 Xiang Li1,2
Xiang Li1,2 Na Li1,2Li Huang3Shi Xu4Xue Zheng1,2Akil Hamsath4Mei Zhang1,2Lijun Dai1,2Hui Zhang1,2
Na Li1,2Li Huang3Shi Xu4Xue Zheng1,2Akil Hamsath4Mei Zhang1,2Lijun Dai1,2Hui Zhang1,2 Justin Jong-Leong Wong5
Justin Jong-Leong Wong5 Ming Xian4
Ming Xian4 Chun-tao Yang1,2*†
Chun-tao Yang1,2*† Jinbao Liu1,2*
Jinbao Liu1,2*- 1Affiliated Cancer Hospital & Institute of Guangzhou Medical University, Guangzhou, China
- 2Guangzhou Municipal and Guangdong Provincial Key Laboratory of Protein Modification and Degradation, School of Basic Medical Science, Guangzhou Medical University, Guangzhou, China
- 3Department of Pancreatobiliary Surgery, The First Affiliated Hospital of Sun Yat-sen University, Guangzhou, China
- 4Department of Chemistry, Washington State University, Pullman, WA, United States
- 5Epigenetics and RNA Biology Program Centenary Institute, The University of Sydney, Camperdown, NSW, Australia
Ammonium tetrathiomolybdate (ATTM) has been used in breast cancer therapy for copper chelation, as elevated copper promotes tumor growth. ATTM is also an identified H2S donor and endogenous H2S facilitates VitB12-induced S-adenosylmethionine (SAM) generation, which have been confirmed in m6A methylation and lung cancer development. The m6A modification was recently shown to participate in lung adenocarcinoma (LUAD) progression. These conflicting analyses of ATTM's anticancer vs. H2S's carcinogenesis suggest that H2S should not be ignored during LUAD's treatment with ATTM. This study was aimed to explore ATTM's effects on LUAD cells and mechanisms associated with H2S and m6A. It was found that treatment with ATTM inhibited cell growth at high concentrations, while enhanced cell growth at low concentrations in three LUAD cell lines (A549, HCC827, and PC9). However, another copper chelator triethylenetetramine, without H2S releasing activity, was not found to induce cell growth. Low ATTM concentrations also elevated m6A content in A549 cells. Analysis of differentially expressed genes in TCGA cohort indicated that m6A writer METTL3 and reader YTHDF1 were upregulated while eraser FTO was downregulated in LUAD tissues, consistent with the findings of protein expression in patient tissues. ATTM treatment of A549 cells significantly increased METTL3/14 and YTHDF1 while decreased FTO expression. Furthermore, inhibition of m6A with shMETTL3 RNA significantly attenuated eukaryotic translation initiation factor (eIF) expressions in A549 cells. Correlation analysis indicated that small nuclear ribonucleic protein PRPF6 was positively expressed with YTHDF1 in LUAD tissues. Knockdown of YTHDF1 partially blocked both basal and ATTM-induced PRPF6 expression, as well as A549 cell growth. Lastly, ATTM treatment not only raised intracellular H2S content but also upregulated H2S-producing enzymes. Exogenous H2S application mimicked ATTM's aforementioned effects, but the effects could be weakened by zinc-induced H2S scavenging. Collectively, H2S impedes ATTM-induced anticancer effects through YTHDF1-dependent PRPF6 m6A methylation in lung adenocarcinoma cells.
Introduction
Ammonium tetrathiomolybdate (ATTM), with the formula (NH4)2MoS4, is a strong copper chelator. It has been clinically used in the treatment of copper toxicosis for Wilson's disease. At high concentrations, copper is also known to promote angiogenesis, metabolism and oxidative phosphorylation, thereby leading to tumor growth (1–3). Consequently, copper-chelating agents, like ATTM and triethylenetetramine (TETA), have been investigated in the treatment of cancers, including breast cancer, thyroid cancer and liver cancer (4–8). Additionally, copper-dependent enzyme, superoxide dismutase (SOD) 1, has been reported to facilitate lung adenocarcinoma (LUAD) development (9). Therefore, ATTM therapy may theoretically be extended to LUAD.
Interestingly, our lab and others have recently discovered ATTM is a pH-dependent hydrogen sulfide (H2S) donor (10, 11). There is no doubt that the development of ATTM as a therapeutic candidate must consider ATTM being both a copper chelator and a H2S releaser. It should be noted that the links between H2S and cancer development is still under debate (12). Some studies showed that elevated H2S can induce angiogenesis and tumor cell proliferation, thereby contributing to cancer development (13–15). H2S was also involved in VitB12-induced S-adenosylmethionine (SAM) generation (16), and high VitB12 has been found to raise the risk of lung cancer (17). Notably, in lung cancer tissues, endogenous H2S and its producing enzymes, like cystathionine beta-synthase (CBS), cystathionine gamma lyase (CSE, also known as CTH) and 3-mercaptopyruvate sulfurtransferase (3-MST), are highly expressed thereby benefiting cancer development (18, 19).
N6-Methyladenosine (m6A) methylation is one major type of mRNA modification, dynamically modulated by the corresponding writers, erasers and readers (20). The m6A methylation can differentially influence all fundamental aspects of mRNA metabolisms, including splicing, stability, and translation efficiency, when read by different m6A readers (21–23). Recently, the m6A methylation has been implicated in cancer pathogenesis, due to its induction of cell proliferation, invasion and immune disorders (24–28). Analysis of the cancer genome atlas (TCGA) cohort and recent studies (29, 30) indicated that m6A writer, methyltransferase like (METTL) 3 is upregulated in LUAD. The eraser, fat mass and obesity-associated gene (FTO), is downregulated. Both increased METTL3 and decreased FTO strongly suggest that LUAD tissues exhibit high m6A levels. However, no direct evidence has shown H2S can regulate m6A methylation, although H2S enhances SAM generation (16), a cofactor of METTL3/14 complex. We therefore hypothesize that m6A methylation is likely to be affected by H2S derived from ATTM and participate in LUAD development.
In the present study, we observed the effects of ATTM on lung adenocarcinoma cells and explored the roles of m6A in this process. Furthermore, we assessed the medication of H2S in ATTM-induced tumor cell proliferation, invasion and growth. Lastly, a purposive strategy was developed to overcome ATTM's side-effects in LUAD therapy.
Materials and Methods
Materials
ATTM and antibodies against METTL3 and METTL14 were purchased from Sigma-Aldrich Co. (St. Louis, MO, US). Antibodies against FTO and YTHDF1 were purchased from Abcam (Plc.Cambridge, MA, US). CuSO4 and Zn(OAc)2 were purchased from Meilun Biotechnology Co. (Dalian, China). TRIzol™ Regent (Invitrogen) was provide by Thermo Fisher Scientific Co. (Shanghai, China). Gemcell™ fetal bovine serum (FBS) was supplied by Gemini Company (Woodland, US).
Cell Culture, Growth, Proliferation, and Invasion Assays
Lung adenocarcinoma cell lines (A549 and HCC827) were purchased from Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China), and PC9 was obtained from ATCC. The cells were maintained in RPMI-1640 medium supplemented with 10% FBS at 37°C under an atmosphere of 5% CO2 and 95% air. They were passaged and harvested with 0.25% trypsin every other day.
Cell number was measured with Cell Counting Kit (CCK)-8 provided by Dojindo Lab (Kyushu, Japan). A549, HCC827, and PC9 cells were plated in 96-well plates at a density of 7, 000 cells/well. When grown to ~60–70% confluence, the cells were treated correspondingly. After the treatments, the CCK-8 solution (100 μL) at a 1:10 dilution with FBS-free medium was added to each well-followed by a 2-h incubation at 37°C. Absorbance (A) was measured at 450 nm with a microplate reader (Molecular Devices, US).
Cell proliferation was tested with BeyoClick™ 5-Ethynyl-2′-deoxyuridine (EdU) kit (Haimen, China). After the treatment of A549 cells with ATTM for 48 h, EdU incorporation assay was performed according to the manufacturer's instructions. TMB-derived color was measured at 630 nm with the microplate reader.
