 Felix Oppel
Felix Oppel Martin Görner2
Martin Görner2 Holger Sudhoff
Holger Sudhoff- 1Department of Otolaryngology, Head and Neck Surgery, Klinikum Bielefeld, Bielefeld, Germany
- 2Department of Hematology and Oncology, Klinikum Bielefeld, Bielefeld, Germany
Tumors may consist of billions of cells, which in malignant cases disseminate and form distant metastases. The large number of tumor cells formed by the high number of cell divisions during tumor progression creates a heterogeneous set of genetically diverse tumor cell clones. For cancer therapy this poses unique challenges, as distinct clones have to be targeted in different tissue locations. Recent research has led to the development of specific inhibitors of defined targets in cellular signaling cascades which promise more effective and more tumor-specific therapy approaches. Many of these molecular targeted therapy (MTT) compounds have already been translated into clinics or are currently being tested in clinical studies. However, the outgrowth of tumor cell clones resistant to such inhibitors is a drawback that affects specific inhibitors in a similar way as classical cytotoxic chemotherapeutics, because additionally acquired genetic alterations can enable tumor cells to circumvent the particular regulators of cellular signaling being targeted. Thus, it might be desirable to reduce genetic heterogeneity prior to molecular targeting, which could reduce the statistical chance of tumor relapse initiated by resistant clones. One way to achieve this is employing unspecific methods to remove as much tumor material as possible before MTT, e.g., by tumor debulking (TD). Currently, this is successfully applied in the clinical treatment of ovarian cancer. We believe that TD followed by treatment with a combination of molecular targeted drugs, optimally guided by biomarkers, might advance survival of patients suffering from various cancer types.
Introduction
Cancer is a life-threatening disease and in western populations, about every third person is projected to suffer from cancer between birth and the age of 74 (1). Between different types of cancer and even among tumors of the same kind in different patients, strong variation can be observed in cell biology and genetics, so that the success of treatment approaches can be unpredictable and some patients fail to respond while others show full remission of the disease. Unraveling oncogenic cell signaling pathways and the underlying genetic alterations has led to the development of specific inhibitors of oncogenic signaling and the establishment of markers that indicate therapy success or failure. Despite initial success with molecular targeted drugs inhibiting oncoproteins or their downstream signaling molecules, resistance is frequently observed (2), as has been well-described for example in non-small-cell lung cancer (NSCLC) (3–5). Thus, advancing treatment regimens in a way to overcome drug resistance reliably is a substantial goal of cancer researchers and clinicians. Decades ago, it became apparent that single drugs or treatments against cancer are less efficient than combined approaches (6–8). Especially for many advanced tumor diseases, it will not be sufficient to use single agents in order to achieve substantial benefit for patients, as in the context of a large tumor burden the intratumoral heterogeneity recurrently creates resistant clones that can reestablish the disease after initial remission (9–11). It is much more promising to hit the neoplastic cells therapeutically from as many different sides as possible, so that the chance of combined resistance against all treatment approaches is reduced (12–14). Here, we discuss tumor debulking (TD) as a method to reduce clonal heterogeneity, which could synergize with the combined application of molecular targeted drugs.
Tumor Size and Clonal Heterogeneity
Tumors in the human body vary strongly in their size, reaching from microscopic lesions to tens of kilograms of tissue. Tumor size has important implications for the genetic heterogeneity within the tumor, as an increase in the amount of neoplastic tissue requires cell divisions. Every new tumor cell, which acquires additional genetic hits and thus becomes genetically distinct from its parental cell, is seen as a new tumor cell clone (15). As tumor size and thus the contained number of cells expands, more and more genetically distinct clones are created. This happens in a statistical manner, as a given number of cell divisions at a given mutation rate has to result in a given average number of new mutations (16). However, this system is more complicated, as distinct tumor cell clones vary in their fitness and proliferation rate due to their distinct genetic conformation affecting tumor cell biology, and their location within the tumor resulting in a different access to oxygen and nutrients. This way, specific clones expand whereas others persist or become lost- a classical selection process of randomly created individuals by their natural characteristics known as “clonal evolution” [reviewed in (17)]. Moreover, cells that acquire new genetic alterations driving them to disseminate from the primary tumor and migrate to other tissue locations form distant metastases. Thus, in metastases, clonal evolution continues from the genetic conformation of the disseminated clone of the primary tumor and it has been possible for scientists to track how tumor cell clones have spread to different sites sequentially and which genetic changes were acquired along the metastatic path (18). This suggests that distant metastases tendentially have a higher mutational burden than the primary tumor.
