 Xing Bian1,2,3,4
Xing Bian1,2,3,4 Xiaolin Wang1,2,3,4Qiuyan Zhang5Liying Ma1,2,3,4Guozhen Cao1,2,3,4Ao Xu6,7Jinhua Han8
Xiaolin Wang1,2,3,4Qiuyan Zhang5Liying Ma1,2,3,4Guozhen Cao1,2,3,4Ao Xu6,7Jinhua Han8 Jun Huang8Wenchu Lin1,3,4*
Jun Huang8Wenchu Lin1,3,4*- 1High Magnetic Field Laboratory, Chinese Academy of Sciences, Hefei, China
- 2University of Science and Technology of China, Hefei, China
- 3Key Laboratory of High Magnetic Field and Ion Beam Physical Biology, Hefei Institutes of Physical Science, Chinese Academy of Sciences, Hefei, China
- 4High Magnetic Field Laboratory of Anhui Province, Hefei, China
- 5The CAS Key Laboratory of Innate Immunity and Chronic Disease, Innovation Center for Cell Signaling Network, School of Life Sciences, University of Science and Technology of China, Hefei, China
- 6The First Affiliated Hospital of University of Science and Technology of China, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, China
- 7Department of Pathology, Anhui Provincial Hospital, Hefei, China
- 8MOE Key Laboratory for Biosystems Homeostasis & Protection and Innovation Center for Cell Signaling Network, Life Sciences Institute, Zhejiang University, Hangzhou, China
Poly (ADP-ribose) polymerase 1 (PARP1) is highly expressed in small cell lung cancer (SCLC) and has emerged as an attractive target for treatment of SCLC. However, the clinical significance of PARP1 expression in SCLC remains elusive. In this study, we showed that high PARP1 expression was associated with better overall survival (OS), and was positively correlated with the expression of MYC paralogs in patients with SCLC. We demonstrated that PARP1 was transcriptionally regulated by MYC paralogs. Integrative analysis of multiple RNA-seq data sets indicated that DNA damage response (DDR) genes involved in the replication stress response (RSR) and homologous recombination (HR) repair pathways were highly enriched in MYC paralog-addicted SCLC cell models and in human SCLC specimens. Targeting the MYC paralog-PARP1 axis with concomitant BET and PARP inhibition resulted in synergistic effects in MYC paralog-activated SCLC. Our study identified a critical PARP1 regulatory pathway, and provided evidence for a rational combination treatment strategy for MYC paralog-activated SCLC.
Introduction
Small cell lung cancer is a high-grade neuroendocrine carcinoma that accounts for over 250,000 annual cases worldwide (1, 2). In the past few decades, platinum-based chemotherapy with or without radiation has been the standard of care for the treatment of patients with SCLC (3). Although the initial response rate to standard treatment is high, patients with SCLC often experience relapse within one year, and the 5-year survival rate is only 7 percent (4). The SCLC treatment landscape had remained unchanged until the recent FDA approval of combined treatment using immunotherapy and chemotherapy (NCT02763579) (5). However, the development of novel therapeutic strategies to expand SCLC treatment options is a pressing need to improve clinical outcomes.
Poly (ADP-ribose) polymerase 1 (PARP1), an enzyme involved in surveillance and maintenance of genome integrity, is highly expressed in SCLC (6). Treatment with PARP inhibitors (PARPi) alone or in combination with chemotherapy has been shown to induce beneficial therapeutic effects against SCLC in preclinical and clinical studies (7–11). However, SCLC cells do not respond uniformly to PARP inhibitors (8). Therefore, investigation of the underlying mechanisms that determine therapeutic sensitivity to PARP inhibitors will allow for the targeted use of PARPi to treat SCLC.
MYC paralogs, including c-MYC, MYCL, and MYCN, play a pivotal role in tumorigenesis and tumor maintenance through regulation of a variety of cellular processes (12–15). Studies have shown that MYC paralogs are often exclusively amplified or overexpressed in SCLC (16, 17). Furthermore, overexpression of c-MYC or MYCL dramatically accelerated SCLC progression in genetically-engineered mouse models, which indicated that MYC paralogs promote oncogenesis in SCLC (18, 19). However, directly targeting MYC paralog has proven challenging due to the unique protein structures of the different paralogs (20). Several studies have modulated MYC paralog signaling through inhibition of BET, which resulted in promising anti-tumor effects against multiple cancer types, including SCLC (21–24). However, the biological significance of MYC paralogs in SCLC development, and the underlying mechanisms of the anti-tumor effects of BET inhibition (BETi) in SCLC, requires further characterization (25).
