 Jiun-I Lai
Jiun-I Lai Yu-Jhen Tseng2
Yu-Jhen Tseng2 Peter Mu-Hsin Chang
Peter Mu-Hsin Chang- 1Division of Medical Oncology, Department of Oncology, Taipei Veterans General Hospital, Taipei, Taiwan
- 2Center for Immuno-Oncology, Department of Oncology, Taipei Veterans General Hospital, Taipei, Taiwan
- 3Institute of Clinical Medicine, School of Medicine, National Yang-Ming University, Taipei, Taiwan
- 4School of Medicine, National Yang-Ming University, Taipei, Taiwan
- 5Institute of Biopharmaceutical Sciences, National Yang-Ming University, Taipei, Taiwan
P-glycoprotein (also known as multidrug resistance protein 1 (MDR1) or ATP-binding cassette sub-family B member 1 (ABCB1) plays a crucial role in determining response against medications, including cancer therapeutics. It is now well established that p-glycoprotein acts as an ATP dependent pump that pumps out small molecules from cells. Ample evidence exist that show p-glycoprotein expression levels correlate with drug efficacy, which suggests the rationale for developing p-glycoprotein inhibitors for treatment against cancer. Preclinical and clinical studies have investigated this possibility, but mostly were limited by substantial toxicities. Repurposing FDA-approved drugs that have p-glycoprotein inhibition activities is therefore a potential alternative approach. In this review, we searched the Drugbank Database (https://www.drugbank.ca/drugs) and identified 98 FDA-approved small molecules that possess p-glycoprotein inhibition properties. Focusing on the small molecules approved with indications against non-cancer diseases, we query the scientific literature for studies that specifically investigate these therapeutics as cancer treatment. In light of this analysis, potential development opportunities will then be thoroughly investigated for future efforts in repositioning of non-cancer p-glycoprotein inhibitors in single use or in combination therapy for clinical oncology treatment.
Introduction
Chemotherapy has been the mainstay of cancer treatment for several decades, and is still an indispensable treatment component in current standard of care. The ability of cancer cells to acquire resistance against cytotoxic chemotherapy is one of the basis for cancer progression. One major mechanism for chemotherapy resistance formation is drug efflux by the ATP-binding cassette (ABC) transporter family of transmembrane proteins (1). These proteins share a common ability to pump chemotherapy agents across the plasma membrane with no prerequisite for structure similarity. The discovery of outward efflux of daunorubicin led to the first report of chemotherapy resistance mechanism by an ABC transporter family protein in 1973 (2). Further work demonstrated that this protein was a cell surface glycoprotein that was later on named P-glycoprotein (p-gp) (3). In the upcoming years, several other proteins of the ABC transporter family were identified that shared a common mechanism of conferring chemotherapy resistance by drug efflux (4). However, p-gp remains the best studied and most potent ABC transporter to induce chemoresistance. The drugs that are amenable to p-gp transport includes many of the most commonly used chemotherapy agents such as anthracyclines, taxenes, vinca alkaloids, and many others (5). In the human body, p-gp is widely expressed in a variety of organs including the liver, kidney and gastrointestinal tract (6), and upregulation of p-gp is closely linked with increased chemoresistance in cancer (7).
Early Development Efforts of First Generation p-gp Inhibitors in Cancer Treatment
With strong mechanistic evidence for p-gp in mediating chemoresistance, a logical therapeutic approach would be to utilize p-gp inhibitors against cancers, with the rationale of enhancing chemotherapy efficacy. Indeed, in the past decades, many efforts have been made to incorporate p-gp inhibitors in clinical trials for cancer patients. Earliest efforts of p-gp inhibitors in cancer included trials using verapamil (8) and cyclosporine (9) but they were unsuccessful and demonstrated many adverse effects (10). Verapamil is a phenylalkylamine that blocks voltage-dependent L-type calcium channels and is used for treatment of hypertension. In a randomized phase II clinical trial in patients diagnosed with small cell lung cancer receiving cyclophosphamide, doxorubicin, vincristine, and etoposide (CAVE), a daily dose of 480 mg was given in combination with chemotherapy, with hypotension as a dose limiting toxicity for verapamil (11). Even at this high dosage, clinical trials failed to demonstrate a benefit for verapamil (11, 12). Further analysis demonstrated that blood levels of verapamil was significantly lower than the dosage proven efficacious in preclinical studies (11), despite already nearing maximum tolerated doses (MTD). Verapamil is representative of the so called “first generation” p-gp inhibitors that utilize drugs with known mechanism of action (MOA). These agents include cyclosporine (13, 14), quinidine (15), tamoxifen (16), and others. In summary, first generation p-gp inhibitors suffer from low therapeutic window, poor efficacy in combination with chemotherapy, and generally low potential for further development (10).
Second Generation p-gp Inhibitors
Second generation p-gp inhibitors aimed to improve the therapeutic window of first generation agents. Dexverapamil is the R-form of verapamil with reduced potency in inhibiting p-gp but also with reduced cardiotoxicity. In a phase I/II trial (17), dexverapamil was combined with vinblastine in treatment of renal cell carcinoma. The toxicity profile was tolerable in comparison to trials using verapamil, with the majority being mild or asymptomatic cardiac related side effects. No partial or complete responses were seen, which is not unexpected given the poor chemosensitivity of renal cell carcinoma. A phase II study tested the hypothesis of dexverapamil in reversing chemoresistance, by adding dexverapamil to cancer patients who had disease progression on anthracyclines (18). Among 21 patients, 2 had partial responses and 2 had stable disease, with a disease control rate (DCR) of 19%, demonstrating a modest activity. It should be highlighted that this trial used the same anthracycline backbone and added dexverapamil upon documented progression, therefore demonstrating the anticipated drug resistance reversal abilities of p-gp inhibition.
S9788 is another p-gp inhibitor that showed MDR reversal in a phase I trial in combination with doxorubicin in colorectal cancer and renal cell carcinoma patients, although cardiac arrhythmia was a major dose limiting toxicity (19). In another phase I trial, also in combination with doxorubicin, arrhythmia including AV-block, QT prolongation, ventricular arrhythmia and torsade de pointes precluded further development of the trial and essentially for the drug altogether (20).
PSC-833, an analog of cyclosporine with a significantly improved toxicity profile and enhanced p-gp inhibitory ability (21, 22), underwent several clinical trials for development as a potent second generation agent. In phase I trials, PSC-833 demonstrated acceptable toxicity profiles (23), and was further developed in combination with chemotherapy, mainly in leukemia patients with the aim to delay ensuing drug resistance from first line MEC (mitoxantrone, etoposide, ara-C) induction chemotherapy (24–26). Some degree of success was observed in these earlier phase trials, leading to several phase III trials utilizing PSC-833 in leukemia (27) and solid tumor or multiple myeloma (28, 29). However, no benefit was seen in any of these phase III trials with addition of PSC-833, dampening the initial excitement and expectations for this drug.
