 Asunción Espinosa-Sánchez
Asunción Espinosa-Sánchez Elisa Suárez-Martínez
Elisa Suárez-Martínez Laura Sánchez-Díaz
Laura Sánchez-Díaz Amancio Carnero
Amancio Carnero- 1Instituto de Biomedicina de Sevilla (IBIS), Hospital Universitario Virgen del Rocío, Universidad de Sevilla, Consejo Superior de Investigaciones Científicas, Seville, Spain
- 2CIBER de Cancer, Madrid, Spain
The theory of cancer stem cells (CSCs) proposes that the different cells within a tumor, as well as metastasis deriving from it, are originated from a single subpopulation of cells with self-renewal and differentiation capacities. These cancer stem cells are supposed to be critical for tumor expansion and metastasis, tumor relapse and resistance to conventional therapies, such as chemo- and radiotherapy. The acquisition of these abilities has been attributed to the activation of alternative pathways, for instance, WNT, NOTCH, SHH, PI3K, Hippo, or NF-κB pathways, that regulate detoxification mechanisms; increase the metabolic rate; induce resistance to apoptotic, autophagic, and senescence pathways; promote the overexpression of drug transporter proteins; and activate specific stem cell transcription factors. The elimination of CSCs is an important goal in cancer therapeutic approaches because it could decrease relapses and metastatic dissemination, which are main causes of mortality in oncology patients. In this work, we discuss the role of these signaling pathways in CSCs along with their therapeutic potential.
Introduction
The understanding on the tumorigenesis process has been explained during decades according to the clonal evolution model. This model postulates that all cells within a tumor contribute to the maintenance of the tumor at different levels (1). In the cell, a number of genetic and epigenetic changes accumulate during time, and, by selection, the most aggressive cancer cells drive tumor progression (1, 2). Therefore, any cancer cell can become highly malignant, contributing to metastases and the resistance against therapies (2). However, currently, the cancer stem cell (CSC) model proposes a more suitable explanation to cancer complexity. The tumors contain a subset of different tumor cells, called cancer stem cells that are crucial for tumor initiation, progression, and recurrence (3, 4). These CSCs, through self-renewal and differentiation, are critical for the generation of most tumor cell types contributing to tumor heterogeneity. However, the rest of mature cells compose the bulk of the tumor, but are not responsible for the tumor generation. Therefore, tumor resistance to therapies and metastases are the direct result of these CSCs (5).
CSCs, or “tumor-initiating cells,” have the ability to self-renew and differentiate as normal stem cells. However, the mechanisms that regulate these processes are deregulated; therefore, CSCs continuously expand and produce differentiated progeny (5–7). Furthermore, CSCs can form new form tumors when grown into animals, but normal stem cells are unable to do (8, 9). CSCs compose a small population of cells within a tumor, share similar surface markers with normal stem cells (10, 11) and share common signaling pathways with normal stem cells (12, 13).
The size of the CSC pool is different in each tumor (14–16) and show, in general, a good correlation with agresivity and response, as well as patient survival (17, 18). CSCs show great heterogeneity, and may be different between grades and/or stages of the same type of tumors. The different pools of CSCs usually coexist in the same tumor, even within the same microenvironment niche. Interestingly, CSC different pools may communicathe physiologically among them thorugh phenotype interconversion (10, 19–21). CSCs are a “status” rather than a fixed “category” of cells. CSCs and non-CSCs can also interconvert in dynamic equilibrium. Non-CSCs can acquire CSC properties through de-differentiation (8, 22–24), and in this process, either genetic or epigenetic alterations, as well as microenvironment may be involved (25–28). Therefore, the CSCs model should be considered to be bidirectional, switching between stem and mature cells within the tumor (8, 22–24). Numerous studies support the concept that mature tumor cells (non-CSCs) are respond to cancer therapy, while CSCs are resistant to treatment [(29) and references therein]. In tumor stem cells (CSCs), different clonal evolution at the genetic and epigenetic levels generate distinct tumorigenic potential and heterogeneity which greatly influences disease progression and response to treatment (27, 29). On the other hand, the dynamic equilibrium between CSCs and differentiated non-CSCs adds another level of complexity. The potential for non-CSCs to revert to CSCs due to genetic, epigenetic, or microenvironment alterations that confer phenotypic plasticity to the tumor cell population is a strong driving force of tumor evolution and resistance to cancer therapy. Moreover, exposure of differentiated cells to therapeutic doses of radiation or many therapeutic compounds increases the CSC pool. To explain this CSC plasticity through cell maturation and dedifferentiating processes, it has been described that acquiring new genetic mutations, epigenetic changes, or microenviromental conditions that are able to activate the epithelial mesenchymal transition (EMT) induces the mature non-CSCs to dedifferentiate and acquire the CSC phenotype. Thus, activating TGF-β activates the EMT transcription factors Twist or Snail, and/or hypoxic conditions seem to influence dedifferentiation processes greatly, activating the SC pathways in tumor mature cells, leading to new CSCs (21, 30–33). Therefore, the phenotypic plasticity inducing conversions between mature non-CSCs and CSCs influences tumor evolution and clinical management.
The capability of one cell type to acquire the phenotype of another or of differentiated somatic and stem cells to interconvert states is denominated cellular plasticity (22, 34). This plasticity may explain the altered gene expression found in different tumor types resembling cell lineages that differ from the true progenitors (22, 35–38). Indeed, the inherent plasticity of stem cell pathways such as Wnt, Notch or Hedgehog, can be modified suggesting that these pathways may be relevant for anticancer research (5, 34, 39–41).
These and other results suggest that some oncogenic signals are able to induce CSCs. These signals are accompanied by an increase in resistance to chemotherapeutic treatments (35, 36) and, in some cases, radiotherapy (42, 43). Therefore, we must take into account the processes involved in the activation of stemness pathways and tumor evolution and evaluate how their influences affect therapy to effectively eliminate a tumor (Figure 1).
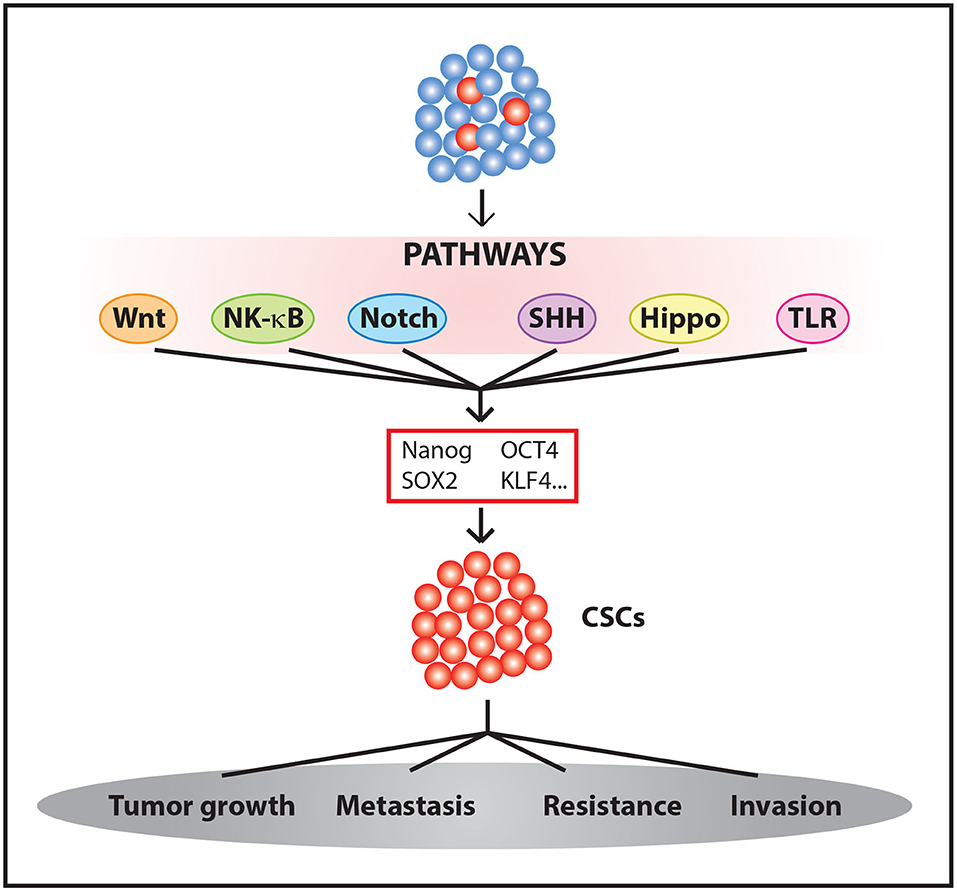
Figure 1. Generation and maintenance CSCs. The activation of different signaling pathways leads to Yamanaka factors expression among other genes, promoting the enrichment of CSC populations within the tumor.
Therefore, cancer cells can move from stem to differentiated states, and viceversa, in response to therapy, transcription changes or signaling in the microenvironment (20, 44, 45). Moreover, inside a single tumor, CSCs can coexist in more than one metabolic and/or pluripotency state. CSCs from breast cancer, for example, can be found in different mesenchymal- and epithelial-like states (24, 46). The transition between these states has been reported to be regulated by epigenetic alterations (47). Phenotypic plasticity contributes to the complexity of the cancer ecosystem and represents a major challenge for tumor eradication since it actively contributes to tumor cell survival and metastasis.
CSC cells present many mechanisms for therapy resistance, such as high-level of drug efflux pumps, reactive oxygen species scavengers, antiapoptotic proteins, DNA repair efficient mechanisms, interactions with the protective microenvironment (37, 48–51) or exosomes loaded with proteins of non-coding RNA prone to modify the environment to favor metastasis (51–54). On the other hand, similar to normal stem cells, CSCs are known to be slow cycling in many tumors and are maintained in the G0 phase (55).
Epigenetic mechanisms may mediate therapeutic resistance in CSCs in many different ways (27, 35, 43, 51, 56–59). The silencing of the epigenome is also involved in maintaining plasticity and the transition of mature tumor non-CSCs to CSCs, as reported for the transition of metabolic states in renal tumor cells by the inactivation of MYBBP1a and the activation of MYB (60–62). For example, epigenetic demethylation of MAP17 driving the resistance against some targeted therapies was observed in lung adenocarcinoma (43). Additionally, studying lung cancer, Sharma and coworkers reported that a reversible drug-tolerant state of EGFR TKi therapy was obtained by chromatin alterations induced by histone demethylase activity (63). These and other results established that CSCs can regulate epigenetic factors to maintain their pool and overcome targeted therapies. However, the reversible nature of these epigenetic alterations suggests that inhibitors of the pathways modifying these epigenetic regulators may hold promise as relevant clinical therapeutic targets, either alone or in combination.
Thus, the CSC hierarchical model explains the failure of treatment and tumor recurrence and promises new targets for anticancer drug discovery. This article does not pretend to be an exhaustive review of all CSC pathways related to plasticity and/or therapeutic approaches. We summarize some evolving treatment strategies related to these pathways with the aim of shedding new light on current therapy development with promising new anticancer agents. Other CSC-related signaling pathways more commonly studied, not reviewed here, but relevant to stemness include MYB, TGF-β, JAK-STAT, FGFs, PI3K, or MEK. Targeting these pathways has been shown to exert anti-CSC effects, and promising agents are currently under investigation, as recently reviewed elsewhere (30, 36, 37, 47, 48, 51, 62, 64–74).
Wnt Pathway
The Wnt pathway is involved in embryonic development and homeostasis. Moreover, this signaling pathway regulates cell proliferation and survival (75). This pathway has two different signaling pathways: canonical (β-catenin dependent) and non-canonical (β-catenin-independent).
According to canonical signaling, Wnt is secreted and binds to Frizzled receptors and/or the low-density lipoprotein-related protein (LRP) 5 and 6 coreceptors. When R-spondin ligands bind to Lgrs, the action of the Wnt pathway is enhanced. In the presence of a Wnt ligand, Wnt interacts with Frizzled and/or LRP5/6 (76). This situation generates a cascade in which the phosphorylation of the cytoplasmic domain in LRP, the recruitment of scaffolding protein Disheveled (Dvl) and the hijack of GSK-3b and Axin are realized, with the last molecule produced through tankyrases. Thus, β-catenin is available for translocation into the nucleus, where it binds to lymphoid enhancer factor (LEF)/T cell factor (TCF) transcription factors and activates target gene transcription (76). In the absence of a Wnt ligand, the level of intracellular β-catenin is very low due to the action of the proteasome degradation complex; this complex is composed of scaffolding protein Axin and adenomatous polyposis coli (APC). Moreover, the kinase proteins glycogen synthase kinase-3b (GSK-3b) and casein kinase 1a (Ck1a) phosphorylate β-catenin triggering its ubiquitination (76) (Figure 2).
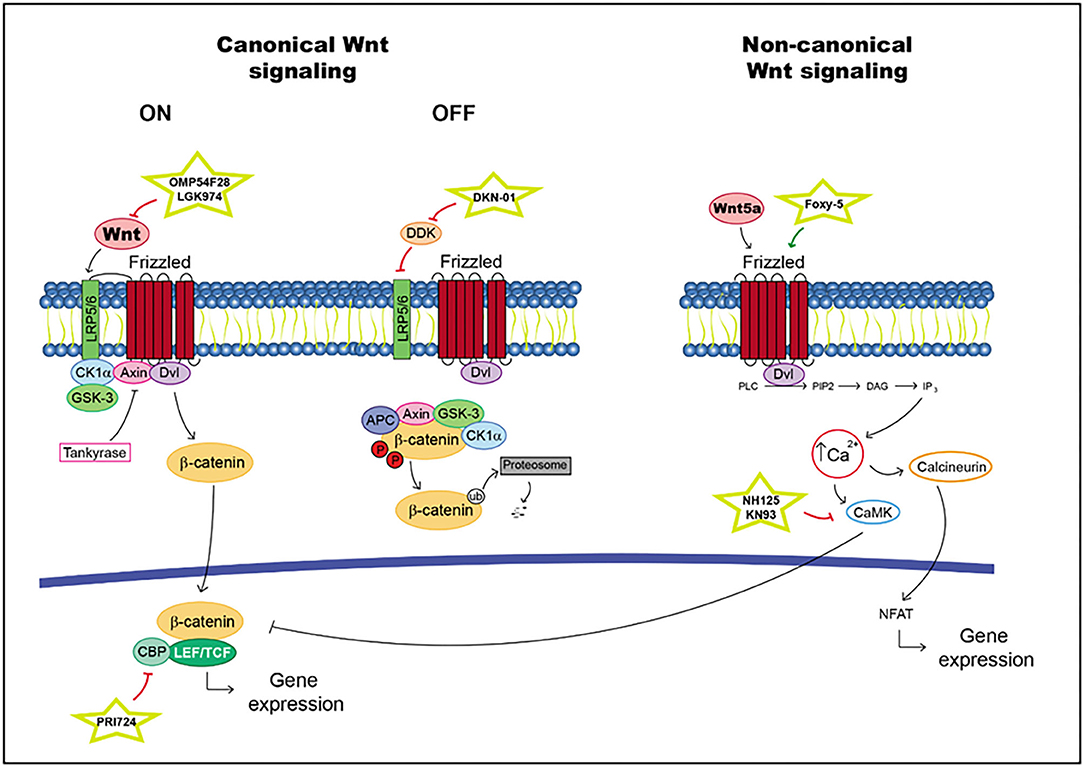
Figure 2. Wnt pathway including targets and drugs that could be employed to inhibit it. (Left) canonical Wnt signaling. (Right) non-canonical Wnt signaling.
In non-canonical signaling, transduction proceeds in some different ways; for example, in the Wnt/Ca2+ pathway. The interaction between Wnt5A and Frizzled FZD2 receptors activates Dvl. Moreover, it promotes the release of Ca2+ into the cytosol as facilitated by FZD2, which cleaves guanine nucleotide-binding protein (G protein). Ca2+ activates CaMKII_AD (Ca2+/calmodulin-dependent protein kinase II) and suppresses the canonical Wnt pathway. This situation increases the differentiation of cells in the neuronal system (77–80) (Figure 2).
Cancer Stem Cells
Some articles suggest that the role of Wnt in the regulation of CSCs differs depending on the types of cancer, such as blood, intestine, lung, mammary, gland, nervous system, skin, and urinary tract. During the development and maintenance of CSCs, the most common alteration is the hyperactivation of Wnt signaling. In skin cancer, the role of the Wnt pathway was discovered using a reporter mouse strain. It was observed that the genetic deletion of β-catenin produced induced tumor regression in chemically induced skin tumors through a decrease in the pool of CD34+ stem cells (81). In human acute leukemia, the crucial role of Wnt signaling activation in cell self-renewal capacity and drug-resistant properties has been suggested (82). Another article reported that the overexpression of miR-582-3p causes Wnt signaling activation by targeting multiple negative regulators of the Wnt pathway, such as AXIN2, DKK3, and SFRP1. Therefore, miR-582-3p promotes tumorigenesis in NSCLC and CSCs in vitro (83). Wnt enhancer R-Spondin-2 (RSPO2) is highly expressed in subpopulations with high intrinsic Wnt activity and with properties indicative of CSCs. Therefore, the employment of promoter inhibitors such as RSPO2 was proposed to block stemness-promoting pathways (84). In metastatic colorectal cancer, an increase in the level of progastrin, a tumor-promoting peptide essential for the self-renewal of colon CSCs, was observed, implicating β-catenin/TCF4 as a direct target gene. Therefore, therapy employing antibodies directed against progastrin was proposed for patients with metastatic colorectal cancer K-RAS-mutations (85). A recent article suggested a role for SOX8 in cancer stem cell properties and therapy resistance. It reported that SOX8 conferred chemoresistance and enhanced stemness properties. Additionally, SOX8 mediated EMT via the FZD7-mediated Wnt/β-catenin pathway (86).
Therapeutic Targets and Drugs
A large number of mutations in the Wnt pathway have been identified. Above all, aberrations in the expression of Wnt ligands, Frizzled receptors, β-catenin and APC were discovered in many different tumors (87–89). According to these results, this pathway has been researched in recent years to identify potential therapeutic targets. Some drugs that may be potential therapeutics to be employed in the treatment of different tumors are detailed.
A fusion protein called OMP-54F28, ipafricept (OncoMed Pharmaceuticals), competes for the FZD8 receptor and antagonizes Wnt signaling. Preclinical models with this drug have shown reduced tumor growth as a single drug in combination with other chemotherapeutic agents (90). OMP-54F28 was studied in phase Ib clinical trials for different types of cancer (ClinicalTrials.gov Identifiers: NCT02050178, NCT02069145, and NCT02092363). In ovarian cancer patients, ipafricept was well-tolerated in combination with sequential carboplatin/paclitaxel, but it caused bone toxicity at efficacy doses; therefore its use is limited (91).
Another drug of interest is SM08502 (Samumed LLC), which is a potent CDC-like kinase (CLK) inhibitor. Moreover, CLKs play important roles in the integral cellular mechanism of mRNA splicing to induce the retention of introns in DVL2, TCF7, ERBB2, and LRP5 and exon skipping in LEF1 and TCF7L2 (92). SM08502 inhibits the growth of gastrointestinal tumors in xenograft mouse models (93). The molecule was recently found in phase 1 (ClinicalTrials.gov Identifier: NCT03355066) to show safety, tolerability and efficacy in patients with advanced solid tumors.
On the other hand, the combination of gedatolisib (a pan-class I PI3K/mTOR isoform inhibitor) and PTK7-ADC (an antibody-drug conjugate used against the cell-surface PTK7 protein) has a double synergistic effect, whereby gedatolisib increases the expression of PTK7-ADC and an auristatin payload causes an effect similar to that of gedatolisib. Currently, this combination is in s phase I trial for patients with metastatic triple-negative breast cancer because the PI3K pathway is dysregulated in most cells of this type of cancer (94).
The inhibition of porcupine, an O-acyltransferase required for Wnt activation and secretion, induced by LGK974 (Novartis, Basel, Switzerland, https://www.novartis.com), reduced the phosphorylation of the LRP6 receptor and the expression of WNT target genes in preclinical models. Furthermore, in vivo studies of LGK974 have demonstrated an antitumoral response in different types of cancer (95). Phase I evaluation of LGK974 alone and in combination with PDR001 (spartalizumab, an anti-PD-1 antibody) is ongoing (ClinicalTrials.gov Identifier: NCT01351103) in patients with lobular or triple negative breast cancer, melanoma or pancreatic cancer (96).
A monoclonal antibody against dickkopf WNT signaling pathway inhibitor 1 (DKK1) and DKN-01 was discovered. The high expression of DKK1 may be a predictive biomarker for effective treatment with DKN-01 and pembrolizumab in gastroesophageal adenocarcinoma (97). A phase 2 study is assessing this molecule as a monotherapy or in combination with paclitaxel in patients with endometrial, uterine or ovarian cancer (ClinicalTrials.gov Identifier: NCT03395080).
PRI-724 (PRISM BioLab) is a small molecule that inhibits the interaction between β-catenin and CBP in the nucleus. Thus, the downregulation of genes involved in symmetric non-differentiation division and increased p300/β-catenin binding were produced. This situation promotes stem cell differentiation and increases the sensitivity of cytotoxic drugs (98). Phase IIb of this drug in combination with gemcitabine in patients with advanced pancreatic adenocarcinoma (APC) has been completed, and the results showed the necessity for more work to find useful predictive and PD markers.
Aberrant activation of signaling from Wnt pathway usually is associated with stem-like phenotypes and EMT, which induce resistance to endocrine therapy. Plumbagin inhibited colony formation and mammosphere formation and decreased the number of cancer stem cell markers by inhibiting the Wnt signaling pathway. Plumbagin also reduced Wnt-dependent genes as well as β-catenin. Moreover, treatment of orthotopic xenografts with Plumbagin inhibited tumor growth, angiogenesis and metastasis without significant adverse effects on body weight or blood coagulation; therefore, Plumbagin may be useful to treat endocrine-resistant breast cancer (99).
On the other hand, the inhibition of the non-canonical Wnt pathway can be achieved through different drugs. Foxy-5 is a mimetic Wnt5a molecule that binds to and activates the Frizzled 2/5 receptor. Increased Wnt-5a signaling may inhibit endothelial tumor cell migration and invasion. This effect may decrease the metastasis of susceptible tumor cells (100). A phase 2 evaluation of it as a neoadjuvant therapy will be performed for subjects with colon cancer who express low levels of Wnt-5a (ClinicalTrials.gov Identifier: NCT03883802). Moreover, other molecules, such as NH125, which is more selective as an eEF-2 kinase inhibitor that is as an PKC, PKA, or CaMKII inhibitor (101), and KN93, which is a selective inhibitor of CaMKII (102), may be potential drugs to regulate the non-canonical Wnt pathway (Table 1).
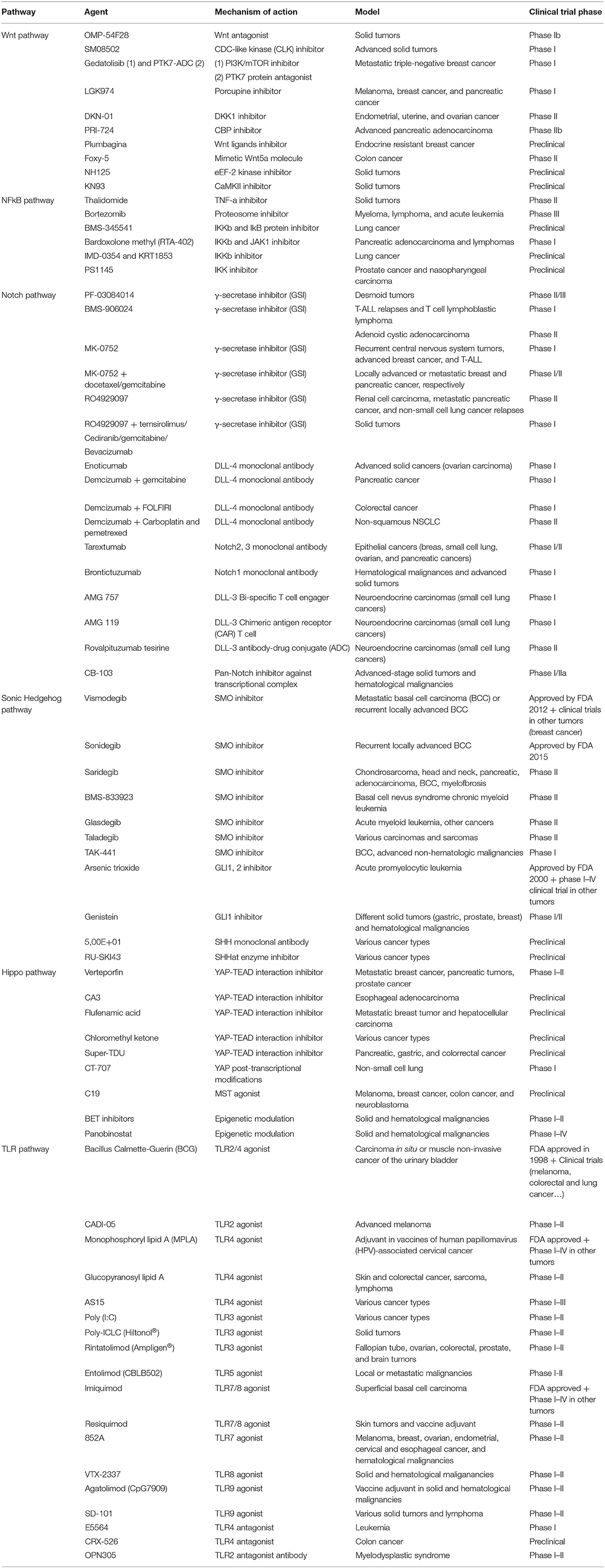
Table 1. Overview of drugs targeting CSC pathways.
