 Luochengling Xiang
Luochengling Xiang Xiao Fu1†
Xiao Fu1† Tao Tian
Tao Tian- 1Department of Oncology, First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, China
- 2Oncology Hospital, Xi’an International Medical Center Hospital, Xi’an, China
Objectives: The Kirsten Rat Sarcoma (KRAS) mutation is the commonest oncogenic drive mutation in lung adenocarcinoma (LUAD) and immunotherapy may be quite promising for KRAS-mutant LUAD. While the effects of tumor mutation burden (TMB) and copy number alteration (CNA) are poorly understood in this illness, our study aimed to explore the roles TMB and CNA play in the prediction of response to immune checkpoint inhibitor (ICI) therapy in advanced KRAS-mutant LUAD.
Methods: Mutation and clinical data were downloaded from cBioPortal. We evaluated KRAS mutation status and divided patients into different subgroups based on TMB and CNA cutoffs to investigate the predictive value of these biomarkers on ICI response.
Results: KRAS mutation with concurrent TP53 or STK11 mutations had higher TMB and CNA compared to KRAS mutation alone. The KRAS G12C and G > T mutation subgroups, with TP53 or STK11 co-mutation, also had higher TMB and CNA. We found that TMB and CNA were independently associated with progression-free survival (PFS) and durable clinical benefits (DCB); TMB was positively correlated with PFS (P = 0.0074) and DCB (P = 0.0008) while low CNA was associated with prolonged PFS (P = 0.0060) and DCB (P = 0.0018). However, TMB alone did not distinguish benefits among KRAS-mutant patients. Notably, when combining TMB and CNA, low TMB and high CNA revealed worse outcomes of ICI therapy (mPFS: 2.20m, P = 0.0023; proportion of DCB: 24%, P = 0.0001).
Conclusion: The combination of TMB and CNA provides more sensible and accurate prediction of ICI response than individual factors in KRAS-mutant LUAD. Moreover, low TMB and high CNA can be utilized as a potential biomarker to predict adverse outcome in KRAS-mutant LUAD.
Introduction
In lung adenocarcinoma (LUAD), the most frequent oncogene driver mutation is Kirsten Rat Sarcoma (KRAS) (1). While patients harboring other driver genes, such as those for Epidermal Growth Factor Receptor (EGFR) and Anaplastic Lymphoma Kinase (ALK), may respond to therapy with tyrosine kinase inhibitors (TKIs), those harboring a KRAS mutation lack efficient treatment regimens. Despite decades of research, the KRAS protein remains a challenging therapeutic target due to the lack of an ideal small molecule binding pocket in the protein and its high affinity toward the abundance of guanosine triphosphate (GTP). While several novel inhibitors targeting the mutant protein KRAS G12C (missense substitution at codon 12; glycine to cysteine) with covalent bonding to the cysteine amino acid have been used in early phase clinical trials, there are many KRAS mutation subtypes, such as G12V (missense substitution at codon 12; glycine to valine) and G12D (missense substitution at codon 12; glycine to aspartic acid) (2). Besides, although the KRAS-MAPK pathway is downstream of EGFR signaling, patients with a KRAS mutation do not respond to EGFR TKIs (3). In addition, patients with KRAS-mutant advanced non-small cell lung cancer (NSCLC) exhibit inferior responses to cytotoxic chemotherapy as well as decreased progression-free survival (PFS) and overall survival (OS) compared to patients harboring native KRAS (4). Recently, immunotherapy has become regarded as most promising for KRAS-mutant LUAD (5).
Immune checkpoint inhibitors (ICIs) have revolutionized the management of NSCLC. Treatment with anti-cytotoxic T lymphocyte antigen 4 (CTLA4) antibody and programmed cell death-1 (PD-1) or PD-1 ligand (PD-L1) inhibitors has greatly improved patient survival. Even though ICIs have emerged as epochal milestones in anti-cancer therapy, only a subset of patients exhibits objective responses and while others show disease progression. Patients treated with ICIs may also suffer life-threatening immune-related adverse effects and even suffer hyper progression of the disease (6). A detailed understanding of key predictive factors necessary to identify patients who may potentially benefit from treatment with ICIs is thus urgent.
To date, among patients with PD-L1-positive disease, tumor-infiltrating lymphocytes have proven to be indicators of ICI therapy (7, 8). Importantly, increasing evidence suggests that the diversity and composition of gut microbiota impacts patient response to ICIs (9, 10). Since the advent of next generation sequencing, an increasing number of genetic tumor features have also been detected, including tumor mutation burden (TMB), microsatellite instability and copy number alteration (CNA), which have been correlated with therapeutic response. The number of non-synonymous single nucleotide variants, or TMB, in a tumor was found to strongly positively correlate with response to ICIs in NSCLC (11, 12). However, Merkel cell carcinoma was reported to respond better than TMB alone expects, while colorectal carcinoma was found to have worse outcomes than that predicted by TMB alone (13). Interestingly, a pan-cancer analysis based on The Cancer Genome Atlas revealed a negative relationship between CNA and immune infiltration. Meanwhile, in the setting of anti-CTLA4 therapy, CNA was reported to be a potential predictive factor of survival, independent of TMB (14).
Here, to evaluate the potential utility of TMB and CNA together in identifying distinct patient subgroups of KRAS-mutant LUAD, we compared the distribution of TMB and CNA among different KRAS mutations and then analyzed efficacy of ICI treatment in subgroups based on TMB and CNA.
Materials and Methods
Clinical Cohorts
Data were collected from published articles. Mutation data of 860 advanced LUAD patients were retrieved from cBioPortal1. From this website, we obtained DNA sequencing data to analyze TMB and CNA distributions among multiple KRAS mutations. Details of samples included were shown as a flowchart in Supplementary Figure 1.
Clinical and mutation data of 240 NSCLC patients were also retrieved from cBioPortal2. We collected 186 advanced LUAD. All patients were treated with anti-PD-1/PD-L1 monotherapy or in combination with anti-CTLA4 blockade between April 2011 and January 2017. Details of these samples were also shown as a flowchart in Supplementary Figure 1. All patients had undergone the MSK-IMPACT assay, a next generation sequencing tumor profile test. Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 was performed to assess efficacy. Efficacy was additionally identified as durable clinical benefit (DCB; complete response (CR) or partial response (PR); or stable disease (SD) that lasted >6 months) or no durable benefit [NDB; progressive disease (PD) or SD that lasted ≤6 months]. Patient PFS was assessed from the date of immunotherapy initiation to the date of disease progression or death for any reason (15).
Tumor Mutation Burden Analysis
Somatic mutation data of advanced LUAD were retrieved from cBioPortal. In the MSK-IMPACT assay, tumor and matched normal data were used to identify somatic variants and optimize mutation calling filters; 100× coverage was needed to defect mutations with true variant frequencies ≥10% with 98% power. All exons and selected introns of custom gene panels of 341 (version 1), 410 (version 2), and 468 (version 3) genes were sequenced and targeted. Patients were classified according to the coding region captured in each panel, thus covering 0.98, 1.06, and 1.22 megabases (Mb) in the 341-, 410-, and 468-gene panels, respectively. The TMB cutoff value was obtained using X-tile, a tool for outcome-based biomarker cut-point optimization (16).
Copy Number Alteration Analysis
Data concerning CNA in the MSKCC database were analyzed by MSK-IMPACT sequencing. Via comparison of sequence coverage of targeted regions in a tumor sample with a standard normal sample, CNA was identified. The Genome Analysis Toolkit (GATK) was used to obtain coverage of targeted regions, and a Loess normalization was applied to adjust guanosine-cytosine content. Log-ratio coverage values were subsequently segmented by circular binary segmentation. Germline cells were removed to ensure somatic final copy number variants. Log2 copy number gain >0.2 or loss <−0.2 (P < 0.05) was used to determine significant whole gene gain or loss events (17).
