 Jia Wang1Ruwen Cui1
Jia Wang1Ruwen Cui1 Cecila G. Clement2
Cecila G. Clement2 Ranjana Nawgiri2
Ranjana Nawgiri2 Don W. Powell3Irina V. Pinchuk3,4
Don W. Powell3Irina V. Pinchuk3,4 Tammara L. Watts1*
Tammara L. Watts1*- 1Department of Otolaryngology, University of Texas Medical Branch, Galveston, TX, United States
- 2Department of Pathology, University of Texas Medical Branch, Galveston, TX, United States
- 3Department of Internal Medicine, Division of Gastroenterology, University of Texas Medical Branch, Galveston, TX, United States
- 4Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, TX, United States
Desmoplasia, a hallmark of a head and neck cancer, has both biologic and physiologic effects on cancer progression and chemotherapeutic response. Mesenchymal stem/stromal cells (MSCs), also known as mesenchymal stromal progenitor cells, have been shown to play a role in cancer progression, alter apoptotic responses, and confer resistance to chemotherapy in various carcinomas. The pathophysiology of MSCs with respect to tumorigenesis is widely reported in other cancers and is sparsely reported in oral squamous cell carcinomas (OSCCs). We previously reported paracrine mediated PDGF-AA/PDGFR-α signaling to underlie MSCs chemotaxis in OSCC. Given the poor clinical response to primary chemotherapy, we hypothesized that MSCs may alter cancer cell sensitivity to cisplatin through activation of PDGFR-α mediated signaling pathways. Co-culture of MSCs with human derived OSCC cell lines, JHU-012 and −019, resulted in a significant increase in the production of PDGF-AA and MCP-1 compared to cancer cells grown alone (p < 0.005) and was accompanied by an increase in the phosphorylation state of PDGFR-α (p < 0.02) and downstream target AKT at S473 (p < 0.025) and T308 (p < 0.02). JHU-012 and −019 cancer cells grown in co-culture were significantly less apoptotic (p < 0.001), expressed significantly higher levels of Bcl-2 (p < 0.04) with a concomitant significant decrease in bid expression (p < 0.001) compared to cancer cells grown alone. There was a significant increase in the cisplatin dose response curve in cancer cell clones derived from JHU-012 and 019 cancer cells grown in co-culture with MSCs compared to clones derived from cancer cells grown alone (p < 0.001). Moreover clones derived from JHU-012 cells grown in co-culture with MSCs were significantly more susceptible to cisplatin following pretreatment with, crenolanib, a PDGFR inhibitor, compared to cancer cells grown alone or in co-culture with MSCs (p < 0.0001). These findings suggest that crosstalk between cancer cells and MSCs is mediated, at least in part, by activation of autocrine PDGF-AA/PDGFR-α loop driving AKT-mediated signaling pathways, resulting in reduced cancer cell sensitivity to cisplatin through alterations in apoptosis.
Introduction
Mesenchymal stem/stromal cells (MSCs) have been recognized to play important roles in the pathogenesis and progression of several solid cancers, including breast, prostate, non-small cell lung cancer, and others (1–5). The robust desmoplastic reaction characteristic of pancreatic cancer, and recent reports that MSCs in the pancreatic cancer stroma have tumor promoting properties, highlights the importance of MSCs in this context (6). Akin to pancreatic cancer, head and neck squamous cell carcinomas (HNSCC), of which the majority arise from the oral cavity and oropharyngx, are characterized as having an extensive desmoplastic response, suggesting that the tumor stroma may play an important role in the pathophysiology of this cancer as well (7). Although the source of mesenchymal cells in the tumor stroma are numerous, several lines of evidence support MSCs as an important source of desmoplastic fibrocytes in the TME (8, 9).
In one of the first reports to identify and detail a mechanism for MSC homing to the TME of HNSCC, we reported that MSCs home to the TME of oral cavity and oropharyngeal squamous cell carcinoma through the chemotactic action of PDGF-AA mediated through PDGFR-α (10). Our work expanded on initial reports by Liotta et al. who isolated CD90+ tumor associated MSCs and showed that they promote tumor suppression by modulating T-cell responsiveness (11). Moreover, Liu et al. reported bone marrow derived MSCs promote oral cavity cancer progression via periostin-mediated activation of PI3K/AKT signaling pathways resulting in reduced cancer cell apoptosis (12).
Activation of AKT-mediated signaling pathways has been shown to play a pivotal role in cancer progression by promoting cancer cell survival by several mechanisms including altering apoptotic response (13). Activation of AKT allows Bcl-2 to outcompete Bax, resulting in an anti-apoptotic response and upregulation of Bcl-2 has been shown to be an important mechanism of chemo-resistance in cancer cells (14, 15). Bcl-2 expression is routinely reported in HNSCC surgical pathology specimens, has been shown to be overexpressed in patients with HNSCC (16), and overexpression in early HNSCC correlates with a significant decrease in 5-year disease free recurrence and overall survival for patients treated with primary radiation (17). Expression of Bcl-2 and activation of AKT are known pathways affecting cisplatin mediated cytotoxicity (18). Park et al. reported overcoming cisplatin resistance by downregulating Bcl-2 in HNSCC by modulating tristetraprolin (TTP) expression (19), the mechanisms underlying linking Bcl-2 overexpression and therefore reduced sensitivity to cisplatin in HNSCC have not been reported.
Resistance to chemotherapy is a multifactorial process. Growing evidence suggests that cancer stem cells (CSCs), cancer associated fibroblasts (CAFs), and MSCs (which differentiate into CAFs in the TME) play an important role in the development of de novo chemo-resistance (4, 20–24). CAFs have been shown to promote decreased sensitivity to gemcitabine in pancreatic cancer (25). Moreover, in non-small cell lung cancer, activation of AKT/Sox2 pathway by CAFs induced cancer cell resistance to chemotherapy (26). Given our recent findings that MSCs home to the TME in oral cavity and oropharyngeal cancer, collectively here referred to as oral squamous cell carcinoma (OSCC) and the recent reports of the role of MSCs in the context of chemotherapy resistance to platinum based agents, we sought to understand if crosstalk between MSCs and oral squamous cell carcinoma cells is mediated by PDGFRα/AKT signaling may be implicated in cisplatin resistance through changes in cancer cell apoptosis.
Methods
Cell Culture
Head and neck cancer cell lines JHU-012, JHU-019 (derived from human oropharyngeal tumors) and OKF-TERT1 human immortalized non-neoplastic oral keratinocyte cells (OKT) were generously provided by Dr. Vicente Resto (Galveston, TX). Cells were maintained in RPMI 1640 medium containing glutamine supplemented with 10% fetal bovine serum at 37°C in 5% CO2. Primary bone marrow-derived human mesenchymal stem cells (MSCs) were obtained from ATCC (Manassas, VA) and maintained according to the manufacturer's recommendations. MSCs were used between passages 2–5 and defined as early passage. The human OPSCC cell lines used in these studies have been extensively characterized both in vitro and in vivo (27, 28). For co-culture conditions, MSCs and HNSCC cell lines JHU-012, JHU-019, and negative OKT controls were grown in a 1:1 and supplemented in 1:1 ratio of appropriate culture media for 6 days.
