 Deborah P. Lavin
Deborah P. Lavin Vijay K. Tiwari
Vijay K. Tiwari- The Wellcome-Wolfson Institute for Experimental Medicine, School of Medicine, Dentistry and Biomedical Science, Queen's University Belfast, Belfast, United Kingdom
Epithelial to mesenchymal transition (EMT) is the process whereby a polarized epithelial cell ceases to maintain cell-cell contacts, loses expression of characteristic epithelial cell markers, and acquires mesenchymal cell markers and properties such as motility, contractile ability, and invasiveness. A complex process that occurs during development and many disease states, EMT involves a plethora of transcription factors (TFs) and signaling pathways. Whilst great advances have been made in both our understanding of the progressive cell-fate changes during EMT and the gene regulatory networks that drive this process, there are still gaps in our knowledge. Epigenetic modifications are dynamic, chromatin modifying enzymes are vast and varied, transcription factors are pleiotropic, and signaling pathways are multifaceted and rarely act alone. Therefore, it is of great importance that we decipher and understand each intricate step of the process and how these players at different levels crosstalk with each other to successfully orchestrate EMT. A delicate balance and fine-tuned cooperation of gene regulatory mechanisms is required for EMT to occur successfully, and until we resolve the unknowns in this network, we cannot hope to develop effective therapies against diseases that involve aberrant EMT such as cancer. In this review, we focus on data that challenge these unknown entities underlying EMT, starting with EMT stimuli followed by intracellular signaling through to epigenetic mechanisms and chromatin remodeling.
Introduction
Epithelial to mesenchymal transition (EMT) is a dynamic reversible process and a fundamental mechanism occurring during embryogenesis and organ development, wound healing and fibrosis, and cancer (1–3). The reverse process (MET; mesenchymal-epithelial transition) is vital for secondary tumor formation and also occurs during embryogenesis with multiple rounds of EMT and MET required for correct formation of complex organs and specialized cellular structures (4, 5). Therefore, understanding EMT is not only beneficial in terms of developing novel cancer therapies, but enables us to fully elucidate the mechanisms behind wound healing and organ fibrosis—processes implicated in diabetic complications (6, 7), heart disease (8), and immunocompromised conditions like cystic fibrosis (9).
Gene regulation controls whether a gene is expressed or silenced, and encompasses transcription factors, epigenetic modifications, chromatin remodeling, higher order chromatin structures (such as looping), and non-coding RNAs e.g. microRNAs (miRNAs) and long non-coding RNAs (lncRNAs). In EMT, transcription factors such as Snail, Zeb1/Zeb2, and Twist, bind to specific promoter and enhancer DNA sequences and work in unison with epigenetic regulators (such as G9a and LSD1) and chromatin remodeling machinery to drive transcription of pro-mesenchymal genes and prevent epithelial gene transcription (3, 10). The chromatin remodelers determine how accessible these DNA sequences are (11); therefore, they determine gene expression during both developmental and disease-related EMT. Like EMT, epigenetic modifications—such as DNA or histone methylation, and histone acetylation—are also reversible, and have been shown to play a pivotal role in EMT regulation, which makes them attractive targets for novel chemotherapeutics (12). Interactions between key EMT transcription factors and enzymes that modify DNA/histones have been reviewed in detail (3, 10, 13, 14).
Nucleosomes (histone octamers) are the basic packaging unit of DNA—the “beads on a string”—which confer 5- to 10-fold compaction of the genome and form arrays that fold hierarchically into higher-order (50-fold and higher) chromatin structures (15). These structures, which include multiple chromatin loops, can repress gene transcription in both stem cells and adult cancer cells, for example the TF GATA4 (16). In addition, PcG proteins and repressive chromatin methylation marks work together to preserve these loops (16). The role of chromatin looping in EMT however is unknown.
Non-coding RNAs represent another layer of epigenetic regulation owing to the susceptibility of their promoters to epigenetic modifications (10, 17). Much is known about the role of non-coding RNAs, such as the miRNA-200s family of microRNAs, in EMT (10, 18–20), with recently published data also showing the involvement of microRNAs [miRNA-151a (21)] and long non-coding RNAs [MYOSLID (22)] in partial EMT. Conversely, the long non-coding RNA NKILA which is upregulated by TGFβ, suppressed TGFβ-induced EMT and tumor metastasis in vivo through negative regulation of downstream TGFβ signaling (23). All of these elements add another layer of complexity and culminate in highly intricate gene regulation.
Unknowns in each area of gene regulation in EMT compromise not only our fundamental understanding of these mechanisms but interfere with our knowledge of EMT pathogenesis. Without this information, we cannot develop critically needed cancer therapeutics targeting EMT, as in addition to driving tumorigenesis and metastasis, EMT confers chemoresistance and helps tumor cells evade destruction by the immune system (24). Advances in the field of regenerative medicine (i.e., cellular reprogramming to restore organ functionality) also rely on deciphering these unknowns. Chemotherapeutics that inhibit DNA methylation (e.g., 5–aza-2′-deoxycytidine, Guadeticabine), histone deacetylation (e.g., Vorinostat, Mocetinostat), and interfere with recognition of acetylated lysine residues (e.g., BRD4 inhibitors such as JQ1, MS417), are promising as they, respectively, restore epithelial phenotypes/reactivate tumor suppressor proteins (10, 13, 25), reduce growth/antagonize Zeb1-mediated miRNA-203 repression (10, 26), and suppress the MYC TF, invasion, and tumorigenicity (10, 25). While these targeted therapies may have a synergistic effect with platinum-based chemotherapies and may sensitize cancer cells to therapies that induce DNA damage (26), these inhibitors are not perfect due to potentially adverse activation of otherwise latent genes, and their somewhat limited effect on solid tumors (10). Here, we discuss unknown epigenetic entities in the gene regulatory network underlying EMT.
Thinking Outside the Cell—Novel EMT Stimuli
Extracellular stimuli are the initiating factors that drive signaling and cellular effects and are often the first point of regulation in disease; pharmacological antagonism of deleterious stimuli or their receptors is often the first treatment option or the only option if the mechanism through which the stimulus mediates its effects are unknown. For example, administration of anti-VEGF is routinely performed in diabetic retinopathy and certain cancers including breast, colorectal, and cervical (27–29). The role of TGFβ in EMT was first shown 25 years ago (30) and is still widely reported; a keyword search for “TGFβ EMT” returns 19,585 results in PMC (27th Sept 2019). Other stimuli known to induce EMT (Figure 1) include epidermal growth factor (EGF) (31, 32), fibroblast growth factor (FGF) (33), hepatocyte growth factor (HGF) (34, 35), vascular endothelial growth factor (VEGF) (36–38), insulin-like growth factor (IGF) (39, 40), WNT (41), Sonic Hedgehog (SHH) (42, 43), BMPs [BMP-2 (44, 45); BMP-4 (46, 47)], TNF-α (48, 49), and hypoxia (32) with the latter thought to promote EMT via epigenetic regulation of DICER; the enzyme involved in miRNA processing (50).
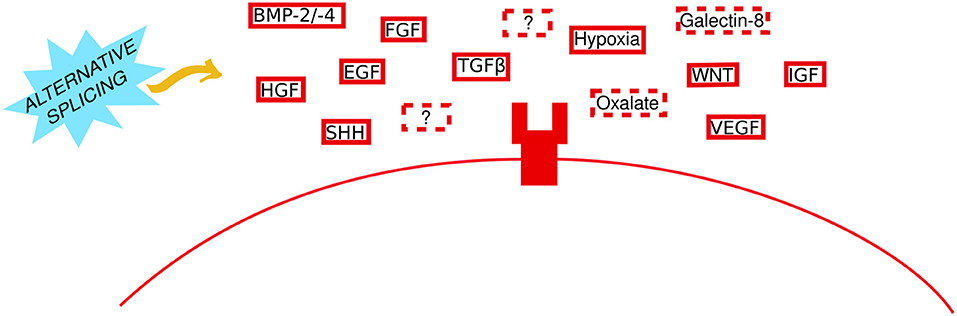
Figure 1. Thinking outside the cell in EMT. Established and novel extracellular stimuli bind to their particular receptors to initiate EMT. A generic transmembrane receptor is shown here for illustrative purposes. Solid and dashed red boxes represent established and novel stimuli, respectively. Question marks represent unknown stimuli. The yellow arrow indicates that the role of alternative splicing (sky blue spiked shape) remains to be investigated.
In recent years, researchers have begun to move away from the idea that only growth factors can stimulate EMT. Novel non-growth factor stimuli, which have been implicated in EMT stimulation, include oxalate and Galectin-8. Oxalate—a routine metabolic by-product—and calcium oxalate—whose deposition (Microcalcification type I) is often seen in benign non-malignant breast tissue (51, 52)—induced EMT both in cultured renal cells and in vivo (53, 54). Similarly, oxalate-treated mice presented with highly aggressive undifferentiated mammary tumors and in vitro oxalate induced breast epithelial cell proliferation and expression of the tumorigenic gene c-fos (55). Calcium oxalate mediates its effect via activation of p38/MAPK (56), and oxalate-induced EMT could be prevented by activation of nuclear factor erythroid-derived 2-related factor 2 (NRF2) signaling (57). Galectin-8—a widely-expressed glycan binding protein—stimulated partial EMT; tumors arising from Galectin-8 overexpression bore a mesenchymal phenotype whilst still expressing E-cadherin and maintaining cell polarity (58). Mechanistically Galectin-8 activated FAK to transactivate the EGFR and increased expression of matrix-degrading enzymes (uPA, MMP-13) (58). Importantly the authors elude to the potential involvement of other signaling pathways in facilitating Galectin-8 EMT-inducing effects. It is possible that Galectin-8 may interact with calcium signaling to increase FAK—similar to novel pro-EMT TRPV4 signaling (59) (discussed further in the next section)—or it may require vimentin expression in order to increase uPA (60), or perhaps it relies on integrin αVβ3 signaling to activate FAK (61). Loss of the receptor Neogenin 1 (a member of the DCC/Frazzled/UNC-40 family) also induced partial EMT in intestinal epithelial cells, via PI3K and MAPK signaling, and extracellular matrix (ECM)-receptor interactions (62).
Despite the novel EMT stimuli above, there are still unanswered questions. It is possible that there are other seemingly harmless metabolic by-products and binding proteins that stimulate EMT. How oxalate promotes pro-cancerous gene expression, and whether it interacts with HATs or other histone modifying enzymes remain to be investigated. Knowing that these stimuli can induce EMT is just the first step in understanding how non-canonical signals can cause EMT. In addition, deciphering the cellular markers and epigenetic signatures associated with partial EMT is crucial. Despite the similarities in the step wise process (the cadherin switch, for example) and with the endpoint ultimately unchanged, it is conceivable that for example, hypoxia-induced EMT may be different from TGFβ-induced EMT in terms of the underlying epigenetic signature and responsiveness to treatment; Cursons et al. showed that where the primary EMT stimulus was hypoxia, treatment directed cells to acquire a more aggressive mesenchymal phenotype (32). Thus, if we were to develop and evaluate therapeutics that selectively target different EMT stimuli as opposed to common downstream targets, perhaps this could enable us to minimize pathological conditions and/or adverse effects which would translate to prognostic differences in patients. Having said this, pre-clinical research of EMT-specific therapeutics is ongoing and yielding promising data. Regorafenib—a multi-kinase inhibitor pre-approved as a second-line treatment in metastatic CRC—displayed potent anti-EMT effects on invasion and metastasis in vitro and in vivo by inhibiting TGFβ-induced STAT3 phosphorylation (required for Twist1 and Zeb1 activation) (63).
Antibody-mediated approaches are among the frontrunners in anti-EMT therapy. For example, an anti-MMP9 antibody—which targets an enzyme involved in EMT-mediated ECM remodeling in cancer—increased survival and reduced metastatic burden and the EMT marker vimentin in an in vivo model of pancreatic cancer (64). Similarly, in an in vivo breast cancer model a monoclonal antibody that blocks active MMP9 inhibited spontaneous and experimentally induced lung metastases (65). Clinical trials using an anti-MMP9 antibody (Adecaliximab) alone or in combination therapies are ongoing. It is hoped that these agents will perform better than previously tested broad-spectrum MMP9 inhibitors, which failed in clinical trials due to dose-limiting side effects (64).
In prostate cancer a monoclonal antibody targeting N-cadherin reduced invasion, metastasis, and proliferation of cancer cells as well as Akt activity (66). However, as N-cadherin is expressed in the heart, peripheral nerves, and liver, adverse off-target effects are a huge concern (66).
It is assumed that upregulation of E-cadherin would be beneficial in cancer, given its downregulation in many cancers. In some instances, this is true; in RCC cell lines small activating RNA stimulated E-cadherin expression and subsequently inhibited cell migration and invasion, and downregulated vital pro-EMT genes such as Zeb1 and vimentin (67). However, E-cadherin was required for invasive ductal carcinoma cell survival and metastasis in multiple in vivo models of breast cancer (68), finally explaining why E-cadherin is often observed in patients with breast cancer. These data also highlight the potential pitfalls associated with assumed pro-/anti-EMT markers. In addition, such therapeutics can be limited by poor tissue penetration and short biological half-lives (69), and antibody therapeutics are often very expensive to produce and may not be covered by an individual's health insurance.
Despite copious research papers that detail microRNAs that promote or inhibit cancer development, the translation of these data to tangible microRNA drugs is lacking. While no Phase 3 trials are ongoing in this area, new candidates (like RGLS5579 for glioblastoma) are entering the early phases of clinical trials (70), and targeted inhibition of microRNA-155 and microRNA-21 has successfully resensitized tumors to chemotherapy in lung and breast cancers, respectively (71, 72).
EMT therapeutics that repurpose existing FDA-approved drugs is most exciting, as it significantly reduces the translational time of such therapeutics. A comprehensive bioinformatics analysis of pre-approved drugs has proposed and validated various drug combinations, like IKBK and SRC kinase inhibition together with HDAC inhibition, to hamper EMT (73). Often, scientists researching novel EMT drug candidates focus on preventing an epithelial cells transition to an invasive mesenchymal cell, or inducing MET to revert to a normal epithelial state. However, MET may in fact enhance metastatic outgrowth (74, 75). Recently, Ishay-Ronen and colleagues demonstrated a novel method of preventing cancer progression that exploits the plasticity of cancer cells (76). By transdifferentiating post-EMT mesenchymal cancer cells into functional post-mitotic adipocytes—using a combination of pre-approved drugs (Rosiglitazone and a MEK inhibitor)—primary tumor growth and metastasis was repressed (76). Importantly, epithelial cells were immune to adipocyte transdifferentiation (76), highlighting the specificity of this targeted therapeutic. Other novel EMT therapeutics include Antrodia salmonea—a fungus indigenous to Taiwan and known for its anti-cancer properties (77). In vitro, Antrodia salmonea significantly reduced invasion and reversed EMT by modulating NFκB and WNT/β-catenin signaling, and in vivo treatment with Antrodia salmonea reduced breast cancer cell-induced lung metastasis (77).
In Tune With EMT Intracellular Signaling
EMT is dependent on the concomitant activity of multiple signaling pathways (Figure 2), which are constantly firing and fluctuating, with turnover time for signaling molecules sometimes infinitesimal [e.g., the half time of HIF-1α clearance is 3–6 min in normoxia (78)]. Positive and negative feedback loops are also incorporated into intracellular signaling pathways, which also rarely work independently and often regulate each other; TGFβ can promote HIF-α expression independent of PI3K by encouraging HIF-1α/HIF-2α translation (79). Downstream TGFβ signaling is the predominant intracellular signaling pathway in EMT. However, as detailed above different stimuli utilize TGFβ signaling and other pathways such as NFκB, YAP/TAZ (Hippo pathway), PI3K/Akt, ERK/JNK, Wnt, Notch, and JAK/STAT3.
