 Jiachen Liu
Jiachen Liu Congcong Xia
Congcong Xia Gaiqing Wang
Gaiqing Wang- 1Clinical Medicine, Xiangya Medical College of Central South University, Changsha, China
- 2Department of Neurology, Sanya Central Hospital (The Third People's Hospital of Hainan Province), Sanya, China
Meningiomas are common intracranial tumors that can be cured by surgical resection in most cases. However, the most disconcerting is high-grade meningiomas, which frequently recur despite initial successful treatment, eventually conferring poor prognosis. Therefore, the early diagnosis and classification of meningioma is necessary for the subsequent intervention and an improved prognosis. A growing body of evidence demonstrates the potential of multi-omics study (including genomics, transcriptomics, epigenomics, proteomics) for meningioma diagnosis and mechanistic links to potential pathological mechanism. This thesis addresses a neglected aspect of recent advances in the field of meningiomas at multiple omics levels, highlighting that the integration of multi-omics can reveal the mechanism of meningiomas, which provides a timely and necessary scientific basis for the treatment of meningiomas.
Introduction
Meningiomas account for 13–36.6% of the primary malignant tumors of the central nervous system (1). Although the reported incidence is around 7.8/100,000 (2), the rate of recurrence increases dramatically to 32% with progressive/higher grade meningiomas (~20% of all meningiomas) (3). Coupled with the high treatment costs (~$83,838 per person) (4), meningioma is increasingly recognized as a serious, worldwide public health concern (5). Since the publication of revised WHO guidelines in 2016, the diagnosis of meningioma is mainly divided into three grades based on the morphological features (6). Unfortunately, this grading system does not ultimately predict the clinical behavior of meningiomas, especially long-term recurrence of atypical meningiomas (7).
Recent advances in omics technologies (genomics, transcriptomics, epigenomics, and proteomics) contribute to large screening of biomarkers for meningioma by tissue microarray to predict biological behavior of meningiomas (8). Notably, integration of multi-omics with clinical data represents an accurate and promising methodology to provide very accurate prediction models for meningioma progression (Figure 1), suggesting the potential of early and accurate diagnosis, effective therapeutic strategies, and favorable prognosis of meningioma (9, 10).
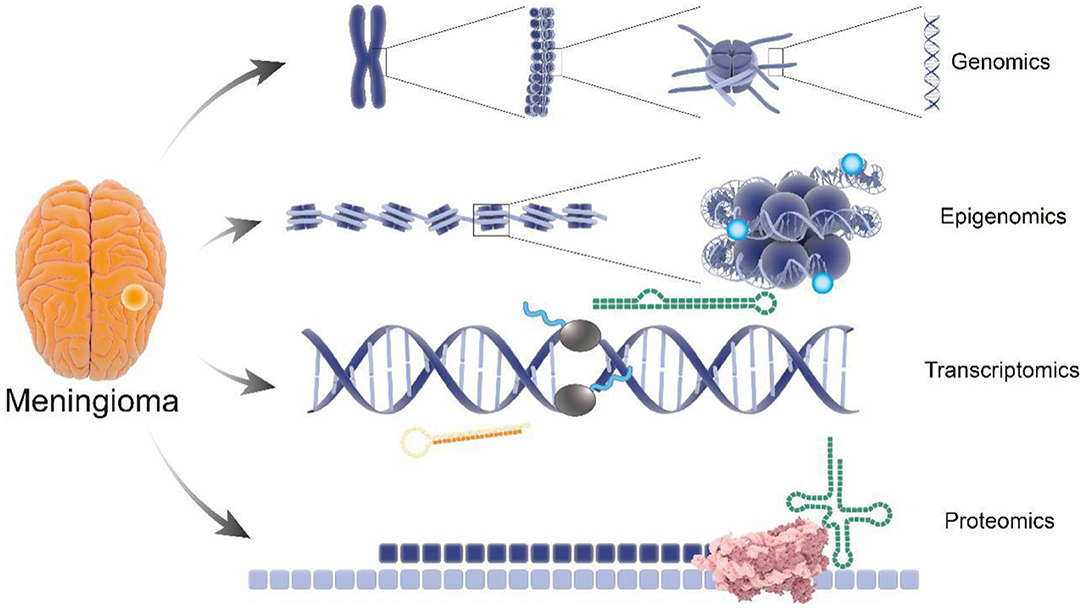
Figure 1. Comprehensive analysis of pathology and molecular genetics of meningioma from multi-omics perspective. Structure of gene with the meningioma pathogenic variants have been revealed by genomics; performance of the epigenomics showing the influence of the genetic modification on meningioma; pathological gene expression in meningioma were analyzed by transcriptomics; applications of proteomics visually show the endocranial shape changes during meningioma. From genomics to proteomics, the pathological process and potential therapeutic targets involved in meningioma progression will be revealed as never before.
Genomics
Accurate and comprehensive sequencing of personal genomes is an important technical advance based on bioinformatics analysis (11), which is crucial to genetic studies of complex human diseases (12). Deep understanding of genetic alterations relating to meningioma development and progression may provide new insights into meningioma classification and personalized treatment (13).
As early as 2011, a sequencing-based genome-wide association study (GWAS) of 859 patients with meningioma and a control group (n = 704) identified MLLT10 as a new susceptibility locus (14). It is worth mentioning that, in the last decades, the role of MLLT10 in the pathogenesis and progression of meningioma has been well-established (15, 16). In that same year, an expanded genome-wide association study of meningioma, including 2,000 patients and 6,000 controls, was initiated by the National Institutes of Health (17), which earned a significant contribution in understanding genetic factors of meningioma. Notably, the results, including an inverse relationship between hormones and allergies, provided a clear framework and direction for further meningioma study as well as the establishment of comparative oncology (18–20). Furthermore, a genotype analysis in 65 samples using high-density single nucleotide polymorphism (SNP) arrays found associations between meningiomas and variation in PIAS2, KATNAL2, TCEB3C, TCEB3CL, and CTNNA3, especially TARDBP mutations with amyotrophic lateral sclerosis (21), which further improves the identification of susceptible sites of meningioma by genomics. Subsequently, a GWAS involving 1,606 meningioma patients and 9,823 controls provided additional support for the link between obesity and risk of recurrence in meningioma (22), which laid a solid foundation for meningioma characteristics, including risk factors and epidemiology (23, 24). To further illustrate the genetic basis and construct a genetic linkage map of meningioma, Claus et al. identified a new meningioma susceptibility site at 11p15.5 through a combined reference panel from UK10K data including a total of 2,138 and 12,081 controls and 1,000 genomic projects in 2018 (25). It is worth pointing out that the susceptible site included a new pathogenic mutation in RIC8A, which is necessary for the development of cranial neural crest-derived structures. Therefore, this study suggests the cytogenetic relationship between meningiomas and nerve sheath structures (26) (Figure 2).