Cell Viability, Apoptosis and Cell Proliferation
Cell viability was measured using the XTT cell viability kit (Cell Signaling Tech., 9095) in 96 well plates at 2 x 103 cells per well following manufacturer's protocol. Apoptosis was measured by flow cytometry analysis with the ANXA5/PE/7-AAD Apoptosis Detection Kit (BD Biosciences) at 1 x 106 cells per falcon tube. Prior to apoptosis detection, cells were stained with APC-anti-human CD326 (EpCAM) Clone:CO17-1A (Biolegend) to detect epithelial cells and PE/Cy7 anti-human CD90 (Thy1) Clone:5E10 to detect human MSCs. Cell acquisition was performed on BD LSRFortessa™ cell analyzer (BD Biosciences) at UTMB Flow Cytometry Core Facility in the Department of Microbiology and Immunology. Cellular proliferation was measured by BrdU - ELISA (colorimetric) assay (Abcam) in a 96 well plate at 2 x 103 cells per well with 4 h BrdU incubation period.
Transwell Studies
To determine if MSCs mediated alterations in cancer cell apoptosis required direct contact transwell co-culture assays were conducted as previously described (2). Briefly, 2.5 x 105 MSCs were seeded on the filter in the upper chamber of the transwell filter with a pore size of 8.0 μm in a final volume of 250 μl and 2.5 x 105 JHU-012 or OKT cells were seeded on the bottom chamber. Following 6 days of co-culture, apoptosis of the epithelial cells was measured as described above.
Clonogenic Assay
Clonogenic assay conditions were established as previously described (29). HNSCC cell line JHU-012 and −019 alone or in co-culture with MSCs, as described above, were seeded in 6-well plates at predetermined densities. Cells were allowed to settle and adhere for 24-h, prior to treatment with cisplatin. Cells were treated with varying doses of cisplatin ranging from 0 to 4 μM for 24 h. After 24-h treatment with cisplatin, the media was exchanged and with basal media and cultured 5 additional days. Cells were fixed with 3% crystal violet/10% formalin, imaged and counted using ImageJ.
Western Immunoblotting
OSCC cells lines JHU-012 and JHU-019 were cultured alone or co-cultured with MSCs at 1:1 ratio in media containing 50% of RPMI 1640 complete medium (Thermo Fisher Scientific, MA) and 50% of MSC Complete Medium (ATCC, VA) for 6 days. Cells were lysed in 1 x RIPA buffer (Cell Signaling Technology, MA) with protease inhibitor cocktail (Sigma-Aldrich, MA). Protein concentrations were measured using BCA protein assay (Thermo Fisher Scientific). Cell lysates (10 μg protein) were separated by NuPAGE 4–12% Bis-Tris protein gels (Thermo Fisher Scientific) and transferred onto PVDF membranes (Bio-Rad, CA). After blocking with 5% (w/v) nonfat dry milk in TBS-T, the membranes were incubated with primary antibodies for anti-PDGFR-α (phosphoY754; abcam #ab5460) p-AKT (Thr308) (#13038, Cell Signaling Technology), p-AKT (Ser473) (#4060, Cell Signaling Technology), total AKT (#4691, Cell Signaling Technology), and β-actin (#A1978, Sigma-Aldrich). Antibody dilution ranged from 1:500 to 1:1000. After three washes with TBS-T, the membranes were incubated with the appropriate secondary antibodies. Reactive protein bands were visualized using Alpha Innotech Fluorchem FC2 imaging system (Cell Bioscience, CA). The intensity of each protein band was quantified by Image J software (NIH).
Quantification of Chemokines
Cytokine production was measured in the supernatants of the conditioned media from OSCC cell lines JHU-012, and −019 Human XL cytokine discovery premixed kit (R and D Systems, FCSTM18, Minneapolis, MN) according to the manufacturer's instructions.
Statistical Analysis
Each experiment was with n ≥ 3 in triplicate, unless otherwise noted. All data were expressed as mean ± standard error of mean (SEM). Statistical analyses were generated using GraphPad Prism (GraphPad Software 7.0) or Excel 2013 (Microsoft Excel Spreadsheet Software 2013). For the comparison between two or more experimental groups, statistical significance was assessed via Student's t-test or two-way ANOVA with p-value < 0.05 being considered statistically significant. Dose response curves were generated using GraphPad Prism set with the following parameters: [drug] vs response variable slope (four parameters). Extra sum of squares F test was used to reject the null hypothesis with p < 0.05 being considered statistically significant. Each dose response curve was done in triplicate with n = 3.
Results
MSCs Activate a PDGF-AA/PDGFR-α Autocrine Loop in Oral Squamous Cell Carcinoma (OSCC)
To investigate whether MSCs induce changes in PDGF-AA/PDFGR-α expression with activation of downstream signaling targets in OSCC cells, JHU-012 and −019 were grown in 1:1 co-culture with early passage human MSCs for 6 days. The conditioned media from OSCC cells grown alone or in co-culture with MSCs was screened using a multiplex approach for cytokines previously shown to be upregulated in human cancers. Of the 45 cytokines assayed, only PDGF-AA (Figure 1A) and MCP-1 (Figure 1B) were found to be significantly increased the conditioned media from OSCC cells grown in co-culture compared to OSCC cells grown alone (p < 0.005). There was no appreciable secretion of PDGF-AA, MCP-1 in MSCs grown alone nor appreciable secretion of PDGF-AB/BB in either MSCs, OSCC cells grown alone, or in co-culture with MSCs (data not shown). To investigate whether increased production of PDGF-AA resulted in activation of PDGFR-α, the expression of phosphor-PDGFR-α was determined by Western immunoblotting. There was a significant increase in phosphorylation of PDGFR-α in JHU-012 (p < 0.02) and −019 (p < 0.006) co-cultured with MSCs compared to cancer cells grown alone (Figure 1C). We did not detect appreciable expression of total PDGFR-α by Western immunoblotting (Supplemental Figure 1).