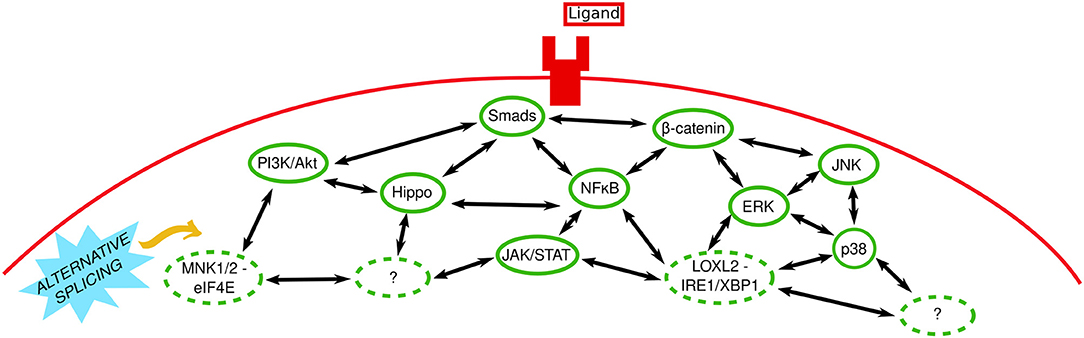
Figure 2. In tune with EMT intracellular signaling. Extracellular stimuli bind to their receptors (shown here as a generic ligand binding a generic transmembrane receptor) and signal via canonical and non-canonical intracellular signaling pathways. Solid and dashed green ovals represent established and novel signaling pathways, respectively. Two-headed black arrows indicate inter-pathway interaction and regulation. Question marks represent unknown pathways involved. The yellow arrow indicates that the role of alternative splicing (sky blue spiked shape) remains to be investigated.
While there are copious research papers and reviews detailing the separate roles of these pathways in EMT [see (24, 80–85)], a systems biology approach—or simply data-sharing between groups working on different pathways—would facilitate further elucidation of the intricate nature of signaling pathways and their molecules in EMT. For example, Akt-mediated NFκB activation has been implicated in EMT (86) and our lab performed comprehensive genome-wide analysis of the active open—transcription facilitating—chromatin mark H3K27ac and subsequent gene expression in mammary epithelial cells undergoing EMT (87). We discovered that ERK signaling was essential for epigenetic reprogramming underlying characteristic EMT-related gene expression and phenotypic changes; ERK inhibition prevented the loss and gain of H3K27ac at epithelial and mesenchymal genes (i.e., repression and expression, respectively) (87).
Unraveling inter-pathway regulation in EMT and deciphering what signaling pathways are required for different stages/steps of EMT (e.g., initiation, progression, maintenance, the cadherin switch, loss of cell polarity and other phenotypic alterations) is of vital importance. Recently, the DEPTOR (DEP domain containing mTOR interacting) protein which normally inhibits mTOR signaling, stimulated partial EMT in HCC cells by activating TGFβ-Smad3/4-Snail signaling through mTOR inhibition (88). Similarly, in vitro mimicking of ECM organization showed that the way the ECM is arranged could trigger partial or full EMT (89). This process was dependent on YAP activation and subsequent YAP-mediated feedback; Wilms Tumor-1-YAP mediated E-cadherin downregulation, and YAP-TRIO-Merlin-mediated Rho GTPase regulation, which, respectively, loosened cell-to-cell contacts and increased cell migration, compromising epithelial sheet integrity, facilitating EMT, and promoting invasion (89). The location of this phenomenon, i.e., several cells away from the sheet edge, or at the sheet edge itself, determined whether EMT was partial or full, respectively (89). Conversely, single-cell RNA sequencing of untreated epithelial cells vs. those treated with TGFβ showed continuous waves of gene regulation in EMT as opposed to discrete “partial” stages (90). The authors showed that deleting/interrupting key signaling moieties or events can cause cells to build-up at regulatory “checkpoints” that mimic “partial” stages, and enrich a particular pattern of gene expression in said cells which creates the impression of a stable intermediate phenotype (90).
It is also essential that the roles of various signaling pathways—like the aforementioned FAK signaling involved downstream of Galectin-8 (58)—in mediating global transcriptional changes through epigenetic regulation, which are required for EMT, be clarified. That said there are publications that are beginning to decipher these complex interactions. Of note, our laboratory was among the first to report TGFβ and JNK signaling in tandem in EMT; TGFβ signaling via canonical Smads was required for EMT initiation whereas JNK signaling was necessary for EMT to progress to fruition (91). Similarly, an Australian group showed that non-canonical ERK1/2 was needed for initiation but not progression of TGFβ-induced EMT in the rodent eye (92).
The importance of calcium signaling in EMT has gained considerable momentum in recent years, not only because of the discovery of calcium oxalate as a novel EMT stimuli, but because the Ca2+-binding protein calreticulin (CRT) was shown to be increased by—and able to regulate—TGFβ-induced EMT in vitro (93). Similarly, the TRPV4 ion channel was shown to mediate calcium-dependent downregulation of E-cadherin in breast cancer via activation of Akt signaling (59). Given the fundamental importance of calcium signaling, there are obvious caveats to widespread calcium chelation/calcium channel blockade—similar to widespread DNA methyltransferase (DNMT) inhibition. That said the development of highly specific calcium chelators has the potential to not only reduce EMT but to potentially do so with minimal adverse effects on the patient. The unknown here is whether the beneficial effects on EMT are (a) due to calcium blockade alone or off-target effects on other signaling pathways, and (b) enough to outweigh any pro-EMT effects that may arise from calcium blockade. For example, in addition to reducing E-cadherin TRPV4 activation also augmented FAK phosphorylation (similar to Galectin-8, above), reduced β-catenin and fibronectin-1, and increased expression of the cytoskeletal protein talin-1 (59), an essential protein in maintaining the structural integrity of focal adhesions (94). How TRPV4 induces EMT when it reduces β-catenin and fibronectin, and the key EMT genes TRPV4 regulates to cause EMT without β-catenin and fibronectin, are still elusive.
Non-canonical interactions and lesser-known signaling pathways in EMT are an additional unknown. For example, interactions between TGFβ and the cytoskeleton are emerging (95, 96), with the histone demethylase (HDM) JMJD5 required for cytoskeletal stabilization and inhibition of TGFβ-induced migration in lung cancer cells (95). In terms of lesser-known signaling pathways that are functional in EMT, MNK1/2-eIF4E signaling (97), LOXL2-IRE1/XBP1 signaling (98), and miRNA-20a-FBXL5/BTG3 signaling (99) are examples of seemingly isolated signaling pathways that have recently been implicated in EMT, yet other remote pathways might be involved. Inhibition of MNK1/2-eIF4E signaling using a novel retinamide yielded an impressive trifecta of anti-growth effects, inhibition of EMT, and androgen receptor signaling (97). Overexpressed LOXL2 (lysyl oxidase-like 2) accumulates in the endoplasmic reticulum (ER) where it forces detachment of heat shock protein 5A (HSP5A) from IRE1/XBP1 to activate the IRE1/XBP1 signaling pathway, which confers expression of notable EMT-TFs such as Snail, Zeb2, and Slug in an XBP1-dependent manner (98). However, given the fact that IRE1 inhibition can block XBP1-mediated TF expression (98), more research is needed to decipher this mechanism. FBXL5/BTG3 are direct miRNA-20a targets that are silenced to enable miRNA-20a-mediated EMT and invasion (99), however the precise role of FBXL5 signaling in EMT requires further research; FBXL5 overexpression reduced Snail protein levels (100), but also supported tumorigenesis by negatively regulated PTEN while promoting PI3K/Akt and mTOR phosphorylation and expression (101). The above data on FBXL5 agrees with previous research in our lab, where we discovered that FBXO32—another F-box protein—was pivotal in upstream EMT regulation not only in tumor metastasis, but in neuronal development also (102). FBXO32 directly ubiquitinated CtBP1 causing its stabilization and nuclear retention, which created a suitable microenvironment for EMT progression by facilitating epigenetic remodeling and transcription of CtBP1 target genes (102). In line with these findings, FBXO32 was amplified in metastatic cancers, and its depletion in vivo inhibited tumor growth and metastasis (102). These data highlight how important it is for scientists today to challenge pathways that were believed to be futile in EMT as well as those that are routine.
Factoring in Transcriptional Regulation in EMT
Transcription factors play important roles in cell-fate decisions, and a hierarchy of TF signals may influence whether a cell is epithelial, intermediate, or mesenchymal. Similarly, TFs may operate in a dose-dependent manner, for example, Twist may be required in nanomolar concentrations whereas Snail may be required in micromolar concentrations; indeed, in an EMT time course Twist was transiently detected whereas neither Snail nor Slug were significantly detected (103). Similarly, SNAIL proteins were shown to promote EMT with different potencies in human mammary epithelial cells (104). This is a novel exciting area of research; however, it is beyond the scope of this review. As the intermediaries between cellular signaling and chromatin, these DNA binding proteins guide the epigenetic machinery to their target sites which facilitates chromatin landscape changes at promoter and enhancer elements to drive downstream activation and repression of mesenchymal and epithelial genes, respectively (3, 10). Much is known about the core TFs involved in EMT (Snail, Zeb1/2, and Twist) but a large number of TFs are required for EMT.
A vast amount of EMT research has focused on TF involvement in the latter stages of EMT, but until recently, little was known about the transcriptional networks that trigger EMT. Newly published data shows that EMT involves a temporal hierarchy of collaborative transcriptional networks (105). This predicted network operates between TFs and between TFs and microRNAs and is composed of 46 (co)transcription factors and 13 miRNAs that were critically required for EMT in NMuMG cells (105). We are also beginning to decipher the requirement of different TFs for EMT initiation, maintenance, and progression (Figure 3). For example, the TF NRF2 delays the transition of a cell to a full mesenchymal phenotype; it maintains the hybrid epithelial/mesenchymal (partial/intermediate EMT) state (106). Similarly, research from our lab has shown that JNK-induced TFs and subsequent signaling are not required for EMT initiation, but was essential for progression of phenotypic hallmarks of EMT (91). Our lab has identified novel JNK-induced TFs that are required for EMT, are highly expressed in invasive cancer cells, and induced during neuronal development (91). These data are pivotal in the fight against EMT because the authors do not examine these novel TFs individually in EMT; they also define their role(s) in EMT that occurs in neuronal development.
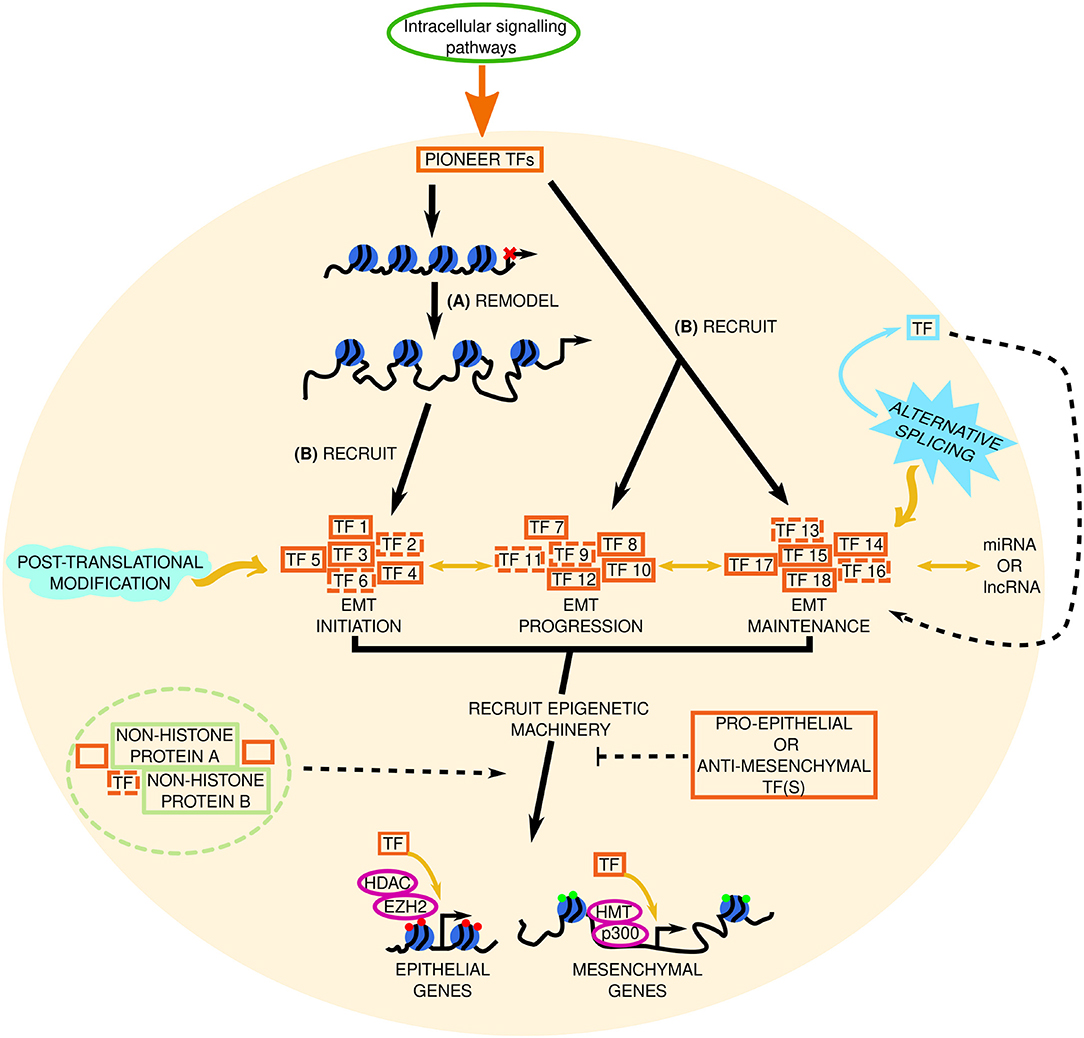
Figure 3. Factoring in transcriptional regulation in EMT. The signal from intracellular signaling pathways is transduced to pioneer transcription factors (TFs) in the nucleus (represented by the beige circle). We hypothesize that in EMT these pioneer TFs: (A) remodel the chromatin to increase accessibility at EMT initiating genes, and (B) recruit a set of TFs needed for EMT initiation (TF 1–TF 6, dashed lines indicate potentially unknown TFs), and recruit sets of TFs involved in EMT progression (TF 7–TF 12) and maintenance (TF 13–TF 18), respectively, to already accessible chromatin. Through subsequent interactions with epigenetic machinery (such as histone methyltransferases [HMT, e.g., EZH2, enhancer of zeste 2; and enzymes involved in histone acetylation (p300) or deacetylation (histone deacetylase, HDAC)] these TFs repress or promote epithelial and mesenchymal gene expression, respectively, to determine the cell phenotype. Red and green circles at these genes denote repressive or active chromatin marks, respectively. Pro- or anti-EMT TFs may inhibit this. Solid and dashed orange boxes represent established and novel TFs, respectively. Two-headed black arrows indicate inter-pathway interaction and regulation. Yellow arrows and black dashed lines indicate key questions that remain to be investigated. The roles of alternative splicing (sky blue spiked shape) and post translational modifications (mint green wave-like shape) in EMT TF regulation are also elusive. Potential TF-non-histone protein complexes are found in the light green dashed circle. lncRNA, long non-coding RNA; miRNA, micro RNA.
An unknown in this area, is the existence of TFs that do more than just control gene expression—perhaps TFs can perform secondary functions, such as chromatin remodeling (Figure 3), or mediate epigenetic regulation in EMT. In terms of TFs that exhibit auxiliary properties, our lab identified NeuroD1 as a pioneer factor; it recognizes and binds to its target sites and drives transcription of neuronal/EMT-related genes (such as Hes6 and Dll3, Snai2 and Twist1, respectively) regardless of the chromatin state (open or closed) or cell type (e.g., ESCs and differentiated fibroblasts) (107, 108). NeuroD1 is not alone in its pioneer TF abilities; FOXA TFs (FOXA1, FOXA2, FOXA3) (109, 110) and GATA3 (109) are also pioneer factors that determine cell fate. Heat shock factor 1 (HSF1) which induces HSPs is another pioneer factor with a secondary function in recruiting DNMT3A to suppress miRNA-137 and promote carcinogenesis (111), and perhaps HSF1 is also involved in HSP5A-mediated regulation of LOXL2-IRE1/XBP1 signaling. Despite these advances, the specific involvement of pioneer TFs in EMT in cancer remains to be seen.