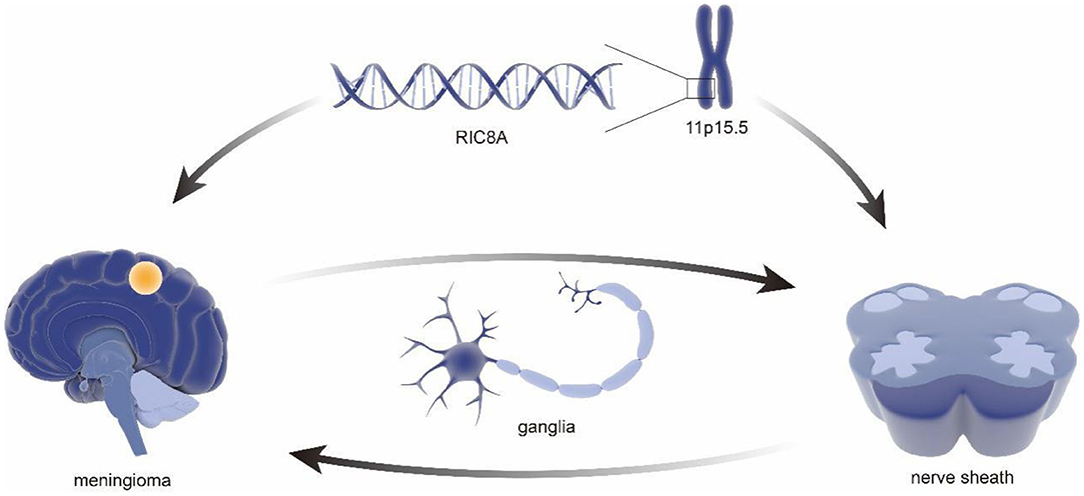
Figure 2. Genetic association of the nerve sheath development and meningioma. The RIC8A located in area 11p15.5 were revealed to be associated with pathological phenotypes in meningioma. It is important to mention that the same genes have been confirmed to be related to cranial neural crest-derived structures. Consider the correlation between nerve sheath and ganglia, which might explain a series of cases of nerve sheath meningioma and ganglia intraparenchymal meningioma.
In addition to the potential role of genetic factors on meningioma, genomics has been applied in the diagnosis and classification of meningioma. Clinically, in the case of a meningioma specimen that contains atypical tumor regions that are difficult to assess, molecular marker techniques for patient genome analysis, such as array comparative genomic hybridization (aCGH) and expression array profiles, can be used for histopathological grading (27). The first instance from a whole genome sequencing project of malignant subtypes revealed mutational signatures and frequently altered genes in malignant meningiomas, including NF2, MN1, ARID1B, SEMA4D, and MUC2, which confirmed the role of pathogenic NF2 mutations in the development of meningiomas, and expression of MN1 may be a valuable diagnostic tool for determining the potential in malignant transformation (28).
So far, genomics has made a tremendous contribution to the criteria for diagnosis, staging, risk stratification, and response assessment of meningiomas (Table 1). Regrettably, some aspects of genetic factors in meningioma have been ignored, and the gene regulatory network leading to meningioma remains unclear. Further pooling research in genomics will advance the field of meningiomas' genetic basis and pathological mechanisms, which may also provide novel research horizons and suggestions for intervention strategies and clinical practice of meningioma.
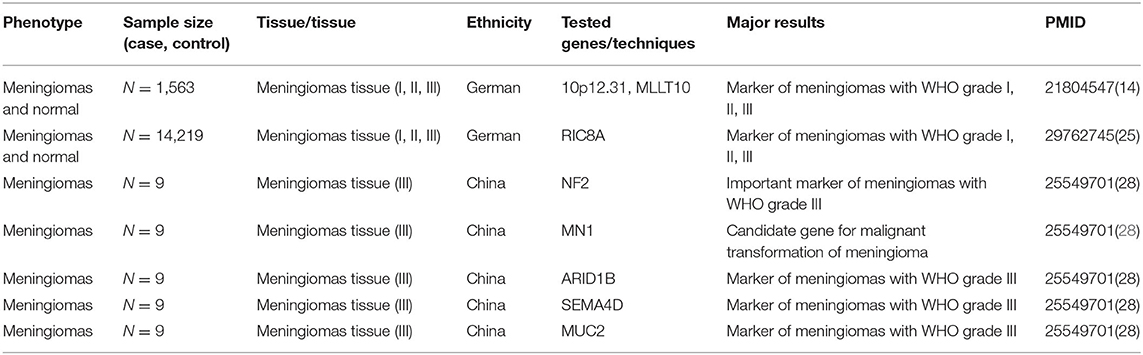
Table 1. Genomics research associated with meningiomas.
Epigenomics
Epigenetic factors, mainly DNA methylation and histone modification, have considerable effects on the pathogenesis of meningioma (29). In the last few years, developments in multi-omics technologies provide tools for high-throughput and high-density molecular analyses, which has provided a novel view regarding the functional organization of the molecular layer. The pathogenic role of chromosome markers in gene regulation and other processes were also inferred by it (30).
WHO classification of meningiomas is based on histologic characteristics. However, part of malignant meningiomas was histologically described to benign meningiomas (31); therefore, novel diagnostic strategies are urgently required while DNA methylation assessment has considerable potential to reconstruct the grade of meningioma. Expression profiles of 10,422 genes at the early stage of meningioma using cDNA microarray indicate hypermethylation of gene subsets are critical in tumor development (32). Further research identified 64-CpG meningioma methylation predictor (64-MMP), which is responsible for tumor recurrence (hazard ratio = 12.16) (33). In 2017, Sahm et al. compiled a genome-wide mapping of differentially methylated regions by DNA methylation profiling from 497 meningioma and 309 extra-axial skull tumors that might histologically mimic meningioma variants. On this basis, six different clinically relevant methylation types of meningioma were distinguished, and they relate to typical mutations, cytogenetics, and gene expression patterns (34). Notably, the classification by methylation provides more precise prognostication of progression-free survival outcomes at 10 years' follow-up compared to WHO grading, which highlights the diagnostic and prognostic implications of malignant meningioma by assessing methylation status.