Cell invasion was observed with Transwell Migration Assay, as described (31) with modifications. RPMI-1640 medium (1% FBS) containing ATTM or Na2S in the absence or presence of Zn(OAc)2 was added to the lower chambers of the 12-well format transwells (8 μm-pore, BD Biosciences). A549 cells were seeded in the upper chambers at 105 cells per well, following a 48 h-culture. After that, the transwells were fixed in methanol, and stained with Giemsa solution. The unmigrated cells were removed from the top of the membranes using cotton swabs. To quantify the number of migrated cells in the bottom of the membrane, four random images of each group were taken at 10× under a light microscope. Migrated cell number was counted with Image J software.
Quantification of m6A RNA Methylation
After treatments of A549 cells with increasing concentrations of ATTM for 24 h, total RNA was extracted using TaKaRa MiniBest kits (Kusatsu, Japan) and quantitated with NanoDrop 1000 spectrophotometer (Thermo Fisher, US). The m6A RNA was detected with EpiQuik m6A RNA Methylation Colorimetric kit (Farmingdale, US). Briefly, 200 ng of fresh extracted RNA sample was added into strip wells with RNA high binding solution, and incubated for 90 min at 37°C. After three washes, capture antibody, detection antibody, and enhancer solution were applied in turn, and incubated for 1 h at 37°C. After washes, color developing solution was added and incubated for 6 min in the dark. When the solution became blue in the m6A positive wells, stop solution was added to turn the color into yellow. Lastly, the absorbance of stable yellow was measured at 450 nm with the microplate reader.
Western Blotting for Protein Expression
After exposed to 60 μM ATTM for 24 h, A549 cells were collected and split at 4°C. Total proteins in the lysate were quantitated with a BCA kit. Thirty micrograms of total protein sample were loaded in SDS-PAGE and electrophoresed. At the end of electrophoresis, the proteins were transferred to PVDF membranes. The membranes were blocked with 5% fat-free milk in Tris-base buffered saline containing 0.1% Tween-20 (TBS-T) for 1 h at room temperature, and then incubated with the primary antibodies against m6A related proteins (METTL3, METTL14, FTO, and YTHDDF1), and H2S-producing enzymes (CSE, CBS, and MPST), respectively, with gentle agitation overnight at 4°C. After three washes, corresponding HRP-conjugated secondary antibodies were applied and incubated for 1.5 h at room temperature. The signal was visualized using an enhanced chemiluminescence detection system. The intensity of bands was quantified with Image J software.
Analysis of TCGA Database and Measurement of Protein Expression of LUAD Patients
Transcriptional expressions of m6A or H2S related genes in primary tumor tissues and adjacent tissues of LUAD patients, as well as correlation analysis in tumor tissues, were performed through TCGA cohort research tool (UALCAN). Furthermore, the protein expressions of METTLE3, METTL14, and FTO in LUAD patients were verified with Western blotting assay as above.
Gene Knockdown
Gene expression microarray data were downloaded at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE76367 (29). METTL3 was knocked down through short hairpin (sh) RNA in A549 cells, and gene expression was profiled by high throughput sequencing. Selected genes of eukaryotic translation initiation factors (eIFs) were shown as the maximum vs. minimum of the ratio shMETTL3 to shGFP.
Small interfering RNA (siRNA) against human YTHDF1 (Gene ID: 54915) was synthesized by GenePharma Co., Ltd (Shanghai, China). YTHDF1 siRNA and control random non-targeting siRNA were transfected into the A549 cells using Lipofectamine 2000 (Invitrogen, USA) (32). To raise the transfection efficiency, the cells were incubated with 20 nM YTHDF1 siRNA or Control siRNA for 6 h followed by a 24-h culture. The silencing ability was evaluated by Western blotting assay.
Quantitative Polymerase Chain Reaction for PRPF6 Gene Expression
Total RNAs were extracted from A549 cells and quantitated as above. First-strand cDNA was synthesized using TaqMan SYBR® Premix Ex TaqTM II (Tli RNase H Plus) virus reverse transcriptase and 2 μg RNA template in 20 μL reaction volume. The cDNA was used for real-time PCR with Prime Script RT reagent kit with gDNA Eraser (Life Technologies, US). Actin was used to normalize the expression of PRPF6. The results were analyzed using the comparative Ct method (2-ΔΔCt with logarithmic transformation). Primer sequences were displayed as below: PRPF6 forward 5-GTCATGCGTGCCGT GATTG-3 and reverse 5-TCCAGGGCATTGTGGGCTA-3, Actin forward 5-TGGCACCCAGCACAATGAA-3 and reverse 5-CTAAGTCATAGTCCGCCTAGAAGCA-3. PCRs were carried out as follows: initial denaturation at 95°C for 30 s, 40 cycles of 95°C for 5 s, 60°C for 34 s, and 95°C for 15 s, 60°C for 1 h, followed by a final extension at 95°C for 15 s.
Measurement of H2S Levels
ATTM-mediated H2S generation in cells was determined with a H2S fluorescent probe WSP-5 (33). A549 cells were inoculated in 24-well plates and grown to 60~70% confluence. After treated with ATTM, the cells were incubated with 10 μM WSP-5 in 1% FBS medium at 37°C for 30 min in the dark. Cell imaging was carried out after a slight wash with PBS. The intracellular H2S-triggered fluorescence was visualized under AMG fluorescence microscopy (Advanced Microscopy Group, US).
For H2S scavenging induced by Zn(OAc)2, CuSO4, FeCl3 or VitB12 was observed through a Unisense H2S micro-sensor (Tueager 1, Denmark) (34). Into 5 mL PBS buffer, fresh Na2S stock solution was added to produce a 100 μM Na2S solution. When the curve reached the peak and kept stable, the same dose of above compounds was immediately added, respectively. The curves were recorded correspondingly with the H2S sensor. For Zn(OAc)2 or CuSO4-induced continuous H2S scavenging from ATTM solution was recorded for 6 h. Into 20 mL of 500 μM ATTM solution (pH 5), fresh Zn(OAc)2 or CuSO4 stock solutions was added, respectively, to produce a 250 μM solution. Real-time H2S content was monitored with the H2S sensor and quantified against a standard curve.
Statistical Analysis
The experiment data are presented as means ± standard deviation (SD). Significance between groups was evaluated by one-way analysis of variance (ANOVA) followed by Student-Newman-Keuls test using GraphPad Prism 8 software (San Diego, US). A probability <0.05 was considered statistically significant.
Results
Low ATTM Levels Enhanced Growth of Lung Adenocarcinoma Cells
As shown in Figure 1A, treatment with high concentrations (≥250 μM) of ATTM for 48 h remarkably reduced cell number, in three types of lung adenocarcinoma cells (A549, HCC827, and PC9). However, at low concentrations (from 60 to 125 μM), the treatment distinctively increased cell number. Notably, another copper chelator TETA, without H2S releasing activity, was not found to elevate cell number at the same treatment profile (Figure 1B). Furthermore, 60–125 μM of ATTM treatment could also induce A549 cell proliferation as evaluated by EdU assay (Figure 1C). The result indicates that low ATTM concentrations are able to promote lung adenocarcinoma growth.
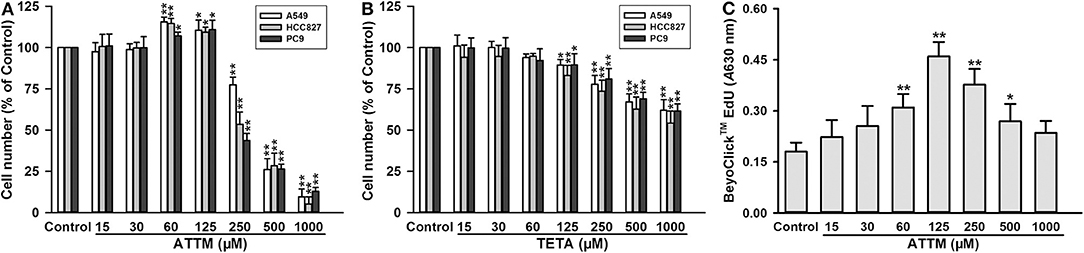
Figure 1. Effects of copper chelators on LUAD cell growth and proliferation. After treatment with increasing concentrations of ATTM (A) or TETA (B) for 48 h, cell number of three cell lines (A549, HCC827, and PC9) was counted with CCK-8 assay. (C) After ATTM treatment for 48 h, the proliferation of A549 cells was tested with BeyoClick™ EdU kit. Data are expressed as mean ± SD of four independent experiments. *P < 0.05, **P < 0.01 vs. Control group.