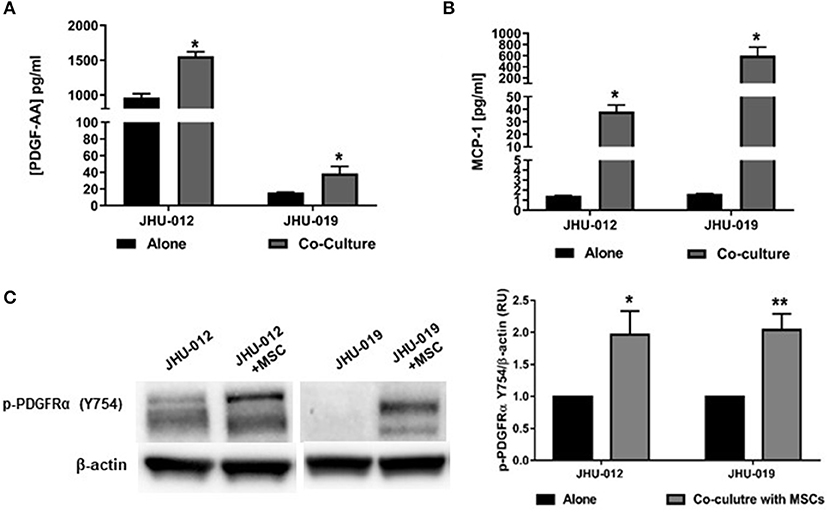
Figure 1. MSCs induce increased production of PDGF-AA and MCP-1 and Autocrine Activation of PDGFR-α in OSCC. JHU-012, and −019 were grown alone or in 1:1 co-culture with MSCs for 6 days and production of PDGF-AA and MCP-1 measured using a multiplex assay approach and activation of PDGFR-α determined by Western immunoblotting. There was a significant increase in the production of PDGFAA (A) and MCP-1 (B) in the conditioned media from cancer cells grown in co-cultured compared to cancer cells grown alone (n = 3, p < 0.005). (C) Following co-culture with MSCs, there was a significant increase in expression of p-PDGFR-α (Y754) in JHU-012 and −019 co-cultures (n = 3; *p < 0.02; **p < 0.006).
Activation of PDGFRs have previously been shown to be an important regulator of Akt activation. Therefore to determine if activation of MSCs induced activation of PDGFR-α in OSCC cells affects AKT expression, p-AKT was assessed by Western immunoblotting. Downstream p-AKT expression was found to be significantly increased in OSCC co-cultures of these same cancer cells lines at S473 (Figure 2A; p < 0.025) and T308 (Figure 2B; p < 0.02); suggesting the presence of MSCs in co-culture with JHU-012 and −019 results in activation of PDGFR-α/AKT mediated signaling pathways. To further confirm the specificity of activation of PDGFR-α in mediating changes in AKT expression, JHU-012 cells were grown alone and in co-culture with MSCs in the presence of PDGFR-α neutralizing antibody and isotype control (Figure 2C). Following treatment with PDGFR-α neutralizing antibodies, there was a significant decrease in p-PDGFR-α expression in JHU-012/MSC co-cultures (p < 0.03) with a concomitant significant decrease in p-AKT at S473 (p < 0.03) and at T308 (p < 0.03) compared to isotype control (Figure 2C). Further, we have preliminary evidence for high expression of levels of PDGFR-α on both tumor epithelial cells and tumor stromal cells in patients with advanced OSCC, further suggesting activation of autocrine PDGF-AA/PDGFR-α loop between MSCs and OSCC (30).
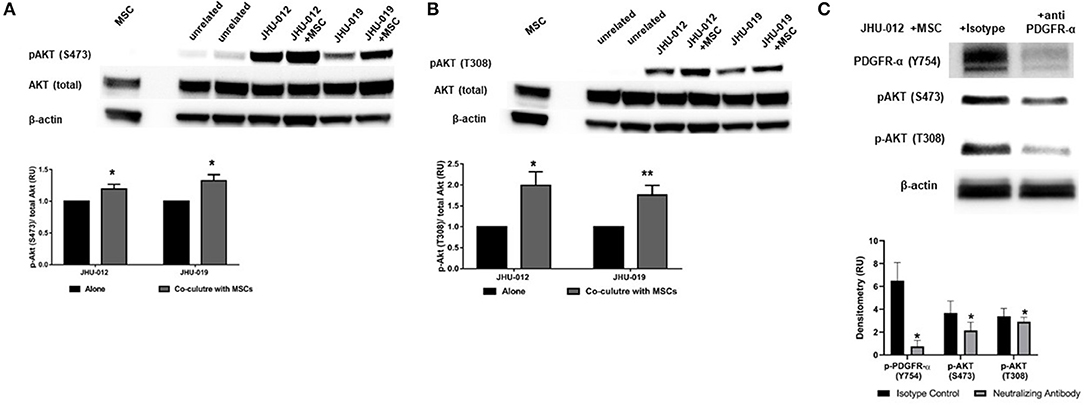
Figure 2. MSCs induce activation (phosphorylation) of PDGFR-α with downstream activation of AKT in OSCC. (A) Downstream activation of p-AKT levels were observed at serine 473 (S473) in co-cultures from JHU-012, and −019 compared to cancer cells grown alone. (n = 3; p < 0.025). (B) A more robust increase in p-AKT levels were also observed at threonine 308 (T308), corresponding to the catalytic site for AKT in JHU-012 and −019 grown in co-culture with MSCs (n = 3; *p < 0.02; **p < 0.007). (C) Inhibition of PDGFR-α results in decreased expression of activated AKT. JHU-012 were grown alone or in 1:1 co-culture with MSCs for 6 days in the presence of PDGFR- α neutralizing antibodies or isotype control and expression of p-PDGFR-α (Y754), pAKT (S473) and p-AKT (T308) measured by Western immunoblotting. There was a significant decrease in p-PDGFR-α, p-AKT (S473), and pAKT (T308) expression following inhibition of PDGFR-α neutralizing antibodies compared to isotype control (n = 3; p < 0.03).
MSCs Mediate Increased Expression of Bcl-2, Decreased Expression Bid and Reduced Apoptosis in OSCC
To determine if activation of AKT by MSCs results in alterations in apoptotic responses in OSCC cells, bcl-2 expression was measured by flow cytometry. Co-cultures were sorted based on expression of the epithelial lineage marker EpCAM and the MSC lineage marker CD90. Following 6-days of co-culture, there was a significant increase in the expression of bcl-2 in EpCAM+ JHU-012 and −019 cells grown in co-culture with MSCs (Figure 3A; p < 0.04). In addition, expression of the pro-apoptotic protein Bid was significantly reduced in JHU-012 and −019 cells co-cultured with MSCs compared to cancer cells grown alone (Figures 3B,C; p < 0.001). Further, there was a significant reduction in apoptosis in EpCAM+/annexin V+ JHU-012 and −019 co-cultures (Figure 3C; p < 0.001). These data suggest that MSCs reduce apoptotic response in OSCC cells in vitro by modulating expression of the Akt target bcl-2 in JHU-012 and -019.