Importantly, epigenetic profiles in meningioma contribute to the construction of an individualized prediction model of early progression and recurrence in meningioma (35). For example, DNA methylation profiles of 282 clinically annotated meningioma samples were used for construction of a prediction model of 5-year recurrence-free survival (RFS) in meningioma. Notably, the recurrence model provides important prognostic information (hazard ratio = 7.7, area under curve = 0.82), which is more accurate than prediction based on clinical factors, including extent of resection and WHO grade (Δ area under curve = 0.25) (36).
In addition to the roles in tumor classification, a comprehensive understanding of epigenetic regulation that has characterized meningioma development and progression may also provide useful guidance for targeted therapies. So far, methylation of TIMP3, CDKN2, and other genes that can regulate the progression of meningiomas have been identified by genome-wide methylation DNA analysis (37); further work reveals the connection between the H3K27me3 signal and hypermethylated phenotype in meningiomas, integrating with microarray analysis of the transcriptional network controlled by E2F2 and FOXM1. This study makes recommendations for potential targets for therapeutic intervention (38).
The progress in epigenetic research on meningioma have proved to be a valuable tool in pathological classification and intervention of meningiomas (Supplementary Table 1) (21). However, recent advances in epigenomics of meningioma have mainly focused on DNA methylation; the role of histone modification and chromosome organization have been neglected. It is also worth noting that chromosomes are associated with homologous recombination repair (HRR) defects, which has been confirmed as a primary causative factor of meningioma (39, 40), suggesting that histone modification has great potential in the development of novel meningioma prevention and intervention measures.
Transcriptomics
By comparing the transcriptome differences between meningioma patients and controls, transcriptomics can screen out the specific expression differences with diagnostic significance, which can be used in the diagnosis and early intervention of meningiomas.
Since the occurrence and development of meningiomas are often caused by the accumulation of multiple gene changes, transcriptome can detect the gene expression differences between normal tissues and meningiomas from the transcriptional level (Supplementary Table 2) (41–45). In 2017, a genome-wide array comparing microRNAs expression in meningioma from 50 patients showed that miRNA-21 expression increased significantly with increasing histopathologic grade with reduction of miRNA-107 (41, 46). Notably, upregulated miR-29c-3p coupled with reduction of its predicted target recombinant pentraxin 3(PTX3) was observed in the same year using whole transcriptome microarray chips, which indicated the level of tumor suppressor PTX3 is inhibited by miR-29c-3p (42). Interestingly, PTX3 overexpression was frequently observed in high-grade gliomas and meningiomas with poor prognosis, which suggests that PTX3 may be an important contributor to meningioma cell proliferation and invasion (47). The conflicting results have been obtained, which remind us that further studies of changes in transcriptome of meningioma is necessary.
As mentioned earlier, due to its high recurrence rate and poor prognosis, a lot of work on the research of malignant meningioma is required (48), and it is associated with shorter progression-free and overall survival after complete resection (49). Fortunately, novel markers of malignant meningiomas identified through differential gene expression analyses can be achieved through transcriptomics. For example, an illumina expression microarray to assess gene expression levels from a sample set of 19 resected meningiomas identified dense coexpression subnetworks in meningioma and detected carcinogenic modules associated with malignant meningioma. Among the 23 identified coexpression modules, a module involving 356 genes is highly correlated with occurrence of meningioma. It should be noted that putative meningioma tumor suppressive meningioma 1 (MN1) in this module was differentially expressed between malignant and benign meningioma (43), indicating it can be used as a predictor of meningioma classification.
In addition to characterization of differentially expressed genes, some RNAs were also found to have potential meaning in classification of benign and malignant meningiomas. In 2013, a tissue microarray indicated reduced expression of miR-145 in WHO grade II/III meningiomas using frozen samples from 42 meningiomas. Notably, the follow-up studies demonstrated the antiproliferation, morphogenesis, and antimigration effects of miR-145 in meningioma cells, suggesting the proposed role of the miR-145 in restraining meningioma progression (44) (Figure 3). Besides, the small nucleolar RNAs(snoRNAs), such as SNORA46 and SNORA48, were also found differentially expressed between grade I and grade II/III meningiomas, which is identified by RNA sequencing (RNA-seq) analysis after numerous genes were found differentially expressed by real time-PCR (45).
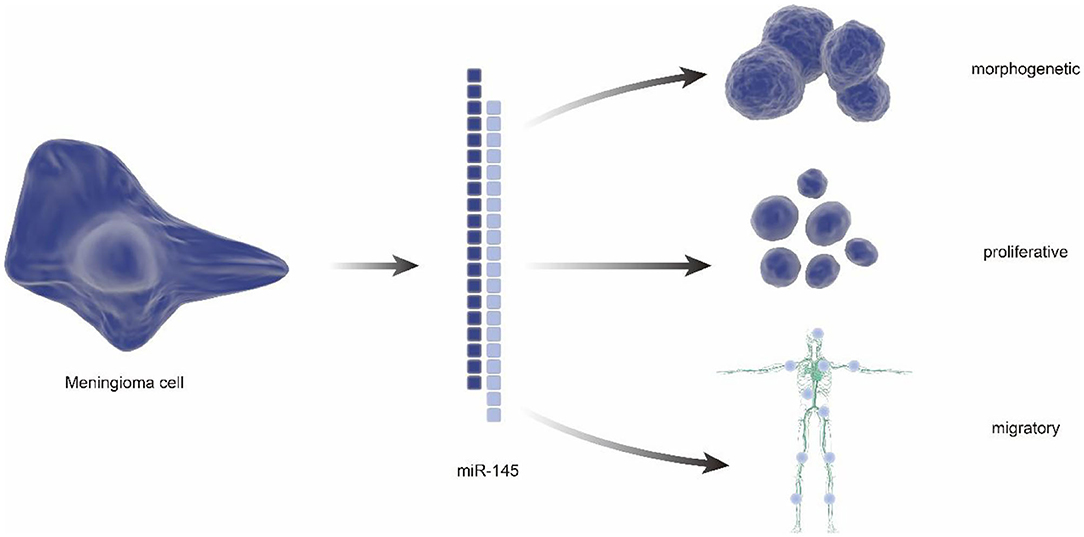
Figure 3. The regulatory role of miRNA in meningiomas. miRNA has a reduced expression in atypical and anaplastic meningiomas, which increases cell proliferation and reduces apoptotic susceptibility. In addition, reduced miRNA reduces migration, invasion, and adhesion of meningioma cells. It can also alter meningioma cell morphology, resulting in low elongation and adhesion.