Third Generation p-gp Inhibitors
With the unsuccessful attempts in clinical development for p-gp inhibitors, medicinal chemistry work developed newer agents with high selectivity and efficacy, including zosuquidar, elaquidar, laniquidar (R101933), and tariquidar (XR9576) (30). Although currently no p-gp inhibitors have been approved for cancer treatment, the clinical development of these novel agents are worth mentioning in detail, which may lead to further understanding of how to utilize MDR reversal in oncology therapy. In the following sections, we discuss the properties of each agent and their clinical development, with a focus on future perspectives for further development.
Zosuquidar
Zosuquidar trihydrochlorid, previously known as LY335979, was developed as a potent and selective inhibitor against p-gp, with minimal inhibitory effect on other MDR proteins (MRP1, MRP2) (31). A phase I trial investigated oral zosuquidar alone and in combination with doxorubicin in advanced malignancies to determine its safety profile (32). Dose limiting toxicities included cerebellar dysfunction, hallucinations, and palinopsia, but overall the drug was well tolerated. Subsequently, another phase I trial investigated the intravenous form of zosuquidar in combination with doxorubicin in cancer (33). In this trial, neurotoxicity was markedly less severe and less frequent, suggesting a different pharmacokinetic profile between oral and intravenous formulation of zosuquidar. It must be noted that although phase I trials were not designed for efficacy, further analysis for these 2 trials did not demonstrate a striking improvement in response. A phase II trial investigated the addition of zosuquidar to docetaxel in metastatic breast cancer. Although the combination was safe, the conclusion of the trial did not demonstrate any difference in progression free survival (PFS), overall survival (OS), or objective response rate (ORR) in patients with metastatic breast cancer.
Zosuquidar was also investigated in the setting of hematological cancers. In a phase I trial with acute leukemia patients, zosuquidar was combined with standard induction chemotherapy using daunorubicin and cytarabine (34). In a small sample size of 16 patients, grade 3/4 adverse effects including respiratory failure, hypokalemia, arrhythmia, and febrile neutropenia were present. Seven patients achieved a complete response (CR), with a CR rate of 43.7%. Considering that standard induction chemotherapy in AML already has a well-established ORR of 50–60% (35), it is difficult to conclude that zosuquidar provided significant additional benefit (although it must be highlighted that this is a relatively small study). In further analysis, 2 out of 4 patients with p-gp negative cancers (according to IHC staining) had CR (50%) while 5 out of the 12 patients with p-gp positive cancers had CR (41.6%), further suggesting that zosuquidar might not provide anticipated efficacy when added to standard induction chemotherapy to AML (acute myeloid leukemia). A confirmatory phase III trial was performed in elderly (over 60 years old) AML patients, receiving standard cytarabine and daunorubicin in addition to either zosuquidar or placebo (36). The response rate and median 2-year OS were similar (20 vs 23%, 7.2 months vs 9.4 months, respectively for with versus without zosuquidar). In an accompanying editorial, the authors rechallenged the validity of using p-gp as the target for leukemia based on this trial and other accumulated trial results (37). They suggest that although p-gp and other MDR proteins including MRP, LRP, and BRCP were indeed expressed by leukemia cells in the majority of patients in the trial, there was no correlation between the expression status of MDR proteins and survival (37). This editorial came to the conclusion that further MDR inhibition in the setting of AML should be highly reconsidered in light of the result from these well designed clinical trials.
Elacridar
Elacridar is a potent and specific inhibitor of p-gp that works by modulating the ATPase activity (38). The efficacy of elacridar in inhibiting p-gp and its effect in plasma concentrations of other drugs have been extensively characterized in multiple studies [for a comprehensive review, please refer to (39)]. Phase I studies for elacridar demonstrated very minor side effects and good pharmacokinetic properties (40). In another early phase trial, elacridar was shown to increase the plasma levels of oral paclitaxel (41), by inhibiting intestinal p-gp activity. Interestingly, when these patients were switched to intravenous paclitaxel in subsequent cycles, the area under curve (AUC) of plasma paclitaxel when using oral paclitaxel plus elacridar was still significantly lower compared to intravenous paclitaxel (41). This suggested a modest effect of elacridar to augment oral paclitaxel levels in patients. Similarly, in another early phase trial, elacridar significantly elevated the oral bioavailability of topotecan to more than 2 fold when used in combination with oral topotecan (42), although blood topotecan levels was still lower than the levels of intravenous topotecan. Nonetheless, this result implicated a potential application for elacridar to be combined with oral chemotherapy for long-term use. Similar conclusions were made in another phase I trial (43). When combining with intravenous chemotherapy Elacridar was demonstrated to be safe with relatively mild side effects in combination with doxorubicin in solid cancers (44). The most common toxicity was neutropenia. Despite the acceptable safety profile of these early phase trials, elacridar was not further developed in later clinical trials.
Tariquidar
Tariquidar is a potent and specific inhibitor of p-gp, acting as an ATPase inhibitor (45). After demonstrating preclinical efficacy, early clinical trials demonstrated good tolerability with minimal side effects in healthy individuals (46). A phase I study combining tariquidar and vinorelbine demonstrated good tolerability, and more importantly, the lack of pharmacokinetic interactions between tariquidar and vinorelbine, as this was found to be a common issue with p-gp inhibitors in human trials (47). However, there was overall a lack of response in this trial. In a phase II study tariquidar was added to the standard chemotherapy (anthracyclines or taxanes) in breast cancer patients’ refractory on these regimens. Tariquidar was added to the current regimen to observe if drug resistance could be reversed. A total of 17 patients were enrolled, and only one patient demonstrated a partial response (ORR: 6%). 5 out of the 17 patients had a positive p-gp staining before treatment, including the patient with response. Therefore, it could be estimated that in p-gp positive patients, the response rate was 20%, albeit a very small sample size (n=5). Interestingly, this trial measured the uptake of technetium-99m (99MTc) as a surrogate for whether the anticipated biology effect of improving drug influx is seen. The strongest increase of 99MTc occurred in the single patient with the response. This suggested that 99MTc might be a viable indicator of patients that could benefit from tariquidar and other p-gp inhibitors. This phenomenon led to an unexpected development of tariquidar into a potential radiotracer when coupled with radioactive [11C] labeling (48). This has become an active area of research for tariquidar and other p-gp inhibitors (49–51). Another phase I trial combined tariquidar with docetaxel in patients with lung, ovarian and cervical cancer (52). This trial confirmed that tariquidar was associated with increased 99MTc sestamibi uptake, however this finding did not translate into a significant increase in ORR as only 10% of patients had a response in this trial. A more recent phase I trial combined tariquidar with doxorubicin, vinorelbine, or docetaxel in children and adolescents with refractory solid tumors (53). An ORR of approximately 10% was observed. Overall, tariquidar clinical trials were similar to other third generation p-gp inhibitors, demonstrating relatively tolerable safety profiles, but with unimpressive anticancer efficacy. We summarize the pg-inhibitors in Table 1.