NF-κB Pathway
The NF-κB pathway is a complex and pathway that has been studied mainly for its controversial roles in inflammation and immune responses. This signaling also plays an important role in cellular proliferation, survival and differentiation (103–105).
The inflammatory effects on cancer development have been studied, above all the modulation of inflammation-associated cancer through the transcription factor NF-κB (106). The excessive innate immunity activation and growth caused by NF-κB are involved in tumor development and progression (107). Moreover, the activation of NF-κB could be caused for genetic alterations such as amplification, mutations, or deletions in different cancer cells. NF-κB binds to promoters of genes such as IL1B, TNF, and IL6 which cause the secretion of cytokines and chemokines (108). For this reason, it is suggested that the exposure to proinflammatory stimuli in tumor microenvironment or genetic alterations in components of IKK—NF-κB pathway could cause the activation of NF-κB in cancer.
The NF-κB family of transcription factors includes five different proteins: p105/p50, p100/p52, p65 (RelA), c-Rel, and RelB, which are inactivated in the cytoplasm upon binding to IκB proteins (109). Two signals are differentiated: canonical and non-canonical.
LPS and proinflammatory cytokines, such as IL-1, Toll-like receptors (TLRs) and tumor necrosis factor alpha (TNF-α), activate the NF-κB signaling pathway. The recruitment of a receptor proximal adaptor protein facilitates the phosphorylation and activation of IκB kinase (IKK) protein complex, which subsequently initiates the phosphorylation of IκB proteins. IκB proteins are degraded, allowing NF-κB translocation into the nucleus to activate specific target genes. This pathway can activate antiapoptotic factors and cytokines as well as proliferation factors (109).
The non-canonical brand of the NF-κB pathway may activated by different factors such as the receptor activator of NF-κB (RANK) and CD40, leading to the stabilization of NF-κB-inducing kinase (NIK). This molecule activates IKKα dimers, promoting p100 phosphorylation and cleavage into p52. The activated RelB/p52 dimer can be translocated into the nucleus regulating specific target gene transcription (109, 110) (Figure 3).
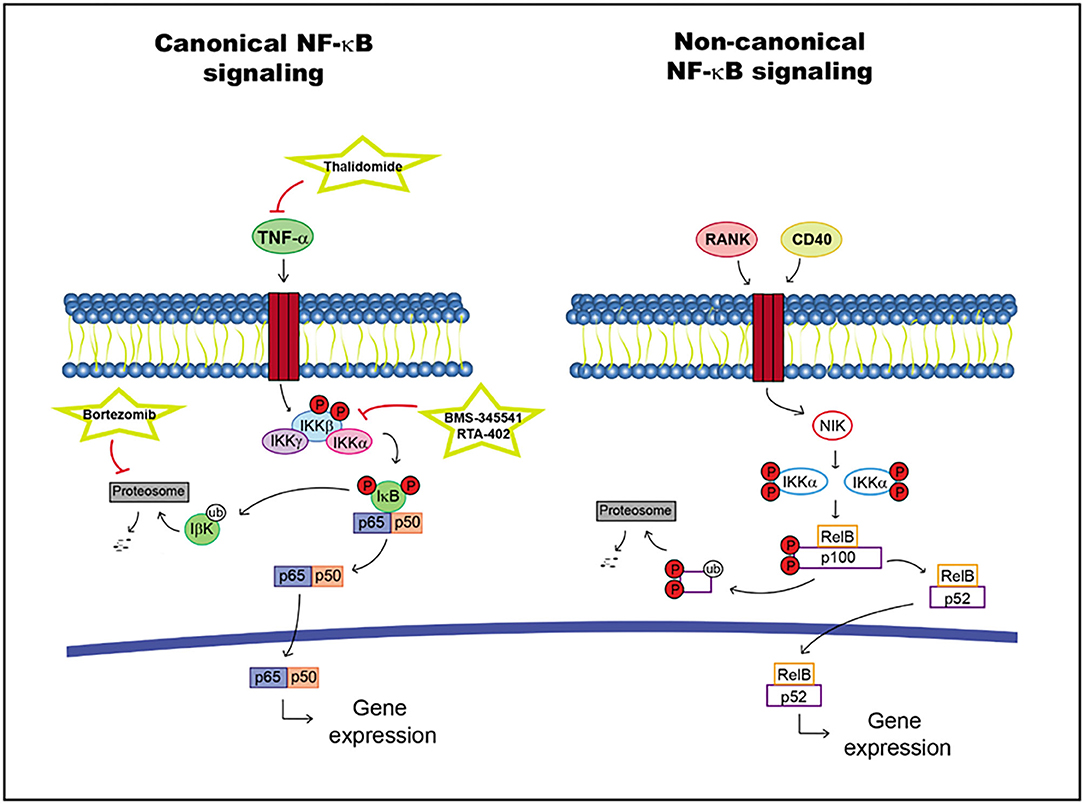
Figure 3. NF-κB pathway including targets and drugs that could be employed to inhibit it. (Left) canonical NF-κB signaling. (Right) non-canonical NF-κB signaling.
Cancer Stem Cells
The link between inflammation and cancer stem cells (CSCs) is found during tumorigenesis and disease progression (111, 112). The involvement of the NF-κB pathway in CSCs was discovered in primary AML samples, in which the DNA in CD34+ cells bind with NF-κB. However, this binding was not observed in normal hematopoietic stem cells (113). In adult murine neurogenesis, TLR2 and TLR4 were found to have opposite functions in adult neural stem/progenitor cell (NPC) proliferation and differentiation. Moreover, the inhibition of the LRPs impacted the self-renewal and the cell fate decision of NPCs (114). A MEC-targeted inducible transgenic inhibitor of NF-κB was developed in mammary oncomice, and it inhibited breast tumor stem cell markers and expanded NANOG and SOX2 expression in vivo and in vitro (115).
Chronic inflammation may be responsible for the accumulation of proinflammatory cytokines, which increase the action of NF-κB. This situation promotes a tumorigenic microenvironment in colon cancer (116). In the basal-like subtype of triple-negative breast cancer, high levels of activated NF-κB pathway components were observed. Inflammatory cytokines or dysregulated NIK expression induced this activation and caused the upregulation of JAG1 expression in normal cancer cells. NOTCH signaling was stimulated in cancer stem cells and induced the expansion of CSC populations (117). The upregulation of NIK caused a decrease in the CSC population, and its reduction led to the results that were opposite those found for breast cancer (118). In addition, mammary stem and progenitor cells were expanded by constitutive RANK signaling, and the overexpression of this activator increased tumorigenesis properties (119).
IKKα and its activator, NF-κB-inducing kinase, were critical for the expansion of tumor-initiating cells (TICs). IKKα is translocated into the nucleus, where it phosphorylates the cyclin-dependent kinase (CDK) inhibitor p27/Kip1 and promotes its nuclear export or exclusion (120).
Therapeutic Targets and Drugs
To determine the efficacy of cancer treatment through the inhibition of the NF-κB pathway, various problems have been found: low efficacy of NF-κB inhibitors in addition to lymphoma and leukemia (121), immunosuppression after long-term systemic administration of these drugs (122) and the appearance of short-term drug resistance (123, 124). However, NF-κB inhibitors can be useful in combination with other chemotherapies, as most anticancer agents can activate this pathway, protecting cancer cells from apoptosis (125).
TNF-α is a target that can be employed to inhibit the NF-kB pathway. Drugs such as thalidomide and its derivatives act in this way (126, 127). However, thalidomide does not show good results in combination with gemcitabine or irinotecan chemotherapy, and its use increases the risk of thrombotic events (128).
On the other hand, bortezomib is an innovative drug with remarkable preclinical and clinical antitumor activity in different types of cancer. The mechanism of action consists of proteasome inhibition, which causes inactivation of the canonical NF-κB pathway (129, 130). Additionally, the prolonged inhibition of this pathway accelerates chemical lung carcinogenesis by perpetuating carcinogen-induced inflammation (131). Currently, many phase III clinical trials are in the recruitment stage for patients with myeloma, lymphoma or acute leukemia who will be treated with bortezomib in combination with other anticancer agents, and the results will be compared to those of conventional treatments.
There is another type of drug that inhibits IKKβ and IκB protein, BMS-345541, which causes the reduction of stemness, self-renewal and migration capacity in lung cancer (132). Another drug in this group, bardoxolone methyl (RTA-402), is a triterpenoid and antioxidant inflammation modulator that inhibits IKKβ and JAK1. Phase I studies have been conducted for various types of cancer: pancreatic adenocarcinoma (133), solid tumors and lymphomas (134). Additionally, IMD-0354 and KRT1853 are IKKβ inhibitors and block IκBα phosphorylation, inactivating its translocation into the nucleus and the activation of NF-κB (135). In lung cancer, it was discovered that these drugs can be potential anticancer agents through suppression of cancer cell invasion, proliferation, and survival (136). In addition, PS1145 reduced the growth of tumorigenic prostate cancer (137) and nasopharyngeal carcinoma (PNC) cell lines (138). Moreover, in the most recent study, it was observed that NF-κBp65 and KLF4 upregulation was involved in drug resistance (138).
These drug groups only partially inhibit NF-κB signaling because IKKα can also activate this pathway. Therefore, inhibitors of IKKα and IKKβ need to be found to generate better effects (139) (Table 1).
Notch Pathway
The Notch pathway is an evolutionarily conserved signaling route involved in a variety of developmental and homeostatic processes, such as proliferation, stem cell maintenance, cell fate specification, differentiation, or angiogenesis, despite the apparent simplicity of its signaling network. The effects of activating this signaling pathway are very diverse, depending on the signal dose and cell context (140–144). Its deregulation contributes to a wide range of disorders and diseases, such as congenital afflictions, viral infections, and/or different types of cancer (142).
The Notch signaling pathway is composed of Notch receptors and Notch ligands, as well as different proteins that serve as posttranslational modifiers, but there is not an amplification cascade induced by different proteins in well-known signaling pathways (142, 145).
There are four Notch receptors encoded by different genes: Notch1–4. The general structure of Notch receptors consists of a single-pass transmembrane protein composed of different protein modules, with some differences: Notch3 and 4 have a shorter extracellular domain and lack the intracellular transcription-activating region, and Notch4 lacks the Notch cytokine response domain. Notch1 and Notch2 are expressed in many tissues during development and in adults, whereas Notch3 is expressed mostly in vascular smooth muscle and pericytes, and Notch4 is expressed in the endothelium (142, 145–149).
There are four Notch ligands in humans: Dll1 and Dll4, from the Delta family of ligands, and Jag1 and Jag2, from the Serrate family. There is a Dll3 ligand, but it is not able to activate Notch receptors. These ligands are also single-pass transmembrane proteins. The expression pattern of these ligands is less established than that of the Notch receptor ligands (142, 145, 150, 151).
Notch receptor activation begins with the binding of a Notch ligand expressed in a neighboring cell upon the release of the Notch receptor negative regulatory region (NRR). The Notch ligand, which has been previously ubiquitinated in the cytoplasmic tail by E3 ligases Neuralized or Mind Bomb to trigger endocytosis, induces a conformational change in the receptor, exposing cleavage sites for ADAM metalloproteases to access to cleave the extracellular receptor domain. Then, the transmembrane domain is cleaved by γ-secretase, releasing the Notch intracellular domain (NICD). The NICD is translocated to the nucleus and, together with the DNA-binding factor RBPJ and coactivators in the Mastermind-like (MAML) family, it forms the Notch transcription complex (NTC). NTC binds to Notch regulatory elements (NREs), and transcriptional coregulators are recruited, activating the transcription of Notch target genes, such as HES and HEY (3, 142, 145, 152, 153) (Figure 4).
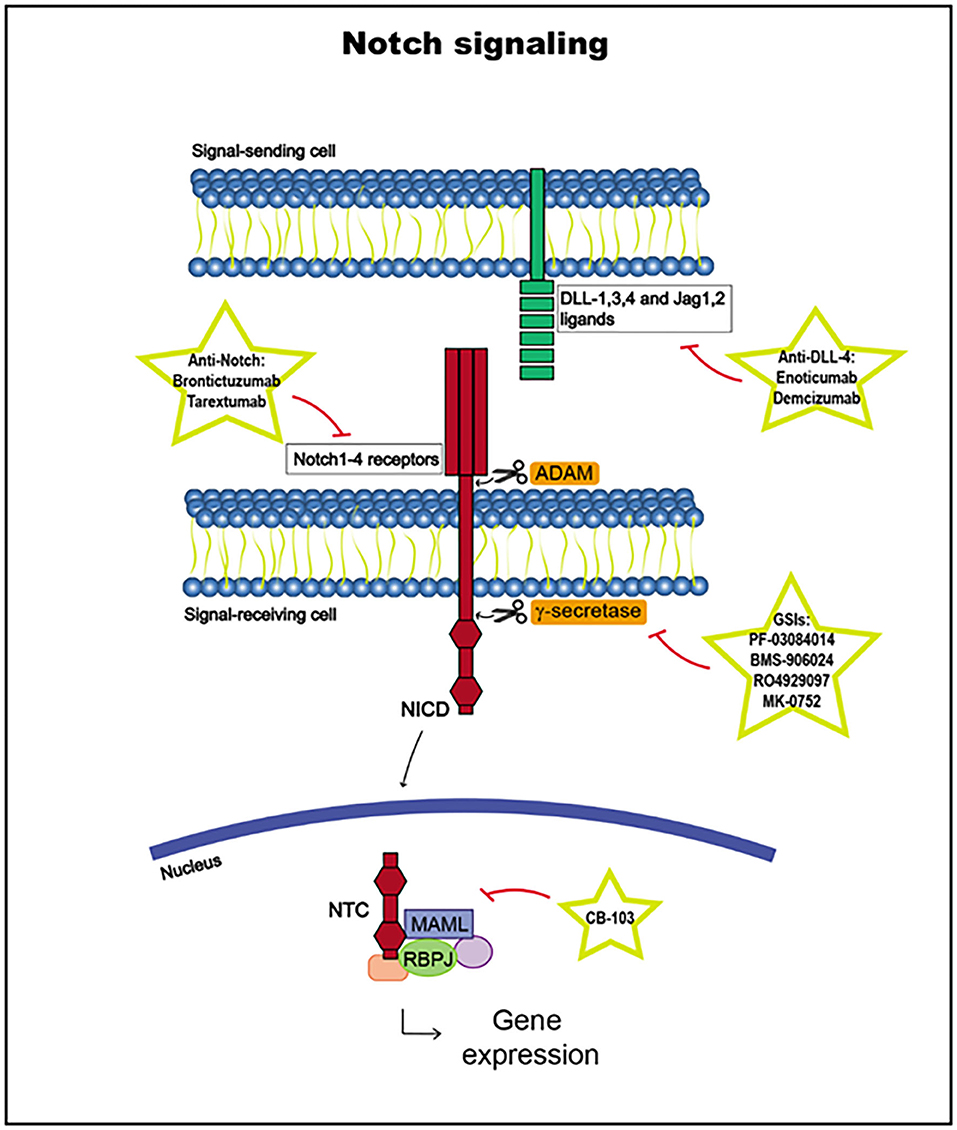
Figure 4. Notch signaling pathway, including targets and drugs that could be employed to inhibit it.
The Notch signaling pathway is highly regulated, and its target genes vary according to different epigenetic contexts, such as different cell subtypes, changing the cellular outcomes. Indeed, NICD function is influenced by other signaling routes and undergoes many posttranslational modifications at different sites, with a variety of consequences, which have not yet been fully elucidated. Notch receptor expression is also closely regulated in normal cells. Notch1 upregulation is crucial in the early stages of T cell development, whereas its downregulation in later stages is important to prevent the transformation in this lineage. Furthermore, there are other levels of regulation, such as inhibitory feedback loops or RBPJ binding to transcriptional repressors, explaining why deregulation of this pathway can lead to different pathologies, such as cancer development (142, 145, 153, 154).
Cancer Stem Cells
Loss of balance in the Notch signaling pathway can lead to tumor formation. Indeed, Notch has been implicated in all of the hallmarks of cancers, including cell growth and survival, the EMT, angiogenesis, and/or metastasis. The most frequent alterations have been found in Notch receptor genes. Notch might act as an oncogene or a tumor suppressor, depending on the context and/or the tumor type (140, 142, 145, 149, 155). Interestingly, in some cancers, such as head and neck squamous cell carcinoma (HNSCC), Notch seems to have a bimodal role, as it might function as an oncogene or a tumor suppressor gene (149).
The Notch pathway can be deregulated by mutational activation or inactivation, overexpression, posttranslational modifications or epigenetic alterations (156). In T cell acute lymphoblastic leukemia, translocation 7; 9 produces a fusion gene consisting of the end of Notch1 and enhancer elements or the TCRβ gene, causing a the removal of the NRR receptor activation inhibitor, and ligand-independent activation of the Notch pathway. Other tumors are characterized by mutations in the Notch PEST domain (C-terminal domain), such as B cell tumors (chronic lymphocytic leukemia, splenic marginal zone lymphoma, etc.) or basal-like breast cancer (142, 157). In these cases, among others, such as cancer of the pancreas, prostate, and lung, hepatocellular cancer, esophageal tumors, and HNSCC (149), Notch functions as an oncogene. On the other hand, several tumors present with mutations in the Notch N-terminal domain, producing a loss of function, such as squamous cell carcinomas (skin, head and neck, esophagus and lung) and small cell lung cancers, urothelial carcinomas, and low-grade gliomas; in these cases, Notch acts as a tumor suppressor (142, 149).
Notch has also been associated with stemness maintenance, important in adult organisms and in pathologies such as cancer. In several cell types, Notch activation seems to maintain or promote expansion of stem cell pools, especially in solid tumors such as glioblastoma, hepatocellular carcinoma, ovarian cancer, breast cancer, and HNSCC. Notch inhibitors can decrease stemness marker expression and sensitize tumor cells to chemo- and radiotherapy (142, 149, 153, 155, 157–160).
Notch signaling mediates the interactions between cancer stem cells and tumor microenvironment cells (endothelial, immune, and mesenchymal cells). The Notch pathway is activated in tumor cells by ligands localized in the vascular niche. Specifically, Jag1 is the main ligand that plays this role. For example, in B cell lymphomas, this interaction induces aggressive behavior and resistance to chemotherapy. In other tumors, Jag1 promotes cancer stem cell self-renewal, proliferation and stemness maintenance. Jag1 can eventually activate the transcription of several genes, such as Klf4 in head and neck cancer cell lines, inducing a stem cell phenotype and chemotherapy resistance (145).
Therapeutic Targets and Drugs
Among the signaling pathways that are activated in cancer cells, the Notch signaling pathway is among the most upregulated, and it is implicated in cancer metastasis, angiogenesis and CSC self-renewal, making it is an important target in cancer therapy (144).
There are two groups of inhibitors that have been developed to target the Notch signaling pathway: γ-secretase inhibitors (GSIs) and monoclonal antibodies against Notch ligand-receptor interactions (mAbs). These therapeutic strategies can be effective in combination with conventional therapies to treat cancer patients with promising results. Furthermore, GSIs and mAbs have been demonstrated to be useful as single agents in the early stage of several tumors, such as thyroid, lung, intracranial and colorectal cancers, and sarcomas. However, Notch signaling is also crucial for normal tissue homeostasis; therefore, its inhibition can induce serious side effects, especially GSIs, such as gastrointestinal toxicity, diarrhea, hepatotoxicity, and nephrotoxicity (3, 140, 144, 149, 161). For this reason, these inhibitors are dose limiting and are administered intermittently (144).
γ-Secretase Inhibitors (GSIs)
γ-Secretase is the enzyme critical for releasing the Notch intracellular domain (NICD) and activating the signaling pathway. Therefore, this enzyme may offer promising results as an inhibiter of the Notch pathway. GSIs constituted the first class of inhibitors developed to be used in cancer patients, and more than 100 GSIs have been developed to date (162).
There are many GSIs currently in clinical trials in several cancer types, as single agents or in combination. For example, PF-03084014 is a small reversible and non-competitive GSI that is being studied alone or in combination in several cancer subtypes (163, 164), but currently, its clinical development has focused on desmoid tumors (phase II/III) (165). BMS-906024 is being studied in relapsed T-ALL and T cell lymphoblastic lymphoma (phase I) (166), as well as adenoid cystic adenocarcinoma (phase II), with promising results (3). Another GSI, MK-0752, is a non-competitive oral inhibitor that is being tested for use against recurrent central nervous system tumors, advanced breast cancer and T-ALL (phase I) and in combination with docetaxel or gemcitabine for use in locally advanced or metastatic breast and pancreatic cancer, respectively (phase I/II) (3, 144, 167, 168). RO4929097 has been studied as a single agent in renal cell carcinoma, metastatic pancreatic cancer, and non-small cell lung cancer relapses (phase II) and in combination with temsirolimus, cediranib, gemcitabine, or bevacizumab in different solid tumors (phase I) (3, 144, 169–171).
All these GSIs have been demonstrated to promote the inhibition of the Notch pathway, tumor growth, angiogenesis, stem cell marker expression, and metastasis, etc. (3, 140, 144). However, some of them, such as MK-0752 or RO4929097, are not being developed for use in active clinical trials because of the limited efficacy observed (3) (Table 1).
Monoclonal Antibodies (mAbs)
Several monoclonal antibodies have been developed to inhibit Notch signaling as in inhibitors of ligands (DLL-4) and receptors (Notch1–3). They impair ligand-receptor interactions or extracellular domain conformational changes, which are crucial for exposing Notch receptor cleavage sites (140, 144).
DLL-4 is one of the ligands that binds Notch receptors and is involved in growth control, stem cell renewal and development. The deletion of its gene has lethal consequences in the vasculature, and its overexpression has been found in cancer cells and tumor vasculature. Furthermore, DLL-4 inhibition impairs the formation of functional capillaries, leading to aberrant angiogenesis and promoting direct effects that inhibit Notch signaling in tumor cells (172–175).
Enoticumab is a humanized mAb against DLL-4 that seems to have a reasonable safety profile and efficacy in advanced solid cancers, such as ovarian carcinoma (phase I) but is no longer in clinical development. Demcizumab is a DLL-4 antibody that is being studied with gemcitabine in pancreatic cancer, with FOLFIRI in colorectal cancer (phase I) and with carboplatin and pemetrexed in non-squamous NSCLC (phase II), but it did not improve efficacy; therefore, it not in clinical development (3, 140, 144).
Monoclonal antibodies against Notch receptors have also been studied. There are two groups of monoclonal antibodies: those directed to the NRR domain, preventing the conformational change necessary to activate Notch signaling, and those targeted to the EGF-repeat region in Notch receptors to impede the ligand-receptor interaction. Both groups promote substantial downregulation of Notch1 signaling (161, 176).
Tarextumab is a humanized monoclonal antibody against Notch2 and 3 that inhibits Notch signaling as a single agent or in combination with chemotherapeutic agents in epithelial cancers, such as breast, small cell lung, ovarian, and pancreatic cancers (phase I/II). It can inhibit tumor cell proliferation, reduce the number of CSCs and prolong the time before tumors recur after chemotherapy. However, the clinical trials did not show survival improvement; therefore, clinical development has been discontinued (3, 140, 144). Brontictuzumab is an anti-Notch1 mAb that is being studied in hematological malignances and advanced solid tumors (phase I) (177, 178) (Table 1).
Other Therapeutic Targets
The DLL-3 Notch ligand is highly expressed in neuroendocrine carcinomas, such as most small cell lung cancers. DLL-3 can promote tumorigenesis through Notch signaling pathway inhibition (in contrast to DLL-4). Therefore, DLL-3 is being studied as a therapeutic target in these cancers. Some other approaches include co-administering AMG 757, a bispecific T cell engager; AMG 119, a chimeric antigen receptor (CAR) T cell (phase I); or as an anti-DLL3 antibody-drug conjugate (ADC), rovalpituzumab tesirine (phase II) (3, 179, 180).
