Abstract
Molecular chaperones have recently emerged as fundamental regulators of salient biological routines, including metabolic adaptations to environmental changes. Yet, many of the molecular mechanisms at the basis of their functions are still unknown or at least uncertain. This is in part due to the lack of chemical tools that can interact with the chaperones to induce measurable functional perturbations. In this context, the use of small molecules as modulators of protein functions has proven relevant for the investigation of a number of biomolecular systems. Herein, we focus on the functions, interactions and signaling pathways of the HSP90 family of molecular chaperones as possible targets for the discovery of new molecular entities aimed at tuning their activity and interactions. HSP90 and its mitochondrial paralog, TRAP1, regulate the activity of crucial metabolic circuitries, making cells capable of efficiently using available energy sources, with relevant implications both in healthy conditions and in a variety of disease states and especially cancer. The design of small-molecules targeting the chaperone cycle of HSP90 and able to inhibit or stimulate the activity of the protein can provide opportunities to finely dissect their biochemical activities and to obtain lead compounds to develop novel, mechanism-based drugs.
Introduction
Chaperones are molecular machines that assist folding, conformational changes and subcellular trafficking of proteins and control their degradation following aggregation, unfolding or misfolding. Fine and orchestrated tuning of these processes is carried out by different chaperone families and leads to maintenance and quality control of the proteome, an extremely complex and vital task for cells (1). Heat Shock Protein 90 (HSP90) proteins are chaperones that exert their regulatory functions on the structure and activity of a variety of diverse client proteins, thus integrating signaling and metabolic circuitries and acting as crucial components in consenting flexible adaptations of cells to environmental changes and stresses (2).
Exposure to harmful environmental stimuli is a common event in the process of tumor growth. Fluctuations in pH, oxygen, or nutrient availability prompt a profound rewiring of the metabolic circuitries of neoplastic cells and major changes in the homeostasis of their proteome (proteostasis) (3, 4). These noxious conditions also affect biochemical functions confined in specific subcellular compartments, such as protein folding in the endoplasmic reticulum (ER) (5, 6) as well as the bioenergetic functions of mitochondria (7, 8). Activation of organelle-restricted signaling pathways and metabolic adaptations can subtly regulate the equilibrium among death, dormancy, and aggressiveness of tumor cells (9–11). In order to cope with these stresses and sustain pro-oncogenic biological routines, including growth, proliferation, invasion, metastasis and evasion from death stimuli, most cancer cells overexpress HSP90 family members (12, 13). The various paralogs of HSP90 proteins can play a key role at the crossroads of these multiple cellular functions in the different cellular districts, namely HSP90 in the cytosol, Grp94 in the ER and TRAP1 in mitochondria (14–16); extracellular HSP90 is also involved in cell-to-cell communication (17). Induction of Hsp90 family protein expression contributes to the adaptations of the metabolic machineries in tumor cells and has been associated with cancer progression, resistance to chemotherapy and poor prognosis (12, 18). Adaptability to stress conditions is linked to specific subsets of clients, and the range of functional flexibility of the chaperone and of potential activities of its interactors are further expanded both by post-translational modifications and by the recruitment of other chaperones and of co-chaperones (19–21). In this context, the development of drugs targeting HSP90 components has emerged as a promising anti-neoplastic strategy.
Here, we report our views on molecular design strategies aimed to act on the circuitries in which HSP90 family members play a key role in cancer cells. In particular, we focus on cytosolic HSP90 and its mitochondrial paralog TRAP1, as Grp94 has a more specialized role in the maturation process of particular secretory and membrane-bound proteins clients, such as immunoglobulins, integrins, and Toll-like receptors (14). We will discuss interventions that range from the use of allosteric modulators of chaperone functions, to the targeting of protein-protein interactions involved in the assembly of functional complexes. We also discuss possible perspectives in combining the use of molecules that target HSP90 complexes with the use of other antineoplastic compounds, with a particular focus on the control of metabolic vulnerabilities in cancer models.
Structure and Function of HSP90 Molecular Chaperones
Molecular chaperones of the HSP90 family are essential cell constituents, making up 1–2% of the proteome. Their expression can be further stimulated by stress, and in tumor cells they can reach up to the 4–7% of the expressed proteome, thus shaping all biological processes required for neoplastic progression (Figure 1). HSP90 exists as a homodimer and each individual chain consists of three globular domains (22). Structures reporting different full length chaperone isoforms can be found at the following pdb codes: 2ioq, 2iop, 2cg9, 2o1v, 2o1u, 4job, 4ipe, 4iyn, 5uls, 5tvu, 5tvx, 6d14, 5tth, 5tvw.
Figure 1
The structures show the common organization in a N-terminal ATP-binding domain (N-domain), a middle domain (M-domain) involved in ATP hydrolysis, and a C-terminal domain (C-domain) responsible for HSP90 dimerization and for interactions with several co-chaperones. HSP90, TRAP1, and Grp94 have a mutual sequence identity of about 30–40%, which reflects in the high structural similarity and alignability of their individual domains (23–25). However, the preferential relative orientation of the domains in the crystal structures solved so far varies significantly depending on the protein, cellular compartment, and organism (26), yielding a global root mean square deviation (RMSD) of atomic positions of at least 7Å.
HSP90 chaperones manifest their functions by promoting the folding and tuning the activity of a plethora of clients endowed with highly diverse structures, cellular localizations and functions. The two main cytosolic HSP90 isoforms, HSP90α and HSP90β, have an interactome that includes more than 400 putative clients (https://www.picard.ch/HSP90Int/index.php), making them central modulators of at least a dozen of important biochemical pathways, including stress regulation, protein folding, DNA repair, kinase signaling, cell survival and metabolism (2, 12). HSP90 effects on clients encompass facilitating the formation of specific protein conformations, as in the case of kinase activation (27), prompting the assembly of multiprotein complexes (28), stabilizing the binding-competent conformation of ligand receptors, and regulating protein dynamics and conformational state ensembles (29). Client stability depends on the chaperone, and its inhibition induces proteasomal degradation of client proteins.
Dimers of HSP90 family proteins undergo a complex functional cycle that might allow them to adapt to different client proteins. ATP binding elicits a series of conformational changes (Figure 2) leading to the “closed conformation” of the chaperone in which ATP hydrolysis occurs. Induction of the closed state is the rate-limiting step of the reaction. ATP binding has a much lower affinity than ADP binding (KD ~400 μM vs. ~10 μM), indicating that under physiological conditions of nucleotide concentrations, cytosolic Hsp90 primarily populates two states that are absent in ATP-regenerating conditions: either ADP bound to both arms, or ATP bound to one arm and ADP bound to the opposite arm (30). A NTD loop termed the “lid” region closes over the ATP-bound active site. NTDs then dimerize and associate with the M-domains, prompting ATP hydrolysis (31). This step is instrumental for dissociation of the two NTDs and the subsequent release of ADP and inorganic phosphate (Pi); eventually, HSP90 returns to the open (apo) conformation.
Figure 2
A dynamic equilibrium exists among the different conformations of HSP90. X-ray crystallography, small-angle X-ray scattering (SAXS) solution data and kinetic measurements have led to the proposal of a general functional mechanism based on global conformational modulations triggered by ATP binding and hydrolysis, which integrates an array of structural information (25). In the absence of nucleotide, various conformations co-exist. ATP binding shifts the chaperone to a partially closed state, and then into a closed conformation; in the case of TRAP1, this is asymmetric and significantly strained, leading to buckling of the MD:CTD interface (32). The hydrolysis of ATP is sequential and deterministic and determines the conformational modulation of the MD:CTD region (32). This region has a key role in client binding (33) and is close to the allosteric C-terminal binding site. Upon ATP hydrolysis, the strain is relieved to yield a symmetric closed state (23). In vitro experiments demonstrate that although the fundamental conformational states are well-conserved among species and paralogs, equilibria and kinetics are unique for every HSP90 homolog (26), suggesting adaptations to the specific needs of clients in each subcellular environment.
In cells, HSP90 acts as a nucleating site for the assembly of networks of stable multiprotein complexes that show tumor-specific traits of physical and functional integration absent in normal cells (34, 35). Such large complexes act to enhance biochemical and metabolic pathways required to bear conditions encountered during malignant transformation. Mechanistically, co-chaperones select stochastically distributed HSP90 conformers that meet functional needs and structurally organize complexes for client activation (e.g., Cdc37 for kinases), or either increase (e.g., Aha1) or slow down (e.g., p23) ATPase rates of HSP90. Some co-chaperones, for instance Aha1 and Cdc37, are overexpressed in cancer and can be post-translationally modified by HSP90 client enzymes, generating reciprocal regulatory mechanisms of the chaperone machinery (36). By using the extraordinary power of state-of-the-art cryoEM, the Agard lab revealed the structures of two very different client HSP90 complexes, namely HSP90:Cdc37:Cdk4 (37) and HSP90:Hsp70:Hop:GR (38). Expectedly, the multiprotein functional assemblies are quite dynamic, which explains why they have eluded crystallization.
The rate of ATP hydrolysis by HSP90 is low, thus the HSP90 cycle may be differently tuned within different tissues or subcellular compartments by complex post-translational modifications (PTMs) that include phosphorylation, sumoylation, acetylation, S-nitrosylation, oxidation, and ubiquitination (20, 36, 39) (Table 1). We are far from understanding the effect of individual PTMs. In general, HSP90 phosphorylation, predominantly on Ser residues, but also on Thr and Tyr residues (68), slows down the chaperone conformational cycle, affecting maturation of clients and interactions with co-chaperones (20, 69). Co-chaperones broaden the functional range of HSP90, either modulating its chaperone cycle or enabling the recruitment of specific subsets of clients, thus providing a suitable folding platform for each client, or even carrying out both activities (12). Co-chaperone binding is also regulated by HSP90 acetylation, whereas S-nitrosylation in the CTD inhibit HSP90 chaperone cycle and activity (65, 68). Furthermore, PTMs can function as allosteric switch points that regulate interdomain communication between the two protomers (65, 69).
Table 1
| Hsp90 residue | Modification | Enzyme | Main effect | References | |
|---|---|---|---|---|---|
| α | β | ||||
| T5, T7 | N/A | Phosphorylation | DNA-PK | Unknown | (40) |
| T36 | T31 | Phosphorylation | CK2 | Decreased ATPase activity, increased inhibitor sensitivity | (41) |
| Y38 | Y33 | Phosphorylation | Swe1/Wee1 | Increased client association, decreased inhibitor binding | (42) |
| Y61, Y284, Y492, Y604 | Y56, Y276, Y484, Y596 | Phosphorylation | Unknown | Unknown | (43–45) |
| S63, T65 | S58, T60 | Phosphorylation | CK2 | Unknown | (46) |
| K69, K100, K292, K327, K478, K546, K558 | K64, K284, K319 | Acetylation | HDAC6, p300 | Increased inhibitor binding, increased extracellular location and association with MMP-2 | (47) |
| T88 | T83 | Phosphorylation | PKA | Increased translocation | (48) |
| T90 | T85 | Phosphorylation | PKA | Increased association with some co-chaperones and client proteins | (49) |
| K112, K283, K292, K407, K546, K558, K615, K631 | K107, K275, K284, K399, K538, K550, K607, K623 | Ubiquitination | CHIP | Inactivation and degradation of HSP90 | (50, 51) |
| T115, T425, T603 | N/A | Phosphorylation | PKCγ, Mps1 | Decreased chaperone activity, increased inhibitor sensitivity | (52, 53) |
| K191 | N/A | Sumoylation | Unknown | Increased association with Aha1, increased inhibitors binding | (54) |
| Y197 | Y192 | Phosphorylation | Yes kinase | Decreased association with Cdc37 | (55) |
| S231, S263 | S226, S255 | Phosphorylation | CK2 | Increased apoptosome formation, decreased client association | (56, 57) |
| K294 | K286 | Acetylation | HDCA6 | Decreased affinity for HSP70, co-chaperones, and client proteins | (58) |
| Y309 | Y301 | Phosphorylation | c-Src | Increased association with eNOS | (59) |
| Y313 | Y305 | Phosphorylation | Yes kinase | Increased association with Aha1 | (55) |
| S391, T624 | T89, S383, T616 | Phosphorylation | Pnck, PKA | Dissociation of client proteins | (60, 61) |
| S460 | S452 | Phosphorylation | PKA | Unknown | (62) |
| C572 | N/A | Oxidation | Unknown | Decreased chaperoning activity | (63) |
| S595 | S587 | Phosphorylation | p38γ | Formation of a ternary complex with mutated K-Ras and p38γ | (64) |
| C597 | C589 | S-nitrosylation | eNOS | Decreased ATPase activity | (65, 66) |
| Y627 | Y619 | Phosphorylation | Yes kinase | Dissociation of clients and co-chaperones | (55) |
| T725, S726 | S718 | Phosphorylation | CK1, CK2, GSK3-β | Decreased CHIP binding, increased HOP binding | (67) |
Summary of the most relevant post-translational modifications of HSP90.