Statistical Analysis
Statistical analysis was conducted by Graph Prism (version 8.0) and SPSS (version 22.0). The Mann–Whitney U test was performed to compare TMB and CNA values; TMB and CNA were presented using box plots that presented mean, interquartile ranges, and ranges. Hazard ratio was determined via univariate and multivariate Cox proportional hazard regression analyses. Kaplan–Meier curve analysis was applied to evaluate PFS and OS using log-rank analysis. Proportional DCB representation was detailed by a 100% stacked column graph. Pearson’s Chi-squared test was applied to evaluate the difference in DCB proportion among different subgroups. All reported P-values were two-tailed, and for all analyses, P ≤ 0.05 was considered statistically significant.
Results
Prognostic Value of KRAS Mutation Status in Advanced Lung Adenocarcinoma
Among the 860 metastatic LUAD patients who underwent genomic analysis in the MSKCC-IMPACT study (1), KRAS mutation was common (Figure 1). As shown in Supplementary Figure 1, we deleted 115 patients without matched survival data. A total of 207 patients with KRAS mutations had statistically shorter OS as compared with 538 patients with wild-type KRAS tumors (HR = 1.515; 95% CI: 1.172–1.960; P = 0.0015, Figure 2A).
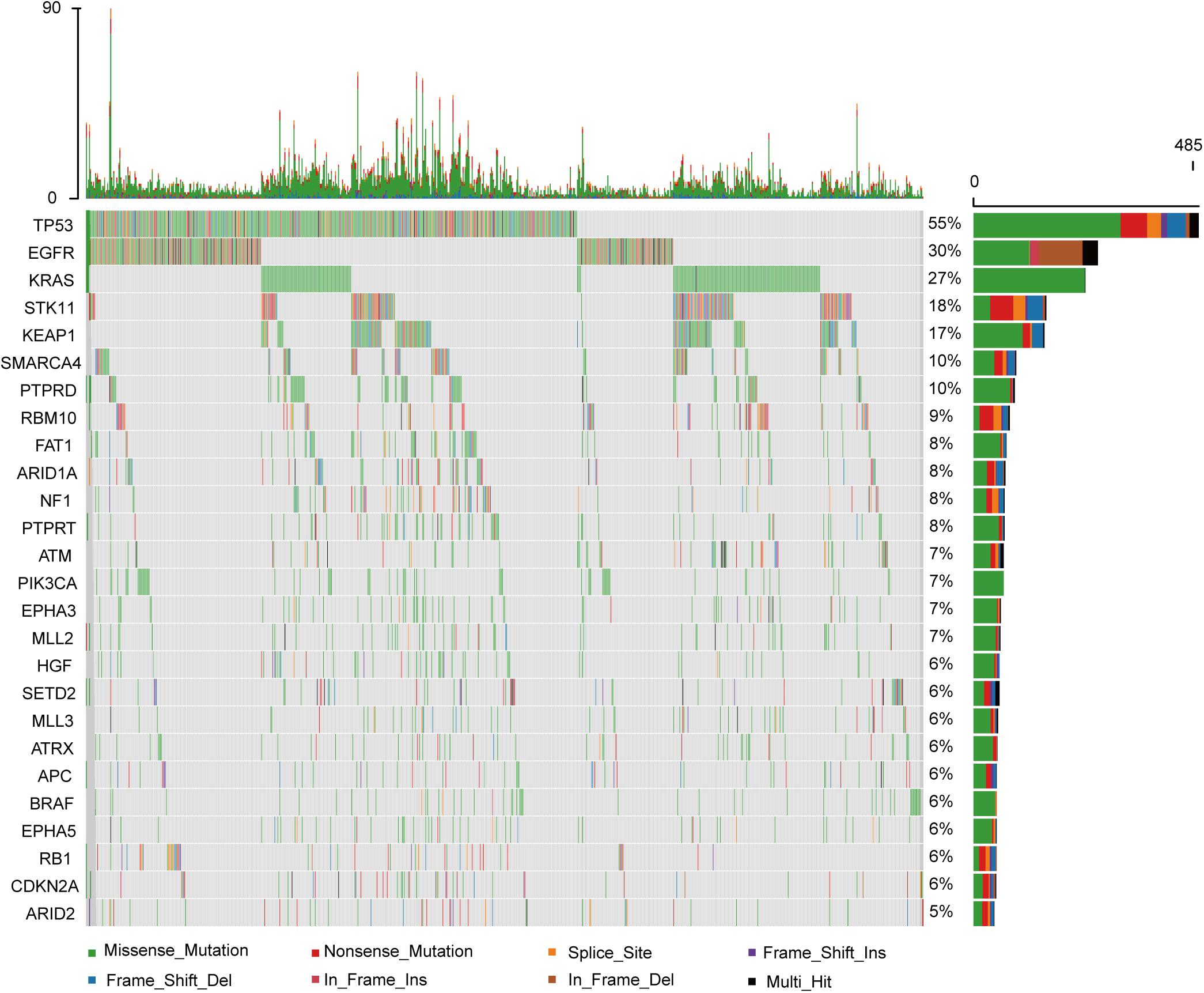
Figure 1. The genomic landscape and the mutation signature of advanced lung adenocarcinoma in the MSKCC database.
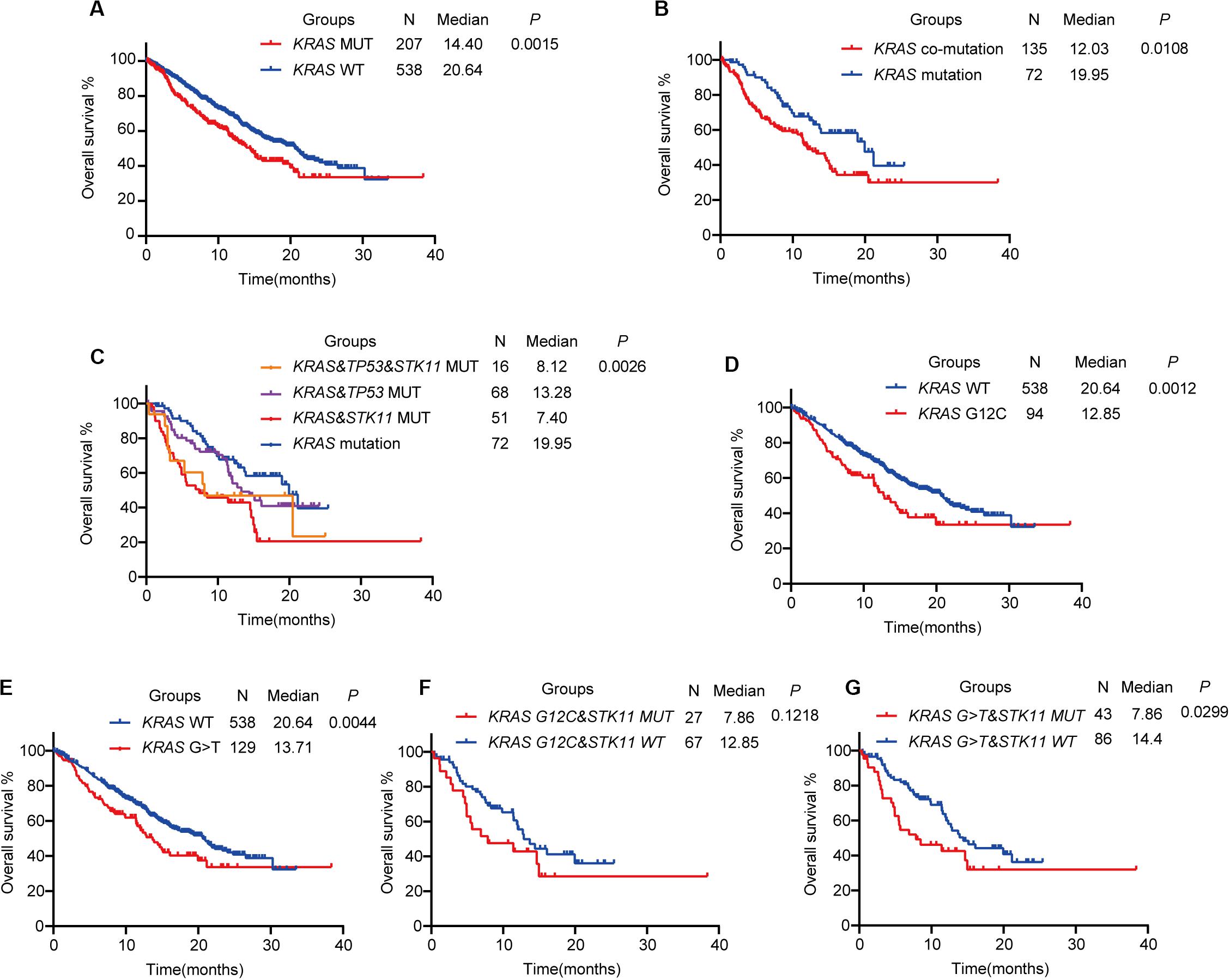
Figure 2. The prognostic value of KRAS mutational status in advanced lung adenocarcinoma. (A) Kaplan–Meier survival analysis based on KRAS mutation status. (B) KRAS-mutant patients with co-mutations have shorter overall survival than those with KRAS mutation alone. (C) Kaplan–Meier survival analysis of KRAS co-mutation subtypes. (D,E) Kaplan–Meier survival analysis of KRAS mutation subtypes G12C (D) or G > T (E) with wild-type. (F,G) Kaplan–Meier survival analysis of KRAS mutation subtypes G12C (F) or G > T (G) with concurrent STK11 mutation. MUT, mutant; WT, wild-type.