ATTM Induced mRNA m6A Methylation in Lung Adenocarcinoma A549 Cells
To understand why ATTM enhanced lung adenocarcinoma growth, A549 cells were selected as a representative in the following experiments. Since mRNA m6A methylation is involved in a variety of cancer growth including lung adenocarcinoma, intracellular m6A mRNA level was then investigated. As shown in Figure 2A, the m6A mRNA content was significantly elevated after the exposure of A549 cells to 60 μM ATTM for 24 h. However, the treatment duration (half of 48 h) did not alter cell number (Figure 2B) or growth status (Figure 2C). Notably, analysis of TCGA cohort shows that LUAD condition significantly upregulates the m6A writer METTL3, while downregulates the m6A eraser FTO (Figure 2D). The result indicates that ATTM can trigger mRNA m6A methylation before cell growth in lung adenocarcinoma cells.
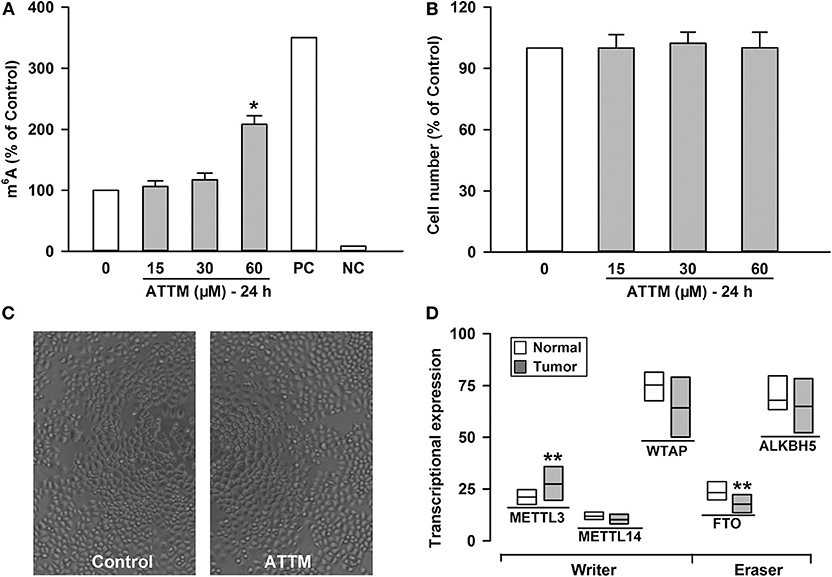
Figure 2. Effects of ATTM on mRNA m6A methylation in A549 cells. The cells were treated with ATTM at concentrations ranging from 0 to 60 μM for 24 h. (A) The content of m6A mRNA was measured with a commercial kit (PC, positive control; NC, negative control). (B) The cell number was tested with CCK-8 assay. Data are presented as mean ± SD. n = 4. *P < 0.01 vs. Control group. (C) Growth of quiescent A549 cells and cells exposed to 60 μM ATTM for 24 h were captured using digital microphotograph. (D) Expressions of the m6A writers (METTL3, METTL14, and WTAP) and erasers (FTO and ALKBH5) were analyzed between LUAD primary tumor tissues (n = 515) and normal tissues (n = 59) in TCGA cohort. Data are showed as median ± quartile. **P < 10−4 vs. Normal tissues.
Upregulated m6A Methylation Was Involved in LUAD Progression and ATTM-Induced mRNA Translation in Lung Adenocarcinoma A549 Cells
To uncover the mechanisms underlying the increased m6A mRNA levels in ATTM-treated cells, m6A related proteins were detected with Western blot. As shown in Figures 3A,C, comparing with the adjacent tissues, the writer (METTL3 and METLL14) expressions were upregulated in tumor tissues of LUAD patients, while the eraser FTO expression was downregulated. Importantly, the writer (METTL3 and METLL14) expressions in A549 cells could also be upregulated, while the eraser FTO could be downregulated under ATTM treatment (Figures 3B,D), supporting the finding of the increased intracellular m6A content. To confirm the roles of m6A methylation, we investigated gene expression profile in METTL3 knockdown A549 cells. As shown in Figure 3E, shRNA-mediated METTL3 knockdown significantly inhibited the expressions of translation initiation factors, including eIFs (2B3, 3B, 3C/CL, 3D, 3IP1, 4A1, and 5/5A). This suggests that m6A methylation is necessary to translation process in A549 cells, and that the elevated m6A levels may be important for LUAD progression and ATTM-induced tumor growth.
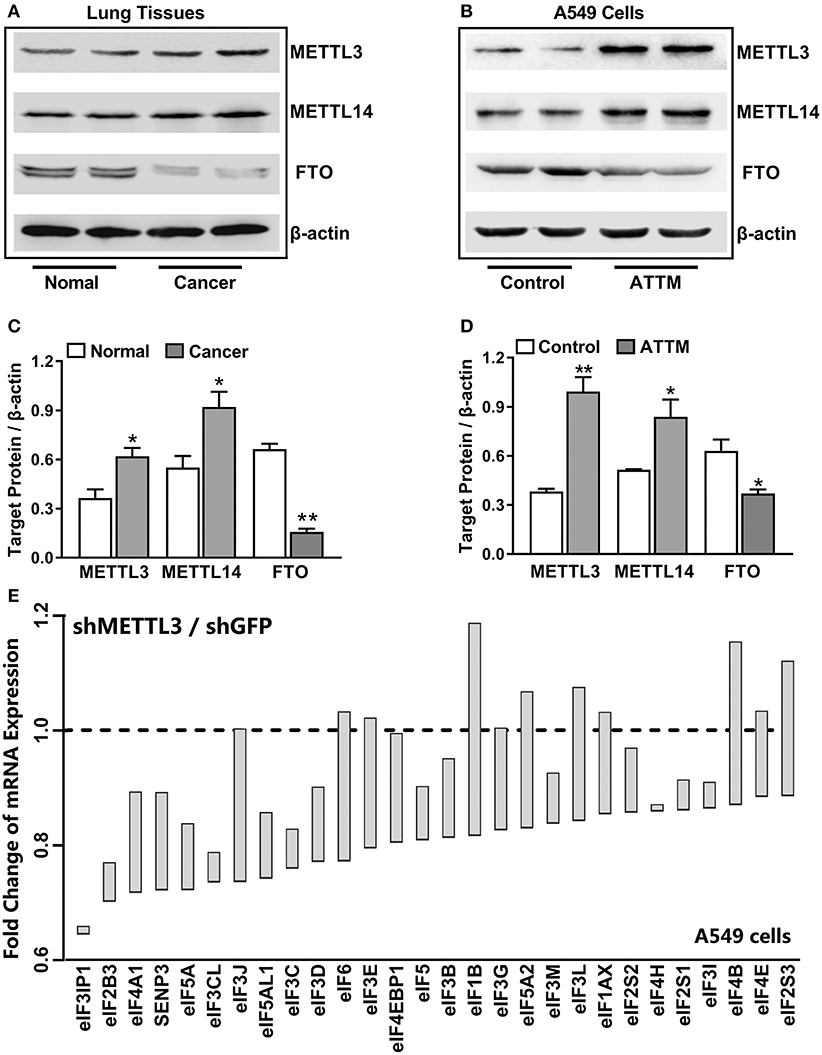
Figure 3. Effects of lung cancer condition and ATTM treatment on m6A related protein expression. (A) Confirmed LUAD patient tumor tissues and normal adjacent tissues, as well as (B) A549 cells treated with 60 μM ATTM for 24 h and normal cells, were collected and split. Total proteins were used for Western blot assay to measure the expressions of METTL3, METTL14, and FTO. Corresponding quantifications were shown as (C,D), respectively. Data are presented as mean ± SD. n = 2. *P < 0.05, **P < 0.01 vs. Control group. (E) A549 cells transfected with shMETTL3 or shGFP were collected and gene expression was profiled by high throughput sequencing. Representative genes of eukaryotic translation initiation factors (eIFs) were analyzed and shown as the maximum vs. minimum of the ratio of shMETTL3 to shGFP (GSE76367).