In addition, miRNAs belong to small ncRNAs (sncRNAs), and small interfering RNA (siRNAs) are functionally similar to miRNA, modulating post-transcriptional gene expression by binding to specific mRNAs (50). But transcriptomics studies focused on miRNAs are much more than that on siRNAs although siRNA has been found to relate to some meaningful molecules in meningiomas. For example, siRNA can decrease the expression of high-mobility group nucleosome-binding protein 5 (HMGN5), which has a positive association with meningioma histological grade (51). As for snoRNA, more and more evidence reveal the importance of snoRNA in tumorigenesis (52, 53), such as SNORD50A/B (C/D box), which can directly bind to and inhibit K-rat sarcoma (K-Ras), is deleted in many cancer types (54). However, the lack of transcriptomic studies pertaining to the expression of siRNAs and snoRNAs or relative pathways suggest that transcriptomic studies taking siRNA into consideration are required in the field of meningioma research.
Proteomics
Proteomics is a large-scale study of protein properties, including protein expression levels, post translation modification, protein–protein interaction, etc., which has been proven to be a useful tool in the identification between varieties of meningeal neoplasms (55).
Proteomics can detect the differential expression of proteins in different grades or types of meningiomas (Supplementary Table 3) (56–58). As early as 2006, the pure meningioma cell population was sequenced to indicate the differentially expressed proteins of each WHO grade meningioma. This study identified the 15 proteins that were significantly related to atypical meningioma, and nine proteins can be used to discriminate atypical from anaplastic meningiomas (57). Similar biomarkers were also reported in 2014; the expression of galectin-3, vimentin was decreased significantly in meningiomas, and the expression of 40S ribosomal protein S12 and glutathione S-transferase was increased significantly (59). It is worth mentioning that the function of galectin-3 was further investigated in 2017; high expression of galectin-3 was observed in meningioma infiltration and recurrence (60). However, the role of galectin-3 in meningioma remains controversial; there is still a need for further studies to confirm the exact mechanism of galectin-3 in meningioma. Recently, with highly sensitive instruments in proteomics, low-abundance proteins could be found to be meaningful in different grades of meningiomas. For example, comparative tissue proteomic analysis was performed by isobaric tags for relative and absolute quantification (ITRAQ)-based quantitative proteomics by using electrospray ionization-quadrupole-time of flight (ESI-Q-TOF) and thermo scientific Q exactive (Q-Exactive MS), which quantified many transmembrane receptors and transcription factors, such as activated RNA polymerase II transcriptional coactivator p15 in pathology of meningioma (61).
In addition, proteomics analysis has also been used to identify different subtypes of meningiomas. To explore the different protein expression patterns of bone-infiltrating and non-invasive meningioma, the researchers used a protein spectrum combined with surface-enhanced laser desorption/ionization time-of-flight mass spectrometry (SELDI), and the results show meaningful differences in fibrous and meningothelial grade I meningiomas that contribute to distinguish the two types of meningiomas (60). Therefore, invasive and non-invasive growth behavior of grade I fibrous and meningothelial meningioma can be distinguished by analyzing the protein profile of benign meningioma. Notably, the early diagnosis of invasive grade I meningioma is thought to contribute to follow-up policies and the issue of radiotherapy (62).
In addition to protein expression, proteomics studies about post-translational modifications have also been conducted to map the mechanisms of aggressiveness of meningiomas. By using two high-throughput technologies: unbiased iTRAQ LCMS/MS and biased Pamchip peptide arrays, it was found that the A-kinase anchor protein 12 (AKAP12) protein (a phosphoprotein) is downregulated in all grades of meningioma (58). Further studies have shown that knocking down AKAP12 in benign meningioma cells promotes proliferation, migration, and invasion, suggesting that AKAP12 is a central regulator of invasive meningioma progression (58). However, although studies have provided increasing evidence that post-translational modification is closely connected with cell-based functional characterization, which has a close connection with function and malignancy of the disease, phospho-proteomes are rarely studied in meningiomas (61, 63, 64).
Multi-Omics Studies in Meningiomas
Despite a valuable contribution, the results from single omics are unable to map the comprehensive meningioma-related signaling pathways and networks. Therefore, advantages of integrated analysis using multi-omics data have been gradually revealed. For example, the FoxM1 target gene in the case of increased FoxM1 mRNA expression was identified by RNA sequencing, DNA methylation sequencing, and target gene expression profile from meningiomas with low survival rate and high local recurrence rate (65). In addition, integration of multi-omics data contributes to the identification of radiation-induced meningioma, an uncommon late risk of cranial irradiation with higher recurrence rate and pathologically malignant features compared to the sporadic meningioma (66). For example, comparative genome hybridization was used for the identification of chromosome 1p loss in radiation-induced meningioma (67). Notably, NF2 rearrangement in radiation-induced meningioma was identified through exome, methylation, and RNA-seq analysis from 31 cases, which can be used for the differentiation of radiation-induced meningioma from sporadic meningioma as neurofibromatosis type 2 (NF2) rearrangement has still not been reported in sporadic meningioma (68).
The target gene identified by multi-omics studies can potentially be used in drug repositioning in meningiomas (Supplementary Table 4), which appeared to be cheaper, quicker, and more effective (69). For example, Fostamatinib, targeting FoxM1, has been approved by the FDA for the treatment of chronic immune thrombocytopenia (ITP). Given the same putative drivers of disease associations, Fostamatinib may improve meningioma via regulating synthesis and secretion of tumor necrosis factor α(TNF-α) (70) (Figure 4).
