact of different genetic testing options in LA to evidence real health benefit costs in the Latin American context; and (vi) engaging health policy makers, insurance companies, political and public health institutions and academia to enforce implementation of national cancer plans and country-specific genetic testing guidelines.
Conclusions
In this work, we established the framework of The Latin American consortium for HBOC-LACAM, a joint regional effort aimed to analyze the genetic component and modifying risk factors of inherited breast cancer in individuals from Latin America. In this first phase of the project, we recruited 403 individuals from Argentina, Colombia, Guatemala, Mexico and Peru and evaluated the germline genetic alterations in an initial cohort of 222 patients from 4 countries. The overall prevalence of pathogenic variants was 17% (38/222), distributed along 14 genes. Even if extended gene panels were used, there was still a high proportion of patients without detectable pathogenic variants, which emphasizes the large, unexplored genetic nature of the disease in these populations. The establishment of a possible associated risk of HBOC with non-BRCA genes still requires additional scientific evidence taking into consideration ancestral components, common genetic variation and non-genetic variables such as lifestyle and other risk factors. These factors will be addressed in the following phases of the LACAM project in a larger number of patients.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The LACAM protocol was approved by the Ethics Committee of each center (Instituto Nacional de Enfermedades Neoplásicas INEN-18-06, Hospital Universitario San Ignacio FM-CIE-0409-17, Hospital Italiano de Buenos Aires HI-2730, Centro Oncológico Estatal de Toluca COE/UEI/PT/02/2018, Instituto Mexicano del Seguro Social Siglo XXI INSP-CI:1065, INSP-341, Instituto para la Investigación Científica y la Educación Acerca de las Enfermedades Genéticas y Metabólicas Humanas CE/INVEGEM 1-2017, Universidad El Bosque UEB 471-2018, IMAT-Oncomédica SA ONC-CEI-801-2018, UPB Clínica Universitaria Bolivariana UPB-2018, Instituto de Salud del Estado de Mexico ISEM 28-09-2015, Instituto Estatal De Cancerología De Colima CEICANCL290515-05GENCMAHER) and it is conducted in accordance with the Declaration of Helsinki.
Author Contributions
AG, EG, CLC, CAC, JO, SP, and FVac: clinical protocol revision. GT, CLC, CAC, CF, EG, AM, and MC: clinical sampling. GT, CLC, CAF, ER, RGi, AA, AM, and MC: questionnaire assessment. RQ, CD, CLC, and CAF: DNA extraction and quality control. RQ, CD, and CAF: NGS Library preparation. JO, CF, LR, RQ, and FVac: bioinformatics analysis. CAF, LR, RQ, JO, SP, and FVac: data curation. CAF, RQ, JO, SP, and FVac: formal analysis and data presentation. RQ, CD, and CM: validation. SP: statistical analysis. CAC, JO, SP, and FVac: funding acquisition. LT, JO, SP, and FVac: resources. JO, SP, and FVac: original draft. SP, JO, and FVac: study conceptualization. All authors are writing, review and editing.
Funding
This work was supported by FNAB foundation grant and PICTO-2016-007 in Argentina. CONACyT (285879, 272573) and PAPIIT-UNAM (IN219217) in Mexico. COLCIENCIAS 130880762841, CTO 697-2018 in Colombia and CONCYTEC #198- 2015-FONDECYT in Peru.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge all individuals, patients and their families who consented to participate in the project.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.01429/full#supplementary-material
Supplementary Figure 1. Global distribution of the mean sequencing depth. Histogram of sequence depth (X) with density of all sequenced samples. Mean depth is indicated.
Supplementary Figure 2. Distribution of the mean sequencing depth per country. Histogram of sequence depth (X) with density of all sequenced samples by country. Mean depth by country is indicated.
Supplementary Table 1. Variants with unknown clínical significance detected.