New genetic alterations in subclones not only drive the metastatic spread, but also can confer drug resistance, e.g., by inhibiting cell death or activating cell signaling downstream of the targeted effectors (19, 20) [reviewed in (21, 22)]. Interestingly, especially small clones (under 10% of the tumor mass) frequently harbor treatment-resistant mutations and were observed more often in tumors with a slower growth rate (23). Thus, clonal heterogeneity within human tumors depends on a complex relation of tumor volume, growth rate, and time.
Molecular Targeted Therapies for Precision Oncology
Standard treatment against most tumor diseases in clinics is mainly composed of surgery, radiotherapy and classical cytotoxic chemotherapy. Standard chemotherapeutic drugs inhibit cell division or damage DNA generally, which harms both normal and cancerous tissues. This causes side effects, mainly tissue defects, affecting quality of live and live-threatening secondary cancers (24). Large cohorts of new information on cancer genetics in the recent two decades have enabled the development of new tumor-specific approaches that inhibit key oncogenic signaling pathways. These so called “molecular targeted therapies1” (MTTs) include but are not limited to epidermal growth factor receptor (EGFR) monoclonal antibodies (e.g., cetuximab, panitumumab, zalutumumab, and nimotuzumab), EGFR tyrosine kinase (TK)-inhibitors (e.g., gefitinib, erlotinib, lapatinib, afatinib, and dacomitinib), vascular endothelial growth factor (VEGF) inhibitors (e.g., bevacizumab), or VEGF receptor (VEGFR) inhibitors (e.g., sorafenib, sunitinib, and vandetanib). Moreover, inhibitors of the PI3K/AKT/mTOR pathway, which is frequently aberrantly activated in various malignancies (e.g., rapamycin, temsirolimus, everolimus), have been developed recently (25). New cancer-targeting compounds also include specific inhibitors of the cell cycle [e.g., cyclin-dependent kinase (CDK)-inhibitors; reviewed in (26)] or inhibitors of epigenetic regulators of cell differentiation and proliferation [e.g., inhibitors of enzymes modifying DNA or histones by methylation/acetylation; reviewed in (27, 28)]. Moreover, PD-1/PD-L1 inhibitors in immunotherapy work as immune checkpoint inhibitors (ICIs) that target the tumor cells' ability to evade immune recognition and recently were described to be the preferable treatment option for a subset of patients with advanced or metastatic tumors (29).
As every individual tumor differs in its genetic alterations, therapies can be tailored to each patient's genetic alterations, which can cause a unique set of vulnerabilities against specific drugs/compounds. This strategy is called “precision oncology” [reviewed in (30)]. Recently, clinical oncologists have examined the relationship between the number of mutations detectable in a tumor disease and the number of mutations that indicate a vulnerability of the tumor toward a specific drug as a prognostic factor (31). This investigation has demonstrated that a patient‘s prognosis is more favorable, the more mutations carried by the individual tumor can be treated by specific drugs. This strikingly demonstrates the advantages of personalized treatment strategies over standard therapies based on general guidelines. However, the selection of molecular targeted drugs solely based on genetic alterations outside their established indications was shown to be ineffective in many cases and led to the conclusion that biomarkers for MTT efficiency have to be established further before individually matched MTT can become standard care (32–34). Thus, the presence of specific mutations verified as biomarkers in one tumor type might not work equivalently in another. For example, BRAF-inhibitors alone work efficiently in BRAF-V600E-positive melanomas as discussed below, but not in BRAF-V600E-positive colon cancer (35). Despite that overall conclusion, there are examples of genetic biomarkers that work universally. For instance, tropomyosin receptor kinase (TRK)-inhibitor larotrectinib inhibits all kinds of TRK-fusion-positive tumors in children and adults (36). As the DNA-sequencing technology further advances, it is highly probable that substantially more genetic markers will be described in the near future, so that precision oncology-based approaches might upgrade traditional treatment regimens in mid-term.