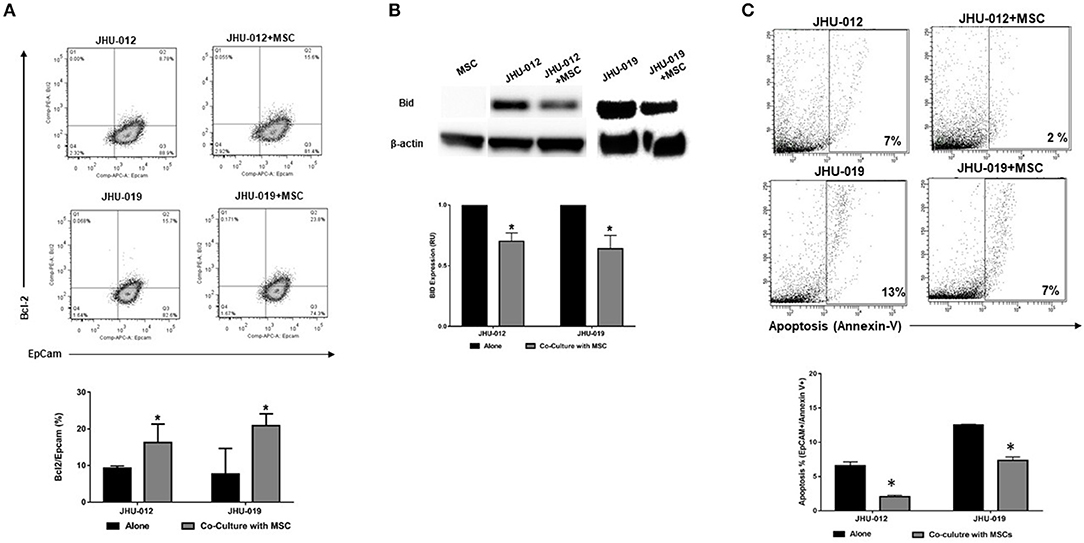
Figure 3. MSCs mediate increased expression of Bcl-2, decreased expression of Bid, and decreased apoptosis in OSCC. JHU-012, and −019 were grown in a 1:1 co-culture with MSCs for 6 days. Bcl-2, Bid and apoptosis expression measured. EpCAM was used to identify cells of epithelial lineage and CD90 used to identify mesenchymal cells. (A) There was a significant increase in the expression of bcl-2/EpCAM+ cells in co-cultures derived from JHU-012 and −019 compared to cancer cells grown alone (n = 3; p < 0.04). (B) There was a significant reduction in bid expression across in JHU-012 and −019 co-cultured with MSCs compared to cancer cells grown alone (n = 3; p < 0.001). (C) There was a significant reduction in the number EpCAM+/annexin V+ cancer cells undergoing apoptosis in the co-cultures derived from JHU-012 and JHU-019 compared to cancer cells grown alone (*p < 0.001, n ≥ 3).
MSCs Mediate Decreased Cytotoxicity to Cisplatin in OSCC
Cisplatin-mediated cytotoxicity occurs through crosslinking of DNA, resulting in loss of DNA repair and the initiation of apoptosis (31). Therefore, we hypothesized that MSC-mediated OSCC cell response to apoptosis may alter cancer cell cytotoxicity to cisplatin. Using a clonogenic assay approach, single clones were isolated from JHU-012 and −019 grown alone or derived from cancer cells grown in 1:1 co-culture with MSCs. Cells were treated with varying doses of cisplatin for 24-h, as previously described (32). There was a significant increase in the dose response curves and IC50 from clones derived from JHU-012 (Figure 4A; p < 0.001) and −019 (Figure 4B; p < 0.001) grown in co-culture with MSCs, signifying MSCs confer decreased cancer cells sensitivity to cisplatin. The IC50, a log-based value, for cisplatin in clones derived from JHU-012 grown in co-culture with MSCs was >2 times higher (0.9 vs. 0.4) than the IC50 in clones derived from JHU-012 grown alone (Figure 4A). While slightly lower at 1.6 times higher (0.5 vs 0.3), a similar significant trend in the IC50 for cisplatin was observed in clones derived from JHU-019 grown in co-culture with MSCs compared to JHU-019 grown alone (Figure 4B). Representative crystal violet staining patterns for cisplatin colongenic assays are depicted for JHU-012 and −019 are provided in Supplemental Figures 2A,B, respectively. In addition, following treatment with 0.5 μM cisplatin, JHU-012 cells grown in co-culture with MSCs were significantly less apoptotic with increased cell viability and proliferation compared to cancer cells grown alone (Figure 5, p < 0.0001). These data support that crosstalk between MSCs OSCC cells as a mechanism mediating altered cisplatin response in oral cancer as seen in other desmoplastic rich cancers (33).
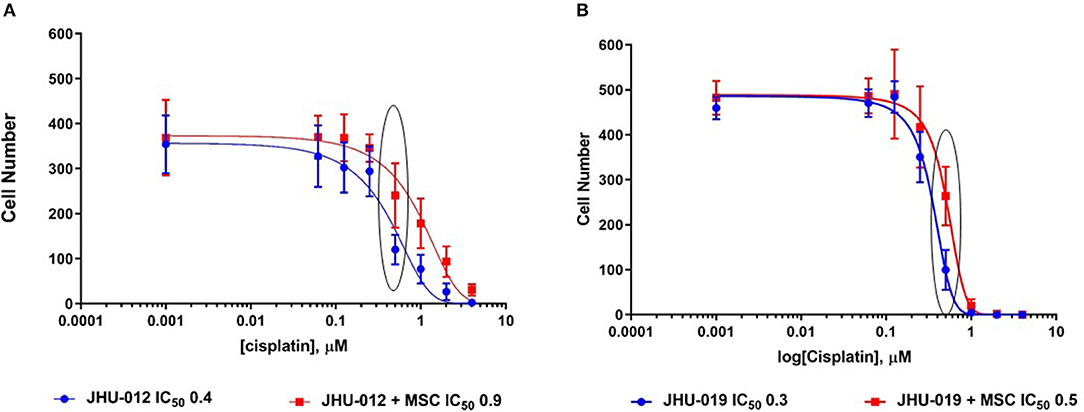
Figure 4. MSCs mediate decreased cytotoxicity to cisplatin in OSCC. Using a clonogenic assay approach, cisplatin dose response curves were generated for clones derived from JHU-012 and −019 cells grown alone and in 1:1 co-culture with MSCs. There was a significant increase in the cisplatin dose response curve and IC50 in clones derived from co-culture with MSCs for JHU-012 (A) and −019 (B; n > 3; p < 0.001).
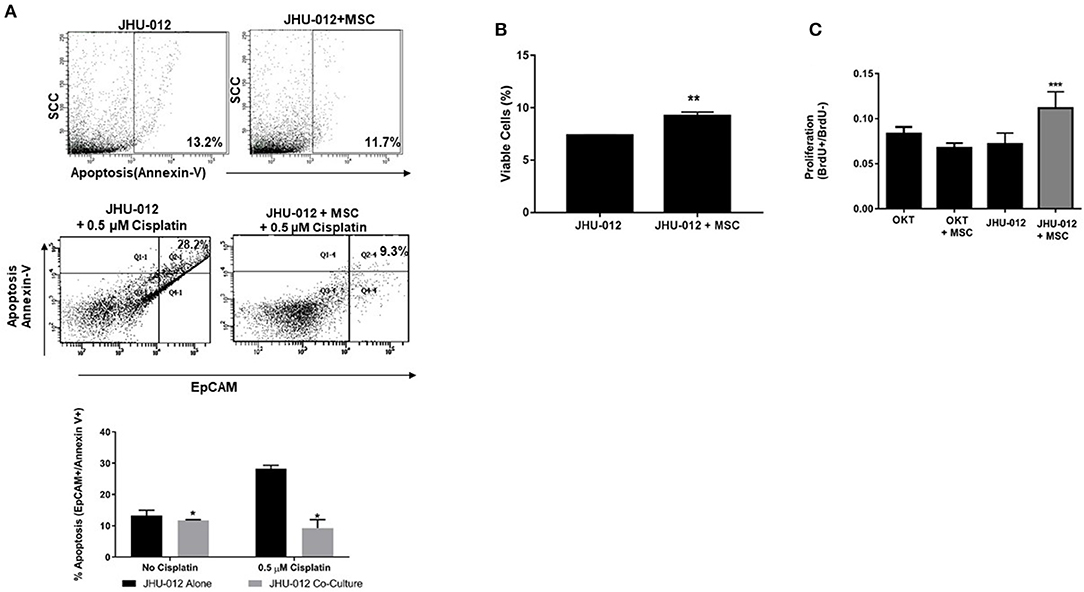
Figure 5. MSCs mediate decreased apoptosis with a concomitant increase cell viability and proliferation following 0.5 μM treatment with cisplatin in OSCC. JHU-012 cells grown alone or in 1:1 co-culture with MSCs were treated with 0.5 μM cisplatin and apoptosis, viability, and proliferation measured. (A) 1:1 co-culture of MSCs with JHU-012 resulted in a significant decrease in EpCAM+/annexin V+ cancer cells undergoing apoptosis compared to JHU-012 cells grown alone (*p < 0.0001, n = 3). (B) JHU-012/MSC co-cultures displayed enhanced viability compared to cancer cells alone (**p < 0.001, n = 3) and (C) cancer cell proliferation was significantly increased following cisplatin treatment in co-culture conditions compared to cancer cells grown alone and OKT negative controls grown alone or in co-culture with MSCs (***p < 0.026, n = 3).