Aha1, HSP90 ATPase homolog 1; Cdc37, cell division cycle 37 homolog; CK1, casein kinase 1; CK2, casein kinase 2; CHIP, C-terminus of Hsp70-interacting protein; DNA-PK, DNA-dependent protein kinase; eNOS, endothelial nitric oxide synthase; GSK3-β, glycogen synthase kinase beta 3; HDAC6, histone deacetylase 6; MMP-2, matrix metalloproteinase-2; Mps1, monopolar spindle-1; PKA, protein kinase A; PKCγ, protein kinase C gamma; Pnck, pregnancy-upregulated non-ubiquitous calmodulin kinase; PP5, serine/threonine-protein phosphatase 5.
Despite this level of depth and sophistication in the knowledge of the roles of various players in the chaperone cycle, the factors that determine whether a protein is a HSP90 client are still elusive. HSP90 might facilitate conformational rearrangements in clients, or it might sequester them, thus avoiding their proteasomal degradation (70, 71).
Furthermore, protein quality control is a compartmentalized process characterized by peculiar features in the various subcellular regions, where different and specific networks of chaperones are present. In mitochondria, which house essential metabolic pathways, such as the tricarboxylic acid (TCA) cycle, the oxidative phosphorylation (OXPHOS) machinery and branches of amino acid, lipid, and nucleotide metabolic pathways, the paralog of the HSP90 chaperone family is TRAP1 (15, 72). TRAP1 shares the same domain structure of HSP90, but lacks a charged linker between middle and C-terminal domains and displays a long N-terminal extension called “strap” that extends between protomers in the closed state and inhibits its function at low temperatures. During TRAP1 chaperone cycle, ATP binding induces a dramatic structural change from the apo, open state to a closed, asymmetric structure, with one protomer buckled and the other one in a straight conformation (25, 73–75). The subsequent hydrolysis of the two ATP molecules bound to the TRAP1 dimer gives off the energy required for client remodeling. Hydrolysis of the first ATP swaps protomer symmetry and rearranges the client-binding site, causing structural changes in client conformation, whereas the second ATP is used to induce the formation of a compact ADP state of the chaperone, which releases the client and eventually the ADP molecules (32). Interestingly, work by the Agard lab has established that the asymmetric theme in the mechanisms of conformational dynamics is a general characteristic of the Hsp90 family (25, 32, 74). We are just beginning to understand PTM regulation of TRAP1 (Table 2), whereas no co-chaperones are known and the number of its known clients remains quite small.
Table 2
| TRAP1 residue | Modification | Enzyme | Main effect | References |
|---|---|---|---|---|
| S501 | S-nitrosylation | NOSs | Decreased ATPase activity, enhanced proteasomal degradation of TRAP1 | (76, 77) |
| S511, S568 | Phosphorylation | ERK1/2 | Formation of a multimeric complex with SDH and ERK1/2, increased SDH inhibition and neoplastic growth | (78) |
| Unknown | Phosphorylation | PINK1 | Prevent oxidative stress-induced apoptosis | (79) |
| Unknown | Deacetylation | SIRT3 | Increased mitochondrial respiration under low glucose conditions | (80) |
| Unknown | Phosphorylation | c-Src | Inhibition of complex IV activity | (81) |
Reported post-translational modifications of TRAP1.
ERK1/2, extracellular signal-regulated kinases 1 and 2; NOSs, nitric oxide synthases; PINK1, PTEN-induced kinase 1; SIRT3, sirtuin 3.
HSP90 Chaperones in Cancer
Tumor cells are exposed to a variety of stresses that can directly hit polypeptide conformation and functionality, including unbalance in redox equilibrium caused by a profound rewiring of their metabolic circuitries and by inconstant oxygen availability (82), thus leading to a potential increase in oxidative stress. Moreover, genomic instability in a framework of relentless proliferation can lead to a high risk of synthesis of misfolded proteins. HSP90 molecular chaperones are central hubs of complex biological pathways and strongly induced by hypoxia, shortage of nutrients, high rate of DNA replication and expression of mutated proteins, thus acting at several levels to block cell death and to promote proliferation under the harsh conditions of neoplastic progression (22).
Indeed, overexpression of HSP90 has been observed in a variety of cancer types, including medulloblastoma, pancreatic, ovarian, breast, lung, and endometrial cancer, oropharyngeal squamous cell carcinoma and multiple myeloma, and high HSP90 levels are associated with poor prognosis in lung, esophageal and bladder cancer, melanoma and in several forms of leukemia (13). Most identified HSP90 clients are proteins related to biological processes dysregulated in cancer, such as signal transduction, survival, growth and invasiveness of cells and include steroid hormone receptors, both wild-type and mutant forms of the tumor suppressor p53, telomerase, hypoxia-inducible factor 1α (HIF1α) (12) and kinases, which display a continuous range of binding affinities for HSP90 (83). Some kinases would require HSP90 to stabilize their open conformation in order to efficiently bind ATP, whilst others seem to only need HSP90 for initial folding (27, 84) and would perform their enzymatic activity without HSP90 assistance. Studies with closely related pairs of client/non-client kinases, like the client v-Src and the non-client c-Src, which share 98% sequence identity, suggest that HSP90 dependence requires a combination of factors, including folding cooperativity and subtle changes in the overall stability and compactness of clients (85). HSP90 can also be secreted in a variety of tumor cells under the regulation of HIF-1α. In the extracellular matrix and on cell surfaces, HSP90 decreases the tumor-suppressing effects of TGFβ and modulates cell migration and invasiveness (17), for instance by interaction with matrix metalloproteases (86–88).
Further layers of complexity exist in the interplay between HSP90 chaperones and cancer. Some HSP90 clients, like p53, may change the chaperone and co-chaperone networks by inducing the expression of co-chaperone subsets (89). The “epichaperome” is a functionally connected network of HSP70 and HSP90 machineries, which includes co-chaperones and is present in more than 50% of tumors (35), where it expands and integrates chaperone activities and promotes tumor survival (12).
Cancer cells incur high level of mitochondrial functional changes in order to maintain the required levels of ATP, reducing equivalents and metabolic intermediates, and exposure to fluctuating levels of oxygen and to high amounts of ROS can hamper proper protein folding and lead to mtDNA mutations, thus damaging mitochondrial structure and function (10, 90). Under these conditions TRAP1, the mitochondrial HSP90 paralog, could contribute to maintain an adequate quality control and to preserve mitochondrial functions. TRAP1 expression is higher in many tumors compared to surrounding non-malignant tissues and was shown to correlate with progression, metastasis and disease recurrence in prostate and breast cancer, hepatocellular and colorectal carcinoma and non-small cell lung cancer (15, 91). In mitochondria, TRAP1 provides resistance to oxidative stress (18, 92), possibly counteracting the effects of several chemotherapeutics, and inhibits opening of the permeability transition pore (PTP), a cell death-inducing mitochondrial channel composed by the ATP synthase holoenzyme and that can be induced by a ROS surge (93). Thus, TRAP1 exerts a pro-neoplastic function by counteracting ROS-induced, PTP-mediated cell death. However, ROS effects on tumor growth are multifaceted, as oxidative stress can favor genetic instability and aggressiveness of tumors in advanced stages. Consequently, the anti-oxidant activity of TRAP1 could hamper growth in specific tumor types or stages as in cervical carcinoma, clear cell renal cell carcinoma and high-grade ovarian cancer, where TRAP1 expression inversely correlates with tumor grade (81, 94). TRAP1 also down-regulates the activity of both cytochrome c oxidase, the complex IV of the respiratory chain (81), and of succinate dehydrogenase (SDH) (95), which oxidizes succinate to fumarate at the crossroad between OXPHOS and the TCA cycle (Figure 3). Hence, TRAP1 participates in the metabolic switch of tumor cells toward aerobic glycolysis, i.e., decreased OXPHOS activity paralleled by enhanced glucose utilization (96). Importantly, SDH inhibition increases intracellular succinate levels, and succinate acts as an oncometabolite in several ways (97). It competitively inhibits α-ketoglutarate–dependent dioxygenases that include prolyl hydroxylases (PHDs), the JmjC domain-containing demethylases (KDMs) and the TET (10–11 translocation) family of 5-methylcytosine hydroxylases (7). PHD inhibition stabilizes the transcription factor HIF1α, increasing invasiveness, angiogenesis and further metabolic changes in tumor cells (98), whereas inhibition of KDMs, which hydroxylate lysine residue on histones, and of TETs, which induce DNA demethylation of CpG islands near gene promoters, prompts complex epigenetic rearrangements in neoplastic cells (Figure 3). Oncogenic kinase pathways directly target TRAP1, as it is both Tyr-phosphorylated in a Src-dependent way and Ser-phosphorylated by ERK1/2, favoring cytochrome oxidase inhibition and enhancing TRAP1 inhibition of SDH activity, respectively (78, 81), whereas S-nitrosylation elicits TRAP1 degradation (76) and decreases its ATPase activity (77) (Table 2).
Figure 3
Inhibiting HSP90 As an Anti-Neoplastic Strategy
To a superficial analysis, HSP90 would appear an unlikely target for anti-neoplastic drugs, as it is highly expressed in all cell types. However, HSP90 inhibitors tend to accumulate in tumors and are more toxic in most cancer cells than in their non-transformed counterparts (99). This could depend on HSP90 induction and/or ectopic localization in many tumor types, where it can undergo selective PTMs and can interact with a specific landscape of co-chaperones and client proteins, creating multimolecular complexes restricted to tumor cells. For instance, HSP90 binds inhibitors more strongly when it is part of epichaperome complexes (35). Some HSP90 clients express oncogenic mutations that can change their association pattern with HSP90. This can create interactions that are selectively druggable and enhance HSP90 affinity for inhibitors (100). Moreover, oncoproteins could become addicted to HSP90 in order to maintain their mutated and potentially unstable conformations, rendering disruption of this interaction particularly toxic for neoplastic cells (101).
To date, more than 50 clinical trials have been performed or are under way with several HSP90 inhibitors, but expectations of evolution toward therapeutic application have been largely frustrated. In most cases, the anti-neoplastic efficacy of HSP90 inhibition has been modest, only inducing a transient growth arrest that is reverted after drug removal, or adverse effects have been recorded, leading to termination or suspension of clinical trials. Possible reasons of these failures include a compensatory induction in the expression of other HSPs, in particular HSP70, off-target effects in patients caused by the multiplicity of biological functions regulated by HSP90 and insufficient stratification of patients enrolled in the studies (12).