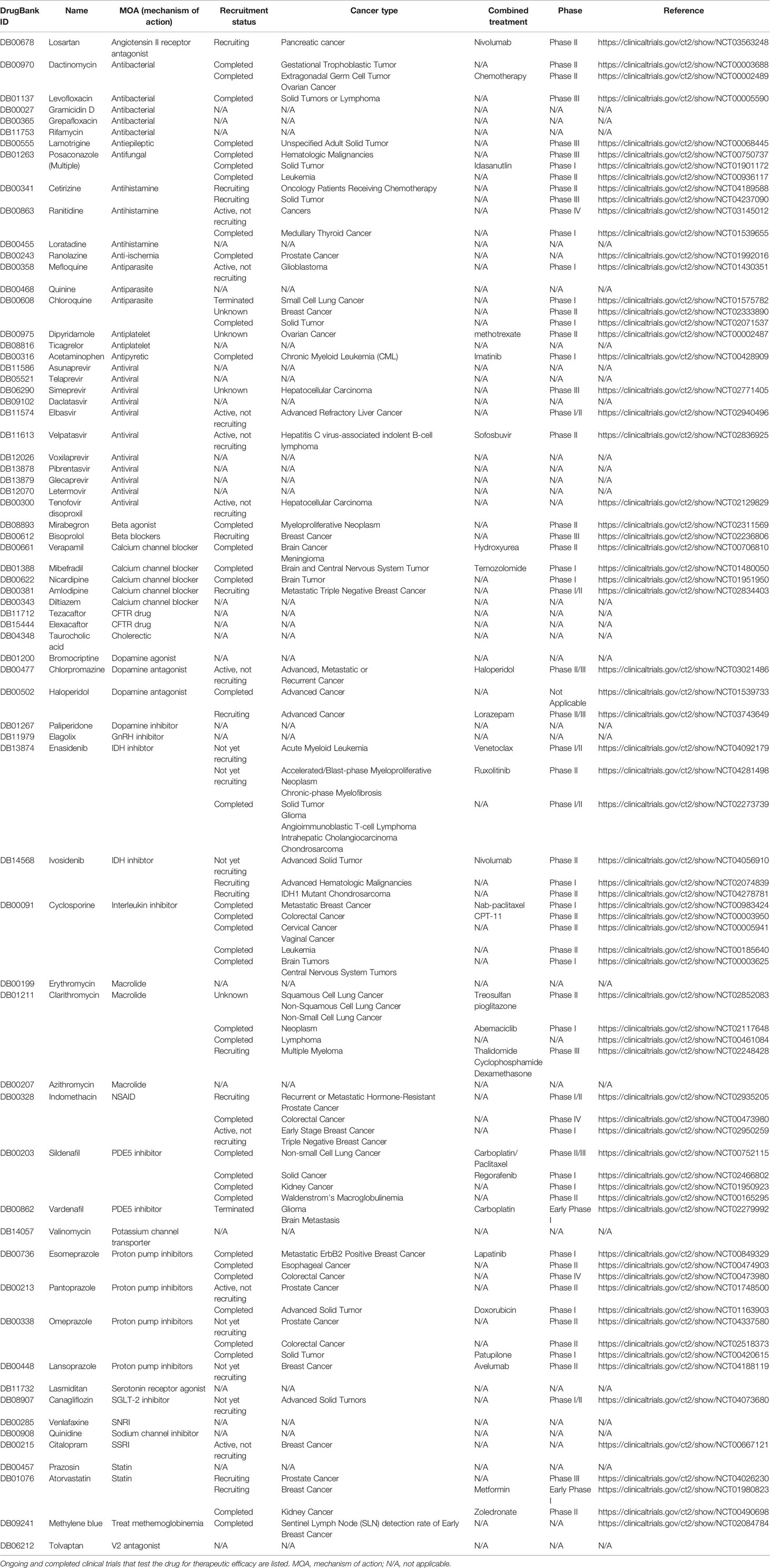
Table 1 FDA approved drugs with p-gp inhibition activities.
Repurposing of FDA Approved Drugs With p-gp Inhibitor Activity in Cancer Treatment
Rationale
In light of previously reviewed clinical development history, small molecules specifically designed for p-gp inhibition were largely unsuccessful in clinical trials. Early generations of p-gp inhibitors were burdened with significant side effect toxicities, while newer generations of p-gp inhibitors generally had a more acceptable safety profile, yet still demonstrated limited efficacy.
We then sought to investigate current FDA approved drugs for their p-gp activity. We propose two rationales for this approach. First, FDA approval as well as real world experience indicates the safety profile of these drugs, therefore minimizing further development issues. Additionally, repurposing of these drugs in combination with current anti-cancer therapeutics may provide an additive effect to enhance efficacy.
Discovery of Candidates for p-gp Inhibitor Repurposing
We queried the Drugbank database (https://www.drugbank.ca/) (54) for FDA approved drugs with demonstrated activity towards p-gp (gene symbol: ABCB1). The search resulted in 508 candidates. We then filtered out drugs without inhibitor activity for p-gp, leaving 116 drugs. We further filtered out drugs that had the “inducer” label, meaning that at least one reference reported upregulation of p-gp by this drug under specific circumstances. The final result was 98 FDA approved drugs with reported p-gp inhibition activity (Figure 1).