Another strategy involves inhibiting the formation of the Notch transcriptional complex, which acts downstream of abnormal Notch receptor activation. CB-103 is an oral pan-Notch inhibitor that belongs to a novel class of small molecules that target the Notch transcriptional complex, inhibiting the expression of target genes. CB-103 is being studied in phase I/IIa clinical trials for patients with advanced-stage solid tumors or hematological malignancies (181) (Table 1).
Sonic Hedgehog Pathway
The Sonic Hedgehog pathway plays a role in embryogenesis and brain development. In adults, it is usually inhibited, although it participates in the maintenance of somatic stem cells and pluripotent cells of many organs, tissue repair, and regeneration of several epithelial cells (182–186). Furthermore, there is evidence that the Hedgehog pathway (Hh) is deregulated in various cancer types, such as pancreatic, gastric, prostate, and esophageal cancer (187–189). The activation of the Hh signaling pathway may have a variety of effects involving cell proliferation, cell fate determination, the epithelial-to-mesenchymal transition, and cell motility or adhesion. Therefore, the deregulated activation of this pathway may lead to the development of tumors or resistance to treatment (190).
There are two different mechanisms by which Hh signaling activated: ligand-receptor binding, known as the canonical pathway, or as a consequence of the activation of another downstream member of the signaling pathway, known as the non-canonical pathway (191).
Canonical
The main receptor in the Hh signaling pathway is Patched (Ptch1), which localized to the base of the primary cilia (PCs), structures that protrude from the cell membrane to sense a variety of stimuli (192). The main ligand that binds Ptch1 is Sonic Hedgehog (SHh), but there are two other ligands: Indian Hedgehog (IHh) and Desert Hedgehog (DHh).
In the absence of Hh binding, Ptch1 does not translocate with the transmembrane protein Smoothened (SMO) to the PCs. When Hh binds to Ptch1, Ptch1 is internalized and degraded, causing the accumulation of SMO in the PC, and as a consequence, the signaling cascade is initiated through a complex formed by kinesin protein (Kif7), suppressor of fused (SUFU) and full-length glioma-associated oncogene (GliFL). This pathway eventually leads to the translocation of the Gli transcription factor to the nucleus.
There are three Gli proteins: Gli1, Gli2, and Gli3. Gli1 is a transcriptional activator, whereas Gli2 and Gli3 could act as activators or repressors. However, Gli2 acts mainly as an activator, and Gli3 acts as a repressor.
SUFU usually inhibits Gli transcription factor translocation to the nucleus by direct binding. SUFU activates the phosphorylation by glycogen synthase kinase 3 beta (GSK3β), casein kinase I (CK1), and protein kinase A (PKA) and promotes the recruitment of β-transducin repeat-containing protein (β-TrCP), which causes the transformation of Gli2 and Gli3 into the Gli repressors Gli2R and Gli3R, which are translocated to the nucleus to inhibit the transcription of Hh pathway target genes (186, 190, 193) (Figure 5).
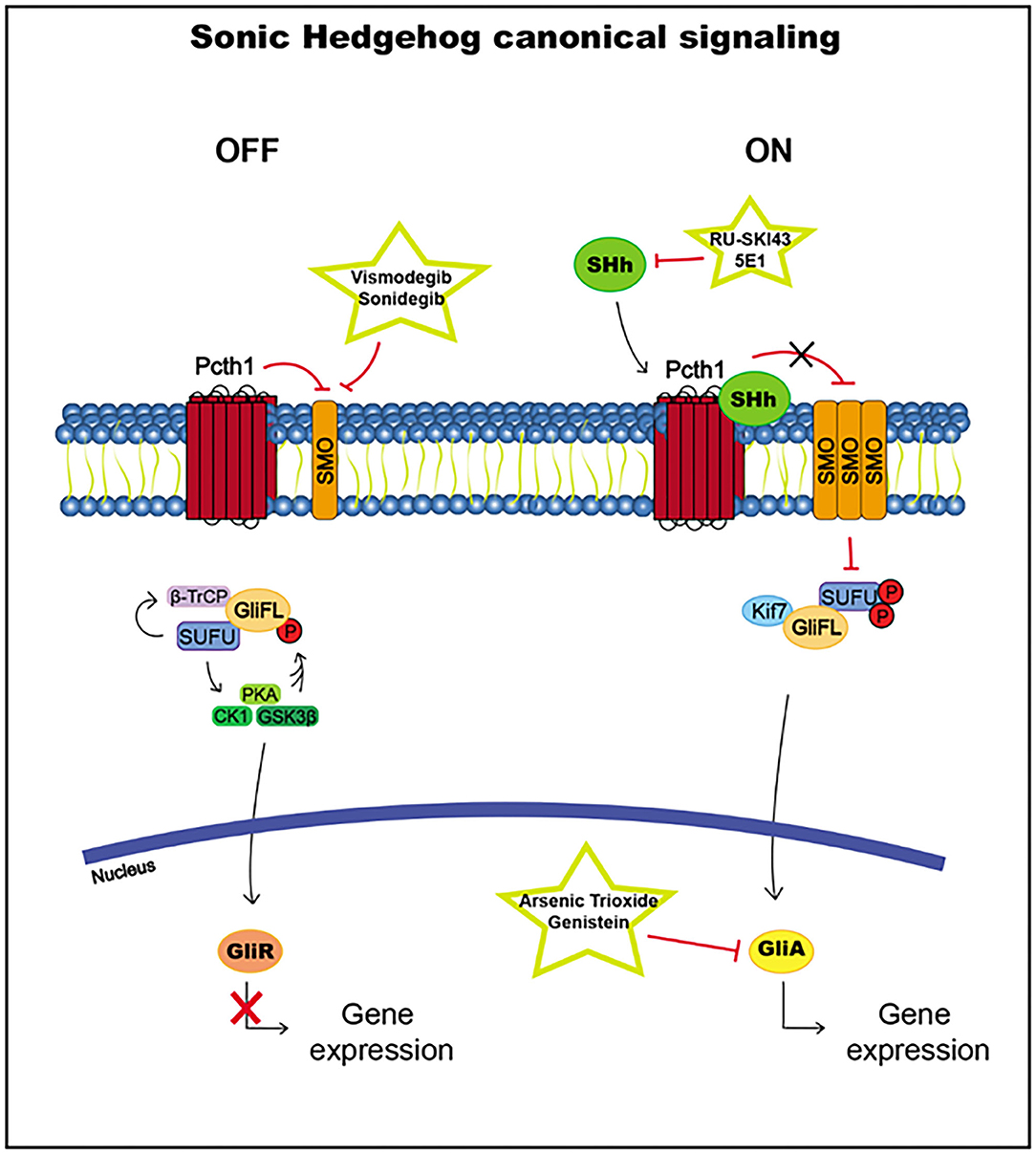
Figure 5. Sonic Hedgehog canonical signaling, including targets and drugs that could be employed to inhibit it. (Left) inactive signaling pathway. (Right) active signaling pathway.
When the signaling pathway is activated by the Hh ligand, the accumulation of SMO leads to the hyperphosphorylation of SUFU, releasing Gli proteins, which translocate to the nucleus and activate the transcription of the target genes Ptch1 and Gli1 (186, 193) (Figure 5).
Non-canonical
The non-canonical signaling pathway is independent of Gli proteins but is not yet fully understood. In the type I non-canonical pathway, SMO appears to be critical for modulating Ca2+ and the actin cytoskeleton, whereas SMO does not participate in the type II non-canonical signaling pathway, which may enable cell proliferation and survival (194).
Cancer Stem Cells
The cancer stem cell (CSC) model explains tumor heterogeneity through the presence of a small group of cells with unlimited self-renewal capacity and the potential to regenerate all cell types in the entire tumor. They seem to be critical for therapy resistance and relapses. The Hh signaling pathway, as well as other signaling pathways, has been implicated in the maintenance of CSCs (190, 195, 196).
The mechanisms by which the Hh signaling pathway can be activated are mutations in Hh signaling members or the deregulation of Hh ligand release. Furthermore, the Hh signaling pathway may participate in the formation of CSCs.
A variety of members of the Hh pathway have been found to be affected by mutations in human cancers, such as the inactivating mutations of Patch1 or SUFU or the activating alterations of SMO, Gli1, and Gli2, which lead to signaling pathway activation independently of ligand binding. Patch1 loss of heterozygosity is usually found in Gorlin syndrome or nervoid basal cell carcinoma syndrome. Furthermore, alterations to the Hh pathway in combination with tumor suppressor mutations are able to generate other sporadic tumors, such as skin, medulloblastoma, gastric, and rhabdomyosarcoma tumors (190, 197–199).
In other cases, the Hh signaling pathway is stimulated by a deregulation in ligand release, which could be autocrine (from the tumor cell to itself, as it seems to occur in gliomas), paracrine (from the tumor cell to the stroma or from the stroma to the tumor cell, as it may happen in multiple myeloma, which has been studied to a lesser extent) or both, as it might take place in esophageal or gastric cancers (182, 190, 195, 196, 199). It is thought that tumor cells can release ligands that may stimulate the Hh signaling pathway in stromal cells via paracrine signaling and then promote a supportive microenvironment for the tumor (190).
Hh signaling pathways have been demonstrated to participate in the formation and maintenance of cancer stem cells in a variety of tumors, such as hematological malignancies and gastric, pancreatic, prostate, and lung cancers. The Hh signaling pathway is able to activate the transcription of key genes that contribute to the stem cell phenotype, such as Nanog, Oct4, Sox2, and Bmi1. Targeting this pathway in CSCs may be a promising strategy to reduce tumor growth, relapse, and metastasis (200–202).
Therapeutic Targets and Drugs
Tumors that present with mutations in signaling components may be ameliorated by some Hh pathway inhibitors, but the efficacy of these treatments is dependent on the level of alterations in the signaling pathway. In addition, ligand-dependent tumors may be treated by inhibitors of the Hh signaling cascade that are directed against any of its signaling components, regardless of the level in the route (190).
Among all proteins that may take part in the signaling cascade, SMO and Gli transcription factors are the main targets on which current research is focused. Although there are currently many SMO inhibitors, spontaneous mutations can arise as a consequence of the treatment, which may cause drug resistance (203).
SMO Inhibitors
Cyclopamine, a natural alkaloid from Veratrum californicum, was the first SMO inhibitor, but the significant side effects in mice did not allow it to be used in humans. Vismodegib, a second-generation cyclopamine derivative approved by the FDA in 2012, is being used in metastatic basal cell carcinoma (BCC) treatment and in recurrent locally advanced BCC, which are not candidates for surgery or radiotherapy. More than 85% of BCC patients have constitutive activation of the SHH pathway, most of which are due to a mutation in PCHT1. Vismodegib binds to SMO and inhibits its function, but continuous exposure can induce mutations in SMO, promoting drug resistance. Furthermore, vismodegib is being studied in many clinical trials in a variety of human cancers, such as breast cancer (204–206).
Sonidegib is another SMO antagonist, approved in 2015 by the FDA, that is used in the treatment of patients with locally advanced BCC that recurred disease after surgery or radiotherapy or patients who are not able to receive surgery or radiation treatments. Sonidegib, similar to vismodegib, binds SMO in the “drug binding pocket,” and mutations at this site lead to resistance (205, 206). Sonidegib and Vismodegib are teratogen, as other Sonic Hedgehog pathway targeting drugs, due to the role of this signaling route in embryogenesis (206).
There are other drugs that are currently in clinical trials for different cancer types, such as Saridegib (phase II), BMS-833923 (phase II), glasdegib (phase II), taladegib (phase II), or TAK-441 (phase I), and they have shown promising results in preclinical models (205, 206). TAK-441 has shown efficacy in cells that have developed adaptive mutations in SMO; therefore, it may be relevant for patients with drug resistance (206) (Table 1).
GLI Inhibitors
Arsenic trioxide, a Gli inhibitor approved by the FDA for the treatment of acute promyelocytic leukemia, binds directly to Gli1 and Gli2, among other actions, causing the inhibition of their activity (205, 206). It has been shown to affect the viability of cancer stem cells in pancreatic and prostate cancer (in the cancer-initiating cells). This drug is being studied in phase I–IV clinical trials for solid tumors and hematological malignancies. Arsenic trioxide could imply some cardiac side effects such as QT interval elongation or tachycardia, as well as circulatory, gastrointestinal and respiratory disorders (205–207).
Genistein (phase I and II), an isoflavone isolated from Genista tinctoria, has been shown to inhibit Gli1, causing the growth suppression of different tumors and the cancer stem niche (206, 208) (Table 1).
SHh Inhibitors
Inhibitors of SHh, which is the most important ligand, have not yet been approved for use in the clinic, but they have been shown to effectively inhibit the SHh pathway in mice. Some of these are SHh monoclonal antibody 5E1, or RU-SKI43, which inhibits the SHHat enzyme, critical for catalyzing the binding of palmitate to SHh, one of the final steps in SHh synthesis (205, 206) (Table 1).
Other Therapeutic Targets
A truncated isoform of the Gli1 transcription factor, which indicates a gain of function, has been discovered: tGli1. This isoform has been detected exclusively in tumor cells, not in normal cells, and promotes tumorigenesis to a greater extent than does Gli1, which makes it a promising drug target. There is no drug directed specifically against tGli1, but it may be possible to target the genes activated by tGli1, such as CD24, VEGF-A, VEGFR2, or HPA1, and inhibit their expression. A phase I clinical trial is testing a CD24 monoclonal antibody. Furthermore, some heparanase (HPA1) inhibitors, such as PI-88 or PG545, are being studied as antiangiogenic anticancer drugs (PI-88 in phase III and PG545 in phase I). Finally, there are several antiangiogenic drugs approved by the FDA and directed against VEGF-A and VEGFR, such as bevacizumab, Ziv-aflibercept, and sorafenib (205).
Hippo Pathway
The highly evolutionarily conserved Hippo signaling pathway regulates biological processes, such as survival, differentiation, cellular proliferation, fate determination, organ size, or tissue homeostasis (209). The core pathway consists of a kinase cassette that is composed of MST1/2 and LATS1/2 (210). In addition to MST1/2, MAP4K, and TAOK also directly phosphorylate LATS1/2 (211–214). NF2 is critical for pathway activation through the phosphorylation of MST1/2. The major target of the Hippo pathway is YAP and its paralog transcriptional coactivator TAZ. Phosphorylation of YAP and TAZ leads to their sequestration in the cytoplasm by 14-3-3 proteins and ubiquitination-dependent proteosomal degradation (215). In the nucleus, YAP/TAZ it can bind and regulate a family of sequence-specific transcription factors called TEA DNA-binding proteins (TEAD1–4) that modulate genes such as CTGF, CRY61, BIRC5, ANKRD1, and AXL, involved in proliferation and survival. In addition to TEADs, the YAP/TAZ complex also cooperates with RUNX1 and 2, TBX5, and SMADs, among others (216–219) (Figure 6). High TEAD expression levels have been correlated with poor clinical outcomes, and therefore, it can serve as a prognostic marker in many solid tumor types (220–231).
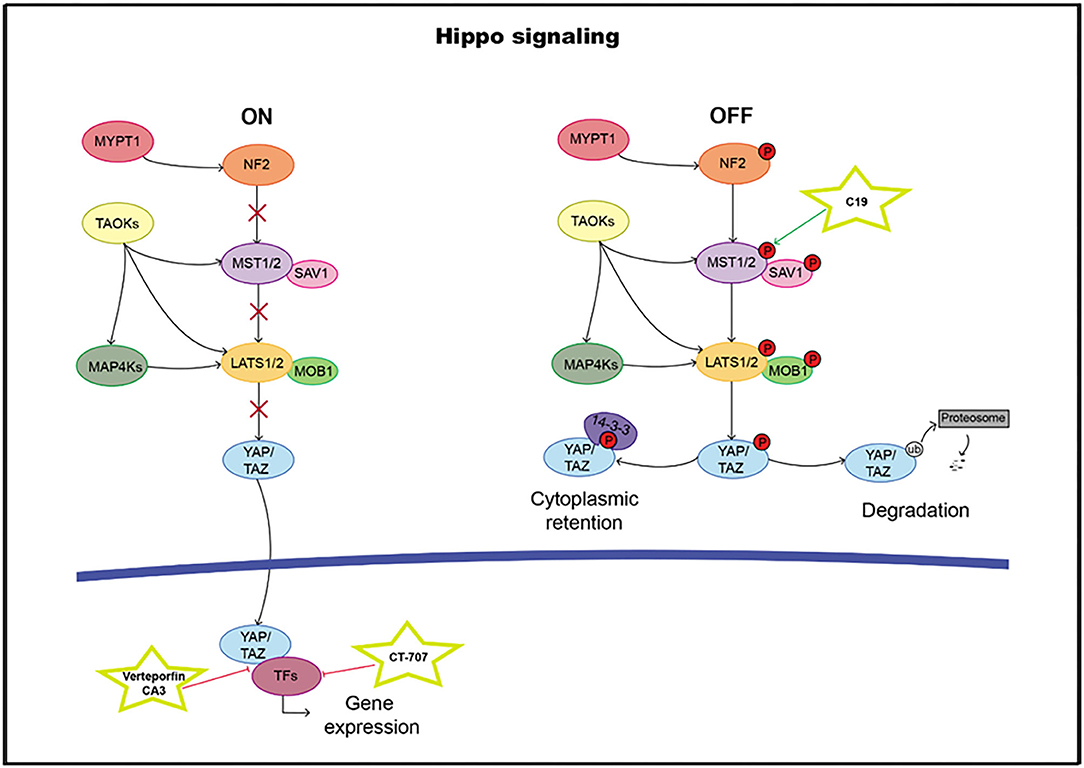
Figure 6. Hippo pathway including targets and drugs that could be employed to inhibit it. (Left) active signaling pathway. (Right) inactive signaling pathway.
Cancer Stem Cells
Hippo pathway effectors activated by YAP/TAZ have been shown to induce cancer stem cell (CSC) properties in a wide range of human cancers, including osteosarcoma, glioblastoma, and chemoresistant breast cancer (232, 233). Moreover, breast cancer tissues with a high content of CSCs show a gene expression profile that overlaps with YAP/TAZ-induced gene expression, and breast CSCs with a CD44+/CD24– phenotype have a relatively high expression of TAZ (234).
YAP/TAZ are known to promote other properties of CSCs, such as the epithelial-to-mesenchymal transition (EMT) and metastasis, via activation of TEAD transcription factors in different tumors, including breast cancer and melanoma (235–241). In the context of these tumors, TEAD activation leads to the disruption of cell–cell junctions, mesenchymal gene expression, increased cell migration, anoikis resistance and cell invasion. TEAD specifically regulates genes such as ZEB1, ZEB2, DNp63, and Slug, which induce an increase in the progression and metastatic potential of tumors such as squamous cell carcinoma, breast cancer, and small-cell lung carcinoma (NSCLC) (242–244). Interestingly, YAP expression was critical for the progression of various KRAS-driven cancers, and YAP/KRAS converged on FOS to promote the EMT, which contributed to oncogenic KRAS oncogenic addiction (245).
It has also been proven that the activation of YAP/TAZ confers resistance to chemotherapy in cancer cells, in part because of the CSC characteristics acquired by the cells. YAP/TAZ has been linked to castration resistance in prostate cancer and paclitaxel and doxorubicin resistance in breast cancer (234, 246, 247). Moreover, different studies suggest that Hippo pathway activation promotes cancer cell survival in the presence of DNA-damaging agents such as UV exposure, radiation, cisplatin, Taxol, and fluorouracil (5-FU) in a wide number of tumor types (248–251).
YAP/TAZ, in conjunction with their target genes of secreted ligands, also promote resistance to targeted therapies, such as the drugs targeting RAF and MEK and BRAF inhibitors and receptor tyrosine inhibitors, such as gefitinib (252–258).
The relevance of the Hippo pathway in cancer and, more specifically, in the biology of CSCs has already been demonstrated in multiple publications. Therefore, the Hippo pathway is currently being studied as an interesting target for use in developing targeted therapies for different types of tumors. There are some approved compounds able to regulate this pathway; however, they have shown to have some bioavailability or toxicity problems. Consequently, extensive effort is being made in the research of new drugs to address these issues.
Therapeutic Targets and Drugs
Regarding therapeutic opportunities for the Hippo pathway, YAP inhibition has been shown to produce promising therapeutic effects in different types of tumors, such as NSCLC, breast cancer and colorectal cancer (259–262). Additionally, the regulation of upstream or downstream molecules of this pathway also show antitumoral effects. However, the inhibition of the upstream regulators of YAP might serve to increase transcriptional activity and thus be counterproductive. This result makes the inhibition of the YAP/TAZ interaction and the suppression of their binding to their targets the most appealing strategies.
Verteporfin, an FDA-approved drug, was identified to inhibit the interaction between YAP and TEAD (263, 264). Therefore, it can decrease the expression of Hippo target genes; however, it has shown low solubility and stability, unfavorable pharmacokinetics, and rapid clearance; complicating its clinical use. Moreover, this compound lacks tumor specificity and have serious Hippo-independent effects, and thus, may originate adverse effects in healthy tissues (265–267). In addition, its mode of inhibition and mechanism of interaction with YAP remain unknown. Considering all, verteporfin might not be the most promising drug, nevertheless, some studies have shown that the loading of verteporfin into microparticles improves its specificity and pharmacokinetics, making it more suitable as a treatment (267–269). Similarly, the CA3 compound has been reported as a modulator of YAP/TEAD transcriptional activity through the inhibition of YAP, but it has drawbacks similar to those of verteporfin (270, 271).
Another possibility for YAP/TEAD inhibition is the lipid pocket at the core of the TEAD family, which is essential for TEAD folding, stability, and YAP binding (272–274). Some compounds targeting this domain in cell assays, such as flufenamic acid or chloromethyl ketone moieties, have been shown to inhibit cell proliferation and several Hippo pathway responsive genes. However, the underlying mechanism remains unclear because, in some cases, YAP/TAZ binding remains unchanged (275, 276). In recent studies, a cyclic YAP-like peptide has been shown to block the YAP-TEAD interaction through competition with endogenous YAP (277). However, this peptide has not yet been converted into a cellularly active compound. “Super-TDU,” a peptide mimicking VGLL4, has also been reported to compete with YAP binding to TEADs. Its mechanism is based on the binding of vestigial-like proteins (VGLL) to TEADs, and it has led to the reduced growth of gastric cells in vitro and in vivo (278). All these antagonistic compounds offer promising strategies; however, their clinical viability and applicability remain unproven.
Due to YAP druggability problems, it has been proposed to target its posttranscriptional modifications, which are critical for its oncogenic properties. In this context, some already approved drugs, including metformin, statins, dasatinib, pazopanib, and nicotinamide (NAM), and some agents being tested in clinical trials, such as the multikinase inhibitor CT-707, have been reported to decrease YAP activation through the regulation of posttranscriptional modifications (279, 280). These findings suggest that they might be plausible cancer therapies, although many more studies are needed.
Another potential approach to inhibiting the Hippo pathway is through the activation of MST and LATS kinases, which phosphorylate and inactivate YAP (281). The small-molecule compound C19 has shown the ability to activate MST and therefore to suppress melanoma cell growth in a mouse xenograft model (282). Moreover, metformin and statins have been known to activate LATS and inhibit YAP activity (283). However, their applicability as cancer therapeutic agents is still controversial and requires more clinical trials (284, 285).
Finally, the downregulation of Hippo pathway regulators is also possible at the transcriptional level. The combination of some epigenetic modulators, including I-BET151 and panobinostat, has been shown to induce the synergistic downregulation of the AKT and Hippo pathways in melanoma cell lines without binding to the cytoplasmic proteins of these pathways (286). Nevertheless, compounds regulating the epigenome show some drawbacks, for instance, off-target effects due to their lack of specificity, low stability and sustainability, and significant toxicity in normal cells (287).
Altogether, there are significant advances in the field of developing targeted therapies for the Hippo pathway. They could be a priceless weapon in the fight against cancer; nevertheless, we need more research in this area. It is important to better understand the mechanism of action of these compounds and to develop clinical trials to test their secondary effects and their applicability in cancer patients (Table 1).
TLR Pathway
Toll-like receptors (TLRs) belong to the pathogen recognition receptor (PRR) group in the innate immune system. They recognize exogenous ligands from invading pathogens (pathogen-associated molecular patterns or PAMPs) and endogenous ligands released from damaged host cells (damage-associated molecular patterns or DAMPs) (288–290). Ten TLRs are found in both human immune cells (T-cell and B-cell subsets, macrophages, and dendritic cells) and non-immune cells (epithelial cells and fibroblasts). Some of these TLRs are localized in intracellular vesicles, for instance, TLR3, TLR7, TLR8, and TLR9, while others are localized on the cell surface (291). TLRs are type I glycoproteins that share some common structural domains: an extracellular domain containing multiple leucine-rich repeats that enable the recognition of the ligand, a transmembrane region, and a highly conserved intracellular Toll-interleukin 1 (IL-1) receptor domain (TIR) necessary for signal transduction (292–294).