ATP-Competitive Inhibitors
Since HSP90 functions depend on its ATPase activity, most drugs have been developed as competitive inhibitors targeting the active site and competing with ATP for binding the protein. The underlying hypothesis is that disrupting the enzymatic activity of HSP90 would reverberate on the chaperone protein folding machinery, simultaneously affecting multiple oncoproteins that are essential to the proliferation and maintenance of cancer cells. The molecular basis is that ATP-ADP exchange regulates and determines a well-balanced, functionally-oriented conformational equilibrium: outcompeting the nucleotide by drug-like ligands will expectedly unbalance conformational dynamics, leading to a blockage of correct biological activities.
The unusual mode of ATP binding to HSP90 allows specific inhibition of HSP90 chaperone activity by chemical compounds. Indeed, the base and the sugar of ATP are lodged in the NTD binding pocket in a “kinked” conformation, with the phosphates pointing outwards and the γ-phosphate becoming buried only when MD and NTD associate (102). Competitive inhibitors in the ATP pocket block ATP hydrolysis and subsequently hamper the closure of the N-terminus of the dimer, thus inhibiting the HSP90 chaperone cycle (103) (Table 3). Prototypical molecules of this class are the benzoquinone geldanamycin (GA) and the macrolide radicicol, whose selectivity has been widely discussed elsewhere (121, 122). Both compounds demonstrate strong toxicity on tumor cells (20). GA was the first HSP90 inhibitor to be evaluated as an antitumor agent. In neoplastic cells GA induces apoptosis, inhibits cell migration associated with FAK and HGF activity, as well as angiogenesis and epithelial-mesenchymal transition by down-regulating VEGF receptor, HIF-1α and NF-κB signaling (123–126). However, GA preclinical trials were discontinued due to its hepatotoxicity, poor solubility and in vivo instability. Similarly, radicicol cannot be used as a drug as it is not stable (13, 19). Therefore, several radicicol and GA derivatives were developed (127, 128). Tanespimycin (17-N-allylamino-17-demethoxygeldanamycin, 17-AAG) is a GA derivative that induces cell cycle arrest and apoptosis in cancer cells. Tanespimycin entered a Phase III clinical trial for multiple myeloma, but its development was halted because of poor solubility and poor oral bioavailability and lapsed patent protection, with a prolonged disease stabilization in several tumor types, without any tumor regression (13). Another GA derivative, Alvespimycin (dimethylaminoethylamino-17-demethoxygeldanamycin, 17-DMAG) demonstrated anti-tumor activity and improved solubility in water, but dose-limiting side effects were recorder during various clinical trials (129, 130) (around 2000 X ray structures can be found at the RCSB protein databank (https://www.rcsb.org/). A new generation of GA derivatives is currently under evaluation, such as retaspimycin hydrochloride (IPI-504) or ganetespib. These molecules are well-tolerated and effective in cells, with reduced liver and cardiovascular toxicity (131, 132). Ganetespib (STA-9090), a small molecule inhibitor containing a triazole moiety that binds to the ATP-binding pocket of HSP90, is the most promising second generation HSP90-targeting compound (133). Its potent anti-tumor activity was translated into several clinical studies, demonstrating efficacy both in monotherapy and in combination with other drugs in various cancer types. Ganetespib produced significant single agent activity in ALK-driven cancer models, however only transient responses were reported in patients with KRAS mutant tumors due to rapid development of resistance (134). Second generation radicicol derivatives, such as NVP-AUY922 (luminespib, VER-2296) or AT13387 (Onalespib) showed strong efficacy both pre-clinically and in clinical trials, some of which are ongoing (103). Other inhibitors targeting the HSP90 ATP-binding pocket include purine analogs that can be administered orally (135), substituted resorcinols and compounds featuring a substituted benzamide substructure. Many of these molecules have entered clinical trials (136).
Table 3
| Inhibitor | Structure | Selected bioactivity indicatorsa | Contact residuesb | PDBc | Clinical trialsd | Other targetse |
|---|---|---|---|---|---|---|
| Geldana mycin | 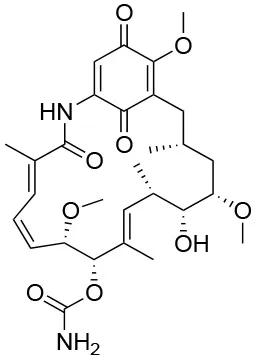 | IC50f = 15.8 nM (104) EC50g = 30 nM (105) Kdh = 1.35 μM (106) | N51, S52, D54, A55, K58, D93, I96, M98, D102, N106, L107, K112, G135, V136, G137, F138, Y139 | 1YET (Hsp90) (107) | (None active or recruiting) | Nitric oxide synthase (104); oncogene product p185erbB−2 protein tyrosine-kinase (104, 108, 109); src protein tyrosine-kinase (110) |
| Radicicol | 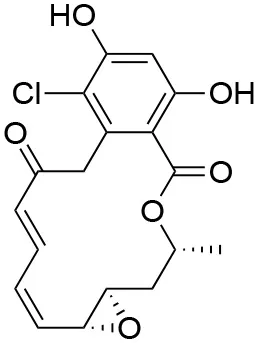 | IC50f = 37.5 nM (104) EC50i = 25 nM (111) Kdj = 2.2 nM (112), 3.04 nM (113) | L48, N51, S52, D54, A55, K58, D93, I96, M98, N106, L107, F138, T184, V186 | 4EGK (Hsp90) (114) | (None active or recruiting) | Topoisomerase VI (115); Pyruvate dehydrogenase kinase (116); Carbonyl reductase 1 (117); |
| PU-H71 | 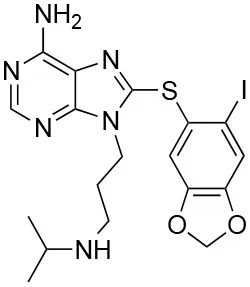 | IC50f = 50 nM (118) EC50k = 16 nM (118) Kdh = 780 nM (106) | N51, S52, A55, K58, D93, I96, G97, M98, L107, F138, Y139, V150, W162, T184 | 2FWZ (Hsp90) (119)4Z1F (Trap1) (120) | Phase I: • metastatic breast cancer (with nab-paclitaxel) • safety and tolerability (with ruxolitinib) • advanced malignancies | (None known) |
Key data concerning orthosteric Hsp90 inhibitors Geldanamycin, Radicicol, and PU-H71.
Chosen bioassays are limited to those that are directly comparable or similar in nature. Only human chaperones and cell lines have been considered. The list is merely indicative: a more complete list is available from the PubChem Database (https://pubchem.ncbi.nlm.nih.gov/compound/5288382; https://pubchem.ncbi.nlm.nih.gov/compound/radicicol; https://pubchem.ncbi.nlm.nih.gov/compound/pu-h71 [accessed May 24, 2020]).
Residues with at least one atom detected to be closer than 3.0 Å to any atom in the ligand. Hydrogen atoms included in the analysis.
Human chaperone targets only.
Active and/or recruiting clinical trials only, as listed in the PubChem Database (https://pubchem.ncbi.nlm.nih.gov/compound/5288382; https://pubchem.ncbi.nlm.nih.gov/compound/radicicol; https://pubchem.ncbi.nlm.nih.gov/compound/pu-h71 [accessed May 24, 2020]).
Documented, non-chaperone human targets only.
Antiproliferation/growth inhibition of SkBr3 breast cancer cell line.
Antiproliferative activity against HCT116 cells (luminescence assay).
Hsp90 competition with fluorescent geldanamycin (fluorescence assay).
Hsp90 binding (Western Blot on MCF-7 cell lysate).
Hsp90 binding (surface plasmon resonance analysis).
Hsp90 competition with fluorescent geldanamycin (fluorescence assay on SkBr3 cell lysate).
The nucleotide-binding pocket of the NTD is considered the most conserved structural component among HSP90 family members, impeding the rational design of paralog-selective inhibitors targeting it. Therefore, a strategy used for inhibiting TRAP1 was to exploit its subcellular localization, and mitochondria-permeable GA derivatives were conceived and synthesized. Gamitrinibs (Geldanamycin mitochondrial matrix inhibitors) are 17-AAG derivatives linked to either guanidinium repeats or triphenylphosphonium (TPP), used as mitochondriotropic moieties (137). These molecules induced mitochondrial PTP opening and tumor cell death in mouse models of prostate cancer (138). Similarly, TPP was linked to the purine-scaffold Hsp90 inhibitor PU-H71PU-H71 (120) to develop a mitochondria-targeting conjugate, SMTIN-P01. Indeed, the co-crystal structures of PU-H71 in complex with either HSP90 or TRAP1 highlighted slight differences in the ATP-binding pocket of the two chaperones. The Leu172-Phe201 sequence is disordered only in the TRAP1 binding site, and the two flanking residues (Asn171 and Gly202) have different configurations (103, 139), providing a molecular basis for the differentiation of ligands to target specifically one isoform. Accordingly, TRAP1 inhibitors without mitochondrial delivery vehicles were also reported, showing a better binding to TRAP1 than to Hsp90 (139).
In spite of this wealth of efforts, so far ATP-competitive HSP90 inhibitors have not met clinical expectations and none of them has been approved for cancer treatment. All compounds showed toxicity (e.g., liver or ocular toxicity) and/or absence of convincing anticancer efficacy (13). Possible reasons include only partial inhibition of the target client proteins, P-glycoprotein-dependent efflux from target cells, requirement for reductive metabolism to reach full activation (17-AAG), off-target effects on biochemical pathways not specific of tumor cells (2). Moreover, the drug concentration required to outcompete ATP and induce client degradation is often the same as that needed to induce the heat shock response (HSR). HSR is based on the activation of the transcription factor HSF1, which leads to overexpression of multiple heat shock proteins, including HSP70, HSP40, and HSP27. As HSR is a survival mechanism, it can be detrimental in an anti-cancer therapy, and trying to avoid it determines dosage, toxicity and tolerance problems (136).
These limitations could be overcome if one were able to disentangle the intricacies in the functional mechanisms of different paralogs together with their relationships to metabolic and/or signaling regulation. Chemical interventions based on using ad hoc designed molecules to perturb a specific aspect of the HSP90 functional spectrum and directly report on the consequences of this perturbation would represent ideal tools. The diversity of conformations, protein-protein interactions, and functions involved in HSP90 mechanisms makes the “one-drug-fits-all” perspective unrealistic. On the other hand, that very diversity may provide a greater number of drug discovery opportunities thanks to the variety of structural and chemical motifs involved in conformational regulation and protein-interaction phenomena.
Allosteric Inhibitors
The perspective of developing different strategies to target various aspects of the multifaceted HSP90 complexes can expectedly generate novel types of chemical intervention, unveiling new chemical tools for the investigation of biological mechanisms and/or novel candidates for therapeutic applications. In this conceptual framework, increasing appreciation is being given to the potential of allosteric modulators (Table 4). Allostery defines the feature of proteins to undergo a modulation of affinity toward a primary binding event caused by binding an “effector” a different distant position called the allosteric site. This modulation may cause increase or decrease of protein activity (ATP in Hsp90 processing for instance) and its downstream effects in the cell. Allosteric modulators represent an interesting opportunity for drug development in HSP90-related metabolic circuitries for several reasons: on the one hand, they permit to finely tune and regulate both the enzymatic functions and the interactions of HSP90, and on the other hand they may facilitate the selective targeting of different chaperone isoforms. The latter option would be highly desirable in the development of drugs that need to perturb the function of one specific paralog of the protein, active in specific pathologic conditions and/or in specific subcellular compartments. This type of chemical tools could selectively regulate/disrupt the functions of paralogs in a controlled way, shedding light on the correlations between the induced perturbation and the consequent biological activities, and laying the bases for novel mechanism-driven therapeutic interventions. Allostery is the prime mechanism by which achieving fine protein regulation via the activation of specific conformational states that meet functional requirements. Modifications at one site, caused for instance by ligand binding, are propagated through the protein, shifting the structural population with the modulation/perturbation of dynamic states that encode specific functions. In this context, the atomistic understanding of allosteric mechanisms provides the basis for the development of new drug candidates. In HSP90, the region at the border between the M-Domain and the C-terminal domain, located at 60Å from the ATP-site, has been shown to host a druggable allosteric site (152–156).