YTHDF1 Mediated ATTM-Induced Growth in Lung Adenocarcinoma A549 Cells
Although the increased m6A writer METTL3/14 and the decreased eraser FTO can result in the enhancement of m6A content, it is the m6A readers that directly determine the outcome of m6A methylated mRNA. With TCGA cohort, we examined the four common m6A readers (YTHDF1, YTHDF2, YTHDF3, and YTHDC1) in LUAD tumor samples and normal samples, and found YTHDF1 highly expressed in tumor tissues (Figure 4A). The increased YTHDF1 continuously expressed within various stages of LUAD (Figure 4B). Importantly, it was found that treatment of A549 cells with 60 μM ATTM for 24 h remarkably upregulated YTHDF1 protein expression (Figure 4C). After knockdown of YTHDF1 with siRNA in A549 cells (Figure 4D), both basal and ATTM-triggered cell growth were attenuated (Figure 4E).
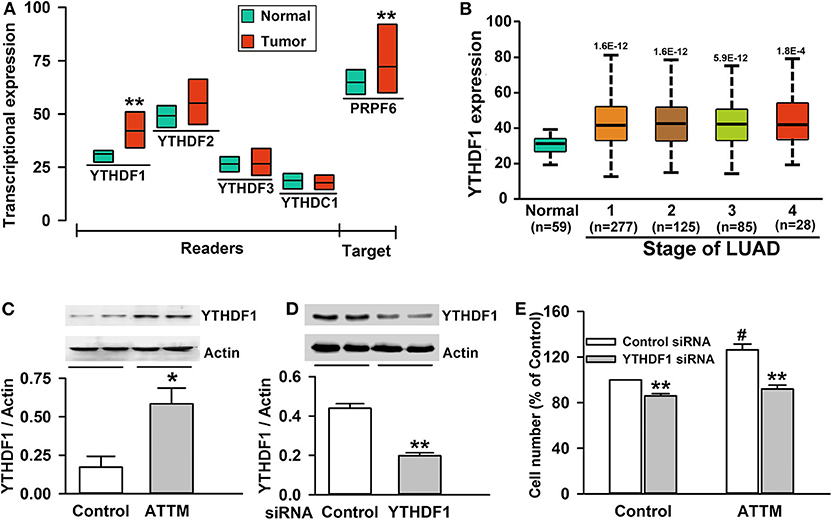
Figure 4. Roles of YTHDF1 in ATTM-induced growth of A549 cells. (A) Expressions of m6A readers (YTHDF1, YTHDF2, YTHDF3, and YTHDC1) and a potential target gene (PRPF6) were compared between LUAD patient primary tumor tissues (n = 515) and normal tissues (n = 59) in TCGA cohort. Data are shown as median ± quartile. **P < 10−7 vs. Normal tissues. (B) YTHDF1 gene expression in normal tissues and various LUAD stage tissues was analyzed in TCGA cohort. Data are shown in the box plot. (C) After treatment of A549 cells with 60 μM ATTM for 24 h, YTHDF1 protein expression was measured with Western blot assay, and the densitometric analysis was performed. Data are shown as mean ± SD. n = 2. *P < 0.05 vs. Control group. (D) A549 cells were incubated with YTHDF1 siRNA or Control siRNA for 6 h followed by a 24-h culture, and the interference efficiency was determined with Western blot assay. (E) Normal A549 cells and YTHDF1-knocked down cells were treated with 60 μM ATTM for 48 h, and cell counting assay was performed. Data are presented as mean ± SD. n = 4. **P < 0.01 vs. Control siRNA cells, #P < 0.01 vs. ATTM-free cells.
YTHDF1-Mediated Cell Growth Was Associated With PRPF6 Induction in ATTM-Treated A549 Cells
To explore targets involved in YTHDF1-mediated A549 cell growth, we screened genes that are positively correlated with YTHDF1 in TCGA LUAD cohort. As shown in Figure 5A, PRPF6 was found to be positively expressed with YTHDF1 in LUAD tissues (r = 0.72). Similar to YTHDF1's expression profile, the increased PRPF6 expression lasted various stages of LUAD (Figure 5B). Furthermore, qPCR analysis showed that PRPF6 mRNA level was significantly reduced after YTHDF1 knockdown in A549 cells. Additionally, the knockdown attenuated ATTM-induced PRPF6 mRNA expression (Figure 5C). The result reveals that PRPF6 may be a potential target gene involved in YTHDF1-mediated A549 cell growth.
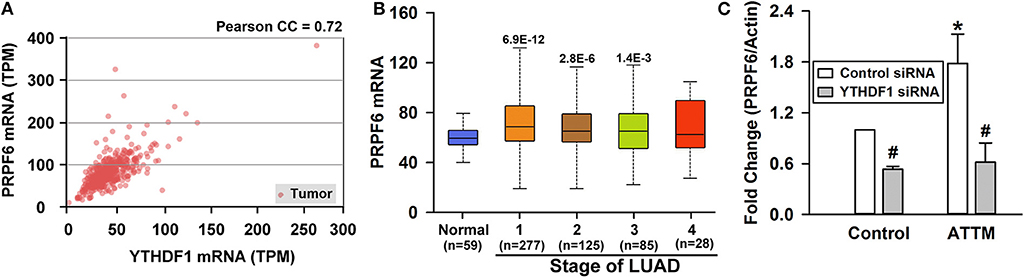
Figure 5. Identification of PRPF6 as a target gene of YTHDF1. (A) TCGA-based correlation analysis between PRPF6 and YTHDF1. (B) PRPF6 gene expression in normal tissues and various LUAD stage tissues was analyzed through TCGA cohort. Data are shown in the box plot. (C) Normal A549 cells and YTHDF1-knocked down cells were treated with 60 μM ATTM and then mRNA was extracted for analysis of PRPF6 expression with qPCR. Data are shown as mean ± SD. n = 4. *P < 0.01 vs. ATTM-free cells. #P < 0.01 vs. Control siRNA cells.
H2S Was Significant to ATTM-Induced A549 Cell Growth and m6A Methylation
ATTM was previously demonstrated to generate H2S on acid conditions in our lab. As shown in Figure 6A, the exposure of A549 cells to ATTM markedly raised intracellular H2S levels. With TCGA cohort, we studied endogenous H2S synthetase expressions and, found that CSE/CTH, CBS and MPST expressions were enhanced in tumor tissues comparing with normal samples (Figure 6B). Additionally, the exposure of A549 cells to 60 μM ATTM for 24 h obviously upregulated CBS and MPST expressions (Figures 6D,E), however, remarkable change of CSE was not found (Figure 6C). Notably, direct H2S donation (Na2S) could also induce m6A methylation (Figure 6F), as well as cell growth (Figure 6G), proliferation (Figure 6H) and invasion (Figures 6I,J), indicating H2S being an effector in ATTM-induced m6A methylation and cell growth.