Inhibition of PDGFR-α With Crenolanib Decreases Expression of Activated PDGFR-α and Improves Responsiveness to Cisplatin in OSCC
PDGFR-α expression was attenuated following treatment with 20 nM crenolanib reaching statistical significance at 200 nM crenolanib (Figures 6A,B; p < 0.04). We did not detect appreciable expression of total PDGFR-α by Western immunoblotting (Supplemental Figure 3). In clinical trials the dosing of crenolanib ranges between ~100 and 600 nM. Prior studies suggest 20 nM crenolanib is sufficient to inhibit FLT3 (type-III tyrosine kinase inhibitor) in acute myeloid leukemia (34). Therefore, we selected 20 nM crenolanib as a more physiologic relevant concentration to determine if pretreated with this monoclonal antibody would attenuate the increase in IC50 in JHU-012 cells grown in co-culture with MSCs. Cancer cell clones derived from JHU-012 co-cultures were treated with and without 20 nM crenolanib for 6 days and the dose response to cisplatin measured, as previously reported. There was a significant downward shift in the cisplatin dose response curve and a reduction in the IC50 by half (0.4 vs. 0.2) in JHU-012 (p < 0.0001) clones derived from co-cultures treated with 20 nM crenolanib compared controls (Figure 6C). Representative crystal violet staining patterns for cisplatin colongenic assays are depicted for JHU-012 are provided in Supplemental Figure 4.
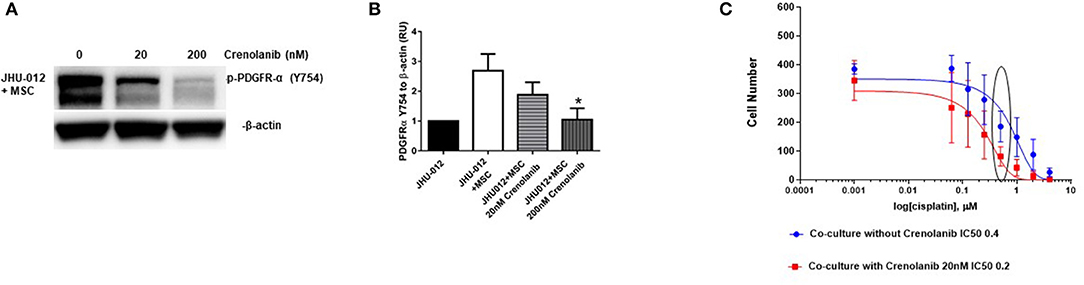
Figure 6. Inhibition of PDGFR-α with crenolanib decreases expression of activated PDGFR-α and improves responsiveness to cisplatin in OSCC. (A,B) JHU-012 were grown in 1:1 co-culture with MSCs in the presence and absence of the receptor tyrosine kinase inhibitor, crenolanib, for 6 days and PDGFR-α expression measured by Western immunoblotting. PDGFR-α expression was decreased following 20 nM treatment and 200 nM treatment with crenolanib. Expression levels were quantified by densitometry which revealed a significant decrease in PDGFR-α expression following treatment with 200 nM crenolanib (n = 3; *p < 0.04). Using a clonogenic assay, cisplatin dose response curves were generated from clones grown from JHU-012 (C) grown in 1:1 co-culture with MSCs with and without 20 nM crenolanib. There was a significant shift in the cisplatin dose response curve and the IC50 was reduced by half in JHU-012 (p < 0.0001) cancer cell clones treated with 20 nM crenolanib compared to untreated cancer cell clones (n ≤ 3).
Discussion
We demonstrate in this study a novel mechanism in OSCC cells induced by MSCs resulting in activation of PDGFR-α/AKT mediated signaling pathways resulting in decreased cancer cell apoptosis and reduced cytotoxicity to cisplatin. Moreover, we further show the activation of PDGFR-α mediated signaling pathways and alterations in cisplatin response induced by MSCs in OSCC cells can be overcome by the PDGFR inhibitor, crenolanib. Autocrine PDGFR-α expression has been shown to be upregulated in ovarian cancer and mammary cancer metastasis and expression levels correlated with tumor progression (35, 36). Matei et al. report 39% of ovarian tumors to be PDGFR-α positive and inhibition of the PDGF-PDGFFR axis with imatinab restricted ovarian cancer cell growth in vitro (35, 37). In addition, ovarian cancer cells transfected to constitutively express AKT were significantly more resistant to imatinab which could only be partially reversed with high concentrations of imatinab (37).
We recently reported that MSC-mediated chemotaxis in oral cancer cells is dependent upon cancer cell secretion of PDGF-AA acting on PDGFR-α+ MSCs (10). In that study, JHU-012 and −019 we did not detect PDFGR-α expression by Western immunoblotting, leading us to conclude paracrine signaling between OSCC cells and MSCs promotes MSC chemotaxis in vitro (10). The physiologic consequence of MSC chemotaxis in this setting was not known. However, in this study using co-culture of MSC with cancer epithelial cells, we conclude that MSCs induce activation of PDGFR-α on OSCC cells resulting in activation of AKT, reduced apoptotic response and decreased sensitivity to cisplatin. Our in vitro data is supported by ex vivo immunohistochemistry studies which show high expression of PDGFR-α on both tumor cells and TME stromal cells in patients with oral cancers (30), which has not been previously reported in this context. Ongkeko et al. describe high expression of PDGFR in head and neck specimens, however in review of their methods, antibodies staining was noted to be for PDGFR-β and the surrounding stromal cells appear to be PDGFR-β negative (38). This distinction is important as we previously reported inhibition of PDGF-β did not affect MSC chemotaxis by OSCC cells (10). Moreover, TME expression of PDGFR-α has been shown to correlate with a worse prognosis in patients with prostate, breast, ovarian, non-small cell lung cancer and osteosarcoma and we have preliminary data that suggests the TME PDGFR-α may bear prognostic significance in OSCC (4, 35, 39, 40), cancers previous described to have reactive stromal microenvironments implicated in disease progression (41, 42). Activation of PDGFR signaling pathways have been shown to play a significant role in desmoplastic response, which is a hallmark feature of OSCC. Studies are beginning to emerge suggesting bone marrow derived mesenchymal cells may be an important source of circulating stromal cells in TME (3, 9, 41) and autocrine activation of PDGFR-α has been shown to promote epithelial ovarian cancer cell proliferation and thought to play a role in metaplastic transformation of mullerian epithelium (35). Our observations are further supported by Ong et al. who recently reported overexpression of PDGFR-α mRNA was associated with advanced disease in patients with oral cavity squamous cell carcinoma (43, 44).
