 Rui Zhang1Yan Zhang1Xianglan Lu2Weihong Xu2He Wang1Wenbin Mo1Hui Pang2Rurong Tang3Shibo Li2Xiaojing Yan1
Rui Zhang1Yan Zhang1Xianglan Lu2Weihong Xu2He Wang1Wenbin Mo1Hui Pang2Rurong Tang3Shibo Li2Xiaojing Yan1 Yan Li1*
Yan Li1*- 1Department of Hematology, The First Affiliated Hospital of China Medical University, Shenyang, China
- 2Department of Pediatrics, University of Oklahoma Health Sciences Center, Oklahoma City, OK, United States
- 3Department of Anesthesiology, The First Affiliated Hospital of China Medical University, Shenyang, China
We report herein that Sprouty-Related EVH1 Domain-Containing Protein1 (SPRED1) is downregulated and a prognostic biomarker in adult acute myeloid leukemia (AML). We determined mRNA levels of SPRED1 in the bone marrow mononuclear cells from adult patients, including 113 AMLs and 22 acute lymphoblastic leukemias (ALLs), as well as in 37 healthy control subjects. Significantly decreased SPRED1 mRNA expression was found in AML patients comparing to those in ALL patients and healthy controls, which was confirmed by immunocytochemistry analysis of SPRED1 protein and ELISA measurement of serum SPRED1 level. Further analysis demonstrated that SPRED1 expression was significantly higher for most patients at complete remission after induction treatment than at diagnosis. Moreover, SPRED1 expression was significantly downregulated in M2 and M3 types. Non-acute promyelocytic leukemia (non-APL) patients with decreased SPRED1 had significantly lower 2-year progression-free survival and event-free survival rates. In vitro, ectopic overexpression of SPRED1 leads to a decrease of extracellular signal-regulated kinase (ERK) phosphorylation, induction of apoptosis and reduction of proliferation of THP-1 cells. Our findings suggest SPRED1 is not only a predictor of treatment response, but also an independent prognostic factor for non-APL, and targeting Ras- Mitogen-activated protein kinase (MAPK) signaling may be a promising strategy for the treatment of AML with downregulation of SPRED1.
Introduction
The SPRED1 gene is located on chromosome 15q14 and encodes SPRED1 protein, a member of the Sprouty-related protein family. The germline loss-of-function mutations of SPRED1 result in Legius syndrome, an autosomal dominant human disorder characterized by multiple cafe'-au-lait macules, axillary freckling, learning disabilities and macrocephaly (1, 2); however, the exact mechanisms of the pathophysiology of SPRED1 are largely unknown. A recent study demonstrates that SPRED1 interacts with neurofibromin (3), a protein encoded by NF1, mutation of which leads to neurofibromatosis type 1 (NF1) syndrome. It has been well-documented that NF1 is a negative regulator of Ras-MAPK signaling, which is deregulated in most cancers (4, 5). These advances suggest that SPRED1 may regulate cell proliferation, differentiation and survival by suppressing Ras-MAPK signaling (6).
Both Legius and NF1 syndromes have various degrees of leukemia predisposition (6). A single-nucleotide polymorphism array analysis has shown that SPRED1 deletion is frequently found in relapsed B cell acute lymphoblastic leukemia (B-ALL) (7). Moreover, a recent study suggests that SPRED1 is a tumor suppressor and is downregulated in pediatric acute myeloid leukemia (AML) (8). Nevertheless, the contribution of SPRED1 to leukemogenesis remains controversial. Several studies have reported the lack of tumorigenesis with SPRED1 mutation in Legius syndrome (5, 9), whereas a child with neuro-cardio-facial-cutaneous (NCFC) syndrome caused by germline SPRED1 mutation was reported to develop AML-M5 (10). We previously found the chromosomal loss of 15q involving the SPRED1 gene in an adult AML-M5 patient using high-resolution array CGH (aCGH) (11). Amounting evidence indicates that NF1 is a tumor suppressor gene of AML and might be a novel therapeutic target for AML (12–14). Considering the shared phenotypes of Legius and NF1 syndromes and the physical interaction of neurofibromin with SPRED1, we hypothesize that, similar to NF1, the SPRED1 gene may be downregulated in adult AML and associated with the disease progression.
In order to define the clinical features as well as the prognostic significance of SPRED1 in leukemia, in the present study, we determined the expression of SPRED1 in adult AML and analyzed the association of SPRED1 expression with clinical parameters, disease status, and survival. We further investigated the proliferation and survival of AML cell lines following ectopic overexpression and silencing of SPRED1 with RNA interference.
Materials and Methods
Study Population
This study was approved by the Ethical Committee of the First Affiliated Hospital of China Medical University (Beijing, China). Before data collection, written informed consent was obtained from each participant. Bone marrow (BM) samples were collected from 135 patients (69 male and 66 female) with de novo acute leukemia (113 AMLs and 22 ALLs). Patients younger than 14 years old were excluded from this study. The median age of patients was 47 years old (range, 14–88 years). The diagnosis was made according to the French-American-British (FAB) Cooperative Group criteria (15). The BM samples were analyzed using flow immunophenotyping, conventional chromosome banding or targeted analyses (reverse transcriptase-PCR and/or FISH). Serum and BM samples of gender-balanced 37 normal donors were collected as a healthy control group (HC).
Therapy and Follow-Up
A total of 68 AML patients, including 26 acute promyelocytic leukemia (APL) patients (M3 subtype of FAB) and 42 non-APL patients have received intensive treatment. All APL patients were included in a national 863 clinical study for APL (Clinical Trial Identifier: 2012AA02A505). The treatment for non-APL patients and the response assessment were based on Chinese guidelines described in a previous study (16). The follow-up time for the patients, calculated from the time of randomization for induction therapy, was 24 months unless death proceeded. Serum and BM samples from patients with AML at both diagnosis and complete remission (CR) were involved for analysis.
Ficoll Gradient Separation
BM mononuclear cells (MNCs) were isolated by Ficoll-paque gradient centrifugation (TAKARA Biotechnology, Dalian, China), aliquoted into fetal calf serum (FCS) with 10% DMSO, and cryopreserved in the liquid phase of a liquid nitrogen tank.
Preparation of RNA and Quantitative Real-Time PCR (qRT-PCR)
Liquid nitrogen-cryopreserved (liquid phase) BM mononuclear cells were rapidly thawed in 37°C water bath, washed with phosphate-buffered saline (PBS) and collected by centrifugation. Total RNA from BM MNC or cultured cells was extracted with Trizol (TAKARA Biotechnology) according to the manufacturer's instructions. Complementary DNA (cDNA) was made from 20 ng of RNA using the SuperScript III first-strand synthesis kit and oligo-dT primer (Promega) according to the manufacturer's instructions. SPRED1 mRNA levels in BM mononuclear cells were determined using the real-time SYBRPCR amplification kit (TAKARA) with β-ACTIN as an internal control. Triplicate amplification reactions were done on an ABI 7500 Real-time PCR System (Applied Biosystems, Foster City, CA, USA) and the data were analyzed by the ABI 7500 system SDS software (1.4 Version). The expression value was calculated with the comparative CT method. SPRED1 gene-specific primers: forward 5′-GATGAGCGAGGAGACGGCGAC-3′ and reverse 5′-GTCTCTGAGTCTCTCTCCACGGA-3′; β-actin: forward 5′-TGGAGATAACACTCTAAGCATAACTAAAGGT-3′ and reverse 5′-GATGTAGTTGCTTGGGACCCA-3′. All the primers were synthesized by Invitrogen. For detection of MIR126, total RNA was isolated using a mirVana miRNA isolation kit (Ambion, Austin, TX, USA). Expression levels of MIR126 were detected using a TaqMan MicroRNA Assay (Applied Biosystems, Foster City, CA, USA) following the manufacturer's protocol.