Supplementary Table 2. Clinical and epidemiological characteristics of individuals recruited in the first phase of the LACAM study by country. DCIS, Ductal carcinoma in situ; IDC, Invasive ductal carcinoma; ILC, Invasive lobular carcinoma; MC, Medullary carcinoma; TN, Triple negative; SC, Serous carcinoma; EC, Endometrioid carcinoma; CCC, Clear cell carcinoma; (*) Breast, Colon, Thyroid, Pancreas, Endometrial. NA, Not applicable.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: Cancer J Clin. (2011) 61:69–90. doi: 10.3322/caac.20107
2. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. (2012) 490:61–70. doi: 10.1038/nature11412
3. Ellis MJ, Perou CM. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Disc. (2013) 3:27–34. doi: 10.1158/2159-8290.CD-12-0462
4. Foulkes WD. Inherited susceptibility to common cancers. N Eng J Med. (2008) 359:2143–53. doi: 10.1056/NEJMra0802968
5. Ruiz-Linares A, Adhikari K, Acuña-Alonzo V, Quinto-Sanchez M, Jaramillo C, Arias W, et al. Admixture in Latin America: geographic structure, phenotypic diversity and self-perception of ancestry based on 7,342 individuals. PLoS Genet. (2014) 10:e1004572. doi: 10.1371/journal.pgen.1004572
6. Adhikari K, Mendoza-Revilla J, Chacón-Duque JC, Fuentes-Guajardo M, Ruiz-Linares A. Admixture in Latin America. Curr Opini Gene Dev. (2016) 41:106–14. doi: 10.1016/j.gde.2016.09.003
7. Norris ET, Wang L, Conley AB, Rishishwar L, Mariño-Ramírez L, Valderrama-Aguirre A, et al. Genetic ancestry, admixture and health determinants in Latin America. BMC Genom. (2018) 19(Suppl. 8):861. doi: 10.1186/s12864-018-5195-7
8. Goss PE, Lee BL, Badovinac-Crnjevic T, Strasser-Weippl K, Chavarri-Guerra Y, Louis JS, et al. Planning cancer control in Latin America and the Caribbean. Lanc Oncol. (2013) 14:391–436. doi: 10.1016/S1470-2045(13)70048-2
9. Di Sibio A, Abriata G, Forman D, Sierra MS. Female breast cancer in Central and South America. Cancer Epidemiol. (2016) 44:S110–20. doi: 10.1016/j.canep.2016.08.010
10. Kehdy FSG, Gouveia MH, Machado M, Magalhães WCS, Horimoto AR, Horta BL, et al. Origin and dynamics of admixture in Brazilians and its effect on the pattern of deleterious mutations. Proc Nat Acad Sci USA. (2015) 112:8696–701. doi: 10.1073/pnas.1504447112
11. Villarreal-Garza C, Aguila C, Magallanes-Hoyos MC, Mohar A, Bargalló E, Meneses A, et al. Breast cancer in young women in Latin America: an unmet, growing burden. Oncologist. (2013) 18:1298–306. doi: 10.1634/theoncologist.2013-0321
12. Nielsen FC, van Overeem Hansen T, Sørensen CS. Hereditary breast and ovarian cancer: new genes in confined pathways. Nat Rev Cancer. (2016) 16:599–612. doi: 10.1038/nrc.2016.72
13. Shimelis H, LaDuca H, Hu C, Hart SN, Na J, Thomas A, et al. Triple-negative breast cancer risk genes identified by multigene hereditary cancer panel testing. J Nat Cancer Instit. (2018) 110:855–62. doi: 10.1093/jnci/djy106
14. Rosenthal ET, Bernhisel R, Brown K, Kidd J, Manley S. Clinical testing with a panel of 25 genes associated with increased cancer risk results in a significant increase in clinically significant findings across a broad range of cancer histories. Cancer Gene. (2017) 218–219:58–68. doi: 10.1016/j.cancergen.2017.09.003
15. Bonache S, Esteban I, Moles-Fernández A, Tenés A, Duran-Lozano L, Montalban G, et al. Multigene panel testing beyond BRCA1/2 in breast/ovarian cancer Spanish families and clinical actionability of findings. J Cancer Res Clin Oncol. (2018) 144:2495–513. doi: 10.1007/s00432-018-2763-9