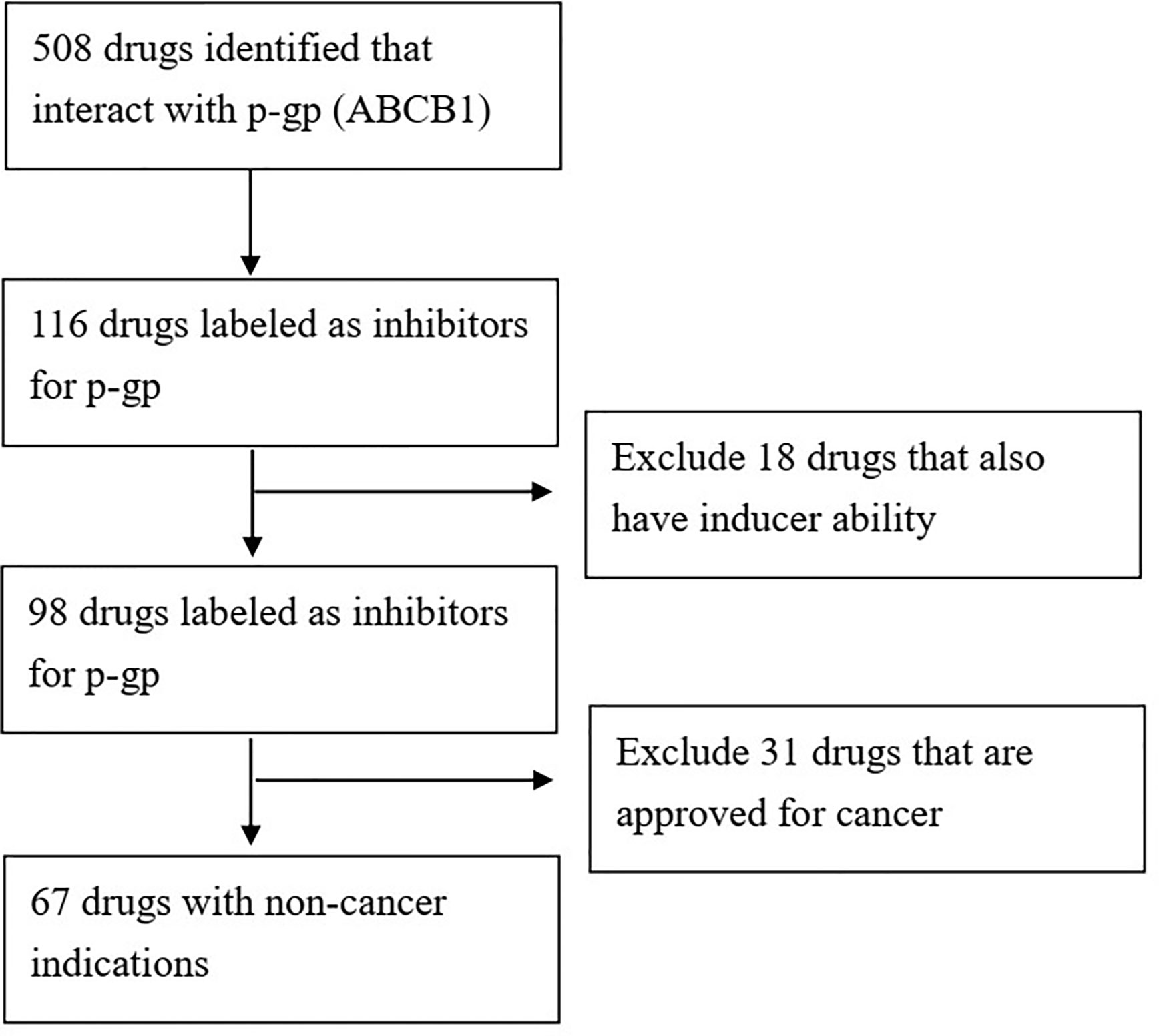
Figure 1 Flowchart of discovery of repurposing FDA approved drugs with p-gp inhibition abilities.
Interestingly, many drugs approved for anti-cancer properties were on this list of p-gp inhibitors. The known anticancer drugs included tyrosine kinase inhibitors (imatinib, sorafenib, dasatinib, gefitinib, nilotinib, erlotinib, and afatinib), PARP inhibitors (olaparib, rucaprib), CDK4/6 inhibitors (palbociclib, abemaciclib), taxanes (paclitaxel, cabazitaxel) and others. This finding leads to several observations: first, multiple agents with the same mechanism concomitantly inhibit p-gp suggest that this is a class effect, with a potential undiscovered mechanism. Secondly, the drugs of the same class that are not reported to possess p-gp inhibitor property (barring undiscovered p-gp inhibitor ability) may therefore possess a novel, unique chemical biological characteristic. For instance, two of the three approved CDK4/6 inhibitors (palbociclib, abemaciclib) inhibit p-gp, while ribociclib was not annotated with this property. Is the p-gp inhibition function not yet discovered in ribociclib, or does ribociclib possess a specific structural aspect that abolishes this possible ability? Third, these drug that intrinsically possess p-gp inhibitory functions suggest that additional p-gp inhibition with specific small molecules might provide limited additional benefit. This may be the case of taxanes, as several p-gp inhibitors were tested in combination with paclitaxel. It must be emphasized that the intrinsic p-gp inhibitory ability is uncharacterized for most of these drugs, so we cannot assess whether they possess strong p-gp inhibition function or otherwise.
We then excluded drugs that were approved for or possessed well established anti-tumor activities, filtering down the list to 67 agents. A summary of the final 67 drugs is shown in Table 1. Again, different agents of the same class appear, including proton pump inhibitors (Pantoprazole, omeprazole, lansoprazole, and esomeprazole), histamine blockers (ranitidine and loratadine), quinolones (levofloxacin and grepafloxacin), calcium channel blockers (amlodipine and nicardipine), phenothiazine (chlorpromazine) and others, hinting at possible class effect of p-gp inhibition. Interestingly, this feature may explain some previous observational studies. For instance, quinolones (ciprofloxacin or ofloxacin) when given in combination with doxorubicin as intravesicular treatment, significantly enhanced the cytotoxicity of doxorubicin towards cancer cells (55). We queried Clinicaltrials.gov database (https://clinicaltrials.gov/ct2/home) to search for ongoing or completed trials that used of these drugs with a cutoff date of July 2020. Specifically, we included the trials that investigate the therapeutic effect of these drugs in cancer, summarized in Table 2. In the following sections, we highlight some of the compounds that have been reported or investigated in combination with chemotherapy for anticancer treatment. We summarize the highlighted compounds in Table 3.
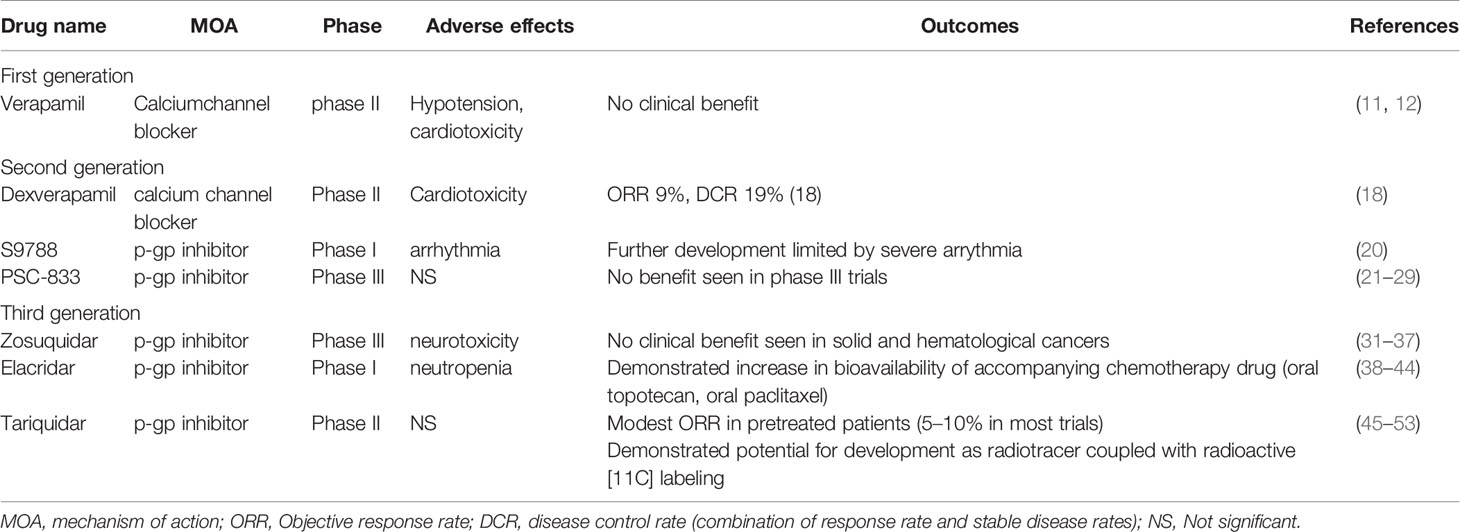
Table 2 Summary of major pg-1 inhibitors in clinical development.