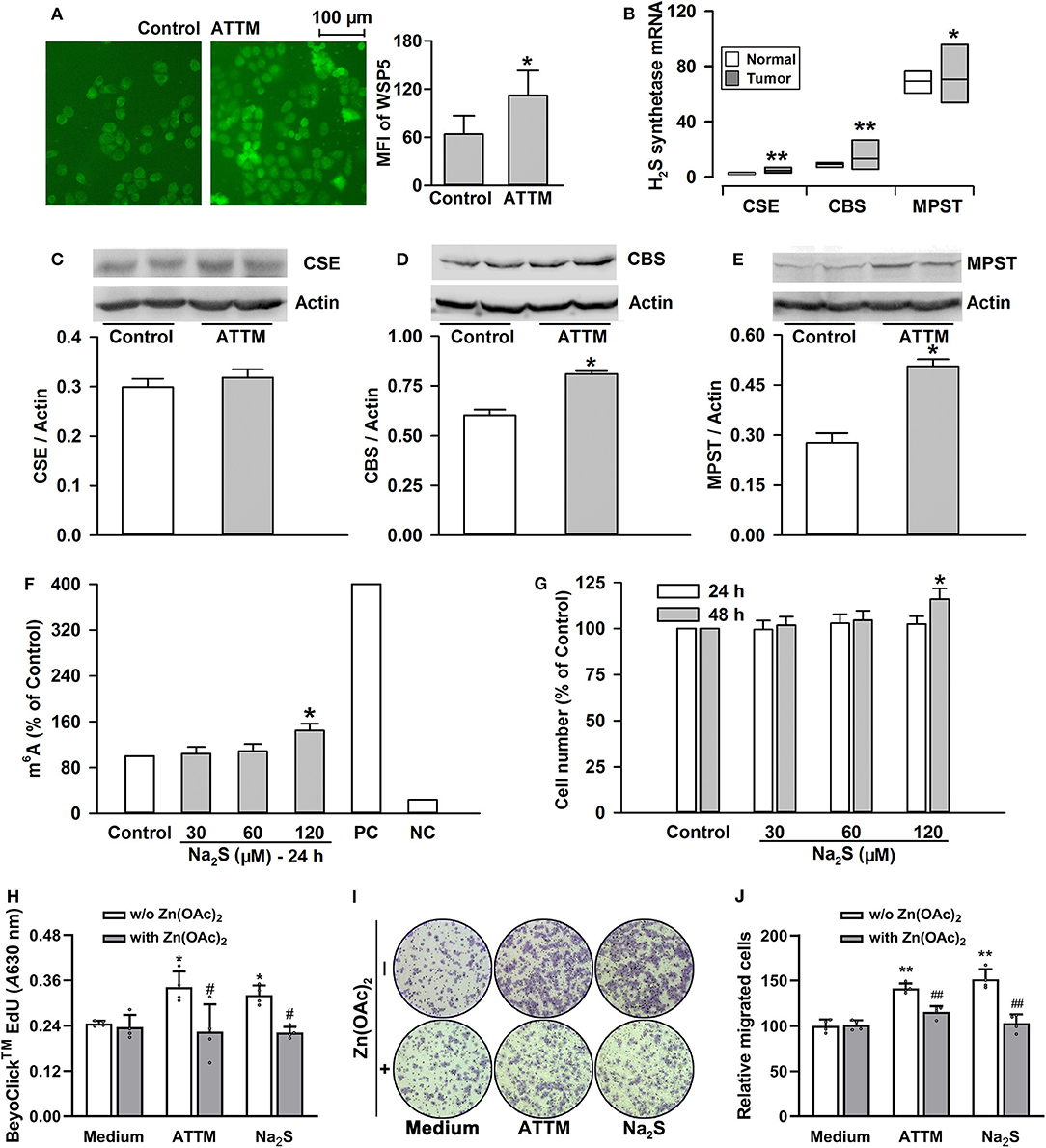
Figure 6. Roles of H2S in ATTM-induced m6A methylation and growth in A549 cells. (A) A549 cells were treated with normal medium (Control) and 60 μM ATTM for 3 h. Intracellular H2S content was observed with H2S fluorescent probe WSP5 staining followed by fluorescence photography. (B) Expressions of H2S synthetases (CSE, CBS and MPST) were compared between LUAD patient primary tumor tissues (n = 515) and normal tissues (n = 59) in TCGA cohort. Data are showed as median ± quartile. *P < 10−6, **P < 10−11 vs. Normal tissues. (C–E) After treatment of A549 cells with 60 μM ATTM for 24 h, the expressions of CSE (C), CBS (D), and MPST (E) were measured with Western blot, and then densitometric analysis were performed. Data are shown as mean ± SD. n = 2. *P < 0.05 vs. Control. (F) A549 cells were treated with increasing concentrations of Na2S for 24 h, intracellular m6A mRNA was tested with a commercial kit. (G) A549 cells were treated with the indicated concentrations of Na2S for 24 and 48 h, respectively. The cell number was measured with CCK-8 assay. (H–J) A549 cells were treated with 60 μM ATTM or 120 μM Na2S in the absence or presence of 25 μM Zn(OAc)2 for 48 h. Cell proliferation was tested with EdU incorporation assay (H). Cell invasion was observed with Transwell Migration Assay (I) and migrated cells were counted using ImageJ software. Data are presented as the mean ± SD. n = 4. *P < 0.05, **P < 0.01 vs. Control/Medium. #P < 0.05, ##P < 0.01 vs. Zn(OAc)2 free group.
H2S Scavenging Attenuated ATTM-Induced A549 Cell Growth and m6A Methylation
Since the enhanced H2S generation was involved in ATTM-induced A549 cell growth and m6A methylation, scavenging H2S might overcome these side-effects of ATTM. Through testing several common H2S scavengers, it was found that Zn(OAc)2 and CuSO4 had powerful ability to remove H2S, while the ability of VitB12 and FeCl3 was weak or even undetectable (Figure 7A). Additional cell viability examination showed that both Zn(OAc)2 and CuSO4 were non-toxic at concentrations <25 μM (Figures 7B,C).
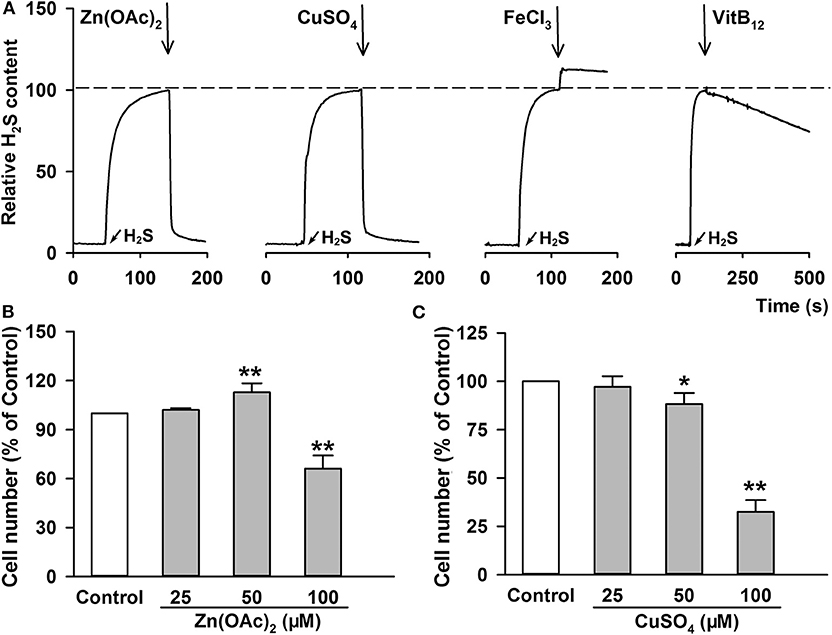
Figure 7. H2S removing efficiency and cell toxicity of the indicated H2S scavengers. (A) After adding 100 μM Na2S, the same amount of indicated H2S scavengers, including Zn(OAc)2, CuSO4, FeCl3 and VitB12, were added, respectively. The content of H2S molecule was monitored with a Unisense H2S microsensor. (B–C) A549 cells were treated with increasing concentration of Zn(OAc)2 (B) or CuSO4 (C) for 24 h. The cell number was measured with CCK-8 assay. Data are shown as the mean ± SD. n = 4. *P < 0.05, **P < 0.01 vs. Control group.
To make sure H2S is necessary to ATTM-induced biological process in A549 cells, we observed the scavenging effect of Zn(OAc)2 or CuSO4 on ATTM-induced H2S release within a period of 6 h. As shown in Figure 8A, adding 25 μM Zn(OAc)2 or CuSO4 time-dependently attenuated ATTM-induced H2S release. Notably, application of 25 μM Zn(OAc)2 inhibited ATTM-induced cell growth (Figure 8B), proliferation (Figure 6H) and invasion (Figures 6I,J). More importantly, Zn(OAc)2 was also able to attenuate ATTM-induced m6A methylation (Figure 8C). Collectively, the result suggests that H2S is necessary and sufficient to ATTM-induced biological changes, and scavenging H2S can potentially overcome ATTM's side-effects in lung cancer treatment.
