Abstract
The cell-microenvironment communication is essential for homing of hematopoietic stem cells in stromal niches. Recent evidences support the involvement of epithelial-to-mesenchymal (EMT) process in hematopoietic stem cell homeostasis as well as in leukemia cells invasiveness and migration capability. Here, we demonstrate that the alteration of iron homeostasis and the consequent increase of redox metabolism, mediated by the stable knock down of ferritin heavy chain (FtH), enhances the expression of CXCR4 in K562 erythroleukemia cells, thus promoting CXCL12-mediated motility. Indeed, addition of the CXCR4 receptor antagonist AMD3100 reverts this effect. Upon FtH knock down K562 cells also acquire an “EMT-like” phenotype, characterized by the increase of Snail, Slug and Vimentin with the parallel loss of E-cadherin. By using fibronectin as substrate, the cell adhesion assay further shows a reduction of cell adhesion capability in FtH-silenced K562 cells. Accordingly, confocal microscopy shows that adherent K562 control cells display a variety of protrusions while FtH-silenced K562 cells remain roundish. These phenomena are largely due to the reactive oxygen species (ROS)-mediated up-regulation of HIF-1α/CXCR4 axis which, in turn, promotes the activation of NF-κB and the enhancement of EMT features. These data are confirmed by treatments with either N-acetylcysteine (NAC) or AMD3100 or NF-κB inhibitor IκB-alpha which revert the FtH-silenced K562 invasive phenotype. Overall, our findings demonstrate the existence of a direct relationship among iron metabolism, redox homeostasis and EMT in the hematological malignancies. The effects of FtH dysregulation on CXCR4/CXCL12-mediated K562 cell motility extend the meaning of iron homeostasis in the leukemia cell microenvironment.
Introduction
The tumor microenvironment (TME) is a major player in cancer progression and several signals, such as oxygen supply, cytokines, and chemokines drive the communication between TME and tumor cells (1–3). The CXCL12/CXCR4 axis promotes tumor cell growth and propagation of distant metastases through the activation of epithelial-to-mesenchymal transition (EMT) (4). During EMT, cancer cells acquire features of mesenchymal-like cells including enhanced migratory and invasive abilities, changes in cellular adhesion and remodeling of the extracellular matrix (5, 6). Cancer cells expressing CXCR4 tend to home to secondary organs where its ligand CXCL12 is actively secreted, mainly by mesenchymal stromal cells (7).
Tumors rapidly exhaust the local oxygen supply creating a hypoxic environment which, in turn, promotes the overproduction of reactive oxygen species (ROS) (8). ROS can induce EMT, but the specificity of their action in the regulation of given EMT markers is dependent on the cellular context and the type of tissue (9–12). Numerous studies have explored the role of ROS in inducing both cell migration and EMT in solid cancer (13, 14). In this regard, we and others have previously reported that the knock down of ferritin heavy chain (FtH), the catalytic subunit of the human ferritin, promotes cell motility through either the activation of CXCR4 signaling or the induction of EMT in a variety of solid cancer in vitro models including breast and lung cancer cell lines (15–17).
The trafficking of tumor cells represents a key process that contributes to progression also of hematological malignancies such as myeloid and lymphoid leukemias or multiple myeloma (18, 19). A common feature of these tumors is the homing and infiltration of hematological cancer cells into the bone marrow (BM) which supports initiation, maintenance and proliferation of the malignant cells (7). Both homing and migration of leukemic stem cells are regulated by niche cells living in the BM through the activation of the CXCL12/CXCR4 axis signaling (20–22). Indeed, blocking CXCL12 binding to CXCR4 with the specific CXCR4 inhibitor AMD3100 disrupts hematological neoplastic cells interaction with the BM microenvironment (21).
In chronic myelogenous leukemia (CML) cells, CXCR4 activates PI3K/AKT signaling pathway and promotes the translocation of NF-κB complexes into nucleus thereby decreasing the expression of pro-apoptotic proteins (23, 24). Moreover, CXCL12 activates pro-survival signal pathways including those mediated by MAPK, S-6-kinase, STAT3 and STAT5, and in vitro treatment with CXCR4 antagonists inhibits cell growth and induces cell death (25, 26). The molecular mechanisms regulating the expression of CXCR4 in hematological malignancies have therefore been largely investigated. Numerous evidences show that hypoxia in BM leads to increased HIF-1α transcriptional activity on CXCR4 expression resulting in enhanced migration and homing of circulating malignant cells to new BM niches (27–29).
During the last decade, EMT has gained increasing attention also in hematological malignancies. Few reports indicate that EMT-transcription factors (TFs), including Twist-1 and Slug, are implicated in hematopoietic stem cell self-renewal by interacting with stemness signaling key factors c-Myc and c-Kit (30, 31) while Slug up-regulation promotes leukemogenesis and confers resistance to apoptosis in leukemia cells (32). In addition, imatinib-resistant CML cells exhibit a so-called “EMT-like” phenotype along with increased invasion and migration properties both in vitro and in vivo (33). Overall these data suggest that EMT might play significant role in inducing tumor dissemination and thus chemoresistance also in hematological malignancies; however, this topic still has remarkable gaps to overwhelm.
In this study, we address for the first time the role of FtH-induced ROS increase in bestowing mesenchymal properties to hematological cells. To achieve this goal, we defined the effects of FtH knock down in the induction of EMT markers, activation of CXCR4/CXCL12 signaling pathway and migration of K562 erythroleukemia cells, and further attempted to understand the molecular mechanisms involved.
Materials and Methods
Cell Culture and Treatment
K562, a human erythroleukemia cell line (ATCC number CCL-243), was cultured as described in Di Sanzo et al. (34). The human stromal cells HS5, were cultured in DMEM medium supplemented with 10% fetal bovine serum and antibiotics at 37°C in an atmosphere of humidified air containing 5% CO2. Lentiviral preparations and transductions were performed as previously described using a shRNA as control (K562shRNA) or a shRNA that targets the 196–210 region of the FtH mRNA (K562shFtH) (35). All the experiments were performed using a puromycin-selected pool of clones (1 μg/mL) (Sigma Aldrich, St. Louis, MI, USA). K562 cells were transfected using the Nucleofector system from Amaxa (Lonza, Basel, Switzerland) according to the manufacturer's optimized protocol. To evaluate the role of NF-κB in inducing EMT-like features, we over-expressed the NF-κB inhibitor IκB-α using a homemade pRc/CMV-HA-IκB-α plasmid and its empty control kindly provided by Prof. Ileana Quinto (Magna Graecia University of Catanzaro, Italy) as previously described by Aversa et al. (36). CXCL12 was added to K562 cell culture medium at a final concentration of 100 ng/ml. N-acetylcysteine (NAC) was added to the K562 cell culture medium at a final concentration of 5 mM for 2 h. Plerixafor (AMD3100) was added to the K562 cell culture medium at a final concentration of 10 μM for 1 h.
Protein Extractions
Protein extractions were performed on K562shRNA, K562shFtH, K562shFtH/pRc/CMV, K562shFtH/pRc/CMV−3HA−IκB and NAC treated cells. Briefly, for total protein extractions, K562 cells were lysed in ice-cold radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitors as described by Zolea et al. (37). For the quantification of nuclear p65 amounts, protein extraction from nucleus was performed as previously described by Aversa et al. (36).
Western Blotting Analysis
A total of 40 μg protein extract was boiled for 10 min in SDS sample buffer, separated by 12% SDS-PAGE and transferred to a nitrocellulose membrane by electroblotting as reported in Di Sanzo et al. (38). The nitrocellulose membranes were incubated overnight at 4°C with the following antibodies: (a) anti-CXCR4 (1:500; Abcam), (b) anti-HIF-1α (H-206) (1:200; Santa Cruz Biotechnology), (c) anti-p65 (C-20) (sc-372, 1:1,000; Santa Cruz Biotechnology), (d) anti-HDAC (1:5,000; Sigma-Aldrich), (e) anti-HA probe (F-7) (1:1,000; Santa Cruz Biotechnology), (f) anti-Vimentin, (g) anti-E-cadherin, (h) anti-Snail, (i) anti-Slug (1:1,000; Cell Signaling Technology, Danvers, MA, USA), (l) anti-FtH (1:200; Santa Cruz Biotechnology), (m) anti-γ-Tubulin (C-20) (1:2,000; Santa Cruz Biotechnology), (n) anti-Nucleolin (D4C7O) (1:1,000; Cell Signaling Technology) over-night at 4°C, followed by incubation with goat anti-rabbit and mouse anti-goat secondary antibodies (1:5,000; Santa Cruz Biotechnology). Membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies and immunoreactive bands were visualized with the ECL Western blotting detection system (BioRad, Hercules, CA, USA).