When a ligand binds to the extracellular domain of a TLR, it induces a conformational change in the receptor allowing for its homodimerization, and therefore, the binding of their TIR domains and the recruitment of different intracellular adaptor molecules. These adaptor proteins include Myd88, TRIF/TICAM-1, TIRAP/Mal, TIRP/TRAM, and SRAM (295, 296). All TLRs except TLR3 initiate signaling through MyD88 (the classical inflammatory pathway), forming a Myd88/IRAK1/IRAK4/TRAF6 axis that activates TAK1. This pathway triggers the activation of transcription factors, such as NF-κB, AP-1, and IFN regulatory factors (IRFs) (297–300). On the other hand, TLR3 and TLR4 can induce signaling through TRIF/TIRAM adaptor molecules instead of Myd88, leading to the activation of IRFs (301). All these activated transcription factors are translocated to the nucleus and interact with their target genes, including inflammatory cytokines, chemokines, and type I interferon (IFNs) (Figure 7).
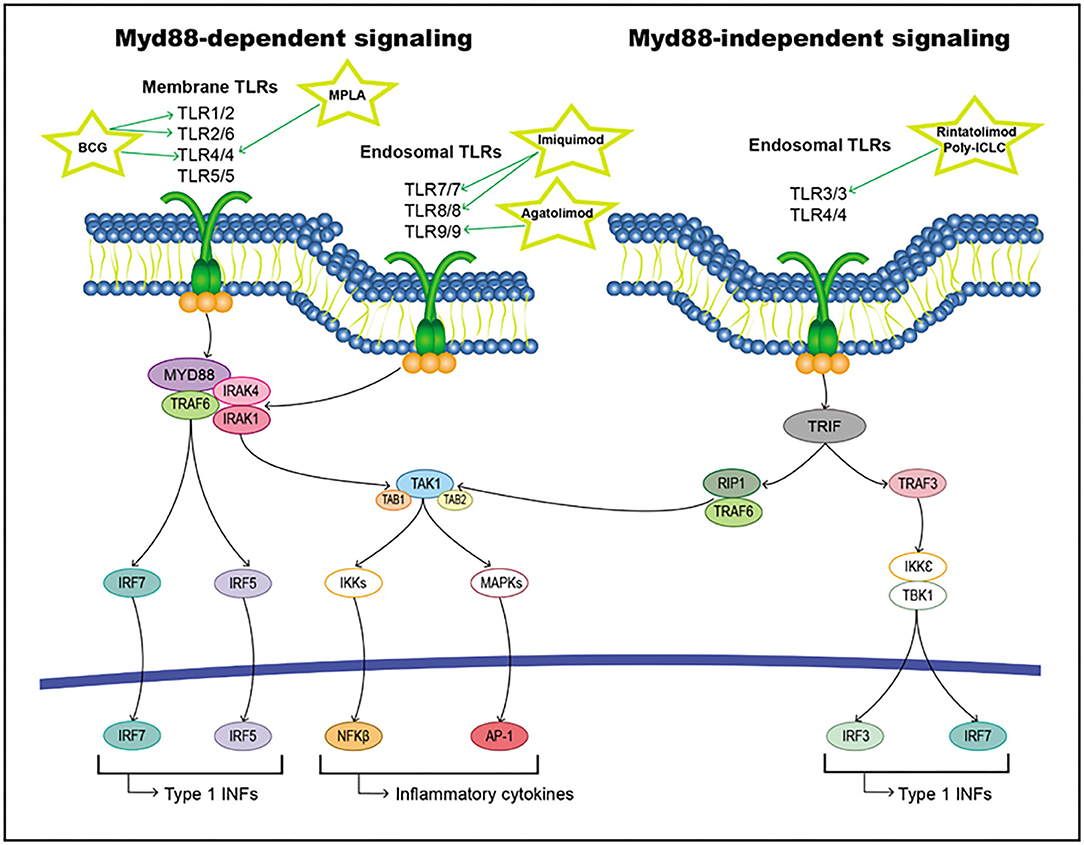
Figure 7. TLRs pathway including targets and drugs that could be employed to inhibit it. (Left) Myd88-dependent signaling. (Right) Myd88-independent signaling.
Alterations in TLR signaling can have both antitumoral and protumoral effects on carcinogenesis and tumor progression. These effects depend on the TLR class, the cell type and the signaling pathway that is triggered in those cells. For instance, TLR agonists have been used as adjuvants in anticancer vaccines to stimulate immune cells to fight the tumor (302, 303). However, TLR expression in some cancer and immune cells is related to the activation of genes related to tumor progression and thus to tumor growth (304). Due to this double action of TLR signaling, it is important to study the role of each type of TLR in cancer individually and to always consider the origin of the tumor being investigated. Additionally, various cells in the tumor and its microenvironment can have different TLR expression patterns, and therefore, they will react differently to TLR modulation (305, 306).
Cancer Stem Cells
Despite their involvement in immunity against tumor invasion, the TLR signaling pathway with cancer stem cell properties have been found in different tumor types. For example, TLR2 activation in epithelial ovarian cancer has been shown to enhance a proinflammatory environment and thus to increase cell self-renewal through the upregulation of stem cell-associated genes (307). Similarly, targeting TLR3 with an agonist in breast cancer cells led to the expression of stem-associated genes, including OCT3/4, NANOG, and SOX2, because of the activation of β-catenin and NF-κB signaling pathways (308). Additionally, in murine models of hepatocellular carcinoma, TLR4 expression was associated with stem-like properties, including the invasion and migration of cells. TLR4 stimulation is suggested to enhance the emergence of stem-like cancer cells through Nanog and STAT3, which cooperate to activate the metastasis regulator TWIST1 (309). Accordingly, there was a strong correlation between TLR4 expression and poor prognosis for patients (310, 311). Moreover, in breast cancer, TLR4 activation has resulted in enhanced stemness and tumorigenicity of cells. It has also been linked to poor prognosis and an increased relapse rate in patients (312). Nevertheless, the downregulation of TLR4 in glioblastoma CSCs has been observed. This result may be considered a mechanism by which these CSCs escape immune surveillance and is thought to be related to an increase in retinoblastoma-binding protein 5 (305).
In the case of TLR7, CD133+ cells in colorectal cancer patients showed increased expression of this marker, which was associated with a poor prognosis (313). Similarly, it has been suggested that TLR9 is able to promote the stem-like prostate cancer cell phenotype through NF-κB and STAT3 upregulation of stem-related genes, including NKX3.1, KLF-4, BMI-1, and COL1A1 (314). It has also been demonstrated that the upregulation of TLR9 in glioma cancer stem-like cells is able to activate STAT3 and thus maintain the quiescent state of tumor-repopulating cells (315).
All this information suggests a feedback loop of inflammation and stemness in tumor cells. The presence of extrinsic stimuli in the tumor microenvironment can activate proinflammatory pathways, for instance, TLR signaling. This activation leads to the upregulation of genes related to stemness and the epithelial-to-mesenchymal transition (EMT), which are able to induce and maintain the dedifferentiation of cells, converting them to CSCs (115, 316–319). On the other hand, CSCs constitutively exhibit deregulation in the expression of NF-kB, increasing the levels of inflammation within the tumor (320). This positive feedback loop might promote malignancy and resistance to treatments. Therefore, targeting TLR signaling may be a promising strategy for reducing CSC expansion and, thus, tumor progression.
Therapeutic Targets and Drugs
Targeting TLRs represents a promising strategy for tumor immunotherapy due to their ability to activate innate immunity and even stimulate adaptative responses for long-lasting defense against tumor antigens. Furthermore, in some cases, the activation of certain TLRs, such as TLR2 or TLR4, can have direct antitumoral effects on cancer cells. These TLRs have been used in different approaches; for example, they have been used as adjuvants for cancer vaccines or in combination with radiotherapy, chemotherapy and other immunotherapies. However, when targeting this pathway, it is important to consider that the same TLR can have different behavior, either protumoral or antitumoral, depending on the tumor type.
Treatments Based on TLR Agonism
There are multiple cancer treatments based on activating the immune response through different TLR receptors. A large number of compounds have been developed and used as monotherapy or in combination with other strategies to treat diverse types of tumors. Some molecules targeting this pathway have already been approved by the Food and Drug Administration (FDA) and are currently being used to treat patients. Many more are being tested in preclinical and clinical trials and might be in the clinic in the near future.
Bacillus Calmette-Guerin (BCG) is an FDA-approved treatment that activates TLR2/4 through its cell wall and TLR9 through its bacterial DNA (321, 322). It is the standard of care for patients with carcinoma in situ or urinary bladder cancer that has not invaded muscle (323). However, it still shows a 50% risk of failure (324) and the difficulty of its manufacturing process and its increasing demand have created a shortage of this treatment (325). Agents similar to BCG and their combination with other therapies are being tested in multiple clinical trials for different tumor types, such as colorectal cancer (326, 327), melanoma (328–330), and small cell lung cancer (331). Other bacteria that activate TLR2, such as Mycobacterium indicus pranii (Immuvac, CADI-05), are also being studied. CADI-05 is an approved treatment for leprosy, and interestingly, it has been shown to reduce myeloma and thymoma tumor size in murine models (332, 333). In addition, monophoryl lipid A (MPLA), currently used as a synthetic adjuvant in vaccines, is a derivative of lipid A and can stimulate TLR4. Some of its analogs are being tested as adjuvants for cancer vaccines, for instance, glucopyranosyl lipid A for skin cancer (334) and AS15 (a combination of MPLA with other immune stimulators) in distinct types of tumors (335, 336). These type of combinations might show a slight increase of mild side effects probability, specially, local injection site reactions (336, 337).
In the case of TLR3, Poly (I:C) is a TLR3 ligand that functions as a potent adjuvant for cancer vaccines (338); however, due to its fast degradation, new alternative agonists are being investigated. Some Poly(I:C) derivatives are promising, for instance, poly-ICLC (Hiltonol®) for solid tumors (339) and rintatolimod (Ampligen®) for fallopian tube, ovarian and brain tumors (NCT03734692 and NCT01312389).
Targeting TLR5 has shown promising effects in murine models, in which flagellin and different nanoparticles have shown antitumoral effects (340–342). A Salmonella flagellin derivative, entolimod (CBLB502), is being tested in clinical trials against advanced local and metastatic malignancies (343). Additionally, it has demonstrated radioprotective effects in animal models (344).
The TLR7/8 agonist imiquimod has been approved by the FDA and is currently being used for treating superficial basal cell carcinoma (345, 346). It has also shown efficacy in the treatment of different cutaneous tumors (347–349), and it is being tested in several clinical trials for skin and other malignancies, such as glioma and breast and prostate cancer (350, 351) (NCT01792505, NCT00899574, and NCT02234921). It might cause some local inflammatory reactions and systemic symptoms, including muscle aches, fatigue, and nausea, but, in general terms, it is well-tolerated (352). Resiquimod is another TLR7/8 agonist and has shown a more intense immune response than imiquimod (353). It is being explored in precancerous and malignant skin tumors (354, 355) and in multiple clinical trials as a vaccine adjuvant. Additionally, 852A, a TLR7 agonist, and VTX-2337, a TLR8 agonist, are being examined in clinical trials against different tumor types, such as ovarian, breast, cervical, endometrial, and head and neck cancers (NCT00319748 and NCT01334177).
Finally, CpG oligodeoxynucleotides are agonists of TLR9 and are being tested in several tumor types and in some clinical trials (356–359). For example, Agatolimod (CpG 7909) and is being studied as a vaccine adjuvant and as a monotherapy for various solid and hematological malignancies (360–362). Although the tolerability and safety of TLR9 ligands in monotherapy have been proven in numerous clinical trials, they have shown scarce antitumoral efficacy (363). The combination with other immune modulating compounds can greatly improve CpG ODNs-based strategies. Hence, Agatolimod is also being tested in combination with monophosphoryl lipid A and MAGE-A3 (a melanoma antigen) in phase II clinical analysis (NCT00085189). Furthermore, an alternative TLR9 agonist, SD-101, has been shown to overcome resistance to checkpoint inhibitors and is being investigated in association with these inhibitors in ongoing clinical trials (NCT02521870). With these types of drugs the primary adverse effects are also related to immunostimulation or systemic-flu like symptoms (364). Nevertheless, they could lead to autoimmune disorders if used as long-term treatments (365) (Table 1).
Treatments Based on TLR Antagonism
In some cellular locations, TLR antagonism, not TLR activation, is needed. For this purpose, inhibiting treatments are being tested, and some have entered phase I and II clinical trials. Molecules derived from LPS, such as E5564 and CRX-526, and antibodies targeting TLRs, such as OPN305, are able to inhibit TLR signaling and reduce inflammation, but they are not currently being used against cancer (366–369) (NCT02363491). Recently, various studies have shown that the blockade of TLR4 might have antitumoral effects in ovarian, breast and prostate cancer (370–372). In another possible approach, bacteria, and/or gut microbiota are modulated by using probiotics or antibiotics. This approach has been shown to reduce TLR tumor-promoting signals and inflammation in several studies (373–376). However, the use of this technique in cancer is still being developed (Table 1).
Future Treatment Opportunities Targeting the TLR Pathway
Immunomodulation in cancer is currently being intensely researched, with some approved innovations showing substantially positive results, such as antibodies targeting T cell-mediated immune checkpoints, adoptive cellular therapies, or cancer vaccines. In this context, numerous trials are combining these novel treatments with TLR agonists to improve their activity (377–380).
Another possible approach using TLR as a clinical cancer treatment is based on targeting its downstream modulators. The MYD88 protein is involved in surface and endosomal TLR pathways and thus is considered an attractive candidate. In addition, some studies suggest that it may be involved in oncogenesis (381) and related to the clinical response to ibrutinib (382, 383). Similarly, IRAK4 is a downstream mediator of the pathway that links multiple TLRs to NF-κB activation. Constitutive activation or increased expression of this gene has been reported for different tumor types and is correlated with chemoresistance (384, 385). Other proteins, including TRAF6 and NF-κB, have been shown to stimulate bortezomib activity in preclinical models (386).
Finally, TLR might serve as a protective agent in cancer treatment. TLR5 agonists have shown chemoprotective and radioprotective effects in numerous studies (344, 387–389). Furthermore, they have been demonstrated to improve the outcome of bone marrow allotransplant and decrease the incidence of graft-vs.-host disease (GVHD) (390, 391).
In summary, TLR-targeted therapies for cancer are currently being successfully used, however, many opportunities remain to develop new compounds to provide better treatment for patients and to minimize the secondary effects of current strategies (Table 1).
Oxidative Stress and CSCs
The generation of reactive oxygen species (ROS) in a high concentration is detrimental for the cell, due to the fact that oxidative stress promotes DNA, RNA, proteins, and lipids modifications. It is well-known that cancer cells, in consequence of its active metabolism and altered oncogenic and tumor suppressor signaling pathways, are usually characterized by a high ROS level. However, cancer cells are able to adapt and proliferate, becoming resistant to oxidative stress (392–394).
Oxidative stress has been associated with a variety of cancer-related effects: cellular proliferation, apoptosis evasion, invasion, metastasis, or angiogenesis. Different signaling pathways mentioned in this review are implicated in these ROS production outcomes. For instance, ROS could regulate EMT through activation of NFkB signaling pathway, or metastasis through Wnt signaling cascade stimulation, among other signaling routes (392, 395–398).
Aerobic glycolysis is energetically more efficient than anaerobic one, but leads to the generation of a big amount of ROS. Tumor cells accomplish adapting to oxidative stress switching aerobic glycolysis for anaerobic one, independently of oxygen available in the microenvironment. This phenomenon is called Warburg effect, and leads to a low ROS level formation, accompanied by a redox potential increase, through NADPH production (399–402). Furthermore, cancer cells possess higher antioxidant efficiency than non-tumoral cells. In order to counteract the less ATP generation in anaerobic glycolysis, tumor cells considerably increase glucose intake. It has been shown that cancer cells also activate the pentose phosphate pathway (PPP) with a prolonged ROS exposure, in order to acquire NADPH and nucleotides for DNA synthesis and repair (402–406).
Cancer stem cells seem to possess a lower ROS level than non-tumoral stem cells, due to different mechanisms useful to reduce oxidative stress and maintain stem cell properties (404, 407–409). However, there are controversial aspects in the metabolism regulation of CSCs. While several studies show that CSCs often present a high glycolytic metabolism, which preserves them from oxidative stress damaging, others highlight that they could be more dependent on oxidative phosphorylation. Furthermore, CSC could switch from anaerobic glycolysis to PPP in order to obtain a stronger antioxidant potential to maintain stemness. Also, increased PPP has been associated with drug resistance (403, 405, 406, 408, 410, 411).
Due to the dual consequences of an augmented ROS levels, the oxidative stress targeted therapies are under discussion. On the one hand, several well-known anticancer treatments increase ROS levels in cancer cells, in order to reach a threshold which causes the cell death, such as radiation, arsenic trioxide, 5-fluorouracil, or paclitaxel. Another strategies try to decrease ROS scavengers in cancer cells. On the other hand, as it has been mentioned above, ROS is related to several cancer-promoting effects, thus it might be advantageous to reduce ROS levels or increase antioxidant molecules, in order to restore de redox balance in the cell. For these reasons, the efficacy of these anticancer strategies depends on the ROS level in the tumor type and the capacity of the tumor to modulate its metabolism routes (404, 409, 412–419).
Conclusions
Currently, many observations have indicated the association between the CSC content and clinicopathological characteristics of tumors upon diagnosis. Additionally, CSC populations are resistant to conventional therapies aimed at the bulk of proliferative cells, and they can be enriched in a posttreatment setting. Therefore, targeting stemness pathways may provide promising strategies to actively eradicate tumors and metastasis. There are many pathways that regulate the CSC phenotype and its pluripotency state, providing mechanistic support for acquired drug resistance, including altered metabolism and oxidation states; phenotypic plasticity; increased membrane extrusion pumps and receptors; altered apoptosis, autophagy, and/or cell cycle regulation; different DNA damage, and/or repair responses; epigenetic regulation and/or differential secretion of proteins or non-coding RNA. Some of these stemness pathways have been reviewed here, and their targeting approaches have been described. However, data on their proper response to the inhibitors and accounts of the extent to which they have antitumor efficacy are scarce. In most cases, the inhibitor activity depends on the extent of the molecular dependence on the specific targeted pathway in the CSCs or its ability to avoid targeting mature cells or normal stem cells to prevent undesirable toxicity.
Importantly, the stemness pathways are interconnected to regulate the CSC phenotype and the transition among different pluripotency states. This cross talk among pathways may drive the resistance to single pathway inhibitors and maintain the CSC phenotype. For example, cross talk between PI3K and Notch may contribute to the resistance to therapeutic PI3K inhibitors of breast cancer (420). Additionally, PIM inhibition might not have the same effect as AKT on MEK targets, which may not be effected by PIM single inhibitors in vivo. This phenomenon may explain in part the negative results obtained in clinical trials with single-agent therapy.
Other important reason for tumor resistance is based on advanced tumors, in general, containing expanded polyclonal CSC populations, which render them resistant to therapies against single CSC signaling pathways. Therefore, ongoing clinical trials using molecular biomarkers may be used to overcome these challenges. Moreover, the incorporation of CSC-based functional assays (for example, 3D organoids, pluripotency assays) may provide a better view of CSCs and therefore of tumor, eradication using these pathway-based therapeutic strategies.
Despite all of the challenges, some inhibitors for these pathways are currently used as a standard of care for patients, and many others are being tested in phases III and IV clinical trials. A huge effort is being made in order to develop more specific and less toxic compounds to target every single pathway known to be involved in CSC establishment and maintenance. Different drugs, antibodies, vaccines and, even, immunotherapeutic approaches are being assayed for this purpose. Therefore, although more research is needed to undercover additional regulatory elements, we expect to see new molecules targeting these pathways approved by the FDA in the following years.
Author Contributions
All authors contributed equally to information and writing the work. AC edited the work.
Funding
This work was supported by grants from the Ministerio de Ciencia, Innovación y Universidades (MCIU) Plan Estatal de I+D+I 2018, a la Agencia Estatal de Investigación (AEI) y al Fondo Europeo de Desarrollo Regional (MCIU/AEI/FEDER, UE): RTI2018-097455-B-I00; grant from AEI-MICIU/FEDER (RED2018-102723-T); from CIBER de Cáncer (CB16/12/00275), co-funded by FEDER from Regional Development European Funds (European Union); from Consejeria de Salud (PI-0397-2017) and Consejeria of Economía, Conocimiento, Empresas y Universidad of the Junta de Andalucia (P18-RT-2501). Also especial thanks to the Fundacion AECC and Fundacion Eugenio Rodriguez Pascual for supporting this work. LS-D and ES-M were funded by a FPU from Ministerio de Ciencia, Innovación y Universidades (MCIU) Plan Estatal de I+D+I 2018, a la Agencia Estatal de Investigación (AEI) y al Fondo Europeo de Desarrollo Regional (MCIU/AEI/FEDER, UE). AE-S was funded by Fundacion AECC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Choi HY, Siddique HR, Zheng M, Kou Y, Yeh D-W, Machida T, et al. p53 destabilizing protein skews asymmetric division and enhances NOTCH activation to direct self-renewal of TICs. Nat Commun. (2020) 11:3084. doi: 10.1038/s41467-020-16616-8
2. Cheng J, Cashman JR. PAWI-2 overcomes tumor stemness and drug resistance via cell cycle arrest in integrin β3-KRAS-dependent pancreatic cancer stem cells. Sci Rep. (2020) 10:9162. doi: 10.1038/s41598-020-65804-5
3. Clara JA, Monge C, Yang Y, Takebe N. Targeting signalling pathways and the immune microenvironment of cancer stem cells - a clinical update. Nat Rev Clin Oncol. (2020) 17:204–32. doi: 10.1038/s41571-019-0293-2
4. Portney BA, Arad M, Gupta A, Brown RA, Khatri R, Lin PN, et al. ZSCAN4 facilitates chromatin remodeling and promotes the cancer stem cell phenotype. Oncogene. (2020) 39:4970–82. doi: 10.1038/s41388-020-1333-1
5. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. (2017) 23:1124–34. doi: 10.1038/nm.4409
6. Borovski T, De Sousa EMF, Vermeulen L, Medema JP. Cancer stem cell niche: the place to be. Cancer Res. (2011) 71:634–9. doi: 10.1158/0008-5472.CAN-10-3220
7. Rycaj K, Tang DG. Cell-of-origin of cancer versus cancer stem cells: assays and interpretations. Cancer Res. (2015) 75:4003–11. doi: 10.1158/0008-5472.CAN-15-0798
8. Cabrera MC, Hollingsworth RE, Hurt EM. Cancer stem cell plasticity and tumor hierarchy. World J Stem Cells. (2015) 7:27–36. doi: 10.4252/wjsc.v7.i1.27
9. Kapoor A, Kumar S. Cancer stem cell: a rogue responsible for tumor development and metastasis. Indian J Cancer. (2014) 51:282–9. doi: 10.4103/0019-509X.146794
10. Fessler E, Dijkgraaf FE, De Sousa EMF, Medema JP. Cancer stem cell dynamics in tumor progression and metastasis: is the microenvironment to blame? Cancer Lett. (2013) 341:97–104. doi: 10.1016/j.canlet.2012.10.015
11. Fulawka L, Donizy P, Halon A. Cancer stem cells - the current status of an old concept: literature review and clinical approaches. Biol Res. (2014) 47:66. doi: 10.1186/0717-6287-47-66
12. Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. (2009) 15:1010–2. doi: 10.1038/nm0909-1010
13. Hamada S, Shimosegawa T. Pancreatic cancer stem cell and mesenchymal stem cell. In: Grippo PJ, Munshi HG, editors. Pancreatic Cancer and Tumor Microenvironment. Trivandrum: Transworld Research Network (2012). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK98934/
14. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. (2008) 8:755–68. doi: 10.1038/nrc2499
15. Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine. (2012) 7:597–615. doi: 10.2217/nnm.12.22
16. Walters Haygood CL, Arend RC, Straughn JM, Buchsbaum DJ. Ovarian cancer stem cells: can targeted therapy lead to improved progression-free survival? World J Stem Cells. (2014) 6:441–7. doi: 10.4252/wjsc.v6.i4.441
17. Kim Y, Joo KM, Jin J, Nam DH. Cancer stem cells and their mechanism of chemo-radiation resistance. Int J Stem Cells. (2009) 2:109–14. doi: 10.15283/ijsc.2009.2.2.109
18. Pylvas-Eerola M, Liakka A, Puistola U, Koivunen J, Karihtala P. Cancer stem cell properties as factors predictive of chemoresistance in neoadjuvantly-treated patients with ovarian cancer. Anticancer Res. (2016) 36:3425–31.