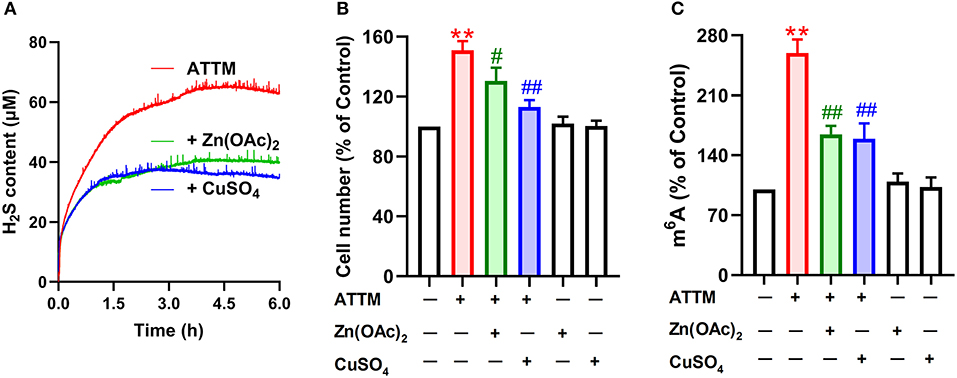
Figure 8. Effects of H2S scavengers on ATTM-induced A549 cell growth and m6A methylation. (A) ATTM solutions were prepared in the presence or absence of 25 μM Zn(OAc)2 or CuSO4. Subsequently, released H2S was recorded for 6 h with a H2S micro-sensor. (B) A549 cells were treated with 60 μM ATTM for 48 h in the absence or presence of 25 μM Zn(OAc)2 or CuSO4. The cell number was measured with CCK-8 assay. (C) A549 cells were treated with 60 μM ATTM for 24 h in the absence or presence of 25 μM Zn(OAc)2 or CuSO4. Intracellular m6A mRNA was tested with a commercial kit. Data are shown as the mean ± SD. n = 4. **P < 0.01 vs. Control. #P < 0.05, ##P < 0.01 vs. ATTM alone group.
Discussion
In the present study, ATTM was found to promote cell growth, proliferation and invasion in lung adenocarcinoma cells, through YTHDF1-dependent PRPF6 m6A methylation. H2S was involved in the above effects of ATTM. Importantly, scavenging H2S was proved to overcome the side-effects of ATTM in lung cancer therapy.
Copper ions are known to essentially maintain organism functions by regulating activity of key enzymes, like cytochrome C oxidase and SOD. However, its aberrant increase in plasma usually leads to pathological consequences, including Wilson's disease and cancers. During cancer development, high contents of copper are believed to enhance angiogenesis and blood supply, as well as activity of mitochondrial cytochrome C oxidase, thereby promoting solid tumor growth and metastasis (1–3, 35). Consequently, chelating agents of copper ions, like ATTM and TETA, have become promising anticancer drugs. The clinical trials of ATTM in breast cancer have recently made great progress (4–6). Notably, SOD1, a copper-dependent enzyme, was reported to participate in lung cancer growth and has become a significant therapeutic target (9). Therefore, ATTM may exert anticancer effects in lung adenocarcinoma through reduction of copper ions. In this study, at high concentrations, ATTM could indeed inhibit the growth of lung adenocarcinoma cells. However, at low concentrations, ATTM distinctively promoted tumor cell growth, as well as proliferation and invasion. The result was different from the reported effects of ATTM in breast cancer (6) and BRAF-driven papillary thyroid cancer (8), which suggests that the findings in other cancers should not be simply transplanted to lung cancer.
To uncover the mechanisms underlying ATTM-induced LUAD cell growth at low concentrations, we investigated mRNA m6A methylation, which has been involved in different cancer growth, like liver cancer (25), endometrial cancer (26), and leukemia (36). The m6A writer METTL3 has also been reported to promote lung cancer survival, growth, and invasion (29). We therefore speculated that the aberrant m6A methylation was involved in ATTM-induced growth of LUAD cells. By measuring m6A content in A549 cells, we found that the treatment with ATTM for 24 h significantly enhanced intracellular m6A content, while it did not significantly alter cell growth, suggesting m6A methylation occurs earlier than cell growth. Generally, m6A methylated mRNA can be synthesized via methyltransferase complex m6A writer, mainly consisting of METTL3, METTL14, and WTAP. Meanwhile, CH3- can also be erased from the RNA using FTO and ALKBH5. Therefore, the process is dynamic and reversible (37). The increased METTL3 and decreased FTO in the present TCGA analysis indicate that the dysregulated writer and/or eraser may be responsible for the increased m6A content in lung adenocarcinoma cells. This finding was supported by protein measurement in LUAD patients. Importantly, we found that the treatment with ATTM significantly augmented METTL3, but reduced FTO protein expression. ATTM could also upregulate METTL14 expression, which was not consistent with the TCGA analysis. We hypothesize that this difference may be due to the uncertainty of gene expression between mRNA level and protein level. Notably, for the increased METTL3 and decreased FTO, the present experiment matched the TCGA analysis. Such unique expressions, we believe, contribute to ATTM-induced m6A increase in A549 cells.
However, the roles of m6A in cancer biology are complicated and conflicting, i.e., tumor growth or anti-tumor (38). In addition, the binding of target mRNA with the writers or erasers is usually instantaneous. Therefore, it may be significant to examine the roles of m6A readers. To date, YT521-B homology (YTH) domain family of proteins, like YTHDF1, YTHDF2, YTHDF3, and YTHDC1, have been identified as m6A readers. YTHDF1-mediated mRNA spicing can increase translation efficiency, whereas YTHDF2-mediated mRNA decay will inhibit gene expression (21). The TCGA analysis indicates that the expression of YTHDF1, instead of YTHDF2/3 or YTHDC1, was markedly increased in LUAD tissues. In A549 cells, the elevated m6A mainly induced gene translation, evidenced by shMETTL3-mediated eIFs' downregulation (29). Western blot test showed that YTHDF1 expression could be upregulated by ATTM. Significantly, YTHDF1 siRNA inhibited basal and ATTM-induced cell growth, confirming the involvement of YTHDF1 in basal and environment-stimulated lung tumorigenesis.
As an m6A reader, it is necessary to discover YTHDF1's target genes. The TCGA analysis shows that PRPF6 (a small nuclear ribonucleic protein) is positively expressed with YTHDF1. The GEO analysis shows that PRPF6 can be attenuated through METTL3 knockdown (shGFP 1023 vs. shMETTL3 848). Additional analysis of MeT V2.0 m6A database indicates that m6A PRPF6 mRNA can be discerned and read by YTHDF1 in Hela cells via GRAC motif (R is G or A) (21). Actually, PRPF6 has been reported to promote lung cancer growth (39, 40). With these bioinformatics analyses and reports, PRPF6 is probably a target gene of YTHDF1. Importantly, the present experiment showed that ATTM treatment not only induced YTHDF1 expression, but also enhanced PRPF6 mRNA abundance. Both basal and ATTM-induced PRPF6 upregulation was significantly reduced by YTHDF1 knockdown, which was supported by previous reports (39, 40). As documented, the increased PRPF6 can alter the constitutive and alternative splicing of ZAK kinase, thereby activating cancer-related pathways, like AP-1, ERK, and JNK (41). In sum, it is believed that ATTM treatment upregulated METTL3/14 and downregulated FTO, raising intracellular m6A mRNA like PRPF6. Furthermore, these specific mRNAs were read by YTHDF1, and the spicing and translation of cancer growth-related genes were induced, thereby promoting LUAD tumor growth.
Significantly, we dissected the cause of ATTM-induced m6A methylation and cell growth. Copper chelating can exert anticancer effects through inhibiting angiogenesis (4, 6, 8). However, apart from copper chelating, ATTM can release H2S (10, 11). Studies have shown that the homeostasis of H2S is essential in organisms (42–46). In this study, we found that intracellular H2S level was markedly raised after ATTM treatment. Nevertheless, another copper chelator without H2S releasing ability, TETA, did not alter tumor cell growth. Direct H2S donation exerted similar effects to ATTM. The effects of ATTM or H2S could be abolished by CuSO4 or Zn(OAc)2. Besides H2S releasing, we found ATTM induced the expressions of H2S-production enzymes, which have been reported to participate in survival and chemoresistance of LUAD cells (18). Therefore, H2S is significant and necessary for ATTM-induced m6A methylation and lung cancer growth.