Immunocytochemistry
To assess the SPRED1 protein expression in leukemia blasts, immunocytochemical staining was performed by the immunoperoxidase technique using the avidin-biotin-peroxidase complex (ABC) as described (17). Cytospin smears of BM cells from AML patients with blast ratio of more than 80% and healthy controls were fixed for 3 min in formalin acetone (3%). The specimens were incubated with peroxidase blocking enzyme for 20 min, followed by incubation with a rabbit anti-human SPRED1 antibody (Abcam, Cambridge, UK) at 4°C overnight. The next day after washing with PBS, the specimens were incubated with a biotinylated donkey anti-goat IgG secondary antibody for 30 min. After washing three times with PBS, the protein was detected using the streptavidin-peroxidase substrate. The specimens were counter-stained with hematoxylin.
Measurement of SPRED1 Level in Serum
Serum SPRED1 protein concentrations were determined in 26 patients with AML and 30 healthy controls using the commercial enzyme-linked immunosorbent assay (ELISA) kit (Ji Jin Chemical Technology Co., Ltd, Shanghai, China) following the manufacturer's protocol. The Optical Density (O.D.) was read at 450 nm using a microtiter plate reader. Sample concentrations were calculated by drawing standard linear regression curves.
Fluorescence in situ Hybridization (FISH)
FISH analysis was performed using SPRED1 probes RP11-644D16 (15q14:36,154,798–36,372,207, SG) and RP11-477P17 (15q14:36, 375, 079–36, 541, 177, SO) (Invitrogen) on 43 cases of AMLs according the manufacturer's instructions. Two hundred interphase nuclei were screened in each case.
Sequence Analysis of SPRED1 Mutation
The exons and the flanking areas of SPRED1 gene were amplified by PCR using the primers as shown in STable 1. The 25 μl PCR mixture contained 50 ng of template DNA, 1X PCR buffer, 0.2 mmol/L dNTP mix, 50 ng of each primer, 2.5 mmol/L MgCl2, and 1 unit of AmpliTaq Gold (Applied Biosystems, Foster City, CA, USA). ddNTP terminator reaction was carried out with the ABI BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems). Sequencing products were loaded and data were collected on ABI 3130 xl genetic analyzer (Applied Biosystems). Mutations were identified via a comparison between the sample sequences and the reference sequences using Mutation Surveyor (SoftGenetics, State College, PA, USA).
Cell Culture
Leukemia cell lines THP-1 (AML-M5), OCI-AML2 (AML-M4), OCI-AML3 (AML-M4), Kasumi-1 (AML-M2), HL-60 (AML-M2), and NB4 (AML-M3) were obtained from American Type Culture Collection (ATCC, USA) and cultured in RPMI-1640 (GIBCO, USA) supplemented with 10% FBS.
Plasmid Constructs and Lentivirus Transduction
The Tet-pLKO-puro vector (Addgene) was used for non-silencing control or for carrying SPRED1 target shRNA. The sequence for SPRED1 shRNA were: GCAGATGACTTACAAGCAA. For SPRED1 expression, the vector was constructed by cloning human SPRED1 cDNA (NM_152594.2) into pLV-EF1a-IRES-NEO vector (Addgene) according to the standard procedure. Lentiviral packaging and transductions were carried out using standard procedure and according to manufacturer's instructions. The cells transfected with virus carrying empty vector was used as a negative control (Control group) and untreated cells were used as blank control (Blank group).
Western Blotting
Cells were lysed on ice with RIPA buffer (Invitrogen) containing protease and phosphatase inhibitors (Sigma). Cell lysates were centrifuged at 12,000 g for 10 min at 4°C, and the supernatants were stored at −80°C. Protein quantification was performed with the BCA protein assay (Pierce). Equal amounts of protein were separated on 10% SDS polyacrylamide gel and transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were blocked with Tris-buffered saline containing 0.1% Tween 20 and 5% dried milk powder. Antibodies to SPRED1 (Abcam, Cambridge, UK), ERK, pERK (Wanleibio, Shenyang, China) and β-actin (Invitrogen) were used to detect the corresponding proteins. Signals were developed with an enhanced chemiluminescence detection system (Pierce). All western analysis were performed for three times.
Cell Proliferation Assay
The viability of THP-1 and OCI-AML3 cells following transfection of shRNA or plasmid was measured using the Cell Counting Kit-8 (CCK-8) (Dojindo Molecular Technologies, Kumamoto, Japan). Cells transfected with shRNA or plasmid were cultured in a 96-well plate at a density of 2 × 104 cells per well for 24, 48, and 72 h. At each time point, cells were incubated in 10% CCK-8 solution for an additional 2 h at 37°C. The absorbance at 450 nm (A450) was examined on a microplate reader (CANY, Shanghai, China).
Cell Cycle and Apoptosis Assay by Flow Cytometry
THP-1 and OCI-AML3 cells following transfection of shRNA or plasmid for 48 h were harvested and re-suspended in PBS containing 2% BSA. After centrifugation at 1,000 rpm for 5 min, cells were re-suspended with 500 μl binding buffer and mixed with 5 μl Annexin V-PE. Cells were then incubated with 5 μl 7-AAD in the dark at room temperature for 5–15 min. All assays were performed in triplicate. Cell apoptosis and cell cycle distribution were assayed by flow cytometry (BD Biosciences, San Jose, CA, USA). Specific steps for flow cytometry were performed according to the manufacturer's instructions. Data were analyzed with FlowJo 10 software.
Statistical Analysis
The relative expression (fold change) level of SPRED1 mRNA was calculated using the 2−ΔΔCt method. For comparison between two groups, we used Student's t-test (when the data is normally distributed) or Mann-Whitney U test (when the data is not normally distributed). Kruskal-Wallis test was used for comparison within multiple groups. For comparison within group at diagnosis vs. post-treatment, we used Paired Student's t-test (when the data is normally distributed) and Wilcoxon signed rank-sum test (when the data is not normally distributed). One-way ANOVA and Bonferroni multiple-comparison tests were used for comparisons in multiple groups. Correlations between SPRED1 and MIR126 expression were estimated with the Spearman rank-order correlation coefficient. The frequency data were compared by chi-squared test. Overall survival (OS) was calculated from diagnosis to the date of mortality. Progression-free survival (PFS) was estimated as the interval between the date of diagnosis to date of progression. Event-free survival (EFS) was measured from the date of entry into study until treatment failure, relapse from CR, or death from any cause. The Kaplan-Meier method was used to perform the survival curve and the log-rank test was used to test statistical significance. All statistical analyses were performed using SPSS. A two-tailed p < 0.05 was considered as statistically significant.
Results
Expression of SPRED1 Significantly Decreases in de novo AML
This study included 113 AML patients, 22 ALL patients, and 37 HC persons. The characteristics of the subjects in these three groups are shown in Table 1. The mRNA expression of the SPRED1 gene in the BM of these 172 subjects was determined by qRT-PCR. Significantly lower expression of SPRED1 was observed in the AML group in comparison with those in the HC (p < 0.05) and ALL groups (p < 0.05). No significant difference was found between ALL and HC groups (p > 0.05; Figure 1A). Comparing to the HC, 99 out of 113 AMLs (87.6%) showed decreased SPRED1 expression (defined as lower than the 25% percentile value in the HC group). We next divided the AML patients into young (age < 35), middle (35 ≤ age < 60) and old (age ≥ 60) groups and found there was no significant difference of SPRED1 mRNA expression among these groups (p > 0.05; Figure 1B). In addition, there was no significant difference in SPRED1 mRNA expression between males and females (p > 0.05; Figure 1C). Further analysis of SPRED1 protein of BM cells from 6 AML patients and 3 HC subjects by immunocytochemistry showed strong brown staining in the three HC subjects while four negative stainings in AML patients (Figure 1D). In addition, measurement of serum SPRED1 level in 27 patients with AML and 30 HC using ELISA demonstrated a significantly lower SPRED1 level in AML than HC group (p < 0.001; Figure 1E).