19. Minami Y. Overview: cancer stem cell and tumor environment. Oncology. (2015) 89(Suppl. 1):22–4. doi: 10.1159/000431060
20. Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. (2015) 16:225–38. doi: 10.1016/j.stem.2015.02.015
21. Carnero A, Lleonart M. The hypoxic microenvironment: a determinant of cancer stem cell evolution. Bioessays. (2016) 38(Suppl. 1):S65–74. doi: 10.1002/bies.201670911
22. Quail DF, Taylor MJ, Postovit LM. Microenvironmental regulation of cancer stem cell phenotypes. Curr Stem Cell Res Ther. (2012) 7:197–216. doi: 10.2174/157488812799859838
23. Mannello F. Understanding breast cancer stem cell heterogeneity: time to move on to a new research paradigm. BMC Med. (2013) 11:169. doi: 10.1186/1741-7015-11-169
24. Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. (2014) 2:78–91. doi: 10.1016/j.stemcr.2013.11.009
25. Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. (2010) 12:468–76. doi: 10.1038/ncb2048
26. Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. (2014) 15:482–96. doi: 10.1038/nrm3823
27. Munoz-Galvan S, Felipe-Abrio B, Garcia-Carrasco M, Dominguez-Pinol J, Suárez-Martínez E, Verdugo-Sivianes EM, et al. New markers for human ovarian cancer that link platinum resistance to the cancer stem cell phenotype and define new therapeutic combinations and diagnostic tools. J Exp Clin Cancer Res. (2019) 38:234. doi: 10.1186/s13046-019-1245-5
28. Munoz-Galvan S, Lucena-Cacace A, Perez M, Otero-Albiol D, Gomez-Cambronero J, Carnero A. Tumor cell-secreted PLD increases tumor stemness by senescence-mediated communication with microenvironment. Oncogene. (2019) 38:1309–23. doi: 10.1038/s41388-018-0527-2
29. Carnero A, Garcia-Mayea Y, Mir C, Lorente J, Rubio IT, LLeonart ME. The cancer stem-cell signaling network and resistance to therapy. Cancer Treat. Rev. (2016) 49:25–36. doi: 10.1016/j.ctrv.2016.07.001
30. Carnero A, Lleonart ME. Epigenetic mechanisms in senescence, immortalisation and cancer. Biol Rev Camb Philos Soc. (2011) 86:443–55. doi: 10.1111/j.1469-185X.2010.00154.x
31. Apostolou P, Toloudi M, Chatziioannou M, Ioannou E, Papasotiriou I. Cancer stem cells stemness transcription factors expression correlates with breast cancer disease stage. Curr Stem Cell Res Ther. (2012) 7:415–9. doi: 10.2174/157488812804484639
32. Biddle A, Mackenzie IC. Cancer stem cells and EMT in carcinoma. Cancer Metastasis Rev. (2012) 31:285–93. doi: 10.1007/s10555-012-9345-0
33. Ansieau S. EMT in breast cancer stem cell generation. Cancer Lett. (2013) 338:63–8. doi: 10.1016/j.canlet.2012.05.014
34. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA. (2011) 108:7950–5. doi: 10.1073/pnas.1102454108
35. Ferrer I, Verdugo-Sivianes EM, Castilla MA, Melendez R, Marin JJ, Munoz-Galvan S, et al. Loss of the tumor suppressor spinophilin (PPP1R9B) increases the cancer stem cell population in breast tumors. Oncogene. (2016) 35:2777–88. doi: 10.1038/onc.2015.341
36. Garcia-Heredia JM, Verdugo Sivianes EM, Lucena-Cacace A, Molina-Pinelo S, Carnero A. Numb-like (NumbL) downregulation increases tumorigenicity, cancer stem cell-like properties and resistance to chemotherapy. Oncotarget. (2016) 7:63611–28. doi: 10.18632/oncotarget.11553
37. Garcia-Heredia JM, Carnero A. The cargo protein MAP17 (PDZK1IP1) regulates the immune microenvironment. Oncotarget. (2017) 8:98580–97. doi: 10.18632/oncotarget.21651
38. Garcia-Heredia JM, Lucena-Cacace A, Verdugo-Sivianes EM, Perez M, Carnero A. The cargo protein MAP17 (PDZK1IP1) regulates the cancer stem cell pool activating the notch pathway by abducting NUMB. Clin Cancer Res. (2017) 23:3871–83. doi: 10.1158/1078-0432.CCR-16-2358
39. Marjanovic ND, Weinberg RA, Chaffer CL. Poised with purpose: cell plasticity enhances tumorigenicity. Cell Cycle. (2013) 12:2713–4. doi: 10.4161/cc.26075
40. Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clin Chem. (2013) 59:168–79. doi: 10.1373/clinchem.2012.184655
41. Ning X, Du Y, Ben Q, Huang L, He X, Gong Y, et al. Bulk pancreatic cancer cells can convert into cancer stem cells (CSCs) in vitro and 2 compounds can target these CSCs. Cell Cycle. (2016) 15:403–12. doi: 10.1080/15384101.2015.1127471
42. Yilmaz T, Hosal S, Ozyar E, Akyol F, Gursel B. Post-operative radiotherapy in advanced laryngeal cancer: effect on local and regional recurrence, distant metastases and second primaries. J Laryngol Otol. (2005) 119:784–90. doi: 10.1258/002221505774481183
43. Ferrer I, Quintanal-Villalonga Á, Molina-Pinelo S, Garcia-Heredia JM, Perez M, Suárez R, et al. MAP17 predicts sensitivity to platinum-based therapy, EGFR inhibitors and the proteasome inhibitor bortezomib in lung adenocarcinoma. J Exp Clin Cancer Res. (2018) 37:195. doi: 10.1186/s13046-018-0871-7
44. Rich JN. Cancer stem cells: understanding tumor hierarchy and heterogeneity. Medicine. (2016) 95(1 Suppl. 1):S2–7. doi: 10.1097/MD.0000000000004764
45. Wiechert A, Saygin C, Thiagarajan PS, Rao VS, Hale JS, Gupta N, et al. Cisplatin induces stemness in ovarian cancer. Oncotarget. (2016) 7:30511–22. doi: 10.18632/oncotarget.8852
46. Liu H, Zhang HW, Sun XF, Guo XH, He YN, Cui SD, et al. Tamoxifen-resistant breast cancer cells possess cancer stem-like cell properties. Chin Med J. (2013) 126:3030–4. doi: 10.3760/cma.j.issn.0366-6999.20130227
47. Saygin C, Matei D, Majeti R, Reizes O, Lathia JD. Targeting cancer stemness in the clinic: from hype to hope. Cell Stem Cell. (2019) 24:25–40. doi: 10.1016/j.stem.2018.11.017
48. Carnero A. MAP17 and the double-edged sword of ROS. Biochim Biophys Acta. (2012) 1826:44–52. doi: 10.1016/j.bbcan.2012.03.004
49. Schulenburg A, Blatt K, Cerny-Reiterer S, Sadovnik I, Herrmann H, Marian B, et al. Cancer stem cells in basic science and in translational oncology: can we translate into clinical application? J Hematol Oncol. (2015) 8:16. doi: 10.1186/s13045-015-0113-9
50. Skvortsova I, Debbage P, Kumar V, Skvortsov S. Radiation resistance: cancer stem cells (CSCs) and their enigmatic pro-survival signaling. Semin Cancer Biol. (2015) 35:39–44. doi: 10.1016/j.semcancer.2015.09.009
51. Garcia-Heredia JM, Carnero A. Dr. Jekyll and Mr. Hyde: MAP17's up-regulation, a crosspoint in cancer and inflammatory diseases. Mol Cancer. (2018) 17:80. doi: 10.1186/s12943-018-0828-7
52. Feng W, Dean DC, Hornicek FJ, Shi H, Duan Z. Exosomes promote pre-metastatic niche formation in ovarian cancer. Mol Cancer. (2019) 18:124. doi: 10.1186/s12943-019-1049-4
53. Mashouri L, Yousefi H, Aref AR, Ahadi AM, Molaei F, Alahari SK. Exosomes: composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol Cancer. (2019) 18:75. doi: 10.1186/s12943-019-0991-5
54. Wu M, Wang G, Hu W, Yao Y, Yu XF. Emerging roles and therapeutic value of exosomes in cancer metastasis. Mol Cancer. (2019) 18:53. doi: 10.1186/s12943-019-0964-8
55. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov. (2014) 13:497–512. doi: 10.1038/nrd4253
56. Magnani L, Stoeck A, Zhang X, Lanczky A, Mirabella AC, Wang TL, et al. Genome-wide reprogramming of the chromatin landscape underlies endocrine therapy resistance in breast cancer. Proc Natl Acad Sci USA. (2013) 110:E1490–9. doi: 10.1073/pnas.1219992110
57. Munoz-Galvan S, Gutierrez G, Perez M, Carnero A. MAP17 (PDZKIP1) Expression determines sensitivity to the proteasomal inhibitor bortezomib by preventing cytoprotective autophagy and NFκB activation in breast cancer. Mol Cancer Ther. (2015) 14:1454–65. doi: 10.1158/1535-7163.MCT-14-1053
58. Rivero M, Peinado-Serrano J, Munoz-Galvan S, Espinosa-Sanchez A, Suárez-Martínez E, Felipe-Abrio B, et al. MAP17 (PDZK1IP1) and pH2AX are potential predictive biomarkers for rectal cancer treatment efficacy. Oncotarget. (2018) 9:32958–71. doi: 10.18632/oncotarget.26010
59. Shao Y, Lv H, Zhong DS, Zhou QH. EGFR-TKI resistance and MAP17 are associated with cancer stem cell like properties. Oncol Lett. (2018) 15:6655–65. doi: 10.3892/ol.2018.8129
60. Felipe-Abrio B, Verdugo-Sivianes EM, Carnero A. c-MYB- and PGC1a-dependent metabolic switch induced by MYBBP1A loss in renal cancer. Mol Oncol. (2019) 13:1519–33. doi: 10.1002/1878-0261.12499
61. Felipe-Abrio B, Verdugo-Sivianes EM, Saez C, Carnero A. Loss of MYBBP1A Induces cancer stem cell activity in renal cancer. Cancers. (2019) 11:235. doi: 10.3390/cancers11020235
62. Felipe-Abrio B, Carnero A. The tumor suppressor roles of MYBBP1A, a major contributor to metabolism plasticity and stemness. Cancers. (2020) 12:254. doi: 10.3390/cancers12010254
63. Sharma SV, Haber DA, Settleman J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat Rev Cancer. (2010) 10:241–53. doi: 10.1038/nrc2820
64. Blasco MA. Telomere length, stem cells and aging. Nat Chem Biol. (2007) 3:640–9. doi: 10.1038/nchembio.2007.38
65. Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JF. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets. (2008) 8:187–98. doi: 10.2174/156800908784293659
66. Carnero A. The PKB/AKT pathway in cancer. Curr Pharm Des. (2010) 16:34–44. doi: 10.2174/138161210789941865
67. Zanella F, Link W, Carnero A. Understanding FOXO, new views on old transcription factors. Curr Cancer Drug Targets. (2010) 10:135–46. doi: 10.2174/156800910791054158
68. Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta. (2011) 1813:1269–78. doi: 10.1016/j.bbamcr.2010.09.019
69. Blanco-Aparicio C, Carnero A. Pim kinases in cancer: diagnostic, prognostic and treatment opportunities. Biochem Pharmacol. (2013) 85:629–43. doi: 10.1016/j.bcp.2012.09.018
70. Bruttel VS, Wischhusen J. Cancer stem cell immunology: key to understanding tumorigenesis and tumor immune escape? Front Immunol. (2014) 5:360. doi: 10.3389/fimmu.2014.00360
71. Narlik-Grassow M, Blanco-Aparicio C, Carnero A. The PIM family of serine/threonine kinases in cancer. Med Res Rev. (2014) 34:136–59. doi: 10.1002/med.21284
72. Carnero A, Blanco-Aparicio C, Kondoh H, Lleonart ME, Martinez-Leal JF, Mondello C, et al. Disruptive chemicals, senescence and immortality. Carcinogenesis. (2015) 36(Suppl. 1):S19–37. doi: 10.1093/carcin/bgv029
73. Garcia-Heredia JM, Carnero A. Decoding Warburg's hypothesis: tumor-related mutations in the mitochondrial respiratory chain. Oncotarget. (2015) 6:41582–99. doi: 10.18632/oncotarget.6057
74. Saydaminova K, Strauss R, Xie M, Bartek J, Richter M, van Rensburg R, et al. Sensitizing ovarian cancer cells to chemotherapy by interfering with pathways that are involved in the formation of cancer stem cells. Cancer Biol Ther. (2016) 17:1079–88. doi: 10.1080/15384047.2016.1219819
75. Holland JD, Klaus A, Garratt AN, Birchmeier W. Wnt signaling in stem and cancer stem cells. Curr Opin Cell Biol. (2013) 25:254–64. doi: 10.1016/j.ceb.2013.01.004
76. Clevers H, Loh KM, Nusse R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science. (2014) 346:1248012. doi: 10.1126/science.1248012
77. Jernigan KK, Cselenyi CS, Thorne CA, Hanson AJ, Tahinci E, Hajicek N, et al. Gβγ activates GSK3 to promote LRP6-mediated β-catenin transcriptional activity. Sci Signal. (2010) 3:ra37. doi: 10.1126/scisignal.2000647
78. MacDonald BT, He X. Frizzled and LRP5/6 receptors for Wnt/β-catenin signaling. Cold Spring Harbor Perspect Biol. (2012) 4:a007880. doi: 10.1101/cshperspect.a007880
79. Okamoto M, Udagawa N, Uehara S, Maeda K, Yamashita T, Nakamichi Y, et al. Noncanonical Wnt5a enhances Wnt/β-catenin signaling during osteoblastogenesis. Sci Rep. (2014) 4:4493. doi: 10.1038/srep04493
80. Jiang X, Charlat O, Zamponi R, Yang Y, Cong F. Dishevelled promotes Wnt receptor degradation through recruitment of ZNRF3/RNF43 E3 ubiquitin ligases. Mol Cell. (2015) 58:522–33. doi: 10.1016/j.molcel.2015.03.015
81. Debaugnies M, Sánchez-Danés A, Rorive S, Raphaël M, Liagre M, Parent M-A, et al. YAP and TAZ are essential for basal and squamous cell carcinoma initiation. EMBO Rep. (2018) 19:e45809. doi: 10.15252/embr.201845809
82. Heidel FH, Bullinger L, Feng Z, Wang Z, Neff TA, Stein L, et al. Genetic and pharmacologic inhibition of β-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell. (2012) 10:412–24. doi: 10.1016/j.stem.2012.02.017
83. Fang L, Cai J, Chen B, Wu S, Li R, Xu X, et al. Aberrantly expressed miR-582-3p maintains lung cancer stem cell-like traits by activating Wnt/β-catenin signalling. Nat Commun. (2015) 6:8640. doi: 10.1038/ncomms9640
84. Ilmer M, Boiles A, Regel I, Yokoi K, Michalski C, Wistuba I, et al. RSPO2 enhances canonical wnt signaling to confer stemness-associated traits to susceptible pancreatic cancer cells. Cancer Res. (2015) 75:1883–96. doi: 10.1158/0008-5472.CAN-14-1327
85. Prieur A, Cappellini M, Habif G, Lefranc M-P, Mazard T, Morency E, et al. Targeting the Wnt pathway and cancer stem cells with anti-progastrin humanized antibodies: a major breakthrough for K-RAS mutated colorectal cancer treatment. Clin Cancer Res. (2017) 23:5267–80. doi: 10.1158/1078-0432.CCR-17-0533
86. Xie SL, Fan S, Zhang SY, Chen WX, Li QX, Pan GK, et al. SOX8 regulates cancer stem-like properties and cisplatin-induced EMT in tongue squamous cell carcinoma by acting on the Wnt/β-catenin pathway. Int J Cancer. (2018) 142:1252–65. doi: 10.1002/ijc.31134
87. Nikuseva Martić T, Pećina-Slaus N, Kusec V, Kokotović T, Musinović H, Tomas D, et al. Changes of AXIN-1 and beta-catenin in neuroepithelial brain tumors. Pathol Oncol Res. (2010) 16:75–9. doi: 10.1007/s12253-009-9190-9
88. Dixit R, Pandey M, Tripathi SK, Dwivedi AND Shukla VK. Genetic mutational analysis of β-catenin gene affecting GSK-3β phosphorylation plays a role in gallbladder carcinogenesis: results from a case control study. Cancer Treat Res Commun. (2020) 23:100173. doi: 10.1016/j.ctarc.2020.100173
89. Worm J, Christensen C, Grønbaek K, Tulchinsky E, Guldberg P. Genetic and epigenetic alterations of the APC gene in malignant melanoma. Oncogene. (2004) 23:5215–26. doi: 10.1038/sj.onc.1207647
90. Le PN, McDermott JD, Jimeno A. Targeting the Wnt pathway in human cancers: therapeutic targeting with a focus on OMP-54F28. Pharmacol Ther. (2015) 146:1–11. doi: 10.1016/j.pharmthera.2014.08.005
91. Moore KN, Gunderson CC, Sabbatini P, McMeekin DS, Mantia-Smaldone G, Burger RA, et al. A phase 1b dose escalation study of ipafricept (OMP54F28) in combination with paclitaxel and carboplatin in patients with recurrent platinum-sensitive ovarian cancer. Gynecol Oncol. (2019) 154:294–301. doi: 10.1016/j.ygyno.2019.04.001
92. Herbst A, Jurinovic V, Krebs S, Thieme SE, Blum H, Göke B, et al. Comprehensive analysis of β-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/β-catenin signaling. BMC Genom. (2014) 15:74. doi: 10.1186/1471-2164-15-74
93. Tam BY, Chiu K, Chung H, Bossard C, Nguyen JD, Creger E, et al. The CLK inhibitor SM08502 induces anti-tumor activity and reduces Wnt pathway gene expression in gastrointestinal cancer models. Cancer Lett. (2020) 473:186–97. doi: 10.1016/j.canlet.2019.09.009
94. Radovich M, Solzak JP, Hancock BA, Storniolo AMV, Schneider BP, Miller KD. Abstract OT3-06-02: An initial safety study of gedatolisib plus PTK7-ADC for metastatic triple-negative breast cancer. Cancer Res. (2019) 79(4 Suppl.):OT3-06-02. doi: 10.1158/1538-7445.SABCS18-OT3-06-02
95. Blagodatski A, Poteryaev D, Katanaev VL. Targeting the Wnt pathways for therapies. Mol Cell Ther. (2014) 2:28. doi: 10.1186/2052-8426-2-28
96. Rodon J, Argilés G, Connolly RM, Vaishampayan U, Jonge Md, Garralda E, et al. Abstract CT175: biomarker analyses from a phase I study of WNT974, a first-in-class Porcupine inhibitor, in patients (pts) with advanced solid tumors. Cancer Res. (2018) 78(13 Suppl.):CT175. doi: 10.1158/1538-7445.AM2018-CT175
97. Klempner SJ, Bendell JC, Villaflor VM, Tenner LL, Stein S, Naik GS, et al. DKN-01 in combination with pembrolizumab in patients with advanced gastroesophageal adenocarcinoma (GEA): Tumoral DKK1 expression as a predictor of response and survival. J Clin Oncol. (2020) 38(4_suppl):357. doi: 10.1200/JCO.2020.38.4_suppl.357
98. El-Khoueiry AB, Ning Y, Yang D, Cole S, Kahn M, Zoghbi M, et al. A phase I first-in-human study of PRI-724 in patients (pts) with advanced solid tumors. J Clin Oncol. (2013) 31(15_suppl):2501. doi: 10.1200/jco.2013.31.15_suppl.2501
99. Sakunrangsit N, Ketchart W. Plumbagin inhibits cancer stem-like cells, angiogenesis and suppresses cell proliferation and invasion by targeting Wnt/β-catenin pathway in endocrine resistant breast cancer. Pharmacol Res. (2019) 150:104517. doi: 10.1016/j.phrs.2019.104517
100. Canesin G, Evans-Axelsson S, Hellsten R, Krzyzanowska A, Prasad CP, Bjartell A, et al. Treatment with the WNT5A-mimicking peptide Foxy-5 effectively reduces the metastatic spread of WNT5A-low prostate cancer cells in an orthotopic mouse model. PLoS ONE. (2017) 12:e0184418. doi: 10.1371/journal.pone.0184418
101. Sheikh S, Saxena D, Tian X, Amirshaghaghi A, Tsourkas A, Brem S, et al. An integrated stress response agent that modulates DR5-dependent TRAIL synergy reduces patient-derived glioma stem cell viability. Mol Cancer Res. (2019) 17:1102. doi: 10.1158/1541-7786.MCR-18-0276
102. Koga T, Mizui M, Yoshida N, Otomo K, Lieberman LA, Crispin JC, et al. KN-93, an inhibitor of calcium/calmodulin-dependent protein kinase IV, promotes generation and function of Foxp3(+) regulatory T cells in MRL/lpr mice. Autoimmunity. (2014) 47:44–50. doi: 10.3109/08916934.2014.915954
103. Courtois G, Gilmore TD. Mutations in the NF-κB signaling pathway: implications for human disease. Oncogene. (2006) 25:6831–43. doi: 10.1038/sj.onc.1209939
104. Karin M. NF-κB and cancer: mechanisms and targets. Mol Carcinog. (2006) 45:355–61. doi: 10.1002/mc.20217
105. Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transduc Target Ther. (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
106. Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol. (2011) 12:715–23. doi: 10.1038/ni.2060
107. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