Table 4
| Name | Structure | Pharmacokinetics | Mechanism | Residues | References |
|---|---|---|---|---|---|
| Novo biocin | 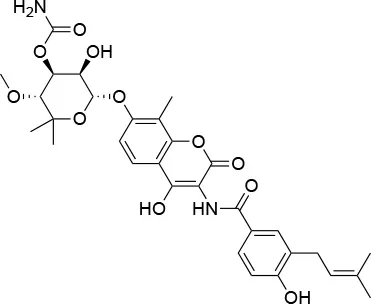 | IC50 (SkBr3) ~700 μMa | Disruption of the interaction with the co-chaperones Hsc70 and p23 | L663—H676 | (140–142) |
| Chloro biocin | 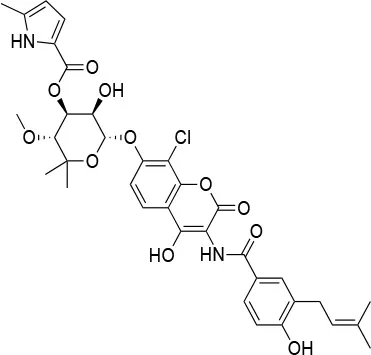 | IC50 (SkBr3) ~60 μMa | Unknown | Unknown | (141, 142) |
| 15a | 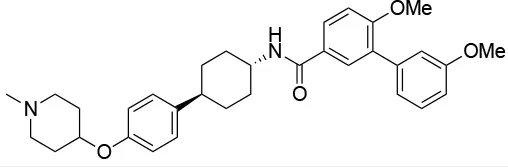 | IC50 (SkBr3) ~0.17 ± 0.02 μMb IC50 (MCF-7) ~0.22 ± 0.01 μMb | Unknown | Unknown | (143) |
| 80c | 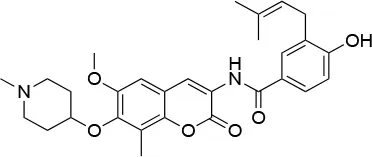 | IC50 (SkBr3) ~0.42 ± 0.01 μMb IC50 (MCF-7) ~0.54 ± 0.02 μMb | Unknown | Unknown | (144) |
| Derrubone | 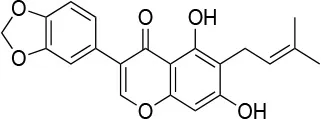 | IC50~ 0.23 ± 0.04 μMc IC50 (SkBr3) ~12 ± 0.3 μMb IC50 (MCF-7) ~9 ± 0.7 μMb | Stabilization of HSP90-client interactions | Unknown | (145) |
| Withaferin A | 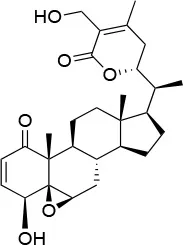 | IC50 (Panc-1) ~1.24 μMb IC50 (MiaPaCa2) ~2.78 μMb | Disruption of the HSP90-Cdc37 complex in an ATP-independent way | Unknown | (146) |
| Celastrol | 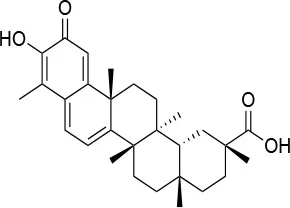 | IC50 (Panc-1) ~3 μMb IC50 (Hep3B) ~0.3 ± 0.08 μMb | Disruption of the association between HSP90 and Cdc37 | T94 – M125 | (147, 148) |
| 19 | 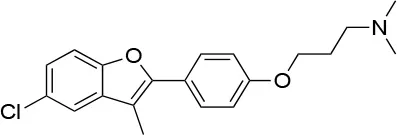 | IC50 (DU145) ~12.7 ± 2.5 μMb IC50 (STO) ~9.1 ± 1.1 μMb | Acceleration of the HSP90 conformational cycle | E477, D503 (protomer A), R591 (protomer B) | (149) |
| 25 | 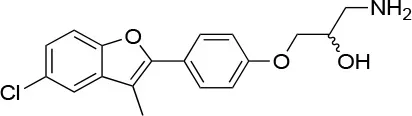 | IC50 (STO) ~22.1 ± 1.1 μMb | Increased HSP90 ATPase activity favoring its active state | E477, R591 (protomer A), K594 (protomer B) | (150) |
| LA1011 |  | Kd (HSP90α) ~3.8 ± 0.7 μMd Kd (HSP90β) ~9.7 ± 0.7 μMd | Alteration of HSP90 chaperoning activity | G675, S677, L678 | (151) |
Summary of the most important compounds identified as allosteric modulators of HSP90.
Expression levels of HSP90 client proteins.
Anti-proliferation assay.
Luciferase refolding.
Isothermal titration calorimetry.
Experimental evidence for the possibility to target this site initially came from the Neckers group, who demonstrated the interaction between HSP90 and coumarin (140, 141). In particular, they observed that the coumarin antibiotics Novobiocin and Chlorobiocin caused the impairment of HSP90 chaperone functions by disrupting the interactions with the large group of TPR-containing co-chaperones. Importantly, despite being used at high concentrations, these molecules did not show the toxicity of ATP-competitive ligands related to the induction of the HSR (12). Novobiocin causes moderate anti-proliferative effects on tumor cell models, down-regulating the expression of important HSP90-dependent clients, including Raf-1, erbB2, mutant p53, and v-Src (141). Since this seminal demonstration of the importance and druggability of alternative binding sites, significant synthetic efforts have been dedicated to improving the activities of coumarin-based allosteric molecules. In this context, the Blagg group demonstrated the possibility of substituting the carbohydrate moiety and the phenyl substituents around the coumarin scaffold by more accessible groups. The coumarin was also substituted by a series of biphenyl- or bi-cyclic scaffolds that permitted to explore the structure-activity relationships of a large number of derivatives. This series of allosteric inhibitors showed anticancer activities reaching the mid/low nanomolar range (143, 144). However, the anti-neoplastic efficacy of this family of inhibitors and their molecular mechanisms of action remain unclear (103), and no carboxy-terminal inhibitor has reached clinical trials up to now (85).
We have recently used a molecular dynamics-based strategy to identify an allosteric pocket distal to the ATPase site of TRAP1, which allowed the rational design and testing of small molecule compounds that target it (157). We have also found that the same allosteric domain can host the bis-dichloroacetate ester of the vegetal derivative honokiol DCA (HDCA) (158). All these molecules inhibit TRAP1 with a high selectivity over HSP90, abolishing TRAP1-dependent down-regulation of SDH activity in cancer cells and their in vitro tumorigenic growth (157, 158). Therefore, they constitute potential leads that can be used to better dissect TRAP1 biochemical functions and to conceive novel anti-neoplastic approaches.
Targeting Protein-Protein Interactions (PPIs) as sources of new leads is another interesting strategy to hit HSP90 chaperone function. PPIs are often less well-conserved than active sites, making easier to achieve selectivity (159). In general, PPIs tend to modulate the activity of the interacting proteins, rather than inducing on/off effects. Thus, PPI targeting compounds could flexibly titrate chaperone activity in the context of specific co-chaperone-client complexes, lowering the possibility of inducing off-target effects. However, not all PPIs are equally druggable, as the surfaces of contact usually have a larger buried surface area than enzyme-active sites, making it difficult to identify small molecules capable to block them (160).
Furthermore, as the pharmacological inhibition of clients or the downregulation of co-chaperone levels was shown to hypersensitize cells to HSP90 inhibitors, the perspective of combining drugs acting on different levels of regulation machineries is gaining increasing traction. Several compounds have been described that lead to the modulation of co-chaperone binding to HSP90. Examples include molecules like derrubone, withaferin A, and celastrol, which block CDC37 binding to HSP90. However, none of these compounds entered therapeutics so far because of insufficient efficacy in clinical studies. This could depend on several factors: variability and flexibility of HSP90 interactome in different cancer types and stages, off-target effects and HSR induction (161). Alternative interventions may involve direct targeting of PPI interfaces, disrupting HSP90-cochaperone interfaces, as recently reported (162).
Allosteric Activators
Together with inhibition, a viable approach to controlling the metabolic implications of HSP90 and its paralogs entails the use of allosteric activators of the ATPase and of the conformational dynamics of the chaperone. If we consider the dynamic nature of the HSP90 chaperone network, whereby different HSP90 conformations are stabilized by interactions with different multi-protein complexes, allosteric activators can expectedly select/favor a subset of HSP90 structures that may have preferential binding to a selected population of interactors (Figure 4). Starting from original methods of molecular dynamics (MD) simulation analysis, we were able to design a series of activators capable of modifying the biochemical properties of the chaperone, as well as its activities in cells (149, 150, 163). Conjugation to mitochondrial targeting moieties showed that designed activators could show activity also on TRAP1, modifying the activities of the client SDH, with an impact on the energy metabolism of targeted cells (150). In the context of activators, Prodromou and coworkers discovered dihydropyridines able to stimulate ATPase activity. Stimulation was shown to reverberate in a compromised ability to chaperone, which consequently induced the HSR in Alzheimer's disease cellular models (151).
Figure 4
The validity of the allosteric approach in studying the metabolic implications of chaperone stems from the fact that the overall activity of HSP90 is not simply shutdown but rather modulated. Ligand binding at a site far from the active site can be propagated through the protein, modifying the dynamic and/or structural populations with the modulation of dynamic states that encode specific interactions. This can in turn translate in the reshaping of surfaces that control interactions with other proteins (co-chaperones and clients), potentially favoring the binding of one partner over other alternatives. In this context, the controlled perturbation of interactions determines an overall modification of the dynamics in the protein networks that underlie signaling and metabolic pathways, providing novel intervention opportunities. Allosteric modulation of the chaperone enzymatic and conformational activities is thus directly coupled to phenotypic effects, through the finely-tuned modification of networks of PPIs.
Coupling HSP90 Targeting with Other Anti-Neoplastic Approaches
The lack of translation to the clinical practice of HSP90 inhibitors clearly indicates the need of novel strategies to exploit cancer cell sensitivity to chaperone targeting. A promising treatment strategy is the combination therapy, in which HSP90-directed molecules are associated with other chemotherapeutics, and the combined action on different targets potently and selectively elicits the death of malignant cells (Table 5). Some examples already exist. A large scale phase III clinical trial (Galaxy-2) in advanced lung cancer evaluated the effects of combining ganetespib and anti-microtubule agent docetaxel in either KRAS mutant or KRAS wild type non-small cell lung cancer (NSCLC) patients. This trial failed to demonstrate any benefit in terms of progression free survival or overall survival (170). It was then demonstrated that resistance to ganetespib and to the combined treatment with docetaxel in KRAS mutant NSCLC patients was caused by hyperactivation of ERK1/2-p90RSK-mTOR signaling pathway and by circumventing the G2-M checkpoint arrest of the cell cycle (171, 172). These observations suggest that combining ganetespib with ERK1/2, p90RSK, or CDC25C inhibitors could overcome the observed resistance (131). In perspective, one would like to define the biochemical features of the tumor and the fine mechanisms of chaperone/client interaction in order to design HSP90 targeting-molecules that modulate specific signaling hubs, such as transcriptional or epigenetic regulation, maintenance of DNA integrity or bioenergetic circuitries. Such a re-shaping of HSP90 inhibition, aimed at avoiding the deleterious effects of a global damage to cellular proteostasis, would imply selecting different agents for combination usage and reconsidering dosing strategies, adapting them to various cancer patient subsets (173). Novel usage options for HSP90 inhibitors are provided by the combination with targeted therapies or with immune therapy approaches, and preliminary data are encouraging [reviewed in (174)]. Combination of the multi-kinase inhibitor sorafenib with tanespimycin demonstrated efficacy in melanoma and renal cancer patients. HER2-positive, metastatic breast cancer patients treated with tanespimycin showed an improved clinical outcome. Ganetespib induced tumor regression in melanoma xenografts when supplied together with the MEK inhibitor TAK-733 and with the BRAF(V600E) inhibitor vemurafenib. Recently, HSP90 inhibitors were also tested in combination with immunotherapy, and approaches combining inhibitors of immune checkpoints and of HSP90 have been assayed. The anti-PD-L1 antibody STI-A1015 elicited higher therapeutic efficacy in melanoma and colon cancer cell models when combined with ganetespib as compared to the monotherapy regimens. In the case of TRAP1, its genetic inhibition prompts an increase in oxygen consumption rate and a decrease in extracellular acidification rate (78, 95), indicative of a metabolic rewiring toward OXPHOS coupled with a down-modulation of glycolysis. Therefore, it would be promising to identify highly selective TRAP1 inhibitors that do not affect Hsp90 activity. In principle, these compounds could be associated with OXPHOS-targeting molecules (175) in order to induce a bioenergetic catastrophe in tumor cells.