TF that mediate epigenetic alterations are coming to the forefront of the field; Zeb2 employs DNA methylation to repress RAB25—a small Rab GTPase with a potential role in epithelial polarity—and E-cadherin, and increased SIRT-1-mediated H3K9 deacetylation at both promoters to maintain this suppression (112). FOXD3 both permits and prevents H3K27ac of regulatory elements and acts as a pioneer factor during neural crest (NC) development to prime NC factors (e.g., snai1b, twist1b), acts upstream of pro-EMT factors, and during NC EMT FOXD3 mediates a cadherin switch (decreased N-cadherin and increased cadherin 7) to modulate cell adhesion (113, 114). BPTF (bromodomain PHD finger TF), a TF which is also the largest subunit of the NURF chromatin remodeling complex [a member of the ISWI remodeling family (11, 115)], was increased in HCC where it inferred poor survival and correlated with high vimentin and low E-cadherin expression (116). These data show us that TFs are more than a “one trick pony”—they can influence the epigenetic signature of a cell. It was accepted that all relevant EMT-TFs had been identified but given the discovery of novel EMT-TFs in recent years and the fact that we still can't resolve EMT, perhaps there are unknown EMT-TFs (capable of altering a cells epigenetic state) and unknown ENT-TF targets that are yet to be discovered. Sox4 was known to play a role in EMT (117), but it was not until the following year that its direct transcriptional targets—like EZH2, fibronectin, N-cadherin—and role as a master regulator of EMT were elucidated (118). EZH2 in turn suppresses the tumor suppressor hepatocyte nuclear factor 1β (HNF1B) which normally represses Slug (119). Similarly, Sox8-mediated chemoresistance and stemness in tongue squamous cell carcinoma was only recently shown to involve Frizzled-7/Wnt/β-catenin signaling (120).
Perhaps alternative splice isoforms of existing TFs are yet to be discovered. For example, splice variant 1 (SV1) is a truncated isoform of KLF6 that has no zinc finger domains, yet it promotes tumor migration, and is thought to stimulate EMT in HCC (121). Hepatocyte nuclear factor 4α (HNF4α) has alternative isoforms that both stimulate (HNF4α7, 8, 9) and repress (HNF4α1, 2, 3) cancer development (121–123). We also cannot rule out the possibility of alternative TF splice isoforms in epithelial and mesenchymal cells. Their existence and how they influence cell signaling in EMT remain elusive. Indeed, extracellular ligands, DNA/histone modifying enzymes, and chromatin remodeling proteins may also be subjected to alternative splicing in EMT. Non-TF proteins could also be subjected to alternative splicing, to enable pro-EMT TFs. Recently, exon skipping in FLNB (an actin-binding protein) was shown to induce EMT by releasing the pro-EMT TF FOXC1 (124). This alternatively spliced isoform of FLNB also correlated with EMT gene signatures in basal breast cancer samples (124).
In addition, there may be TFs that we believe are fully elucidated, and so are not examined in the context of EMT. For example, GLI TFs are the primary mediators of developmental Hedgehog-GLI signaling pathway, which is mostly inactive in adults (except for in stem cells) (125), but GLI inhibition was found to block EMT in pancreatic cancer stem cells through reversion of the typical EMT cadherin switch and blockade of EMT-TFs (126). Together these data make the HH-GLI pathway an attractive—previously unthought-of—target in EMT therapy and add to the rationale for examining TFs in other developmental pathways, but it has taken over a decade for such pathways to become a focus of therapeutics—which is perplexing given that TFs necessary for development are often implicated in EMT.
We also cannot rule out the possibility of TFs forming fixed complexes with epigenetic proteins or non-histone proteins in EMT. For example, the non-histone chromatin protein HMGA1 forms a complex with the TF FOXM1 which stabilizes FOXM1 in the nucleus and increases expression of shared target genes like VEGFA to promote angiogenesis, inferring a negative prognosis (127). This is not the only HMG protein implicated in EMT; the pro-EMT role of HMBG-1 is discussed below. Perhaps TFs are epigenetically altered in EMT to silence or amplify their effect. Take MYC-GATA3/ESR1 TFs for instance; MYC overexpression increased MYC enrichment and reduced active histone marks (H3K27ac, H3K4me1/3) at regulatory elements for GATA3 and ESR1, resulting in cellular dedifferentiation i.e., a stem cell-like state (128). These data were validated in breast cancer patients where there was an inverse relationship between MYC overexpression and GATA3/ESR1 gene levels (128). It is plausible that there are other TFs that interact like this in EMT.
Epigenetic Mechanisms Involved in EMT—Room to Explore
Histones are the primary component of chromatin. Positioning DNA around histone octamers—nucleosomes—to form chromatin is essential to maintain the integrity of the genome; chromatin prevents DNA strand entanglement, but also dynamically regulates DNA replication and gene expression (129, 130). Extracellular stimuli, intracellular pathways, and transcription factors are subject to variation between patients and between cancers, and chromosomal instability promotes cancer (131), but the DNA is the same in all cells of the organism (132) and mechanisms and regulators of chromatin condensation are well-known (133). This makes chromatin the ultimate platform for action. Epigenetic—“above genetic”—modifications alter the genetic read-out of a cell, and include DNA methylation, histone modifications, and chromatin higher order structures (such as looping), which in collaboration with chromatin remodelers determine chromatin accessibility.
Establishing the epigenetic state involves epigenetic readers, writers, and erasers which recognise, create, or remove epigenetic modifications respectively (134). A patient's epigenetic state is therefore both dynamic and stable, and is involved not only in disease pathogenesis but in their response to treatment and their rates of disease-free survival and recurrence (135–138). Therefore, epigenetic regulators are prime therapeutic targets, and understanding epigenetic mechanisms of regulation would pave the way for novel therapeutics and personalized cancer medicine. We know that EMT-TFs guide the epigenetic machinery to target promoters/enhancers, but we do not know the mechanism behind this; i.e., how cells decide which loci are targeted during EMT initiation, progression, and maintenance.
Single-cell analysis has shown that during development progenitor cells display epigenetic heterogeneity (139). Specifically, the pre-EMT and delaminating crest (undergoing EMT) generate migrating progenitor cells whose heterogeneity is associated with transcriptional properties of early genes from competing downstream cell fates, which are activated when NC cells undergo EMT (139). The authors also note that mesenchymal potential may be established as early as delamination; lone overexpression of Twist1 was able to drive trunk NC toward a mesenchymal fate rather than the traditional neuronal fate (139). These data provide us with a better understanding of developmental NC EMT which in turn aids our understanding of NC-derived cancers such as melanoma and glioma, and perhaps such heterogeneity is present in other cancers (139, 140).
Epigenetic regulation of EMT (Figure 4) has mainly focused on histone methylation/acetylation, with additional studies detailing the roles of other histone modifications such as phosphorylation (141–143), ubiquitination (144–146), citrullination (85, 147–149), SUMOylation (150–152), and biotinylation (153) in EMT. Histone modifying enzymes are dynamic and diverse. Established enzymes [reviewed in (3, 10)] are involved in DNA methylation (DNMTs), histone (de)methylation (HMTs, HDMs), histone (de)acetylation (HATs, HDACs), and ubiquitination (E3 ubiquitin ligases). Despite the wealth of knowledge regarding these enzymes, we have much to learn. For example, class I HDAC enzymes generally promote tumorigenesis, and class IIA may promote or impede cancer development; however it was only recently discovered that HDAC5 induced anti-proliferative or pro-EMT effects depending on the cell line in which it was overexpressed (154).
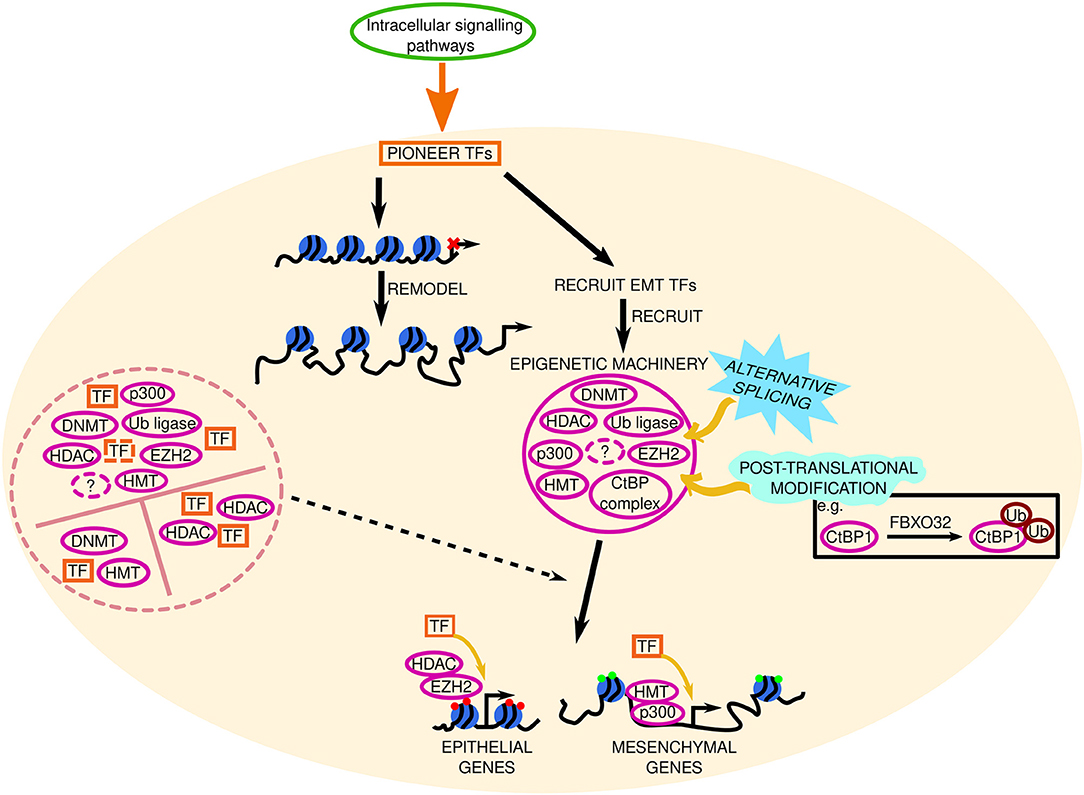
Figure 4. Epigenetic mechanisms involved in EMT. The signal from intracellular signaling pathways is transduced to pioneer transcription factors (TFs) in the nucleus (represented by the beige circle). These pioneer TFs remodel the chromatin and recruit EMT TFs to accessible chromatin, which subsequently recruit and interact with epigenetic machinery (solid pink circle), such as DNA methyltransferases (DNMT), histone methyltransferases (HMT, e.g., EZH2, enhancer of zeste 2; ubiquitinating enzymes, Ub ligase; CtBP, C-terminal binding protein; complex, and enzymes involved in histone acetylation (p300) or deacetylation (histone deacetylase, HDAC) to target loci. Together, TFs and epigenetic machinery repress or promote epithelial and mesenchymal gene expression, respectively, to determine the cell phenotype. Red and green circles at these genes denote repressive or active chromatin marks, respectively. Solid and dashed pink circles represent established and novel epigenetic machinery, respectively. Black dashed lines indicate key questions that remain to be investigated. The roles of alternative splicing (sky blue spiked shape) and post translational modifications (mint green wave-like shape) in EMT epigenetic machinery are also elusive. FBXO32 is an ubiquitin ligase. Question marks represent unknown epigenetic machinery. Potential TF-epigenetic machinery complexes are contained within the pink dashed circle.
Unsurprisingly a number of these enzymes contain methyl-lysine binding motifs, which curiously differ between enzymes; DNMT3A contains a PWWP domain to read H3K36me3 (155), ankyrin repeats in the histone methyltransferase (HMT) G9a enable H3K9me1/2 binding (156), and a double Tudor domain exists in the HDM JMJD2A for H3K4me3 and H4K20me3 binding (157). Even though these binding motifs are crucial for correct chromatin recognition—needed for enzymatic effects on chromatin methylation state—the importance of these different domains in EMT remains unknown; we do not know why there are three distinct methyl-lysine binding motifs among histone modifying enzymes, or whether they play a causal role in EMT development or not.
PWWP domains are distinct from the catalytic domain in DNMT3A (155). Missense mutations in the PWWP domain (W330R) abolish H3K36me2/3 binding and cause DNA hypermethylation at Polycomb-regulated regions in patients and in vivo (158). This is associated with reduced H3K27me3 and H3K4me3 bivalent marks, and modified chromatin accessibility at key developmental regulators (e.g., SOX and HOX family members, FOXA1, NEUROG1/2) (158). The implications for such mutations in EMT are still unknown. Perhaps mutations in the PWWP domain of DNMT3A confer increased methylase activity and allow the mutant to methylate polycomb regions, thus preventing PRC2 binding and EZH2-mediated H3K27me3 at epithelial genes as suggested by Heyn et al. (158). Alternatively, maybe methylase or other DNA recognition/binding motif activity is decreased upon PWWP mutation, allowing mesenchymal gene expression. The PWWP domain might even interact with non-histone proteins in EMT; however whether this is strengthened or weakened in mutant DNMT3 remains to be investigated. Whether mutant DNMT3A has enhanced or diminished recruitment by the aforementioned pioneer factor HSF1 also remains to be seen. Studies that examine human cancers for the presence of these and other PWWP mutations would be worthwhile, as the PWWP binding motif is highly conserved in human hepatoma-derived growth factor (HDGF) which is overexpressed in a number of human cancers and is involved in PI3K signaling (159). We cannot rule out a causative—or at the very least a contributing—role for PWWP domain mutations in EMT development in cancer, thus deciphering the unknowns that still surround the PWWP domain is of great importance.
The methyl-lysine binding motif in the HMT G9a contains ankyrin repeats; the rationale behind ankyrin repeats over a PWWP domain is unknown. Perhaps each motif bestows different effects on epigenetic regulation of EMT. In EMT G9a interacts with Snail which recruits it to the CDH1 promoter (160). Enhanced in hypoxia, G9a activity is beneficial to cancer cell survival and G9a can methylate non-histone proteins (161), like the ATPases Pontin and Reptin (162). The functional importance of the ankyrin repeats was only recently discovered. Levels of H3K9 mono/di/tri-methylation were analogous in ESCs expressing either wild-type G9a, G9a bearing a mutation in ankyrin repeats, or G9a lacking ankyrin repeats (163). However, the HMT activity of G9a/GLP is dependent on the ankyrin repeats (164). These data illustrate that these enzymes are more than just their catalytic activity. In terms of G9a-Snail interactions in EMT, the role of the ankyrin repeats in facilitating non-histone protein (e.g., E-cadherin) methylation required elucidation. It is plausible that there are G9a variants—like the variant that skips exon 10 (165)—that are alternatively spliced in exons pertaining to the ankyrin repeats, which may be present and causal in EMT. In line with this, a novel splice isoform of the arginine methyltransferase PRMT1 which lacks exons 8/9 (that encode for the dimerization arm, essential for enzymatic activity) was increased in cancer cell lines and induced by Snail (166). Similarly, PRMT9 elicited pro-invasive and metastatic effects via Snail and the PI3K/Akt pathway, which was validated in clinical HCC samples (167). Given the high stability of histone modifications in bodily fluids (168), methylated arginine residues could be developed as a novel prognostic biomarkers/therapeutic targets.