Quantification of CXCR4 Surface Expression
K562 cells (2 × 105) were harvested and rinsed once. Then, the cells were incubated with anti-CXCR4 antibody (1:400; Abcam) for 1 h at 4°C. After primary antibody incubation, the cells were rinsed with 1X PBS and incubated for 30 min with Alexa Fluor 633 donkey anti-goat antibody (H+L) (1:400) resuspended in 5 mg/ml BSA and 0.76 mg/ml EDTA. The cells were rinsed with 1X PBS, resuspended in 300 μl 1X PBS and evaluated by a FACS BD LSRFortessaTM X-20 cytofluorometer (BD Biosciences, San Jose, CA, USA).
Immunofluorescence
K562 cells were cultured on cover slip coated with fibronectin for 24 h. Samples preparation was performed as reported in Biamonte et al. (39). Thereafter, these cover slips were incubated for 1 h with primary antibodies anti-Vimentin (clone V9, ready to use, Dako) and 1 h with secondary antibody FITC-conjugated anti-mouse diluted in blocking buffer. For E-cadherin staining, the cover slips were incubated overnight at 4°C with primary antibody (clone 24E10, 1:200, Cell Signaling) in a humidified room, and for 1 h with Alexa Fluor 488-conjugated anti-rabbit, both diluted in blocking buffer. To stain actin filaments, cells were incubated for 30 min in this buffer containing Alexa Fluor 488 phalloidin at 1:40 dilution (Thermo Fisher Scientific, Waltham, Massachusetts, USA). After 3 washes with PBS, nuclear DAPI (1:500, Invitrogen, Carlsbad, CA) was added for 20 min. The samples were mounted on microscope slides using a mounting solution ProLong Gold antifade reagent (Thermo Fisher Scientific). Images were collected using a Leica DM-IRB/TC-SP2 confocal microscopy system (63× objective).
Cell Adhesion Assay
K562shRNA, K562shFtH and K562shFtH NAC-treated cells were subjected to adhesion assays using Fibronectin as an adhesion substrate. Briefly, 6-well flat-bottom plates were incubated overnight at 37°C with 5 μg/cm2 of Fibronectin in PBS. After gentle washing with PBS and incubation with 1% BSA for 1 h at room temperature, cells (3 × 104 cells/well) were added and allowed to adhere for 24 h at 37°C. Non-adherent cells were then removed by washing each well with PBS and adherent cells were counted using the cell count function in Image J 1.42 software, on ten fields per well. Each field consisted of a photo obtained at 200× magnification.
Migration Assay
K562shRNA, K562shFtH and K562shFtH NAC-treated cells were used to the migration assay using as chemoattractant CXCL12 chemokine as already described by Aversa et al. (15). After 18 h of incubation, the upper chambers were removed, and the cells in the lower chambers were counted using an optical microscope. Migration assay using the conditioned media derived from stromal cells HS5 (HS5-CM) was assessed in 24-well plate and polycarbonate filters with an 8.0 μm pore size. First, the supernatant media of HS5 cells was collected after 12 h and passed through a 0.45 μm filter. Briefly, 2 × 105 K562 cells were harvested, suspended in 200 μl serum-free RPMI with 1% BSA and placed in the upper chamber. The lower chambers contained 500 μl of HS5-CM. The plates were incubated at 37°C in 5 % CO2 for 8 h. The upper chambers were removed, and the cells in the lower chambers were counted using an optical microscope. The cell migration is expressed as the percentage of increase compared with the corresponding control.
ROS Detection
ROS were determined by incubating 2 × 105 K562shRNA, K562shFtH and K562shFtH cells NAC-treated with 1μM redox-sensitive probe 2′-7′-DCF (CM-H2CFDA; MolecularProbes, Eugene, OR, USA) for 30 min at 37°C. Afterward, pellet was washed twice with 1X PBS, then the pellet was resuspended in 1X PBS and analyzed using a FACS BD LSRFortessaTM X-20 cytofluorometer (BD Biosciences).
RNA Isolation and qPCR Analysis
Total RNA isolation was performed as previously reported in Sottile et al. (40). Gene expression analysis was assessed by real-time PCR using the cDNA obtained from K562shRNA, K562shFtH, K562shFtH/pRc/CMV, K562shFtH/pRc/CMV−3HA−IκB cells and K562shFtH cells treated with NAC.
Real time PCR was performed ad reported in Biamonte et al. (41). Briefly, 50 ng of cDNA was amplified in 20 μl of reaction mix containing Power SYBR Green PCR Master mix (Thermo Fisher Scientific) and the expression of CXCR4, FtH, E-cadherin, HIF-1α, Snail, Slug and Vimentin were analyzed. The human GAPDH cDNA fragment was amplified as the internal control. Data analysis was performed using the 2−ΔΔCt.
Statistical Analysis
All experiments were conducted at least three times, and the results are reported as mean values ± standard deviations (SD). Data analysis was performed by Student's-t-test assuming equal variances. p-values ≤ 0.05 were considered statistically significant.
Results
FtH Knock Down Promotes CXCL12/CXCR4 Axis Activation and Motility in K562 Cells
Earlier studies by others and us indicate that FtH interacts with internalized olo-CXCR4 receptor thereby suppressing the downstream signaling pathway in a variety of epithelial tumor cells (15, 16). As experimental model we used a pool of erythroleukemia K562 cell clones stably silenced for FtH (K562shFtH). FtH mRNA and protein expression levels in K562shFtH and in K562shRNA control cells are reported in Figure S1A. To assess the role of FtH on CXCR4 signaling in hematological tumors, we first measured CXCR4 levels in K562shFtH and in K562shRNA and we found that FtH knock down significantly enhanced CXCR4 expression at both mRNA and protein levels (Figure 1A). Flow cytometry analysis revealed an increase of about 25% in CXCR4 cell surface expression in K562shFtH cells compared to K562shRNA control cells (Figure 1B). Next, to determine whether the CXCR4 increase modulates the cell migration ability, we cultured both K562shRNA and K562shFtH cells either in RPMI medium supplemented with 100 ng/ml CXCL12 or in a HS5 mesenchymal stromal cells-conditioned medium for 8 h. As controls, we prepared parallel cultures of K562 cells with RPMI complete medium alone (data not shown). Cell migration assays demonstrate that K562shFtH cells exhibit a higher migration ability of about 5-fold compared to K562 control cells upon exposure to both CXCL12 or HS5-conditioned medium. Accordingly, treatment with the specific CXCR4 inhibitor AMD3100 (10 μM for 8 h) significantly decreased the migration of K562shFtH in both modified culture media (Figures 1C,D).