Table 5
| Hsp90 inhibitor | Drugs in combination | Cancer type | Outcome | References |
|---|---|---|---|---|
| Tanespimycin (17-AAG) | Sorafenib (multi-kinase inhibitor) | Melanoma, renal and colorectal cancer | Clinical efficacy in melanoma and renal cancer patients | (164) |
| Tanespimycin | Trastuzumab (anti-HER2 antibody) | HER-2 positive, metastatic breast cancer | Significant anti-cancer activity | (165) |
| Ganetespib (STA-9090) | Vemurafenib (BRAF(V600E) inhibitor) | Melanoma | Inhibition of tumor growth in BRAF-inhibitor sensitive melanoma | (166) |
| Ganetespib | TAK-733 (MEK inhibitor) | Melanoma | Tumor regression in vemurafenib-resistant xenografts | (166) |
| Ganetespib | Capecitabine (DNA synthesis inhibitor) | Colorectal cancer | Improved anti-tumor activity of combinatorial therapy | (167) |
| Ganetespib | STI-A1015 (anti-PD-L1 antibody) | MC38 colon carcinoma and B16 melanoma | Enhanced anti-tumor efficacy of the combinatorial regimen | (168) |
| Luminespib (NVP-AUY922) | Omipalisib (PI3K inhibitor) | KRAS mutant NSCLC | Synergistic anti-tumor effect | (169) |
Preclinical and clinical studies where HSP90 inhibitors were combined with other anti-cancer drugs.
Design of new molecules, such as conjugated compounds that link established chemotherapeutic drugs with HSP90 inhibitors, may open further therapeutic windows. This strategy exploits the observation that HSP90 inhibitors highly accumulate in cancer cells, and could therefore act as cargoes for chemotherapeutics, thereby increasing their efficacy while reducing toxicity. The bifunctional compound STA-8666 consists of an HSP90 inhibitor and a topoisomerase inhibitor (SN-38) and demonstrated antitumor effects and lower systemic toxicity in preclinical studies on different cancer models (176, 177). Finally, the simultaneous inhibition of HSP90 and specific protein kinases appears to be another promising avenue to reduce drug resistance (12).
Conclusions
Understanding the fine details that regulate the function of hub proteins as central as HSP90 and TRAP1 for biochemical and metabolic pathways is a highly challenging task. In this context, chemical biology approaches based on the use of small molecules represent attractive means to expand the reach of our investigations of the complex biology of this chaperone. Designed molecules have the potential to induce variable functional responses by inhibiting or stimulating a certain activity and thus directly focus on the molecular mechanism they are perturbing. In terms of investigation of biological processes, such data must be combined to complementary methods rooted in molecular biology such as pull-downs, genetic screens, CRISPRi, biochemical assays, proteomic and interactomic analyses, expression of mutant proteins where strategically positioned residues are modified. This integrated approach would improve our understanding of the roles of HSP90 in cancer metabolism at different levels. In a more complex scenario, regulation of chaperone networks should also be molecularly investigated during orchestrated responses of the cell to a variety of noxious stimuli such as compartmentalized unfolded protein responses.
Overall, we propose that a molecular understanding of the biology of chaperones through small molecule compounds is important for both fundamental and practical reasons. On the fundamental side, they would illuminate the determinants of biochemical mechanisms. On the practical side, selective chemical tools can expectedly be evolved into compounds that might modulate selected chaperones (and their isoforms) under specific conditions of stress. Finally, combinatorial therapies could aim at simultaneously exposing tumors to specific damaging agents while blunting the activity of protecting chaperones, thus setting the stage for the definition of innovative anti-neoplastic strategies.
Statements
Author contributions
CS-M, GC, and AR contributed conception and design of the paper. CS-M and SS wrote sections of the manuscript and prepared figures. All authors contributed to manuscript revision, read, and approved the submitted version.
Acknowledgments
GC and AR gratefully acknowledge the financial support of Associazione Italiana Ricerca Cancro (AIRC grant IG 2017/20019 to GC and IG 2017/20749 to AR) and of Neurofibromatosis Therapeutic Acceleration Program (NTAP). AR was also supported by grants from Piano for Life OdV and Linfa OdV.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
KimYEHippMSBracherAHayer-HartlMHartlFU. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem. (2013) 82:323–55. 10.1146/annurev-biochem-060208-092442
2.
NeckersLWorkmanP. Hsp90 molecular chaperone inhibitors: are we there yet?Clin Cancer Res. (2012) 18:64–76. 10.1158/1078-0432.CCR-11-1000
3.
JoshiSWangTAraujoTLSSharmaSBrodskyJLChiosisG. Adapting to stress - chaperome networks in cancer. Nat Rev Cancer. (2018) 18:562–75. 10.1038/s41568-018-0020-9
4.
Vander HeidenMGDeBerardinisRJ. Understanding the intersections between metabolism and cancer biology. Cell. (2017) 168:657–69. 10.1016/j.cell.2016.12.039
5.
HetzCChevetEOakesSA. Proteostasis control by the unfolded protein response. Nat Cell Biol. (2015) 17:829–38. 10.1038/ncb3184
6.
LuoBLeeAS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. (2013) 32:805–18. 10.1038/onc.2012.130
7.
CanninoGCiscatoFMasgrasISanchez-MartinCRasolaA. Metabolic plasticity of tumor cell mitochondria. Front Oncol. (2018) 8:333. 10.3389/fonc.2018.00333
8.
LinYFHaynesCM. Metabolism and the UPR(mt). Mol Cell. (2016) 61:677–82. 10.1016/j.molcel.2016.02.004
9.
ChevetEHetzCSamaliA. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. (2015) 5:586–97. 10.1158/2159-8290.CD-14-1490
10.
VyasSZaganjorEHaigisMC. Mitochondria and cancer. Cell. (2016) 166:555–66. 10.1016/j.cell.2016.07.002
11.
ZongWXRabinowitzJDWhiteE. Mitochondria and cancer. Mol Cell. (2016) 61:667–76. 10.1016/j.molcel.2016.02.011
12.
SchopfFHBieblMMBuchnerJ. The HSP90 chaperone machinery. Nat Rev Mol Cell Biol. (2017) 18:345–60. 10.1038/nrm.2017.20
13.
WuJLiuTRiosZMeiQLinXCaoS. Heat shock proteins and cancer. Trends Pharmacol Sci. (2017) 38:226–56. 10.1016/j.tips.2016.11.009
14.
LeeAS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. (2014) 14:263–76. 10.1038/nrc3701
15.
RasolaANeckersLPicardD. Mitochondrial oxidative phosphorylation TRAP(1)ped in tumor cells. Trends Cell Biol. (2014) 24:455–63. 10.1016/j.tcb.2014.03.005
16.
TaipaleMJaroszDFLindquistS. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. (2010) 11:515–28. 10.1038/nrm2918
17.
WongDSJayDG. Emerging roles of extracellular Hsp90 in cancer. Adv Cancer Res. (2016) 129:141–63. 10.1016/bs.acr.2016.01.001
18.
MasgrasISanchez-MartinCColomboGRasolaA. The chaperone TRAP1 as a modulator of the mitochondrial adaptations in cancer cells. Front Oncol. (2017) 7:58. 10.3389/fonc.2017.00058
19.
LiJBuchnerJ. Structure, function and regulation of the hsp90 machinery. Biomed J. (2013) 36:106–17. 10.4103/2319-4170.113230
20.
MollapourMNeckersL. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim Biophys Acta. (2012) 1823:648–55. 10.1016/j.bbamcr.2011.07.018
21.
ProdromouC. The ‘active life’ of Hsp90 complexes. Biochim Biophys Acta. (2012) 1823:614–23. 10.1016/j.bbamcr.2011.07.020
22.
LanneauDBrunetMFrisanESolaryEFontenayMGarridoC. Heat shock proteins: essential proteins for apoptosis regulation. J Cell Mol Med. (2008) 12:743–61. 10.1111/j.1582-4934.2008.00273.x
23.
AliMMRoeSMVaughanCKMeyerPPanaretouBPiperPWet al. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. (2006) 440:1013–7. 10.1038/nature04716
24.
DollinsDEWarrenJJImmorminoRMGewirthDT. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol Cell. (2007) 28:41–56. 10.1016/j.molcel.2007.08.024
25.
LaveryLAPartridgeJRRamelotTAElnatanDKennedyMAAgardDA. Structural asymmetry in the closed state of mitochondrial Hsp90 (TRAP1) supports a two-step ATP hydrolysis mechanism. Mol Cell. (2014) 53:330–43. 10.1016/j.molcel.2013.12.023
26.
SouthworthDRAgardDA. Species-dependent ensembles of conserved conformational states define the Hsp90 chaperone ATPase cycle. Mol Cell. (2008) 32:631–40. 10.1016/j.molcel.2008.10.024
27.
BoczekEEReefschlagerLGDehlingMStrullerTJHauslerESeidlAet al. Conformational processing of oncogenic v-Src kinase by the molecular chaperone Hsp90. Proc Natl Acad Sci USA. (2015) 112:E3189–98. 10.1073/pnas.1424342112
28.
KitagawaKSkowyraDElledgeSJHarperJWHieterP. SGT1 encodes an essential component of the yeast kinetochore assembly pathway and a novel subunit of the SCF ubiquitin ligase complex. Mol Cell. (1999) 4:21–33. 10.1016/S1097-2765(00)80184-7
29.
EchtenkampFJGvozdenovZAdkinsNLZhangYLynch-DayMWatanabeSet al. Hsp90 and p23 molecular chaperones control chromatin architecture by maintaining the functional pool of the RSC chromatin remodeler. Mol Cell. (2016) 64:888–99. 10.1016/j.molcel.2016.09.040
30.
HalpinJCStreetTO. Hsp90 sensitivity to ADP reveals hidden regulation mechanisms. J Mol Biol. (2017) 429:2918–30. 10.1016/j.jmb.2017.08.005
31.
CunninghamCNKrukenbergKAAgardDA. Intra- and intermonomer interactions are required to synergistically facilitate ATP hydrolysis in Hsp90. J Biol Chem. (2008) 283:21170–8. 10.1074/jbc.M800046200
32.
ElnatanDBetegonMLiuYRamelotTKennedyMAAgardDA. Symmetry broken and rebroken during the ATP hydrolysis cycle of the mitochondrial Hsp90 TRAP1. Elife. (2017) 6:e25235. 10.7554/eLife.25235
33.
GenestOReidyMStreetTOHoskinsJRCambergJLAgardDAet al. Uncovering a region of heat shock protein 90 important for client binding in E. coli and chaperone function in yeast. Mol Cell. (2013) 49:464–73. 10.1016/j.molcel.2012.11.017
34.
JoshiSTolkunovDAvivHHakimiAAYaoMHsiehJJet al. The genomic landscape of renal oncocytoma identifies a metabolic barrier to tumorigenesis. Cell Rep. (2015) 13:1895–908. 10.1016/j.celrep.2015.10.059
35.