The role of Tudor domains (found in the HDM JMJD2A) in epigenetic regulation of EMT is yet another unknown. In addition to recognizing and binding H3K4me3 and H4K20me3, the two tandem Tudor domains in JMJD2A bind H3K9me3 and H4K20me2 which confer roles for JMJD2A in initiating and preventing transcription (169). As there are two Tudor domains, these conflicting functions might be localized to a specific domain, or the two Tudor domains might have opposing effects on EMT. Further work is required to determine whether JMJD2A has a pro- or anti-EMT effect given its ability to silence and active genes. Given the aforementioned involvement of fellow Jumonji family member JMJD5 in inhibition of TGFβ-induced migration in lung cancer cells (95), it is plausible that JMJD2A has a similar anti-EMT effect, however this claim warrants further research. In addition, to stabilize chromatin binding and mediate its epigenetic effects—and oncogenic effects—JMJD2A must be SUMOylated at K471 (170). We know that lysine residues can accept different epigenetic modifications, but there is no hard and fast rule that the enzymes that perform these histone/DNA modifications are exempt from modification themselves; For example, phosphorylation of G9a on Ser569 is crucial for its recruitment to damaged DNA (171).
Remodeling Our Understanding of Chromatin Machinery in EMT
Chromatin remodeling can involve nucleosome repositioning, swapping, displacement or even translocation to a fragment on a neighboring strand of DNA, as well as histone replacement (11, 172). In addition to roles in nucleosome positioning and DNA dependent biological processes such as repair and replication (173), remodelers are crucial determinants of chromatin accessibility and subsequent gene expression (11). Four major chromatin remodeling families are characterized; SWI/SNF, ISWI, CHD, and INO80 (11). Genes encoding components of SWI/SNF (e.g., BRG1) are among the most common targets of mutation in cancer; ~20% of human tumors were shown to contain mutations in at least one member of this complex (11). Despite this alarming statistic, mechanisms of epigenetic regulation by chromatin remodeling proteins and histone chaperones—in particular, how such regulation may be detrimental to our health, as is the case in EMT-related tumorigenesis—are largely overlooked.
Known chromatin remodelers with functional roles in EMT (Figure 5) have been reviewed see (174) and include the NuRD subunits MTA1 and MTA3 within the CHD family, the BPTF subunit of the ISWI family member NURF (116), and the BAF250/ARID1, BRG1, and hBRM subunits of the mammalian SWI/SNF complex (174, 175). Interestingly, the combined loss of BAF250A/ARID1A and gain of expressed mutated PI3K subunit PIK3CAH1047R results in partial EMT in the endometrial epithelium (176). Resistance to ER antagonists in breast cancer was recently attributed to loss of ARID1A, which reduced HDAC1 activity and increased H4K acetylation—sensitizing the cancer cells to BRD4 inhibition (177). Nucleophosmin 1 (NPM1) is another well-studied histone chaperone [reviewed by (178)] with a role in EMT/invasion (179–181).
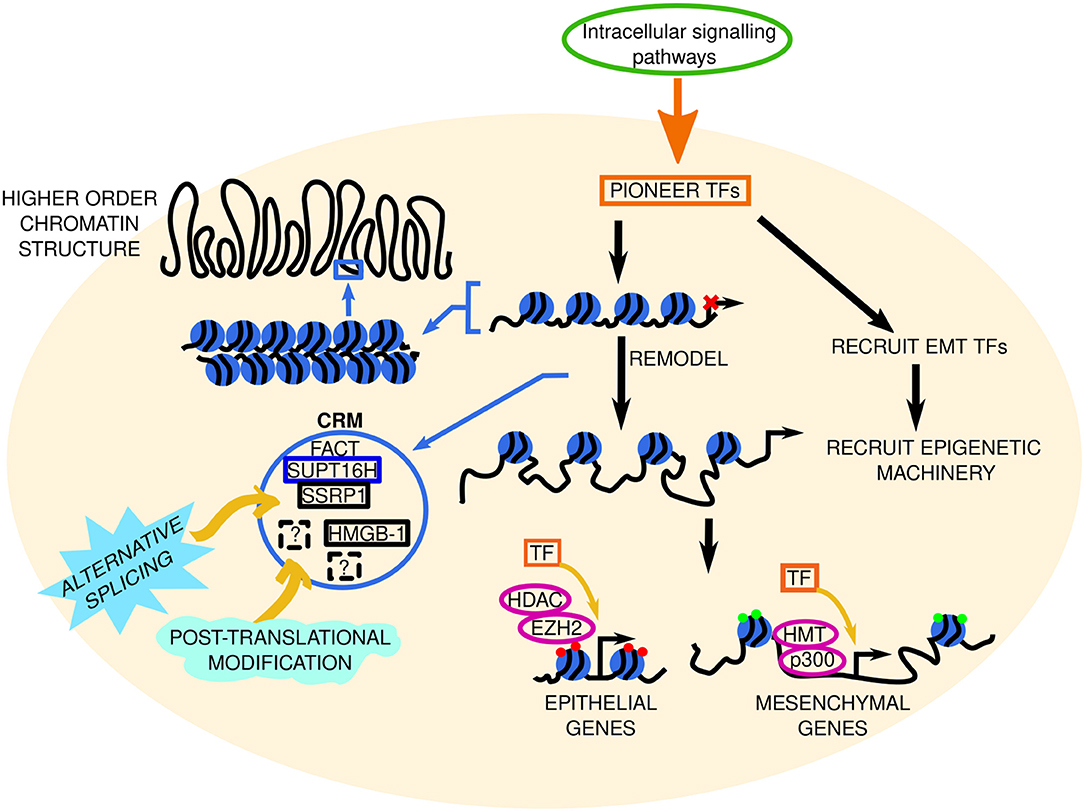
Figure 5. Chromatin remodeling machinery in EMT. The signal from intracellular signaling pathways is transduced to pioneer transcription factors (TFs) in the nucleus (represented by the beige circle). These pioneer TFs remodel the chromatin and recruit EMT TFs to accessible chromatin, which subsequently recruit and interact with epigenetic machinery such as DNA methyltransferases, DNMT; histone methyltransferases, HMT; e.g., EZH2, enhancer of zeste 2; ubiquitinating enzymes, Ub ligase; CtBP, C-terminal binding protein; complex, and enzymes involved in histone acetylation (p300) or deacetylation (histone deacetylase, HDAC). Together, TFs and epigenetic machinery repress or promote epithelial and mesenchymal gene expression, respectively, to determine the cell phenotype. Red and green circles at these genes denote repressive or active chromatin marks, respectively. The function of higher order chromatin structures in EMT phenotype determination remains elusive. Chromatin remodeling machinery (CRM) within the blue circle control chromatin accessibility and gene expression. Solid and dashed black boxes represent established and novel CRM, respectively. FACT, facilitates chromatin transcription; composed of SSRP1, structure specific recognition protein 1; and SUPT16H (SPT16 homolog, facilitates chromatin remodeling subunit); HMGB-1, high mobility group box 1. Question marks represent unknown CRM. The solid blue box around SUPT16H signifies that this CRM has PH domains, whose role in EMT is unknown. The roles of alternative splicing (sky blue spiked shape) and post translational modifications (mint green wave-like shape) in EMT CRM are also elusive.
In terms of nucleosome structure, the FACT (FAcilitates Chromatin Transcription) histone chaperone complex is a heterodimer consisting of SSRP1 and SUPT16H that has established roles not only in nucleosome destabilization and formation, but in the replacement of histones with histone variants, and FACT also prevents unwanted histone movement during transcription (182). FACT subunits were shown to be overexpressed in breast cancer and pharmacological entrapment of FACT within chromatin—which reduced FACT activity—had positive (dose-dependent) effects on murine survival, prevented tumor onset and delayed progression in vivo (183). Given these data, our understanding of the EMT-promoting effects of the chromatin remodeling machinery is lacking. In human intestinal cell lines the FACT subunit SUPT16H positively correlated with epithelial markers such as E-cadherin and showed an inverse relationship with the expression of classical mesenchymal markers like Zeb1 (3). Crystallographic examination of the FACT subunit Spt16 [isolated from yeast, the middle domain (-M) in particular] showed that a double pleckstrin homology (PH) domain exists in Spt16-M (184). Although analogous studies with SUPT16H are required, these data suggest that like its homolog, SUPT16H may contain PH domain(s) to bind to proteins other than histones or to facilitate intracellular signaling. Elucidating the role of PH domains in chromatin remodeling proteins could uncover previously unknown functions of histone chaperone proteins in EMT—their role in regulating chromatin accessibility may be just one avenue through which they promote EMT. Spt16-M could bind to H3-H4 (184), and therefore may influence the methylation/acetylation state of key lysine residues on H3-H4, possibly through interacts with DNMT3A, G9a, JMJD2A, or other epigenetic modifying enzymes, which may increase chromatin accessibility at epithelial gene promoters/regulatory elements and vice versa for mesenchymal genes. The second FACT subunit SSRP1-M was initially thought to lack histone binding (185), however it was later shown to bind H3-H4 as well as H2A-H2B (186). In vivo the SSRP1 subunit enhanced xenograft tumor growth/proliferation and SSRP1 overexpression in vitro promoted EMT (187). Mechanistically, miRNA-28-5p was implicated in upstream negative regulation of SSRP1 (187), however given the discovery of PH domains within Spt16 and SUPT16H-H3/H4 binding, it is reasonable to suggest that SSRP1 promotes EMT via epigenetic regulation of key genes, albeit further research is required.
The histone chaperone SUPT6H was found to be indispensable in estrogen-dependent breast cancer cells in terms of maintaining chromatin structure, facilitating estrogen-mediated transcription, and suppressing H3K27me3 on lineage-specific genes (188). However, in vitro murine Spt6 was shown to offset H3K27me3 by facilitating the H3K27 demethylase KDM6A (a.k.a. UTX, another Jumonji family member) (189). Consequently, Spt6 promoted differentiation and muscle gene expression in skeletal muscle cells (189). UTX was later shown to epigenetically repress the EMT-TFs Snail and Zeb1/2—independent of H3K27 demethylation—via decreased H3K4me2 and H3 acetylation at their promoters (190), with these anti-EMT effects in agreement with previously described data on JMJD5. In contrast, KDM5B overexpression increased cell proliferation, positively correlated with EMT markers, and promoted aggressive tumors in lung cancer (191). These data highlight just one facet of the intricate relationship between chromatin remodeling machinery and epigenetic regulation and illustrate the need for further research into how chromatin remodeling proteins may contribute to EMT.
HMGB-1 (high-mobility group box 1); a well-studied chromatin-binding nuclear protein that acts as a chromosome guardian/DNA chaperone and has immune/inflammatory functions [all of which have been extensively reviewed by (192)], was shown to promote EMT by upregulating MMPs (-1/-3/-10) via RAGE/NFκB pathway (193), and activating the TLR4/p38/NRF2 pathway to facilitate HMGB-1-mediated downstream EMT signaling (194). However, the epigenetics regarding HMGB-1 are unknown. Perhaps HMGB-1 itself epigenetically regulated, given the fact that its promoter coincides with a CpG island (195). Maybe HMGB-1 influences the epigenetic regulation of EMT target genes (epithelial or mesenchymal), or its DNA chaperone activity could promote/prohibit potential epigenetic regulation of target genes by competing with other chromatin proteins for the same chromosomal DNA sites as suggested by Spada et al. (196). Its EMT-promoting effects are somewhat localized to targeted interaction with its 3′ untranslated region (197, 198)—whether the crucial roles of 3′ UTRs in translation efficiency, mRNA stability and subcellular localization (199) play a role in HMGB-1-induced EMT remains to be studied. Given its ubiquitous expression it is possible that HMGB-1 is merely a ticking time bomb which can silently instigate EMT via a multitude of signaling pathways under the guise of other stimuli; HMGB-1 can signal via numerous receptors including but not limited to TLRs, RAGE, and integrins (192), so perhaps the EMT-inducing effects of HMGB-1 can be mirrored by other stimuli that activate these receptors and/or these downstream signaling cascades.
Chromatin Assembly Factor 1 (CAF1) consists of three proteins—p150, p60, and p48—and is necessary for preserving chromatin structure in our cells (200, 201). A novel role for CAF1 in EMT was recently uncovered, where CAF1 was implicated in cellular invasion and motility, and siRNA-mediated CAF1 depletion (specifically the p150 subunit) promoted the development of a Slug and/or claudin-guided EMT-like phenotype (202). Given its primary role as a histone chaperone, CAF1 may facilitate EMT via alterations in chromatin remodeling at key EMT genes, i.e., its depletion may enhance accessibility at mesenchymal genes. While these data implicate CAF1 depletion in cancer development, in a protein complex each subunit needs to be studied separately first and then as a whole. The p150 subunit was highly expressed in HCC tissue and epithelial ovarian cancer where it was associated a poor prognosis (203, 204). Similar data were obtained regarding the p60 subunit in prostate and lung cancers (205, 206), however virtually no information is available on p48, as it has not yet been identified in humans. As informative as these data may be, they do not address the gaping hole of how and why these CAF1 subunits are involved. Surely, there are epigenetic mechanisms involved, but as of right now these are unknown.
Conclusion
EMT is a dynamic multifaceted process occurring during development, tissue/organ repair, and disease, involving a vast network of signaling molecules working in unison and/or against one another (Figure 6). Just because there are different sub-types of EMT, does not necessarily mean that the stimuli, intracellular signaling pathways, TFs, epigenetic mechanisms, and chromatin remodeling machinery underlying developmental EMT, wound healing/fibrotic EMT, and cancer-related EMT are dissimilar. There must be some common ground between the sub-types and between partial and full EMT. Moreover, the partial/intermediate phenotype is of particular interest given recent single-cell sequencing data depicting continuous waves of gene regulation in EMT as opposed to discrete “partial” stages.