Drug Specificity and Resistance
An important challenge for the clinical use of chemotherapeutics and new targeted drugs is the resistance of tumor cells. Tumor cells can be inherently resistant or acquire resistance. Even after a drug initially caused effective remission, single resistant tumor cell clones can survive and reestablish the tumor which then will be entirely resistant toward repeated treatment with the same drug. This phenomenon was observed recurrently [reviewed in (37)], especially when a single drug is used to treat a highly malignant type of cancer in an advanced stage. One reason for drug resistance is that above a certain tumor size, there will be clones that have the matching genetic alterations to resist the given drug (Figure 1). The important influence of resistant subclones on MTT has previously been discussed (38). How likely a cell can acquire resistance mainly depends on the drug‘s chemical structure, mode-of-action, and the target specificity (21, 39). It is obvious that a more specific drug which exclusively blocks one target protein, e.g., a single kinase, will be easier to resist than a compound with a broader impact, e.g., inhibiting all kinases, shutting down mitochondrial energy production, or blocking mRNA translation. Tumor cells can bypass a defined point in cellular signaling, e.g., by acquiring new mutations that activate downstream effectors. For this reason, resistance against single inhibitors used for MTT approaches can be observed frequently (4, 39). Even though it has been demonstrated that combined treatment with separate inhibitors can overcome tumor cell resistance (37, 40–42), a combined acquired resistance against several MTT drugs has been described as well (14).
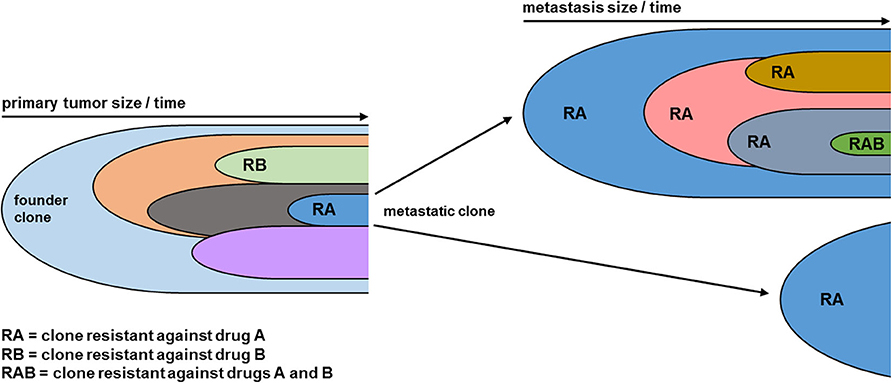
Figure 1. Clonal evolution leads to acquired combined drug resistance. As the tumor grows over time, new genetic alterations lead to the formation of distinct clones (indicated by distinct colors), each representing a subpopulation of the tumor mass. Clones vary in their topology within the tumor and their biology, ultimately leading to the formation of metastatic clones which initiate metastases at different locations. Moreover, new genetic hits lead to drug resistance against single drugs. As the tumor disease progresses, further individual clones can acquire a combined resistance against several drugs. As the genetic diversity statistically correlates with tumor volume, larger metastases (Top Right) are genetically more diverse than smaller metastases (Left Right). Within metastases the clonal evolution continues from the metastatic clone of the primary tumor, leading to a higher mutational burden.
Tumor Debulking to Support Precision Oncology
As outlined above, clonal heterogeneity is positively linked to tumor burden and increases the chance of drug resistance. This indicates that reduction of tumor volume will elevate the likelihood of successful drug treatment. Clonal heterogeneity also plays an important role in acquired resistance against immunotherapy [reviewed in (43)]. However, as various cell types in the microenvironment participate in the complex interaction between tumor cells and the immune system, resistance against this kind of therapy is influenced by many more determinants [reviewed in (44)]. As TD surgery can reduce clonal heterogeneity by removing large tumor bodies, but cannot eliminate dispersed tumor cells, and MTT can target dispersed cells, but might find resistant clones in large tumor bodies, both treatments could work synergistically together when TD is performed prior to MTT (Figure 2).