108. Taniguchi K, Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. (2018) 18:309–24. doi: 10.1038/nri.2017.142
109. Hayden MS, Ghosh S. Shared principles in NF-κB signaling. Cell. (2008) 132:344–62. doi: 10.1016/j.cell.2008.01.020
110. Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. (2013) 12:86. doi: 10.1186/1476-4598-12-86
111. Karin M. NF-κB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. (2009) 1:a000141. doi: 10.1101/cshperspect.a000141
112. Kaltschmidt C, Banz-Jansen C, Benhidjeb T, Beshay M, Forster C, Greiner J, et al. A Role for NF-κB in organ specific cancer and cancer stem cells. Cancers. (2019) 11:655: doi: 10.3390/cancers11050655
113. Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA, et al. Nuclear factor-κB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. (2001) 98:2301–7. doi: 10.1182/blood.V98.8.2301
114. Sallustio F, Curci C, Stasi A, De Palma G, Divella C, Gramignoli R, et al. Role of toll-like receptors in actuating stem/progenitor cell repair mechanisms: different functions in different cells. Stem Cells Int. (2019) 2019:6795845. doi: 10.1155/2019/6795845
115. Liu M, Sakamaki T, Casimiro MC, Willmarth NE, Quong AA, Ju X, et al. The canonical NF-κB pathway governs mammary tumorigenesis in transgenic mice and tumor stem cell expansion. Cancer Res. (2010) 70:10464–73. doi: 10.1158/0008-5472.CAN-10-0732
116. Terzi ć J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. (2010) 138:2101–14.e2105. doi: 10.1053/j.gastro.2010.01.058
117. Yamamoto M, Taguchi Y, Ito-Kureha T, Semba K, Yamaguchi N, Inoue J. NF-κB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nat Commun. (2013) 4:2299. doi: 10.1038/ncomms3299
118. Vazquez-Santillan K, Melendez-Zajgla J, Jimenez-Hernandez LE, Gaytan-Cervantes J, Muñoz-Galindo L, Piña-Sanchez P, et al. NF-κB-inducing kinase regulates stem cell phenotype in breast cancer. Sci Rep. (2016) 6:37340. doi: 10.1038/srep37340
119. Pellegrini P, Cordero A, Gallego MI, Dougall WC, Munoz P, Pujana MA, et al. Constitutive activation of RANK disrupts mammary cell fate leading to tumorigenesis. Stem Cells. (2013) 31:1954–65. doi: 10.1002/stem.1454
120. Zhang W, Tan W, Wu X, Poustovoitov M, Strasner A, Li W, et al. A NIK-IKKα module expands ErbB2-induced tumor-initiating cells by stimulating nuclear export of p27/Kip1. Cancer Cell. (2013) 23:647–59. doi: 10.1016/j.ccr.2013.03.012
121. Andresen V, Gjertsen BT. Drug repurposing for the treatment of acute myeloid leukemia. Front Med. (2017) 4:211. doi: 10.3389/fmed.2017.00211
122. Li Q, Verma IM. NF-κB regulation in the immune system. Nat Rev Immunol. (2002) 2:725–34. doi: 10.1038/nri910
123. Xue W, Meylan E, Oliver TG, Feldser DM, Winslow MM, Bronson R, et al. Response and resistance to NF-κB inhibitors in mouse models of lung adenocarcinoma. Cancer Discov. (2011) 1:236–47. doi: 10.1158/2159-8290.CD-11-0073
124. Xia Y, Yeddula N, Leblanc M, Ke E, Zhang Y, Oldfield E, et al. Reduced cell proliferation by IKK2 depletion in a mouse lung-cancer model. Nat Cell Biol. (2012) 14:257–65. doi: 10.1038/ncb2428
125. Wu Z-H, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-κB signaling in response to genotoxic stimuli. Science. (2006) 311:1141. doi: 10.1126/science.1121513
126. Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. (2014) 343:301–5. doi: 10.1126/science.1244851
127. Stewart AK. Medicine. How thalidomide works against cancer. Science. (2014) 343:256–7. doi: 10.1126/science.1249543
128. Jazieh AR, Komrokji R, Gupta A, Patil S, Flora D, Knapp M, et al. Phase II trial of thalidomide, irinotecan and gemcitabine in chemonaive patients with advanced non-small cell lung cancer. Cancer Invest. (2009) 27:932–6. doi: 10.3109/07357900801944856
129. Vaeteewoottacharn K, Kariya R, Matsuda K, Taura M, Wongkham C, Wongkham S, et al. Perturbation of proteasome function by bortezomib leading to ER stress-induced apoptotic cell death in cholangiocarcinoma. J Cancer Res Clin Oncol. (2013) 139:1551–62. doi: 10.1007/s00432-013-1473-6
130. Markovina S, Callander NS, O'Connor SL, Kim J, Werndli JE, Raschko M, et al. Bortezomib-resistant nuclear factor-κB activity in multiple myeloma cells. Mol Cancer Res. (2008) 6:1356–64. doi: 10.1158/1541-7786.MCR-08-0108
131. Karabela SP, Psallidas I, Sherrill TP, Kairi CA, Zaynagetdinov R, Cheng DS, et al. Opposing effects of bortezomib-induced nuclear factor-κB inhibition on chemical lung carcinogenesis. Carcinogenesis. (2012) 33:859–67. doi: 10.1093/carcin/bgs024
132. Zakaria N, Mohd Yusoff N, Zakaria Z, Widera D, Yahaya BH. Inhibition of NF-κB signaling reduces the stemness characteristics of lung cancer stem cells. Front Oncol. (2018) 8:166. doi: 10.3389/fonc.2018.00166
133. Nagaraj S, Youn J-I, Weber H, Iclozan C, Lu L, Cotter MJ, et al. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res. (2010) 16:1812–23. doi: 10.1158/1078-0432.CCR-09-3272
134. Hong DS, Kurzrock R, Supko JG, He X, Naing A, Wheler J, et al. A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin Cancer Res. (2012) 18:3396–406. doi: 10.1158/1078-0432.CCR-11-2703
135. Tanaka A, Konno M, Muto S, Kambe N, Morii E, Nakahata T, et al. A novel NF-κB inhibitor, IMD-0354, suppresses neoplastic proliferation of human mast cells with constitutively activated c-kit receptors. Blood. (2005) 105:2324–31. doi: 10.1182/blood-2004-08-3247
136. Kim S, Ko D, Lee Y, Jang S, Lee Y, Lee IY, et al. Anti-cancer activity of the novel 2-hydroxydiarylamide derivatives IMD-0354 and KRT1853 through suppression of cancer cell invasion, proliferation, and survival mediated by TMPRSS4. Sci Rep. (2019) 9:10003. doi: 10.1038/s41598-019-46447-7
137. Yemelyanov A, Gasparian A, Lindholm P, Dang L, Pierce J, Kisseljov F, et al. Effects of IKK inhibitor PS1145 on NF-B function, proliferation, apoptosis and invasion activity in prostate carcinoma cells. Oncogene. (2006) 25:387–98. doi: 10.1038/sj.onc.1209066
138. Lung HL, Kan R, Chau WY, Man OY, Mak NK, Fong CH, et al. The anti-tumor function of the IKK inhibitor PS1145 and high levels of p65 and KLF4 are associated with the drug resistance in nasopharyngeal carcinoma cells. Sci Rep. (2019) 9:12064. doi: 10.1038/s41598-019-48590-7
139. Yu M, Yeh J, Van Waes C. Protein kinase casein kinase 2 mediates inhibitor-κB kinase and aberrant nuclear factor-κB activation by serum factor(s) in head and neck squamous carcinoma cells. Cancer Res. (2006) 66:6722–31. doi: 10.1158/0008-5472.CAN-05-3758
140. Previs RA, Coleman RL, Harris AL, Sood AK. Molecular pathways: translational and therapeutic implications of the Notch signaling pathway in cancer. Clin Cancer Res. (2015) 21:955–61. doi: 10.1158/1078-0432.CCR-14-0809
141. Bray SJ. Notch signalling in context. Nat Rev Mol Cell Biol. (2016) 17:722–35. doi: 10.1038/nrm.2016.94
142. Aster JC, Pear WS, Blacklow SC. The varied roles of notch in cancer. Annu Rev Pathol. (2017) 12:245–75. doi: 10.1146/annurev-pathol-052016-100127
143. Li L, Tang P, Li S, Qin X, Yang H, Wu C, et al. Notch signaling pathway networks in cancer metastasis: a new target for cancer therapy. Med Oncol. (2017) 34:180. doi: 10.1007/s12032-017-1039-6
144. Venkatesh V, Nataraj R, Thangaraj GS, Karthikeyan M, Gnanasekaran A, Kaginelli SB, et al. Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investig. (2018) 5:5. doi: 10.21037/sci.2018.02.02
145. Meurette O, Mehlen P. Notch signaling in the tumor microenvironment. Cancer Cell. (2018) 34:536–48. doi: 10.1016/j.ccell.2018.07.009
146. Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. (2003) 161:1163–77. doi: 10.1083/jcb.200302047
147. Dufraine J, Funahashi Y, Kitajewski J. Notch signaling regulates tumor angiogenesis by diverse mechanisms. Oncogene. (2008) 27:5132–7. doi: 10.1038/onc.2008.227
148. Boucher JM, Peterson SM, Urs S, Zhang C, Liaw L. The miR-143/145 cluster is a novel transcriptional target of Jagged-1/Notch signaling in vascular smooth muscle cells. J Biol Chem. (2011) 286:28312–21. doi: 10.1074/jbc.M111.221945
149. Fukusumi T, Califano JA. The NOTCH pathway in head and neck squamous cell carcinoma. J Dent Res. (2018) 97:645–53. doi: 10.1177/0022034518760297
150. Ladi E, Nichols JT, Ge W, Miyamoto A, Yao C, Yang LT, et al. The divergent DSL ligand Dll3 does not activate Notch signaling but cell autonomously attenuates signaling induced by other DSL ligands. J Cell Biol. (2005) 170:983–92. doi: 10.1083/jcb.200503113
151. Geffers I, Serth K, Chapman G, Jaekel R, Schuster-Gossler K, Cordes R, et al. Divergent functions and distinct localization of the Notch ligands DLL1 and DLL3 in vivo. J Cell Biol. (2007) 178:465–76. doi: 10.1083/jcb.200702009
152. Gordon WR, Arnett KL, Blacklow SC. The molecular logic of Notch signaling–a structural and biochemical perspective. J Cell Sci. (2008) 121(Pt 19):3109–19. doi: 10.1242/jcs.035683
153. Bigas A, Porcheri C. Notch stem cells. Adv Exp Med Biol. (2018) 1066:235–63. doi: 10.1007/978-3-319-89512-3_12
154. Yashiro-Ohtani Y, He Y, Ohtani T, Jones ME, Shestova O, Xu L, et al. Pre-TCR signaling inactivates Notch1 transcription by antagonizing E2A. Genes Dev. (2009) 23:1665–76. doi: 10.1101/gad.1793709
155. Wang R, Sun Q, Wang P, Liu M, Xiong S, Luo J, et al. Notch and Wnt/beta-catenin signaling pathway play important roles in activating liver cancer stem cells. Oncotarget. (2016) 7:5754–68. doi: 10.18632/oncotarget.6805
156. Ntziachristos P, Lim JS, Sage J, Aifantis I. From fly wings to targeted cancer therapies: a centennial for notch signaling. Cancer Cell. (2014) 25:318–34. doi: 10.1016/j.ccr.2014.02.018
157. Kontomanolis EN, Kalagasidou S, Pouliliou S, Anthoulaki X, Georgiou N, Papamanolis V, et al. The Notch pathway in breast cancer progression. ScientificWorldJournal. (2018) 2018:2415489. doi: 10.1155/2018/2415489
158. Lin X, Sun B, Zhu D, Zhao X, Sun R, Zhang Y, et al. Notch4+ cancer stem-like cells promote the metastatic and invasive ability of melanoma. Cancer Sci. (2016) 107:1079–91. doi: 10.1111/cas.12978
159. Xiao W, Gao Z, Duan Y, Yuan W, Ke Y. Notch signaling plays a crucial role in cancer stem-like cells maintaining stemness and mediating chemotaxis in renal cell carcinoma. J Exp Clin Cancer Res. (2017) 36:41. doi: 10.1186/s13046-017-0507-3
160. Dittmer J. Breast cancer stem cells: features, key drivers and treatment options. Semin Cancer Biol. (2018) 53:59–74. doi: 10.1016/j.semcancer.2018.07.007
161. Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, et al. Therapeutic antibody targeting of individual Notch receptors. Nature. (2010) 464:1052–7. doi: 10.1038/nature08878
162. Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, Miele L. Rational targeting of Notch signaling in cancer. Oncogene. (2008) 27:5124–31. doi: 10.1038/onc.2008.226
163. Messersmith WA, Shapiro GI, Cleary JM, Jimeno A, Dasari A, Huang B, et al. A Phase I, dose-finding study in patients with advanced solid malignancies of the oral γ-secretase inhibitor PF-03084014. Clin Cancer Res. (2015) 21:60–7. doi: 10.1158/1078-0432.CCR-14-0607
164. Papayannidis C, DeAngelo DJ, Stock W, Huang B, Shaik MN, Cesari R, et al. A Phase 1 study of the novel γ-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. (2015) 5:e350. doi: 10.1038/bcj.2015.80
165. Kummar S, O'Sullivan Coyne G, Do KT, Turkbey B, Meltzer PS, Polley E, et al. Clinical activity of the γ-secretase inhibitor PF-03084014 in adults with desmoid tumors (aggressive fibromatosis). J Clin Oncol. (2017) 35:1561–9. doi: 10.1200/JCO.2016.71.1994
166. Zweidler-McKay PA, DeAngelo DJ, Douer D, Dombret H, Ottmann OG, Vey N, et al. The safety and activity of BMS-906024, a γ secretase inhibitor (GSI) with anti-notch activity, in patients with relapsed T-cell acute lymphoblastic leukemia (T-ALL): initial results of a phase 1 trial. Blood. (2014) 124:968. doi: 10.1182/blood.V124.21.968.968
167. Schott AF, Landis MD, Dontu G, Griffith KA, Layman RM, Krop I, et al. Preclinical and clinical studies of γ secretase inhibitors with docetaxel on human breast tumors. Clin Cancer Res. (2013) 19:1512–24. doi: 10.1158/1078-0432.CCR-11-3326
168. Cook N, Basu B, Smith DM, Gopinathan A, Evans J, Steward WP, et al. A phase I trial of the γ-secretase inhibitor MK-0752 in combination with gemcitabine in patients with pancreatic ductal adenocarcinoma. Br J Cancer. (2018) 118:793–801. doi: 10.1038/bjc.2017.495
169. Strosberg JR, Yeatman T, Weber J, Coppola D, Schell MJ, Han G, et al. A phase II study of RO4929097 in metastatic colorectal cancer. Eur J Cancer. (2012) 48:997–1003. doi: 10.1016/j.ejca.2012.02.056
170. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. (2015) 12:445–64. doi: 10.1038/nrclinonc.2015.61
171. Yuan X, Wu H, Xu H, Xiong H, Chu Q, Yu S, et al. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett. (2015) 369:20–7. doi: 10.1016/j.canlet.2015.07.048
172. Duarte A, Hirashima M, Benedito R, Trindade A, Diniz P, Bekman E, et al. Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. (2004) 18:2474–8. doi: 10.1101/gad.1239004
173. Patel NS, Li JL, Generali D, Poulsom R, Cranston DW, Harris AL. Up-regulation of delta-like 4 ligand in human tumor vasculature and the role of basal expression in endothelial cell function. Cancer Res. (2005) 65:8690–7. doi: 10.1158/0008-5472.CAN-05-1208
174. Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. (2006) 444:1083–7. doi: 10.1038/nature05313
175. Kuhnert F, Kirshner JR, Thurston G. Dll4-Notch signaling as a therapeutic target in tumor angiogenesis. Vasc Cell. (2011) 3:20. doi: 10.1186/2045-824X-3-20
176. Phng LK, Gerhardt H. Angiogenesis: a team effort coordinated by notch. Dev Cell. (2009) 16:196–208. doi: 10.1016/j.devcel.2009.01.015
177. Casulo C, Ruan J, Dang NH, Gore L, Diefenbach C, Beaven AW, et al. Safety and preliminary efficacy results of a phase I first-in-human study of the novel Notch-1 targeting antibody brontictuzumab (OMP-52M51) administered intravenously to patients with hematologic malignancies. Blood. (2016) 128:5108. doi: 10.1182/blood.V128.22.5108.5108
178. Ferrarotto R, Eckhardt G, Patnaik A, LoRusso P, Faoro L, Heymach JV, et al. A phase I dose-escalation and dose-expansion study of brontictuzumab in subjects with selected solid tumors. Ann Oncol. (2018) 29:1561–8. doi: 10.1093/annonc/mdy171
179. Rudin CM, Pietanza MC, Bauer TM, Ready N, Morgensztern D, Glisson BS, et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol. (2017) 18:42–51. doi: 10.1016/S1470-2045(16)30565-4
180. Morgensztern D, Besse B, Greillier L, Santana-Davila R, Ready N, Hann CL, et al. Efficacy and safety of rovalpituzumab tesirine in third-line and beyond patients with DLL3-expressing, relapsed/refractory small-cell lung cancer: results from the phase II TRINITY study. Clin Cancer Res. (2019) 25:6958–66. doi: 10.1158/1078-0432.CCR-19-1133
181. Weber D, Lehal R, Frismantas V, Bourquin JP, Bauer M, Murone M, et al. Pharmacological activity of CB-103: An oral pan-NOTCH inhibitor targeting the NOTCH transcription complex. Ann Oncol. (2018) 29:iii14. doi: 10.1093/annonc/mdy047.015
182. Sims-Mourtada J, Izzo JG, Apisarnthanarax S, Wu TT, Malhotra U, Luthra R, et al. Hedgehog: an attribute to tumor regrowth after chemoradiotherapy and a target to improve radiation response. Clin Cancer Res. (2006) 12:6565–72. doi: 10.1158/1078-0432.CCR-06-0176
183. Le H, Kleinerman R, Lerman OZ, Brown D, Galiano R, Gurtner GC, et al. Hedgehog signaling is essential for normal wound healing. Wound Repair Regen. (2008) 16:768–73. doi: 10.1111/j.1524-475X.2008.00430.x
184. Lowry WE, Richter L, Yachechko R, Pyle AD, Tchieu J, Sridharan R, et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci USA. (2008) 105:2883–8. doi: 10.1073/pnas.0711983105
185. Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. (2008) 22:2454–72. doi: 10.1101/gad.1693608
186. Skoda AM, Simovic D, Karin V, Kardum V, Vranic S, Serman L. The role of the Hedgehog signaling pathway in cancer: a comprehensive review. Bosn J Basic Med Sci. (2018) 18:8–20. doi: 10.17305/bjbms.2018.2756
187. Sheng T, Li C, Zhang X, Chi S, He N, Chen K, et al. Activation of the hedgehog pathway in advanced prostate cancer. Mol Cancer. (2004) 3:29. doi: 10.1186/1476-4598-3-29
188. Garcia MA, Collado M, Munoz-Fontela C, Matheu A, Marcos-Villar L, Arroyo J, et al. Antiviral action of the tumor suppressor ARF. EMBO J. (2006) 25:4284–92. doi: 10.1038/sj.emboj.7601302
189. Bailey JM, Mohr AM, Hollingsworth MA. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene. (2009) 28:3513–25. doi: 10.1038/onc.2009.220
190. Cochrane CR, Szczepny A, Watkins DN, Cain JE. Hedgehog signaling in the maintenance of cancer stem cells. Cancers. (2015) 7:1554–85. doi: 10.3390/cancers7030851
191. Blotta S, Jakubikova J, Calimeri T, Roccaro AM, Amodio N, Azab AK, et al. Canonical and noncanonical Hedgehog pathway in the pathogenesis of multiple myeloma. Blood. (2012) 120:5002–13. doi: 10.1182/blood-2011-07-368142
192. Plotnikova OV, Golemis EA, Pugacheva EN. Cell cycle-dependent ciliogenesis and cancer. Cancer Res. (2008) 68:2058–61. doi: 10.1158/0008-5472.CAN-07-5838
193. Carballo GB, Honorato JR, de Lopes GPF, Spohr T. A highlight on Sonic hedgehog pathway. Cell Commun Signal. (2018) 16:11. doi: 10.1186/s12964-018-0220-7
194. Robbins DJ, Fei DL, Riobo NA. The Hedgehog signal transduction network. Sci Signal. (2012) 5:re6. doi: 10.1126/scisignal.2002906
195. Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. (2007) 25:2524–33. doi: 10.1634/stemcells.2007-0166
196. Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci USA. (2007) 104:4048–53. doi: 10.1073/pnas.0611682104
197. Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. (1996) 85:841–51. doi: 10.1016/S0092-8674(00)81268-4
198. Xie J, Johnson RL, Zhang X, Bare JW, Waldman FM, Cogen PH, et al. Mutations of the PATCHED gene in several types of sporadic extracutaneous tumors. Cancer Res. (1997) 57:2369–72.