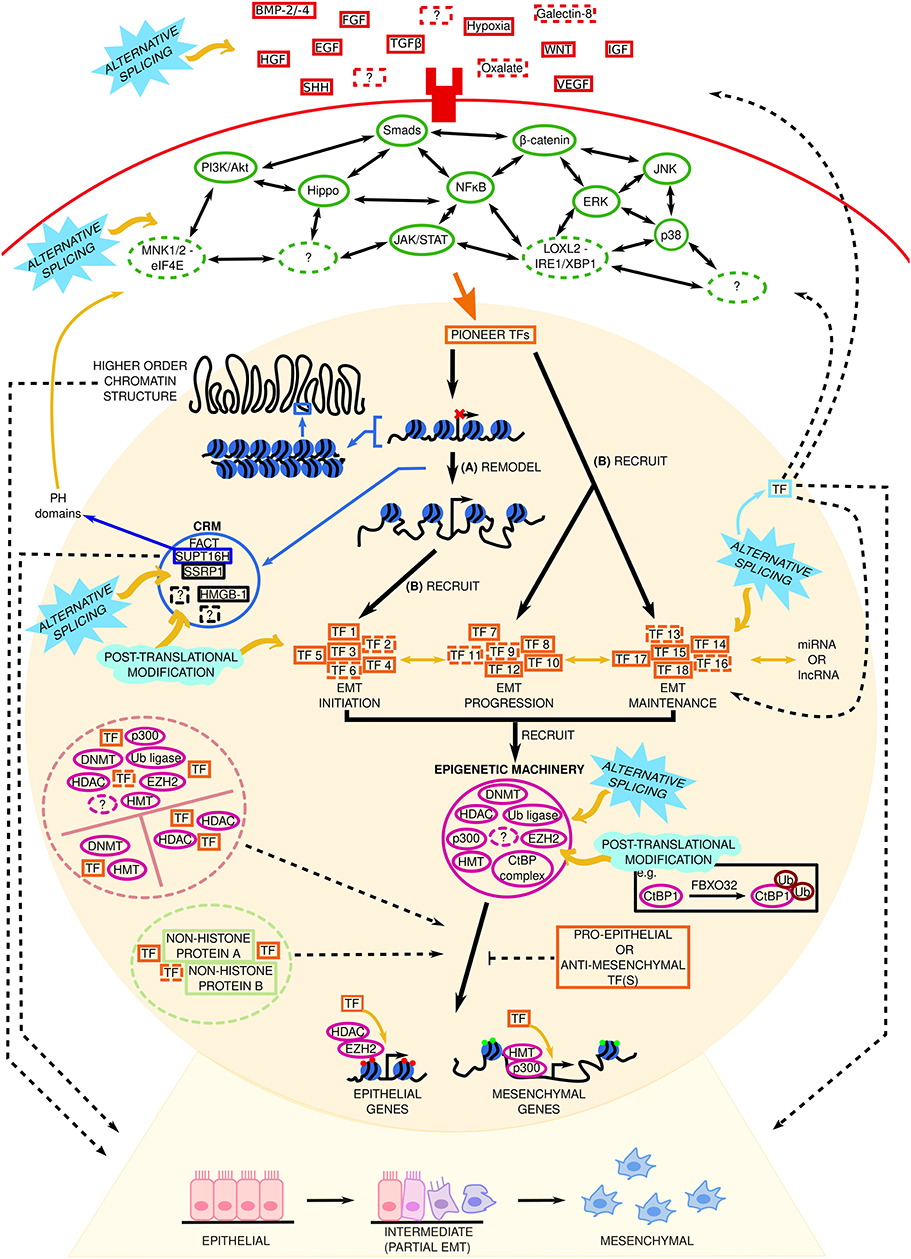
Figure 6. Complexity surrounding the gene regulatory network in Epithelial Mesenchymal Transition (EMT). Established (solid red boxes) and novel (dashed red boxes) extracellular stimuli bind to their particular receptors to initiate EMT. A generic transmembrane receptor is shown here for illustrative purposes. Extracellular stimuli subsequently signal via canonical and non-canonical intracellular signaling pathways. Solid and dashed green ovals represent established and novel signaling pathways, respectively. Two-headed black arrows indicate inter-pathway interaction and regulation. The signal from these signaling pathways is transduced to pioneer transcription factors (TFs) in the nucleus (represented by the beige circle). We hypothesize that in EMT these pioneer TFs: (A) remodel the chromatin to increase accessibility at EMT initiating genes, and (B) recruit a set of TFs needed for EMT initiation (TF 1–TF 6, dashed lines indicate potentially unknown TFs), and recruit sets of TFs involved in EMT progression (TF 7–TF 12) and maintenance (TF 13–TF 18), respectively, to already accessible chromatin. Solid and dashed orange boxes represent established and novel TFs, respectively. Through subsequent interactions with epigenetic machinery such as histone methyltransferases, HMT; e.g., EZH2, enhancer of zeste 2; and enzymes involved in histone acetylation (p300) or deacetylation (histone deacetylase, HDAC) at target loci, these TFs repress or promote epithelial and mesenchymal gene expression, respectively, to determine the cell phenotype (epithelial, intermediate, or mesenchymal). Red and green circles at these genes denote repressive or active chromatin marks, respectively. Pro- or anti-EMT TFs may inhibit this. Potential TF-non-histone protein complexes are found in the light green dashed circle. lncRNA, long non-coding RNA; miRNA, micro RNA. Solid and dashed pink circles represent established and novel epigenetic machinery, respectively. Black dashed lines indicate key questions that remain to be investigated. FBXO32 is a ubiquitin ligase. Potential TF-epigenetic machinery complexes are contained within the pink dashed circle. The function of higher order chromatin structures in EMT phenotype determination remains elusive. Chromatin remodeling machinery (CRM) within the blue circle control chromatin accessibility and gene expression. Solid and dashed black boxes represent established and novel CRM, respectively. FACT, facilitates chromatin transcription, composed of SSRP1 (structure specific recognition protein 1) and SUPT16H (SPT16 homolog, facilitates chromatin remodeling subunit); HMGB-1, high mobility group box 1. The solid blue box around SUPT16H signifies that this CRM has PH domains, whose role in EMT is unknown. The roles of alternative splicing (sky blue spiked shape) and post translational modifications (mint green wave-like shape) at the indicated steps/components remains to be investigated Question marks represent unknowns at each stage of EMT regulation. Yellow arrows and black dashed lines indicate key questions that remain to be investigated (see main text for more information).
Novel stimuli and intracellular pathways, undiscovered TFs and DNA/histone modifying enzymes, and the role of chromatin remodelers in EMT are huge unknowns in EMT. Auxiliary TF functions and the role of different sets of TFs involved in EMT (initiation versus progression etc.) warrant further investigation, and inter- and intra-tumor epigenetic heterogeneity and the impact of a cancer patient's epigenetic state are other compelling areas of research. To date, the majority of EMT research has focused on individual sub-types; data examining them as one process that occurs at three distinct “time points” is lacking. The challenge for future research therefore lies in (a) examining EMT concomitantly in disease and non-disease states, (b) looking at the interplay between canonical and non-canonical stimuli, TFs, and pathways involved in EMT, and (c) elucidating the roles the chromatin remodeling machinery and alternative splicing in phenotype determination during EMT, all with the aim of unraveling the complexity of the gene regulatory network underlying EMT. The data highlighted in these sections illustrate that researchers have begun to challenge these unknowns, but more work is needed to expand our knowledge and ultimately advance our ability to cure cancer.
Author Contributions
DL and VT worked on the conceptual framework and generated the final version for submission.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AKT, Protein kinase B; ARID1, AT-rich interaction domain 1; BMP, bone morphogenetic protein; BPTF, bromodomain PHD finger TF; BRD4, bromodomain containing 4; BRG1, Brahma-related gene 1, a.k.a. SMARCA4, SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member; BTG3, BTG anti-proliferation factor 3; CAF1, chromatin assembly factor 1; CHD(1), chromodomain helicase DNA-binding (protein 1); CRT, calreticulin; DCC, deleted in colorectal cancer; a netrin receptor, ; DEPTOR, DEP domain containing mTOR interacting protein; DNA, deoxyribonucleic acid; DNMT, DNA methyltransferase; ECM, extracellular matrix; EGF, epidermal growth factor; EMT, epithelial to mesenchymal transition; EGFR, epidermal growth factor receptor; EIF4E, eukaryotic initiation factor 4E; ER, endoplasmic reticulum; ERK, extracellular signal-regulated kinase; ESR1, estrogen receptor alpha; EZH2, enhancer of zeste 2, PRC2 subunit; FACT, facilitates chromatin transcriptionFAK, focal adhesion kinase; FBXL5, F-box and leucine rich repeat protein 5; FGF, fibroblast growth factor; FLNB, filamin B; FOX, Forkhead box (A1, C1, D3, M1 denote individual proteins); GATA3/4, GATA binding protein (3, 4 denote different proteins); GLI, glioma-associated oncogene; HAT(s), histone acetyltransferase(s); hBRM, human paralog of Drosophila Brahma, a.k.a. SMARCA2 SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 2; HCC, hepatocellular carcinoma; HDAC(s), histone deacetylase(s); HDGF, hepatoma-derived growth factor; HDM(s), histone demethylase(s); HGF, hepatocyte growth factor; HH, hedgehog; HIF-1α, hypoxia inducible factor 1α; HIF-2α, hypoxia inducible factor 2α; HOX, homeobox; HMGA1, High Mobility Group AT-Hook 1; HMGB-1, high mobility group box 1; HMT(s), histone methyltransferase(s); HNF4α, hepatocyte nuclear factor 4α; HSF1, heat shock factor 1; HSP5A, heat shock protein 5A; IGF, insulin-like growth factor; IRE1, Inositol-requiring enzyme 1; INO80, INO80 complex ATPase subunit; ISWI, Imitation SWItch; JAK, Janus kinase; JMJD2A, Jumonji-C (JmjC) domain-containing protein 2A, a.k.a. KMD4A; JMJD5, Jumonji-C (JmjC) domain-containing protein 5, a.k.a. KDM8; JNK, c-Jun N-terminal kinase; KDM4A, lysine demethylase 4Aα; KDM6A, lysine demethylase 6A, a.k.a. UTX histone demethylase; KDM8, lysine demethylase 8; KLF6, Krueppel-like factor 6; lncRNA, long non-coding RNA; LOXL2, lysyl oxidase-like 2; MET, mesenchymal-epithelial transition; miRNA(s), micro RNA(s); MNK1/2, mitogen-activated protein kinase-interacting kinases 1 and 2; MTOR, mechanistic/mammalian target of rapamycin; NC, neural crest; NEUROD1, neuronal differentiation 1; NEUROG1, neurogenin 1; NEUROG 2, neurogenin 2; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; NPM1, nucleophosmin 1; NRF2, nuclear factor erythroid-derived 2-related factor 2; NuRD, nucleosome remodeling and deacetylase; NURF, nucleosome remodeling factor; MYOSLID, myocardin-induced smooth muscle lncRNA, inducer of differentiation; MAPK, mitogen activated protein kinase; MMP, matrix metalloproteinase; MRNA, messenger ribonucleic acid; MTA1, metastasis-associated antigen 1; MTA3, metastasis-associated antigen 3; PAF, PCNA clamp associated factor; PCG, Polycomb group; PH, pleckstrin homology; PI3K, phosphoinositide 3-kinase; PIK3CA, phosphatidylinositol-4, 5-bisphosphate 3-kinase catalytic subunit alpha; PTEN, phosphatase and tensin homolog; PRC2, polycomb repressive complex 2; RAGE, a.k.a. AGER, advanced glycosylation end-product specific receptor; RNA, ribonucleic acid; SHH, sonic hedgehog; SIRT-1, Sirtuin 1; SOX4, SRY-box transcription factor 4; SSRP1, structure specific recognition protein 1; STAT3, signal transducer and activator of transcription 3; SUPT6H, SPT6 homolog, histone chaperone and transcription elongation factor; SUPT16H, SPT16 homolog, facilitates chromatin remodeling subunit; SWI/SNF, SWItch/Sucrose Non-Fermentable; TAZ, transcriptional coactivator with PDZ binding motif; TF(S), transcription factor(s); TGFβ, transforming growth factor beta; TLR4, Toll-like receptor 4; TRIO, Rho guanine nucleotide exchange factor; TRPV4, transient receptor potential vanilloid 4; UNC-40, un-coordinated 40, a netrin receptor. C. elegans homolog of DCC; UPA, urokinase-type plasminogen activator; UTRs, untranslated regions; VEGF(A), vascular endothelial growth factor (A); WNT, fusion of the name of the Drosophila segment polarity gene wingless and the name of the vertebrate homolog, integrated or int-1; XBP1, x-box–binding protein 1; YAP, yes-associated protein; ZEB1, zinc finger E-box binding homeobox 1; ZEB2, zinc finger E-box binding homeobox 2.
References
1. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. (2009) 119:1420–8. doi: 10.1172/JCI39104
2. Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. (2009) 119:1427–37. doi: 10.1172/JCI36183
3. Bedi U, Mishra VK, Wasilewski D, Scheel C, Johnsen SA. Epigenetic plasticity: a central regulator of epithelial-to-mesenchymal transition in cancer. Oncotarget. (2014) 5:2016–29. doi: 10.18632/oncotarget.1875
4. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. (2009) 139:871–90. doi: 10.1016/j.cell.2009.11.007
5. Yang J, Du X, Wang G, Sun Y, Chen K, Zhu X, et al. Mesenchymal to epithelial transition in sarcomas. Eur J Cancer. (2014) 50:593–601. doi: 10.1016/j.ejca.2013.11.006
6. Ban CR, Twigg SM. Fibrosis in diabetes complications: pathogenic mechanisms and circulating and urinary markers. Vasc Health Risk Manag. (2008) 4:575–96. doi: 10.2147/VHRM.S1991
7. Patel S, Srivastava S, Singh MR, Singh D. Mechanistic insight into diabetic wounds: pathogenesis, molecular targets and treatment strategies to pace wound healing. Biomed Pharmacother. (2019) 112:108615. doi: 10.1016/j.biopha.2019.108615
8. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac fibrosis: the fibroblast awakens. Circ Res. (2016) 118:1021–40. doi: 10.1161/CIRCRESAHA.115.306565
9. Rout-Pitt N, Farrow N, Parsons D, Donnelley M. Epithelial mesenchymal transition (EMT): a universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir Res. (2018) 19:136. doi: 10.1186/s12931-018-0834-8
10. Sun L, Fang J. Epigenetic regulation of epithelial-mesenchymal transition. Cell Mol Life Sci. (2016) 73:4493–515. doi: 10.1007/s00018-016-2303-1
11. Längst G, Manelyte L. Chromatin remodelers: from function to dysfunction. Genes. (2015) 6:299–324. doi: 10.3390/genes6020299
12. Kiesslich T, Pichler M, Neureiter D. Epigenetic control of epithelial-mesenchymal-transition in human cancer. Mol Clin Oncol. (2013) 1:3–11. doi: 10.3892/mco.2012.28
13. Serrano-Gomez SJ, Maziveyi M, Alahari SK. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol Cancer. (2016) 15:18. doi: 10.1186/s12943-016-0502-x
14. Lachat C, Boyer-Guittaut M, Peixoto P, Hervouet E. Epigenetic regulation of EMT (epithelial to mesenchymal transition) and tumor aggressiveness: a view on paradoxical roles of KDM6B and EZH2. Epigenomes. (2018) 3:1. doi: 10.3390/epigenomes3010001
15. Bell O, Tiwari VK, Thoma NH, Schubeler D. Determinants and dynamics of genome accessibility. Nat Rev Genet. (2011) 12:554–64. doi: 10.1038/nrg3017
16. Tiwari VK, McGarvey KM, Licchesi JDF, Ohm JE, Herman JG, Schübeler D, et al. PcG proteins, DNA methylation, and gene repression by chromatin looping. PLoS Biol. (2008) 6:2911–27. doi: 10.1371/journal.pbio.0060306
17. Llinàs-Arias P, Esteller M. Epigenetic inactivation of tumour suppressor coding and non-coding genes in human cancer: an update. Open Biol. (2017) 7:170152. doi: 10.1098/rsob.170152
18. Xiong M, Jiang L, Zhou Y, Qiu W, Fang L, Tan R, et al. The miR-200 family regulates TGF-beta1-induced renal tubular epithelial to mesenchymal transition through smad pathway by targeting ZEB1 and ZEB2 expression. Am J Physiol Renal Physiol. (2012) 302:F369–79. doi: 10.1152/ajprenal.00268.2011
19. Dhamija S, Diederichs S. From junk to master regulators of invasion: lncRNA functions in migration, EMT and metastasis. Int J Cancer. (2016) 139:269–80. doi: 10.1002/ijc.30039
20. Cao M-X, Jiang Y-P, Tang Y-L, Liang X-H. The crosstalk between lncRNA and microRNA in cancer metastasis: orchestrating the epithelial-mesenchymal plasticity. Oncotarget. (2017) 8:12472–83. doi: 10.18632/oncotarget.13957
21. Daugaard I, Sanders KJ, Idica A, Vittayarukskul K, Hamdorf M, Krog JD, et al. miR-151a induces partial EMT by regulating E-cadherin in NSCLC cells. Oncogenesis. (2017) 6:e366. doi: 10.1038/oncsis.2017.66