Table 3 Summary of highlighted compounds that have been reported or investigated in combination with chemotherapy for anticancer treatment.
Chlorpromazine
Chlorpromazine is a phenothiazine drug with p-gp inhibiting abilities. Clinically it possesses anti-emetic properties as well as antipsychotic properties. Interestingly, antipsychotics were another class of drug that were overrepresented in our p-gp inhibitor list (including paliperidone, haloperidol). Chlorpromazine is a p-gp inhibitor with an EC50 10 fold weaker than PSC833, but nevertheless is in low micromolar range (58). There has been much active research relating to chlorpromazine and its anticancer properties, as it has been reported to suppress the Hippo pathway related YAP signaling and cancer stemness (59), suppress chemoresistant cancer growth (60, 61), epigenetic anticancer mechanisms (62), and many other abilities. The ability to inhibit cancer stem cells seems to be a class effect of phenothiazines, and many novel derivatives are currently being developed towards this goal (63). It remains to be seen whether the ability to inhibit p-gp of phenothaizaines such as chlorpromazine will be a positive factor in enhancing antitumor activity either in monodrug use or in combination with other cytotoxic agents.
Macrolides: Erythromycin, Azithromycin, and Clarithromycin
The FDA approved macrolide antibiotics (erythromycin, azithromycin, and clarithromycin) were all reported as p-gp inhibitors. This has been supported by many studies in the literature (64–66). Macrolides are well known CYP3A4 inducers (67, 68), thus it may interfere with pharmacokinetics with various results, since CYP3A4 and p-gp would both play a role in drug-drug interaction. Indeed, investigations regarding macrolides upon anticancer drugs have been made to further clarify whether efficacious drug effect can be adequately delivered (69). The macrolide clarithromycin has been studied in multiple clinical trials with cancer subjects, in combination with metronomic chemotherapy, abemaciclib, etc. (see Tables 2, 3). In these trials, the rationale was mainly to study the CYP3A inhibitor activity of clarithromycin and its impact on the accompanying drug, but some have shown promising efficacy with possible potential for further follow up clinical trials.
Phosphodiesterase Inhibitors: Sildenafil and Vardenafil
Two FDA approved phosphodiesterase inhibitors, sildenafil and vardenafil were reported to possess p-gp inhibition activities. Both sildenafil and vardenafil are potent PDE5 inhibitors that are widely used in erectile dysfunction. Interestingly, sildenafil has been actively studied for its role in anticancer treatment (70, 71), and has been proposed to potentiate cisplatin mediated tumor killing (71–73). While multiple mechanisms have been proposed for this phenomenon, the role of p-gp inhibition remain a very likely explanation. The anticancer roles of PDE5 inhibitors in single use and in combination with chemotherapy remains an active area of research and we anticipate more results to be reported on this topic.
Conclusions
The clinical development of bona fide p-gp inhibitors has been rather disappointing in the past two decades. Although much progress has been made in medicinal chemistry to develop newer generation p-gp inhibitors, failure in clinical trials suggest that solely targeting p-gp may not be an efficacious strategy for cancer. A lot of the early generation p-gp inhibitors suffered from toxicity, while newer generation agents largely failed from lack of clinical response. A silver lining from these failed clinical trial is the development of the p-gp inhibitor tariquidar in nuclear tracer uptake enhancing agents for improvement in medical imaging. As we have demonstrated here, this is a promising field of research that may lead to clinical usefulness of p-gp inhibitors in the near future.
Repurposing current FDA approved drugs as a strategy for cancer development offers the benefit of well-known toxicity profile and mechanism of action, minimizing unexpected setbacks in clinical development. From the brief review of FDA approved drugs that possess p-gp inhibition activity, we highlight several candidates such as chlorpromazine and sildenafil that could have potential for further development, utilizing its role of p-gp inhibitor to enhance the potency of tumor cytotoxic agents. As highlighted in Table 2, many of these drugs have underwent clinical trials, either as monotherapy or in combination for cancer patients. Repurposing “old” currently common drugs for cancer treatment is an area of heightened interest, and many drugs with long history of clinical use have been discovered for novel mechanisms. One example is metformin (FDA approved in 1998), which has recently been reported to have promising roles for anticancer therapies in both preclinical and clinical studies. Metformin is now being investigated in numerous active clinical trials for discovering a positive role in cancer. We are hopeful that future research would define a role of repurposed p-gp inhibitors in cancer treatment and in improving patient survival.
Author Contributions
J-IL performed the literature search and drafted the manuscript. Y-JT and M-HC performed the data curation and construction of the Tables. C-YH contributed to study design, insight, and manuscript drafting. PC designed the study, assisted in the literature search, revised and refined the completed manuscript and the Figure and Tables. All authors contributed to the article and approved the submitted version.