Figure 1
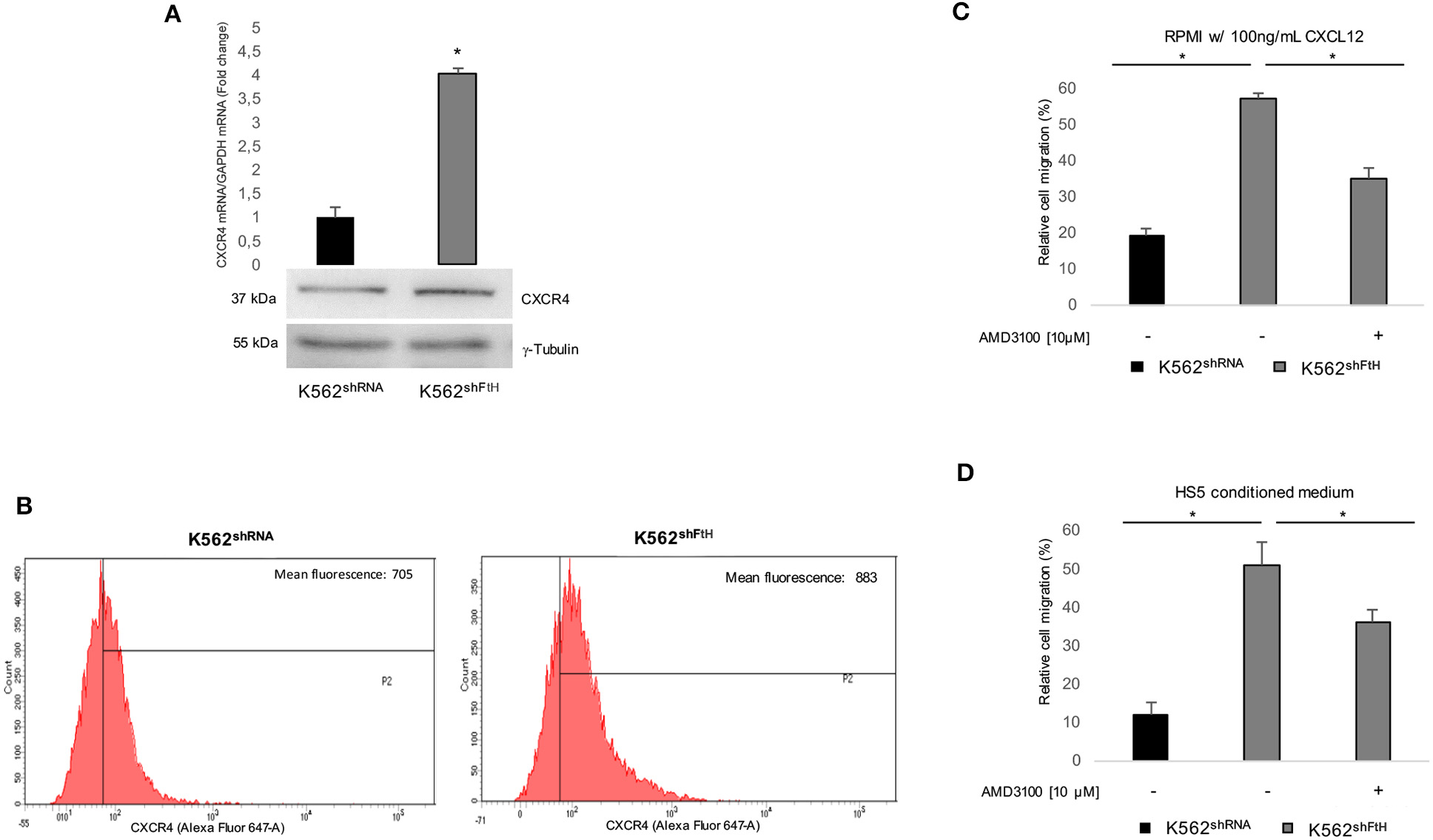
CXCR4/CXCL12 axis is activated in K562shFtH cells. (A) qPCR and western blot analysis of CXCR4 in K562shRNA and K562shFtH cells. Final results represent mean ± SD of three independent experiments, *p < 0.05. γ-Tubulin was used as loading control in WB experiments (B) Flow cytometry analysis of CXCR4 in K562shRNA and K562shFtH cells. Data are representative of three independent experiments (C) CXCL12 dependent-cell migration was examined in 24-well plates. Cells were placed in the upper chamber (8 μm) in nude RPMI. After 8 h, cells migrated toward medium containing CXCL12 (100 ng/ml) were counted using an optical microscope. Data are reported percentage of migrated cells. Final results represent mean ± SD of three independent experiments, *p < 0.05. (D) Migration analysis of K562shRNA and K562shFtH cells performed in 24-well plates. Cells were allowed to migrate for 8 h into the lower chamber containing conditioned medium of HS5 cells. Migrated cells were counted, and the percentage of migrated cells was calculated. Final results represent mean ± SD of three independent experiments, *p < 0.05.
FtH Silencing Induces Mesenchymal-Like Features in K562 Cells
We then explored the effect of FtH knock down on the classic EMT markers E-cadherin and Vimentin, as well as on the two EMT-transcription factors (EMT-Tfs) Snail and Slug. Real-time PCR and WB analyses clearly indicate a consistent increase in the steady-state amounts of both EMT-Tfs in K562shFtH cells compared to K562shRNA cells (Figures 2A,B). Accordingly, upon FtH silencing, the expression of Vimentin appears roughly triplicated in parallel with a significant decrease of E-cadherin amounts either at mRNA or protein level (Figures 2C–F).
Figure 2
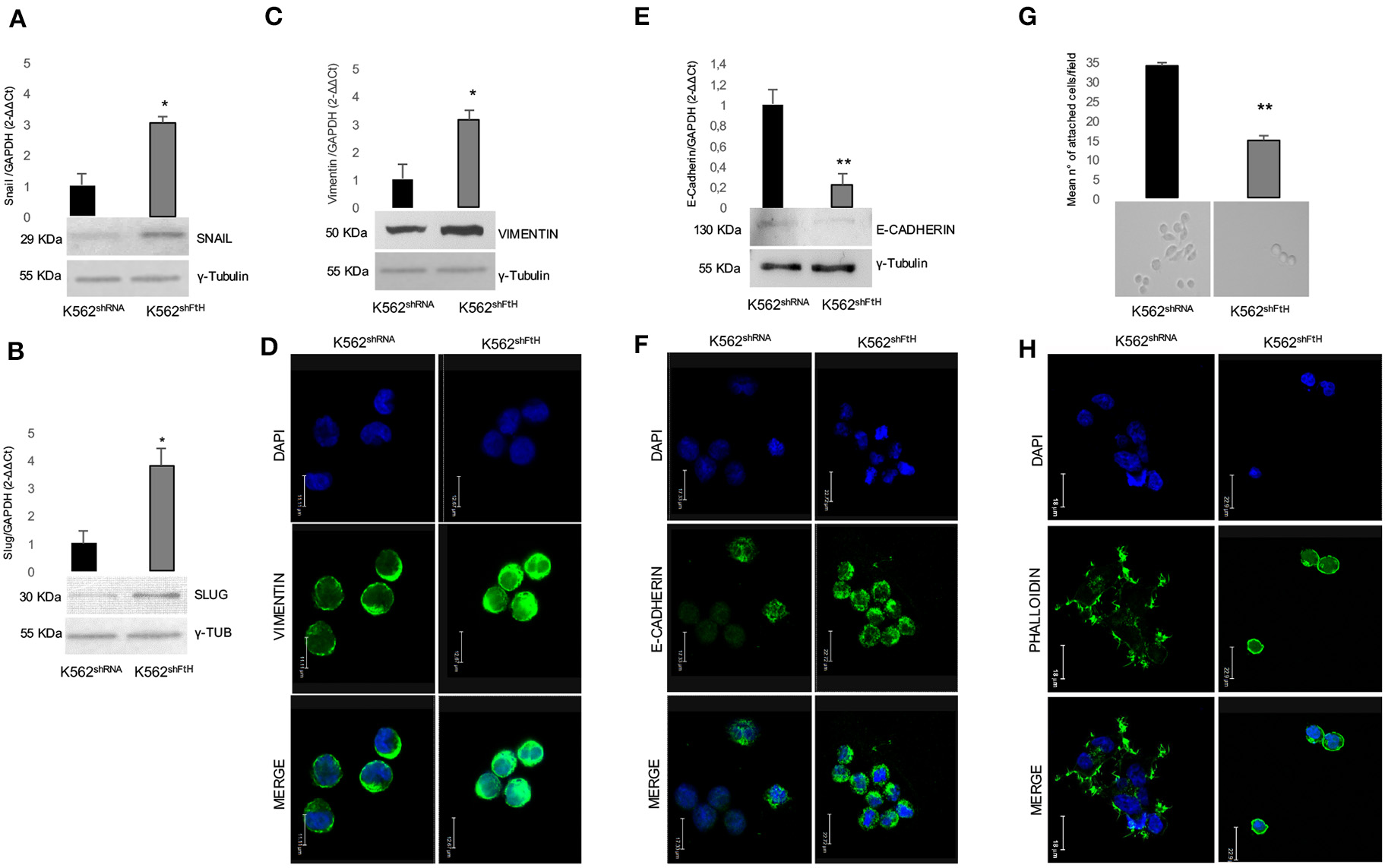
Analysis of EMT markers in K562 cells upon FtH silencing. qPCR and Western Blot analysis of (A) Snail, (B) Slug, (C) Vimentin and (E) E-Cadherin in K562shRNA and K562shFtH cells. Final results are reported as mean ± SD of three independent experiments, *p < 0.05. γ-Tubulin was used as loading control in WB experiments. Immunofluorescence staining for (D) Vimentin and (F) E-Cadherin in K562shRNA and K562shFtH cells. Images were collected using a Leica TCS SP2 confocal microscopy system (63X). Data are representative of three independent experiments (E). (G) Cell adhesion assay in K562shRNA and K562shFtH cells performed in a 6-well plate coated with 5 μg/mL fibronectin. Results are reported as the mean number of adherent cells counted per field upon 24 h through the optical microscope, **p < 0.01 (upper panel). A representative image of adherent K562shRNA and K562shFtH cells obtained from optical microscope (lower panel). (H) Representative images of immunofluorescence staining for Phalloidin in K562shRNA and K562shFtH cells. Images were collected using a Leica TCS SP2 confocal microscopy system (63X).