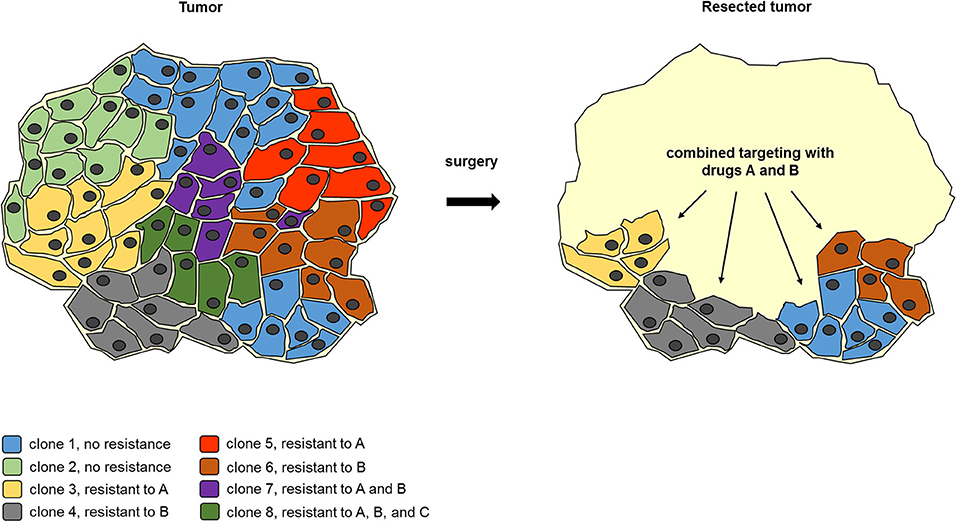
Figure 2. Example for surgery to reduce clonal heterogeneity synergizes with subsequent combined molecular targeting. A tumor body consists of a genetically heterogeneous set of tumor cell clones with a differential drug resistance spectrum. Upon partial removal of the tumor body, the number of tumor cell clones is reduced, so that a combined treatment with drugs A and B can eliminate the remaining tumor cells. This demonstrates how TD can facilitate subsequent molecular targeting even when residual tumor tissue remains.
Both treatments are already applied to treat ovarian cancer (OC) (45) and metastatic renal cell carcinoma (mRCC) (46). In OC targeted therapy, the VEGF-inhibitor bevacizumab or the VEGFR-inhibitors pazopanib (47) and cediranib (48) are used to inhibit angiogenesis. Similarly, PARP inhibitors are used to inhibit DNA-repair in tumor cells combined with chemotherapy which leads to the death of tumor cells (49, 50). Moreover, OC patients frequently undergo TD, as this has been shown to prolong the survival of OC patients if the residual nodules are <0.5 cm in size (51). For example, bevacizumab was shown to efficiently increase patient survival after TD surgery (45). In patients with mRCC, immunotherapy was shown to be much more effective in combination with TD than when applied alone (52).
Besides OC and mRCC, there is preclinical data indicating potential for the use of TD with MTT or chemotherapy in other tumor types as well. In a mouse model of malignant mesothelioma, TD was found to support anti-tumor memory when it was combined with chemotherapy and adjuvant immunotherapy (53, 54). In particular, one study demonstrated that a partial TD induced a long-term anti-tumor memory which was not observed when a complete resection was performed (53). Moreover, a partial tumor removal stimulated an anti-tumor immune reaction in a mouse model of NSCLC and it was shown that the excision of one tumor body increased the efficiency of anti-PD1 immunotherapy in another tumor location within the same individual (55). The reason for this might be the release of tumor antigens stimulating the immune system due to the destruction of tumor material along surgery and the decreased release of immunosuppressive cytokines from the reduced tumor tissue burden. This indicates that TD could be beneficial in combination with immunotherapy in different tumor types, even if a large proportion of the tumor burden remains. In addition to immunotherapy, TD was successfully combined with a cisplatin-loaded polymer platform in a mouse model of head and neck squamous cell carcinoma (56).
These examples indicate that TD could support therapy against a variety of cancer types. NSCLC, for example, is a cancer type with many molecular targeted drugs available in clinical practice. MTT for NSCLC includes drugs blocking receptor tyrosine kinases (RTKs) like EGFR, hepatocyte growth factor receptor (HGFR), and anaplastic lymphoma kinase (ALK), as these oncoproteins are frequently aberrantly activated. As described above, resistance against these new approaches in NSCLC therapy is frequently observed (3–5) and thus new strategies to overcome resistance are needed. Surgery is regularly used for locoregionally advanced lung cancers, but usually not combined with MTT in clinical routine even if clear biomarkers for targeted drugs are detected. Beneficial results in applying MTT after NSCLC surgery have been observed in initial clinical trials [reviewed in (57)]. In a single-arm phase II trial, the EGFR-inhibitor erlotinib was applied in patients with stage IA-IIIA NSCLC with EGFR-mutation after surgery, which resulted in an increased 2-year survival and a block of recurrence during the period of drug application, so that recurrence was delayed in most cases until treatment was discontinued (58). However, this indicates that erlotinib was not sufficient to kill all remaining tumor cells, but inhibited the surviving fraction to repopulate the tumor while being administered. Using an even more effective MTT, e.g., a combination of several targeted drugs, TD could support MTT against NSCLCs, maybe even in advanced stages, and minimize the chance of relapse by reducing the number of potentially resistant tumor cells. This could be further tested in clinical trials with combinations of targeted drugs, dividing patients with NSCLC who have an indication for MTT into two groups. One group would receive MTT in the conventional way, whereas the patients in the second group undergo TD prior to drug administration. By setting the proportion of residual tumor material after debulking in correlation to the therapy success, this study could reveal how reasonable an incomplete tumor resection is for this tumor type.