Funding
This study was partially funded by the grants: V109C-009 and MOST-109-2314-B-075-080 (to PC), and 108DHA0100627 (to PC, J-IL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. (2013) 13(10):714–26. doi: 10.1038/nrc3599
2. Dano K. Active outward transport of daunomycin in resistant Ehrlich ascites tumor cells. Biochim Biophys Acta (1973) 323(3):466–83. doi: 10.1016/0005-2736(73)90191-0
3. Chen CJ, Chin JE, Ueda K, Clark DP, Pastan I, Gottesman MM, et al. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell (1986) 47(3):381–9. doi: 10.1016/0092-8674(86)90595-7
4. Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discovery (2006) 5(3):219–34. doi: 10.1038/nrd1984
5. Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol (1999) 39:361–98. doi: 10.1146/annurev.pharmtox.39.1.361
6. Cordon-Cardo C, O’Brien JP, Boccia J, Casals D, Bertino JR, Melamed MR. Expression of the multidrug resistance gene product (P-glycoprotein) in human normal and tumor tissues. J Histochem Cytochem (1990) 38(9):1277–87. doi: 10.1177/38.9.1974900
7. Nanayakkara AK, Follit CA, Chen G, Williams NS, Vogel PD, Wise JG. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci Reports (2018) 8(1):967. doi: 10.1038/s41598-018-19325-x
8. Giaccone G, Pinedo HM. Drug Resistance. Oncologist (1996) 1(1 2):82–7. doi: 10.1634/theoncologist.1-1-82
9. Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathology (2005) 205(2):275–92. doi: 10.1002/path.1706
10. Ferry DR, Traunecker H, Kerr DJ. Clinical trials of P-glycoprotein reversal in solid tumours. Eur J Cancer (1996) 32A(6):1070–81. doi: 10.1016/0959-8049(96)00091-3
11. Milroy R. A randomised clinical study of verapamil in addition to combination chemotherapy in small cell lung cancer. West of Scotland Lung Cancer Research Group, and the Aberdeen Oncology Group. Br J Cancer (1993) 68(4):813–8. doi: 10.1038/bjc.1993.433
12. Saltz L, Murphy B, Kemeny N, Bertino J, Tong W, Keefe D, et al. A phase I trial of intrahepatic verapamil and doxorubicin. Regional therapy to overcome multidrug resistance. Cancer (1994) 74(10):2757–64. doi: 10.1002/1097-0142(19941115)74:10<2757::AID-CNCR2820741004>3.0.CO;2-O
13. Bartlett NL, Lum BL, Fisher GA, Brophy NA, Ehsan MN, Halsey J, et al. Phase I trial of doxorubicin with cyclosporine as a modulator of multidrug resistance. J Clin Oncol Off J Am Soc Clin Oncol (1994) 12(4):835–42. doi: 10.1200/JCO.1994.12.4.835
14. List AF, Spier C, Greer J, Wolff S, Hutter J, Dorr R, et al. Phase I/II trial of cyclosporine as a chemotherapy-resistance modifier in acute leukemia. J Clin Oncol Off J Am Soc Clin Oncol (1993) 11(9):1652–60. doi: 10.1200/JCO.1993.11.9.1652
15. Jones RD, Kerr DJ, Harnett AN, Rankin EM, Ray S, Kaye SB. A pilot study of quinidine and epirubicin in the treatment of advanced breast cancer. Br J Cancer (1990) 62(1):133–5. doi: 10.1038/bjc.1990.244
16. Chang SM, Barker FG,2, Huhn SL, Nicholas MK, Page M, Rabbitt J, et al. High dose oral tamoxifen and subcutaneous interferon alpha-2a for recurrent glioma. J Neurooncol (1998) 37(2):169–76. doi: 10.1023/A:1005826323652
17. Motzer RJ, Lyn P, Fischer P, Lianes P, Ngo RL, Cordon-Cardo C, et al. Phase I/II trial of dexverapamil plus vinblastine for patients with advanced renal cell carcinoma. J Clin Oncol Off J Am Soc Clin Oncol (1995) 13(8):1958–65. doi: 10.1200/JCO.1995.13.8.1958
18. Warner E, Hedley D, Andrulis I, Myers R, Trudeau M, Warr D, et al. Phase II study of dexverapamil plus anthracycline in patients with metastatic breast cancer who have progressed on the same anthracycline regimen. Clin Cancer Res an Off J Am Assoc Cancer Res (1998) 4(6):1451–7. doi: 10.1038/bjc.1997.563
19. Punt CJ, Voest EE, Tueni E, Van Oosterom AT, Backx A, De Mulder PH, et al. Phase IB study of doxorubicin in combination with the multidrug resistance reversing agent S9788 in advanced colorectal and renal cell cancer. Br J Cancer (1997) 76(10):1376–81. doi: 10.1038/bjc.1997.563
20. Stupp R, Bauer J, Pagani O, Gerard B, Cerny T, Sessa C, et al. Ventricular arrhythmia and torsade de pointe: dose limiting toxicities of the MDR-modulator S9788 in a phase I trial. Ann Oncol (1998) 9(11):1233–42. doi: 10.1023/A:1008495919071
21. Twentyman PR, Bleehen NM. Resistance modification by PSC-833, a novel non-immunosuppressive cyclosporin [corrected]. Eur J Cancer (1991) 27(12):1639–42. doi: 10.1016/0277-5379(91)90435-G
22. Boesch D, Gaveriaux C, Jachez B, Pourtier-Manzanedo A, Bollinger P, Loor F. In vivo circumvention of P-glycoprotein-mediated multidrug resistance of tumor cells with SDZ PSC 833. Cancer Res (1991) 51(16):4226–33.
23. Boote DJ, Dennis IF, Twentyman PR, Osborne RJ, Laburte C, Hensel S, et al. Phase I study of etoposide with SDZ PSC 833 as a modulator of multidrug resistance in patients with cancer. J Clin Oncol Off J Am Soc Clin Oncol (1996) 14(2):610–8. doi: 10.1200/JCO.1996.14.2.610
24. Visani G, Milligan D, Leoni F, Chang J, Kelsey S, Marcus R, et al. Combined action of PSC 833 (Valspodar), a novel MDR reversing agent, with mitoxantrone, etoposide and cytarabine in poor-prognosis acute myeloid leukemia. Leukemia (2001) 15(5):764–71. doi: 10.1038/sj.leu.2402117
25. Advani R, Saba HI, Tallman MS, Rowe JM, Wiernik PH, Ramek J, et al. Treatment of refractory and relapsed acute myelogenous leukemia with combination chemotherapy plus the multidrug resistance modulator PSC 833 (Valspodar). Blood (1999) 93(3):787–95. doi: 10.1182/blood.V93.3.787
26. Advani R, Visani G, Milligan D, Saba H, Tallman M, Rowe JM, et al. Treatment of poor prognosis AML patients using PSC833 (valspodar) plus mitoxantrone, etoposide, and cytarabine (PSC-MEC). Adv Exp Med Biol (1999) 457:47–56. doi: 10.1007/978-1-4615-4811-9_6
27. Greenberg PL, Lee SJ, Advani R, Tallman MS, Sikic BI, Letendre L, et al. Mitoxantrone, etoposide, and cytarabine with or without valspodar in patients with relapsed or refractory acute myeloid leukemia and high-risk myelodysplastic syndrome: a phase III trial (E2995). J Clin Oncol Off J Am Soc Clin Oncol (2004) 22(6):1078–86. doi: 10.1200/JCO.2004.07.048
28. Friedenberg WR, Rue M, Blood EA, Dalton WS, Shustik C, Larson RA, et al. Phase III study of PSC-833 (valspodar) in combination with vincristine, doxorubicin, and dexamethasone (valspodar/VAD) versus VAD alone in patients with recurring or refractory multiple myeloma (E1A95): a trial of the Eastern Cooperative Oncology Group. Cancer (2006) 106(4):830–8. doi: 10.1002/cncr.21666
29. Lhomme C, Joly F, Walker JL, Lissoni AA, Nicoletto MO, Manikhas GM, et al. Phase III study of valspodar (PSC 833) combined with paclitaxel and carboplatin compared with paclitaxel and carboplatin alone in patients with stage IV or suboptimally debulked stage III epithelial ovarian cancer or primary peritoneal cancer. J Clin Oncol Off J Am Soc Clin Oncol (2008) 26(16):2674–82. doi: 10.1200/JCO.2007.14.9807
30. Palmeira A, Sousa E, Vasconcelos MH, Pinto MM. Three decades of P-gp inhibitors: skimming through several generations and scaffolds. Curr Med Chem (2012) 19(13):1946–2025. doi: 10.2174/092986712800167392
31. Shepard RL, Cao J, Starling JJ, Dantzig AH. Modulation of P-glycoprotein but not MRP1- or BCRP-mediated drug resistance by LY335979. Int J Cancer (2003) 103(1):121–5. doi: 10.1002/ijc.10792
32. Rubin EH, de Alwis DP, Pouliquen I, Green L, Marder P, Lin Y, et al. A phase I trial of a potent P-glycoprotein inhibitor, Zosuquidar.3HCl trihydrochloride (LY335979), administered orally in combination with doxorubicin in patients with advanced malignancies. Clin Cancer Res an Off J Am Assoc Cancer Res (2002) 8(12):3710–7.