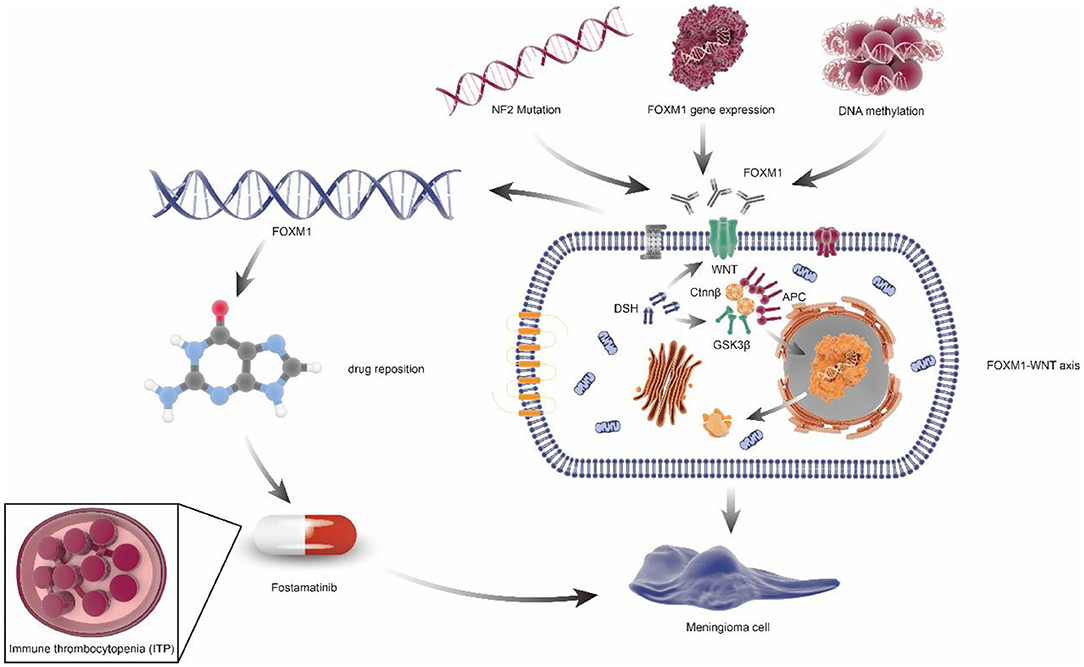
Figure 4. FOXM1/Wnt signaling axis drives meningioma prolife ratio and tumor growth. NF2 mutation, FOXM1 gene expression, and DNA methylation can cause the increase of FOXM1 expression or activity, which would activate the FOXM1/WNT signaling axis, resulting in primary or aggressive meningioma cell proliferation. In addition, through the principle of drug repositioning, fostamatinib, a kind of medicine aimed at chronic immune thrombocytopenia (ITP), which targets the FoxM1, may also be used in the treatment of meningiomas.
Conclusion
In 2016, the World Health Organization included the molecular standards into the classification of meningiomas (71). Soon after this, accurate pathological diagnosis and treatment decisions at the molecular level depend on powerful clinical molecular detection using genome, epigenome, and transcriptome tools is highly applied in clinical studies (72). Although it is necessary to carry out molecular detection of brain tumors in medicine, there are still great differences in the acquisition and utilization of molecular diagnosis technology in various institutions, and the lack of compensation for such detection is still a major obstacle (72). Notably, the important role of omics studies in the molecular level pathological study and grading of meningiomas has potential value in clinical diagnosis and treatment. Therefore, there is no doubt that multi-omics studies will shed further light on the novel strategies for the prediction, prevention, and treatment of meningiomas.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank all clinical personnel, epidemiologists, and county public health institute personnel for their contribution to 2019-COVID outbreak control.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.01491/full#supplementary-material
Supplementary Table 1. Summary of epigenomics studies for meningiomas.
Supplementary Table 2. Research on meningiomas in transcriptomics.
Supplementary Table 3. Proteomics research on meningiomas.
Supplementary Table 4. Meningioma driver genes-based drug-repositioning.
Abbreviations
WHO, World Health Organization; GWAS, Genome Wide Association Study; UK, United Kingdom; aCGH, array comparative genomic hybridization; 64-MMP, 64-CpG meningiomas methylation predictor; HRR, homologous recombination repair; PTX3, Recombinant Pentraxin 3; MN1, meningioma 1; miRNA, microRNA; snoRNA, small nucleolar RNA; sncRNA, small interfering RNA; siRNA, small interfering RNA; HMGN5, high-mobility group nucleosome-binding protein 5; K-Ras, K-rat sarcoma; SELDI-TOF MS, surface-enhanced laser desorption/ionization time-of-flight mass spectrometry; ITRAQ, isobaric tags for relative and absolute quantification; ESI-Q-TOF, electrospray ionization- quadrupole-time of flight; Q-Exactive MS, thermo scientific Q exactive; SELDI, surface-enhanced laser desorption/ionization time-of-flight mass spectrometry; AKAP12, a-kinase anchor protein 12; FoxM1, Forkhead Box M1; ITP, immune thrombocytopenia; TNF-α, tumor necrosis factor α; NF2, Neurofibromatosis Type 2.