16. Lhota F, Zemankova P, Kleiblova P, Soukupova J, Vocka M, Stranecky V, et al. Hereditary truncating mutations of DNA repair and other genes in BRCA1/BRCA2/PALB2-negatively tested breast cancer patients. Clin Gene. (2016) 90:324–33. doi: 10.1111/cge.12748
17. Cock-Rada AM, Ossa CA, Garcia HI, Gomez LR. A multi-gene panel study in hereditary breast and ovarian cancer in Colombia. Famil Cancer. (2018) 17:23–30. doi: 10.1007/s10689-017-0004-z
18. Quezada Urban R, Díaz Velásquez CE, Gitler R, Rojo Castillo MP, Sirota Toporek M, Figueroa Morales A, et al. Comprehensive analysis of germline variants in Mexican patients with hereditary breast and ovarian cancer susceptibility. Cancers. (2018) 10:361. doi: 10.3390/cancers10100361
19. Torrezan GT, de Almeida FGDSR, Figueiredo MCP, Barros BDF, de Paula CAA, Valieris R, et al. Complex landscape of germline variants in Brazilian patients with hereditary and early onset breast cancer. Front Gene. (2018) 9:161. doi: 10.3389/fgene.2018.00161
20. Felix GES, Zheng Y, Guindalini RSC, Machado-Lopes TMB, Zhang J, Côrtes J, et al. BROCA gene panel testing in African descendants from northeastern Brazil: genetic susceptibility profile of an admixed population. J Clin Oncol. (2017) 35:1572. doi: 10.1200/JCO.2017.35.15_suppl.1572
21. Adaniel C, Salinas F, Donaire JM, Bravo ME, Peralta O, Paredes H, et al. Non-BRCA1/2 variants detected in a high-risk chilean cohort with a history of breast and/or ovarian cancer. J Glob Oncol. (2019) 5:1–14. doi: 10.1200/JGO.18.00163
22. National Comprehensive Cancer Network (NCCN). Genetic/Familial High-Risk Assessment: Breast and Ovarian (Version 2.2018) (2018).
23. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)–a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. (2009) 42:377–81. doi: 10.1016/j.jbi.2008.08.010
24. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
25. Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Gene. (2017) 100:267–80. doi: 10.1016/j.ajhg.2017.01.004
26. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical Genetics and genomics and the association for molecular pathology. Gene Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
27. Weitzel JN, Lagos VI, Herzog JS, Judkins T, Hendrickson B, Ho JS, et al. Evidence for common ancestral origin of a recurring BRCA1 Genomic rearrangement identified in high-risk hispanic families. Cancer Epidemiol Biomark Preven. (2007) 16:1615–20. doi: 10.1158/1055-9965.EPI-07-0198
28. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
29. Ossa CA, Torres D. Founder and recurrent mutations in BRCA1 and BRCA2 Genes in Latin American Countries: State of the art and literature review. Oncologist. (2016) 21:832–9. doi: 10.1634/theoncologist.2015-0416
30. Popejoy AB, Fullerton SM. Genomics is failing on diversity. Nature. (2016) 538:161–4. doi: 10.1038/538161a
31. Judkins T, Rosenthal E, Arnell C, Burbidge LA, Geary W, Barrus T, et al. Clinical significance of large rearrangements in BRCA1 and BRCA2. Cancer. (2012) 118:5210–6. doi: 10.1002/cncr.27556
32. Torres D, Bermejo JL, Rashid MU, Briceño I, Gil F, Beltran A, et al. Prevalence and penetrance of BRCA1 and BRCA2 germline mutations in Colombian breast cancer patients. Sci Rep. (2017) 7:4713. doi: 10.1038/s41598-017-05056-y
33. Briceño-Balcázar I, Gómez-Gutiérrez A, Díaz-Dussán NA, Noguera-Santamaría MC, Díaz-Rincón D, Casas-Gómez MC. Mutational spectrum in breast cancer associated BRCA1 and BRCA2 genes in Colombia. Colomb Medica. (2017) 48:58–63. doi: 10.25100/cm.v48i2.1867