The most common concurrent pathogenic mutations were TP53 (84 patients, 40.6%) and STK11 (67 patients, 32.4%), consistent with previous studies (18). We divided KRAS-mutant patients into two groups based on concurrent TP53 and STK11 mutation status. One group was the KRAS co-mutation group (KRAS-mutant patients with either TP53 or STK11 mutation) and the other was the KRAS mutation group (KRAS-mutant patients without TP53 or STK11 mutation). We found that patients in the KRAS co-mutation group had shorter OS than those in the KRAS mutation group (HR = 1.618; 95% CI: 1.128–2.505; P = 0.0108, Figure 2B). Further analysis revealed that KRAS-mutant patients with co-occurring STK11 mutation had shorter OS than those with either co-occurring TP53 (HR = 1.864; 95% CI: 1.115–3.117; P = 0.0176) or both TP53 and STK11 (HR = 2.856; 95% CI: 1.645–4.958; P = 0.0002) mutations. No significant difference between KRAS-mutant patients with and without co-occurring TP53 and STK11 mutations was noted (HR = 2.219; 95% CI: 0.886–5.555; P = 0.0234), likely because KRAS-mutant patients with co-occurring TP53 and STK11 mutations only totaled 16 (Figure 2C).
The KRAS G12C mutation (missense substitution at codon 12; glycine to cysteine) has been previously reported to be oncogenic and potentially targetable; several novel KRAS G12C inhibitors, such as AMG150 and MRTX849, are being studied (2). In advanced LUAD, the KRAS G12C mutation was the most common, accounting for 45.4% of all KRAS-mutant advanced LUAD (G12C: N = 94, 45.4%; G12V, missense substitution at codon 12; glycine to valine: N = 31, 15.0%; G12D, missense substitution at codon 12; glycine to aspartic acid: N = 28, 13.5%). At the same time, G > T substitution (nucleotide substitution in sequences coding for amino acids in protein; G is substituted by T, N = 129, 62.3%) was the most common nucleotide substitution in KRAS-mutant advanced LUAD. On Kaplan–Meier analysis, the KRAS G12C mutation subtype was associated with shorter OS than wild-type KRAS (HR = 1.741; 95% CI: 1.209–2.509; P = 0.0012, Figure 2D), as was the KRAS G > T mutation subtype (HR = 1.583; 95% CI: 1.154–2.170; P = 0.0044, Figure 2E). In further analysis of the effect of concurrent STK11 mutation, the KRAS G12C mutation subtype with or without concurrent STK11 mutation was not found to have significantly different OS (HR = 1.668; 95% CI: 0.872–3.190; P = 0.1218, Figure 2F). The KRAS G > T mutation subtype with co-occurring STK11 mutation, however, was found to have a much shorter OS when compared to the co-occurring STK11 mutation alone (HR = 1.869; 95% CI: 1.063–3.286; P = 0.0299, Figure 2G).
Correlation Between KRAS Mutation and Tumor Mutation Burden in Advanced Lung Adenocarcinoma
Investigation of whether KRAS mutation status impacted TMB revealed significant differences in TMB among KRAS mutation and wild-type patients (P < 0.0001, Figure 3A). Moreover, patients with either TP53 or STK11 co-mutation had higher TMB than those with KRAS mutation alone (P < 0.0001, Figure 3B). Interestingly, each concurrent mutation was found to have higher TMB than KRAS mutation alone (KRAS&TP53&STK11 vs. KRAS, P = 0.0023; KRAS&TP53 vs. KRAS, P < 0.0001; KRAS&STK11 vs. KRAS, P = 0.0005; Figure 3C).
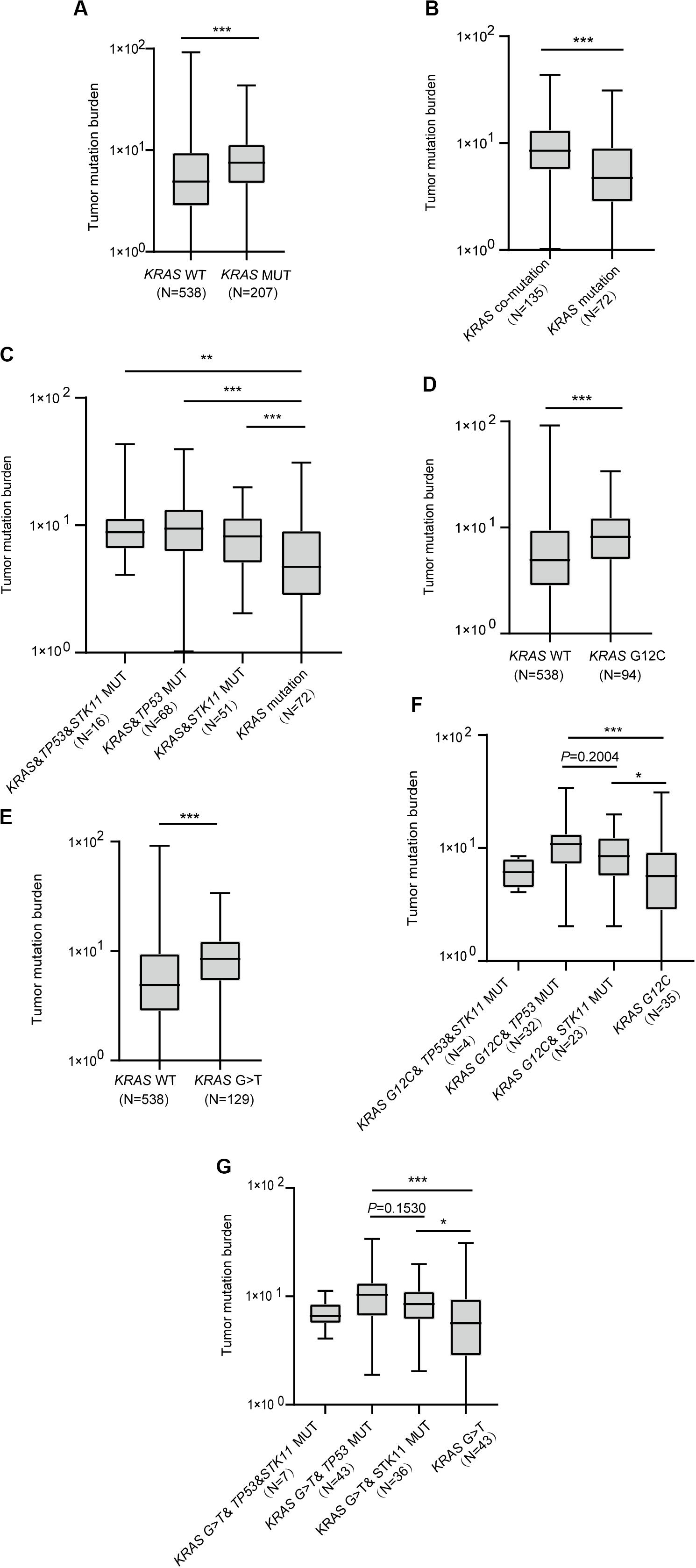
Figure 3. The correlation between KRAS mutational status and tumor mutation burden. (A) Patients with KRAS mutation have greater tumor mutation burden. (B) Patients with KRAS mutation and concurrent mutations have greater tumor mutation burden than those with KRAS mutation alone. (C) Comparison of tumor mutation burden in KRAS co-mutation subtypes. (D,E) Comparison of tumor mutation burden in KRAS G12C (D) and G > T (E) subtypes. (F,G) Comparison of tumor mutation burden in G12C (F) and G > T (G) subtypes with co-mutations. MUT, mutant; WT, wild-type. Box plot data are presented as mean, interquartile ranges, and ranges. ***P < 0.001; **P < 0.01; *P < 0.05.