Besides increased motility, reduced cell-cell adhesion capability is a key feature of the EMT process. By using fibronectin as substrate, we analyzed the adhesion ability of K562shFtH and control cells, founding that the FtH-silenced cells halved their capability to adhere to the substrate (Figure 2G, upper panel). Optical imaging of K562shRNA cells reveals that they behave as adherent cells stucking to fibronectin through cell protrusion, while the fraction of adherent K562shFtH remains roundish (Figure 2G, lower panel). Confocal microscopy shows F-actin aggregates, cell-surface protrusions and/or extensions with a complex network of actin filaments (pseudopodia, lamellipodia) and actin bundles (filopodia) in K562shRNA cells. In K562shFtH cells, instead, F-actin is organized in a three-dimensional network beneath the plasma membrane, which likely accounts for the rounded shape of these cells (Figure 2H).
ROS Increase Induces CXCR4 Signaling and EMT Trans-Differentiation Process in K562shFtH Cells
We have already reported that FtH-silencing induces, in a variety of cell types including K562, a dysregulation of redox homeostasis ending in a consistent ROS overproduction (15, 36, 42, 43). Given the role of ROS in mediating the communication between tumor cell and tumor microenvironment (TME), we sought to explore the effects of the antioxidant agent NAC on both CXCR4 activation and EMT trans-differentiation process of K562shFtH. To this, we first re-determined the intracellular levels of ROS by using the DCF-DA assay. As shown in Figure 3A, a 4 h treatment with 5 mM NAC strongly reduced the amounts of ROS in the silenced cells. The decrease of ROS is accompanied by the reversal of the majority of the phenomena induced by FtH-silencing; indeed, the intracellular protein amount of CXCR4 is consistently reduced (Figure 3B) as well as its messenger RNA (Figure 3C) while its cell surface expression is only slightly affected (data not shown). Accordingly, EMT markers expression is down-regulated, with the exception of E-cadherin (Figure 3D), and the migratory ability is also consistently impaired (Figure 3E). The percentage of K562shFtH cells adherent to fibronectin substrate is increased (Figure 3F, upper panel), together with the lack of roundish morphology and with a partial recovery of cell protrusions (Figure 3F, lower panel).
Figure 3
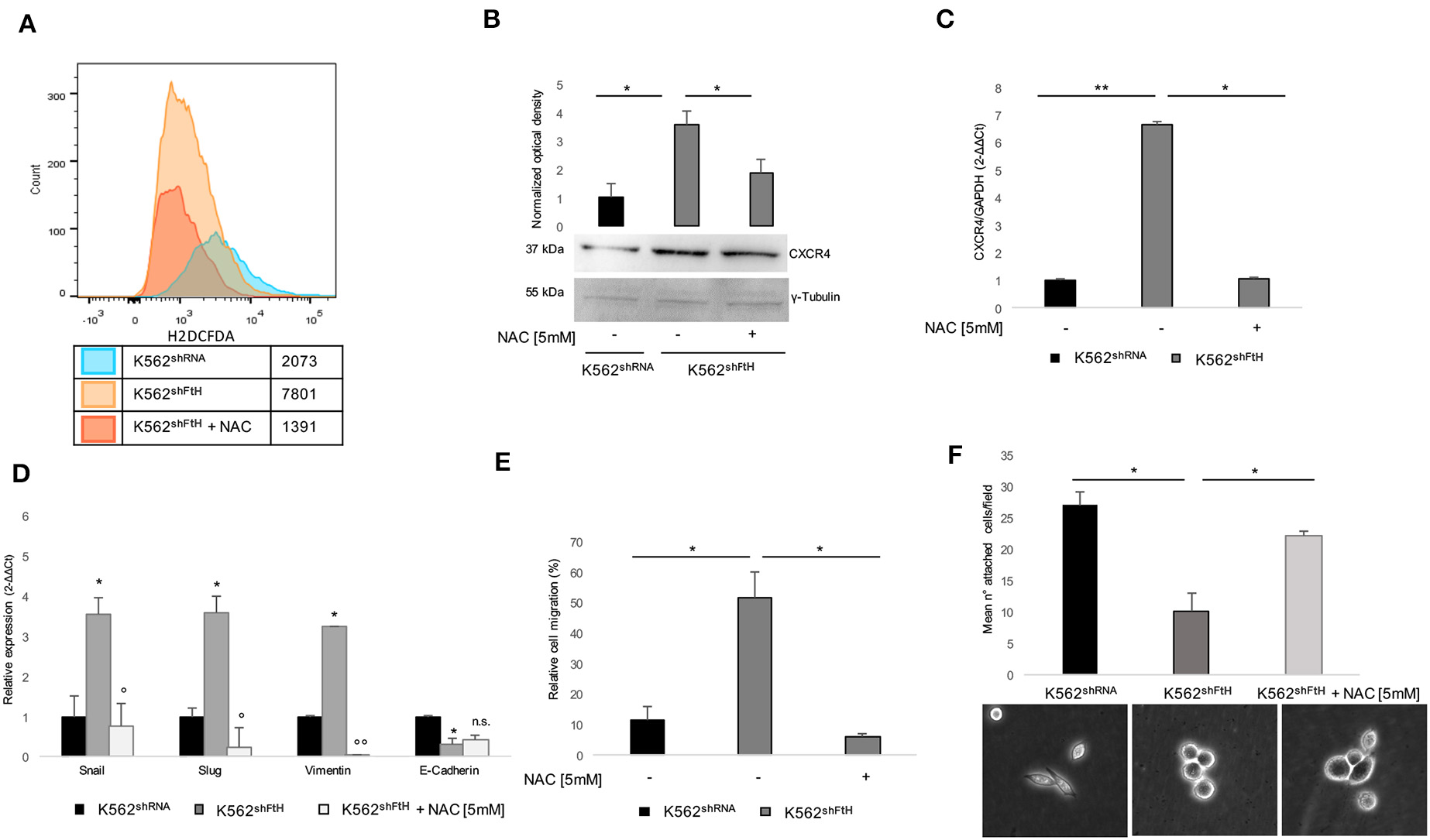
Effects of NAC treatment on EMT transdifferentiation in K562shFtH cells. (A) Flow cytometry analysis of ROS in K562shRNA and K562shFtH cells treated and not treated with 5 mM NAC for 2 h. Cells were labeled with H2DCFDA and the assay was performed in triplicate. (B) Western blot analysis (lower panel) and optical densitometry (upper panel) of CXCR4 in K562shRNA and K562shFtH cells treated and not treated with 5 mM NAC. Final results are reported as mean ± SD of three independent experiments, *p < 0.05. γ-Tubulin was used as loading control. (C) qPCR analysis of CXCR4 in K562shRNA and K562shFtH cells treated and not treated with 5 mM NAC. Final results represent mean ± SD of three independent experiments, *p < 0.05; **p < 0.01. (D) qPCR analysis of Snail, Slug, Vimentin, and E-Cadherin mRNA expression in K562shRNA, K562shFtH and K562shFtH cells treated with NAC. Final results represent mean ± SD of three independent experiments, *p < 0.05 compared to K562shRNA cells. °p < 0.05, °°p < 0.01 compared to K562shFtH cells. n.s., not significant. (E) Migration analysis in K562shRNA and K562shFtH cells treated and not treated with NAC was examined in 24-well plates. After 8 h, cells migrated toward CXCL12 (100 ng/ml) were counted using an optical microscope. Data are reported as the percentage of migrated cells. Final results represent mean ± SD of three independent experiments, *p < 0.05. (F) Cell adhesion assay in K562shRNA, K562shFtH and K562shFtH cells treated with NAC were cultured in a 6-well plate coated with 5 μg/mL fibronectin. After 24 h adherent cells were counted using an optical microscope; data are reported as mean of the number of adherent cells per field (upper panel), *p < 0.05. A representative image of adherent K562shRNA, K562shFtH and K562shFtH cells treated with NAC obtained from optical microscope (lower panel).