Personalized Application of Tumor Debulking
As TD represents an unspecific mechanical method, the reduction of genetically distinct tumor cell clones by TD could significantly support all therapy approaches, which are limited in their efficacy by tumor cell resistance mediated by genetic variation within the tumor. If this is true, TD will support MTT regardless of the specific drug's mode-of-action by lowering the chance of resistance. Newer MTT approaches consider the genetics of individual tumor diseases and match molecular targeted drugs to the genetic profile of the tumor (31). This indicates that if it was possible to measure the effect that TD has on genetic heterogeneity, e.g., using a liquid biopsy or similar procedures, the MTT design could be created after TD to benefit maximally from the improved tumor genetics. Liquid biopsies are designed to detect circulating tumors cells, circulating tumor DNA (ctDNA), and other tumor-derived components in a patient's blood sample (59). In a clinical study ctDNA detection rates of >75% were observed for many cancer types, with ~50–75% even in cases with localized disease (60). Interestingly, the authors detected driver mutations in the KRAS oncogene in ctDNA as well as mutations related to the development of resistance toward EGFR blockade in 23 of 24 patients that initially responded but later relapsed. This indicates that liquid biopsy is a sensitive method for analyzing the tumor genome and tailoring MTT to each patient individually. Liquid biopsy might even enable a comparison of the tumor genome before and after TD, so that the impact of TD on clonal heterogeneity could be monitored.
Of course, the application of TD in order to decrease clonal heterogeneity would make sense especially if promising MTT options can be identified for the particular patient. An example of a highly potent MTT is BRAF-inhibition in melanoma which initially works highly effectively and can eradicate even large tumors, but in most cases induces resistance due to alternative activation of MAPK/Erk signaling or activation of PI3K/Akt signaling [reviewed in (61)]. Even combined inhibition of BRAF and MEK was followed by relapse, despite a significantly longer survival compared to single BRAF-inhibitor treatment (62). This indicated that effective treatment, even in combination, most frequently faces resistant tumor cell clones in advanced diseases. Thus, TD prior to BRAF/MEK-inhibitor application might be effective in melanoma treatment. This hypothesis is supported by a clinical phase III trial that reported a significantly decreased recurrence of completely resected, stage III melanoma with BRAF-V600E or -V600K mutations treated with a combination of BRAF and MEK inhibitors after surgery (63).
Due to higher tumor volumes and the resulting higher genetic heterogeneity, advanced stage tumor patients might benefit more likely from TD (29). However, surgery-related mortality and morbidity have to be considered to estimate for every patient individually whether the expected benefits of the planned MTT are high enough to justify the operation risks and negative impact on life quality. In the scenario when TD is not possible to perform due to excessive risks, MTT might be combined with other treatments like chemotherapy, radiotherapy, hyperthermia, or others to achieve a cytoreductive effect that will reduce the chance of resistance against MTT. However, in our view, TD is not primarily supposed to change how MTT is performed, but rather support it whenever possible. Hence, TD to support MTT must be performed as intensely as reasonably safe.
Conclusions
Preclinical and clinical studies indicate that TD might cooperate well with MTT approaches. Immunotherapy approaches in particular have been shown to benefit from tumor resection in a large variety of tumor types. The reduction of as many genetically distinct tumor cell clones as possible could be used to reduce the ability of tumors to resist MTT for precision oncology. In order to create synergy effects, unspecific non-mutagenic treatment options like TD should precede genetics-guided combined molecular targeting for a variety of tumor types. Depending on the individual patient's characteristics, tumor type, stage, and genetic profile, oncologists could design a personalized strategy to support specific treatment options like MTT with cytoreductive methods like TD to outsmart the tumor's intrinsic compulsion to resistance. In future, clinical treatment guidelines might be adapted this way to facilitate an effective patient-specific MTT.