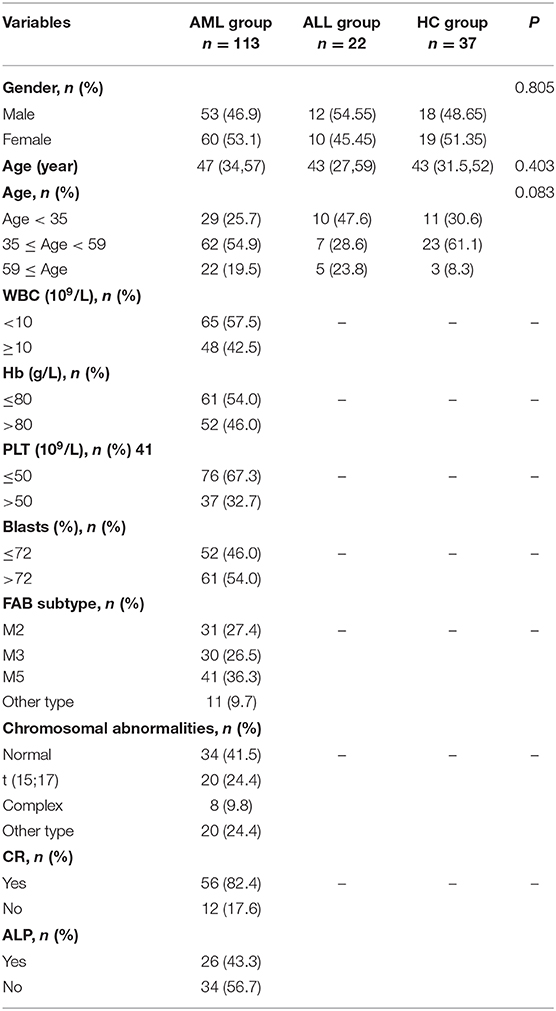
Table 1. Characteristics of the subjects in this study.
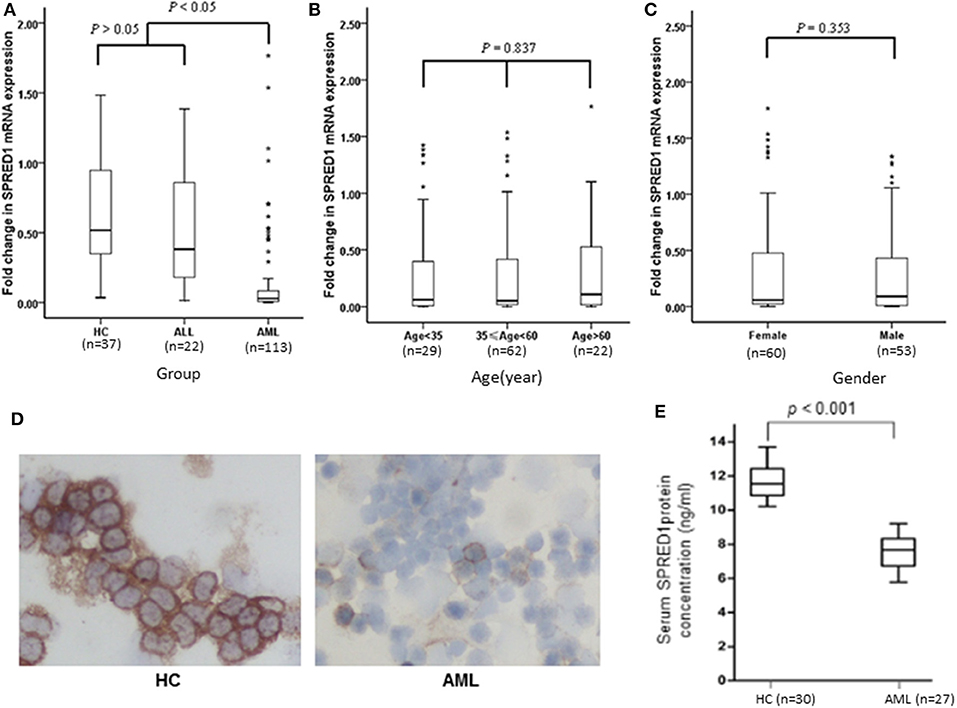
Figure 1. Box plots of SPRED1 mRNA expression in all subjects. SPRED1 mRNA expression (A) in healthy controls (HC), ALL and AML groups (Kruskal–Wallis test); (B) by different age groups (Kruskal–Wallis test); (C) between female and male (Wilcoxon tests); (D) representative immunocytochemical staining of SPRED1 protein in BM cells. Positive expression of SPRED1 was stained brown in BM of HC while negative expression of SPRED1 in BM of AML patient. (E) Serum SPRED1 level in AML patients (7.52 ± 0.93 ng/ml) and HC (11.68 ± 0.94 ng/ml) (Wilcoxon tests).
SPRED1 Expression Is Significantly Decreased in AML of M2 and M3 Types
We next analyzed the correlations of SPRED1 mRNA expression in BMs of 113 AML patients with a series of prognostic relevant clinical and laboratory parameters of AML including, white blood cell (WBC) count, hemoglobin (Hb) concentration, platelet (PLT) count and higher percent of blast cells in bone marrow (>72% comparing to the average blast ratio in bone marrow), FAB subtypes, and chromosomal abnormalities. No significant difference of SPRED1 expressions was found between subgroups with different WBC (p = 0.838; Figure 2A), Hb concentration (p = 0.254; Figure 2B), PLT count (p = 0.582; Figure 2C), and the percentage of blast cells in BM (p = 0.975; Figure 2D). Different expression of SPRED1 was found among the FAB subtypes of AMLs (Figure 2E). In M2, M3, and M5 subtypes with similar sample sizes, a significantly decreased expression of SPRED1 was found in M2 and M3 compared to that of M5 and other types (p < 0.05). There was no significant difference of SPRED1 expression between M2 and M3 (p > 0.05) and between M5 and other types (p > 0.05). However, no significant difference of SPRED1 expressions was found between subgroups with different chromosomal abnormalities (p > 0.05; Figure 2F and STable 2).
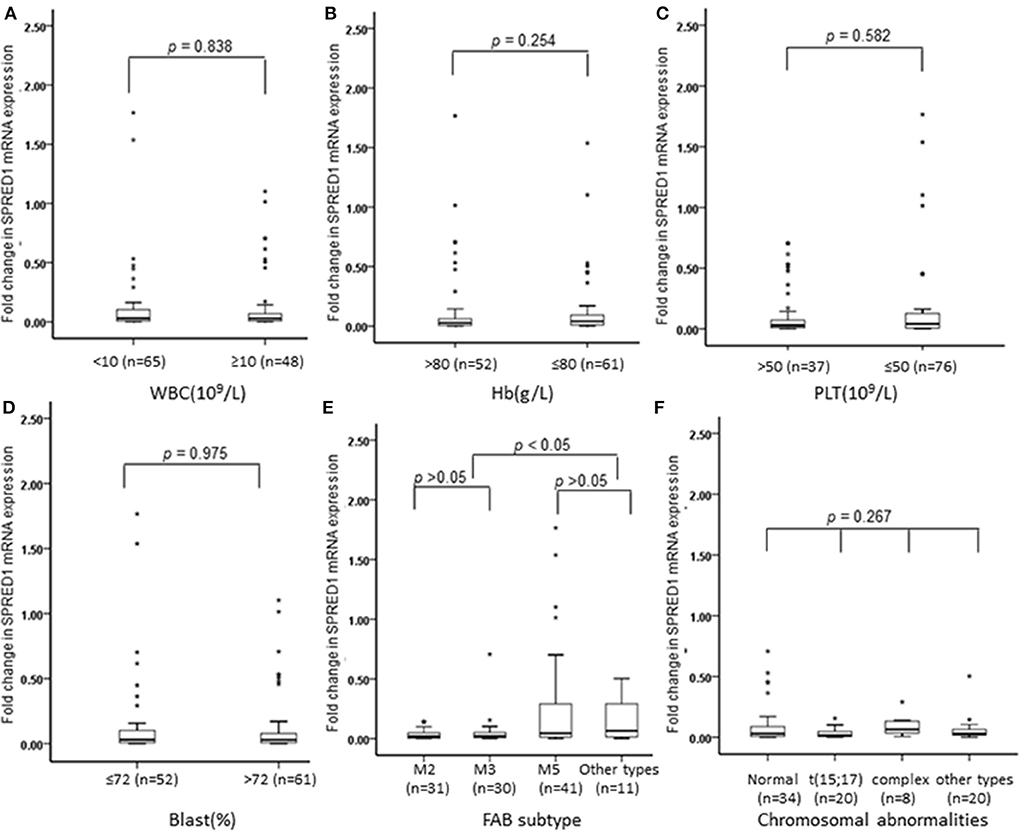
Figure 2. Box plots of SPRED1 mRNA expression in AML patients with different characteristics. SPRED1 mRNA expression in AML patients with different (A) WBC (Wilcoxon tests); (B) Hb (Wilcoxon tests); (C) PLT (Wilcoxon tests); (D) blast (Wilcoxon tests); (E) FAB subtype (Kruskal–Wallis test); (F) chromosomal abnormalities (Kruskal–Wallis test).