ROS Orchestrate EMT Trans-Differentiation Process by Acting on HIF-1α/CXCR4/NF-κB Axis in K562shFtH Cells
Next, we analyzed HIF-1α expression levels in the FtH silenced cells, given its function as transcriptional factor of CXCR4. As shown in Figures 4A,B, FtH silencing induced HIF-1α up-regulation at both mRNA and protein levels and this effect was significantly attenuated by treatment with 5 mM NAC for 2 h. It has been recently found that CXCR4 modulates PI3K/Akt/NF-κB signaling pathway and that both NF-κB and CXCR4 belong to a regulatory network driving the migration of cancer stem cells (24). Moreover, NF-κB is currently considered a master regulator of cancer cells aggressive phenotype through the direct transcriptional activation of EMT genes in solid tumors (10, 44, 45), and we recently demonstrated its ROS-mediated activation in K562shFtH cells (36).
Figure 4
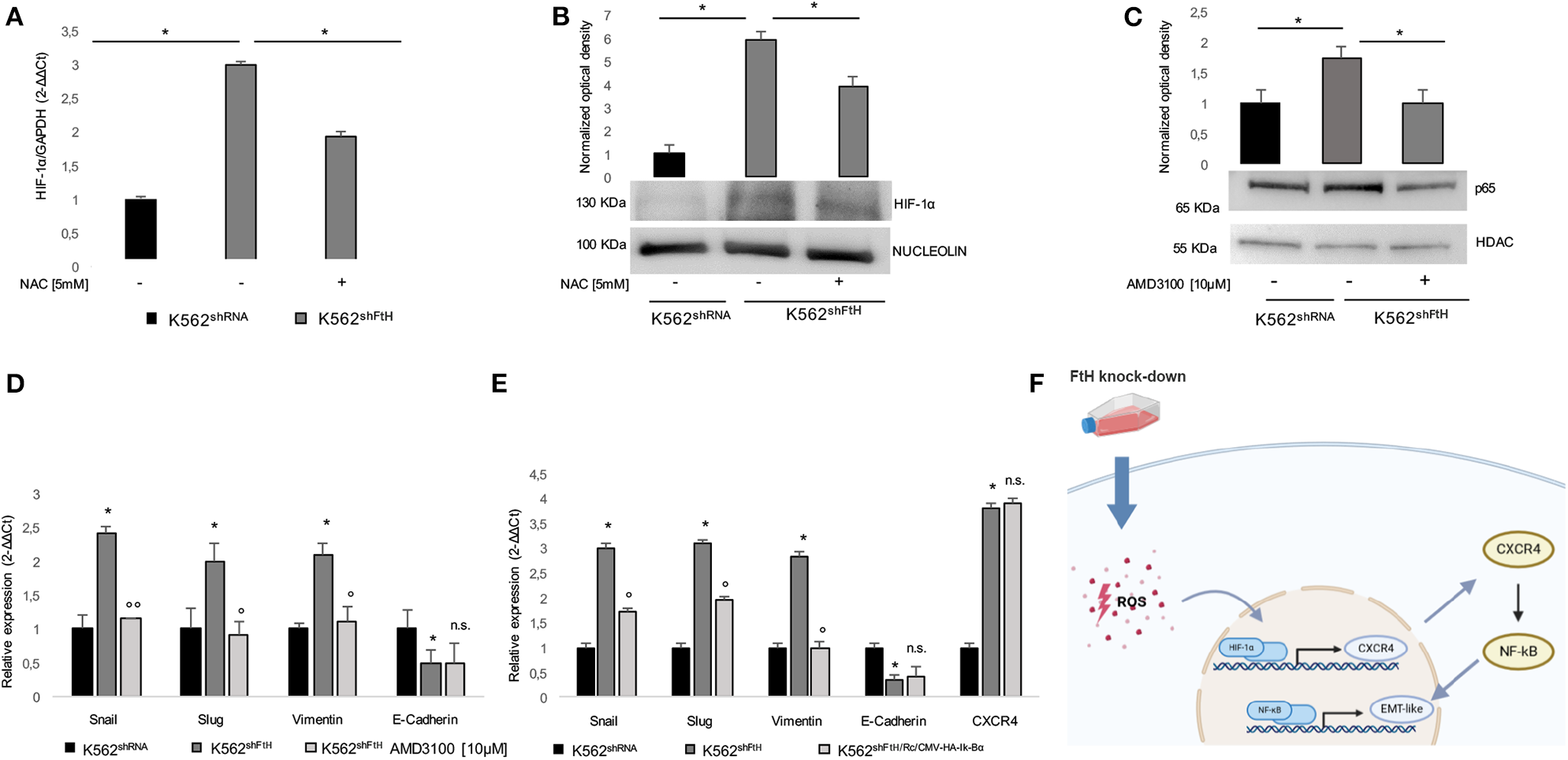
ROS/ HIF-1α/ CXCR4/ NF-kB axis activates EMT in K562shFtH cells. (A) qPCR analysis of HIF-1α in K562shRNA and K562shFtH cells treated and not treated with NAC. Final results represent mean ± SD of three independent experiments, *p < 0.05. (B) Western Blot analysis of HIF-1α (lower panel) and optical densitometry (upper panel) in K562shRNA and K562shFtH cells treated and not treated with NAC. Final results are reported as mean ± SD of three independent experiments, *p < 0.05. γ-Tubulin was used as loading control. (C) Western Blot analysis of p65 (lower panel) and optical densitometry (upper panel) in K562shRNA and K562shFtH cells treated and not treated with AMD3100. Final results are reported as mean ± SD of three independent experiments, *p < 0.05. HDAC was used as loading control of nuclear protein fraction. (D) qPCR analysis of Snail, Slug, Vimentin, and E-Cadherin mRNA expression in K562shRNA and K562shFtH cells treated and not treated with AMD3100. Final results represent mean ± SD of three independent experiments, *p < 0.05, compared to K562shRNA cells. °p < 0.05, °°p < 0.01, compared to KK562shFtH cells. n.s., not significant. (E) qPCR analysis of Snail, Slug, Vimentin, E-Cadherin, and CXCR4 mRNA expression in K562shRNA, K562shFtH and K562shFtH/Rc/CMV−HA−Ik−Bα cells. Final results represent mean ± SD of three independent experiments, *p < 0.05, compared to K562shRNA cells. °p < 0.05, compared to K562shFtH cells. n.s., not significant. (F) Cartoon of ROS/ HIF-1α/ CXCR4/ NF-kB axis activating EMT in K562shFtH cells.
Therefore, we assessed the effects of the CXCR4 inhibitor AMD3100 on the phenotype acquired by K562 cells upon FtH silencing. As shown in Figures 4C,D, p65 nuclear accumulation in K562shFtH cells was significantly reduced upon AMD3100 treatment and this was accompanied by a consistent decrease of all the EMT markers except for E-Cadherin that appeared unaffected. This effect was dependent on NF-κB transcriptional activity since blocking NF-κB with the specific inhibitor Ik-Bα suppressed Snail, Slug and Vimentin upregulation in K562shFtH cells (Figure 4E). The expression of Ik-Bα in K562shFtH cells is reported in Figure S1B.
These results strongly suggest that FtH is involved in bestowing K562 cells with more migratory and more mesenchymal-like features through the hypoxia-induced activation of CXCR4 and p65 transcriptional activation of selected EMT markers (Figure 4F).
Discussion
Tumor cell migration is a critical process that contributes to the development and progression of both solid and hematological malignancies (46, 47). In solid tumors, the EMT process enhances the metastatic potential converting polarized epithelial cells into non-polarized mesenchymal cells thus promoting cell mobility, invasion and resistance to apoptotic stimuli (48–50). In hematological malignancies, the blasts move from BM into peripheral blood and colonize distant sites such as liver and spleen, a process reminiscent of EMT in metastatic solid tumors (51–53). Moreover, it has been recently demonstrated that EMT transcription factors are critical in promoting leukemia and lymphoma progression (6, 18).