22. Xiong H-G, Li H, Xiao Y, Yang Q-C, Yang L-L, Chen L, et al. Long noncoding RNA MYOSLID promotes invasion and metastasis by modulating the partial epithelial-mesenchymal transition program in head and neck squamous cell carcinoma. J Exp Clin Cancer Res. (2019) 38:278. doi: 10.1186/s13046-019-1254-4
23. Wu W, Chen F, Cui X, Yang L, Chen J, Zhao J, et al. LncRNA NKILA suppresses TGF-β-induced epithelial-mesenchymal transition by blocking NF-κB signaling in breast cancer. Int J Cancer. (2018) 143:2213–24. doi: 10.1002/ijc.31605
24. Burger GA, Danen EHJ, Beltman JB. Deciphering epithelial-mesenchymal transition regulatory networks in cancer through computational approaches. Front Oncol. (2017) 7:162. doi: 10.3389/fonc.2017.00162
25. Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. (2013) 19:1438–49. doi: 10.1038/nm.3336
26. Damaskos C, Valsami S, Spartalis E, Antoniou EA, Tomos P, Karamaroudis S, et al. Histone deacetylase inhibitors: a novel therapeutic weapon against medullary thyroid cancer? Anticancer Res. (2016) 36:5019–24. doi: 10.21873/anticanres.11070
27. Meadows KL, Hurwitz HI. Anti-VEGF therapies in the clinic. Cold Spring Harb Perspect Med. (2012) 2:a006577. doi: 10.1101/cshperspect.a006577
28. Tewari KS, Sill MW, Long HJ, Penson RT, Huang H, Ramondetta LM, et al. Improved survival with bevacizumab in advanced cervical cancer. New Eng J Med. (2014) 370:734–43. doi: 10.1056/NEJMoa1309748
29. Zirlik K, Duyster J. Anti-angiogenics: current situation and future perspectives. Oncol Res Treat. (2018) 41:166–71. doi: 10.1159/000488087
30. Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. (1994) 127:2021–36. doi: 10.1083/jcb.127.6.2021
31. Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, et al. Epithelial–mesenchymal and mesenchymal–epithelial transitions in carcinoma progression. J Cell Physiol. (2007) 213:374–83. doi: 10.1002/jcp.21223
32. Cursons J, Leuchowius K-J, Waltham M, Tomaskovic-Crook E, Foroutan M, Bracken CP, et al. Stimulus-dependent differences in signalling regulate epithelial-mesenchymal plasticity and change the effects of drugs in breast cancer cell lines. Cell Commun Signal. (2015) 13:26. doi: 10.1186/s12964-015-0106-x
33. Iwano M, Pleith D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. (2002) 110:341–9. doi: 10.1172/JCI0215518
34. Cañadas I, Taus A, González I, Villanueva X, Gimeno J, Pijuan L, et al. High circulating hepatocyte growth factor levels associate with epithelial to mesenchymal transition and poor outcome in small cell lung cancer patients. Oncotarget. (2014) 5:5246–56. doi: 10.18632/oncotarget.2124
35. Sagi Z, Hieronymus T. The impact of the epithelial-mesenchymal transition regulator hepatocyte growth factor receptor/met on skin immunity by modulating langerhans cell migration. Front Immunol. (2018) 9:517. doi: 10.3389/fimmu.2018.00517
36. Mak P, Leav I, Pursell B, Bae D, Yang X, Taglienti CA, et al. ERβ impedes prostate cancer EMT by destabilizing HIF-1α and inhibiting VEGF-mediated snail nuclear localization: implications for gleason grading. Cancer Cell. (2010) 17:319–32. doi: 10.1016/j.ccr.2010.02.030
37. Fantozzi A, Gruber DC, Pisarsky L, Heck C, Kunita A, Yilmaz M, et al. VEGF-mediated angiogenesis links EMT-induced cancer stemness to tumor initiation. Cancer Res. (2014) 74:1566. doi: 10.1158/0008-5472.CAN-13-1641
38. Li C, Wang Q, Shen S, Wei X, Li G. HIF-1α/VEGF signaling-mediated epithelial–mesenchymal transition and angiogenesis is critically involved in anti-metastasis effect of luteolin in melanoma cells. Phytother Res. (2019) 33:798–807. doi: 10.1002/ptr.6273
39. Graham TR, Zhau HE, Odero-Marah VA, Osunkoya AO, Kimbro KS, Tighiouart M, et al. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. (2008) 68:2479–88. doi: 10.1158/0008-5472.CAN-07-2559
40. Li H, Batth IS, Qu X, Xu L, Song N, Wang R, et al. IGF-IR signaling in epithelial to mesenchymal transition and targeting IGF-IR therapy: overview and new insights. Mol Cancer. (2017) 16:6. doi: 10.1186/s12943-016-0576-5
41. Zhang J, Tian X-J, Xing J. Signal transduction pathways of EMT induced by TGF-β, SHH, and WNT and their crosstalks. J Clin Med. (2016) 5:41. doi: 10.3390/jcm5040041
42. Gupta S, Takebe N, Lorusso P. Targeting the hedgehog pathway in cancer. Ther Adv Med Oncol. (2010) 2:237–50. doi: 10.1177/1758834010366430
43. Yoo YA, Kang MH, Lee HJ, Kim BH, Park JK, Kim HK, et al. Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP-9 pathway in gastric cancer. Cancer Res. (2011) 71:7061–70. doi: 10.1158/0008-5472.CAN-11-1338
44. Katsuno Y, Hanyu A, Kanda H, Ishikawa Y, Akiyama F, Iwase T, et al. Bone morphogenetic protein signaling enhances invasion and bone metastasis of breast cancer cells through smad pathway. Oncogene. (2008) 27:6322–33. doi: 10.1038/onc.2008.232
45. Kang MH, Kang HN, Kim JL, Kim JS, Oh SC, Yoo YA. Inhibition of PI3 kinase/Akt pathway is required for BMP2-induced EMT and invasion. Oncol Rep. (2009) 22:525–34. doi: 10.3892/or_00000467
46. Park K-S, Dubon MJ, Gumbiner BM. N-cadherin mediates the migration of MCF-10A cells undergoing bone morphogenetic protein 4-mediated epithelial mesenchymal transition. Tumour Biol. (2015) 36:3549–56. doi: 10.1007/s13277-014-2991-9
47. Zabkiewicz C, Resaul J, Hargest R, Jiang WG, Ye L. Bone morphogenetic proteins, breast cancer, and bone metastases: striking the right balance. Endocr Relat Cancer. (2017) 24:R349–66. doi: 10.1530/ERC-17-0139
48. Li C-W, Xia W, Huo L, Lim S-O, Wu Y, Hsu JL, et al. Epithelial–mesenchymal transition induced by TNF-α requires NF-κB–mediated transcriptional upregulation of twist1. Cancer Res. (2012) 72:1290–300. doi: 10.1158/0008-5472.CAN-11-3123
49. Peixoto P, Etcheverry A, Aubry M, Missey A, Lachat C, Perrard J, et al. EMT is associated with an epigenetic signature of ECM remodeling genes. Cell Death Dis. (2019) 10:205. doi: 10.1038/s41419-019-1397-4
50. van den Beucken T, Koch E, Chu K, Rupaimoole R, Prickaerts P, Adriaens M, et al. Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER. Nat Commun. (2014) 5:5203. doi: 10.1038/ncomms6203
51. Sharma T, Radosevich JA, Pachori G, Mandal CC. A molecular view of pathological microcalcification in breast cancer. J Mammary Gland Biol Neoplasia. (2016) 21:25–40. doi: 10.1007/s10911-015-9349-9
52. Wilkinson L, Thomas V, Sharma N. Microcalcification on mammography: approaches to interpretation and biopsy. Br J Radiol. (2017) 90:20160594. doi: 10.1259/bjr.20160594
53. Convento MB, Pessoa EA, Cruz E, da Glória MA, Schor N, Borges FT. Calcium oxalate crystals and oxalate induce an epithelial-to-mesenchymal transition in the proximal tubular epithelial cells: contribution to oxalate kidney injury. Sci Rep. (2017) 7:45740. doi: 10.1038/srep45740
54. Convento M, Pessoa E, Aragão A, Schor N, Borges F. Oxalate induces type II epithelial to mesenchymal transition (EMT) in inner medullary collecting duct cells (IMCD) in vitro and stimulate the expression of osteogenic and fibrotic markers in kidney medulla in vivo. Oncotarget. (2019) 10:1102–18. doi: 10.18632/oncotarget.26634
55. Castellaro AM, Tonda A, Cejas HH, Ferreyra H, Caputto BL, Pucci OA, et al. Oxalate induces breast cancer. BMC Cancer. (2015) 15:761. doi: 10.1186/s12885-015-1747-2
56. Peerapen P, Thongboonkerd V. p38 MAPK mediates calcium oxalate crystal-induced tight junction disruption in distal renal tubular epithelial cells. Sci Rep. (2013) 3:1041. doi: 10.1038/srep01041
57. Kanlaya R, Khamchun S, Kapincharanon C, Thongboonkerd V. Protective effect of epigallocatechin-3-gallate (EGCG) via Nrf2 pathway against oxalate-induced epithelial mesenchymal transition (EMT) of renal tubular cells. Sci Rep. (2016) 6:30233. doi: 10.1038/srep30233
58. Oyanadel C, Holmes C, Pardo E, Retamal C, Shaughnessy R, Smith P, et al. Galectin-8 induces partial epithelial-mesenchymal transition with invasive tumorigenic capabilities involving a FAK/EGFR/proteasome pathway in madin-darby canine kidney cells. Mol Biol Cell. (2018) 29:557–74. doi: 10.1091/mbc.E16-05-0301
59. Lee WH, Choong LY, Jin TH, Mon NN, Chong S, Liew CS, et al. TRPV4 plays a role in breast cancer cell migration via Ca(2+)-dependent activation of AKT and downregulation of E-cadherin cell cortex protein. Oncogenesis. (2017) 6:e338. doi: 10.1038/oncsis.2017.39
60. Nalluri SM, O'Connor JW, Gomez EW. Cytoskeletal signaling in TGFbeta-induced epithelial-mesenchymal transition. Cytoskeleton. (2015) 72:557–69. doi: 10.1002/cm.21263
61. Shah PP, Fong MY, Kakar SS. PTTG induces EMT through integrin αVβ3-focal adhesion kinase signaling in lung cancer cells. Oncogene. (2012) 31:3124–35. doi: 10.1038/onc.2011.488
62. Chaturvedi V, Fournier-Level A, Cooper HM, Murray MJ. Loss of neogenin1 in human colorectal carcinoma cells causes a partial EMT and wound-healing response. Sci Rep. (2019) 9:4110. doi: 10.1038/s41598-019-40886-y
63. Fan L-C, Teng H-W, Shiau C-W, Tai W-T, Hung M-H, Yang S-H, et al. Regorafenib (stivarga) pharmacologically targets epithelial-mesenchymal transition in colorectal cancer. Oncotarget. (2016) 7:64136–47. doi: 10.18632/oncotarget.11636
64. Awasthi N, Mikels-Vigdal AJ, Stefanutti E, Schwarz MA, Monahan S, Smith V, et al. Therapeutic efficacy of anti-MMP9 antibody in combination with nab-paclitaxel-based chemotherapy in pre-clinical models of pancreatic cancer. J Cell Mol Med. (2019) 23:3878–87. doi: 10.1111/jcmm.14242
65. Owyong M, Chou J, van den Bijgaart RJ, Kong N, Efe G, Maynard C, et al. MMP9 modulates the metastatic cascade and immune landscape for breast cancer anti-metastatic therapy. Life Sci Alliance. (2019) 2:e201800226. doi: 10.26508/lsa.201800226
66. Tanaka H, Kono E, Tran CP, Miyazaki H, Yamashiro J, Shimomura T, et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med. (2010) 16:1414–20. doi: 10.1038/nm.2236
67. Dai J, He H, Lin D, Wang C, Zhu Y, Xu D. Up-regulation of E-cadherin by saRNA inhibits the migration and invasion of renal carcinoma cells. Int J Clin Exp Pathol. (2018) 11:5792–800.
68. Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS, et al. E-cadherin is required for metastasis in multiple models of breast cancer. Nature. (2019) 573:439–44. doi: 10.1038/s41586-019-1526-3
69. Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. (2009) 157:220–33. doi: 10.1111/j.1476-5381.2009.00190.x
70. Hanna J, Hossain GS, Kocerha J. The potential for microRNA therapeutics and clinical research. Front Genet. (2019) 10:478. doi: 10.3389/fgene.2019.00478
71. Mei M, Ren Y, Zhou X, Yuan XB, Han L, Wang GX, et al. Downregulation of miR-21 enhances chemotherapeutic effect of taxol in breast carcinoma cells. Technol Cancer Res Treat. (2010) 9:77–86. doi: 10.1177/153303461000900109
72. Van Roosbroeck K, Fanini F, Setoyama T, Ivan C, Rodriguez-Aguayo C, Fuentes-Mattei E, et al. Combining Anti-Mir-155 with chemotherapy for the treatment of lung cancers. Clin Cancer Res. (2017) 23:2891. doi: 10.1158/1078-0432.CCR-16-1025
73. Barneh F, Mirzaie M, Nickchi P, Tan TZ, Thiery JP, Piran M, et al. Integrated use of bioinformatic resources reveals that co-targeting of histone deacetylases, IKBK and SRC inhibits epithelial-mesenchymal transition in cancer. Brief Bioinform. (2018) 20:717–31. doi: 10.1093/bib/bby030
74. Brabletz T. To differentiate or not—routes towards metastasis. Nat Rev Cancer. (2012) 12:425–36. doi: 10.1038/nrc3265
75. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells—what challenges do they pose? Nat Rev Drug Discov. (2014) 13:497–512. doi: 10.1038/nrd4253
76. Ishay-Ronen D, Diepenbruck M, Kalathur RKR, Sugiyama N, Tiede S, Ivanek R, et al. Gain fat - lose metastasis: converting invasive breast cancer cells into adipocytes inhibits cancer metastasis. Cancer Cell. (2019) 35:17–32.e16. doi: 10.1016/j.ccell.2018.12.002
77. Hseu Y-C, Lin Y-C, Rajendran P, Thigarajan V, Mathew DC, Lin K-Y, et al. Antrodia salmonea suppresses invasion and metastasis in triple-negative breast cancer cells by reversing EMT through the NF-κB and Wnt/β-catenin signaling pathway. Food Chem Toxicol. (2019) 124:219–30. doi: 10.1016/j.fct.2018.12.009
78. Moroz E, Carlin S, Dyomina K, Burke S, Thaler HT, Blasberg R, et al. Real-time imaging of HIF-1alpha stabilization and degradation. PLoS ONE. (2009) 4:e5077. doi: 10.1371/journal.pone.0005077
79. Hanna C, Hubchak SC, Liang X, Rozen-Zvi B, Schumacker PT, Hayashida T, et al. Hypoxia-inducible factor-2α and TGF-β signaling interact to promote normoxic glomerular fibrogenesis. Am J Physiol Renal Physiol. (2013) 305:F1323. doi: 10.1152/ajprenal.00155.2013
80. Xie M, Zhang L, He CS, Xu F, Liu JL, Hu ZH, et al. Activation of notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J Cell Biochem. (2012) 113:1501–13. doi: 10.1002/jcb.24019
81. Ishida T, Hijioka H, Kume K, Miyawaki A, Nakamura N. Notch signaling induces EMT in OSCC cell lines in a hypoxic environment. Oncol Lett. (2013) 6:1201–6. doi: 10.3892/ol.2013.1549
82. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. (2014) 15:178–96. doi: 10.1038/nrm3758
83. Xu W, Yang Z, Lu N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh Migr. (2015) 9:317–24. doi: 10.1080/19336918.2015.1016686
84. Liu X, Li Z, Song Y, Wang R, Han L, Wang Q, et al. AURKA induces EMT by regulating histone modification through Wnt/β-catenin and PI3K/Akt signaling pathway in gastric cancer. Oncotarget. (2016) 7:33152–64. doi: 10.18632/oncotarget.8888
85. Qin H, Liu X, Li F, Miao L, Li T, Xu B, et al. PAD1 promotes epithelial-mesenchymal transition and metastasis in triple-negative breast cancer cells by regulating MEK1-ERK1/2-MMP2 signaling. Cancer Lett. (2017) 409:30–41. doi: 10.1016/j.canlet.2017.08.019
86. Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, van Roy F, et al. Activation of NF-kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene. (2007) 26:7445–56. doi: 10.1038/sj.onc.1210546
87. Navandar M, Garding A, Sahu SK, Pataskar A, Schick S, Tiwari VK. ERK signalling modulates epigenome to drive epithelial to mesenchymal transition. Oncotarget. (2017) 8:29269–81. doi: 10.18632/oncotarget.16493
88. Chen J, Zhu H, Liu Q, Ning D, Zhang Z, Zhang L, et al. DEPTOR induces a partial epithelial-to-mesenchymal transition and metastasis via autocrine TGFβ1 signaling and is associated with poor prognosis in hepatocellular carcinoma. J Exp Clin Cancer Res. (2019) 38:273. doi: 10.1186/s13046-019-1220-1
89. Park J, Kim D-H, Shah SR, Kim H-N, Kshitiz, Kim P, et al. Switch-like enhancement of epithelial-mesenchymal transition by YAP through feedback regulation of WT1 and Rho-family GTPases. Nat Commun. (2019) 10:2797. doi: 10.1038/s41467-019-10729-5
90. McFaline-Figueroa JL, Hill AJ, Qiu X, Jackson D, Shendure J, Trapnell C. A pooled single-cell genetic screen identifies regulatory checkpoints in the continuum of the epithelial-to-mesenchymal transition. Nat Genet. (2019) 51:1389–98. doi: 10.1038/s41588-019-0489-5
91. Sahu SK, Garding A, Tiwari N, Thakurela S, Toedling J, Gebhard S, et al. JNK-dependent gene regulatory circuitry governs mesenchymal fate. EMBO J. (2015) 34:2162–81. doi: 10.15252/embj.201490693
92. Wojciechowski MC, Mahmutovic L, Shu DY, Lovicu FJ. ERK1/2 signaling is required for the initiation but not progression of TGFβ-induced lens epithelial to mesenchymal transition (EMT). Exp Eye Res. (2017) 159:98–113. doi: 10.1016/j.exer.2017.03.012
93. Wu Y, Xu X, Ma L, Yi Q, Sun W, Tang L. Calreticulin regulates TGF-β1-induced epithelial mesenchymal transition through modulating smad signaling and calcium signaling. Int J Biochem Cell Biol. (2017) 90:103–13. doi: 10.1016/j.biocel.2017.07.023
94. Zemljic-Harpf A, Manso AM, Ross RS. Vinculin and talin: focus on the myocardium. J Investig Med. (2009) 57:849–55. doi: 10.2310/JIM.0b013e3181c5e074
95. Wu J, He Z, Yang XM, Li KL, Wang DL, Sun FL. RCCD1 depletion attenuates TGF-β-induced EMT and cell migration by stabilizing cytoskeletal microtubules in NSCLC cells. Cancer Lett. (2017) 400:18–29. doi: 10.1016/j.canlet.2017.04.021
96. Gladilin E, Ohse S, Boerries M, Busch H, Xu C, Schneider M, et al. TGFβ-induced cytoskeletal remodeling mediates elevation of cell stiffness and invasiveness in NSCLC. Sci Rep. (2019) 9:7667. doi: 10.1038/s41598-019-43409-x
97. Ramamurthy VP, Ramalingam S, Gediya LK, Njar VCO. The retinamide VNLG-152 inhibits f-AR/AR-V7 and MNK-eIF4E signaling pathways to suppress EMT and castration-resistant prostate cancer xenograft growth. FEBS J. (2018) 285:1051–63. doi: 10.1111/febs.14383
98. Cuevas EP, Eraso P, Mazón MJ, Santos V, Moreno-Bueno G, Cano A, et al. LOXL2 drives epithelial-mesenchymal transition via activation of IRE1-XBP1 signalling pathway. Sci Rep. (2017) 7:44988. doi: 10.1038/srep44988
99. Xiong Y, Sun F, Dong P, Watari H, Yue J, Yu M-F, et al. iASPP induces EMT and cisplatin resistance in human cervical cancer through miR-20a-FBXL5/BTG3 signaling. J Exp Clin Cancer Res. (2017) 36:48. doi: 10.1186/s13046-017-0520-6
100. Wu W, Ding H, Cao J, Zhang W. FBXL5 inhibits metastasis of gastric cancer through suppressing snail1. Cell Physiol Biochem. (2015) 35:1764–72. doi: 10.1159/000373988
101. Yao H, Su S, Xia D, Wang M, Li Z, Chen W, et al. F-box and leucine-rich repeat protein 5 promotes colon cancer progression by modulating PTEN/PI3K/AKT signaling pathway. Biomed Pharmacother. (2018) 107:1712–9. doi: 10.1016/j.biopha.2018.08.119
102. Sahu SK, Tiwari N, Pataskar A, Zhuang Y, Borisova M, Diken M, et al. FBXO32 promotes microenvironment underlying epithelial-mesenchymal transition via CtBP1 during tumour metastasis and brain development. Nat Commun. (2017) 8:1523. doi: 10.1038/s41467-017-01366-x
103. Karacosta LG, Anchang B, Ignatiadis N, Kimmey SC, Benson JA, Shrager JB, et al. Mapping lung cancer epithelial-mesenchymal transition states and trajectories with single-cell resolution. Nat Commun. (2019) 10:5587. doi: 10.1038/s41467-019-13441-6
104. Gras B, Jacqueroud L, Wierinckx A, Lamblot C, Fauvet F, Lachuer J, et al. Snail Family members unequally trigger EMT and thereby differ in their ability to promote the neoplastic transformation of mammary epithelial cells. PLoS ONE. (2014) 9:e92254. doi: 10.1371/journal.pone.0092254
105. Meyer-Schaller N, Cardner M, Diepenbruck M, Saxena M, Tiede S, Lüönd F, et al. A hierarchical regulatory landscape during the multiple stages of EMT. Dev Cell. (2019) 48:539–53.e536. doi: 10.1016/j.devcel.2018.12.023
106. Bocci F, Tripathi SC, Vilchez Mercedes SA, George JT, Casabar JP, Wong PK, et al. NRF2 activates a partial epithelial-mesenchymal transition and is maximally present in a hybrid epithelial/mesenchymal phenotype. Integr Biol. (2019) 11:251–63. doi: 10.1093/intbio/zyz021
107. Iwafuchi-Doi M, Zaret KS. Pioneer transcription factors in cell reprogramming. Genes Dev. (2014) 28:2679–92. doi: 10.1101/gad.253443.114
108. Pataskar A, Jung J, Smialowski P, Noack F, Calegari F, Straub T, et al. NeuroD1 reprograms chromatin and transcription factor landscapes to induce the neuronal program. EMBO J. (2016) 35:24–45. doi: 10.15252/embj.201591206
109. Manavathi B, Samanthapudi VSK, Gajulapalli VNR. Estrogen receptor coregulators and pioneer factors: the orchestrators of mammary gland cell fate and development. Front Cell Dev Bio. (2014) 2:34. doi: 10.3389/fcell.2014.00034
110. Guo R, Tang W, Yuan Q, Hui L, Wang X, Xie X. Chemical cocktails enable hepatic reprogramming of mouse fibroblasts with a single transcription factor. Stem Cell Rep. (2017) 9:499–512. doi: 10.1016/j.stemcr.2017.06.013
111. Li J, Song P, Jiang T, Dai D, Wang H, Sun J, et al. Heat shock factor 1 epigenetically stimulates glutaminase-1-dependent mTOR activation to promote colorectal carcinogenesis. Mol Ther. (2018) 26:1828–39. doi: 10.1016/j.ymthe.2018.04.014
112. Skrypek N, Bruneel K, Vandewalle C, De Smedt E, Soen B, Loret N, et al. ZEB2 stably represses RAB25 expression through epigenetic regulation by SIRT1 and DNMTs during epithelial-to-mesenchymal transition. Epigenetics Chromatin. (2018) 11:70. doi: 10.1186/s13072-018-0239-4
113. Simões-Costa M, Bronner ME. Establishing neural crest identity: a gene regulatory recipe. Development. (2015) 142:242–57. doi: 10.1242/dev.105445
114. Lukoseviciute M, Gavriouchkina D, Williams RM, Hochgreb-Hagele T, Senanayake U, Chong-Morrison V, et al. From pioneer to repressor: bimodal foxd3 activity dynamically remodels neural crest regulatory landscape in vivo. Dev Cell. (2018) 47:608–28.e606. doi: 10.1016/j.devcel.2018.11.009
115. Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, et al. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. (2006) 442:91–5. doi: 10.1038/nature04802
116. Xiao S, Liu L, Fang M, Zhou X, Peng X, Long J, et al. BPTF associated with EMT indicates negative prognosis in patients with hepatocellular carcinoma. Dig Dis Sci. (2015) 60:910–8. doi: 10.1007/s10620-014-3411-0
117. Zhang J, Liang Q, Lei Y, Yao M, Li L, Gao X, et al. SOX4 induces epithelial-mesenchymal transition and contributes to breast cancer progression. Cancer Res. (2012) 72:4597–608. doi: 10.1158/0008-5472.CAN-12-1045
118. Tiwari N, Tiwari VK, Waldmeier L, Balwierz PJ, Arnold P, Pachkov M, et al. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell. (2013) 23:768–83. doi: 10.1016/j.ccr.2013.04.020
119. Wang J, He C, Gao P, Wang S, Lv R, Zhou H, et al. HNF1B-mediated repression of SLUG is suppressed by EZH2 in aggressive prostate cancer. Oncogene. (2019) 39:1335–46. doi: 10.1038/s41388-019-1065-2
120. Xie SL, Fan S, Zhang SY, Chen WX, Li QX, Pan GK, et al. SOX8 regulates cancer stem-like properties and cisplatin-induced EMT in tongue squamous cell carcinoma by acting on the Wnt/β-catenin pathway. Int J Cancer. (2018) 142:1252–65. doi: 10.1002/ijc.31134
121. Krivtsova O, Makarova A, Lazarevich N. Aberrant expression of alternative isoforms of transcription factors in hepatocellular carcinoma. World J Hepatol. (2018) 10:645–61. doi: 10.4254/wjh.v10.i10.645
122. Lazarevich NL, Cheremnova OA, Varga EV, Ovchinnikov DA, Kudrjavtseva EI, Morozova OV, et al. Progression of HCC in mice is associated with a downregulation in the expression of hepatocyte nuclear factors. Hepatology. (2004) 39:1038–47. doi: 10.1002/hep.20155
123. Vuong LM, Chellappa K, Dhahbi JM, Deans JR, Fang B, Bolotin E, et al. Differential effects of hepatocyte nuclear factor 4α isoforms on tumor growth and t-cell factor 4/AP-1 interactions in human colorectal cancer cells. Mol Cell Biol. (2015) 35:3471–90. doi: 10.1128/MCB.00030-15
124. Li J, Choi PS, Chaffer CL, Labella K, Hwang JH, Giacomelli AO, et al. An alternative splicing switch in FLNB promotes the mesenchymal cell state in human breast cancer. Elife. (2018) 7:e37184. doi: 10.7554/eLife.37184
125. Sabol M, Trnski D, Musani V, Ozretić P, Levanat S. Role of GLI transcription factors in pathogenesis and their potential as new therapeutic targets. Int J Mol Sci. (2018) 19:2562. doi: 10.3390/ijms19092562
126. Fu J, Rodova M, Roy SK, Sharma J, Singh KP, Srivastava RK, et al. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. (2013) 330:22–32. doi: 10.1016/j.canlet.2012.11.018
127. Zanin R, Pegoraro S, Ros G, Ciani Y, Piazza S, Bossi F, et al. HMGA1 promotes breast cancer angiogenesis supporting the stability, nuclear localization and transcriptional activity of FOXM1. J Exp Clin Cancer Res. (2019) 38:313. doi: 10.1186/s13046-019-1307-8
128. Poli V, Fagnocchi L, Fasciani A, Cherubini A, Mazzoleni S, Ferrillo S, et al. MYC-driven epigenetic reprogramming favors the onset of tumorigenesis by inducing a stem cell-like state. Nat Commun. (2018) 9:1024. doi: 10.1038/s41467-018-03264-2
129. Widom J. Structure, dynamics, and function of chromatin in vitro. Annu Rev Biophys Biomol Struct. (1998) 27:285–327. doi: 10.1146/annurev.biophys.27.1.285
130. Mercer TR, Mattick JS. Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol. (2013) 20:300–7. doi: 10.1038/nsmb.2480
131. Kagami Y, Yoshida K. The functional role for condensin in the regulation of chromosomal organization during the cell cycle. Cell Mol Life Sci. (2016) 73:4591–8. doi: 10.1007/s00018-016-2305-z
132. Teif VB, Bohinc K. Condensed DNA: condensing the concepts. Prog Biophys Mol Biol. (2011) 105:208–22. doi: 10.1016/j.pbiomolbio.2010.07.002
133. Ravi M, Ramanathan S, Krishna K. Factors, mechanisms and implications of chromatin condensation and chromosomal structural maintenance through the cell cycle. J Cell Physiol. (2019) 235:758–75. doi: 10.1002/jcp.29038
134. Biswas S, Mallikarjuna Rao C. Epigenetic tools (the writers, the readers and the rasers) and their implications in cancer therapy. Eur J Pharmacol. (2018) 837:8–24. doi: 10.1016/j.ejphar.2018.08.021
135. Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, et al. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. (2005) 435:1262–6. doi: 10.1038/nature03672
136. Stott-Miller M, Zhao S, Wright JL, Kolb S, Bibikova M, Klotzle B, et al. Validation study of genes with hypermethylated promoter regions associated with prostate cancer recurrence. Cancer Epidemiol Biomarkers Prev. (2014) 23:1331–9. doi: 10.1158/1055-9965.EPI-13-10000
137. Terry MB, McDonald JA, Wu HC, Eng S, Santella RM. Epigenetic biomarkers of breast cancer risk: across the breast cancer prevention continuum. Adv Exp Med Biol. (2016) 882:33–68. doi: 10.1007/978-3-319-22909-6_2
138. Schutte M, Ogilvie LA, Rieke DT, Lange BMH, Yaspo ML, Lehrach H. Cancer precision medicine: why more is more and DNA is not enough. Public Health Genomics. (2017) 20:70–80. doi: 10.1159/000477157
139. Soldatov R, Kaucka M, Kastriti ME, Petersen J, Chontorotzea T, Englmaier L, et al. Spatiotemporal structure of cell fate decisions in murine neural crest. Science. (2019) 364:eaas9536. doi: 10.1126/science.aas9536
140. Powell DR, O'Brien JH, Ford HL, Artinger KB. Chapter 16 - neural crest cells and cancer: insights into tumor progression. In: Trainor PA, editor. Neural Crest Cells. Boston: Academic Press (2014). p. 335–57.