Author Contributions
FO developed the idea and created the design of the article. HS and MG contributed to the idea. All authors participated in writing and editing the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
1. ^https://www.cancer.gov/about-cancer/treatment/types/targeted-therapies/targeted-therapies-fact-sheet
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. Sabnis AJ, Bivona TG. Principles of resistance to targeted cancer therapy: lessons from basic and translational cancer biology. Trends Mol Med. (2019) 25:185–97. doi: 10.1016/j.molmed.2018.12.009
3. Govindan R. Overcoming resistance to targeted therapy for lung cancer. N Engl J Med. (2015) 372:1760–1. doi: 10.1056/NEJMe1500181
4. Schrank Z, Chhabra G, Lin L, Iderzorig T, Osude C, Khan N, et al. Current molecular-targeted therapies in NSCLC and their mechanism of resistance. Cancers. (2018) 10:224. doi: 10.3390/cancers10070224
5. Spaans JN, Goss GD. Drug resistance to molecular targeted therapy and its consequences for treatment decisions in non-small-cell lung cancer. Front Oncol. (2014) 4:190. doi: 10.3389/fonc.2014.00190
6. Mouridsen HT, Palshof T, Brahm M, Rahbek I. Evaluation of single-drug versus multiple-drug chemotherapy in the treatment of advanced breast cancer. Cancer Treat Rep. (1977) 61:47–50.
7. DeVita VT Jr. Single agent versus combination chemotherapy. CA Cancer J Clin. (1975) 25:152–8. doi: 10.3322/canjclin.25.3.152
8. DeVita VT Jr, Young RC, Canellos GP. Combination versus single agent chemotherapy: a review of the basis for selection of drug treatment of cancer. Cancer. (1975) 35:98–110. doi: 10.1002/1097-0142(197501)35:1<98::AID-CNCR2820350115>3.0.CO;2-B
9. Somasundaram R, Villanueva J, Herlyn M. Intratumoral heterogeneity as a therapy resistance mechanism: role of melanoma subpopulations. Adv Pharmacol. (2012) 65:335–59. doi: 10.1016/B978-0-12-397927-8.00011-7
10. Lim ZF, Ma PC. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J Hematol Oncol. (2019) 12:134.
11. Stanta G, Bonin S. Overview on clinical relevance of intra-tumor heterogeneity. Front Med. (2018) 5:85. doi: 10.3389/fmed.2018.00085
12. Palmer AC, Sorger PK. Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell. (2017) 171:1678–91.e13. doi: 10.1016/j.cell.2017.11.009
13. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev. Clin Oncol. (2018) 15:81–94. doi: 10.1038/nrclinonc.2017.166
14. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. (2012) 367:1694–703. doi: 10.1056/NEJMoa1210093
15. Nowell PC. The clonal evolution of tumor cell populations. Science. (1976) 194:23–8. doi: 10.1126/science.959840
16. Baer CF, Miyamoto MM, Denver DR. Mutation rate variation in multicellular eukaryotes: causes and consequences. Nat Rev Genet. (2007) 8:619–31. doi: 10.1038/nrg2158
17. Greaves M, Maley CC. Clonal evolution in cancer. Nature. (2012) 481:306–13. doi: 10.1038/nature10762
18. Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. (2010) 467:1114–7. doi: 10.1038/nature09515
19. Brady SW, McQuerry JA, Qiao Y, Piccolo SR, Shrestha G, Jenkins DF, et al. Combating subclonal evolution of resistant cancer phenotypes. Nat Commun. (2017) 8:1231. doi: 10.1038/s41467-017-01174-3
20. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. (2012) 366:883–92. doi: 10.1056/NEJMoa1113205
21. Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, et al. Drug resistance in cancer: an overview. Cancers. (2014) 6:1769–92. doi: 10.3390/cancers6031769
22. Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer. (2013) 108:479–85. doi: 10.1038/bjc.2012.581
23. Chowell D, Napier J, Gupta R, Anderson KS, Maley CC, M.Sayres AW. Modeling the subclonal evolution of cancer cell populations. Cancer Res. (2018) 78:830–9. doi: 10.1158/0008-5472.CAN-17-1229
24. Brower V. Tracking chemotherapy's effects on secondary cancers. J Natl Cancer Inst. (2013) 105:1421–2. doi: 10.1093/jnci/djt273
25. Kozakiewicz P, Grzybowska-Szatkowska L. Application of molecular targeted therapies in the treatment of head and neck squamous cell carcinoma. Oncol Lett. (2018) 15:7497–505. doi: 10.3892/ol.2018.8300
26. Law ME, Corsino PE, Narayan S, Law BK. Cyclin-dependent kinase inhibitors as anticancer therapeutics. Mol Pharmacol. (2015) 88:846–52. doi: 10.1124/mol.115.099325
27. Wee S, Dhanak D, Li H, Armstrong SA, Copeland RA, Sims R, et al. Targeting epigenetic regulators for cancer therapy. Ann N Y Acad Sci. (2014) 1309:30–6. doi: 10.1111/nyas.12356