Many of the molecules driving homing and retention of leukemic cells in tissues have been identified (54, 55); among them, the CXCL12/CXCR4 axis has been shown to be essential for hematopoietic stem cell (HSC) migration and homing and also for cancer cell migration and metastasis (3, 20, 56). In particular, CXCR4 expression is necessary to keep the leukemic cells in the CXCL12-enriched BM microenvironment, and the efficient blockade of CXCR4 mobilizes the cells from the BM into the circulation (3, 19, 22).
In solid cancers, a tumor microenvironment that is rich in reactive oxygen species (ROS) promotes the binding of the hypoxia inducible factor subunit HIF-1α to its response element (HRE) in the promoter region of CXCR4, thus critically influencing CXCR4-mediated expression and functions, and ultimately encouraging cancer metastasis (27, 28). Hypoxia represents a key driver of metabolic reprograming also in the leukemic BM niche where it is often associated with increased production of ROS (27, 57). This feature has been observed in numerous leukemic cell lines and also in cells from patients with CML and AML (58–60). Growing evidences suggest the role of ROS-mediated metabolic alterations in triggering hematopoietic cancer cell mobilization (54, 61).
In the last years, we and others have demonstrated that the knock down of ferritin heavy chain (FtH) induces EMT in epithelial derived cell lines, mainly though not exclusively by increasing ROS production (15, 17). The role of ferritin in hematological malignancies has been explored as well (42, 62, 63). In chronic myelogenous leukemia K562 cells FtH-silencing, by altering the redox metabolism, triggers p65 nuclear activation and resistance to doxorubicin (36).
In this study, we demonstrate that FtH knock-down promotes a quasi-mesenchymal phenotype and enhances mobility in K562 cells through the activation of a molecular axis arising from ROS mediated-induction of HIF-1α/CXCR4 and ending in p65-mediated transcriptional activation of the mesenchymal markers Snail, Slug and Vimentin.
In detail, K562 cells react to FtH knock down-induced oxidative stress by enhancing the expression of the regulatory HIF-1α subunit that, in turn, acts as a transcription factor for CXCR4. According to these results, ROS attenuation with NAC specifically reduced CXCR4 mRNA and protein levels induced by hypoxia. In K562shFtH cells, CXCR4 up-regulation promotes the nuclear translocation of p65 subunit belonging to the transcriptional complex NF-κB. There is increasing evidence suggesting a reciprocal interplay between CXCR4 and NF-κB signaling in fine tuning cancer cellular signaling pathways (23, 24). Although the vast majority of data report that NF-κB contributes to the increase in CXCR4 expression (64), few recent reports suggest the possible existence of a regulatory feedback loop (24). Our results highlight that, besides being redox sensitive, the activation of NF-κB in K562shFtH cells is also dependent on CXCR4 increase, being reversible upon AMD3100 inhibitor treatment.
A positive correlation between NF-κB activation and EMT has been described in several human solid tumors including breast cancer (44), prostate cancer (65), renal carcinoma (66) and head and neck squamous cell carcinomas (67). A number of studies have also recently demonstrated that NF-κB regulates the transcription of EMT-inducing factors Slug, Twist and Sip1 (68). Our results provide further evidence regarding NF-κB's involvement in EMT regulation also in the hematological malignancies since, in K562shFtH cells, p65 nuclear translocation is accompanied by the over expression of the two key EMT-TF Snail and Slug and of the major mesenchymal marker Vimentin in association with the break-down of the epithelial marker E-cadherin. These molecular rearrangements are mirrored by cytoskeletal remodeling along with increased cell motility and reduced cell adhesion capability to fibronectin substrate. The assessment of the EMT transdifferentiation highlights that either NAC or AMD3100 or IκB alpha treatment is able to attenuate the increase of Snail, Slug and Vimentin as well as the migratory and the adhesion abilities of K562 lacking of FtH expression. On the contrary, none of the above mentioned treatments restore E-cadherin levels in K562shFtH cells suggesting the non-involvement of ROS/CXCR4/NF-κB molecular axis in the regulation of this marker but rather the existence of other underlying molecular regulatory mechanisms such as the previously reported epigenetic imprinting (69).
To the best of our knowledge this is one of the few reports highlighting the role of ROS in the acquisition of characteristics ascribable to EMT phenotype in cells of hematological origins. Moreover, we describe a link between iron metabolism and CXCR4 in the hematological malignancies which may suggest a potential mechanism through which leukemic cells acquire a metastatic phenotype and a tendency to move to a distal organ. Finally, we believe that iron metabolism might be considered as part of the dynamic crosstalk between hematopoietic cancer cells and their microenvironment and that a perturbation of this crosstalk affects the metastatic potential in the hematological malignancies. Clearly, the cellular context of iron/redox metabolism in the modulation of this phenotype is important; hence, cell- or leukemia-subtype specific dependence of this new molecular axis would be the focus of future studies.
Statements
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
RC, IA, FB, and FC conceived and designed the study. RC, IA, AD, ASal, AB, ASac, MD, and FB performed the experiments. RC, IA, MF, BQ, CP, FB, and FC analyzed the data. RC, IA, FB, and FC wrote the first draft of the manuscript. All authors contributed to manuscritp revision, read and approved the submitted version.
Acknowledgments
We thank Caterina Alessi for the administrative support. We thank the Interdipartimental Centre of Services (CIS) for the flow cytometry facilities.
Conflict of interest
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The handling editor declared a past co-authorship with one of the authors AD.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.00698/full#supplementary-material
References
1.
Bissell MJ Radisky DC Rizki A Weaver VM Petersen OW . The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation. (2002) 70:537–46. 10.1046/j.1432-0436.2002.700907.x
2.
Bardaweel SK Gul M Alzweiri M Ishaqat A Alsalamat HA Bashatwah RM . Reactive oxygen species: the dual role in physiological and pathological conditions of the human body. Eurasian J Med. (2018) 50:193–201. 10.5152/eurasianjmed.2018.17397
3.
Weisberg E Azab AK Manley PW Kung AL Christie AL Bronson R et al . Inhibition of CXCR4 in CML cells disrupts their interaction with the bone marrow microenvironment and sensitizes them to nilotinib. Leukemia. (2012) 26:985–90. 10.1038/leu.2011.360
4.
Hu TH Yao Y Yu S Han LL Wang WJ Guo H et al . SDF-1/CXCR4 promotes epithelial-mesenchymal transition and progression of colorectal cancer by activation of the Wnt/??-catenin signaling pathway. Cancer Lett. (2014) 354:417–26. 10.1016/j.canlet.2014.08.012
5.
Yang J Mani SA Weinberg RA . Exploring a new twist on tumor metastasis. Cancer Res. (2006) 66:4549–52. 10.1158/0008-5472.CAN-05-3850
6.
Kahlert UD Joseph JV Kruyt FAE . EMT- and MET-related processes in nonepithelial tumors: importance for disease progression, prognosis, and therapeutic opportunities. Mol Oncol. (2017) 11:860–77. 10.1002/1878-0261.12085
7.
Singh P Pelus LM . CXCR4 expression in the bone marrow microenvironment is required for hematopoietic stem and progenitor cell maintenance and early hematopoietic regeneration after myeloablation. Stem Cells. (2020) [Epub ahead of print]. 1–15. 10.1002/stem.3174.
8.
Yu LM Zhang WH Han XX Li YY Lu Y Pan J et al . Hypoxia-Induced ROS contribute to myoblast pyroptosis during obstructive sleep apnea via the NF-κ B/HIF-1 signaling pathway. Oxid Med Cell Longev. (2019) 2019:4596368. 10.1155/2019/4596368
9.
Wang Z Li Y Sarkar F . Signaling mechanism(S) of reactive oxygen species in epithelial-mesenchymal transition reminiscent of cancer stem cells in tumor progression. Curr Stem Cell Res Ther. (2010) 5:74–80. 10.2174/157488810790442813
10.
Cichon MA Radisky DC . ROS-induced epithelial-mesenchymal transition in mammary epithelial cells is mediated by NF-κB-dependent activation of Snail. Oncotarget. (2014) 5:2827–38. 10.18632/oncotarget.1940
11.
Li W Cao L Han L Xu Q Ma Q . Superoxide dismutase promotes the epithelial-mesenchymal transition of pancreatic cancer cells via activation of the H2O2ERK/NF-κB axis. Int J Oncol. (2015) 46:2613–20. 10.3892/ijo.2015.2938
12.