References
1. Poulen G, Vignes JR, Corre ML, Loiseau H, Bauchet L. WHO Grade II Meningioma: epidemiology, survival and interest of post-operative radiotherapy in a multicenter cohort of 88 patients. Neurochirurgie. (2020) 66:73–9. doi: 10.1016/j.neuchi.2019.12.008
2. Baldi I, Engelhardt J, Bonnet C, Bauchet L, Berteaud E, Grüber A, et al. Epidemiology of meningiomas. Neurochirurgie. (2018) 64:5–14. doi: 10.1016/j.neuchi.2014.05.006
3. Holleczek B, Zampella D, Urbschat S, Sahm F, von Deimling A, Oertel J, et al. Incidence, mortality and outcome of meningiomas: a population-based study from Germany. Cancer Epidemiol. (2019) 62:101562. doi: 10.1016/j.canep.2019.07.001
4. Gandhoke GS, Pease M, Smith KJ, Sekula RF Jr. Supraorbital versus endoscopic endonasal approaches for olfactory groove meningiomas: a cost-minimization study. World Neurosurg. (2017) 105:126–36. doi: 10.1016/j.wneu.2017.03.148
5. Al-Rashed M, Foshay K, Abedalthagafi M. Recent advances in meningioma immunogenetics. Front Oncol. (2019) 9:1472. doi: 10.3389/fonc.2019.01472
6. Goldbrunner R, Minniti G, Preusser M, Jenkinson MD, Sallabanda K, Houdart E, et al. EANO guidelines for the diagnosis and treatment of meningiomas. Lancet Oncol. (2016) 17:e383–91. doi: 10.1016/S1470-2045(16)30321-7
7. Lee YS, Lee YS. Molecular characteristics of meningiomas. J Pathol Transl Med. (2020) 54:45–63. doi: 10.4132/jptm.2019.11.05
8. Chen D, Zhao X, Sui Z, Niu H, Chen L, Hu C, et al. A multi-omics investigation of the molecular characteristics and classification of six metabolic syndrome relevant diseases. Theranostics. (2020) 10:2029–46. doi: 10.7150/thno.41106
9. Ramroach S, Joshi A, John M. Optimisation of cancer classification by machine learning generates an enriched list of candidate drug targets and biomarkers. Mol Omics. (2020) 16:113–25. doi: 10.1039/C9MO00198K
10. Wang Q, Shen Q, Zhang Z, Cai C, Lu H, Zhou X, et al. [Prediction of gene mutation in lung cancer based on deep learning and histomorphology analysis]. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi. (2020) 37:10–8. doi: 10.7507/1001-5515.201904018
11. Hindorff LA, Bonham VL, Brody LC, Ginoza MEC, Hutter CM, Manolio TA, et al. Prioritizing diversity in human genomics research. Nat Rev Genet. (2018) 19:175–85. doi: 10.1038/nrg.2017.89
12. Oti M, Sammeth M. Comparative Genomics in Homo sapiens. Methods Mol Biol. (2018) 1704:451–72. doi: 10.1007/978-1-4939-7463-4_18
13. Pham MH, Zada G, Mosich GM, Chen TC, Giannotta SL, Wang K, et al. Molecular genetics of meningiomas: a systematic review of the current literature and potential basis for future treatment paradigms. Neurosurg Focus. (2011) 30:E7. doi: 10.3171/2011.2.FOCUS1117
14. Dobbins SE, Broderick P, Melin B, Feychting M, Johansen C, Andersson U, et al. Common variation at 10p12.31 near MLLT10 influences meningioma risk. Nat Genet. (2011) 43:825–7. doi: 10.1038/ng.879
15. Egan KM, Baskin R, Nabors LB, Thompson RC, Olson JJ, Browning JE, et al. Brain tumor risk according to germ-line variation in the MLLT10 locus. Eur J Hum Genet. (2015) 23:132–4. doi: 10.1038/ejhg.2014.70
16. Han XY, Wang W, Wang LL, Wang XR, Li G. Genetic variants and increased risk of meningioma: an updated meta-analysis. Onco Targets Ther. (2017) 10:1875–88. doi: 10.2147/OTT.S130147
17. Vanchieri C. New data on nonmalignant brain tumors could spur research efforts. J Natl Cancer Inst. (2011) 103:706–7, 13. doi: 10.1093/jnci/djr166
18. Pouchieu C, Raherison C, Piel C, Migault L, Carles C, Fabbro-Perray P, et al. Allergic conditions and risk of glioma and meningioma in the CERENAT case-control study. J Neurooncol. (2018) 138:271–81. doi: 10.1007/s11060-018-2816-6
19. Swerdlow AJ, Cooke R, Beckers D, Butler G, Carel JC, Cianfarani S, et al. Risk of Meningioma in European patients treated with growth hormone in childhood: results from the SAGhE cohort. J Clin Endocrinol Metab. (2019) 104:658–64. doi: 10.1210/jc.2018-01133
20. Schiffman JD, Breen M. Comparative oncology: what dogs and other species can teach us about humans with cancer. Philos Trans R Soc Lond B Biol Sci. (2015) 370:20140231. doi: 10.1098/rstb.2014.0231
21. Hosking FJ, Feldman D, Bruchim R, Olver B, Lloyd A, Vijayakrishnan J, et al. Search for inherited susceptibility to radiation-associated meningioma by genomewide SNP linkage disequilibrium mapping. Br J Cancer. (2011) 104:1049–54. doi: 10.1038/bjc.2011.61
22. Takahashi H, Cornish AJ, Sud A, Law PJ, Disney-Hogg L, Calvocoressi L, et al. Mendelian randomization provides support for obesity as a risk factor for meningioma. Sci Rep. (2019) 9:309. doi: 10.1038/s41598-018-36186-6
23. Modzelewska P, Chludzinska S, Lewko J, Reszec J. The influence of leptin on the process of carcinogenesis. Contemp Oncol. (2019) 23:63–8. doi: 10.5114/wo.2019.85877
24. Muskens IS, Wu AH, Porcel J, Cheng I, Le Marchand L, Wiemels JL. Body mass index, comorbidities, and hormonal factors in relation to meningioma in an ethnically diverse population: the Multiethnic Cohort. Neuro Oncol. (2019) 21:498–507. doi: 10.1093/neuonc/noz005
25. Claus EB, Cornish AJ, Broderick P, Schildkraut JM, Dobbins SE, Holroyd A, et al. Genome-wide association analysis identifies a meningioma risk locus at 11p15.5. Neuro Oncol. (2018) 20:1485–93. doi: 10.1093/neuonc/noy077
26. Singh S, Kumar A, Mehrotra A, Rao RN, Behari S. Nonsecretory paraganglioma in cavernous sinus masquerading as meningioma. World Neurosurg. (2019) 126:399–404. doi: 10.1016/j.wneu.2019.02.111
27. Rohilla S, Garg HK, Singh I, Yadav RK, Dhaulakhandi DB. rCBV- and ADC-based grading of meningiomas with glimpse into emerging molecular diagnostics. Basic Clin Neurosci. (2018) 9:417–28. doi: 10.32598/bcn.9.6.417
28. Zhang X, Jia H, Lu Y, Dong C, Hou J, Wang Z, et al. Exome sequencing on malignant meningiomas identified mutations in neurofibromatosis type 2 (NF2) and meningioma 1 (MN1) genes. Discov Med. (2014) 18:301–11.
29. Drakos SS, Anifantaki F, Zarros A, Liapi C. The role of folate metabolism-related gene polymorphisms in the development of meningiomas. Cancer Genomics Proteomics. (2010) 7:105–9.