The clinicopathologic response for patients with advanced OSCC to conventional protocols including surgical excision, chemotherapy, and radiation is poor, suggesting that treatment strategies directed at tumor cells alone are inadequate. Single use biologic agents such as cetuximab have shown poor clinical efficacy, and recently, Braig et al. reported that cetuximab resistance is linked to a single polymorphism in the EGFR (k-allele), which is harbored by >40% of patients with HNSCC (45). Immunotherapy based targeted treatment for oral cancers is in its infancy, with pembrolizumab and nivolumab gaining approval in late 2017 for platinum refractory patients with advanced HNSCC (46).
Immunotherapy and treatments that target non-cancerous cells in the TME, such as MSCs and desmoplastic fibrocytes (i.e., CAFs), may improve clinical outcomes. Crenolanib is a small molecule tyrosine kinase inhibitor with that is effective against PDGFR-α and presently in clinical trials for patients with gastrointestinal stromal tumors (GIST) in which PDGFRA activation mutations confer resistance to imatinab (47). Use of receptor tyrosine kinase inhibitors like imatinib have been shown to improve responsiveness to doxorubicin through modulation of AKT signaling in head and neck cancer cells (48), suggesting small molecules like crenolanib may offer therapeutic value in patients with PDGFR-α+ oral cancers.
Activation of PDGR's leads to activation of several downstream events including the PI3K/AKT signaling pathway (49). The AKT pathway is frequently dysregulated in the setting of cancer, and activation of AKT results in an anti-apoptotic phenotype with increased expression of bcl-2 (50). We provide strong evidence that co-culture with MSCs results in increased expression of bcl-2 in oral cancer cells (Figure 2A), and these events appear to be mediated via activation of AKT at T308 and S473 (Figures 1B,C). Vincent et al. report that increased p-AKT at T308 bears prognostic significance in non-small cell lung cancer (51). Phosphorylation of T308 has been found to be essential for catalytic activity and S473 site required for maximal activation (52). In our model, we have found increase p-PDGFR-α and p-AKT at both sites for JHU-012 and -019.
Therefore, we hypothesized that disruption of the PDGF-AA/PDGFR-α signaling between OSCC cells and MSCs with the receptor tyrosine kinase inhibitor crenolanib, may restore cancer cell sensitivity to cisplatin. Crenolanib is presently in active clinical trials for both hematologic and solid tumors and has been shown to be 100-times more selective for PDGFR-α compared to other agents (34, 47, 53, 54). We show here that treatment with crenolanib blocks activation of PDGFR-α (Figure 6A) and pretreated with 20 nM crenolanib significantly reduces the cisplatin dose response OSCCC cells grown in co-culture with MSCs compared to untreated MSC oral cancer cell co-cultures (Figure 6C; p < 0.0001). Pretreatment with of MSC oral cancer co-cultures with crenolanib restored cisplatin sensitivity to that of cancer cells grown alone.
Given the widespread use of cisplatin in the management of advanced OSCC, understanding how MSCs mediate resistance to this agent has translational relevance. In addition to these novel findings with respect to MSCs contribution to cisplatin resistance through anti-apoptosis in oral cancers, we also demonstrate this resistance can be overcome by a small molecule PDGFR-α inhibitor crenolanib. The emerging role of MSCs in pathophysiology of cancer, metastasis, and drug resistance has translational relevance and may affect patient outcomes. We have demonstrated here that MSCs contribute to anti-apoptosis and resistance to cisplatin in OSCC in vitro. Our findings suggest selective targeting of TME MSCs may provide a viable treatment strategy to combat cisplatin resistance and warrants further mechanistic and translational study in OSCC.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
JW and RC: study design, collection and/or assembly of data, and data analysis. CC and RN: blinded pathologic grading of specimens. IP and DP: study design, data analysis and interpretation, manuscript review. TW: conception and study design, collection and assembly of data, data analysis and interpretation, manuscript preparation, and final approval.
Funding
The project described was supported by Award Numbers UL1TR000071/KL2TR000072, UL1TR001439 from the National Center for Advancing Translational Sciences, The Gromada Head and Neck Cancer Foundation, Cincinnati, OH, and K08 CA184915-01A1 from the NIH National Cancer Institute.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Jeffrey Myers, University of Texas MD Anderson Cancer Center, for his thoughtful review of this manuscript and mentorship on the project.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.00552/full#supplementary-material
Supplemental Figure 1. Total PDFGR-α expression. JHU-012, and −019 were grown alone or in 1:1 co-culture with MSCs for 6 days activation of PDGFR-α determined by Western immunoblotting. Total PDGFR-α was not detected by Western immunoblotting (n = 2).
Supplemental Figure 2. MSCs confer decreased cytotoxicity to cisplatin in OSCC. Representative photographs demonstrate crystal violet staining for JHU-012 (A) and JHU-019 (B) clonogenic assays as previously described and reported in Figure 4. Note the number of blue stained colonies within the wells. The black/violet staining clumps outside the wells are artifacts: crystallized crystal violet.
Supplemental Figure 3. MSC-induced cisplatin resistance is overcome by the PDGFR inhibitor, crenolanib in OSCC. Representative photographs demonstrate crystal violet staining for JHU-012 clonogenic assays following pretreatment with and without crenolanib as previously described and reported in Figure 5C.
Supplemental Figure 4. Total PDFGR-α expression. JHU-012 were grown alone or in 1:1 co-culture with MSCs were treated with either 0, 20, or 200 nM crenolanib for 6 days and activation of p-PDGFR-α determined by Western immunoblotting. Total PDGFR-α expression was not detected by Western immunoblotting at 0 and 20 nM crenolanib treatment (n = 2).