Sioutas A Vainikka LK Kentson M Dam-Larsen S Wennerström U Jacobson P et al . Oxidant-induced autophagy and ferritin degradation contribute to epithelial–mesenchymal transition through lysosomal iron. J Inflamm Res. (2017) 10:29–39. 10.2147/JIR.S128292
13.
Cannito S Novo E di Bonzo LV Busletta C Colombatto S Parola M . Epithelial-mesenchymal transition: from molecular mechanisms, redox regulation to implications in human health and disease. Antioxid Redox Signal. (2010) 12:1383–430. 10.1089/ars.2009.2737
14.
Storz P . Reactive oxygen species in tumor progression. Front Biosci. (2005) 10:1881–96. 10.2741/1667
15.
Aversa I Zolea F Ieranò C Bulotta S Trotta AM Faniello MC et al . Epithelial-to-mesenchymal transition in FHC-silenced cells: The role of CXCR4/CXCL12 axis. J Exp Clin Cancer Res. (2017) 36:1–15. 10.1186/s13046-017-0571-8
16.
Li R Luo C Mines M Zhang J Fan GH . Chemokine CXCL12 induces binding of ferritin heavy chain to the chemokine receptor CXCR4, alters CXCR4 signaling, and induces phosphorylation and nuclear translocation of ferritin heavy chain. J Biol Chem. (2006) 281:37616–27. 10.1074/jbc.M607266200
17.
Zhang KH Tian HY Gao X Lei WW Hu Y Wang DM et al . Ferritin heavy chain-mediated iron homeostasis and subsequent increased reactive oxygen species production are essential for epithelial-mesenchymal transition. Cancer Res. (2009) 69:5340–48. 10.1158/0008-5472.CAN-09-0112
18.
Chen SC Liao TT Yang MH . Emerging roles of epithelial-mesenchymal transition in hematological malignancies. J Biomed Sci. (2018) 25:37. 10.1186/s12929-018-0440-6
19.
Roccaro AM Mishima Y Sacco A Moschetta M Tai YT Shi J et al . CXCR4 regulates extra-medullary myeloma through epithelial-mesenchymal-transition-like transcriptional activation. Cell Rep. (2015) 12:622–35. 10.1016/j.celrep.2015.06.059
20.
Dewan MZ Ahmed S Iwasaki Y Ohba K Toi M Yamamoto N . Stromal cell-derived factor-1 and CXCR4 receptor interaction in tumor growth and metastasis of breast cancer. Biomed Pharmacother. (2006) 60:273–6. 10.1016/j.biopha.2006.06.004
21.
D'Alterio C Barbieri A Portella L Palma G Polimeno M Riccio A et al . Inhibition of stromal CXCR4 impairs development of lung metastases. Cancer Immunol Immunother. (2012) 61:1713–20. 10.1007/s00262-012-1223-7
22.
Peled A Klein S Beider K Burger JA Abraham M . Role of CXCL12 and CXCR4 in the pathogenesis of hematological malignancies. Cytokine. (2018) 109:11–6. 10.1016/j.cyto.2018.02.020
23.
Jiang C Ma S Hu R Wang X Li M Tian F et al . Effect of CXCR4 on apoptosis in osteosarcoma cells via the PI3K/Akt/NF-κβ signaling pathway. Cell Physiol Biochem. (2018) 46:2250–60. 10.1159/000489593
24.
Es-haghi M Soltanian S Dehghani H . Perspective: cooperation of nanog, NF-κB, and CXCR4 in a regulatory network for directed migration of cancer stem cells. Tumor Biol. (2016) 37:1559–65. 10.1007/s13277-015-4690-6
25.
Liu X Xiao Q Bai X Yu Z Sun M Zhao H et al . Activation of STAT3 is involved in malignancy mediated by CXCL12-CXCR4 signaling in human breast cancer. Oncol Rep. (2014) 32:2760–8. 10.3892/or.2014.3536
26.
Delgado-Martín C Escribano C Pablos JL Riol-Blanco L Rodríguez-Fernández JL . CXCL12 uses CXCR4 and a signaling core formed by bifunctional Akt, Erk1/2 and mTORC1 to control simultaneously chemotaxis and survival in mature dendritic cells. J Biol Chem. (2011) 286:37222–36. 10.1074/jbc.M111.294116
27.
Gu Q He Y Ji J Yao Y Shen W Luo J et al . Hypoxia-inducible factor 1α (HIF-1α) and reactive oxygen species (ROS) mediates radiation-induced invasiveness through the SDF-1α/CXCR4 pathway in non-small cell lung carcinoma cells. Oncotarget. (2015) 6:10893–907. 10.18632/oncotarget.3535
28.
Barriga EH Maxwell PH Reyes AE Mayor R . The hypoxia factor Hif-1α controls neural crest chemotaxis and epithelial to mesenchymal transition. J Cell Biol. (2013) 201:759–76. 10.1083/jcb.201212100
29.
Chetram MA Hinton CV . ROS-mediated regulation of CXCR4 in cancer. Front Biol. (2013) 8273-8. 10.1007/s11515-012-1204-4
30.
Dong CY Liu XY Wang N Wang LN Yang BX Ren Q et al . Twist-1, A novel regulator of hematopoietic stem cell self-renewal and myeloid lineage development. Stem Cells. (2014) 32:3173–82. 10.1002/stem.1803
31.
Zhang Z Zhu P Zhou Y Sheng Y Hong Y Xiang D et al . A novel slug-containing negative-feedback loop regulates SCF/c-Kit-mediated hematopoietic stem cell self-renewal. Leukemia. (2017) 31:403–13. 10.1038/leu.2016.201
32.
Wei CR Liu J Yu XJ . Targeting SLUG sensitizes leukemia cells to ADR-induced apoptosis. Int J Clin Exp Med. (2015) 8:22139−48.
33.
Puissant A Dufies M Fenouille N Ben Sahra I Jacquel A Robert G et al . Imatinib triggers mesenchymal-like conversion of CML cells associated with increased aggressiveness. J Mol Cell Biol. (2012) 4:207–20. 10.1093/jmcb/mjs010
34.
Di Sanzo M Aversa I Santamaria G Gagliardi M Panebianco M Biamonte F et al . FTH1P3, a novel H-ferritin pseudogene transcriptionally active, is ubiquitously expressed and regulated during cell differentiation. PLoS ONE. (2016) 11:e0151359. 10.1371/journal.pone.0151359
35.
Zolea F Battaglia AM Chiarella E Malanga D De Marco C Bond HM et al . Ferritin heavy subunit silencing blocks the erythroid commitment of K562 cells via miR-150 up-regulation and GATA-1 repression. Int J Mol Sci. (2017) 18:2167. 10.3390/ijms18102167
36.
Aversa I Chirillo R Chiarella E Zolea F Di Sanzo M Biamonte F et al . Chemoresistance in H-ferritin silenced cells: The role of NF-κB. Int J Mol Sci. (2018) 19:2969. 10.3390/ijms19102969
37.
Zolea F Biamonte F Battaglia AM Faniello MC Cuda G Costanzo F . Caffeine positively modulates ferritin heavy chain expression in H460 cells: effects on cell proliferation. PLoS ONE. (2016) 11:e0163078. 10.1371/journal.pone.0163078
38.
Di Sanzo M Gaspari M Misaggi R Romeo F Falbo L De Marco C et al . H ferritin gene silencing in a human metastatic melanoma cell line: a proteomic analysis. J Proteome Res. (2011) 10:5444–53. 10.1021/pr200705z
39.
Biamonte F Santamaria G Sacco A Perrone FM Di Cello A Battaglia AM et al . MicroRNA let-7g acts as tumor suppressor and predictive biomarker for chemoresistance in human epithelial ovarian cancer. Sci Rep. (2019) 9:1–12. 10.1038/s41598-019-42221-x
40.
Sottile R Federico G Garofalo C Tallerico R Faniello MC Quaresima B et al . Iron and ferritin modulate MHC class I expression and NK cell recognition. (2019) 10:1–12. 10.3389/fimmu.2019.00224
41.
Biamonte F Battaglia AM Zolea F Oliveira DM Aversa I Santamaria G et al . Ferritin heavy subunit enhances apoptosis of non-small cell lung cancer cells through modulation of miR-125b/p53 axis. Cell Death Dis. (2018) 9:1174. 10.1038/s41419-018-1216-3
42.