SPRED1 Expression Increases in AML Patients With CR After Induction Treatment
The mRNA levels in the BM samples from 37 AML patients (M0 n = 1, M1 n = 2, M2 n = 9, M3 n = 12, M5 n = 12, M6 n = 1) and serum SPRED1 protein concentrations in the serum samples from 27 AML patients were compared both at diagnosis and after induction treatment. Both the BM and the serum samples were matched. For patients with CR achievement, the median levels of SPRED1 mRNA (0.9558 ± 0.5166, n = 34) and SPRED1 protein (0.0961 ± 0.0074 ng/ml, n = 26) at CR were significantly higher than those at diagnosis (0.1416 ± 0.3536, 0.0745 ± 0.0091 ng/ml, all p < 0.001; Figures 3A,B). The majority of the patients showed increased SPRED1 either in BM (91.9%) or in serum (96.3%) at CR. Three patients who did not achieve CR after one induction course showed significantly reduced SPRED1 mRNA in BM (p = 0.044) and one of the patients had the same trend in serum (Figure 3C).
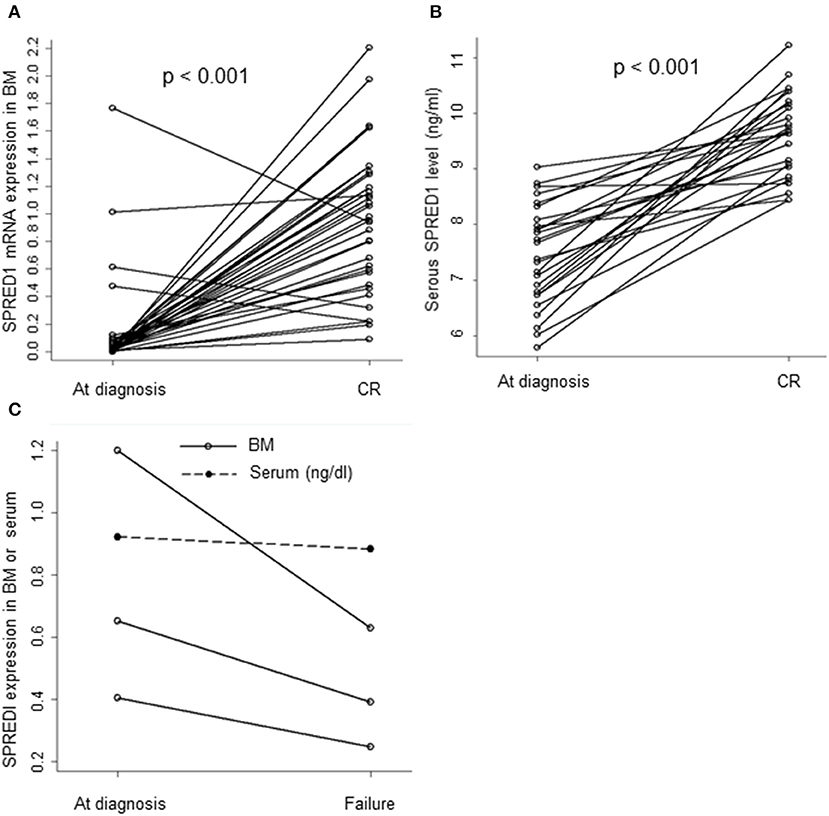
Figure 3. SPRED1 in AML patients at diagnosis and post-treatment. SPRED1 in AML patients with CR at (A) mRNA (Paired Student's t-test); (B) protein level (Paired Student's t-test); (C) SPRED1 in AML patients induction failure at diagnosis and post-treatment.
Lower SPRED1 Expression at Diagnosis Is Associated With Worse Survival of AML Patients
We performed a follow-up (median 17 months, range 2–24 months) for a total of 68 AML patients, including 26 of APL and 42 of non-APL. Fifty-six of these patients achieved CR, 25 relapsed, and 29 died. Both the non-APL patients and APL patients were further grouped based on the median of SPRED1 expression in BM at diagnosis and rates of survival parameters were compared between the two groups. There was no significant difference in SPRED1 mRNA expression between APL and non-APL patients (p > 0.05; Figure 4A), and patients with and without CR (p > 0.05; Figure 4B). In non-APL, significantly lower EFS (p = 0.026; Figure 5A) and PFS (p = 0.041; Figure 5B) rate at 2 years were demonstrated in the group with lower SPRED1 mRNA; however, there was no significant difference of OS of non-APL patients with lower or higher SPRED1 mRNA (p = 0.348; Figure 5C). In APL, the group with lower SPRED1 had comparable 2-year OS, 2-year EFS and 2-year PFS (p = 0.194; Figure 5D) in contrast to those of group with higher SPRED1 expression.
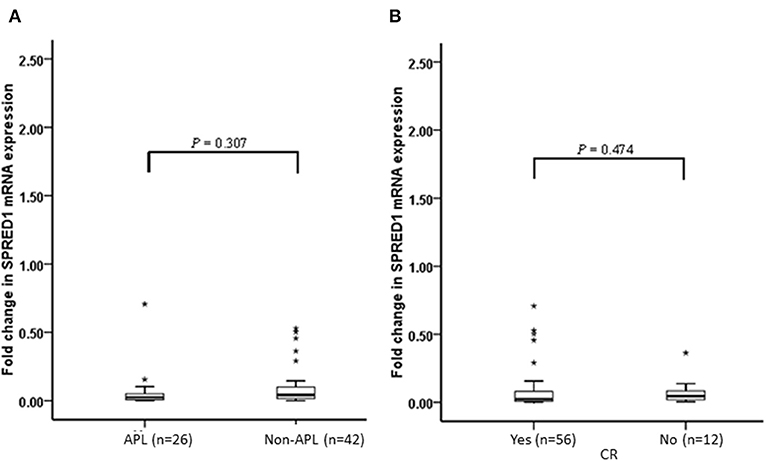
Figure 4. Fold change of SPRED1 mRNA expression in AML patients at follow-up. Fold change in AML patients (Wilcoxon tests); (A) with APL and without APL; (B) with and without CR (Wilcoxon tests).