30. Wang KC, Chang HY. Epigenomics: technologies and applications. Circ Res. (2018) 122:1191–9. doi: 10.1161/CIRCRESAHA.118.310998
31. Streitberger KJ, Lilaj L, Schrank F, Braun J, Hoffmann KT, Reiss-Zimmermann M, et al. How tissue fluidity influences brain tumor progression. Proc Natl Acad Sci USA. (2020) 117:128–34. doi: 10.1073/pnas.1913511116
32. Kishida Y, Natsume A, Kondo Y, Takeuchi I, An B, Okamoto Y, et al. Epigenetic subclassification of meningiomas based on genome-wide DNA methylation analyses. Carcinogenesis. (2012) 33:436–41. doi: 10.1093/carcin/bgr260
33. Olar A, Wani KM, Wilson CD, Zadeh G, DeMonte F, Jones DT, et al. Global epigenetic profiling identifies methylation subgroups associated with recurrence-free survival in meningioma. Acta Neuropathol. (2017) 133:431–44. doi: 10.1007/s00401-017-1678-x
34. Sahm F, Schrimpf D, Stichel D, Jones DTW, Hielscher T, Schefzyk S, et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol. (2017) 18:682–94. doi: 10.1016/S1470-2045(17)30155-9
35. Suppiah S, Nassiri F, Bi WL, Dunn IF, Hanemann CO, Horbinski CM, et al. Molecular and translational advances in meningiomas. Neuro Oncol. (2019) 21(Suppl. 1):i4–17. doi: 10.1093/neuonc/noy178
36. Nassiri F, Mamatjan Y, Suppiah S, Badhiwala JH, Mansouri S, Karimi S, et al. DNA methylation profiling to predict recurrence risk in meningioma: development and validation of a nomogram to optimize clinical management. Neuro Oncol. (2019) 21:901–10. doi: 10.1093/neuonc/noz061
37. He S, Pham MH, Pease M, Zada G, Giannotta SL, Wang K, et al. A review of epigenetic and gene expression alterations associated with intracranial meningiomas. Neurosurg Focus. (2013) 35:E5. doi: 10.3171/2013.10.FOCUS13360
38. Harmanci AS, Youngblood MW, Clark VE, Coskun S, Henegariu O, Duran D, et al. Integrated genomic analyses of de novo pathways underlying atypical meningiomas. Nat Commun. (2017) 8:14433. doi: 10.1038/ncomms14433
39. Sievers P, Stichel D, Hielscher T, Schrimpf D, Reinhardt A, Wefers AK, et al. Chordoid meningiomas can be sub-stratified into prognostically distinct DNA methylation classes and are enriched for heterozygous deletions of chromosomal arm 2p. Acta Neuropathol. (2018) 136:975–8. doi: 10.1007/s00401-018-1924-x
40. Paramasivam N, Hubschmann D, Toprak UH, Ishaque N, Neidert M, Schrimpf D, et al. Mutational patterns and regulatory networks in epigenetic subgroups of meningioma. Acta Neuropathol. (2019) 138:295–308. doi: 10.1007/s00401-019-02008-w
41. Katar S, Baran O, Evran S, Cevik S, Akkaya E, Baran G, et al. Expression of miRNA-21, miRNA-107, miRNA-137 and miRNA-29b in meningioma. Clin Neurol Neurosurg. (2017) 156:66–70. doi: 10.1016/j.clineuro.2017.03.016
42. Dalan AB, Gulluoglu S, Tuysuz EC, Kuskucu A, Yaltirik CK, Ozturk O, et al. Simultaneous analysis of miRNA-mRNA in human meningiomas by integrating transcriptome: a relationship between PTX3 and miR-29c. BMC Cancer. (2017) 17:207. doi: 10.1186/s12885-017-3198-4
43. Chang X, Shi L, Gao F, Russin J, Zeng L, He S, et al. Genomic and transcriptome analysis revealing an oncogenic functional module in meningiomas. Neurosurg Focus. (2013) 35:E3. doi: 10.3171/2013.10.FOCUS13326
44. Kliese N, Gobrecht P, Pachow D, Andrae N, Wilisch-Neumann A, Kirches E, et al. miRNA-145 is downregulated in atypical and anaplastic meningiomas and negatively regulates motility and proliferation of meningioma cells. Oncogene. (2013) 32:4712–20. doi: 10.1038/onc.2012.468
45. Viaene AN, Zhang B, Martinez-Lage M, Xiang C, Tosi U, Thawani JP, et al. Transcriptome signatures associated with meningioma progression. Acta Neuropathol Commun. (2019) 7:67. doi: 10.1186/s40478-019-0690-x
46. Dalan AB, Gulluoglu S, Tuysuz EC, Kuskucu A, Yaltirik CK, Ozturk O, et al. Simultaneous analysis of miRNA-mRNA in human meningiomas by integrating transcriptome: A relationship between PTX3 and miR-29c. BMC Cancer. (2017) 17:207. doi: 10.1186/s12885-017-3198-4
47. Ke HH, Hueng DY, Tsai WC. Low expression of pentraxin 3 and nuclear factor-like 2 implying a relatively longer overall survival time in gliomas. Chin J Physiol. (2019) 62:35–43. doi: 10.4103/CJP.CJP_3_19
48. Weber DC, Ares C, Villa S, Peerdeman SM, Renard L, Baumert BG, et al. Adjuvant postoperative high-dose radiotherapy for atypical and malignant meningioma: a phase-II parallel non-randomized and observation study (EORTC 22042–26042). Radiother Oncol. 128:260–5. doi: 10.1016/j.radonc.2018.06.018
49. Riemenschneider MJ, Perry A, Reifenberger G. Histological classification and molecular genetics of meningiomas. Lancet Neurol. (2006) 5:1045–54. doi: 10.1016/S1474-4422(06)70625-1
50. Raghavachari N, Garcia-Reyero N. Overview of Gene Expression Analysis: Transcriptomics. Methods Mol Biol. (2018) 1783:1–6. doi: 10.1007/978-1-4939-7834-2_1
51. He J, Liu C, Wang B, Li N, Zuo G, Gao D. HMGN5 blockade by siRNA enhances apoptosis, suppresses invasion and increases chemosensitivity to temozolomide in meningiomas. Int J Oncol. (2015) 47:1503–11. doi: 10.3892/ijo.2015.3131