Next, we sought to confirm the association between KRAS mutation subtypes and TMB. Results revealed that both KRAS G12C and G > T substitution mutations had higher TMB than did wild-type KRAS (P < 0.0001, Figure 3D; P < 0.0001, Figure 3E). We further found that KRAS G12C with either TP53 or STK11 co-mutation had higher TMB (KRAS G12C&TP53 vs. KRAS G12C, P = 0.0005; KRAS G12C&STK11 vs. KRAS G12C, P = 0.0264; Figure 3F). Similarly, KRAS G > T substitution mutation with either TP53 or STK11 co-mutation had higher TMB (KRAS G > T&TP53 vs. KRAS G > T, P = 0.0004; KRAS G > T&STK11 vs. KRAS G > T, P = 0.0129; Figure 3G).
KRAS Mutation Status and Copy Number Alteration in Advanced Lung Adenocarcinoma
Recent studies have reported CNA to be useful in the construction of predictive models concerning response to ICI treatment (13, 14). Our analysis revealed that KRAS mutation with concurrent mutations had higher CNA compared with KRAS mutation alone (P < 0.0001, Figure 4A). We further found that KRAS mutation with either TP53 or STK11 co-mutation significantly differed in CNA (KRAS&TP53 vs. KRAS mutation, P = 0.0021; KRAS&STK11 vs. KRAS mutation, P = 0.0002; Figure 4B). Analysis of the relationship between the common KRAS G12C and G > T substitution mutation subtypes and CNA revealed similar findings; both subtypes with either TP53 or STK11 co-mutation had significant differences in CNA (KRAS G12C&TP53 vs. KRAS G12C, P = 0.0014; KRAS G12C&STK11 vs. KRAS G12C, P = 0.0029; Figure 4C; KRAS G > T&TP53 vs. KRAS G > T, P = 0.0022; KRAS G > T&STK11 vs. KRAS G > T, P = 0.0015; Figure 4D).
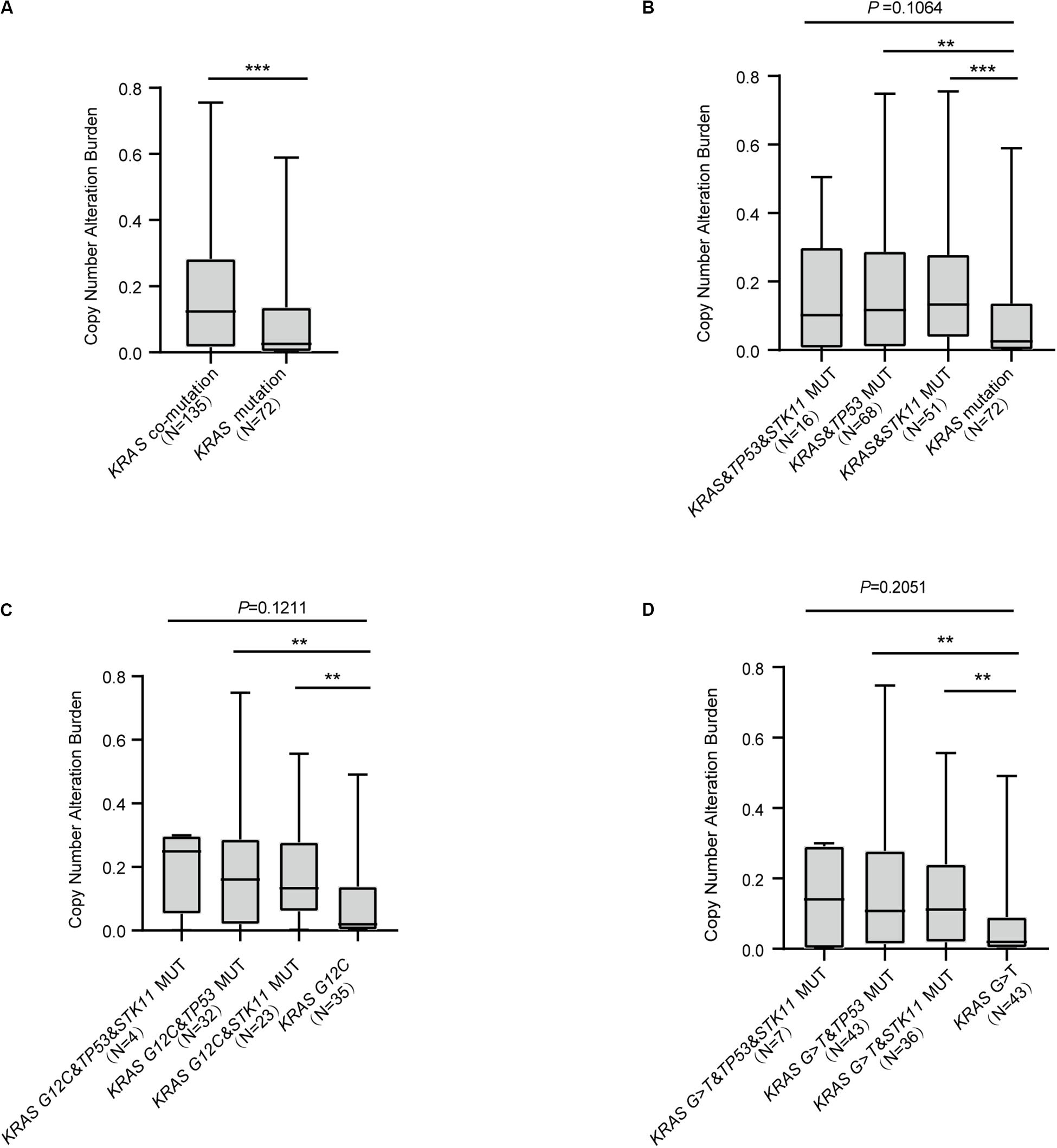
Figure 4. The correlation between KRAS mutational status and copy number alteration burden. (A) Patients with KRAS concurrent mutations have greater copy number alteration burden with only KRAS mutation. (B) Comparison of copy number alteration burden in KRAS co-mutation subtypes. (C,D) Comparison of copy number alteration burden in KRAS G12C (C) or G > T (D) subtypes with concurrent mutations. MUT, mutant; WT, wild-type. Box plot data are presented as mean, interquartile ranges, and ranges. ***P < 0.001; **P < 0.01.