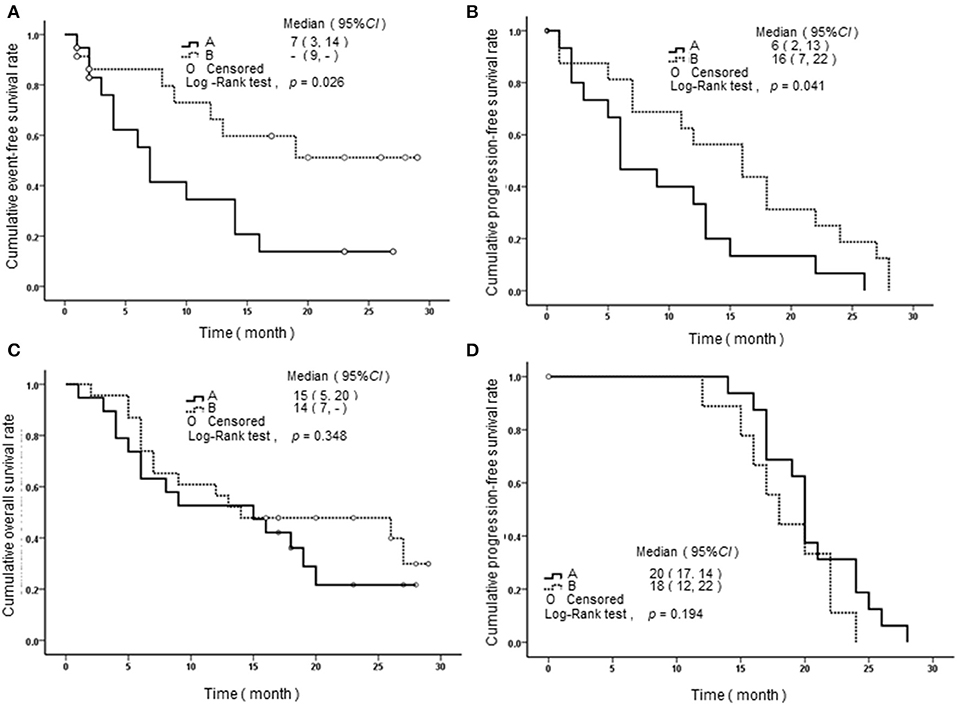
Figure 5. Survival curves of AML patients with different SPRED1 mRNA expression at diagnosis. (A) Event-free survival curves of patients with non-APL; (B) progression-free survival curves of patients with non-APL; (C) overall survival curves of patients with non-APL; (D) progression-Free survival curves of patients with APL. A line, fold change in SPRED1 mRNA <0.03; B line, fold change in SPRED1 mRNA ≥0.03 (log-rank test).
SPRED1 Gene Is Not Mutated or Deleted in de novo AML With Decreased SPRED1 Expression
To explore the potential mechanisms of the downregulation of SPRED1 in AML, we performed FISH analysis of SPRED1 in 48 AML patients, who had decreased SPRED1 mRNA expression comparing to the healthy control. We observed that all of the cases showed normal signals in 200 interphase nuclei, suggesting no deletion of the SPRED1 gene (SFigure 1). Furthermore, we determined the gene sequence of SPRED1 by direct DNA sequencing in 35 of the AML patients who had a normal FISH signal. Again, we found that none of the patients carried SPRED1 mutation.
SPRED1 Downregulation Is Not Related to the Expression of MIR126 in AML
SPRED1 is a potential target of miR126. To test whether the reduction of SPRED1 results from the deregulation of miR126 in AML, we determined MIR126 levels in 47 AMLs and nine controls. The MIR126 level in AMLs (0.7321 ± 1.0318) showed a higher trend than that of the healthy controls (0.5111 ± 0.3976) with no statistical significance though (p = 0.532; Figure 6A). Although all except one (97.9%) of the 17 patients had decreased SPRED1 expression, no correlation was shown between MIR126 and SPRED1 expression (r = 0.132, p = 0.344; Figure 6B).
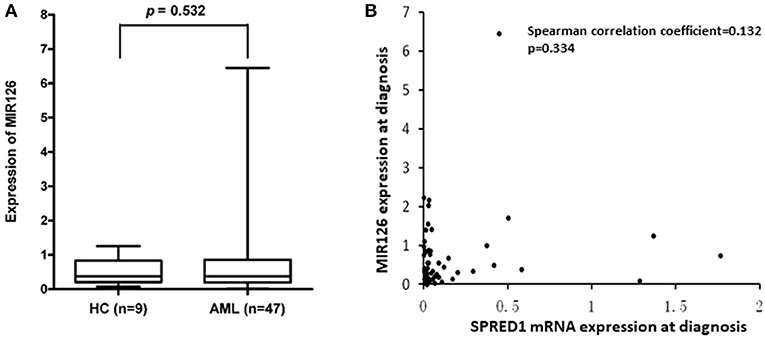
Figure 6. Correlation between SPRED1 and MIR126 mRNA expression level at diagnosis. (A) Comparison on MIR126 expression in HC and AML (Wilcoxon tests); (B) spearman correlation analysis between SPRED1 mRNA expression MIR126 expression.
Overexpression of SPRED1 Suppresses ERK Signaling and the Proliferation of AML Cells
To assess SPRED1 in the proliferation and survival of AML cells, the mRNA expression levels of SPRED1 in AML cell lines THP-1, OCI-AML2, OCI-AML3, Kasumi-1, HL-60, and NB4 were determined by RT-PCR. THP-1 had the lowest while OCI-AML3 had the highest SPRED1 mRNA levels (SFigure 2A). Corresponding protein expression of SPRED1 was confirmed in cell lines THP-1, HL-60 and OCI-AML3 (SFigure 2B). In the following experiments, we used THP-1 cells to overexpress SPRED1 and OCI-AML3 cells to downregulate SPRED1. Transfection of THP-1 cells with a plasmid containing SPRED1 significantly increased the SPRED1 mRNA level (0.91 ± 0.01) comparing to blank (0.54 ± 0.01) and control cells (0.67 ± 0.37) (p < 0.05; Figure 7A). Immunoblotting demonstrated that the overexpression of SPRED1 reduced the phosphorylation of ERK (Figure 7B). Compared to the empty vector, transfection SPRED1 plasmid significantly inhibited the proliferation of THP-1 cells as demonstrated by CCK-8 assay. The 450 nm optical density (OD) value in blank, control and SPRED1 overexpression cells was 0.64 ± 0.03, 0.63 ± 0.01 and 0.51 ± 0.03, respectively (p < 0.05; Figure 7C). The increase of apoptosis by overexpression of SPRED1 was proved by cell apoptosis assay. It showed that apoptotic cells ratio (%) in the blank, control, and SPRED1 overexpression groups was 2.73 ± 0.21, 3.00 ± 0.36, and 25.21 ± 0.59, respectively (p < 0.05; Figures 7D,E). Accumulation of cells at the S phase of the cell cycle in SPRED1 overexpression cells was demonstrated in Figures 7F,G. The cell ratio of G0/G1 phase, S phase and G2/M phase was 51.26 ± 5.31, 34.37 ± 4.80, and 9.76 ± 0.95% in the blank group; 67.39 ± 2.35, 16.95 ± 3.10, and 13.35 ± 2.72% in control group; 62.63 ± 2.34, 29.14 ± 1.42, and 6.07 ± 2.71% in SPRED1 overexpression group.