References
1. Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. (2007) 449:557–63. doi: 10.1038/nature06188
2. Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F, et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. (2011) 71:614–24. doi: 10.1158/0008-5472.CAN-10-0538
3. Ruscetti M, Quach B, Dadashian EL, Mulholland DJ, Wu H. Tracking and functional characterization of epithelial-mesenchymal transition and mesenchymal tumor cells during prostate cancer metastasis. Cancer Res. (2015) doi: 10.1158/0008-5472.CAN-14-3476
4. Bremnes RM, Dønnem T, Al-Saad S, Al-Shibli K, Andersen S, Sirera R, et al. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-Associated fibroblasts and non-small cell lung cancer. J Thor Oncol. (2011) 6:209–17. doi: 10.1097/JTO.0b013e3181f8a1bd
5. Kabashima-Niibe A, Higuchi H, Takaishi H, Masugi Y, Matsuzaki Y, Mabuchi Y, et al. Mesenchymal stem cells regulate epithelial-mesenchymal transition and tumor progression of pancreatic cancer cells. Cancer Sci. (2013) 104:157–64. doi: 10.1111/cas.12059
6. Mathew E, Brannon AL, Del Vecchio A, Garcia PE, Penny MK, Kane KT, et al. Mesenchymal stem cells promote pancreatic tumor growth by inducing alternative polarization of macrophages()(). Neoplasia. (2016) 18:142–51. doi: 10.1016/j.neo.2016.01.005
7. Curry JM, Sprandio J, Cognetti D, Luginbuhl A, Bar-ad V, Pribitkin E, et al. Tumor microenvironment in head and neck squamous cell carcinoma. Sem Oncol. (2014) 41:217–34. doi: 10.1053/j.seminoncol.2014.03.003
8. Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. (2008) 68:4331–9. doi: 10.1158/0008-5472.CAN-08-0943
9. Worthley DL, Si Y, Quante M, Churchill M, Mukherjee S, Wang TC. Bone marrow cells as precursors of the tumor stroma. Exp Cell Res. (2013) 319:1650–6. doi: 10.1016/j.yexcr.2013.03.006
10. Watts TL, Cui R, Szaniszlo P, Resto VA, Powell DW, Pinchuk IV. PDGF-AA mediates mesenchymal stromal cell chemotaxis to the head and neck squamous cell carcinoma tumor microenvironment. J Trans Med. (2016) 14:337. doi: 10.1186/s12967-016-1091-6
11. Liotta F, Querci V, Mannelli G, Santarlasci V, Maggi L, Capone M, et al. Mesenchymal stem cells are enriched in head neck squamous cell carcinoma, correlates with tumour size and inhibit t-cell proliferation. Br J Cancer. (2015) 112:745–54. doi: 10.1038/bjc.2015.15
12. Liu C, Feng X, Wang B, Wang X, Wang C, Yu M, et al. Bone marrow mesenchymal stem cells promote head and neck cancer progression through periostin-mediated phosphoinositide 3-kinase/Akt/mammalian target of rapamycin. Cancer Sci. (2018) 109:688–98. doi: 10.1111/cas.13479
13. Altomare DA, Testa JR. Perturbations of the aKT signaling pathway in human cancer. Oncogene. (2005) 24:7455–64. doi: 10.1038/sj.onc.1209085
14. Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, et al. Involvement of pI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. (2003) 17:590–603. doi: 10.1038/sj.leu.2402824
15. Maji S, Panda S, Samal SK, Shriwas O, Rath R, Pellecchia M, et al. Bcl-2 antiapoptotic family proteins and chemoresistance in cancer. Adv Cancer Res. (2018) 137:37–75. doi: 10.1016/bs.acr.2017.11.001
16. Reed JC. Dysregulation of apoptosis in cancer. J Clin Oncol. (1999) 17:2941–53. doi: 10.1200/JCO.1999.17.9.2941
17. Gallo O, Boddi V, Calzolari A, Simonetti L, Trovati M, Bianchi S. bcl-2 protein expression correlates with recurrence and survival in early stage head and neck cancer treated by radiotherapy. Clin Cancer Res. (1996) 2:261–7.
18. Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. (2003) 22:7265–79. doi: 10.1038/sj.onc.1206933
19. Park SB, Lee JH, Jeong WW, Kim YH, Cha HJ, Joe Y, et al. TTP mediates cisplatin-induced apoptosis of head and neck cancer cells by down-regulating the expression of bcl-2. J Chemother. (2015) 27:174–80. doi: 10.1179/1973947814Y.0000000234
20. Adorno-Cruz V, Kibria G, Liu X, Doherty M, Junk DJ, Guan D, et al. Cancer stem cells: targeting the roots of cancer, seeds of metastasis, and sources of therapy resistance. Cancer Res. (2015) 75:924–9. doi: 10.1158/0008-5472.CAN-14-3225
21. Hagiwara M, Kikuchi E, Tanaka N, Kosaka T, Mikami S, Saya H, et al. Variant isoforms of cD44 involves acquisition of chemoresistance to cisplatin and has potential as a novel indicator for identifying a cisplatin-resistant population in urothelial cancer. BMC Cancer. (2018) 18:113. doi: 10.1186/s12885-018-3988-3
22. Prince ME, Ailles LE. Cancer stem cells in head and neck squamous cell cancer. J Clin Oncol. (2008) 26:2871–2875. doi: 10.1200/JCO.2007.15.1613
23. Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. (2011) 19:257–72. doi: 10.1016/j.ccr.2011.01.020
24. Roodhart JM, Daenen LG, Stigter EC, Prins HJ, Gerrits J, Houthuijzen JM, et al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell. (2011) 20:370–83. doi: 10.1016/j.ccr.2011.08.010
25. Li XY, Hu SQ, Xiao L. The cancer-associated fibroblasts and drug resistance. Eur Rev Med Pharmacol Sci. (2015) 19:2112–9.
26. Zhang Q, Yang J, Bai J, Ren J. Reverse of non-small cell lung cancer drug resistance induced by cancer-associated fibroblasts via a paracrine pathway. Cancer Sci. (2018) 109:944–55. doi: 10.1111/cas.13520
27. Szaniszlo P, Fennewald SM, Qiu S, Kantara C, Shilagard T, Vargas G, et al. Temporal characterization of lymphatic metastasis in an orthotopic mouse model of oral cancer. Head Neck. (2014) 36:1638–47. doi: 10.1002/hed.23500
28. Moser R, Xu C, Kao M, Annis J, Lerma LA, Schaupp CM, et al. Functional kinomics identifies candidate therapeutic targets in head and neck cancer. Clin Cancer Res. (2014) 20:4274–88. doi: 10.1158/1078-0432.CCR-13-2858
29. Skinner HD, Sandulache VC, Ow TJ, Meyn RE, Yordy JS, Beadle BM, et al. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin Cancer Res. (2012) 18:290–300. doi: 10.1158/1078-0432.CCR-11-2260
30. Clement C, Nawigi R, Watts TL. Does the tumor stroma bear prognostic significance in oral cancers. In: 10th International Conference on Head and Neck Cancer. Chicago, IL (2020).
31. Pendleton KP, Grandis JR. Cisplatin-Based chemotherapy options for recurrent and/or metastatic squamous cell cancer of the head and neck. Clin Med Insights Ther. (2013) 2013:10409. doi: 10.4137/CMT.S10409
32. Sano D, Matsumoto F, Valdecanas DR, Zhao M, Molkentine DP, Takahashi Y, et al. Vandetanib restores head and neck squamous cell carcinoma cells' sensitivity to cisplatin and radiation in vivo and in vitro. Clin Cancer Res. (2011) 17:1815–827. doi: 10.1158/1078-0432.CCR-10-2120
33. Izumiya M, Kabashima A, Higuchi H, Igarashi T, Sakai G, Iizuka H, et al. Chemoresistance is associated with cancer stem cell-like properties and epithelial-to-mesenchymal transition in pancreatic cancer cells. Anticancer Res. (2012) 32:3847–53.