28. Cheng Y, He C, Wang M, Ma X, Mo F, Yang S, et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Trans Targeted Ther. (2019) 4:62. doi: 10.1038/s41392-019-0095-0
29. Sun L, Zhang L, Yu J, Zhang Y, Pang X, Ma C, et al. Clinical efficacy and safety of anti-PD-1/PD-L1 inhibitors for the treatment of advanced or metastatic cancer: a systematic review and meta-analysis. Sci Rep. (2020) 10:2083. doi: 10.1038/s41598-020-58674-4
30. Nadauld LD, Ford JM, Pritchard D, Brown T. Strategies for clinical implementation: precision oncology at three distinct institutions. Health Aff . (2018) 37:751–6. doi: 10.1377/hlthaff.2017.1575
31. Sicklick JK, Kato S, Okamura R, Schwaederle M, Hahn ME, Williams CB, et al. Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat Med. (2019) 25:744–50. doi: 10.1038/s41591-019-0407-5
32. Le Tourneau C, Delord JP, Goncalves A, Gavoille C, Dubot C, Isambert N, et al. investigators, molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. (2015) 16:1324–34. doi: 10.1016/S1470-2045(15)00188-6
33. Galot R, Le Tourneau C, Guigay J, Licitra L, Tinhofer I, Kong A, et al. Personalized biomarker-based treatment strategy for patients with squamous cell carcinoma of the head and neck: EORTC position and approach. Ann Oncol. (2018) 29:2313–27. doi: 10.1093/annonc/mdy452
34. Moscow JA, Fojo T, Schilsky RL. The evidence framework for precision cancer medicine. Nat Rev Clin Oncol. (2018) 15:183–92. doi: 10.1038/nrclinonc.2017.186
35. Kopetz S, Desai J, Chan E, Hecht JR, O'Dwyer PJ, Maru D, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol. (2015) 33:4032–8. doi: 10.1200/JCO.2015.63.2497
36. Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. (2018) 378:731–9. doi: 10.1056/NEJMoa1714448
37. Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. (2019) 575:299–309. doi: 10.1038/s41586-019-1730-1
38. Schmitt MW, Loeb LA, Salk JJ. The influence of subclonal resistance mutations on targeted cancer therapy. Nat Rev Clin Oncol. (2016) 13:335–47. doi: 10.1038/nrclinonc.2015.175
39. Groenendijk FH, Bernards R. Drug resistance to targeted therapies: deja vu all over again. Mol Oncol. (2014) 8:1067–83. doi: 10.1016/j.molonc.2014.05.004
40. Corcoran RB, Andre T, Atreya CE, J.Schellens HM, Yoshino T, Bendell JC, et al. Combined BRAF, EGFR, and MEK inhibition in patients with BRAF(V600E)-mutant colorectal cancer. Cancer Discov. (2018) 8:428–43. doi: 10.1158/2159-8290.CD-17-1226
41. Sato H, Yamamoto H, Sakaguchi M, Shien K, Tomida S, Shien T, et al. Combined inhibition of MEK and PI3K pathways overcomes acquired resistance to EGFR-TKIs in non-small cell lung cancer. Cancer Sci. (2018) 109:3183–96. doi: 10.1111/cas.13763
42. Tricker EM, Xu C, Uddin S, Capelletti M, Ercan D, Ogino A, et al. Combined EGFR/MEK inhibition prevents the emergence of resistance in EGFR-mutant lung cancer. Cancer Discov. (2015) 5:960–71. doi: 10.1158/2159-8290.CD-15-0063
43. Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. (2002) 3:999–1005. doi: 10.1038/ni1102-999
44. Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S. Mechanisms of resistance to immune checkpoint blockade: why does checkpoint inhibitor immunotherapy not work for all patients? Am Soc Clin Oncol Educ Book. (2019) 39:147–64. doi: 10.1200/EDBK_240837
45. Perren TJ, Swart AM, Pfisterer J, Ledermann JA, Pujade-Lauraine E, Kristensen G, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. (2011) 365:2484–96. doi: 10.1056/NEJMoa1103799
46. Aslam MZ, Matthews PN. Cytoreductive nephrectomy for metastatic renal cell carcinoma: a review of the historical literature and its role in the era of targeted molecular therapy. ISRN Urol. (2014) 2014:717295. doi: 10.1155/2014/717295
47. Davidson BA, Secord AA. Profile of pazopanib and its potential in the treatment of epithelial ovarian cancer. Int J Women's Health. (2014) 6:289–300. doi: 10.2147/IJWH.S49781
48. Orbegoso C, Marquina G, George A, Banerjee S. The role of cediranib in ovarian cancer. Expert Opin Pharmacother. (2017) 18:1637–48. doi: 10.1080/14656566.2017.1383384
49. Meehan RS, Chen AP. New treatment option for ovarian cancer: PARP inhibitors. Gynecol Oncol Res Pract. (2016) 3:3. doi: 10.1186/s40661-016-0024-7
50. Ray-Coquard I, Pautier P, Pignata S, Perol D, Gonzalez-Martin A, Berger R, et al. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N Engl J Med. (2019) 381:2416–28. doi: 10.1056/NEJMoa1911361
51. Hacker NF, Berek JS, Lagasse LD, Nieberg RK, Elashoff RM. Primary cytoreductive surgery for epithelial ovarian cancer. Obstet Gynecol. (1983) 61:413–20.