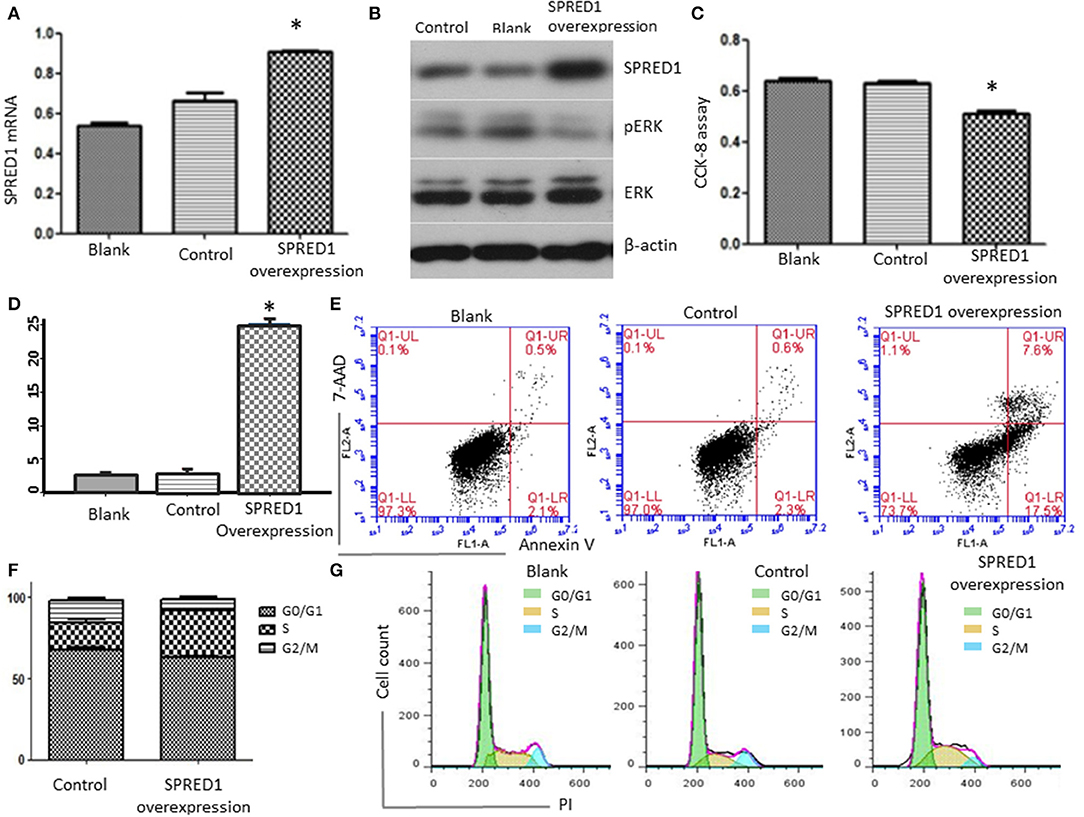
Figure 7. THP-1 cells were transfected with SPRED1 plasmid for 48 h. (A) SPRED1 mRNA was determined by RT-PCR in the blank; (B) SPRED1, pERK, ERK, and β-actin) were assessed by immunoblotting; (C) cell proliferation was determined by CCK-8 assay; (D,E) cell apoptosis was assessed by using annexin V–PE/7-AAD staining and (F,G) cell cycle distribution was assayed by flow cytometry. *p < 0.05 vs. control (Student's t-test).
Silencing of SPRED1 Activates ERK Signaling and Enhances the Proliferation of AML Cells
Finally, we downregulated SPRED1 in OCI-AML3 cells by shRNA and assessed cell proliferation and ERK activity. Transfection of OCI-AML3 cells with a SPRED1 shRNA significantly decreased the SPRED1 mRNA level (1.30 ± 0.17) in contrast to blank (3.19 ± 0.08) and control cells (3.95 ± 0.52) (p < 0.05; Figure 8A). Immunoblotting analysis demonstrated the silencing of SPRED1 by shRNA increased the phosphorylation of ERK (Figure 8B). In comparison that of scrambled control shRNA, transfection SPRED1 shRNA significantly promoted the proliferation of OCI-AML3 cells, as demonstrated by CCK-8 assay. The 450 nm optical density (OD) value in blank, control and SPRED1 shRNA cells was 0.49 ± 0.04, 0.46 ± 0.04, and 0.53 ± 0.04 (p < 0.05; Figure 8C). The silencing of SPRED1 by shRNA suppressed apoptosis. Apoptotic cells ratio (%) in blank, control and SPRED1 shRNA groups was 2.93 ± 0.18, 2.92 ± 0.16, and 2.01 ± 0.15 (p < 0.05; Figures 8D,E). The reduction of the cells at the S phase was identified by cell cycle assay. The cell ratio of G0/G1 phase, S phase and G2/M phase was 49.84 ± 6.63, 30.64 ± 8.28, and 19.74 ± 1.80% in the blank group; 49.19 ± 5.70, 33.64 ± 4.04, and 17.33 ± 1.62% in control group; 45.87 ± 6.99, 27.26 ± 2.46, and 26.25 ± 4.21% in SPRED1 shRNA group (Figures 8F,G).
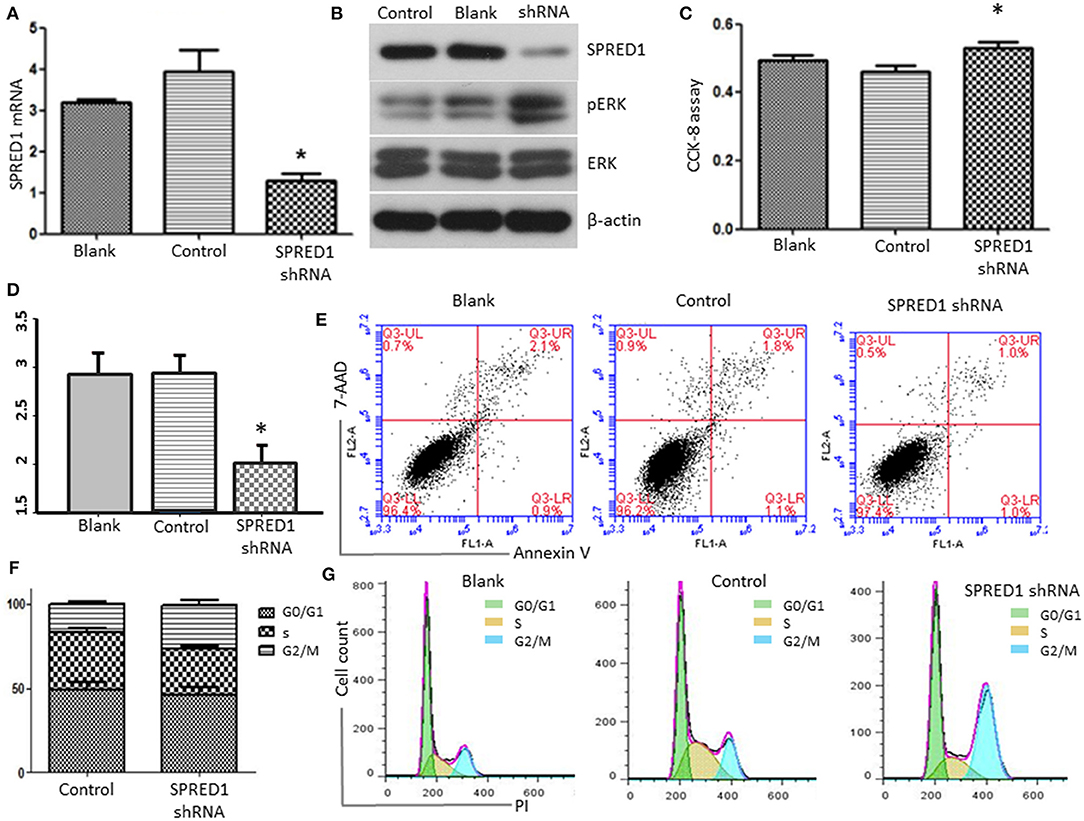
Figure 8. OCI-AML3 cells were transfected with SPRED1 shRNA for 48 h. (A) SPRED1 mRNA was determined by RT-PCR; (B) SPRED1, pERK, ERK, and β-actin were assessed by immunoblotting; (C) cell proliferation was determined by CCK-8 assay; (D,E) cell apoptosis and (F,G) cell cycle distribution was assessed by flow cytometry. *p < 0.05 vs. control (Student's t-test).
Discussion
In the present study, we demonstrated that expression of SPRED1 was significantly downregulated in AML, particularly M2 and M3 types. In a subset of non-APL patients, we observed lower SPRED1 expression at diagnosis was associated with worse PFS of AML patients. Moreover, SPRED1 expression was upregulated in AML patients with CR after induction treatment. In vitro, ectopic overexpression of SPRED1 suppressed ERK signaling and the proliferation of AML cells, which was accompanied the accumulation of cells at S phase and induction of cell apoptosis; in contrast, silencing of SPRED1 activated ERK signaling, enhanced the proliferation and reduced the apoptosis of AML cells.