Independent Predictive Value of Tumor Mutational Burden and Copy Number Alteration for Immune Checkpoint Inhibitor Response in Advanced Lung Adenocarcinoma
To estimate the predictive value of TMB and CNA in patient response to ICI treatment, available data in the MSKCC database were analyzed. A total of 240 patients with advanced NSCLC who underwent PD-1/PD-L1 inhibitor treatment alone or in combination with anti-CTLA-4 treatment were identified (15). We chose 186 patients with advanced LUAD for further analysis. For this particular population with ICI (PD-1/PD-L1 inhibitor alone or in combination with anti-CTLA-4), optimal cutoff points for TMB (13.27 mut/Mb) and CNA (0.05) were acquired using X-tile software. This population was subsequently divided into high (TMB ≥ 13.27 mut/Mb) and low (TMB < 13.27 mut/Mb) TMB groups; high TMB group patients were found to have significantly prolonged PFS (HR = 0.596; 95% CI: 0.408–0.870; P = 0.0074, Figure 5A) as well as an increased proportion of DCB (50 vs. 27%, P = 0.0008, Figure 5E). Analysis of patients classified into high (CNA ≥ 0.05) and low (CNA < 0.05) CNA groups revealed high CNA to be associated with shortened PFS (HR = 1.578; 95% CI: 1.140–2.184; P = 0.0060, Figure 5B) and a decreased proportion of DCB (24 vs. 45%, P = 0.0018, Figure 5F). Cox proportional hazard regression analysis revealed, after multivariate adjustment, TMB and CNA to be independent biomarkers for ICI response (TMB, HR = 0.46, P = 0.0011; CNA, HR = 1.86, P = 0.0007, Table 1).
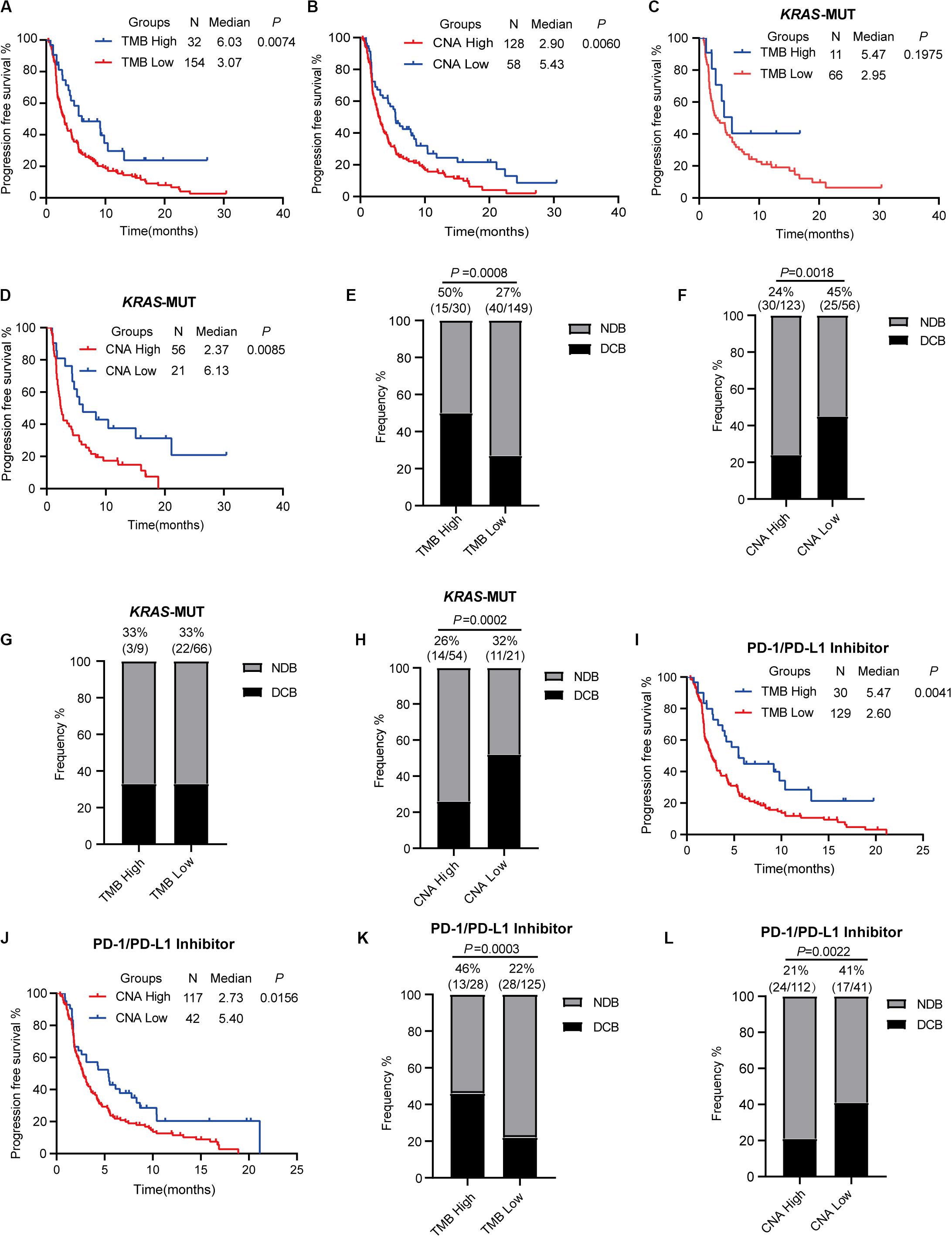
Figure 5. Tumor mutation burden and copy number alteration burden correlated with clinical response to immune checkpoint inhibitor treatment. (A,B) Progression-free survival curve for patients receiving ICI (PD-1/L1 inhibitor or in combination with anti-CTLA-4) based on tumor mutation burden (A) or copy number alteration burden (B). (C,D) Progression-free survival curve for KRAS-mutant patients receiving ICI (PD-1/L1 inhibitor or in combination with anti-CTLA-4) based on tumor mutation burden (C) and copy number alteration burden (D). (E,F) Proportional representation of durable clinical benefits in advanced lung adenocarcinoma patients receiving ICI (PD-1/L1 inhibitor or in combination with anti-CTLA-4). (G,H) Proportional representation of durable clinical benefits in advanced KRAS-mutant lung adenocarcinoma patients receiving ICI (PD-1/L1 inhibitor or in combination with anti-CTLA-4). (I,J) Progression-free survival curve for patients receiving PD-1/PD-L1 inhibitor alone based on tumor mutation burden (I) and copy number alteration burden (J). (K,L) Proportional representation of durable clinical benefits in advanced KRAS-mutant lung adenocarcinoma patients receiving PD-1/PD-L1 inhibitor alone. MUT, mutant; WT, wild-type; DCB, durable clinical benefit; NDB, no durable clinical benefit.
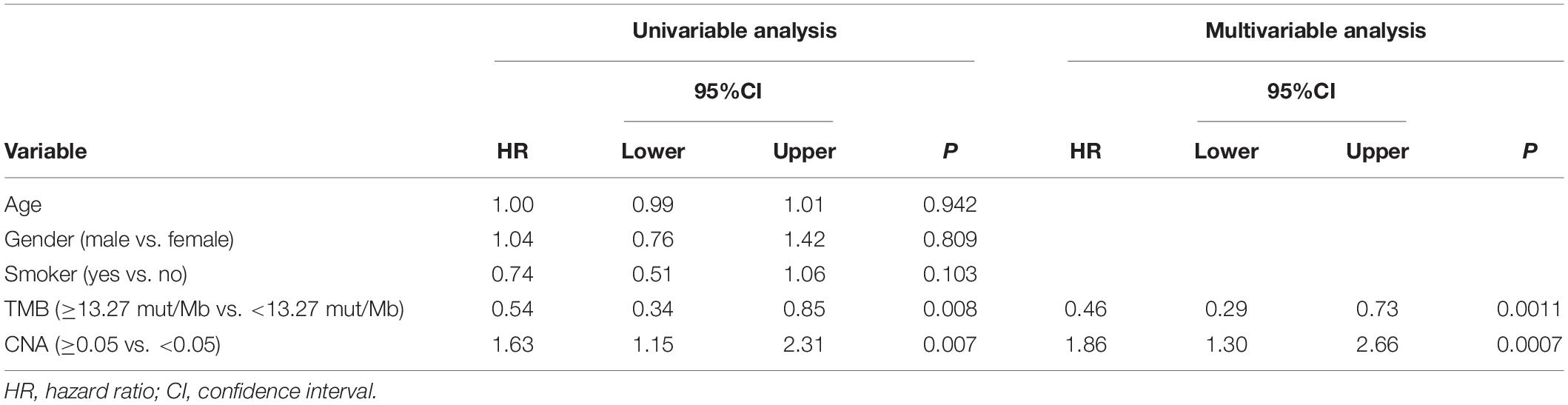
Table 1. Univariable and multivariable Cox proportional hazards regression.