Zolea F Biamonte F Candeloro P Di Sanzo M Cozzi A Di Vito A et al . H ferritin silencing induces protein misfolding in K562 cells: A Raman analysis. Free Radic Biol Med. (2015) 89:614–23. 10.1016/j.freeradbiomed.2015.07.161
43.
Di Sanzo M Chirillo R Aversa I Biamonte F Santamaria G Giovannone ED et al . shRNA targeting of ferritin heavy chain activates H19/miR-675 axis in K562 cells. Gene. (2018) 657:92–9. 10.1016/j.gene.2018.03.027
44.
Huber MA Azoitei N Baumann B Grünert S Sommer A Pehamberger H et al . NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. (2004) 114:569–81. 10.1172/JCI200421358
45.
Nomura A Majumder K Giri B Dauer P Dudeja V Roy S et al . Inhibition of NF-kappa B pathway leads to deregulation of epithelial – mesenchymal transition and neural invasion in pancreatic cancer. Lab Invest. (2016) 96:1268–78. 10.1038/labinvest.2016.109
46.
Redondo-muñoz J García-pardo A Teixidó J . Molecular players in hematologic tumor cell trafficking. Front Immunol. (2019) 10:156. 10.3389/fimmu.2019.00156
47.
Kuznetsov M Kolobov A . Mathematical biology investigation of solid tumor progression with account of proliferation / migration dichotomy via Darwinian mathematical model. J Math Biol. (2019) 80:601–26. 10.1007/s00285-019-01434-4
48.
Chen CC You JY Gau JP Huang CE Chen YY Tsai YH et al . Favorable clinical outcome and unique characteristics in association with Twist1 overexpression in de novo acute myeloid leukemia. Blood Cancer J. (2015) 5:e339. 10.1038/bcj.2015.67
49.
Jordan NV Johnson GL Abell AN . Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle. (2011) 10:2865–73. 10.4161/cc.10.17.17188
50.
Kalluri R Weinberg RA . The basics of epithelial-mesenchymal transition. J Clin Invest. (2009) 119:1420–28. 10.1172/JCI39104
51.
Percival M Lai C Estey E Hourigan CS Hutchinson F Section M et al . Bone marrow evaluation for diagnosis and monitoring of acute myeloid leukemia. HHS Public Access Blood Rev. (2018) 31:185–92. 10.1016/j.blre.2017.01.003
52.
Al-katari MS . Acute lymphoblastic leukemia presenting with liver infiltration and severe lactic acidosis. Am J Case Rep. (2018) 19:453–7. 10.12659/AJCR.907383
53.
Fracchiolla NS Fattizzo B Cortelezzi A . Review article mesenchymal stem cells in myeloid malignancies : a focus on immune escaping and therapeutic implications. Stem Cells Int. (2017) 2017::6720594. 10.1155/2017/6720594
54.
Schroeder MA Dipersio JF . Mobilization of hematopoietic stem and leukemia cells. J Leukoc Biol. (2012) 91:47–57. 10.1189/jlb.0210085
55.
Burger JA . Mechanisms of leukemia cell trafficking, homing, and tissue retention in chronic lymphocytic leukemia. Chronic Lymph Leuk. (2011) 5:91–9.
56.
Tavor S Petit I Porozov S Avigdor A Dar A Leider-Trejo L et al . CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. (2004) 64:2817–24. 10.1158/0008-5472.CAN-03-3693
57.
Testa U Labbaye C Castelli G Pelosi E . Oxidative stress and hypoxia in normal and leukemic stem cells. Exp Hematol. (2016) 44:540–60. 10.1016/j.exphem.2016.04.012
58.
Validity RM . The influence of hypoxia on CML trafficking through modulation of CXCR4 and E-cadherin expression. Leukemia. (2013) 27:961–4. 10.1038/leu.2012.353
59.
Prieto-bermejo R Romo-gonzález M Pérez-fernández A Ijurko C . Reactive oxygen species in haematopoiesis : leukaemic cells take a walk on the wild side. J Exp Clin Cancer Res. (2018) 37:125. 10.1186/s13046-018-0797-0
60.
Muz B Puente P De Azab F Luderer M Azab AK . The role of hypoxia and exploitation of the hypoxic environment in hematologic malignancies. Mol Cancer Res. (2014) 12:1347–55. 10.1158/1541-7786.MCR-14-0028
61.
Self-renewal SC Gur-cohen S Golan K Kaufmann KB Itkin T . Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development,as well as their bone marrow microenvironment. Antioxid Redox Signal. (2014) 21:1605–19. 10.1089/ars.2014.5941
62.
Biamonte F Zolea F Bisognin A Di Sanzo M Saccoman C Scumaci D et al . H-ferritin-regulated microRNAs modulate gene expression in K562 cells. PLoS ONE. (2015) 10:e0122105. 10.1371/journal.pone.0122105
63.
Bertoli S Paubelle E Bérard E Saland E Thomas X et al . Ferritin heavy/light chain (FTH1/FTL) expression, serum ferritin levels, and their functional as well as prognostic roles in acute myeloid leukemia. Eur J Haematol. (2019) 102:131–42. 10.1111/ejh.13183
64.
Penzo M Habiel DM Ramadass M Kew RR Marcu KB . Cell migration to CXCL12 requires simultaneous IKK α and IKK β -dependent NF- κ B signaling?. Biochim Biophys Acta. (2014) 1843:1796–804. 10.1016/j.bbamcr.2014.04.011
65.
Zhang Q Helfand BT Jang TL Zhu LJ Chen L Yang XJ et al . Nuclear factor-κB-mediated transforming growth factor-beta-induced expression of vimentin is an independent predictor of biochemical recurrence after radical prostatectomy nuclear factor- κ b-mediated transforming growth factor- β -induced expression of vimentin is an independent predictor of biochemical recurrence after radical prostatectomy. Clin Cancer Res. (2009) 15:3557–67. 10.1158/1078-0432.CCR-08-1656
66.
Pantuck AJ An J Liu H Rettig MB . NF- κ B – dependent plasticity of the epithelial to mesenchymal transition induced by von hippel-lindau inactivation in renal cell carcinomas. Cancer Res. (2010) 70:752–62. 10.1158/0008-5472.CAN-09-2211
67.
Chung CH Parker JS Ely K Carter J Yi Y Murphy BA et al . Gene expression profiles identify epithelial-to-mesenchymal transition and activation of nuclear factor- K B signaling as characteristics of a high-risk head and neck squamous cell carcinoma. Cancer Res. (2006) 66:8210–9. 10.1158/0008-5472.CAN-06-1213
68.
Pires BRB Mencalha AL Ferreira GM Souza WF De . NF-kappaB is involved in the regulation of EMT genes in breast cancer cells. PLoS ONE. (2017) 12:e0169622. 10.1371/journal.pone.0169622
69.
Zha L Cao Q Cui X Li F Liang H . Epigenetic regulation of E-cadherin expression by the histone demethylase UTX in colon cancer cells. Med Oncol. (2016) 33:21. 10.1007/s12032-016-0734-z
Summary
Keywords
ferritin heavy chain, ROS, CXCR4, EMT, NF-κB, leukemia, tumor microenvironment, hematological malignancies
Citation
Chirillo R, Aversa I, Di Vito A, Salatino A, Battaglia AM, Sacco A, Di Sanzo MA, Faniello MC, Quaresima B, Palmieri C, Biamonte F and Costanzo F (2020) FtH-Mediated ROS Dysregulation Promotes CXCL12/CXCR4 Axis Activation and EMT-Like Trans-Differentiation in Erythroleukemia K562 Cells. Front. Oncol. 10:698. doi: 10.3389/fonc.2020.00698
Received
21 March 2020
Accepted
14 April 2020
Published
05 May 2020
Volume
10 - 2020
Edited by
Cirino Botta, Cosenza Hospital, Italy
Reviewed by
Margarida Coucelo, Centro Hospitalar e Universitário de Coimbra, Portugal; Sonia Levi, Vita-Salute San Raffaele University, Italy
Updates
Copyright
© 2020 Chirillo, Aversa, Di Vito, Salatino, Battaglia, Sacco, Di Sanzo, Faniello, Quaresima, Palmieri, Biamonte and Costanzo.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Flavia Biamonte flavia.biamonte.fb@gmail.com
This article was submitted to Cancer Molecular Targets and Therapeutics, a section of the journal Frontiers in Oncology
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.