34. Castéra L, Krieger S, Rousselin A, Legros A, Baumann J, Bruet O, et al. Next-generation sequencing for the diagnosis of hereditary breast and ovarian cancer using genomic capture targeting multiple candidate genes. Eur J Hum Gene. (2014) 22:1305–13. doi: 10.1038/ejhg.2014.16
35. Tedaldi G, Tebaldi M, Zampiga V, Danesi R, Arcangeli V, Ravegnani M, et al. Multiple-gene panel analysis in a case series of 255 women with hereditary breast and ovarian cancer. Oncotarget. (2017) 8:47064–75. doi: 10.18632/oncotarget.16791
36. Girard E, Eon-Marchais S, Olaso R, Renault A, Damiola F, Dondon M, et al. Familial breast cancer and DNA repair genes: insights into known and novel susceptibility genes from the GENESIS study, and implications for multigene panel testing. Int J Cancer. (2019) 144:1962–74. doi: 10.1002/ijc.31921
37. Lin P, Kuo W, Huang A, Lu Y, Lin C, Kuo S, et al. Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer. Oncotarget. (2016) 7:8310–20. doi: 10.18632/oncotarget.7027
38. Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. (2002) 108:781–94. doi: 10.1016/S0092-8674(02)00671-2
39. Riballo E, Kühne M, Rief N, Doherty A, Smith GCM, Recio M, et al. A Pathway of double-strand break rejoining dependent upon ATM, artemis, and proteins locating to γ-H2AX foci. Mol Cell. (2004) 16:715–24. doi: 10.1016/j.molcel.2004.10.029
40. Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. (2001) 105:177–86. doi: 10.1016/S0092-8674(01)00309-9
41. Woodbine L, Brunton H, Goodarzi AA, Shibata A, Jeggo PA. Endogenously induced DNA double strand breaks arise in heterochromatic DNA regions and require ataxia telangiectasia mutated and artemis for their repair. Nucleic Acids Res. (2011) 39:6986–97. doi: 10.1093/nar/gkr331
42. Li L, Moshous D, Zhou Y, Wang J, Xie G, Salido E, et al. A founder mutation in artemis, an SNM1-like protein, causes SCID in athabascan-speaking Native Americans. J Immunol. (2002) 168:6323. doi: 10.4049/jimmunol.168.12.6323
43. Ege M, Ma Y, Manfras B, Kalwak K, Lu H, Lieber MR, et al. Omenn syndrome due to ARTEMIS mutations. Blood. (2005) 105:4179–86. doi: 10.1182/blood-2004-12-4861
44. Zhang X, Succi J, Feng Z, Prithivirajsingh S, Story MD, Legerski RJ. Artemis is a phosphorylation target of ATM and ATR and is involved in the G2/M DNA damage checkpoint response. Mol Cell Biol. (2004) 24:9207–20. doi: 10.1128/MCB.24.20.9207-9220.2004
45. Poinsignon C, Moshous D, Callebaut I, de Chasseval R, Villey I, de Villartay J. The metallo-beta-lactamase/beta-CASP domain of artemis constitutes the catalytic core for V(D)J recombination. J Exp Med. (2004) 199:315–21. doi: 10.1084/jem.20031142
46. Bolger GB, Stamberg J, Kirsch IR, Hollis GF, Schwarz DF, Thomas GH. Chromosome translocation t(14;22) and oncogene (c-sis) variant in a pedigree with familial meningioma. New Eng J Med. (1985) 312:564–7. doi: 10.1056/NEJM198502283120907
47. Smidt M, Kirsch I, Ratner L. Deletion of Alu sequences in the fifth c-sis intron in individuals with meningiomas. J Clin Invest. (1990) 86:1151–7. doi: 10.1172/JCI114820
48. Keller A, Westenberger A, Sobrido MJ, García-Murias M, Domingo A, Sears RL, et al. Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nat Gene. (2013) 45:1077–82. doi: 10.1038/ng.2723
49. Kamath-Loeb AS, Lan L, Nakajima S, Yasui A, Loeb LA. Werner syndrome protein interacts functionally with translesion DNA polymerases. Proc Nat Acad Sci USA. (2007) 104:10394–9. doi: 10.1073/pnas.0702513104