Recent studies have suggested that SPRED1 is attenuated, and SPRED1 is a potential tumor suppressor in pediatric ALL or AML (7, 8). However, the association of SPRED1 expression status with the clinical features as well as the prognostic significance of SPRED1 in acute leukemia, especially adult AML, remains to be determined. In addition, the previous study of pediatric leukemia showed that SPRED1 expression appeared to be significantly decreased in AMLs and T-ALL, but normal in B-ALL (8). Therefore, further investigation is warranted in ALL with different immunophenotypes since most ALL cases in this study were B-ALL. Our results showed that expression of SPRED1 was significantly downregulated in adult AML, particularly M2 and M3 types. More strikingly, the decrease in SPRED1 expression was not associated with recurrent chromosomal abnormalities of AML, including t (8;21) and t (15;17), which are common in M2 and M3 (18). The cause of this contradictory result might be that the classification of chromosomal abnormalities adopted in this study was not completely consistent with the WHO classification by the limitation of the small case number of recurrent chromosomal abnormalities. Considering that NF1 deletion frequently occurs in AML-M4eo and AML with t (16;16)/inv(16) (13), the association of SPRED1 downregulation and AMLs with particular recurrent chromosomal abnormalities still needs further investigation.
The mechanism of decreased SPRED1 expression AML is mostly unknown. Although SPRED1 germline mutation was reported to develop AML-M5 (10), no mutation of SPRED1 was discovered in 23 juvenile myelomonocytic leukemia (JMML) patients (19). Regarding deletion, both Olsson et al. and our group previously did detect SPRED1 deletion with high-resolution array analysis (7, 11). In this study, however, using an equally sensitive assay of FISH analysis, we demonstrate that the deletion of SPRED1 is a rare event in AML.
Li and his colleague proposed that SPRED1 was the potential target of MIR126 in AML (20). MIR126 is entirely complementary to 3'UTR of SPRED1 and is required for vascular integrity and angiogenesis. Recently, Lechman et al. demonstrated that MIR126 targets the PI3K/AKT/MTOR signaling pathway, preserving leukemia stem cell quiescence and promoting chemotherapy resistance (21). Overexpression of MIR126 can relieve the repressive influence of SPRED1 on the signaling pathways activated by VEGF and FGF (22, 23). Prior study also reported the overexpression of MIR126 in core-binding factor (CBF) AMLs (20). However, neither a significant elevation of MIR126 nor its negative correlation with SPRED1 was observed in our study. The discrepancy may come from the fact that our study focused on general AML instead of specific subgroup identified on cytogenetic abnormalities and gene mutations. As reduction of SPRED1 occurred much more frequently than did elevation of MIR126 in this study (87.6 vs. 36.2%) and no distinction of SPRED1 expression was observed between CBF AML and AML with other recurrent chromosomal abnormalities, we speculate that upregulation of MIR126 is not the major underlying mechanism of SPRED1 reduction even if it does impact the SPRED1 expression in certain subgroup of AML.
Currently, there is a lack of reports on the treatment response and survival duration of AML with abnormal expression of SPRED1. Our study indicates that SPRED1 serves not only as a marker of treatment response but also as an independent prognostic factor for non-APL. We found that decreased SPRED1 expression was significantly related to the inferior EFS and PFS of non-APL. It suggests that SPRED1 plays a vital role in the progression of leukemia. Therefore, it is necessary to investigate the contribution of SPRED1 to leukemogenesis. SPRED1 regulates the proliferation and differentiation of hematopoietic cells, modulates angiogenesis and mediates cell metastasis (24–27). SPRED1 interacts with multiple factors such as NF1, c-KIT and VEGFR thereby suppressing Ras-MAPK signaling, which promotes leukemogenesis (5, 28–32). Consistent with these previous studies, we demonstrated that ectopic overexpression of SPRED1 lead to a decrease of pERK, induction of apoptosis and reduction of proliferation of THP-1 cells; in sharp contrast, silencing of SPRED1 activated ERK signaling, enhanced the proliferation and reduced the apoptosis of OCI-AML3 cells, which highly express SPRED1. Therefore, one of the mechanisms by which SPRED1 functions as a tumor suppressor in AML is to antagonizing Ras-MAPK signaling. However, more functional analysis in cells and animals are needed to explore the underlying mechanism of SPRED1 in AML.
Conclusion
SPRED1 is downregulated in adult AML, and SPRED1 is not only a predictor of treatment response but also an independent prognostic factor for non-APL. Mechanistically, SPRED1 may suppress AML transformation by antagonizing Ras-MAPK signaling (Figure 9). With the advance of precision medicine coupled with next-generation sequencing, targeting Ras-MAPK signaling may be a promising strategy for the treatment of AML with the downregulation of SPRED1.
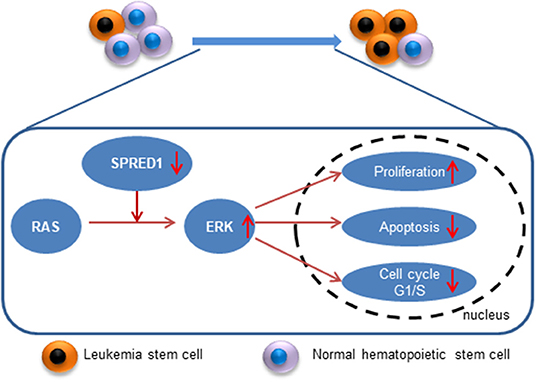
Figure 9. A schematic diagram of the down-regulation of SPRED1 in leukemia transformation.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
This study was approved by Ethical Committee of the First Affiliated Hospital of China Medical University (No. AF-SOP-07-1. 0-01, date Feb 25, 2016) and conducted in accordance with the Declaration of Helsinki. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
YL designed the study. RZ and YZ prepared the manuscript. XL, WX, and HW performed the experiments. WM, HP, and RT collected the data. SL and XY made the data analysis. All the authors made the final approval.
Funding
This work was supported by the Science and Technology Plan Projects of Liaoning Province grant (Grant No. 2013225079), the National Natural Science Foundation of China (Grant No. 81600117), and the National Youth Top-notch Talent of Ten Thousand Talent Program.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Liu XP (Facultad de Farmacia, USA) for her assistance in data analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.00204/full#supplementary-material
SFigure 1. FISH hybridization with home-brewed SPRED1 probes named RP11-644D16 (SG, 15q14) and RP11-477P17 (SO, 15q14). Two green signals and two orange signals were observed, suggesting no deletion of SPRED1.
SFigure 2. The expression of SPRED1 in AML cell lines. (A) The mRNA expression levels of SPRED1 in AML cell lines THP-1, OCI-AML2, OCI-AML3, Kasumi-1, HL-60, and NB4 were determined by RT-PCR; (B) the protein expression of SPRED1 in AML cell lines THP-1, OCI-AML3, and HL-60 were assayed by Western blot.
STable 1. Primers used to amplify SPRED1 exons (coding regions).
STable 2. The results of FAB subtypes, Karyotypes and FISH using SPRED1 probes for 43 de novo AML patients with reduced SPRED1 expression.
Abbreviations
SPRED1, Sprouty-Related EVH1 Domain-Containing Protein1; AML, acute myeloid leukemia; ALLs, acute lymphoblastic leukemias; non-APL, Non-acute promyelocytic leukemia; ERK, extracellular signal-regulated kinase; MAPK, Mitogen-activated protein kinase; FISH, fluorescence in situ hybridization; NCFC, neuro-cardio-facial-cutaneous; FAB, French-American-British; CR, complete remission; FCS, fetal calf serum; PBS, phosphate buffered saline; ELISA, enzyme-linked immunosorbent assay.