We evaluated the data of 77 KRAS-mutant patients from the population outlined above to further confirm our findings, but no significant differences in PFS (HR = 0.636; 95% CI: 0.319–1.266; P = 0.1975, Figure 5C) and proportion of DCB (high vs. low TMB; 33 vs. 33%, Figure 5G) were noted in the KRAS-mutant population. Significantly prolonged PFS (HR = 0.497; 95% CI: 0.293–0.837; P = 0.0085, Figure 5D) and higher proportion of DCB (high vs. low CNA; 26 vs. 52%, P = 0.0002, Figure 5H) were observed in KRAS-mutant patients of the low CNA group as compared to those in the high CNA group.
Recent studies revealed high TMB to be correlated with combination PD-1 and CTLA-4 inhibitor treatment efficacy in NSCLC (11, 19). However, the predictive value of TMB in PD-1/PD-L1 inhibitor efficacy in patients with advanced NSCLC remains uncertain. We classified 159 advanced LUAD patients treated with anti-PD-1/PD-L1 monotherapy into two (high and low TMB) groups using a TMB cutoff value of 13.27 mut/Mb. Our findings revealed that high TMB was significantly correlated with prolonged PFS and greater DCB (HR = 0.564; 95% CI: 0.382–0.834; P = 0.0041, Figure 5I; DCB, 46 vs. 22%, P = 0.0003, Figure 5K). We found that low CNA was also associated with prolonged PFS and greater DCB (median PFS in high vs. low CNA group patients, 2.73 vs. 5.40 months, P = 0.0156, Figure 5J; DCB, 21 vs. 41%, P = 0.0022, Figure 5L).
Low Tumor Mutational Burden and High Copy Number Alteration Together Predict a Poor Response to Immune Checkpoint Inhibitor Therapy
As TMB and CNA were established independent predictive factors of ICI response, we conjectured that combined use of both TMB and CNA would better predict ICI efficacy. In advanced LUAD patients with ICI (PD-1/PD-L1 inhibitor alone or in combination with anti-CTLA-4), low TMB and high CNA were found to have significantly shorter PFS compared to patients with high TMB and high CNA, high TMB and low CNA, and low TMB and low CNA (low TMB and high CNA vs. high TMB and high CNA: HR = 1.803, 95% CI: 1.199–2.712, P = 0.0047; low TMB and high CNA vs. high TMB and low CNA: HR = 2.693, 95% CI: 1.276–5.683, P = 0.0094; low TMB and high CNA vs. low TMB and low CNA: HR = 1.752, 95% CI: 1.240–2.476, P = 0.0015; Figure 6A). Patients with low TMB and high CNA had the significantly lowest proportion of DCB as compared to those in the three aforementioned subgroups (low TMB and high CNA vs. high TMB and high CNA vs. high TMB and low CNA vs. low TMB and low CNA; 19 vs. 46 vs. 75 vs. 42%, P < 0.0001, Figure 6C). Our analysis revealed findings consistent with those above in advanced LUAD patients with PD-1/PD-L1 inhibitor alone; patients with low TMB and high CNA were confirmed to have the significantly shortest PFS (low TMB and high CNA vs. high TMB and high CNA: HR = 1.771, 95% CI: 1.156–2.713, P = 0.0086; low TMB and high CNA vs. high TMB and low CNA: HR = 2.851, 95% CI: 1.385–5.872, P = 0.0045; low TMB and high CNA vs. low TMB and low CNA: HR = 1.608, 95% CI: 1.095–2.363, P = 0.0154, Figure 6B) and lowest proportion of DCB (low TMB and high CNA vs. high TMB and high CNA vs. high TMB and low CNA vs. low TMB and low CNA: 16 vs. 42 vs. 75 vs. 38%, P < 0.0001, Figure 6D).
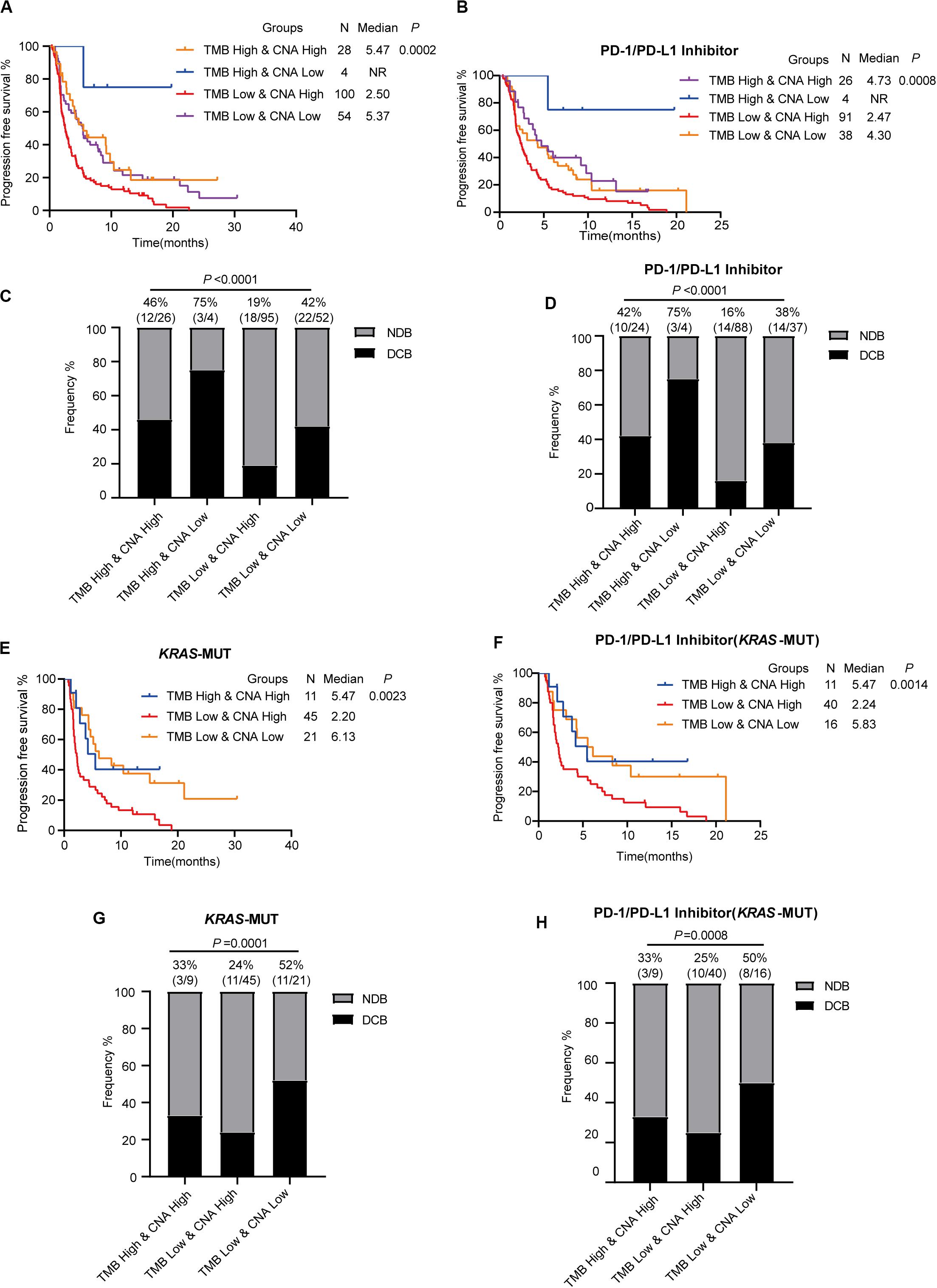
Figure 6. Low tumor mutational burden and high copy number alteration together predict a poor response to immune checkpoint inhibitor therapy. (A,B) Low TMB and high CNA show shorter progression-free survival in patients receiving ICI (PD-1/L1 inhibitor or in combination with anti-CTLA-4) (A) and patients receiving PD-1/PD-L1 inhibitor alone (B). (C,D) Low TMB and high CAN show decreased proportion of DCB in patients receiving ICI (PD-1/L1 inhibitor or in combination with anti-CTLA-4) (C) and patients receiving PD-1/PD-L1 inhibitor alone (D). (E,F) Low TMB and high CNA show shorter progression-free survival in KRAS-mutant patients receiving ICI (PD-1/L1 inhibitor or in combination with anti-CTLA-4) (E) and KRAS-mutant patients receiving PD-1/L1 inhibitor alone (F). (G,H) Low TMB and high CNA show decreased proportion of DCB in KRAS-mutant patients receiving ICI (PD-1/L1 inhibitor or in combination with anti-CTLA-4) (G) and KRAS-mutant patients receiving PD-1/L1 inhibitor alone (H). MUT, mutant; WT, wild-type; DCB, durable clinical benefit; NDB, no durable clinical benefit.