RodinaAWangTYanPGomesEDDunphyMPPillarsettyNet al. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature. (2016) 538:397–401. 10.1038/nature19807
36.
RohlARohrbergJBuchnerJ. The chaperone Hsp90: changing partners for demanding clients. Trends Biochem Sci. (2013) 38:253–62. 10.1016/j.tibs.2013.02.003
37.
VerbaKAWangRYArakawaALiuYShirouzuMYokoyamaSet al. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science. (2016) 352:1542–7. 10.1126/science.aaf5023
38.
KirschkeEGoswamiDSouthworthDGriffinPRAgardDA. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell. (2014) 157:1685–97. 10.1016/j.cell.2014.04.038
39.
MayerMPLe BretonL. Hsp90: breaking the symmetry. Mol Cell. (2015) 58:8–20. 10.1016/j.molcel.2015.02.022
40.
Lees-MillerSPAndersonCW. The human double-stranded DNA-activated protein kinase phosphorylates the 90-kDa heat-shock protein, hsp90 alpha at two NH2-terminal threonine residues. J Biol Chem. (1989) 264:17275–80.
41.
MollapourMTsutsumiSKimYSTrepelJNeckersL. Casein kinase 2 phosphorylation of Hsp90 threonine 22 modulates chaperone function and drug sensitivity. Oncotarget. (2011) 2:407–17. 10.18632/oncotarget.272
42.
MollapourMTsutsumiSDonnellyACBeebeKTokitaMJLeeMJet al. Swe1Wee1-dependent tyrosine phosphorylation of Hsp90 regulates distinct facets of chaperone function. Mol Cell. (2010) 37:333–43. 10.1016/j.molcel.2010.01.005
43.
BaiYLiJFangBEdwardsAZhangGBuiMet al. Phosphoproteomics identifies driver tyrosine kinases in sarcoma cell lines and tumors. Cancer Res. (2012) 72:2501–11. 10.1158/0008-5472.CAN-11-3015
44.
MertinsPManiDRRugglesKVGilletteMAClauserKRWangPet al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. (2016) 534:55–62. 10.1038/nature18003
45.
TsaiCFWangYTYenHYTsouCCKuWCLinPYet al. Large-scale determination of absolute phosphorylation stoichiometries in human cells by motif-targeting quantitative proteomics. Nat Commun. (2015) 6:6622. 10.1038/ncomms7622
46.
RoseDWWettenhallREKudlickiWKramerGHardestyB. The 90-kilodalton peptide of the heme-regulated eIF-2 alpha kinase has sequence similarity with the 90-kilodalton heat shock protein. Biochemistry. (1987) 26:6583–7. 10.1021/bi00395a003
47.
YangYRaoRShenJTangYFiskusWNechtmanJet al. Role of acetylation and extracellular location of heat shock protein 90alpha in tumor cell invasion. Cancer Res. (2008) 68:4833–42. 10.1158/0008-5472.CAN-08-0644
48.
LeiHVenkatakrishnanAYuSKazlauskasA. Protein kinase A-dependent translocation of Hsp90 alpha impairs endothelial nitric-oxide synthase activity in high glucose and diabetes. J Biol Chem. (2007) 282:9364–71. 10.1074/jbc.M608985200
49.
WangXLuXASongXZhuoWJiaLJiangYet al. Thr90 phosphorylation of Hsp90alpha by protein kinase A regulates its chaperone machinery. Biochem J. (2012) 441:387–97. 10.1042/BJ20110855
50.
KundratLReganL. Identification of residues on Hsp70 and Hsp90 ubiquitinated by the cochaperone CHIP. J Mol Biol. (2010) 395:587–94. 10.1016/j.jmb.2009.11.017
51.
MurtaghJLuHSchwartzEL. Taxotere-induced inhibition of human endothelial cell migration is a result of heat shock protein 90 degradation. Cancer Res. (2006) 66:8192–9. 10.1158/0008-5472.CAN-06-0748
52.
LuXAWangXZhuoWJiaLJiangYFuYet al. The regulatory mechanism of a client kinase controlling its own release from Hsp90 chaperone machinery through phosphorylation. Biochem J. (2014) 457:171–83. 10.1042/BJ20130963
53.
WoodfordMRTrumanAWDunnDMJensenSMCotranRBullardRet al. Mps1 mediated phosphorylation of Hsp90 confers renal cell carcinoma sensitivity and selectivity to Hsp90 inhibitors. Cell Rep. (2016) 14:872–84. 10.1016/j.celrep.2015.12.084
54.
MollapourMBourbouliaDBeebeKWoodfordMRPolierSHoangAet al. Asymmetric Hsp90 N domain SUMOylation recruits Aha1 and ATP-competitive inhibitors. Mol Cell. (2014) 53:317–29. 10.1016/j.molcel.2013.12.007
55.
XuWMollapourMProdromouCWangSScrogginsBTPalchickZet al. Dynamic tyrosine phosphorylation modulates cycling of the HSP90-P50(CDC37)-AHA1 chaperone machine. Mol Cell. (2012) 47:434–43. 10.1016/j.molcel.2012.05.015
56.
OgisoHKagiNMatsumotoENishimotoMAraiRShirouzuMet al. Phosphorylation analysis of 90 kDa heat shock protein within the cytosolic arylhydrocarbon receptor complex. Biochemistry. (2004) 43:15510–9. 10.1021/bi048736m
57.
ZhangFZHoDHWongRH. Triptolide, a HSP90 middle domain inhibitor, induces apoptosis in triple manner. Oncotarget. (2018) 9:22301–15. 10.18632/oncotarget.24737
58.
ScrogginsBTRobzykKWangDMarcuMGTsutsumiSBeebeKet al. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol Cell. (2007) 25:151–9. 10.1016/j.molcel.2006.12.008
59.
DuvalMLe BoeufFHuotJGrattonJP. Src-mediated phosphorylation of Hsp90 in response to vascular endothelial growth factor (VEGF) is required for VEGF receptor-2 signaling to endothelial NO synthase. Mol Biol Cell. (2007) 18:4659–68. 10.1091/mbc.e07-05-0467
60.
DagarMSinghJPDagarGTyagiRKBagchiG. Phosphorylation of HSP90 by protein kinase A is essential for the nuclear translocation of androgen receptor. J Biol Chem. (2019) 294:8699–710. 10.1074/jbc.RA119.007420
61.
DebTBZuoAHWangYBarndtRJCheemaAKSenguptaSet al. Pnck induces ligand-independent EGFR degradation by probable perturbation of the Hsp90 chaperone complex. Am J Physiol Cell Physiol. (2011) 300:C1139–54. 10.1152/ajpcell.00167.2010
62.
HuangSYTsaiMLChenGYWuCJChenSH. A systematic MS-based approach for identifying in vitro substrates of PKA and PKG in rat uteri. J Proteome Res. (2007) 6:2674–84. 10.1021/pr070134c
63.
CarboneDLDoornJAKieblerZIckesBRPetersenDR. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. J Pharmacol Exp Ther. (2005) 315:8–15. 10.1124/jpet.105.088088
64.
QiXXieCHouSLiGYinNDongLet al. Identification of a ternary protein-complex as a therapeutic target for K-Ras-dependent colon cancer. Oncotarget. (2014) 5:4269–82. 10.18632/oncotarget.2001
65.
RetzlaffMStahlMEberlHCLaglederSBeckJKesslerHet al. Hsp90 is regulated by a switch point in the C-terminal domain. EMBO Rep. (2009) 10:1147–53. 10.1038/embor.2009.153
66.
Martinez-RuizAVillanuevaLGonzalez de OrdunaCLopez-FerrerDHiguerasMATarinCet al. S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc Natl Acad Sci USA. (2005) 102:8525–30. 10.1073/pnas.0407294102
67.
MullerPRuckovaEHaladaPCoatesPJHrstkaRLaneDPet al. C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene. (2013) 32:3101–10. 10.1038/onc.2012.314
68.
ScrogginsBTNeckersL. Post-translational modification of heat-shock protein 90: impact on chaperone function. Expert Opin Drug Discov. (2007) 2:1403–14. 10.1517/17460441.2.10.1403
69.
SorokaJWandingerSKMausbacherNSchreiberTRichterKDaubHet al. Conformational switching of the molecular chaperone Hsp90 via regulated phosphorylation. Mol Cell. (2012) 45:517–28. 10.1016/j.molcel.2011.12.031
70.
PearlLH. Review: the HSP90 molecular chaperone-an enigmatic ATPase. Biopolymers. (2016) 105:594–607. 10.1002/bip.22835
71.
ScaltritiMDawoodSCortesJ. Molecular pathways: targeting hsp90–who benefits and who does not. Clin Cancer Res. (2012) 18:4508–13. 10.1158/1078-0432.CCR-11-2138
72.
KangBH. TRAP1 regulation of mitochondrial life or death decision in cancer cells and mitochondria-targeted TRAP1 inhibitors. BMB Rep. (2012) 45:1–6. 10.5483/BMBRep.2012.45.1.1
73.
LeskovarAWegeleHWerbeckNDBuchnerJReinsteinJ. The ATPase cycle of the mitochondrial Hsp90 analog Trap1. J Biol Chem. (2008) 283:11677–88. 10.1074/jbc.M709516200
74.
PartridgeJRLaveryLAElnatanDNaberNCookeRAgardDA. A novel N-terminal extension in mitochondrial TRAP1 serves as a thermal regulator of chaperone activity. Elife. (2014) 3:e03487. 10.7554/eLife.03487
75.
SungNLeeJKimJHChangCJoachimiakALeeSet al. Mitochondrial Hsp90 is a ligand-activated molecular chaperone coupling ATP binding to dimer closure through a coiled-coil intermediate. Proc Natl Acad Sci USA. (2016) 113:2952–7. 10.1073/pnas.1516167113
76.
RizzaSMontagnaCCardaciSMaianiEDi GiacomoGSanchez-QuilesVet al. S-nitrosylation of the mitochondrial chaperone trap1 sensitizes hepatocellular carcinoma cells to inhibitors of succinate dehydrogenase. Cancer Res. (2016) 76:4170–82. 10.1158/0008-5472.CAN-15-2637
77.
FaienzaFLambrughiMRizzaSPecorariCGiglioPSalamanca ViloriaJet al. S-nitrosylation affects TRAP1 structure and ATPase activity and modulates cell response to apoptotic stimuli. Biochem Pharmacol. (2020) 176:113869. 10.1016/j.bcp.2020.113869
78.
MasgrasICiscatoFBrunatiAMTibaldiEIndraccoloSCurtarelloMet al. Absence of neurofibromin induces an oncogenic metabolic switch via mitochondrial ERK-mediated phosphorylation of the chaperone TRAP1. Cell Rep. (2017) 18:659–72. 10.1016/j.celrep.2016.12.056
79.
PridgeonJWOlzmannJAChinLSLiL. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. (2007) 5:e172. 10.1371/journal.pbio.0050172
80.
ParkHKHongJHOhYTKimSSYinJLeeAJet al. Interplay between TRAP1 and sirtuin-3 modulates mitochondrial respiration and oxidative stress to maintain stemness of glioma stem cells. Cancer Res. (2019) 79:1369–82. 10.1158/0008-5472.CAN-18-2558
81.
YoshidaSTsutsumiSMuhlebachGSourbierCLeeMJLeeSet al. Molecular chaperone TRAP1 regulates a metabolic switch between mitochondrial respiration and aerobic glycolysis. Proc Natl Acad Sci USA. (2013) 110:E1604–12. 10.1073/pnas.1220659110
82.
CairnsRAHarrisISMakTW. Regulation of cancer cell metabolism. Nat Rev Cancer. (2011) 11:85–95. 10.1038/nrc2981
83.
TaipaleMKrykbaevaIKoevaMKayatekinCWestoverKDKarrasGIet al. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell. (2012) 150:987–1001. 10.1016/j.cell.2012.06.047
84.