141. Vicent GP, Ballare C, Nacht AS, Clausell J, Subtil-Rodriguez A, Quiles I, et al. Induction of progesterone target genes requires activation of Erk and Msk kinases and phosphorylation of histone H3. Mol Cell. (2006) 24:367–81. doi: 10.1016/j.molcel.2006.10.011
142. Zhu P, Zhang C-B, Yang P, Chen J, Liu Y-Q, Hu H-M, et al. Phosphohistone H3 (pHH3) is a prognostic and epithelial to mesenchymal transition marker in diffuse gliomas. Oncotarget. (2016) 7:45005–14. doi: 10.18632/oncotarget.7154
143. Ju J, Chen A, Deng Y, Liu M, Wang Y, Wang Y, et al. NatD promotes lung cancer progression by preventing histone H4 serine phosphorylation to activate slug expression. Nat Commun. (2017) 8:928. doi: 10.1038/s41467-017-00988-5
144. Kalb R, Mallery DL, Larkin C, Huang JTJ, Hiom K. BRCA1 is a histone-H2A-specific ubiquitin ligase. Cell Rep. (2014) 8:999–1005. doi: 10.1016/j.celrep.2014.07.025
145. Benitz S, Regel I, Reinhard T, Popp A, Schäffer I, Raulefs S, et al. Polycomb repressor complex 1 promotes gene silencing through H2AK119 mono-ubiquitination in acinar-to-ductal metaplasia and pancreatic cancer cells. Oncotarget. (2015) 7:11424–33. doi: 10.18632/oncotarget.6717
146. Jeusset LMP, McManus KJ. Developing targeted therapies that exploit aberrant histone ubiquitination in cancer. Cells. (2019) 8:165. doi: 10.3390/cells8020165
147. Stadler SC, Vincent CT, Fedorov VD, Patsialou A, Cherrington BD, Wakshlag JJ, et al. Dysregulation of PAD4-mediated citrullination of nuclear GSK3β activates TGF-β signaling and induces epithelial-to-mesenchymal transition in breast cancer cells. Proc Natl Acad Sci USA. (2013) 110:11851–6. doi: 10.1073/pnas.1308362110
148. DeVore SB, Young CH, Li G, Sundararajan A, Ramaraj T, Mudge J, et al. Histone citrullination represses MicroRNA expression, resulting in increased oncogene mRNAs in somatolactotrope cells. Mol Cell Biol. (2018) 38:e00084. doi: 10.1128/MCB.00084-18
149. Grilz E, Mauracher LM, Posch F, Konigsbrugge O, Zochbauer-Muller S, Marosi C, et al. Citrullinated histone H3, a biomarker for neutrophil extracellular trap formation, predicts the risk of mortality in patients with cancer. Br J Haematol. (2019) 186:311–20. doi: 10.1055/s-0039-1680128
150. Shiio Y, Eisenman RN. Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci USA. (2003) 100:13225–30. doi: 10.1073/pnas.1735528100
151. Bawa-Khalfe T, Lu L-S, Zuo Y, Huang C, Dere R, Lin F-M, et al. Differential expression of SUMO-specific protease 7 variants regulates epithelial-mesenchymal transition. Proc Natl Acad Sci USA. (2012) 109:17466–71. doi: 10.1073/pnas.1209378109
152. Dhall A, Weller CE, Chu A, Shelton PMM, Chatterjee C. Chemically sumoylated histone H4 stimulates intranucleosomal demethylation by the LSD1-CoREST complex. ACS Chem Biol. (2017) 12:2275–80. doi: 10.1021/acschembio.7b00716
153. So AKC, Kaur J, Kak I, Assi J, MacMillan C, Ralhan R, et al. Biotinidase is a novel marker for papillary thyroid cancer aggressiveness. PLoS ONE. (2012) 7:e40956. doi: 10.1371/journal.pone.0040956
154. Jaguva Vasudevan AA, Hoffmann MJ, Beck MLC, Poschmann G, Petzsch P, Wiek C, et al. HDAC5 expression in urothelial carcinoma cell lines inhibits long-term proliferation but can promote epithelial-to-mesenchymal transition. Int J Mol Sci. (2019) 20:E2135. doi: 10.3390/ijms20092135
155. Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, et al. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J Biol Chem. (2010) 285:26114–20. doi: 10.1074/jbc.M109.089433
156. Collins RE, Northrop JP, Horton JR, Lee DY, Zhang X, Stallcup MR, et al. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat Struct Mol Biol. (2008) 15:245–50. doi: 10.1038/nsmb.1384
157. Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science. (2006) 312:748–51. doi: 10.1126/science.1125162
158. Heyn P, Logan CV, Fluteau A, Challis RC, Auchynnikava T, Martin C-A, et al. Gain-of-function DNMT3A mutations cause microcephalic dwarfism and hypermethylation of Polycomb-regulated regions. Nat Genet. (2019) 51:96–105. doi: 10.1038/s41588-018-0274-x
159. Chen L-Y, Huang Y-C, Huang S-T, Hsieh Y-C, Guan H-H, Chen N-C, et al. Domain swapping and SMYD1 interactions with the PWWP domain of human hepatoma-derived growth factor. Sci Rep. (2018) 8:287. doi: 10.1038/s41598-017-18510-8
160. Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG, et al. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J Clin Invest. (2012) 122:1469–86. doi: 10.1172/JCI57349
161. Casciello F, Windloch K, Gannon F, Lee JS. Functional role of G9a histone methyltransferase in cancer. Front Immunol. (2015) 6:487. doi: 10.3389/fimmu.2015.00487
162. Matias PM, Baek SH, Bandeiras TM, Dutta A, Houry WA, Llorca O, et al. The AAA+ proteins pontin and reptin enter adult age: from understanding their basic biology to the identification of selective inhibitors. Front Mol Biosci. (2015) 2:17. doi: 10.3389/fmolb.2015.00017
163. Zhang T, Termanis A, Özkan B, Bao XX, Culley J, de Lima Alves F, et al. G9a/GLP complex maintains imprinted DNA methylation in embryonic stem cells. Cell Rep. (2016) 15:77–85. doi: 10.1016/j.celrep.2016.03.007
164. Liu N, Zhang Z, Wu H, Jiang Y, Meng L, Xiong J, et al. Recognition of H3K9 methylation by GLP is required for efficient establishment of H3K9 methylation, rapid target gene repression, and mouse viability. Genes Dev. (2015) 29:379–93. doi: 10.1101/gad.254425.114
165. Mauger O, Klinck R, Chabot B, Muchardt C, Allemand E, Batsche E. Alternative splicing regulates the expression of G9A and SUV39H2 methyltransferases, and dramatically changes SUV39H2 functions. Nucleic Acids Res. (2015) 43:1869–82. doi: 10.1093/nar/gkv013
166. Patounas O, Papacharalampous I, Eckerich C, Markopoulos GS, Kolettas E, Fackelmayer FO. A novel splicing isoform of protein arginine methyltransferase 1 (PRMT1) that lacks the dimerization arm and correlates with cellular malignancy. J Cell Biochem. (2018) 119:2110–23. doi: 10.1002/jcb.26373
167. Jiang H, Zhou Z, Jin S, Xu K, Zhang H, Xu J, et al. PRMT9 promotes hepatocellular carcinoma invasion and metastasis via activating PI3K/Akt/GSK-3beta/Snail signaling. Cancer Sci. (2018) 109:1414–27. doi: 10.1111/cas.13598
168. Beltrán-García J, Osca-Verdegal R, Mena-Mollá S, García-Giménez JL. Epigenetic IVD tests for personalized precision medicine in cancer. Front Genet. (2019) 10:621. doi: 10.3389/fgene.2019.00621
169. Ozboyaci M, Gursoy A, Erman B, Keskin O. Molecular recognition of H3/H4 histone tails by the tudor domains of JMJD2A: a comparative molecular dynamics simulations study. PLoS ONE. (2011) 6:e14765. doi: 10.1371/journal.pone.0014765
170. Yang W-S, Campbell M, Chang P-C. SUMO modification of a heterochromatin histone demethylase JMJD2A enables viral gene transactivation and viral replication. PLoS Pathog. (2017) 13:e1006216. doi: 10.1371/journal.ppat.1006216
171. Ginjala V, Rodriguez-Colon L, Ganguly B, Gangidi P, Gallina P, Al-Hraishawi H, et al. Protein-lysine methyltransferases G9a and GLP1 promote responses to DNA damage. Sci Rep. (2017) 7:16613. doi: 10.1038/s41598-017-16480-5
172. Chen W, Zhu Q, Liu Y, Zhang Q. Chapter nine - chromatin remodeling and plant immunity. In: Donev R, editor. Advances in Protein Chemistry and Structural Biology. Swansea: Academic Press (2017). p. 243–60.
173. Tyagi M, Imam N, Verma K, Patel AK. Chromatin remodelers: we are the drivers!! Nucleus. (2016) 7:388–404. doi: 10.1080/19491034.2016.1211217
174. Warns JA, Davie JR, Dhasarathy A. Connecting the dots: chromatin and alternative splicing in EMT. Biochem Cell Biol. (2016) 94:12–25. doi: 10.1139/bcb-2015-0053
175. Marquez-Vilendrer SB, Rai SK, Gramling SJ, Lu L, Reisman DN. Loss of the SWI/SNF ATPase subunits BRM and BRG1 drives lung cancer development. Oncoscience. (2016) 3:322–36. doi: 10.18632/oncoscience.323
176. Wilson MR, Reske JJ, Holladay J, Wilber GE, Rhodes M, Koeman J, et al. ARID1A and PI3-kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. Nat Commun. (2019) 10:3554. doi: 10.1038/s41467-019-11403-6
177. Nagarajan S, Rao SV, Sutton J, Cheeseman D, Dunn S, Papachristou EK, et al. ARID1A influences HDAC1/BRD4 activity, intrinsic proliferative capacity and breast cancer treatment response. Nat Genet. (2020) 52:187–97. doi: 10.1038/s41588-019-0541-5
178. Box JK, Paquet N, Adams MN, Boucher D, Bolderson E, O'Byrne KJ, et al. Nucleophosmin: from structure and function to disease development. BMC Mol Biol. (2016) 17:19. doi: 10.1186/s12867-016-0073-9
179. Liu Y, Zhang F, Zhang X-F, Qi L-S, Yang L, Guo H, et al. Expression of nucleophosmin/NPM1 correlates with migration and invasiveness of colon cancer cells. J Biomed Sci. (2012) 19:53. doi: 10.1186/1423-0127-19-53
180. Kaowinn S, Kim J, Lee J, Shin DH, Kang C-D, Kim D-K, et al. Cancer upregulated gene 2 induces epithelial-mesenchymal transition of human lung cancer cells via TGF-β signaling. Oncotarget. (2017) 8:5092–110. doi: 10.18632/oncotarget.13867
181. Lin J, Kato M, Nagata K, Okuwaki M. Efficient DNA binding of NF-κB requires the chaperone-like function of NPM1. Nucleic Acids Res. (2017) 45:3707–23. doi: 10.1093/nar/gkw1285
182. Formosa T. The role of FACT in making and breaking nucleosomes. Biochim Biophys Acta. (2012) 1819:247–55. doi: 10.1016/j.bbagrm.2011.07.009
183. Koman IE, Commane M, Paszkiewicz G, Hoonjan B, Pal S, Safina A, et al. Targeting FACT complex suppresses mammary tumorigenesis in Her2/neu transgenic mice. Cancer Prev Res. (2012) 5:1025–35. doi: 10.1158/1940-6207.CAPR-11-0529
184. Kemble DJ, Whitby FG, Robinson H, McCullough LL, Formosa T, Hill CP. Structure of the Spt16 middle domain reveals functional features of the histone chaperone FACT. J Biol Chem. (2013) 288:10188–94. doi: 10.1074/jbc.C113.451369
185. Zhang W, Zeng F, Liu Y, Shao C, Li S, Lv H, et al. Crystal structure of human SSRP1 middle domain reveals a role in DNA binding. Sci Rep. (2015) 5:18688. doi: 10.1038/srep18688
186. Marcianò G, Da Vela S, Tria G, Svergun DI, Byron O, Huang DT. Structure specific recognition protein-1 (SSRP1) is an elongated homodimer that binds histones. J Biol Chem. (2018) 293:10071–83. doi: 10.1074/jbc.RA117.000994
187. Wu W, He K, Guo Q, Chen J, Zhang M, Huang K, et al. SSRP1 promotes colorectal cancer progression and is negatively regulated by miR-28-5p. J Cell Mol Med. (2019) 23:3118–29. doi: 10.1111/jcmm.14134
188. Bedi U, Scheel AH, Hennion M, Begus-Nahrmann Y, Ruschoff J, Johnsen SA. SUPT6H controls estrogen receptor activity and cellular differentiation by multiple epigenomic mechanisms. Oncogene. (2015) 34:465–73. doi: 10.1038/onc.2013.558
189. Wang AH, Zare H, Mousavi K, Wang C, Moravec CE, Sirotkin HI, et al. The histone chaperone Spt6 coordinates histone H3K27 demethylation and myogenesis. EMBO J. (2013) 32:1075–86. doi: 10.1038/emboj.2013.54
190. Choi H-J, Park J-H, Park M, Won H-Y, Joo H-S, Lee CH, et al. UTX inhibits EMT-induced breast CSC properties by epigenetic repression of EMT genes in cooperation with LSD1 and HDAC1. EMBO Rep. (2015) 16:1288–98. doi: 10.15252/embr.201540244
191. Kuo KT, Huang WC, Bamodu OA, Lee WH, Wang CH, Hsiao M, et al. Histone demethylase JARID1B/KDM5B promotes aggressiveness of non-small cell lung cancer and serves as a good prognostic predictor. Clin Epigenetics. (2018) 10:107. doi: 10.1186/s13148-018-0533-9
192. Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, et al. HMGB1 in health and disease. Mol Aspects Med. (2014) 40:1–116. doi: 10.1016/j.mam.2014.05.001
193. Zhang J, Shao S, Han D, Xu Y, Jiao D, Wu J, et al. High mobility group box 1 promotes the epithelial-to-mesenchymal transition in prostate cancer PC3 cells via the RAGE/NF-κB signaling pathway. Int J Oncol. (2018) 53:659–71. doi: 10.3892/ijo.2018.4420
194. Li H, Li J, Zhang G, Da Q, Chen L, Yu S, et al. HMGB1-induced p62 overexpression promotes snail-mediated epithelial-mesenchymal transition in glioblastoma cells via the degradation of GSK-3β. Theranostics. (2019) 9:1909–22. doi: 10.7150/thno.30578
195. Ferrari S, Ronfani L, Calogero S, Bianchi ME. The mouse gene coding for high mobility group 1 protein (HMG1). J Biol Chem. (1994) 269:28803–8.
196. Spada F, Brunet A, Mercier Y, Renard JP, Bianchi ME, Thompson EM. High mobility group 1 (HMG1) protein in mouse preimplantation embryos. Mech Dev. (1998) 76:57–66. doi: 10.1016/S0925-4773(98)00095-1
197. Luo J, Chen J, Li H, Yang Y, Yun H, Yang S, et al. LncRNA UCA1 promotes the invasion and EMT of bladder cancer cells by regulating the miR-143/HMGB1 pathway. Oncol Lett. (2017) 14:5556–62. doi: 10.3892/ol.2017.6886
198. Wang L, Kang F-B, Wang J, Yang C, He D-W. Downregulation of miR-205 contributes to epithelial-mesenchymal transition and invasion in triple-negative breast cancer by targeting HMGB1-RAGE signaling pathway. Anticancer Drugs. (2019) 30:225–32. doi: 10.1097/CAD.0000000000000705
199. Mignone F, Gissi C, Liuni S, Pesole G. Untranslated regions of mRNAs. Genome Biol. (2002) 3:1–10 doi: 10.1186/gb-2002-3-3-reviews0004
200. Kaufman PD, Kobayashi R, Kessler N, Stillman B. The p150 and p60 subunits of chromatin assembly factor I: a molecular link between newly synthesized histones and DNA replication. Cell. (1995) 81:1105–14. doi: 10.1016/S0092-8674(05)80015-7
201. Verreault A, Kaufman PD, Kobayashi R, Stillman B. Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell. (1996) 87:95–104. doi: 10.1016/S0092-8674(00)81326-4
202. Endo A, Ly T, Pippa R, Bensaddek D, Nicolas A, Lamond AI. The chromatin assembly factor complex 1 (CAF1) and 5-azacytidine (5-AzaC) affect cell motility in src-transformed human epithelial cells. J Biol Chem. (2017) 292:172–84. doi: 10.1074/jbc.M116.751024
203. Xu M, Jia Y, Liu Z, Ding L, Tian R, Gu H, et al. Chromatin assembly factor 1, subunit A (P150) facilitates cell proliferation in human hepatocellular carcinoma. Onco Targets Ther. (2016) 9:4023–35. doi: 10.2147/OTT.S107050
204. Xia D, Yang X, Liu W, Shen F, Pan J, Lin Y, et al. Over-expression of CHAF1A in epithelial ovarian cancer can promote cell proliferation and inhibit cell apoptosis. Biochem Biophys Res Commun. (2017) 486:191–7. doi: 10.1016/j.bbrc.2017.03.026
205. Staibano S, Mascolo M, Mancini FP, Kisslinger A, Salvatore G, Di Benedetto M, et al. Overexpression of chromatin assembly factor-1 (CAF-1) p60 is predictive of adverse behaviour of prostatic cancer. Histopathology. (2009) 54:580–9. doi: 10.1111/j.1365-2559.2009.03266.x
Keywords: EMT, cancer, gene regulation, epigenetics, signaling, transcription factors, chromatin
Citation: Lavin DP and Tiwari VK (2020) Unresolved Complexity in the Gene Regulatory Network Underlying EMT. Front. Oncol. 10:554. doi: 10.3389/fonc.2020.00554
Received: 11 January 2020; Accepted: 27 March 2020;
Published: 12 May 2020.
Edited by:
Katarína Smolková, Institute of Physiology (ASCR), CzechiaReviewed by:
Eric Hervouet, Université Bourgogne Franche-Comté, FranceParames C. Sil, Bose Institute, India
Copyright © 2020 Lavin and Tiwari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vijay K. Tiwari, di50aXdhcmlAcXViLmFjLnVr