Next, we further analyzed the predictive value of low TMB and high CNA in KRAS-mutant LUAD. In those patients with ICI (PD-1/PD-L1 inhibitor alone or in combination with anti-CTLA-4), although there were no KRAS-mutant LUAD patients in the high TMB and low CNA subgroup, patients with low TMB and high CNA were found to have shortened PFS (low TMB and high CNA vs. high TMB and high CNA: HR = 1.977, 95% CI: 1.025–3.814, P = 0.0420; low TMB and high CNA vs. low TMB and low CNA: HR = 2.338, 95% CI: 1.368–3.995, P = 0.0019, Figure 6E) and a smaller proportion of DCB (low TMB and high CNA vs. high TMB and high CNA vs. low TMB and low CNA: 24 vs. 33 vs. 52%, P = 0.0001, Figure 6G). Significant differences in PFS (low TMB and high CNA vs. high TMB and high CNA: HR = 1.994, 95% CI: 1.021–3.894, P = 0.0433; low TMB and high CNA vs. low TMB and low CNA: HR = 2.022, 95% CI: 1.131–3.616, P = 0.0176, Figure 6F) and DCB (low TMB and high CNA vs. high TMB and high CNA vs. low TMB and low CNA: 25 vs. 33 vs. 50%, P = 0.0008, Figure 6H) in patients with low TMB and high CNA receiving anti-PD-1/PD-L1 monotherapy were noted compared with those of the other two groups. Thus, the combination of TMB and CNA was confirmed to increase the sensitivity of ICI efficacy prediction in advanced KRAS-mutant LUAD. In addition, the combination of low TMB and high CNA was confirmed to predict poor ICI response in advanced KRAS-mutant LUAD.
Discussion
Among lung cancer patients, KRAS mutation is the commonest mutation and 27% of LUAD patients harbor it (20). Patients suffering KRAS-mutant NSCLC continue to have a poor prognosis and lack efficient treatment strategies. Effective pharmacologic targeting of KRAS mutations also remains an unprecedented challenge. Recent studies, however, have reported that patients suffering KRAS-mutant NSCLC treated with ICI therapy had improved OS and PFS compared to those treated with chemotherapy (21, 22). In addition, TMB and CNA have been reported to be features of the genomic landscape that affect ICI efficacy (13). Here, we found that combined use of TMB and CNA increased the predictive sensitivity for ICI response in patients suffering KRAS-mutant advanced LUAD. Importantly, we found that low TMB and high CNA were associated with a poor prognosis, and TMB level positively correlated with response to anti-PD-1/PD-L1 monotherapy.
Recent studies have reported KRAS-mutant tumors to show greater PD-L1 expression (23) and T-cell infiltration (24). Here, our analysis of the correlation between KRAS mutation status and TMB revealed TMB to be associated with tumor immunogenicity and greater benefit of ICI therapy (25). We found that KRAS–mutant tumors showed higher TMB than did wild-type tumors. In further analysis of mutation subtypes and co-mutations, we demonstrated that KRAS with either co-occurring TP53 or STK11 mutation had greater TMB as compared to KRAS mutation alone. In KRAS-mutant LUAD, KRAS with STK11 co-mutation was reported to facilitate immune escape and resistance to anti-PD-1 therapy and to mostly be an “immune desert” phenotype (26, 27). Interestingly, TP53 inactivation in KRAS-mutant LUAD was reported to increase inflammatory marker levels and improve PFS (21, 27).
Tumor CNA burden has been reported to be a pan-cancer prognostic factor for recurrence and death (28). Here, we found that KRAS with either co-occurring TP53 or STK11 mutation had higher CNA. Furthermore, high CNA was a potential predictor of poor ICI efficacy in KRAS-mutant advanced LUAD. This finding was in agreement with prior evidence of CNA as a biomarker predictive for ICI response. Recently, CNA was reported to improve cell proliferation, reduce immune infiltration, and at lower levels correlate with poor ICI response (14). Of note, CNA likely is involved in the suppression of antigen presentation in cancer cells (29).
Although TMB and CNA have been reported to impact immune infiltration and predict ICI response, there have been few studies exploring associations among the combined application of TMB and CNA and clinical benefits of ICI. Multivariate Cox proportional hazard regression analysis of TMB and CNA confirmed that these two biomarkers were independent predictive factors for ICI response. Thus, while CNA provides complementary analysis of clinical ICI response, combining TMB and CNA improves the predictive sensitivity and accuracy of ICI response compared to use of these biomarkers independently. We divided patients into subgroups based on the cutoff value of TMB (13.27 mut/Mb) and CNA (0.05) from X-tile software. Previous studies have revealed that a cut-off value for TMB of 14.31 mut/Mb was used to predict survival in patients who underwent immunotherapy for advanced gastric cancer (30), while intermediate CNA was found to discriminate for recurrence in a prostate cancer population (31). Therefore, more researches are needed to speculate the optimal cutoff for clinical practices. We found that patients with low TMB and high CNA suffered significantly worse outcomes in the setting of ICI therapy. In KRAS-mutant LUAD, combination of TMB and CNA revealed that patients with low TMB and high CNA suffered a significantly worse prognosis. Thus, combined application of TMB and CNA values can be used to accurately select patients who would benefit from ICI treatment.
Our research had several limitations. First, all of our data were obtained from open databases, and patient characteristics were limited. As such, we were confined to analyzing data that was available. For example, patients receiving ICI treatment had PFS but lacked OS data; thus we could only analyze differences in PFS. In addition, we were only able to obtain genomic and clinical data; as PD-L1 mRNA expression and TPS data were unavailable, we could not compare any difference among them across KRAS-mutant LUAD subgroups. Finally, as our analysis was retrospective in nature, prospective and multi-center clinical trials should further be performed prior to utilization of combined TMB and CNA in the prediction of patient outcomes to ICI therapy.
Conclusion
In conclusion, we here detailed that combining TMB and CNA provides a potential biomarker that effectively predicts patient response to ICI therapy. We found that TMB and CNA were higher in KRAS-mutant tumors as compared to wild-type tumors. Furthermore, KRAS with either TP53 or STK11 co-mutations had higher TMB and CNA as compared with KRAS alone. Our findings highlight that low TMB and high CNA is useful in predicting adverse patient outcomes for ICI therapy.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/ Supplementary Material.