52. Flanigan RC, Mickisch G, Sylvester R, Tangen C, Van Poppel H, Crawford ED. Cytoreductive nephrectomy in patients with metastatic renal cancer: a combined analysis. J Urol. (2004) 171:1071–6. doi: 10.1097/01.ju.0000110610.61545.ae
53. Broomfield S, Currie A, van der Most RG, Brown M, van Bruggen I, Robinson BW, et al. Partial, but not complete, tumor-debulking surgery promotes protective antitumor memory when combined with chemotherapy and adjuvant immunotherapy. Cancer Res. (2005) 65:7580–4. doi: 10.1158/0008-5472.CAN-05-0328
54. Khong A, Cleaver AL, Fahmi Alatas M, Wylie BC, Connor T, Fisher SA, et al. The efficacy of tumor debulking surgery is improved by adjuvant immunotherapy using imiquimod and anti-CD40. BMC Cancer. (2014) 14:969. doi: 10.1186/1471-2407-14-969
55. Guisier F, Cousse S, Jeanvoine M, Thiberville L, Salaun M. A rationale for surgical debulking to improve anti-PD1 therapy outcome in non small cell lung cancer. Sci Rep. (2019) 9:16902. doi: 10.1038/s41598-019-52913-z
56. Hu D, Lau OD, Wang L, Wang G, Schaue D, Zhu L, et al. A novel modular polymer platform for the treatment of head and neck squamous cell carcinoma in an animal model. Arch Otolaryngol Head Neck Surg. (2012) 138:412–7. doi: 10.1001/archoto.2012.20
57. Yuan M, Huang LL, Chen JH, Wu J, Xu Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal Trans Targeted Ther. (2019) 4:61. doi: 10.1038/s41392-019-0099-9
58. Pennell NA, Neal JW, Chaft JE, Azzoli CG, Janne PA, Govindan R, et al. SELECT: a phase II trial of adjuvant erlotinib in patients with resected epidermal growth factor receptor-mutant non-small-cell lung cancer. J Clin Oncol. (2019) 37:97–104. doi: 10.1200/JCO.18.00131
59. Bai Y, Zhao H. Liquid biopsy in tumors: opportunities and challenges. Ann Transl Med. (2018) 6:S89. doi: 10.21037/atm.2018.11.31
60. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. (2014) 6:224ra24. doi: 10.1126/scitranslmed.3007094
61. Luebker SA, Koepsell SA. Diverse mechanisms of BRAF inhibitor resistance in melanoma identified in clinical and preclinical studies. Front Oncol. (2019) 9:268. doi: 10.3389/fonc.2019.00268
62. Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. (2015) 386:444–51. doi: 10.1016/S0140-6736(15)60898-4
Keywords: precision oncology, molecular targeting, clonal heterogeneity, therapy resistance, cancer therapy, cancer genetics
Citation: Oppel F, Görner M and Sudhoff H (2020) The Potential of Tumor Debulking to Support Molecular Targeted Therapies. Front. Oncol. 10:801. doi: 10.3389/fonc.2020.00801
Received: 05 February 2020; Accepted: 23 April 2020;
Published: 18 June 2020.
Edited by:
Triantafyllos Stylianopoulos, University of Cyprus, CyprusReviewed by:
Scott Anderw Fisher, University of Western Australia, AustraliaMassimo Fantini, Precision Biologics, Inc., United States
Copyright © 2020 Oppel, Görner and Sudhoff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Felix Oppel, ZmVsaXgub3BwZWxAa2xpbmlrdW1iaWVsZWZlbGQuZGU=; Holger Sudhoff, aG9sZ2VyLnN1ZGhvZmZAcnViLmRl