MitraSGhoshBGayenNRoyJMandalAK. Bipartite role of heat shock protein 90 (Hsp90) keeps CRAF kinase poised for activation. J Biol Chem. (2016) 291:24579–93. 10.1074/jbc.M116.746420
85.
BieblMMBuchnerJ. Structure, function, and regulation of the Hsp90 machinery. Cold Spring Harb Perspect Biol. (2019) 11:a034017. 10.1101/cshperspect.a034017
86.
ClaytonATurkesANavabiHMasonMDTabiZ. Induction of heat shock proteins in B-cell exosomes. J Cell Sci. (2005) 118:3631–8. 10.1242/jcs.02494
87.
EustaceBKSakuraiTStewartJKYimlamaiDUngerCZehetmeierCet al. Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nat Cell Biol. (2004) 6:507–14. 10.1038/ncb1131
88.
LiWLiYGuanSFanJChengCFBrightAMet al. Extracellular heat shock protein-90alpha: linking hypoxia to skin cell motility and wound healing. EMBO J. (2007) 26:1221–33. 10.1038/sj.emboj.7601579
89.
MattisonSABlatchGLEdkinsAL. HOP expression is regulated by p53 and RAS and characteristic of a cancer gene signature. Cell Stress Chaperones. (2017) 22:213–23. 10.1007/s12192-016-0755-8
90.
NakazawaMSKeithBSimonMC. Oxygen availability and metabolic adaptations. Nat Rev Cancer. (2016) 16:663–73. 10.1038/nrc.2016.84
91.
KowalikMAGuzzoGMorandiAPerraAMenegonSMasgrasIet al. Metabolic reprogramming identifies the most aggressive lesions at early phases of hepatic carcinogenesis. Oncotarget. (2016) 7:32375–93. 10.18632/oncotarget.8632
92.
GuzzoGSciacovelliMBernardiPRasolaA. Inhibition of succinate dehydrogenase by the mitochondrial chaperone TRAP1 has anti-oxidant and anti-apoptotic effects on tumor cells. Oncotarget. (2014) 5:11897–908. 10.18632/oncotarget.2472
93.
BernardiPRasolaAForteMLippeG. The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol Rev. (2015) 95:1111–55. 10.1152/physrev.00001.2015
94.
AmorosoMRMatassaDSAgliaruloIAvolioRLuHSisinniLet al. TRAP1 downregulation in human ovarian cancer enhances invasion and epithelial-mesenchymal transition. Cell Death Dis. (2016) 7:e2522. 10.1038/cddis.2016.400
95.
SciacovelliMGuzzoGMorelloVFrezzaCZhengLNanniniNet al. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab. (2013) 17:988–99. 10.1016/j.cmet.2013.04.019
96.
PavlovaNNThompsonCB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. 10.1016/j.cmet.2015.12.006
97.
AdamJYangMSogaTPollardPJ. Rare insights into cancer biology. Oncogene. (2014) 33:2547–56. 10.1038/onc.2013.222
98.
SemenzaGL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. (2013) 123:3664–71. 10.1172/JCI67230
99.
ChiosisGNeckersL. Tumor selectivity of Hsp90 inhibitors: the explanation remains elusive. ACS Chem Biol. (2006) 1:279–84. 10.1021/cb600224w
100.
KamalAThaoLSensintaffarJZhangLBoehmMFFritzLCet al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. (2003) 425:407–10. 10.1038/nature01913
101.
BarrottJJHaysteadTA. Hsp90, an unlikely ally in the war on cancer. FEBS J. (2013) 280:1381–96. 10.1111/febs.12147
102.
ProdromouCRoeSMO'BrienRLadburyJEPiperPWPearlLH. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. (1997) 90:65–75. 10.1016/S0092-8674(00)80314-1
103.
LiLWangLYouQDXuXL. Heat shock protein 90 inhibitors: an update on achievements, challenges, and future directions. J Med Chem. (2019) 63:1798–822. 10.1021/acs.jmedchem.9b00940
104.
JolyGAAyresMKilbournRG. Potent inhibition of inducible nitric oxide synthase by geldanamycin, a tyrosine kinase inhibitor, in endothelial, smooth muscle cells, and in rat aorta. FEBS Lett. (1997) 403:40–4. 10.1016/S0014-5793(97)00004-5
105.
ZapfCWBloomJDMcBeanJLDushinRGGolasJMLiuHet al. Discovery of a macrocyclic o-aminobenzamide Hsp90 inhibitor with heterocyclic tether that shows extended biomarker activity and in vivo efficacy in a mouse xenograft model. Bioorg Med Chem Lett. (2011) 21:3627–31. 10.1016/j.bmcl.2011.04.102
106.
BeebeKMollapourMScrogginsBProdromouCXuWTokitaMet al. Posttranslational modification and conformational state of heat shock protein 90 differentially affect binding of chemically diverse small molecule inhibitors. Oncotarget. (2013) 4:1065–74. 10.18632/oncotarget.1099
107.
StebbinsCERussoAASchneiderCRosenNHartlFUPavletichNP. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell. (1997) 89:239–50. 10.1016/S0092-8674(00)80203-2
108.
MimnaughEGChavanyCNeckersL. Polyubiquitination and proteasomal degradation of the p185c-erbB-2 receptor protein-tyrosine kinase induced by geldanamycin. J Biol Chem. (1996) 271:22796–801. 10.1074/jbc.271.37.22796
109.
SchnurRCCormanMLGallaschunRJCooperBADeeMFDotyJLet al. Inhibition of the oncogene product p185erbB-2 in vitro and in vivo by geldanamycin and dihydrogeldanamycin derivatives. J Med Chem. (1995) 38:3806–12. 10.1021/jm00019a010
110.
HallTJSchaeublinMMissbachM. Evidence that c-src is involved in the process of osteoclastic bone resorption. Biochem Biophys Res Commun. (1994) 199:1237–44. 10.1006/bbrc.1994.1363
111.
ZhangLFanJVuKHongKLe BrazidecJYShiJet al. 7'-substituted benzothiazolothio- and pyridinothiazolothio-purines as potent heat shock protein 90 inhibitors. J Med Chem. (2006) 49:5352–62. 10.1021/jm051146h
112.
TerraccianoSChiniMGPiazFDVassalloARiccioRBrunoIet al. Dimeric and trimeric triazole based molecules as a new class of Hsp90 molecular chaperone inhibitors. Eur J Med Chem. (2013) 65:464–76. 10.1016/j.ejmech.2013.05.016
113.
MussoLDallavalleSMerliniLBavaANasiniGPencoSet al. Natural and semisynthetic azaphilones as a new scaffold for Hsp90 inhibitors. Bioorg Med Chem. (2010) 18:6031–43. 10.1016/j.bmc.2010.06.068
114.
AustinCPettitSNMagnoloSKSanvoisinJChenWWoodSPet al. Fragment screening using capillary electrophoresis (CEfrag) for hit identification of heat shock protein 90 ATPase inhibitors. J Biomol Screen. (2012) 17:868–76. 10.1177/1087057112445785
115.
CorbettKDBergerJM. Structural basis for topoisomerase VI inhibition by the anti-Hsp90 drug radicicol. Nucleic Acids Res. (2006) 34:4269–77. 10.1093/nar/gkl567
116.
KatoMLiJChuangJLChuangDT. Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure. (2007) 15:992–1004. 10.1016/j.str.2007.07.001
117.
ZimmermannTJNiesenFHPilkaESKnappSOppermannUMaierME. Discovery of a potent and selective inhibitor for human carbonyl reductase 1 from propionate scanning applied to the macrolide zearalenone. Bioorg Med Chem. (2009) 17:530–6. 10.1016/j.bmc.2008.11.076
118.
HeHZatorskaDKimJAguirreJLlaugerLSheYet al. Identification of potent water soluble purine-scaffold inhibitors of the heat shock protein 90. J Med Chem. (2006) 49:381–90. 10.1021/jm0508078
119.
ImmorminoRMKangYChiosisGGewirthDT. Structural and quantum chemical studies of 8-aryl-sulfanyl adenine class Hsp90 inhibitors. J Med Chem. (2006) 49:4953–60. 10.1021/jm060297x
120.
LeeCParkHKJeongHLimJLeeAJCheonKYet al. Development of a mitochondria-targeted Hsp90 inhibitor based on the crystal structures of human TRAP1. J Am Chem Soc. (2015) 137:4358–67. 10.1021/ja511893n
121.
WangYKoayYCMcAlpineSR. How selective are Hsp90 inhibitors for cancer cells over normal cells?ChemMedChem. (2017) 12:353–7. 10.1002/cmdc.201600595
122.
WangYKoayYCMcAlpineSR. Redefining the phenotype of heat shock protein 90 (Hsp90) inhibitors. Chemistry. (2017) 23:2010–3. 10.1002/chem.201604807
123.
KogaFTsutsumiSNeckersLM. Low dose geldanamycin inhibits hepatocyte growth factor and hypoxia-stimulated invasion of cancer cells. Cell Cycle. (2007) 6:1393–402. 10.4161/cc.6.11.4296
124.
NagarajuGPLongTEParkWLandryJCTaliaferro-SmithLFarrisABet al. Heat shock protein 90 promotes epithelial to mesenchymal transition, invasion, and migration in colorectal cancer. Mol Carcinog. (2015) 54:1147–58. 10.1002/mc.22185
125.
NomuraMNomuraNNewcombEWLukyanovYTamasdanCZagzagD. Geldanamycin induces mitotic catastrophe and subsequent apoptosis in human glioma cells. J Cell Physiol. (2004) 201:374–84. 10.1002/jcp.20090
126.
WangXZhangYZhaoYLiangYXiangCZhouHet al. CD24 promoted cancer cell angiogenesis via Hsp90-mediated STAT3/VEGF signaling pathway in colorectal cancer. Oncotarget. (2016) 7:55663–76. 10.18632/oncotarget.10971
127.
KhandelwalACrowleyVMBlaggBSJ. Natural product inspired N-terminal Hsp90 inhibitors: from bench to bedside?Med Res Rev. (2016) 36:92–118. 10.1002/med.21351
128.
SideraKPatsavoudiE. HSP90 inhibitors: current development and potential in cancer therapy. Recent Pat Anticancer Drug Discov. (2014) 9:1–20. 10.2174/15748928113089990031
129.
KummarSGutierrezMEGardnerERChenXFiggWDZajac-KayeMet al. Phase I trial of 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG), a heat shock protein inhibitor, administered twice weekly in patients with advanced malignancies. Eur J Cancer. (2010) 46:340–7. 10.1016/j.ejca.2009.10.026
130.
LancetJEGojoIBurtonMQuinnMTigheSMKerseyKet al. Phase I study of the heat shock protein 90 inhibitor alvespimycin (KOS-1022, 17-DMAG) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia. (2010) 24:699–705. 10.1038/leu.2009.292
131.
ChatterjeeSBurnsTF. Targeting heat shock proteins in cancer: a promising therapeutic approach. Int J Mol Sci. (2017) 18:1978. 10.3390/ijms18091978
132.
LianosGDAlexiouGAManganoAManganoARauseiSBoniLet al. The role of heat shock proteins in cancer. Cancer Lett. (2015) 360:114–8. 10.1016/j.canlet.2015.02.026
133.
ProiaDABatesRC. Ganetespib and HSP90: translating preclinical hypotheses into clinical promise. Cancer Res. (2014) 74:1294–300. 10.1158/0008-5472.CAN-13-3263
134.
SocinskiMAGoldmanJEl-HariryIKoczywasMVukovicVHornLet al. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clin Cancer Res. (2013) 19:3068–77. 10.1158/1078-0432.CCR-12-3381
135.
ChiosisGKangYSunW. Discovery and development of purine-scaffold Hsp90 inhibitors. Expert Opin Drug Discov. (2008) 3:99–114. 10.1517/17460441.3.1.99
136.