Ethics Statement
The studies involving human participants were reviewed and approved by the Medical Ethics Committee of Xi’an Jiaotong University. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
LX, XF, KN, and TT designed the study and wrote the manuscript. XW, WL, and XZ downloaded and analyzed the data. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by Youths Program of the Natural Science Foundation of Shaanxi Province (2020JQ-512).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.559896/full#supplementary-material
Supplementary Figure 1 | Flowchart of study. TMB, tumor mutation burden; CNA, copy number alteration; MUT, mutant; WT, wild-type; DCB, durable clinical benefit; NDB, no durable clinical benefit.
Footnotes
- ^ https://www.cbioportal.org/study/summary?id=lung_msk_2017
- ^ https://www.cbioportal.org/study/summary?id=nsclc_pd1_msk_2018
References
1. Jordan EJ, Kim HR, Arcila ME, Barron D, Chakravarty D, Gao J, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. (2017) 7:596–609. doi: 10.1158/2159-8290.Cd-16-1337
2. Nagasaka M, Li Y, Sukari A, Ou SI, Al-Hallak MN, Azmi AS. KRAS G12C Game of Thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treat Rev. (2020) 84:101974. doi: 10.1016/j.ctrv.2020.101974
3. Murray S, Dahabreh IJ, Linardou H, Manoloukos M, Bafaloukos D, Kosmidis P. Somatic mutations of the tyrosine kinase domain of epidermal growth factor receptor and tyrosine kinase inhibitor response to TKIs in non-small cell lung cancer: an analytical database. J Thorac Oncol. (2008) 3:832–9. doi: 10.1097/JTO.0b013e31818071f3
4. Roman M, Baraibar I, Lopez I, Nadal E, Rolfo C, Vicent S, et al. KRAS oncogene in non-small cell lung cancer: clinical perspectives on the treatment of an old target. Mol Cancer. (2018) 17:33. doi: 10.1186/s12943-018-0789-x
5. Ferrer I, Zugazagoitia J, Herbertz S, John W, Paz-Ares L, Schmid-Bindert G. KRAS-mutant non-small cell lung cancer: from biology to therapy. Lung Cancer. (2018) 124:53–64. doi: 10.1016/j.lungcan.2018.07.013
6. Ma K, Jin Q, Wang M, Li X, Zhang Y. Research progress and clinical application of predictive biomarker for immune checkpoint inhibitors. Expert Rev Mol Diagn. (2019) 19:517–29. doi: 10.1080/14737159.2019.1617702
7. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. (2016) 16:275–87. doi: 10.1038/nrc.2016.36
8. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. (2012) 12:298–306. doi: 10.1038/nrc3245
9. Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. (2019) 30:2012. doi: 10.1093/annonc/mdz224
10. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. (2018) 359:91–7. doi: 10.1126/science.aan3706
11. Hellmann MD, Nathanson T, Rizvi H, Creelan BC, Sanchez-Vega F, Ahuja A, et al. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell. (2018) 33:843–52.e4. doi: 10.1016/j.ccell.2018.03.018
12. Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. (2018) 378:2093–104. doi: 10.1056/NEJMoa1801946
13. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. (2019) 19:133–50. doi: 10.1038/s41568-019-0116-x
14. Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science. (2017) 355:eaaf8399. doi: 10.1126/science.aaf8399
15. Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. (2018) 36:633–41. doi: 10.1200/jco.2017.75.3384
16. Camp RL, Dolled-Filhart M, Rimm DL. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res. (2004) 10:7252–9. doi: 10.1158/1078-0432.Ccr-04-0713
17. Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. (2015) 17:251–64. doi: 10.1016/j.jmoldx.2014.12.006
18. Aredo JV, Padda SK, Kunder CA, Han SS, Neal JW, Shrager JB, et al. Impact of KRAS mutation subtype and concurrent pathogenic mutations on non-small cell lung cancer outcomes. Lung Cancer. (2019) 133:144–50. doi: 10.1016/j.lungcan.2019.05.015
19. Ready N, Hellmann MD, Awad MM, Otterson GA, Gutierrez M, Gainor JF, et al. First-line nivolumab plus ipilimumab in advanced non-small-cell lung cancer (CheckMate 568): outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J Clin Oncol. (2019) 37:992–1000. doi: 10.1200/jco.18.01042
20. El Osta B, Behera M, Kim S, Berry LD, Sica G, Pillai RN, et al. Characteristics and outcomes of patients with metastatic KRAS-mutant lung adenocarcinomas: the lung cancer mutation consortium experience. J Thorac Oncol. (2019) 14:876–89. doi: 10.1016/j.jtho.2019.01.020
21. Dong ZY, Zhong WZ, Zhang XC, Su J, Xie Z, Liu SY, et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res. (2017) 23:3012–24. doi: 10.1158/1078-0432.Ccr-16-2554
22. Adderley H, Blackhall FH, Lindsay CR. KRAS-mutant non-small cell lung cancer: converging small molecules and immune checkpoint inhibition. EBioMedicine. (2019) 41:711–6. doi: 10.1016/j.ebiom.2019.02.049
23. Calles A, Liao X, Sholl LM, Rodig SJ, Freeman GJ, Butaney M, et al. Expression of PD-1 and its ligands, PD-L1 and PD-L2, in smokers and never smokers with KRAS-mutant lung cancer. J Thorac Oncol. (2015) 10:1726–35. doi: 10.1097/jto.0000000000000687
24. Liu C, Zheng S, Jin R, Wang X, Wang F, Zang R, et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. (2020) 470:95–105. doi: 10.1016/j.canlet.2019.10.027
25. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. (2015) 348:124–8. doi: 10.1126/science.aaa1348
26. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. (2018) 8:822–35. doi: 10.1158/2159-8290.Cd-18-0099
27. Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. (2015) 5:860–77. doi: 10.1158/2159-8290.Cd-14-1236
28. Hieronymus H, Murali R, Tin A, Yadav K, Abida W, Moller H, et al. Tumor copy number alteration burden is a pan-cancer prognostic factor associated with recurrence and death. eLife. (2018) 7:e37294. doi: 10.7554/eLife.37294
29. Ock CY, Hwang JE, Keam B, Kim SB, Shim JJ, Jang HJ, et al. Genomic landscape associated with potential response to anti-CTLA-4 treatment in cancers. Nat Commun. (2017) 8:1050. doi: 10.1038/s41467-017-01018-0
30. Kim J, Kim B, Kang SY, Heo YJ, Park SH, Kim ST, et al. Tumor mutational burden determined by panel sequencing predicts survival after immunotherapy in patients with advanced gastric cancer. Front Oncol. (2020) 10:314. doi: 10.3389/fonc.2020.00314
Keywords: KRAS mutation, lung adenocarcinoma, tumor mutation burden, copy number of alteration, biomarker
Citation: Xiang L, Fu X, Wang X, Li W, Zheng X, Nan K and Tian T (2020) A Potential Biomarker of Combination of Tumor Mutation Burden and Copy Number Alteration for Efficacy of Immunotherapy in KRAS-Mutant Advanced Lung Adenocarcinoma. Front. Oncol. 10:559896. doi: 10.3389/fonc.2020.559896
Received: 07 May 2020; Accepted: 03 September 2020;
Published: 24 September 2020.
Edited by:
Paweł Adam Krawczyk, Medical University of Lublin, PolandReviewed by:
Reyes Bernabé, Spanish National Health System, SpainZoltan Lohinai, National Koranyi Institute of TB and Pulmonology, Hungary
Copyright © 2020 Xiang, Fu, Wang, Li, Zheng, Nan and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Tian, dGlhbnRhbzA2MDdAMTYzLmNvbQ==; Kejun Nan, bmFua2pAMTYzLmNvbQ==
†These authors have contributed equally to this work