In healthy individuals, plasma free copper ion content is <20 μM (47), but for cancer patients, the recommended daily ATTM dosage was 90 mg (48). Therefore, the initial plasma ATTM should be much higher than that of free copper ions, so the free ATTM will release H2S during the clinical application (11). Of course, zinc application maybe overcomes ATTM's side-effects in LUAD therapy, while copper should not be recommended because of its tumorigenesis risk.
Interestingly, during the examination of VitB12 roles, it was found that VitB12 enhanced ATTM-induced LUAD cell growth (Supplementary Figure 1), unlike zinc or copper's inhibitory effects. However, both our study and previous reports suggest VitB12 is a H2S scavenger (34, 49). In fact, it has been documented that high VitB12 can raise the risk of lung cancer (17). H2S can also facilitate VitB12-induced SAM generation (16) that is the first substrate of METTTL3/14 complex. These findings further support H2S-induced m6A methylation promotes LUAD tumor growth.
In conclusion, the present study demonstrated that ATTM can induce m6A methylation through upregulation of METTL3 and downregulation of FTO in LUAD cells. YTHDF1-mediated PRPF6 expression is probably a pivotal reason. The effects of ATTM are closely associated with H2S generation (Figure 9). For the first time, this work reveals a potential risk and mechanism for ATTM application in LUAD treatment. Meanwhile, this is the first report on the roles of H2S in m6A methylation.
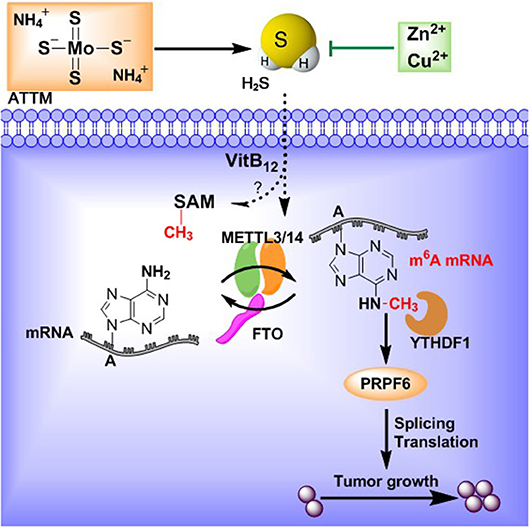
Figure 9. ATTM-produced H2S induces mRNA m6A methylation and promotes lung adenocarcinoma growth.
Data Availability Statement
The datasets generated for this study can be found in TCGA LUAD (http://ualcan.path.uab.edu/analysis.html), GSE76367 (https://www.ncbi.nlm.nih.gov/geo/).
Ethics Statement
The studies involving human participants were reviewed and approved by Medical Ethics Committee of Guangzhou Medical University. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
CY, MX, JL, and JW designed the experiments and wrote the manuscript. XL, NL, SX, and XZ performed all the experiments and statistical analyses. LH and HZ analyzed the clinical data and provided the patient tissues. LD, MZ, and AH provided the critical suggestions. All the authors reviewed the final manuscript.
Funding
This work was supported by Natural Science Foundation of Guangdong Province Grant (2017A030313892), Guangzhou Key Medical Discipline Construction Project.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank R. Gregory and S. Lin in Harvard Medical School for their mRNA expression array. We also thank the reviewers for the careful and valuable work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.00234/full#supplementary-material
Supplementary Figure 1. Effects of VitB12 on ATTM-induced A549 cell growth.
References
1. Raju KS, Alessandri G, Ziche M, Gullino PM. Ceruloplasmin, copper ions, and angiogenesis. J Natl Cancer Inst. (1982) 69:1183–8.
2. Ishida S, Andreux P, Poitry-Yamate C, Auwerx J, Hanahan D. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc Natl Acad Sci USA. (2013) 110:19507–12. doi: 10.1073/pnas.1318431110
3. Hyvonen MT, Ucal S, Pasanen M, Peraniemi S, Weisell J, Khomutov M, et al. Triethylenetetramine modulates polyamine and energy metabolism and inhibits cancer cell proliferation. Biochem J. (2016) 473:1433–41. doi: 10.1042/BCJ20160134
4. Yoshii J, Yoshiji H, Kuriyama S, Ikenaka Y, Noguchi R, Okuda H, et al. The copper-chelating agent, trientine, suppresses tumor development and angiogenesis in the murine hepatocellular carcinoma cells. Int J Cancer. (2001) 94:768–73. doi: 10.1002/ijc.1537
5. Sproull M, Brechbiel M, Camphausen K. Antiangiogenic therapy through copper chelation. Expert Opin Ther Targets. (2003) 7:405–9. doi: 10.1517/14728222.7.3.405
6. Garber K. BIOMEDICINE. Targeting copper to treat breast cancer. Science. (2015) 349:128–9. doi: 10.1126/science.349.6244.128
7. Chan N, Willis A, Kornhauser N, Ward MM, Lee SB, Nackos E, et al. Influencing the tumor microenvironment: a phase II study of copper depletion using tetrathiomolybdate in patients with breast cancer at high risk for recurrence and in preclinical models of lung metastases. Clin Cancer Res. (2017) 23:666–76. doi: 10.1158/1078-0432.CCR-16-1326
8. Xu M, Casio M, Range DE, Sosa JA, Counter CM. Copper chelation as targeted therapy in a mouse model of oncogenic BRAF-driven papillary thyroid cancer. Clin Cancer Res. (2018) 24:4271–81. doi: 10.1158/1078-0432.CCR-17-3705
9. Somwar R, Erdjument-Bromage H, Larsson E, Shum D, Lockwood WW, Yang G, et al. Superoxide dismutase 1 (SOD1) is a target for a small molecule identified in a screen for inhibitors of the growth of lung adenocarcinoma cell lines. Proc Natl Acad Sci USA. (2011) 108:16375–80. doi: 10.1073/pnas.1113554108
10. Xu S, Yang CT, Meng FH, Pacheco A, Chen L, Xian M. Ammonium tetrathiomolybdate as a water-soluble and slow-release hydrogen sulfide donor. Bioorg Med Chem Lett. (2016) 26:1585–8. doi: 10.1016/j.bmcl.2016.02.005
11. Dyson A, Dal-Pizzol F, Sabbatini G, Lach AB, Galfo F, Dos Santos Cardoso J, et al. Ammonium tetrathiomolybdate following ischemia/reperfusion injury: chemistry, pharmacology, and impact of a new class of sulfide donor in preclinical injury models. PLoS Med. (2017) 14:e1002310. doi: 10.1371/journal.pmed.1002310
12. Hellmich MR, Szabo C. Hydrogen sulfide and cancer. Handb Exp Pharmacol. (2015) 230:233–41. doi: 10.1007/978-3-319-18144-8_12
13. Szabo C, Coletta C, Chao C, Modis K, Szczesny B, Papapetropoulos A, et al. Tumor-derived hydrogen sulfide, produced by cystathionine-beta-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc Natl Acad Sci USA. (2013) 110:12474–9. doi: 10.1073/pnas.1306241110
14. Untereiner AA, Olah G, Modis K, Hellmich MR, Szabo C. H2S-induced S-sulfhydration of lactate dehydrogenase a (LDHA) stimulates cellular bioenergetics in HCT116 colon cancer cells. Biochem Pharmacol. (2017) 136:86–98. doi: 10.1016/j.bcp.2017.03.025
15. Das A, Huang GX, Bonkowski MS, Longchamp A, Li C, Schultz MB, et al. Impairment of an endothelial NAD(+)-H2S signaling network is a reversible cause of vascular aging. Cell. (2018) 173:74–89e20. doi: 10.1016/j.cell.2018.02.008
16. Toohey JI. Possible involvement of hydrosulfide in B12-dependent methyl group transfer. Molecules. (2017) 22:E582. doi: 10.3390/molecules22040582
17. Fanidi A, Carreras-Torres R, Larose TL, Yuan JM, Stevens VL, Weinstein SJ, et al. Is high vitamin B12 status a cause of lung cancer? Int J Cancer. (2019) 145:1499–503. doi: 10.1002/ijc.32033
18. Szczesny B, Marcatti M, Zatarain JR, Druzhyna N, Wiktorowicz JE, Nagy P, et al. Inhibition of hydrogen sulfide biosynthesis sensitizes lung adenocarcinoma to chemotherapeutic drugs by inhibiting mitochondrial DNA repair and suppressing cellular bioenergetics. Sci Rep. (2016) 6:36125. doi: 10.1038/srep36125
19. Yang CT, Devarie-Baez NO, Hamsath A, Fu XD, Xian M. S-persulfidation: chemistry, chemical biology, and significance in health and disease. Antioxid Redox Signal. (2019). doi: 10.1089/ars.2019.7889. [Epub ahead of print].
20. Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell. (2017) 169:1187–200. doi: 10.1016/j.cell.2017.05.045
21. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N6-methyladenosine modulates messenger RNA translation efficiency. Cell. (2015) 161:1388–99. doi: 10.1016/j.cell.2015.05.014
22. Yue Y, Liu J, He C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. (2015) 29:1343–55. doi: 10.1101/gad.262766.115
23. Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, et al. m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. (2017) 548:338–42. doi: 10.1038/nature23450
24. Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet. (2014) 15:293–306. doi: 10.1038/nrg3724
25. Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. (2018) 67:2254–70. doi: 10.1002/hep.29683
26. Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, et al. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. (2018) 20:1074–83. doi: 10.1038/s41556-018-0174-4
27. Liu ZX, Li LM, Sun HL, Liu SM. Link between m6A modification and cancers. Front Bioeng Biotechnol. (2018) 6:89. doi: 10.3389/fbioe.2018.00089
28. Pan Y, Ma P, Liu Y, Li W, Shu Y. Multiple functions of m6A RNA methylation in cancer. J Hematol Oncol. (2018) 11:48. doi: 10.1186/s13045-018-0590-8
29. Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m6A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. (2016) 62:335–45. doi: 10.1016/j.molcel.2016.03.021
30. Zeng Y, Wang S, Gao S, Soares F, Ahmed M, Guo H, et al. Refined RIP-seq protocol for epitranscriptome analysis with low input materials. PLoS Biol. (2018) 16:e2006092. doi: 10.1371/journal.pbio.2006092
31. Jiang L, Sun Y, Wang J, He Q, Chen X, Lan X, et al. Proteasomal cysteine deubiquitinase inhibitor b-AP15 suppresses migration and induces apoptosis in diffuse large B cell lymphoma. J Exp Clin Cancer Res. (2019) 38:453. doi: 10.1186/s13046-019-1446-y
32. Yang C, Ling H, Zhang M, Yang Z, Wang X, Zeng F, et al. Oxidative stress mediates chemical hypoxia-induced injury and inflammation by activating NF-κb-COX-2 pathway in HaCaT cells. Mol Cells. (2011) 31:531–8. doi: 10.1007/s10059-011-1025-3
33. Peng B, Chen W, Liu C, Rosser EW, Pacheco A, Zhao Y, et al. Fluorescent probes based on nucleophilic substitution-cyclization for hydrogen sulfide detection and bioimaging. Chemistry. (2014) 20:1010–6. doi: 10.1002/chem.201303757
34. Yang CT, Wang Y, Marutani E, Ida T, Ni X, Xu S, et al. Data-driven identification of hydrogen sulfide scavengers. Angew Chem Int Ed Engl. (2019) 58:10898–902. doi: 10.1002/anie.201905580
35. Alvarez HM, Xue Y, Robinson CD, Canalizo-Hernandez MA, Marvin RG, Kelly RA, et al. Tetrathiomolybdate inhibits copper trafficking proteins through metal cluster formation. Science. (2010) 327:331–4. doi: 10.1126/science.1179907
36. Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m6A modification. Cell Stem Cell. (2018) 22:191–205e199. doi: 10.1016/j.stem.2017.11.016
37. Wu D, Li M, Tian W, Wang S, Cui L, Li H, et al. Hydrogen sulfide acts as a double-edged sword in human hepatocellular carcinoma cells through EGFR/ERK/MMP-2 and PTEN/AKT signaling pathways. Sci Rep. (2017) 7:5134. doi: 10.1038/s41598-017-05457-z
38. Wang S, Chai P, Jia R, Jia R. Novel insights on m6A RNA methylation in tumorigenesis: a double-edged sword. Mol Cancer. (2018) 17:101. doi: 10.1186/s12943-018-0847-4
39. Adler AS, McCleland ML, Yee S, Yaylaoglu M, Hussain S, Cosino E, et al. An integrative analysis of colon cancer identifies an essential function for PRPF6 in tumor growth. Genes Dev. (2014) 28:1068–84. doi: 10.1101/gad.237206.113
40. Pan Y, Liu H, Wang Y, Kang X, Liu Z, Owzar K, et al. Associations between genetic variants in mRNA splicing-related genes and risk of lung cancer: a pathway-based analysis from published GWASs. Sci Rep. (2017) 7:44634. doi: 10.1038/srep44634
41. Yang JJ, Lee YJ, Hung HH, Tseng WP, Tu CC, Lee H, et al. ZAK inhibits human lung cancer cell growth via ERK and JNK activation in an AP-1-dependent manner. Cancer Sci. (2010) 101:1374–81. doi: 10.1111/j.1349-7006.2010.01537.x
42. Du J, Hui Y, Cheung Y, Bin G, Jiang H, Chen X, et al. The possible role of hydrogen sulfide as a smooth muscle cell proliferation inhibitor in rat cultured cells. Heart Vessels. (2004) 19:75–80. doi: 10.1007/s00380-003-0743-7
43. Akaike T, Ida T, Wei FY, Nishida M, Kumagai Y, Alam MM, et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat Commun. (2017) 8:1177. doi: 10.1038/s41467-017-01311-y
44. Zheng Y, Yu B, De La Cruz LK, Roy Choudhury M, Anifowose A, Wang B. Toward hydrogen sulfide based therapeutics: Critical drug delivery and developability issues. Med Res Rev. (2018) 38:57–100. doi: 10.1002/med.21433
45. Cui Q, Yang Y, Ji N, Wang JQ, Ren L, Yang DH, et al. Gaseous signaling molecules and their application in resistant cancer treatment: from invisible to visible. Future Med. Chem. (2019) 11:323–36. doi: 10.4155/fmc-2018-0403
46. Shinkai Y, Kumagai Y. Sulfane sulfur in toxicology: a novel defense system against electrophilic stress. Toxicol Sci. (2019) 170:3–9. doi: 10.1093/toxsci/kfz091
47. Twomey PJ, Viljoen A, House IM, Reynolds TM, Wierzbicki AS. Relationship between serum copper, ceruloplasmin, and non-ceruloplasmin-bound copper in routine clinical practice. Clin Chem. (2005) 51:1558–9. doi: 10.1373/clinchem.2005.052688
48. Brewer GJ, Dick RD, Grover DK, LeClaire V, Tseng M, Wicha M, et al. Treatment of metastatic cancer with tetrathiomolybdate, an anticopper, antiangiogenic agent: phase I study. Clin Cancer Res. (2000) 6:1–10.
Keywords: H2S, m6A methylation, Ammonium tetrathiomolybdate, lung cancer, PRPF6
Citation: Li X, Li N, Huang L, Xu S, Zheng X, Hamsath A, Zhang M, Dai L, Zhang H, Wong JJ-L, Xian M, Yang C and Liu J (2020) Is Hydrogen Sulfide a Concern During Treatment of Lung Adenocarcinoma With Ammonium Tetrathiomolybdate? Front. Oncol. 10:234. doi: 10.3389/fonc.2020.00234
Received: 03 December 2019; Accepted: 11 February 2020;
Published: 28 February 2020.
Edited by:
Olivier Feron, Université Catholique de Louvain, BelgiumReviewed by:
Qi Zeng, Xidian University, ChinaWen Zhou, Guangzhou University of Chinese Medicine, China
Copyright © 2020 Li, Li, Huang, Xu, Zheng, Hamsath, Zhang, Dai, Zhang, Wong, Xian, Yang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chun-tao Yang, Y3lhbmdAZ3pobXUuZWR1LmNu; Jinbao Liu, amxpdUBnemhtdS5lZHUuY24=
†ORCID: Chun-tao Yang orcid.org/0000-0002-5640-0930