33. Sandler A, Gordon M, De Alwis DP, Pouliquen I, Green L, Marder P, et al. A Phase I trial of a potent P-glycoprotein inhibitor, zosuquidar trihydrochloride (LY335979), administered intravenously in combination with doxorubicin in patients with advanced malignancy. Clin Cancer Res an Off J Am Assoc Cancer Res (2004) 10(10):3265–72. doi: 10.1158/1078-0432.CCR-03-0644
34. Lancet JE, Baer MR, Duran GE, List AF, Fielding R, Marcelletti JF, et al. A phase I trial of continuous infusion of the multidrug resistance inhibitor zosuquidar with daunorubicin and cytarabine in acute myeloid leukemia. Leukemia Res (2009) 33(8):1055–61. doi: 10.1016/j.leukres.2008.09.015
35. Steensma DP, Ebert BL. Clonal Hematopoiesis after Induction Chemotherapy for Acute Myeloid Leukemia. New Engl J Medicine (2018) 378(13):1244–5. doi: 10.1056/NEJMe1802610
36. Cripe LD, Uno H, Paietta EM, Litzow MR, Ketterling RP, Bennett JM, et al. Zosuquidar, a novel modulator of P-glycoprotein, does not improve the outcome of older patients with newly diagnosed acute myeloid leukemia: a randomized, placebo-controlled trial of the Eastern Cooperative Oncology Group 3999. Blood (2010) 116(20):4077–85. doi: 10.1182/blood-2010-04-277269
37. Libby E, Hromas R. Dismounting the MDR horse. Blood. (2010) 116(20):4037–8. doi: 10.1182/blood-2010-09-304311
38. Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control (2003) 10(2):159–65. doi: 10.1177/107327480301000207
39. Dash RP, Jayachandra Babu R, Srinivas NR. Therapeutic Potential and Utility of Elacridar with Respect to P-glycoprotein Inhibition: An Insight from the Published In Vitro, Preclinical and Clinical Studies. Eur J Drug Metab Pharmacokinet (2017) 42(6):915–33. doi: 10.1007/s13318-017-0411-4
40. Sparreboom A, Planting AS, Jewell RC, van der Burg ME, van der Gaast A, de Bruijn P, et al. Clinical pharmacokinetics of doxorubicin in combination with GF120918, a potent inhibitor of MDR1 P-glycoprotein. Anticancer Drugs (1999) 10(8):719–28. doi: 10.1097/00001813-199909000-00005
41. Malingre MM, Beijnen JH, Rosing H, Koopman FJ, Jewell RC, Paul EM, et al. Co-administration of GF120918 significantly increases the systemic exposure to oral paclitaxel in cancer patients. Br J Cancer (2001) 84(1):42–7. doi: 10.1054/bjoc.2000.1543
42. Kruijtzer CM, Beijnen JH, Rosing H, ten Bokkel Huinink WW, Schot M, Jewell RC, et al. Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-glycoprotein inhibitor GF120918. J Clin Oncol Off J Am Soc Clin Oncol (2002) 20(13):2943–50. doi: 10.1200/JCO.2002.12.116
43. Kuppens IE, Witteveen EO, Jewell RC, Radema SA, Paul EM, Mangum SG, et al. A phase I, randomized, open-label, parallel-cohort, dose-finding study of elacridar (GF120918) and oral topotecan in cancer patients. Clin Cancer Res an Off J Am Assoc Cancer Res (2007) 13(11):3276–85. doi: 10.1158/1078-0432.CCR-06-2414
44. Planting AS, Sonneveld P, van der Gaast A, Sparreboom A, van der Burg ME, Luyten GP, et al. A phase I and pharmacologic study of the MDR converter GF120918 in combination with doxorubicin in patients with advanced solid tumors. Cancer Chemotherapy Pharmacol (2005) 55(1):91–9. doi: 10.1007/s00280-004-0854-6
45. Fox E, Bates SE. Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther (2007) 7(4):447–59. doi: 10.1586/14737140.7.4.447
46. Stewart A, Steiner J, Mellows G, Laguda B, Norris D, Bevan P. Phase I trial of XR9576 in healthy volunteers demonstrates modulation of P-glycoprotein in CD56+ lymphocytes after oral and intravenous administration. Clin Cancer Res an Off J Am Assoc Cancer Res (2000) 6(11):4186–91.
47. Abraham J, Edgerly M, Wilson R, Chen C, Rutt A, Bakke S, et al. A phase I study of the P-glycoprotein antagonist tariquidar in combination with vinorelbine. Clin Cancer Res an Off J Am Assoc Cancer Res (2009) 15(10):3574–82. doi: 10.1158/1078-0432.CCR-08-0938
48. Bauer F, Kuntner C, Bankstahl JP, Wanek T, Bankstahl M, Stanek J, et al. Synthesis and in vivo evaluation of [11C]tariquidar, a positron emission tomography radiotracer based on a third-generation P-glycoprotein inhibitor. Bioorg Med Chem (2010) 18(15):5489–97. doi: 10.1016/j.bmc.2010.06.057
49. Bauer M, Karch R, Zeitlinger M, Stanek J, Philippe C, Wadsak W, et al. Interaction of 11C-tariquidar and 11C-elacridar with P-glycoprotein and breast cancer resistance protein at the human blood-brain barrier. J Nucl Med (2013) 54(8):1181–7. doi: 10.2967/jnumed.112.118232
50. Kawamura K, Konno F, Yui J, Yamasaki T, Hatori A, Yanamoto K, et al. Synthesis and evaluation of [11C]XR9576 to assess the function of drug efflux transporters using PET. Ann Nucl Med (2010) 24(5):403–12. doi: 10.1007/s12149-010-0373-y
51. Bauer M, Zeitlinger M, Karch R, Matzneller P, Stanek J, Jager W, et al. Pgp-mediated interaction between (R)-[11C]verapamil and tariquidar at the human blood-brain barrier: a comparison with rat data. Clin Pharmacol Ther (2012) 91(2):227–33. doi: 10.1038/clpt.2011.217
52. Kelly RJ, Draper D, Chen CC, Robey RW, Figg WD, Piekarz RL, et al. A pharmacodynamic study of docetaxel in combination with the P-glycoprotein antagonist tariquidar (XR9576) in patients with lung, ovarian, and cervical cancer. Clin Cancer Res an Off J Am Assoc Cancer Res (2011) 17(3):569–80. doi: 10.1158/1078-0432.CCR-10-1725
53. Fox E, Widemann BC, Pastakia D, Chen CC, Yang SX, Cole D, et al. Pharmacokinetic and pharmacodynamic study of tariquidar (XR9576), a P-glycoprotein inhibitor, in combination with doxorubicin, vinorelbine, or docetaxel in children and adolescents with refractory solid tumors. Cancer chemotherapy Pharmacol (2015) 76(6):1273–83. doi: 10.1007/s00280-015-2845-1
54. Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res (2006) 34(Database issue):D668–72.