BickelDGohlkeH. C-terminal modulators of heat shock protein of 90kDa (HSP90): state of development and modes of action. Bioorg Med Chem. (2019) 27:115080. 10.1016/j.bmc.2019.115080
137.
BattogtokhGChoiYSKangDSParkSJShimMSHuhKMet al. Mitochondria-targeting drug conjugates for cytotoxic, anti-oxidizing and sensing purposes: current strategies and future perspectives. Acta Pharm Sin B. (2018) 8:862–80. 10.1016/j.apsb.2018.05.006
138.
SiegelinMD. Inhibition of the mitochondrial Hsp90 chaperone network: a novel, efficient treatment strategy for cancer?Cancer Lett. (2013) 333:133–46. 10.1016/j.canlet.2013.01.045
139.
ParkHKJeongHKoELeeGLeeJELeeSKet al. Paralog specificity determines subcellular distribution, action mechanism, and anticancer activity of TRAP1 inhibitors. J Med Chem. (2017) 60:7569–78. 10.1021/acs.jmedchem.7b00978
140.
MarcuMGChadliABouhoucheICatelliMNeckersLM. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J Biol Chem. (2000) 275:37181–6. 10.1074/jbc.M003701200
141.
MarcuMGSchulteTWNeckersL. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J Natl Cancer Inst. (2000) 92:242–8. 10.1093/jnci/92.3.242
142.
DonnellyABlaggBS. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr Med Chem. (2008) 15:2702–17. 10.2174/092986708786242895
143.
GargGForsbergLKZhaoHBlaggBSJ. Development of phenyl cyclohexylcarboxamides as a novel class of Hsp90 C-terminal inhibitors. Chemistry. (2017) 23:16574–85. 10.1002/chem.201703206
144.
ZhaoHDonnellyACKusumaBRBrandtGEBrownDRajewskiRAet al. Engineering an antibiotic to fight cancer: optimization of the novobiocin scaffold to produce anti-proliferative agents. J Med Chem. (2011) 54:3839–53. 10.1021/jm200148p
145.
HaddenMKGalamLGestwickiJEMattsRLBlaggBS. Derrubone, an inhibitor of the Hsp90 protein folding machinery. J Nat Prod. (2007) 70:2014–8. 10.1021/np070190s
146.
YuYHamzaAZhangTGuMZouPNewmanBet al. Withaferin A targets heat shock protein 90 in pancreatic cancer cells. Biochem Pharmacol. (2010) 79:542–51. 10.1016/j.bcp.2009.09.017
147.
WeiWWuSWangXSunCKYangXYanXet al. Novel celastrol derivatives inhibit the growth of hepatocellular carcinoma patient-derived xenografts. Oncotarget. (2014) 5:5819–31. 10.18632/oncotarget.2171
148.
ZhangTHamzaACaoXWangBYuSZhanCGet al. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol Cancer Ther. (2008) 7:162–70. 10.1158/1535-7163.MCT-07-0484
149.
SattinSTaoJVettorettiGMoroniEPennatiMLopergoloAet al. Activation of Hsp90 enzymatic activity and conformational dynamics through rationally designed allosteric ligands. Chemistry. (2015) 21:13598–608. 10.1002/chem.201502211
150.
D'AnnessaISattinSTaoJPennatiMSanchez-MartinCMoroniEet al. Design of allosteric stimulators of the Hsp90 ATPase as new anticancer leads. Chemistry. (2017) 23:5188–92. 10.1002/chem.201700169
151.
RoeMSWahabBTorokZHorvathIVighLProdromouC. Dihydropyridines allosterically modulate Hsp90 providing a novel mechanism for heat shock protein co-induction and neuroprotection. Front Mol Biosci. (2018) 5:51. 10.3389/fmolb.2018.00051
152.
D'AnnessaIRanioloSLimongelliVDi MarinoDColomboG. Ligand binding, unbinding, and allosteric effects: deciphering small-molecule modulation of HSP90. J Chem Theory Comput. (2019) 15:6368–81. 10.1021/acs.jctc.9b00319
153.
MorraGNevesMAPlesciaCJTsustsumiSNeckersLVerkhivkerGet al. Dynamics-based discovery of allosteric inhibitors: selection of new ligands for the C-terminal domain of Hsp90. J Chem Theory Comput. (2010) 6:2978–89. 10.1021/ct100334n
154.
MorraGPotestioRMichelettiCColomboG. Corresponding functional dynamics across the Hsp90 Chaperone family: insights from a multiscale analysis of MD simulations. PLoS Comput Biol. (2012) 8:e1002433. 10.1371/journal.pcbi.1002433
155.
MorraGVerkhivkerGColomboG. Modeling signal propagation mechanisms and ligand-based conformational dynamics of the Hsp90 molecular chaperone full-length dimer. PLoS Comput Biol. (2009) 5:e1000323. 10.1371/journal.pcbi.1000323
156.
VettorettiGMoroniESattinSTaoJAgardDABernardiAet al. Molecular dynamics simulations reveal the mechanisms of allosteric activation of Hsp90 by designed ligands. Sci Rep. (2016) 6:23830. 10.1038/srep23830
157.
Sanchez-MartinCMoroniEFerraroMLaquatraCCanninoGMasgrasIet al. Rational design of allosteric and selective inhibitors of the molecular Chaperone TRAP1. Cell Rep. (2020) 31:107531. 10.1016/j.celrep.2020.107531
158.
Sanchez-MartinCMenonDMoroniEFerraroMMasgrasIElseyJet al. Honokiol bis-dichloroacetate is a selective allosteric inhibitor of the mitochondrial Chaperone TRAP1. Antioxid Redox Signal. (2020) 10.1089/ars.2019.7972. [Epub ahead of print].
159.
ArkinMRWhittyA. The road less traveled: modulating signal transduction enzymes by inhibiting their protein-protein interactions. Curr Opin Chem Biol. (2009) 13:284–90. 10.1016/j.cbpa.2009.05.125
160.
GestwickiJEShaoH. Inhibitors and chemical probes for molecular chaperone networks. J Biol Chem. (2019) 294:2151–61. 10.1074/jbc.TM118.002813
161.
GargGKhandelwalABlaggBS. Anticancer inhibitors of Hsp90 function: beyond the usual suspects. Adv Cancer Res. (2016) 129:51–88. 10.1016/bs.acr.2015.12.001
162.
D'AnnessaIHurwitzNPirotaVBerettaGLTinelliSWoodfordMRet al. Design of disruptors of the Hsp90-Cdc37 interface. Molecules. (2020) 25:360. 10.3390/molecules25020360
163.
FerraroMD'AnnessaIMoroniEMorraGPaladinoARinaldiSet al. Allosteric modulators of HSP90 and HSP70: dynamics meets function through structure-based drug design. J Med Chem. (2019) 62:60–87. 10.1021/acs.jmedchem.8b00825
164.
VaishampayanUNBurgerAMSausvilleEAHeilbrunLKLiJHoribaMNet al. Safety, efficacy, pharmacokinetics, and pharmacodynamics of the combination of sorafenib and tanespimycin. Clin Cancer Res. (2010) 16:3795–804. 10.1158/1078-0432.CCR-10-0503
165.
ModiSStopeckATGordonMSMendelsonDSolitDBBagatellRet al. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: a phase I dose-escalation study. J Clin Oncol. (2007) 25:5410–7. 10.1200/JCO.2007.11.7960
166.
AcquavivaJSmithDLJimenezJPZhangCSequeiraMHeSet al. Overcoming acquired BRAF inhibitor resistance in melanoma via targeted inhibition of Hsp90 with ganetespib. Mol Cancer Ther. (2014) 13:353–63. 10.1158/1535-7163.MCT-13-0481
167.
HeSSmithDLSequeiraMSangJBatesRCProiaDA. The HSP90 inhibitor ganetespib has chemosensitizer and radiosensitizer activity in colorectal cancer. Invest New Drugs. (2014) 32:577–86. 10.1007/s10637-014-0095-4
168.
ProiaDAKaufmannGF. Targeting Heat-Shock Protein 90 (HSP90) as a complementary strategy to immune checkpoint blockade for cancer therapy. Cancer Immunol Res. (2015) 3:583–9. 10.1158/2326-6066.CIR-15-0057
169.
ParkKSYangHChoiJSeoSKimDLeeCHet al. The HSP90 inhibitor, NVP-AUY922, attenuates intrinsic PI3K inhibitor resistance in KRAS-mutant non-small cell lung cancer. Cancer Lett. (2017) 406:47–53. 10.1016/j.canlet.2017.07.028
170.
RamalingamSGossGRosellRSchmid-BindertGZaricBAndricZet al. A randomized phase II study of ganetespib, a heat shock protein 90 inhibitor, in combination with docetaxel in second-line therapy of advanced non-small cell lung cancer (GALAXY-1). Ann Oncol. (2015) 26:1741–8. 10.1093/annonc/mdv220
171.
ChatterjeeSHuangEHChristieIBurnsTF. Reactivation of the p90RSK-CDC25C pathway leads to bypass of the ganetespib-induced G2-M arrest and mediates acquired resistance to ganetespib in KRAS-mutant NSCLC. Mol Cancer Ther. (2017) 16:1658–68. 10.1158/1535-7163.MCT-17-0114
172.
ChatterjeeSHuangEHChristieIKurlandBFBurnsTF. Acquired resistance to the Hsp90 inhibitor, ganetespib, in KRAS-mutant NSCLC is mediated via reactivation of the ERK-p90RSK-mTOR signaling network. Mol Cancer Ther. (2017) 16:793–804. 10.1158/1535-7163.MCT-16-0677
173.
ZuehlkeADMosesMANeckersL. Heat shock protein 90: its inhibition and function. Philos Trans R Soc Lond B Biol Sci. (2018) 373:20160527. 10.1098/rstb.2016.0527
174.
ShevtsovMMulthoffGMikhaylovaEShibataAGuzhovaIMargulisB. Combination of anti-cancer drugs with molecular chaperone inhibitors. Int J Mol Sci. (2019) 20:5284. 10.3390/ijms20215284
175.
Martinez-OutschoornUEPeiris-PagesMPestellRGSotgiaFLisantiMP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. (2017) 14:11–31. 10.1038/nrclinonc.2016.60
176.
HeskeCMMendozaAEdessaLDBaumgartJTLeeSTrepelJet al. STA-8666, a novel HSP90 inhibitor/SN-38 drug conjugate, causes complete tumor regression in preclinical mouse models of pediatric sarcoma. Oncotarget. (2016) 7:65540–52. 10.18632/oncotarget.11869
177.
ProiaDASmithDLZhangJJimenezJPSangJOgawaLSet al. HSP90 inhibitor-SN-38 conjugate strategy for targeted delivery of topoisomerase I inhibitor to tumors. Mol Cancer Ther. (2015) 14:2422–32. 10.1158/1535-7163.MCT-15-0455
Summary
Keywords
chaperones, HSP90, TRAP1, mitochondria, tumor metabolism, allosteric inhibitor, ATP-competitive inhibitors, anti-neoplastic strategies
Citation
Sanchez-Martin C, Serapian SA, Colombo G and Rasola A (2020) Dynamically Shaping Chaperones. Allosteric Modulators of HSP90 Family as Regulatory Tools of Cell Metabolism in Neoplastic Progression. Front. Oncol. 10:1177. doi: 10.3389/fonc.2020.01177
Received
27 January 2020
Accepted
10 June 2020
Published
16 July 2020
Volume
10 - 2020
Edited by
Alessandra Castegna, University of Bari Aldo Moro, Italy
Reviewed by
Didier Picard, Université de Genève, Switzerland; Ciro Leonardo Pierri, University of Bari Aldo Moro, Italy
Updates
Copyright
© 2020 Sanchez-Martin, Serapian, Colombo and Rasola.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giorgio Colombo g.colombo@unipv.itAndrea Rasola andrea.rasola@unipd.it
This article was submitted to Cancer Metabolism, a section of the journal Frontiers in Oncology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.