References
1. Spurlock G, Bennett E, Chuzhanova N, Thomas N, Jim HP, Side L, et al. SPRED1 mutations (legius syndrome): another clinically useful genotype for dissecting the neurofibromatosis type 1 phenotype. J Med Gene. (2009) 46:431–7. doi: 10.1136/jmg.2008.065474
2. Wakioka T, Sasaki A, Kato R, Shouda T, Matsumoto A, Miyoshi K, et al. Spred is a sprouty-related suppressor of ras signalling. Nature. (2001) 412:647–51. doi: 10.1038/35088082
3. Stowe IB, Mercado EL, Stowe TR, Bell EL, Oses-Prieto JA, Hernández H, et al. A shared molecular mechanism underlies the human rasopathies legius syndrome and neurofibromatosis-1. Genes Dev. (2012) 26:1421–6. doi: 10.1101/gad.190876.112
4. Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. (2001) 104:593–604. doi: 10.1016/S0092-8674(01)00245-8
5. Denayer E, de Ravel T, Legius E. Clinical and molecular aspects of RAS related disorders. J Med Gene. (2008) 45:695–703. doi: 10.1136/jmg.2007.055772
6. Lauchle JO, Braun BS, Loh ML, Shannon K. Inherited predispositions and hyperactive ras in myeloid leukemogenesis. Pediatr Blood Cancer. (2006) 46:579–85. doi: 10.1002/pbc.20644
7. Olsson L, Castor A, Behrendtz M, Biloglav A, Forestier E, Paulsson K, et al. Deletions of IKZF1 and SPRED1 are associated with poor prognosis in a population-based series of pediatric B-cell precursor acute lymphoblastic leukemia diagnosed between 1992 and 2011. Leukemia. (2014) 28:302–10. doi: 10.1038/leu.2013.206
8. Pasmant E, Gilbert-Dussardier B, Petit A, de Laval B, Luscan A, Gruber A, et al. SPRED1, a RAS MAPK pathway inhibitor that causes legius syndrome, is a tumour suppressor downregulated in paediatric acute myeloblastic leukaemia. Oncogene. (2015) 34:631–8. doi: 10.1038/onc.2013.587
9. Muram-Zborovski TM, Stevenson DA, Viskochil DH, Dries DC, Wilson AR, Rong M. SPRED 1 mutations in a neurofibromatosis clinic. J Child Neurol. (2010) 25:1203–9. doi: 10.1177/0883073809359540
10. Pasmant E, Ballerini P, Lapillonne H, Perot C, Vidaud D, Leverger G, et al. SPRED1 disorder and predisposition to leukemia in children. Blood. (2009) 114:1131. doi: 10.1182/blood-2009-04-218503
11. Zhang R, Lee JY, Wang X, Xu W, Hu X, Lu X, et al. Identification of novel genomic aberrations in AML-M5 in a level of array CGH. PLoS ONE. (2014) 9:e87637. doi: 10.1371/journal.pone.0087637
12. Parkin B, Ouillette P, Wang Y, Liu Y, Wright W, Roulston D, et al. NF1 inactivation in adult acute myelogenous leukemia. Clin Cancer Res. (2010) 16:4135–47. doi: 10.1158/1078-0432.CCR-09-2639
13. Haferlach C, Dicker F, Kohlmann A, Schindela S, Weiss T, Kern W, et al. AML with CBFB-MYH11 rearrangement demonstrate RAS pathway alterations in 92% of all cases including a high frequency of NF1 deletions. Leukemia. (2010) 24:1065–9. doi: 10.1038/leu.2010.22
14. Lauchle JO, Kim D, Le DT, Akagi K, Crone M, Krisman K, et al. Response and resistance to MEK inhibition in leukaemias initiated by hyperactive ras. Nature. (2009) 461:411–4. doi: 10.1038/nature08279
15. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. (1976) 33:451–8. doi: 10.1111/j.1365-2141.1976.tb03563.x
16. Chinese Society of Hematology, Chinese Medical Association. Expert consensus on treatment of acute myeloid leukemia (part I). Chin J Hematol. (2009) 30:429–31. doi: 10.3760/cma.j.issn.0253-2727.2009.06.020
17. Hsu SM, Raine L, Fanger H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem. (1981) 29:577–80. doi: 10.1177/29.4.6166661
18. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the world health organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
19. Batz C, Hasle H, Bergstrasser E, van den Heuvel-Eibrink MM, Zecca M, Niemeyer CM, et al. Does SPRED1 contribute to leukemogenesis in juvenile myelomonocytic leukemia (JMML)? Blood. (2010) 115:2557–8. doi: 10.1182/blood-2009-12-260901
20. Li Z, Lu J, Sun M, Mi S, Zhang H, Luo RT, et al. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc Natl Acad Sci USA. (2008) 105:15535–40. doi: 10.1073/pnas.0808266105
21. Lechman ER, Gentner B, Ng SW, Schoof EM, van Galen P, Kennedy JA, et al. miR-126 Regulates distinct self-renewal outcomes in normal and malignant hematopoietic stem cells. Cancer Cell. (2016) 29:214–28. doi: 10.1016/j.ccell.2015.12.011
22. Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, et al. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. (2008) 15:261–71. doi: 10.1016/j.devcel.2008.07.002
23. Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, et al. miR-126 Regulates angiogenic signaling and vascular integrity. Dev Cell. (2008) 15:272–84. doi: 10.1016/j.devcel.2008.07.008
24. Nonami A, Kato R, Taniguchi K, Yoshiga D, Taketomi T, Fukuyama S, et al. Spred-1 negatively regulates interleukin-3-mediated ERK/mitogen-activated protein (MAP) kinase activation in hematopoietic cells. J Biol Chem. (2004) 279:52543–51. doi: 10.1074/jbc.M405189200
25. Taniguchi K, Kohno R, Ayada T, Kato R, Ichiyama K, Morisada T, et al. Spreds are essential for embryonic lymphangiogenesis by regulating vascular endothelial growth factor receptor 3 signaling. Mol Cell Biol. (2007) 27:4541–50. doi: 10.1128/MCB.01600-06
26. Miyoshi K, Wakioka T, Nishinakamura H, Kamio M, Yang L, Inoue M, et al. The sprouty-related protein, spred, inhibits cell motility, metastasis, and rho-mediated actin reorganization. Oncogene. (2004) 23:5567–76. doi: 10.1038/sj.onc.1207759
27. Bundschu K, Walter U, Schuh K. Getting a first clue about SPRED functions. BioEssays. (2007) 29:897–907. doi: 10.1002/bies.20632
28. Boissel N, Leroy H, Brethon B, Philippe N, de Botton S, Auvrignon A, et al. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia. (2006) 20:965–70. doi: 10.1038/sj.leu.2404188
29. Scholl C, Gilliland DG, Frohling S. Deregulation of signaling pathways in acute myeloid leukemia. Semin Oncol. (2008) 35:336–45. doi: 10.1053/j.seminoncol.2008.04.004
30. Bacher U, Haferlach T, Schoch C, Kern W, Schnittger S. Implications of NRAS mutations in AML: a study of 2502 patients. Blood. (2006) 107:3847–53. doi: 10.1182/blood-2005-08-3522
31. McClatchey AI, Cichowski K. SPRED proteins provide a NF-ty link to ras suppression. Genes Dev. (2012) 26:1515–9. doi: 10.1101/gad.197434.112
32. Chiu YE, Dugan S, Basel D, Siegel DH. Association of piebaldism, multiple cafe-au-lait macules, and intertriginous freckling: clinical evidence of a common pathway between KIT and sprouty-related, ena/vasodilator-stimulated phosphoprotein homology-1 domain containing protein 1 (SPRED1). Pediatr Dermatol. (2013) 30:379–82. doi: 10.1111/j.1525-1470.2012.01858.x
Keywords: SPRED1, AML, non-APL, MAPK, MIR126
Citation: Zhang R, Zhang Y, Lu X, Xu W, Wang H, Mo W, Pang H, Tang R, Li S, Yan X and Li Y (2020) SPRED1 Is Downregulated and a Prognostic Biomarker in Adult Acute Myeloid Leukemia. Front. Oncol. 10:204. doi: 10.3389/fonc.2020.00204
Received: 18 September 2019; Accepted: 06 February 2020;
Published: 27 February 2020.
Edited by:
Timothy I. Shaw, St. Jude Children's Research Hospital, United StatesReviewed by:
Eric Pasmant, Université Paris Descartes, FranceDora Visnjic, University of Zagreb, Croatia
Copyright © 2020 Zhang, Zhang, Lu, Xu, Wang, Mo, Pang, Tang, Li, Yan and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Li, bGl5YW4yQG1lZG1haWwuY29tLmNu