50. Li B, Comai L. Displacement of DNA-PKcs from DNA ends by the werner syndrome protein. Nucleic Acids Res. (2002) 30:3653–61. doi: 10.1093/nar/gkf488
51. Beitsch PD, Whitworth PW, Hughes K, Patel R, Rosen B, Compagnoni G, et al. Underdiagnosis of hereditary breast cancer: are genetic testing guidelines a tool or an obstacle? J Clin Oncol. (2018) 37:453–60. doi: 10.1200/JCO.18.01631
52. Faucz FR, Horvath A, Rothenbuhler A, Almeida MQ, Libé R, Raffin-Sanson M, et al. Phosphodiesterase 11A (PDE11A) genetic variants may increase susceptibility to prostatic cancer. J Clin Endocrinol Metab. (2011) 96:E135–40. doi: 10.1210/jc.2010-1655
53. Pathak A, Stewart DR, Faucz FR, Xekouki P, Bass S, Vogt A, et al. Rare inactivating PDE11A variants associated with testicular germ cell tumors. Endocr Relat Cancer. (2015) 22:909–17. doi: 10.1530/ERC-15-0034
54. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. (2009) 461:747–53. doi: 10.1038/nature08494
55. Torres Á, Oliver J, Frecha C, Montealegre AL, Quezada-Urbán R, Díaz-Velásquez CE, et al. Cancer genomic resources and present needs in the Latin American region. Pub Health Genom. (2017) 20:194–201. doi: 10.1159/000479291
56. Chavarri-Guerra Y, Blazer KR, Weitzel JN. Genetic Cancer risk assessment for breast cancer in Latin America. Rev Invest Clin. (2017) 69:94–102. doi: 10.24875/RIC.17002195
Keywords: breast cancer susceptibility, massively parallel sequencing, germline pathogenic variants, Latin America, HBOC
Citation: Oliver J, Quezada Urban R, Franco Cortés CA, Díaz Velásquez CE, Montealegre Paez AL, Pacheco-Orozco RA, Castro Rojas C, García-Robles R, López Rivera JJ, Gaitán Chaparro S, Gómez AM, Suarez Obando F, Giraldo G, Maya MI, Hurtado-Villa P, Sanchez AI, Serrano N, Orduz Galvis AI, Aruachan S, Nuñez Castillo J, Frecha C, Riggi C, Jauk F, Gómez García EM, Carranza CL, Zamora V, Torres Mejía G, Romieu I, Castañeda CA, Castillo M, Gitler R, Antoniano A, Rojas Jiménez E, Romero Cruz LE, Vallejo Lecuona F, Delgado Enciso I, Martínez Rizo AB, Flores Carranza A, Benites Godinez V, Méndez Catalá CF, Herrera LA, Chirino YI, Terrazas LI, Perdomo S and Vaca Paniagua F (2019) Latin American Study of Hereditary Breast and Ovarian Cancer LACAM: A Genomic Epidemiology Approach. Front. Oncol. 9:1429. doi: 10.3389/fonc.2019.01429
Received: 17 July 2019; Accepted: 02 December 2019;
Published: 20 December 2019.
Edited by:
Maria Paula Curado, ACCamargo Cancer Center, BrazilReviewed by:
Mohammad Reza Akbari, University of Toronto, CanadaHamidullah Khan, University of Wisconsin-Madison, United States
Julie Dutil, Ponce Health Sciences University, Puerto Rico
Alvaro Monteiro, Moffitt Cancer Center, United States
Copyright © 2019 Oliver, Quezada Urban, Franco Cortés, Díaz Velásquez, Montealegre Paez, Pacheco-Orozco, Castro Rojas, García-Robles, López Rivera, Gaitán Chaparro, Gómez, Suarez Obando, Giraldo, Maya, Hurtado-Villa, Sanchez, Serrano, Orduz Galvis, Aruachan, Nuñez Castillo, Frecha, Riggi, Jauk, Gómez García, Carranza, Zamora, Torres Mejía, Romieu, Castañeda, Castillo, Gitler, Antoniano, Rojas Jiménez, Romero Cruz, Vallejo Lecuona, Delgado Enciso, Martínez Rizo, Flores Carranza, Benites Godinez, Méndez Catalá, Herrera, Chirino, Terrazas, Perdomo and Vaca Paniagua. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra Perdomo, cGVyZG9tb3NhbmRyYUB1bmJvc3F1ZS5lZHUuY28=; Felipe Vaca Paniagua, ZmVsaXBlLnZhY2FAZ21haWwuY29t
†These authors have contributed equally to this work