199. Akyala AI, Peppelenbosch MP. Gastric cancer and Hedgehog signaling pathway: emerging new paradigms. Genes Cancer. (2018) 9:1–10. doi: 10.18632/genesandcancer.168
200. Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. (2007) 17:165–72. doi: 10.1016/j.cub.2006.11.033
201. Po A, Ferretti E, Miele E, De Smaele E, Paganelli A, Canettieri G, et al. Hedgehog controls neural stem cells through p53-independent regulation of Nanog. EMBO J. (2010) 29:2646–58. doi: 10.1038/emboj.2010.131
202. Justilien V, Walsh MP, Ali SA, Thompson EA, Murray NR, Fields AP. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell. (2014) 25:139–51. doi: 10.1016/j.ccr.2014.01.008
203. Sharpe HJ, Pau G, Dijkgraaf GJ, Basset-Seguin N, Modrusan Z, Januario T, et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell. (2015) 27:327–41. doi: 10.1016/j.ccell.2015.02.001
204. Benvenuto M, Masuelli L, De Smaele E, Fantini M, Mattera R, Cucchi D, et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the Hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget. (2016) 7:9250–70. doi: 10.18632/oncotarget.7062
205. Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW. Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers. (2016) 8:22. doi: 10.3390/cancers8020022
206. Carpenter RL, Ray H. Safety and Tolerability of sonic hedgehog pathway inhibitors in cancer. Drug Saf . (2019) 42:263–79. doi: 10.1007/s40264-018-0777-5
207. Han JB, Sang F, Chang JJ, Hua YQ, Shi WD, Tang LH, et al. Arsenic trioxide inhibits viability of pancreatic cancer stem cells in culture and in a xenograft model via binding to SHH-Gli. Onco Targets Ther. (2013) 6:1129–38. doi: 10.2147/OTT.S49148
208. Yu D, Shin HS, Lee YS, Lee D, Kim S, Lee YC. Genistein attenuates cancer stem cell characteristics in gastric cancer through the downregulation of Gli1. Oncol Rep. (2014) 31:673–8. doi: 10.3892/or.2013.2893
209. Calses PC, Crawford JJ, Lill JR, Dey A. Hippo pathway in cancer: aberrant regulation and therapeutic opportunities. Trends Cancer. (2019) 5:297–307. doi: 10.1016/j.trecan.2019.04.001
210. Nishio M, Otsubo K, Maehama T, Mimori K, Suzuki A. Capturing the mammalian Hippo: elucidating its role in cancer. Cancer Sci. (2013) 104:1271–7. doi: 10.1111/cas.12227
211. Li S, Cho YS, Yue T, Ip YT, Jiang J. Overlapping functions of the MAP4K family kinases Hppy and Msn in Hippo signaling. Cell Discov. (2015) 1:15038. doi: 10.1038/celldisc.2015.38
212. Meng Z, Moroishi T, Mottier-Pavie V, Plouffe SW, Hansen CG, Hong AW, et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat Commun. (2015) 6:8357. doi: 10.1038/ncomms9357
213. Zheng Y, Wang W, Liu B, Deng H, Uster E, Pan D. Identification of Happyhour/MAP4K as alternative Hpo/Mst-like kinases in the hippo kinase cascade. Dev Cell. (2015) 34:642–55. doi: 10.1016/j.devcel.2015.08.014
214. Plouffe SW, Meng Z, Lin KC, Lin B, Hong AW, Chun JV, et al. Characterization of hippo pathway components by gene inactivation. Mol Cell. (2016) 64:993–1008. doi: 10.1016/j.molcel.2016.10.034
215. Hong W, Guan KL. The YAP and TAZ transcription co-activators: key downstream effectors of the mammalian Hippo pathway. Semin Cell Dev Biol. (2012) 23:785–93. doi: 10.1016/j.semcdb.2012.05.004
216. Harvey K, Tapon N. The Salvador-Warts-Hippo pathway - an emerging tumour-suppressor network. Nat Rev Cancer. (2007) 7:182–91. doi: 10.1038/nrc2070
217. Liu CY, Zha ZY, Zhou X, Zhang H, Huang W, Zhao D, et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{β}−TrCP E3 ligase. J Biol Chem. (2010) 285:37159–69. doi: 10.1074/jbc.M110.152942
218. Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCFβ-TRCP. Genes Dev. (2010) 24:72–85. doi: 10.1101/gad.1843810
219. Totaro A, Panciera T, Piccolo S. YAP/TAZ upstream signals and downstream responses. Nat Cell Biol. (2018) 20:888–99. doi: 10.1038/s41556-018-0142-z
220. Korkola JE, Houldsworth J, Chadalavada RS, Olshen AB, Dobrzynski D, Reuter VE, et al. Down-regulation of stem cell genes, including those in a 200-kb gene cluster at 12p13.31, is associated with in vivo differentiation of human male germ cell tumors. Cancer Res. (2006) 66:820–7. doi: 10.1158/0008-5472.CAN-05-2445
221. Skotheim RI, Autio R, Lind GE, Kraggerud SM, Andrews PW, Monni O, et al. Novel genomic aberrations in testicular germ cell tumors by array-CGH, and associated gene expression changes. Cell Oncol. (2006) 28:315–26. doi: 10.1155/2006/219786
222. Han W, Jung EM, Cho J, Lee JW, Hwang KT, Yang SJ, et al. DNA copy number alterations and expression of relevant genes in triple-negative breast cancer. Genes Chromosomes Cancer. (2008) 47:490–9. doi: 10.1002/gcc.20550
223. Knight JF, Shepherd CJ, Rizzo S, Brewer D, Jhavar S, Dodson AR, et al. TEAD1 and c-Cbl are novel prostate basal cell markers that correlate with poor clinical outcome in prostate cancer. Br J Cancer. (2008) 99:1849–58. doi: 10.1038/sj.bjc.6604774
224. Fernandez LA, Northcott PA, Dalton J, Fraga C, Ellison D, Angers S, et al. YAP1 is amplified and up-regulated in hedgehog-associated medulloblastomas and mediates Sonic hedgehog-driven neural precursor proliferation. Genes Dev. (2009) 23:2729–41. doi: 10.1101/gad.1824509
225. Zhou GX, Li XY, Zhang Q, Zhao K, Zhang CP, Xue CH, et al. Effects of the hippo signaling pathway in human gastric cancer. Asian Pac J Cancer Prev. (2013) 14:5199–205. doi: 10.7314/APJCP.2013.14.9.5199
226. Liang K, Zhou G, Zhang Q, Li J, Zhang C. Expression of hippo pathway in colorectal cancer. Saudi J Gastroenterol. (2014) 20:188–94. doi: 10.4103/1319-3767.133025
227. Schütte U, Bisht S, Heukamp LC, Kebschull M, Florin A, Haarmann J, et al. Hippo signaling mediates proliferation, invasiveness, and metastatic potential of clear cell renal cell carcinoma. Transl Oncol. (2014) 7:309–21. doi: 10.1016/j.tranon.2014.02.005
228. Wang C, Nie Z, Zhou Z, Zhang H, Liu R, Wu J, et al. The interplay between TEAD4 and KLF5 promotes breast cancer partially through inhibiting the transcription of p27Kip1. Oncotarget. (2015) 6:17685–97. doi: 10.18632/oncotarget.3779
229. Liu Y, Wang G, Yang Y, Mei Z, Liang Z, Cui A, et al. Increased TEAD4 expression and nuclear localization in colorectal cancer promote epithelial-mesenchymal transition and metastasis in a YAP-independent manner. Oncogene. (2016) 35:2789–800. doi: 10.1038/onc.2015.342
230. Zhou Y, Huang T, Zhang J, Wong CC, Zhang B, Dong Y, et al. TEAD1/4 exerts oncogenic role and is negatively regulated by miR-4269 in gastric tumorigenesis. Oncogene. (2017) 36:6518–30. doi: 10.1038/onc.2017.257
231. Zhang W, Li J, Wu Y, Ge H, Song Y, Wang D, et al. TEAD4 overexpression promotes epithelial-mesenchymal transition and associates with aggressiveness and adverse prognosis in head neck squamous cell carcinoma. Cancer Cell Int. (2018) 18:178. doi: 10.1186/s12935-018-0675-z
232. Basu-Roy U, Bayin NS, Rattanakorn K, Han E, Placantonakis DG, Mansukhani A, et al. Sox2 antagonizes the Hippo pathway to maintain stemness in cancer cells. Nat Commun. (2015) 6:6411. doi: 10.1038/ncomms7411
233. Sorrentino G, Ruggeri N, Zannini A, Ingallina E, Bertolio R, Marotta C, et al. Glucocorticoid receptor signalling activates YAP in breast cancer. Nat Commun. (2017) 8:14073. doi: 10.1038/ncomms14073
234. Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. (2011) 147:759–72. doi: 10.1016/j.cell.2011.09.048
235. Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, et al. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci USA. (2006) 103:12405–10. doi: 10.1073/pnas.0605579103
236. Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, et al. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol. (2008) 28:2426–36. doi: 10.1128/MCB.01874-07
237. Varelas X, Sakuma R, Samavarchi-Tehrani P, Peerani R, Rao BM, Dembowy J, et al. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat Cell Biol. (2008) 10:837–48. doi: 10.1038/ncb1748
238. Zhao B, Ye X, Yu J, Li L, Li W, Li S, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. (2008) 22:1962–71. doi: 10.1101/gad.1664408
239. Zhang H, Liu CY, Zha ZY, Zhao B, Yao J, Zhao S, et al. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J Biol Chem. (2009) 284:13355–62. doi: 10.1074/jbc.M900843200
240. Lamar JM, Stern P, Liu H, Schindler JW, Jiang ZG, Hynes RO. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc Natl Acad Sci USA. (2012) 109:E2441–50. doi: 10.1073/pnas.1212021109
241. Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. (2012) 26:54–68. doi: 10.1101/gad.173435.111
242. Gao Y, Zhang W, Han X, Li F, Wang X, Wang R, et al. YAP inhibits squamous transdifferentiation of Lkb1-deficient lung adenocarcinoma through ZEB2-dependent DNp63 repression. Nat Commun. (2014) 5:4629. doi: 10.1038/ncomms5629
243. Lehmann W, Mossmann D, Kleemann J, Mock K, Meisinger C, Brummer T, et al. ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat Commun. (2016) 7:10498. doi: 10.1038/ncomms10498
244. Yu M, Chen Y, Li X, Yang R, Zhang L, Huangfu L, et al. YAP1 contributes to NSCLC invasion and migration by promoting Slug transcription via the transcription co-factor TEAD. Cell Death Dis. (2018) 9:464. doi: 10.1038/s41419-018-0515-z
245. Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. (2014) 158:171–84. doi: 10.1016/j.cell.2014.06.004
246. Bartucci M, Dattilo R, Moriconi C, Pagliuca A, Mottolese M, Federici G, et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene. (2015) 34:681–90. doi: 10.1038/onc.2014.5
247. Guo Y, Cui J, Ji Z, Cheng C, Zhang K, Zhang C, et al. miR-302/367/LATS2/YAP pathway is essential for prostate tumor-propagating cells and promotes the development of castration resistance. Oncogene. (2017) 36:6336–47. doi: 10.1038/onc.2017.240
248. Fernandez LA, Squatrito M, Northcott P, Awan A, Holland EC, Taylor MD, et al. Oncogenic YAP promotes radioresistance and genomic instability in medulloblastoma through IGF2-mediated Akt activation. Oncogene. (2012) 31:1923–37. doi: 10.1038/onc.2011.379
249. Mao B, Hu F, Cheng J, Wang P, Xu M, Yuan F, et al. SIRT1 regulates YAP2-mediated cell proliferation and chemoresistance in hepatocellular carcinoma. Oncogene. (2014) 33:1468–74. doi: 10.1038/onc.2013.88
250. Cheng H, Zhang Z, Rodriguez-Barrueco R, Borczuk A, Liu H, Yu J, et al. Functional genomics screen identifies YAP1 as a key determinant to enhance treatment sensitivity in lung cancer cells. Oncotarget. (2016) 7:28976–88. doi: 10.18632/oncotarget.6721
251. Ciamporcero E, Shen H, Ramakrishnan S, Yu Ku S, Chintala S, Shen L, et al. YAP activation protects urothelial cell carcinoma from treatment-induced DNA damage. Oncogene. (2016) 35:1541–53. doi: 10.1038/onc.2015.219
252. Zhang J, Ji JY, Yu M, Overholtzer M, Smolen GA, Wang R, et al. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat Cell Biol. (2009) 11:1444–50. doi: 10.1038/ncb1993
253. Busser B, Sancey L, Josserand V, Niang C, Favrot MC, Coll JL, et al. Amphiregulin promotes BAX inhibition and resistance to gefitinib in non-small-cell lung cancers. Mol Ther. (2010) 18:528–35. doi: 10.1038/mt.2009.226
254. O'Connell MP, Marchbank K, Webster MR, Valiga AA, Kaur A, Vultur A, et al. Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov. (2013) 3:1378–93. doi: 10.1158/2159-8290.CD-13-0005
255. Anastas JN, Kulikauskas RM, Tamir T, Rizos H, Long GV, von Euw EM, et al. WNT5A enhances resistance of melanoma cells to targeted BRAF inhibitors. J Clin Invest. (2014) 124:2877–90. doi: 10.1172/JCI70156
256. Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E, et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet. (2015) 47:250–6. doi: 10.1038/ng.3218
257. Park HW, Kim YC, Yu B, Moroishi T, Mo JS, Plouffe SW, et al. Alternative Wnt signaling activates YAP/TAZ. Cell. (2015) 162:780–94. doi: 10.1016/j.cell.2015.07.013
258. Tung SL, Huang WC, Hsu FC, Yang ZP, Jang TH, Chang JW, et al. miRNA-34c-5p inhibits amphiregulin-induced ovarian cancer stemness and drug resistance via downregulation of the AREG-EGFR-ERK pathway. Oncogenesis. (2017) 6:e326. doi: 10.1038/oncsis.2017.25
259. Chen Q, Zhang N, Gray RS, Li H, Ewald AJ, Zahnow CA, et al. A temporal requirement for Hippo signaling in mammary gland differentiation, growth, and tumorigenesis. Genes Dev. (2014) 28:432–7. doi: 10.1101/gad.233676.113
260. Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. (2016) 29:783–803. doi: 10.1016/j.ccell.2016.05.005
261. Lo Sardo F, Forcato M, Sacconi A, Capaci V, Zanconato F, Di Agostino S, et al. MCM7 and its hosted miR-25, 93 and 106b cluster elicit YAP/TAZ oncogenic activity in lung cancer. Carcinogenesis. (2017) 38:64–75. doi: 10.1093/carcin/bgw110
262. Lo Sardo F, Strano S, Blandino G. YAP and TAZ in lung cancer: oncogenic role and clinical targeting. Cancers. (2018) 10:137. doi: 10.3390/cancers10050137
263. Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. (2012) 26:1300–5. doi: 10.1101/gad.192856.112
264. Liu K, Du S, Gao P, Zheng J. Verteporfin suppresses the proliferation, epithelial-mesenchymal transition and stemness of head and neck squamous carcinoma cells via inhibiting YAP1. J Cancer. (2019) 10:4196–207. doi: 10.7150/jca.34145
265. Brodowska K, Al-Moujahed A, Marmalidou A, Meyer Zu Horste M, Cichy J, Miller JW, et al. The clinically used photosensitizer Verteporfin (VP) inhibits YAP-TEAD and human retinoblastoma cell growth in vitro without light activation. Exp Eye Res. (2014) 124:67–73. doi: 10.1016/j.exer.2014.04.011
266. Gibault F, Corvaisier M, Bailly F, Huet G, Melnyk P, Cotelle P. Non-photoinduced biological properties of verteporfin. Curr Med Chem. (2016) 23:1171–84. doi: 10.2174/0929867323666160316125048
267. Shah SR, Kim J, Schiapparelli P, Vazquez-Ramos CA, Martinez-Gutierrez JC, Ruiz-Valls A, et al. Verteporfin-loaded polymeric microparticles for intratumoral treatment of brain cancer. Mol Pharm. (2019) 16:1433–43. doi: 10.1021/acs.molpharmaceut.8b00959
268. Michy T, Massias T, Bernard C, Vanwonterghem L, Henry M, Guidetti M, et al. Verteporfin-loaded lipid nanoparticles improve ovarian cancer photodynamic therapy in vitro and in vivo. Cancers. (2019) 11:1760. doi: 10.3390/cancers11111760
269. Shamul JG, Shah SR, Kim J, Schiapparelli P, Vazquez-Ramos CA, Lee BJ, et al. Verteporfin-loaded anisotropic poly(β-amino ester)-based micelles demonstrate brain cancer-selective cytotoxicity and enhanced pharmacokinetics. Int J Nanomed. (2019) 14:10047–60. doi: 10.2147/IJN.S231167
270. Song S, Xie M, Scott AW, Jin J, Ma L, Dong X, et al. A novel YAP1 inhibitor targets CSC-enriched radiation-resistant cells and exerts strong antitumor activity in esophageal adenocarcinoma. Mol Cancer Ther. (2018) 17:443–54. doi: 10.1158/1535-7163.MCT-17-0560
271. Kandasamy S, Adhikary G, Rorke EA, Friedberg JS, Mickle MB, Alexander HR, et al. The YAP1 signaling inhibitors, verteporfin and CA3, suppress the mesothelioma cancer stem cell phenotype. Mol Cancer Res. (2020) 18:343. doi: 10.1158/1541-7786.MCR-19-0914
272. Chan P, Han X, Zheng B, DeRan M, Yu J, Jarugumilli GK, et al. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat Chem Biol. (2016) 12:282–9. doi: 10.1038/nchembio.2036
273. Noland CL, Gierke S, Schnier PD, Murray J, Sandoval WN, Sagolla M, et al. Palmitoylation of TEAD transcription factors is required for their stability and function in Hippo pathway signaling. Structure. (2016) 24:179–86. doi: 10.1016/j.str.2015.11.005
274. Mesrouze Y, Meyerhofer M, Bokhovchuk F, Fontana P, Zimmermann C, Martin T, et al. Effect of the acylation of TEAD4 on its interaction with co-activators YAP and TAZ. Protein Sci. (2017) 26:2399–409. doi: 10.1002/pro.3312
275. Pobbati AV, Han X, Hung AW, Weiguang S, Huda N, Chen GY, et al. Targeting the central pocket in human transcription factor TEAD as a potential cancer therapeutic strategy. Structure. (2015) 23:2076–86. doi: 10.1016/j.str.2015.09.009
276. Bum-Erdene K, Zhou D, Gonzalez-Gutierrez G, Ghozayel MK, Si Y, Xu D, et al. Small-molecule covalent modification of conserved cysteine leads to allosteric inhibition of the TEAD·yap protein-protein interaction. Cell Chem Biol. (2019) 26:378–89.e313. doi: 10.1016/j.chembiol.2018.11.010
277. Zhang Z, Lin Z, Zhou Z, Shen HC, Yan SF, Mayweg AV, et al. Structure-based design and synthesis of potent cyclic peptides inhibiting the YAP-TEAD protein-protein interaction. ACS Med Chem Lett. (2014) 5:993–8. doi: 10.1021/ml500160m
278. Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X, et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell. (2014) 25:166–80. doi: 10.1016/j.ccr.2014.01.010
279. Gibault F, Sturbaut M, Bailly F, Melnyk P, Cotelle P. Targeting transcriptional enhanced associate domains (TEADs). J Med Chem. (2018) 61:5057–72. doi: 10.1021/acs.jmedchem.7b00879
280. Yan F, Qian M, He Q, Zhu H, Yang B. The posttranslational modifications of Hippo-YAP pathway in cancer. Biochim Biophys Acta Gen Subj. (2020) 1864:129397. doi: 10.1016/j.bbagen.2019.07.006
281. Nakatani K, Maehama T, Nishio M, Goto H, Kato W, Omori H, et al. Targeting the Hippo signalling pathway for cancer treatment. J Biochem. (2017) 161:237–44. doi: 10.1093/jb/mvw074
282. Basu D, Lettan R, Damodaran K, Strellec S, Reyes-Mugica M, Rebbaa A. Identification, mechanism of action, and antitumor activity of a small molecule inhibitor of hippo, TGF-β, and Wnt signaling pathways. Mol Cancer Ther. (2014) 13:1457–67. doi: 10.1158/1535-7163.MCT-13-0918
283. Zhou TY, Zhuang LH, Hu Y, Zhou YL, Lin WK, Wang DD, et al. Inactivation of hypoxia-induced YAP by statins overcomes hypoxic resistance tosorafenib in hepatocellular carcinoma cells. Sci Rep. (2016) 6:30483. doi: 10.1038/srep30483
284. Morales DR, Morris AD. Metformin in cancer treatment and prevention. Annu Rev Med. (2015) 66:17–29. doi: 10.1146/annurev-med-062613-093128
285. Chimento A, Casaburi I, Avena P, Trotta F, De Luca A, Rago V, et al. Cholesterol and its metabolites in tumor growth: therapeutic potential of statins in cancer treatment. Front Endocrinol. (2019) 9:807. doi: 10.3389/fendo.2018.00807
286. Heinemann A, Cullinane C, De Paoli-Iseppi R, Wilmott JS, Gunatilake D, Madore J, et al. Combining BET and HDAC inhibitors synergistically induces apoptosis of melanoma and suppresses AKT and YAP signaling. Oncotarget. (2015) 6:21507–21. doi: 10.18632/oncotarget.4242
287. Patnaik S, Anupriya. Drugs targeting epigenetic modifications and plausible therapeutic strategies against colorectal cancer. Front Pharmacol. (2019) 10:588. doi: 10.3389/fphar.2019.00588
288. Hoebe K, Jiang Z, Georgel P, Tabeta K, Janssen E, Du X, et al. TLR signaling pathways: opportunities for activation and blockade in pursuit of therapy. Curr Pharm Des. (2006) 12:4123–34. doi: 10.2174/138161206778743466
289. Chen K, Huang J, Gong W, Iribarren P, Dunlop NM, Wang JM. Toll-like receptors in inflammation, infection and cancer. Int Immunopharmacol. (2007) 7:1271–85. doi: 10.1016/j.intimp.2007.05.016
290. Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediat Inflamm. (2010) 2010:672395. doi: 10.1155/2010/672395
291. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. (2006) 124:783–801. doi: 10.1016/j.cell.2006.02.015
292. Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF. A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci USA. (1998) 95:588–93. doi: 10.1073/pnas.95.2.588
293. Martin MU, Wesche H. Summary and comparison of the signaling mechanisms of the Toll/interleukin-1 receptor family. Biochim Biophys Acta. (2002) 1592:265–80. doi: 10.1016/S0167-4889(02)00320-8
294. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. (2010) 11:373–84. doi: 10.1038/ni.1863
295. O'Neill LA, Fitzgerald KA, Bowie AG. The Toll-IL-1 receptor adaptor family grows to five members. Trends Immunol. (2003) 24:286–90. doi: 10.1016/S1471-4906(03)00115-7
296. O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. (2007) 7:353–64. doi: 10.1038/nri2079
297. Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-β signal transduction. Science. (1995) 270:2008–11. doi: 10.1126/science.270.5244.2008
298. Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. The kinase TAK1 can activate the NIK-I κB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. (1999) 398:252–6. doi: 10.1038/18465
299. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. (2001) 412:346–51. doi: 10.1038/35085597
300. Pandey S, Agrawal DK. Immunobiology of Toll-like receptors: emerging trends. Immunol Cell Biol. (2006) 84:333–41. doi: 10.1111/j.1440-1711.2006.01444.x
301. Basith S, Manavalan B, Lee G, Kim SG, Choi S. Toll-like receptor modulators: a patent review (2006-2010). Expert Opin Ther Pat. (2011) 21:927–44. doi: 10.1517/13543776.2011.569494
302. Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. (2004) 5:987–95. doi: 10.1038/ni1112
303. Boudreau JE, Bonehill A, Thielemans K, Wan Y. Engineering dendritic cells to enhance cancer immunotherapy. Mol Ther. (2011) 19:841–53. doi: 10.1038/mt.2011.57
304. Fukata M, Abreu MT. Role of Toll-like receptors in gastrointestinal malignancies. Oncogene. (2008) 27:234–43. doi: 10.1038/sj.onc.1210908
305. Alvarado AG, Thiagarajan PS, Mulkearns-Hubert EE, Silver DJ, Hale JS, Alban TJ, et al. Glioblastoma cancer stem cells evade innate immune suppression of self-renewal through reduced TLR4 expression. Cell Stem Cell. (2017) 20:450–61.e454. doi: 10.1016/j.stem.2016.12.001
306. Takizawa H, Fritsch K, Kovtonyuk LV, Saito Y, Yakkala C, Jacobs K, et al. Pathogen-Induced TLR4-TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell. (2017) 21:225–40.e225. doi: 10.1016/j.stem.2017.06.013
307. Chefetz I, Alvero AB, Holmberg JC, Lebowitz N, Craveiro V, Yang-Hartwich Y, et al. TLR2 enhances ovarian cancer stem cell self-renewal and promotes tumor repair and recurrence. Cell Cycle. (2013) 12:511–21. doi: 10.4161/cc.23406
308. Jia D, Yang W, Li L, Liu H, Tan Y, Ooi S, et al. β-Catenin and NF-κB co-activation triggered by TLR3 stimulation facilitates stem cell-like phenotypes in breast cancer. Cell Death Differ. (2015) 22:298–310. doi: 10.1038/cdd.2014.145
309. Uthaya Kumar DB, Chen CL, Liu JC, Feldman DE, Sher LS, French S, et al. TLR4 Signaling via NANOG cooperates with STAT3 to activate Twist1 and promote formation of tumor-initiating stem-like cells in livers of mice. Gastroenterology. (2016) 150:707–19. doi: 10.1053/j.gastro.2015.11.002
310. Chen CL, Tsukamoto H, Liu JC, Kashiwabara C, Feldman D, Sher L, et al. Reciprocal regulation by TLR4 and TGF-β in tumor-initiating stem-like cells. J Clin Invest. (2013) 123:2832–49. doi: 10.1172/JCI65859
311. Liu WT, Jing YY, Yu GF, Han ZP, Yu DD, Fan QM, et al. Toll like receptor 4 facilitates invasion and migration as a cancer stem cell marker in hepatocellular carcinoma. Cancer Lett. (2015) 358:136–43. doi: 10.1016/j.canlet.2014.12.019
312. Zhao XL, Lin Y, Jiang J, Tang Z, Yang S, Lu L, et al. High-mobility group box 1 released by autophagic cancer-associated fibroblasts maintains the stemness of luminal breast cancer cells. J Pathol. (2017) 243:376–89. doi: 10.1002/path.4958
313. Grimm M, Kim M, Rosenwald A, Heemann U, Germer CT, Waaga-Gasser AM, et al. Toll-like receptor (TLR) 7 and TLR8 expression on CD133+ cells in colorectal cancer points to a specific role for inflammation-induced TLRs in tumourigenesis and tumour progression. Eur J Cancer. (2010) 46:2849–57. doi: 10.1016/j.ejca.2010.07.017
314. Moreira D, Zhang Q, Hossain DM, Nechaev S, Li H, Kowolik CM, et al. TLR9 signaling through NF-κB/RELA and STAT3 promotes tumor-propagating potential of prostate cancer cells. Oncotarget. (2015) 6:17302–13. doi: 10.18632/oncotarget.4029
315. Herrmann A, Cherryholmes G, Schroeder A, Phallen J, Alizadeh D, Xin H, et al. TLR9 is critical for glioma stem cell maintenance and targeting. Cancer Res. (2014) 74:5218–28. doi: 10.1158/0008-5472.CAN-14-1151
316. Chiou SH, Yu CC, Huang CY, Lin SC, Liu CJ, Tsai TH, et al. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin Cancer Res. (2008) 14:4085–95. doi: 10.1158/1078-0432.CCR-07-4404
317. Kumar SM, Liu S, Lu H, Zhang H, Zhang PJ, Gimotty PA, et al. Acquired cancer stem cell phenotypes through Oct4-mediated dedifferentiation. Oncogene. (2012) 31:4898–911. doi: 10.1038/onc.2011.656
318. Zhou C, Liu J, Tang Y, Liang X. Inflammation linking EMT and cancer stem cells. Oral Oncol. (2012) 48:1068–75. doi: 10.1016/j.oraloncology.2012.06.005
319. Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. (2014) 511:246–50. doi: 10.1038/nature13305
320. Gu J, Liu Y, Xie B, Ye P, Huang J, Lu Z. Roles of toll-like receptors: from inflammation to lung cancer progression. Biomed Rep. (2018) 8:126–32. doi: 10.3892/br.2017.1034
322. Dias LP, Luzo ÂCM, Volpe BB, Durán M, Galdames SEM, Ferreira LAB, et al. Effects of intravesical therapy with platelet-rich plasma (PRP) and Bacillus Calmette-Guérin (BCG) in non-muscle invasive bladder cancer. Tissue and Cell. (2018) 52:17–27. doi: 10.1016/j.tice.2018.03.011
323. Babjuk M, Burger M, Compérat EM, Gontero P, Mostafid AH, Palou J, et al. European Association of urology guidelines on non-muscle-invasive bladder cancer (TaT1 and Carcinoma in situ) - 2019 update. Eur Urol. (2019) 76:639–57. doi: 10.1016/j.eururo.2019.08.016
324. Packiam VT, Lamm DL, Barocas DA, Trainer A, Fand B, Davis RL, et al. An open label, single-arm, phase II multicenter study of the safety and efficacy of CG0070 oncolytic vector regimen in patients with BCG-unresponsive non–muscle-invasive bladder cancer: interim results. Urol Oncol. (2018) 36:440–7. doi: 10.1016/j.urolonc.2017.07.005
325. Golla V, Lenis AT, Faiena I, Chamie K. Intravesical Therapy for Non-muscle invasive bladder cancer-current and future options in the age of bacillus calmette-guerin shortage. Rev Urol. (2019) 21:145–53.