52. Krishnan P, Ghosh S, Wang B, Heyns M, Graham K, Mackey JR, et al. Profiling of small nucleolar RNAs by next generation sequencing: potential new players for breast cancer prognosis. PLoS ONE. (2016) 11:e0162622. doi: 10.1371/journal.pone.0162622
53. Williams GT, Farzaneh F. Are snoRNAs and snoRNA host genes new players in cancer?. Nat Rev Cancer. (2012) 2:84–8. doi: 10.1038/nrc3195
54. Siprashvili Z, Webster DE, Johnston D, Shenoy RM, Ungewickell AJ, Bhaduri A, et al. The noncoding RNAs SNORD50A and SNORD50B bind K-Ras and are recurrently deleted in human cancer. Nat Genet. (2016) 48:53–8. doi: 10.1038/ng.3452
55. Bouamrani A, Ternier J, Ratel D, Benabid Al, Issartel JP, Brambilla E, et al. Direct-tissue SELDI-TOF mass spectrometry analysis: a new application for clinical proteomics. Clin Chem. (2006) 52:2103–6. doi: 10.1373/clinchem.2006.070979
56. Cui GQ, Jiao AH, Xiu CM, Wang YB, Sun P, Zhang LM, et al. Proteomic analysis of meningiomas. Acta Neurol Belg. (2014) 114:187–94. doi: 10.1007/s13760-013-0253-z
57. Okamoto H, Li J, Vortmeyer AO, Jaffe H, Lee YS, Glasker S, et al. Comparative proteomic profiles of meningioma subtypes. Cancer Res. (2006) 66:10199–204. doi: 10.1158/0008-5472.CAN-06-0955
58. Parada CA, Osbun J, Kaur S, Yakkioui Y, Shi M, Pan C, et al. Kinome and phosphoproteome of high-grade meningiomas reveal AKAP12 as a central regulator of aggressiveness and its possible role in progression. Sci Rep. (2018) 8:2098. doi: 10.1038/s41598-018-19308-y
59. Cui GQ, Jiao AH, Xiu CM, Wang YB, Sun P, et al. Proteomic analysis of meningiomas. Neuro Oncol. (2019) 21:1028–38. doi: 10.1093/neuonc/noz084
60. Wibom C, Mörén L, Aarhus M, Knappskog PM, Lund-Johansen M, Antti H, et al. Proteomic profiles differ between bone invasive and noninvasive benign meningiomas of fibrous and meningothelial subtype. J Neurooncol. (2009) 94:321–31. doi: 10.1007/s11060-009-9865-9
61. Sharma S, Ray S, Mukherjee S, Moiyadi A, Moiyadi AE, Sridhar E, et al. Multipronged quantitative proteomic analyses indicate modulation of various signal transduction pathways in human meningiomas. Proteomics. (2015) 15:394–407. doi: 10.1002/pmic.201400328
62. Commins DL, Atkinson RD, Burnett ME. Review of meningioma histopathology. Neurosurg Focus. (2007) 23:E3. doi: 10.3171/FOC-07/10/E3
63. Barkhoudarian G, Whitelegge JP, Kelly DF, Simonian M. Proteomics analysis of brain meningiomas in pursuit of novel biomarkers of the aggressive behavior. J Proteomics Bioinform. (2016) 9:53–7. doi: 10.4172/jpb.1000389
64. Osbun JW, Tatman PD, Kaur S, Parada C, Busald T, Gonzalez-Cuyar L, et al. Comparative proteomic profiling using two-dimensional Gel electrophoresis and identification via LC-MS/MS reveals novel protein biomarkers to identify aggressive subtypes of WHO Grade I meningioma. J Neurol Surg B Skull Base. (2017) 78:371–9. doi: 10.1055/s-0037-1601889
65. Vasudevan HN, Braunstein SE, Phillips JJ, Pekmezci M, Tomlin BA, Wu A, et al. Comprehensive molecular profiling identifies FOXM1 as a key transcription factor for meningioma proliferation. Cell Rep. (2018) 22:3672–83. doi: 10.1016/j.celrep.2018.03.013
66. Raheja A, Satyarthee GD. Sphenoid wing en plaque meningioma development following craniopharyngioma surgery and radiotherapy: radiation-induced after three decades. Asian J Neurosurg. (2017) 12:358–61. doi: 10.4103/1793-5482.180946
67. Brassesco MS, Valera ET, Neder L, Castro-Gamero AM, de Oliveira FM, Santos AC, et al. Childhood radiation-associated atypical meningioma with novel complex rearrangements involving chromosomes 1 and 12. Neuropathology. (2009) 29:585–90. doi: 10.1111/j.1440-1789.2008.00991.x
68. Agnihotri S, Suppiah S, Tonge PD, Jalali S, Danesh A, Bruce JP, et al. Therapeutic radiation for childhood cancer drives structural aberrations of NF2 in meningiomas. Nat Commun. (2017) 8:186. doi: 10.1038/s41467-017-00174-7
69. Armando RG, Mengual Gómez DL, Gomez DE. New drugs are not enough-drug repositioning in oncology: an update. Int J Oncotl. (2020) 56:651–84. doi: 10.3892/ijo.2020.4966
70. Karsy M, Azab MA, Abou-Al-Shaar H, Guan J, Eli I, Jensen RL, et al. Clinical potential of meningioma genomic insights: a practical review for neurosurgeons. Neurosurg Focus. (2018) 44:E10. doi: 10.3171/2018.2.FOCUS1849
71. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
Keywords: meningiomas, biomarker, genomics, epigenomics, transcriptomics, proteomics
Citation: Liu J, Xia C and Wang G (2020) Multi-Omics Analysis in Initiation and Progression of Meningiomas: From Pathogenesis to Diagnosis. Front. Oncol. 10:1491. doi: 10.3389/fonc.2020.01491
Received: 14 April 2020; Accepted: 13 July 2020;
Published: 28 August 2020.
Edited by:
Hailiang Tang, Huashan Hospital Affiliated to Fudan University, ChinaReviewed by:
Dusten Unruh, Northwestern Medicine, United StatesJason M. Miska, Northwestern University, United States
Subhas K. Konar, National Institute of Mental Health and Neurosciences (NIMHANS), India
Copyright © 2020 Liu, Xia and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gaiqing Wang, d2FuZ2dxMDhAMTYzLmNvbQ==