55. Kamat AM, DeHaven JI, Lamm DL. Quinolone antibiotics: a potential adjunct to intravesical chemotherapy for bladder cancer. Urology (1999) 54(1):56–61. doi: 10.1016/s0090-4295(99)00064-3
56. Heudobler SC, Fischer JR, Staib P, Wehler T, Südhoff T, Schichtl T, et al. Pioglitazone and clarithromycin combined with metronomic low-dose chemotherapy versus nivolumab in patients with advanced non-small cell lung cancer treated in 2nd-line and beyond: Outcomes from a randomized phase II trial (ModuLung). Ann Oncol (2019) 30(suppl_5):v602–60. doi: 10.1093/annonc/mdz260
57. Patterson CJ, Soumerai J, Hunter Z, Leleu X, Ghobrial I, Treon SP. Sildenafil citrate suppresses disease progression in patients with Waldenstrom’s macroglobulinemia. J Clin Oncol (2006) 24:7556–6:18_suppl. doi: 10.1093/annonc/mdz260.092
58. Wang JS, Zhu HJ, Markowitz JS, Donovan JL, DeVane CL. Evaluation of antipsychotic drugs as inhibitors of multidrug resistance transporter P-glycoprotein. Psychopharmacol (Berl) (2006) 187(4):415–23. doi: 10.1007/s00213-006-0437-9
59. Yang CE, Lee WY, Cheng HW, Chung CH, Mi FL, Lin CW. The antipsychotic chlorpromazine suppresses YAP signaling, stemness properties, and drug resistance in breast cancer cells. Chem Biol Interact (2019) 302:28–35. doi: 10.1016/j.cbi.2019.01.033
60. Oliva CR, Zhang W, Langford C, Suto MJ, Griguer CE. Repositioning chlorpromazine for treating chemoresistant glioma through the inhibition of cytochrome c oxidase bearing the COX4-1 regulatory subunit. Oncotarget. (2017) 8(23):37568–83. doi: 10.18632/oncotarget.17247
61. Lee WY, Lee WT, Cheng CH, Chen KC, Chou CM, Chung CH, et al. Repositioning antipsychotic chlorpromazine for treating colorectal cancer by inhibiting sirtuin 1. Oncotarget (2015) 6(29):27580–95. doi: 10.18632/oncotarget.4768
62. Kurita JI, Hirao Y, Nakano H, Fukunishi Y, Nishimura Y. Sertraline, chlorprothixene, and chlorpromazine characteristically interact with the REST-binding site of the corepressor mSin3, showing medulloblastoma cell growth inhibitory activities. Sci Reports (2018) 8(1):13763. doi: 10.1038/s41598-018-31852-1
63. Gao Y, Sun TY, Bai WF, Bai CG. Design, synthesis and evaluation of novel phenothiazine derivatives as inhibitors of breast cancer stem cells. Eur J Med Chem (2019) 183:111692. doi: 10.1016/j.ejmech.2019.111692
64. Munic V, Kelneric Z, Mikac L, Erakovic Haber V. Differences in assessment of macrolide interaction with human MDR1 (ABCB1, P-gp) using rhodamine-123 efflux, ATPase activity and cellular accumulation assays. Eur J Pharm Sci (2010) 41(1):86–95. doi: 10.1016/j.ejps.2010.05.016
65. Parasrampuria DA, Mendell J, Shi M, Matsushima N, Zahir H, Truitt K. Edoxaban drug-drug interactions with ketoconazole, erythromycin, and cyclosporine. Br J Clin Pharmacol (2016) 82(6):1591–600. doi: 10.1111/bcp.13092
66. Pachot JI, Botham RP, Haegele KD, Hwang K. Experimental estimation of the role of P-Glycoprotein in the pharmacokinetic behaviour of telithromycin, a novel ketolide, in comparison with roxithromycin and other macrolides using the Caco-2 cell model. J Pharm Pharm Sci (2003) 6(1):1–12. doi: 10.3389/fphar.2017.00202
67. Westphal JF. Macrolide - induced clinically relevant drug interactions with cytochrome P-450A (CYP) 3A4: an update focused on clarithromycin, azithromycin and dirithromycin. Br J Clin Pharmacol (2000) 50(4):285–95. doi: 10.1046/j.1365-2125.2000.00261.x
68. Perez-Del Palacio J, Diaz C, Vergara N, Algieri F, Rodriguez-Nogales A, de Pedro N, et al. Exploring the Role of CYP3A4 Mediated Drug Metabolism in the Pharmacological Modulation of Nitric Oxide Production. Front Pharmacol (2017) 8:202. doi: 10.3389/fphar.2017.00202
69. Agarwal SK, Tong B, Bueno OF, Menon RM, Salem AH. Effect of Azithromycin on Venetoclax Pharmacokinetics in Healthy Volunteers: Implications for Dosing Venetoclax with P-gp Inhibitors. Adv Ther (2018) 35(11):2015–23. doi: 10.1007/s12325-018-0793-y
70. Keats T, Rosengren RJ, Ashton JC. The Rationale for Repurposing Sildenafil for Lung Cancer Treatment. Anticancer Agents Med Chem (2018) 18(3):367–74. doi: 10.2174/1871520617666171103100959
71. El-Naa MM, Othman M, Younes S. Sildenafil potentiates the antitumor activity of cisplatin by induction of apoptosis and inhibition of proliferation and angiogenesis. Drug Des Devel Ther (2016) 10:3661–72. doi: 10.2147/DDDT.S107490
72. Domvri K, Zarogoulidis K, Zogas N, Zarogoulidis P, Petanidis S, Porpodis K, et al. Potential synergistic effect of phosphodiesterase inhibitors with chemotherapy in lung cancer. J Cancer (2017) 8(18):3648–56. doi: 10.7150/jca.21783
Keywords: p-glycoprotein, repurposing, cancer, drug resistance, chemotherapy
Citation: Lai J-I, Tseng Y-J, Chen M-H, Huang C-YF and Chang PM-H (2020) Clinical Perspective of FDA Approved Drugs With P-Glycoprotein Inhibition Activities for Potential Cancer Therapeutics. Front. Oncol. 10:561936. doi: 10.3389/fonc.2020.561936
Received: 14 May 2020; Accepted: 01 October 2020;
Published: 16 November 2020.
Edited by:
Sungpil Yoon, Sungkyunkwan University, South KoreaReviewed by:
Helena Kuivaniemi, Stellenbosch University, South AfricaShinobu Ohnuma, Tohoku University, Japan
Copyright © 2020 Lai, Tseng, Chen, Huang and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Mu-Hsin Chang, cHRjaGFuZ0B2Z2h0cGUuZ292LnR3