326. Moradi-Marjaneh R, Hassanian SM, Fiuji H, Soleimanpour S, Ferns GA, Avan A, et al. Toll like receptor signaling pathway as a potential therapeutic target in colorectal cancer. J Cell Physiol. (2018) 233:5613–22. doi: 10.1002/jcp.26273
327. Lemdani K, Mignet N, Boudy V, Seguin J, Oujagir E, Bawa O, et al. Local immunomodulation combined to radiofrequency ablation results in a complete cure of local and distant colorectal carcinoma. Oncoimmunology. (2019) 8:1550342. doi: 10.1080/2162402X.2018.1550342
328. Lotem M, Merims S, Frank S, Hamburger T, Nissan A, Kadouri L, et al. Adjuvant autologous melanoma vaccine for macroscopic stage III disease: survival, biomarkers, and improved response to CTLA-4 blockade. J Immunol Res. (2016) 2016:8121985. doi: 10.1155/2016/8121985
329. Faries MB, Mozzillo N, Kashani-Sabet M, Thompson JF, Kelley MC, DeConti RC, et al. Long-term survival after complete surgical resection and adjuvant immunotherapy for distant melanoma metastases. Ann Surg Oncol. (2017) 24:3991–4000. doi: 10.1245/s10434-017-6072-3
330. Nishida S, Tsuboi A, Tanemura A, Ito T, Nakajima H, Shirakata T, et al. Immune adjuvant therapy using Bacillus Calmette-Guérin cell wall skeleton (BCG-CWS) in advanced malignancies: a phase 1 study of safety and immunogenicity assessments. Medicine. (2019) 98:e16771. doi: 10.1097/MD.0000000000016771
331. Qiu YF, Liu ZG, Yang WJ, Zhao Y, Tang J, Tang WZ, et al. Research progress in the treatment of small cell lung cancer. J Cancer. (2017) 8:29–38. doi: 10.7150/jca.16822
332. Rakshit S, Ponnusamy M, Papanna S, Saha B, Ahmed A, Nandi D. Immunotherapeutic efficacy of Mycobacterium indicus pranii in eliciting anti-tumor T cell responses: critical roles of IFNγ. Int J Cancer. (2012) 130:865–75. doi: 10.1002/ijc.26099
333. Belani CP, Chakraborty BC, Modi RI, Khamar BM. A randomized trial of TLR-2 agonist CADI-05 targeting desmocollin-3 for advanced non-small-cell lung cancer††This study was previously presented as: an oral presentation at the annual meeting of the American Society of Clinical Oncology (ASCO), June 3–6, 2011, Chicago, IL, USA, and at the 14th World conference on Lung Cancer, July 3–7, 2011, Amsterdam, Netherlands. Ann Oncol. (2017) 28:298–304. doi: 10.1093/annonc/mdw608
334. Bhatia S, Miller NJ, Lu H, Longino NV, Ibrani D, Shinohara MM, et al. Intratumoral G100, a TLR4 agonist, induces antitumor immune responses and tumor regression in patients with merkel cell carcinoma. Clin Cancer Res. (2019) 25:1185–95. doi: 10.1158/1078-0432.CCR-18-0469
335. Cluff CW. Monophosphoryl lipid A (MPL) as an adjuvant for anti-cancer vaccines: clinical results. Adv Exp Med Biol. (2010) 667:111–23. doi: 10.1007/978-1-4419-1603-7_10
336. Slingluff CL Jr, Petroni GR, Olson WC, Smolkin ME, Chianese-Bullock KA, Mauldin IS. A randomized pilot trial testing the safety and immunologic effects of a MAGE-A3 protein plus AS15 immunostimulant administered into muscle or into dermal/subcutaneous sites. Cancer Immunol Immunother. (2016) 65:25–36. doi: 10.1007/s00262-015-1770-9
337. Casella CR, Mitchell TC. Putting endotoxin to work for us: monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell Mol Life Sci. (2008) 65:3231–40. doi: 10.1007/s00018-008-8228-6
338. Ammi R, De Waele J, Willemen Y, Van Brussel I, Schrijvers DM, Lion E, et al. Poly(I:C) as cancer vaccine adjuvant: knocking on the door of medical breakthroughs. Pharmacol Ther. (2015) 146:120–31. doi: 10.1016/j.pharmthera.2014.09.010
339. Salazar AM, Erlich RB, Mark A, Bhardwaj N, Herberman RB. Therapeutic in situ autovaccination against solid cancers with intratumoral poly-ICLC: case report, hypothesis, clinical trial. Cancer Immunol Res. (2014) 2:720–4. doi: 10.1158/2326-6066.CIR-14-0024
340. Garaude J, Kent A, van Rooijen N, Blander JM. Simultaneous targeting of toll- and nod-like receptors induces effective tumor-specific immune responses. Sci. Transl. Med. (2012) 4:120ra116. doi: 10.1126/scitranslmed.3002868
341. Müller E, Christopoulos PF, Halder S, Lunde A, Beraki K, Speth M, et al. Toll-like receptor ligands and interferon-γ synergize for induction of antitumor M1 macrophages. Front Immunol. (2017) 8:1383. doi: 10.3389/fimmu.2017.01383
342. Zheng JH, Nguyen VH, Jiang S-N, Park S-H, Tan W, Hong SH, et al. Two-step enhanced cancer immunotherapy with engineered Salmonella typhimurium secreting heterologous flagellin. Sci Transl Med. (2017) 9:eaak9537. doi: 10.1126/scitranslmed.aak9537
343. Bakhribah H, Dy GK, Ma WW, Zhao Y, Opyrchal M, Purmal A, et al. A phase I study of the toll-like receptor 5 (TLR5) agonist, entolimod in patients (pts) with advanced cancers. J Clin Oncol. (2015) 33(15_suppl):3063. doi: 10.1200/jco.2015.33.15_suppl.3063
344. Toshkov IA, Gleiberman AS, Mett VL, Hutson AD, Singh AK, Gudkov AV, et al. Mitigation of radiation-induced epithelial damage by the TLR5 agonist entolimod in a mouse model of fractionated head and neck irradiation. Radiat Res. (2017) 187:570–80. doi: 10.1667/RR14514.1
345. Bichakjian C, Armstrong A, Baum C, Bordeaux JS, Brown M, Busam KJ, et al. Guidelines of care for the management of basal cell carcinoma. J Am Acad Dermatol. (2018) 78:540–59. doi: 10.1016/j.jaad.2017.10.006
346. Jansen MHE, Mosterd K, Arits AHMM, Roozeboom MH, Sommer A, Essers BAB, et al. Five-year results of a randomized controlled trial comparing effectiveness of photodynamic therapy, topical imiquimod, and topical 5-fluorouracil in patients with superficial basal cell carcinoma. J Invest Dermatol. (2018) 138:527–33. doi: 10.1016/j.jid.2017.09.033
347. Rajpar SF, Marsden JR. Imiquimod in the treatment of lentigo maligna. Br J Dermatol. (2006) 155:653–6. doi: 10.1111/j.1365-2133.2006.07476.x
348. Martínez-González MC, Verea-Hernando MM, Yebra-Pimentel MT, Del Pozo J, Mazaira M, Fonseca E. Imiquimod in mycosis fungoides. Eur J Dermatol. (2008) 18:148–52
349. Salazar LG, Lu H, Reichow JL, Childs JS, Coveler AL, Higgins DM, et al. Topical imiquimod plus Nab-paclitaxel for breast cancer cutaneous metastases: a phase 2 clinical trial. JAMA Oncol. (2017) 3:969–73. doi: 10.1001/jamaoncol.2016.6007
350. Adams S, O'Neill DW, Nonaka D, Hardin E, Chiriboga L, Siu K, et al. Immunization of malignant melanoma patients with full-length NY-ESO-1 protein using TLR7 agonist imiquimod as vaccine adjuvant. J Immunol. (2008) 181:776–84. doi: 10.4049/jimmunol.181.1.776
351. Mauldin IS, Wages NA, Stowman AM, Wang E, Olson WC, Deacon DH. Topical treatment of melanoma metastases with imiquimod, plus administration of a cancer vaccine, promotes immune signatures in the metastases. Cancer Immunol Immunother. (2016) 65:1201–12. doi: 10.1007/s00262-016-1880-z
352. Papanikolaou M, Lawrence CM. Long-term outcomes of imiquimod-treated lentigo maligna. Clin Exp Dermatol. (2019) 44:631–6. doi: 10.1111/ced.13896
353. Meyer T, Surber C, French LE, Stockfleth E. Resiquimod, a topical drug for viral skin lesions and skin cancer. Expert Opin Investig Drugs. (2013) 22:149–59. doi: 10.1517/13543784.2013.749236
354. Rook AH, Gelfand JM, Wysocka M, Troxel AB, Benoit B, Surber C, et al. Topical resiquimod can induce disease regression and enhance T-cell effector functions in cutaneous T-cell lymphoma. Blood. (2015) 126:1452–61. doi: 10.1182/blood-2015-02-630335
355. Sabado RL, Pavlick A, Gnjatic S, Cruz CM, Vengco I, Hasan F, et al. Resiquimod as an immunologic adjuvant for NY-ESO-1 protein vaccination in patients with high-risk melanoma. Cancer Immunol Res. (2015) 3:278–87. doi: 10.1158/2326-6066.CIR-14-0202
356. Frank MJ, Reagan PM, Bartlett NL, Gordon LI, Friedberg JW, Czerwinski DK, et al. In situ vaccination with a TLR9 agonist and local low-dose radiation induces systemic responses in untreated indolent lymphoma. Cancer Discov. (2018) 8:1258–69. doi: 10.1158/2159-8290.CD-18-0743
357. Kell SA, Kachura MA, Renn A, Traquina P, Coffman RL, Campbell JD. Preclinical development of the TLR9 agonist DV281 as an inhaled aerosolized immunotherapeutic for lung cancer: pharmacological profile in mice, non-human primates, and human primary cells. Int Immunopharmacol. (2019) 66:296–308. doi: 10.1016/j.intimp.2018.11.019
358. Ishihara M, Tono Y, Miyahara Y, Muraoka D, Harada N, Kageyama S, et al. First-in-human phase I clinical trial of the NY-ESO-1 protein cancer vaccine with NOD2 and TLR9 stimulants in patients with NY-ESO-1-expressing refractory solid tumors. Cancer Immunol Immunother. (2020) 69:663–75. doi: 10.1007/s00262-020-02483-1
359. Wang D, Jiang W, Zhu F, Mao X, Agrawal S. Modulation of the tumor microenvironment by intratumoral administration of IMO-2125, a novel TLR9 agonist, for cancer immunotherapy. Int J Oncol. (2018) 53:1193–203. doi: 10.3892/ijo.2018.4456
360. Zent CS, Smith BJ, Ballas ZK, Wooldridge JE, Link BK, Call TG, et al. Phase I clinical trial of CpG oligonucleotide 7909. (PF-03512676) in patients with previously treated chronic lymphocytic leukemia. Leuk Lymphoma. (2012) 53:211–7. doi: 10.3109/10428194.2011.608451
361. Xing N, Qiao T, Zhuang X, Yuan S, Zhang Q, Xu G. CpG oligodeoxyribonucleotide 7909 enhances radiosensitivity via downregulating Oct-4 expression in radioresistant lung cancer cells. Onco Targets Ther. (2015) 8:1443–9. doi: 10.2147/OTT.S84467
362. Chen X, Zhang Q, Luo Y, Gao C, Zhuang X, Xu G, et al. High-dose irradiation in combination with toll-like receptor 9 agonist CpG oligodeoxynucleotide 7909 downregulates PD-L1 expression via the NF-κB signaling pathway in non-small cell lung cancer cells. Onco Targets Ther. (2016) 9:6511–8. doi: 10.2147/OTT.S116629
363. Adamus T, Kortylewski M. The revival of CpG oligonucleotide-based cancer immunotherapies. Contemporary oncology. (2018) 22:56–60. doi: 10.5114/wo.2018.73887
364. Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov. (2006) 5:471–84. doi: 10.1038/nrd2059
365. Krieg AM. CpG still rocks! Update on an accidental drug. Nucl Acid Ther. (2012) 22:77–89. doi: 10.1089/nat.2012.0340
366. Fort MM, Mozaffarian A, Stöver AG, Correia Jda S, Johnson DA, Crane RT, et al. A synthetic TLR4 antagonist has anti-inflammatory effects in two murine models of inflammatory bowel disease. J Immunol. (2005) 174:6416–23. doi: 10.4049/jimmunol.174.10.6416
367. Sha T, Sunamoto M, Kitazaki T, Sato J, Ii M, Iizawa Y. Therapeutic effects of TAK-242, a novel selective Toll-like receptor 4 signal transduction inhibitor, in mouse endotoxin shock model. Eur J Pharmacol. (2007) 571:231–9. doi: 10.1016/j.ejphar.2007.06.027
368. Arslan F, Houtgraaf JH, Keogh B, Kazemi K, de Jong R, McCormack WJ, et al. Treatment with OPN-305, a humanized anti-Toll-Like receptor-2 antibody, reduces myocardial ischemia/reperfusion injury in pigs. Circ Cardiovasc Interv. (2012) 5:279–87. doi: 10.1161/CIRCINTERVENTIONS.111.967596
369. Huang HY, Zhang ZJ, Cao CB, Wang N, Liu FF, Peng JQ, et al. The TLR4/NF-κB signaling pathway mediates the growth of colon cancer. Eur Rev Med Pharmacol Sci. (2014) 18:3834–43.
370. Wang W, Wang J. Toll-like receptor 4 (TLR4)/cyclooxygenase-2 (COX-2) regulates prostate cancer cell proliferation, migration, and invasion by NF-κB activation. Med Sci Monit. (2018) 24:5588–97. doi: 10.12659/MSM.906857
371. Kashani B, Zandi Z, Karimzadeh MR, Bashash D, Nasrollahzadeh A, Ghaffari SH. Blockade of TLR4 using TAK-242 (resatorvid) enhances anti-cancer effects of chemotherapeutic agents: a novel synergistic approach for breast and ovarian cancers. Immunol Res. (2019) 67:505–16. doi: 10.1007/s12026-019-09113-8
372. Zandi Z, Kashani B, Poursani EM, Bashash D, Kabuli M, Momeny M, et al. TLR4 blockade using TAK-242 suppresses ovarian and breast cancer cells invasion through the inhibition of extracellular matrix degradation and epithelial-mesenchymal transition. Eur J Pharmacol. (2019) 853:256–63. doi: 10.1016/j.ejphar.2019.03.046
373. Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. (2012) 21:504–16. doi: 10.1016/j.ccr.2012.02.007
374. Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. (2012) 491:254–8. doi: 10.1038/nature11465
375. Li R, Zhou R, Wang H, Li W, Pan M, Yao X, et al. Gut microbiota-stimulated cathepsin K secretion mediates TLR4-dependent M2 macrophage polarization and promotes tumor metastasis in colorectal cancer. Cell Death Differ. (2019) 26:2447–63. doi: 10.1038/s41418-019-0312-y
376. Chang CW, Lee HC, Li LH, Chiang Chiau JS, Wang TE, Chuang WH, et al. Fecal microbiota transplantation prevents intestinal injury, Upregulation of Toll-like receptors, and 5-fluorouracil/oxaliplatin-induced toxicity in colorectal cancer. Int J Mol Sci. (2020) 21:386. doi: 10.3390/ijms21020386
377. Kyi C, Sabado RL, Blazquez A, Posner MR, Genden EM, Miles BA, et al. A phase I study of the safety and immunogenicity of a multipeptide personalized genomic vaccine in the adjuvant treatment of solid cancers. J Clin Oncol. (2017) 35(15_suppl):TPS3114. doi: 10.1200/JCO.2017.35.15_suppl.TPS3114
378. Sato-Kaneko F, Yao S, Ahmadi A, Zhang SS, Hosoya T, Kaneda MM, et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight. (2017) 2:e93397. doi: 10.1172/jci.insight.93397
379. Takeda Y, Kataoka K, Yamagishi J, Ogawa S, Seya T, Matsumoto M. A TLR3-specific adjuvant relieves innate resistance to PD-L1 blockade without cytokine toxicity in tumor vaccine immunotherapy. Cell Rep. (2017) 19:1874–87. doi: 10.1016/j.celrep.2017.05.015
380. Lai Y, Weng J, Wei X, Qin L, Lai P, Zhao R, et al. Toll-like receptor 2 costimulation potentiates the antitumor efficacy of CAR T Cells. Leukemia. (2018) 32:801–8. doi: 10.1038/leu.2017.249
381. Wang JQ, Jeelall YS, Ferguson LL, Horikawa K. Toll-like receptors and cancer: MYD88 mutation and inflammation. Front Immunol. (2014) 5:367. doi: 10.3389/fimmu.2014.00367
382. Treon SP, Xu L, Hunter Z. MYD88 mutations and response to ibrutinib in Waldenström's macroglobulinemia. N Engl J Med. (2015) 373:584–6. doi: 10.1056/NEJMc1506192
383. Phelan JD, Young RM, Webster DE, Roulland S, Wright GW, Kasbekar M, et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature. (2018) 560:387–91. doi: 10.1038/s41586-018-0290-0
384. Zhang D, Li L, Jiang H, Knolhoff BL, Lockhart AC, Wang-Gillam A, et al. Constitutive IRAK4 activation underlies poor prognosis and chemoresistance in pancreatic ductal adenocarcinoma. Clin Cancer Res. (2017) 23:1748–59. doi: 10.1158/1078-0432.CCR-16-1121
385. Smith MA, Choudhary GS, Pellagatti A, Choi K, Bolanos LC, Bhagat TD, et al. U2AF1 mutations induce oncogenic IRAK4 isoforms and activate innate immune pathways in myeloid malignancies. Nat Cell Biol. (2019) 21:640–50. doi: 10.1038/s41556-019-0314-5
386. Fang J, Rhyasen G, Bolanos L, Rasch C, Varney M, Wunderlich M, et al. Cytotoxic effects of bortezomib in myelodysplastic syndrome/acute myeloid leukemia depend on autophagy-mediated lysosomal degradation of TRAF6 and repression of PSMA1. Blood. (2012) 120:858–67. doi: 10.1182/blood-2012-02-407999
387. Burdelya LG, Brackett CM, Kojouharov B, Gitlin II, Leonova KI, Gleiberman AS, et al. Central role of liver in anticancer and radioprotective activities of Toll-like receptor 5 agonist. Proc Natl Acad Sci USA. (2013) 110:E1857–66. doi: 10.1073/pnas.1222805110
388. Kojouharov BM, Brackett CM, Veith JM, Johnson CP, Gitlin II, Toshkov IA, et al. Toll-like receptor-5 agonist Entolimod broadens the therapeutic window of 5-fluorouracil by reducing its toxicity to normal tissues in mice. Oncotarget. (2014) 5:802–14. doi: 10.18632/oncotarget.1773
389. Haderski GJ, Kandar BM, Brackett CM, Toshkov IM, Johnson CP, Paszkiewicz GM, et al. TLR5 agonist entolimod reduces the adverse toxicity of TNF while preserving its antitumor effects. PLoS ONE. (2020) 15:e0227940. doi: 10.1371/journal.pone.0227940
390. Taylor PA, Ehrhardt MJ, Lees CJ, Panoskaltsis-Mortari A, Krieg AM, Sharpe AH, et al. TLR agonists regulate alloresponses and uncover a critical role for donor APCs in allogeneic bone marrow rejection. Blood. (2008) 112:3508–16. doi: 10.1182/blood-2007-09-113670
391. Kornblit B, Müller K. Sensing danger: toll-like receptors and outcome in allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. (2017) 52:499–505. doi: 10.1038/bmt.2016.263
392. Matsui A, Ikeda T, Enomoto K, Hosoda K, Nakashima H, Omae K, et al. Increased formation of oxidative DNA damage, 8-hydroxy-2'-deoxyguanosine, in human breast cancer tissue and its relationship to GSTP1 and COMT genotypes. Cancer Lett. (2000) 151:87–95. doi: 10.1016/S0304-3835(99)00424-3
393. Sosa V, Moline T, Somoza R, Paciucci R, Kondoh H, LLeonart ME. Oxidative stress and cancer: an overview. Ageing Res Rev. (2013) 12:376–90. doi: 10.1016/j.arr.2012.10.004
394. Ghanbari Movahed Z, Rastegari-Pouyani M, Mohammadi MH, Mansouri K. Cancer cells change their glucose metabolism to overcome increased ROS: One step from cancer cell to cancer stem cell? Biomed Pharmacother. (2019) 112:108690. doi: 10.1016/j.biopha.2019.108690
395. Lee K, Esselman WJ. Inhibition of PTPs by H(2)O(2) regulates the activation of distinct MAPK pathways. Free Radic Biol Med. (2002) 33:1121–32. doi: 10.1016/S0891-5849(02)01000-6
396. Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. (2005) 436:123–7. doi: 10.1038/nature03688
397. Rhyu DY, Yang Y, Ha H, Lee GT, Song JS, Uh ST, et al. Role of reactive oxygen species in TGF-β1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol. (2005) 16:667–75. doi: 10.1681/ASN.2004050425
398. Vallee A, Lecarpentier Y. Crosstalk between peroxisome proliferator-activated receptor γ and the canonical WNT/β-catenin pathway in chronic inflammation and oxidative stress during carcinogenesis. Front Immunol. (2018) 9:745. doi: 10.3389/fimmu.2018.00745
399. Warburg O. On the origin of cancer cells. Science. (1956) 123:309–14. doi: 10.1126/science.123.3191.309
400. Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, et al. Glycolytic enzymes can modulate cellular life span. Cancer Res. (2005) 65:177–85.
401. Kondoh H, Lleonart ME, Bernard D, Gil J. Protection from oxidative stress by enhanced glycolysis; a possible mechanism of cellular immortalization. Histol Histopathol. (2007) 22:85–90. doi: 10.14670/HH-22.85
402. Vaughn AE, Deshmukh M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat Cell Biol. (2008) 10:1477–83. doi: 10.1038/ncb1807
403. Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. (2011) 334:1278–83. doi: 10.1126/science.1211485
404. Piao LS, Hur W, Kim TK, Hong SW, Kim SW, Choi JE, et al. CD133+ liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Lett. (2012) 315:129–37. doi: 10.1016/j.canlet.2011.10.012
405. Jiang P, Du W, Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell. (2014) 5:592–602. doi: 10.1007/s13238-014-0082-8
406. Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. (2014) 39:347–54. doi: 10.1016/j.tibs.2014.06.005
407. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. (2009) 458:780–3. doi: 10.1038/nature07733
408. Chang CW, Chen YS, Tsay YG, Han CL, Chen YJ, Yang CC, et al. ROS-independent ER stress-mediated NRF2 activation promotes warburg effect to maintain stemness-associated properties of cancer-initiating cells. Cell Death Dis. (2018) 9:194. doi: 10.1038/s41419-017-0250-x
409. Zhong G, Qin S, Townsend D, Schulte BA, Tew KD, Wang GY. Oxidative stress induces senescence in breast cancer stem cells. Biochem Biophys Res Commun. (2019) 514:1204–9. doi: 10.1016/j.bbrc.2019.05.098
410. Shen YA, Wang CY, Hsieh YT, Chen YJ, Wei YH. Metabolic reprogramming orchestrates cancer stem cell properties in nasopharyngeal carcinoma. Cell Cycle. (2015) 14:86–98. doi: 10.4161/15384101.2014.974419
411. Chang KY, Huang CT, Hsu TI, Hsu CC, Liu JJ, Chuang CK, et al. Stress stimuli induce cancer-stemness gene expression via Sp1 activation leading to therapeutic resistance in glioblastoma. Biochem Biophys Res Commun. (2017) 493:14–9. doi: 10.1016/j.bbrc.2017.09.095
412. Benhar M, Engelberg D, Levitzki A. ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep. (2002) 3:420–5. doi: 10.1093/embo-reports/kvf094
413. Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. (2005) 11:1306–13. doi: 10.1038/nm1320
414. Block KI, Koch AC, Mead MN, Tothy PK, Newman RA, Gyllenhaal C. Impact of antioxidant supplementation on chemotherapeutic toxicity: a systematic review of the evidence from randomized controlled trials. Int J Cancer. (2008) 123:1227–39. doi: 10.1002/ijc.23754
415. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. (2009) 8:579–91. doi: 10.1038/nrd2803
416. Mateescu B, Batista L, Cardon M, Gruosso T, de Feraudy Y, Mariani O, et al. miR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat Med. (2011) 17:1627–35. doi: 10.1038/nm.2512
417. Afzal S, Jensen SA, Sørensen JB, Henriksen T, Weimann A, Poulsen HE. Oxidative damage to guanine nucleosides following combination chemotherapy with 5-fluorouracil and oxaliplatin. Cancer Chemother Pharmacol. (2012) 69:301–7. doi: 10.1007/s00280-011-1700-2
418. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. (2013) 12:329–41. doi: 10.1016/j.stem.2012.12.013
419. Glasauer A, Chandel NS. Targeting antioxidants for cancer therapy. Biochem Pharmacol. (2014) 92:90–101. doi: 10.1016/j.bcp.2014.07.017
Keywords: CSC, cancer, EMT, stem cell-like pathways, therapy
Citation: Espinosa-Sánchez A, Suárez-Martínez E, Sánchez-Díaz L and Carnero A (2020) Therapeutic Targeting of Signaling Pathways Related to Cancer Stemness. Front. Oncol. 10:1533. doi: 10.3389/fonc.2020.01533
Received: 22 May 2020; Accepted: 16 July 2020;
Published: 26 August 2020.
Edited by:
Hiroshi Kondoh, Kyoto University, JapanReviewed by:
Parames C. Sil, Bose Institute, IndiaDaniel Pereira Bezerra, Oswaldo Cruz Foundation (Fiocruz), Brazil
Copyright © 2020 Espinosa-Sánchez, Suárez-Martínez, Sánchez-Díaz and Carnero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amancio Carnero, YWNhcm5lcm9AdXMuZXM=; YWNhcm5lcm8taWJpc0B1cy5lcw==
†These authors have contributed equally to this work