34. Galanis A, Ma H, Rajkhowa T, Ramachandran A, Small D, Cortes J, et al. Crenolanib is a potent inhibitor of fLT3 with activity against resistance-conferring point mutants. Blood. (2014) 123:94–100. doi: 10.1182/blood-2013-10-529313
35. Matei D, Emerson RE, Lai YC, Baldridge LA, Rao J, Yiannoutsos C, et al. Autocrine activation of pDGFRalpha promotes the progression of ovarian cancer. Oncogene. (2006) 25:2060–2069. doi: 10.1038/sj.onc.1209232
36. Jechlinger M, Sommer A, Moriggl R, Seither P, Kraut N, Capodiecci P, et al. Autocrine pDGFR signaling promotes mammary cancer metastasis. J Clin Invest. (2006) 116:1561–70. doi: 10.1172/JCI24652
37. Matei D, Chang DD, Jeng MH. Imatinib mesylate (Gleevec) inhibits ovarian cancer cell growth through a mechanism dependent on platelet-derived growth factor receptor alpha and akt inactivation. Clin Cancer Res. (2004) 10:681–90. doi: 10.1158/1078-0432.CCR-0754-03
38. Ongkeko WM, Altuna X, Weisman RA, Wang-Rodriguez J. Expression of protein tyrosine kinases in head and neck squamous cell carcinomas. Am J Clin Pathol. (2005) 124:71–6. doi: 10.1309/BTLN5WTMJ3PCNRRC
39. Shinagawa K, Kitadai Y, Tanaka M, Sumida T, Onoyama M, Ohnishi M, et al. Stroma-directed imatinib therapy impairs the tumor-promoting effect of bone marrow-derived mesenchymal stem cells in an orthotopic transplantation model of colon cancer. Int J Cancer. (2013) 132:813–23. doi: 10.1002/ijc.27735
40. Yu YC, Yang PM, Chuah QY, Huang YH, Peng CW, Lee YJ, et al. Radiation-induced senescence in securin-deficient cancer cells promotes cell invasion involving the iL-6/STAT3 and pDGF-BB/PDGFR pathways. Sci Rep. (2013) 3:1675. doi: 10.1038/srep01675
41. Barron DA, Rowley DR. The reactive stroma microenvironment and prostate cancer progression. Endocr Relat Cancer. (2012) 19:R187–204. doi: 10.1530/ERC-12-0085
42. Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, et al. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE. (2009) 4:e4992. doi: 10.1371/journal.pone.0004992
43. Ong HS, Gokavarapu S, Tian Z, Li J, Xu Q, Cao W, et al. PDGFRA mRNA is overexpressed in oral cancer patients as compared to normal subjects with a significant trend of overexpression among tobacco users. J Oral Pathol Med. (2017) 46:591–7. doi: 10.1111/jop.12571
44. Ong HS, Gokavarapu S, Tian Z, Li J, Xu Q, Zhang CP, et al. PDGFRA mRNA overexpression is associated with regional metastasis and reduced survival in oral squamous cell carcinoma. J Oral Pathol Med. (2018) 47:652–9. doi: 10.1111/jop.12713
45. Braig F, Kriegs M, Voigtlaender M, Habel B, Grob T, Biskup K, et al. Cetuximab resistance in head and neck cancer is mediated by eGFR-K521 polymorphism. Cancer Res. (2017) 77:1188–99. doi: 10.1158/0008-5472.CAN-16-0754
46. Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, et al. Nivolumab for recurrent squamous-Cell carcinoma of the head and neck. N Engl J Med. (2016) 375:1856–67. doi: 10.1056/NEJMoa1602252
47. Heinrich MC, Griffith D, McKinley A, Patterson J, Presnell A, Ramachandran A, et al. Crenolanib inhibits the drug-resistant pDGFRA d842V mutation associated with imatinib-resistant gastrointestinal stromal tumors. Clin Cancer Res. (2012) 18:4375–84. doi: 10.1158/1078-0432.CCR-12-0625
48. Chu TS, Chen JS, Lopez JP, Pardo FS, Aguilera J, Ongkeko WM. Imatinib-mediated inactivation of akt regulates aBCG2 function in head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. (2008) 134:979–84. doi: 10.1001/archotol.134.9.979
49. Yokota JUN, Chosa N, Sawada S, Okubo N, Takahashi N, Hasegawa T, et al. PDGF-induced pI3K-mediated signaling enhances the tGF-β-induced osteogenic differentiation of human mesenchymal stem cells in a tGF-β-activated mEK-dependent manner. Int J Mol Med. (2014) 33:534–42. doi: 10.3892/ijmm.2013.1606
50. Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase aKT pathway in human cancer. Nat Rev Cancer. (2002) 2:489–501. doi: 10.1038/nrc839
51. Vincent EE, Elder DJ, Thomas EC, Phillips L, Morgan C, Pawade J, et al. Akt phosphorylation on thr308 but not on ser473 correlates with akt protein kinase activity in human non-small cell lung cancer. Br J Cancer. (2011) 104:1755–61. doi: 10.1038/bjc.2011.132
52. Hart JR, Vogt PK. Phosphorylation of aKT: a mutational analysis. Oncotarget. (2011) 2:467–76. doi: 10.18632/oncotarget.293
53. Ekpe-Adewuyi E, Lopez-Campistrous A, Tang X, Brindley DN, McMullen TP. Platelet derived growth factor receptor alpha mediates nodal metastases in papillary thyroid cancer by driving the epithelial-mesenchymal transition. Oncotarget. (2016) 7:83684–700. doi: 10.18632/oncotarget.13299
Keywords: PDGFR-α, AKT, oral cancer, crenolanib, apoptosis, mesenchymal stem cells, cisplatin
Citation: Wang J, Cui R, Clement CG, Nawgiri R, Powell DW, Pinchuk IV and Watts TL (2020) Activation PDGFR-α/AKT Mediated Signaling Pathways in Oral Squamous Cell Carcinoma by Mesenchymal Stem/Stromal Cells Promotes Anti-apoptosis and Decreased Sensitivity to Cisplatin. Front. Oncol. 10:552. doi: 10.3389/fonc.2020.00552
Received: 26 November 2019; Accepted: 27 March 2020;
Published: 28 April 2020.
Edited by:
Changshun Shao, Soochow University, ChinaReviewed by:
Hamid Morjani, Université de Reims Champagne-Ardenne, FranceAnantha Koteswararao Kanugula, Northeast Ohio Medical University, United States
Copyright © 2020 Wang, Cui, Clement, Nawgiri, Powell, Pinchuk and Watts. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tammara L. Watts, dGFtbWFyYS53YXR0c0BkdWtlLmVkdQ==