MYC paralog and PARP1 are both amplified or overexpressed in SCLC, but the association between MYC paralog and PARP1 has not been investigated in SCLC. Recent studies showed that PARP1 transcriptionally regulated c-MYC in quiescent cells (26), and MYCN transcriptionally regulated PARP1 and several other DNA damage response genes in neuroendocrine prostate cancer cells (27). However, whether MYC paralogs activate PARP1 in SCLC is unknown. We hypothesized that MYC paralogs transcriptionally activate PARP1, which might contribute to increased expression of PARP1 in SCLC.
In this study, we showed that patients with SCLC with high expression of PARP1 had better prognoses than patients with low PARP1 expression, and PARP1 expression correlated positively with the expression of MYC paralogs. We also uncovered that genes related to the DDR pathway were highly enriched in MYC paralog-activated SCLC cells through evaluation of multiple SCLC gene expression datasets. Targeting of the MYC paralog-PARP1-DDR signaling pathway using the combination of BETi JQ1 and PARPi BMN673 demonstrated excellent anti-tumor efficacy in MYC paralog-dependent SCLC cells. In contrast, MYC paralog-independent SCLC cells did not respond well to this combination treatment. Finally, we showed that JQ1 and BMN673 induced synergistic effects in SCLC xenograft models and in ex vivo cultured PDX tumor explants. Our findings showed that inhibition of PARP and BET resulted in synergistic effects, and MYC paralogs were identified as possible determinants of treatment response.
Materials and Methods
Cell Lines and Reagents
All human small cell lung cancer cell lines were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (PS) at 37°C in a 5% CO2 incubator. BMN673 was purchased from Biochempartner (Shanghai, China), JQ1 was purchased from Selleck Chemical (Shanghai, China), and all drugs were dissolved in DMSO (Sigma-Aldrich, Saint Louis, MO, USA).
SCLC Cell Line Data Processing and Unsupervised Clustering Analysis
Sequencing data (RNA-seq) from 50 SCLC cell lines, and general information for these cell lines, was downloaded from https://portals.broadinstitute.org/ccle/data. Transcriptome sequencing data from 77 human primary SCLC tumors and sample information were obtained from George et al, 2015. Sequencing data (RNA-seq) from 14 murine SCLC tumors were downloaded from GSE89660 (18). Expression data for RSR, HR repair, NHEJ pathway genes, and MYC paralogs were extracted, analyzed, and displayed in scatter plots or subjected to unsupervised cluster analysis and displayed in a heatmap.
Immunohistochemistry Staining of Human SCLC Tumor Tissues
Paraffin-embedded tumor tissues were subjected to immunohistochemical staining. Four-micrometer slices were deparaffinized in xylene, then rehydrated. Then, antigen retrieval was performed for 30 min. Endogenous peroxidase activity was blocked with 30% hydrogen peroxide in methanol solution at room temperature for 30 min. Then, the slices were blocked against non-specific binding for 30 min using goat serum, and the sections were incubated with primary antibodies against PARP1 (Affinity, DF7198) and c-MYC (Abcam, ab32072) overnight at 4°C. The sections were stained using a DAB kit (Vector, SK4100). The sections were then counterstained with hematoxylin, dehydrated, and mounted. Images were captured using a Leica microscope (Leica Microsystems). All immunohistochemical staining of PARP1 and c-MYC was evaluated and quantified as the percentage of nuclear-positive cells.
Chromatin Immunoprecipitation and PCR
Chromatin immunoprecipitation (ChIP) assay was performed as previously described (28). Cells were cross-linked using a UV cross-linker, lysed in SDS lysis buffer (1% SDS, 10 mM EDTA, and 50 mM Tris-HCl) containing complete protease-inhibitor cocktail (Roche), then incubated for 20 min on ice. The cells were sonicated for 5 min using a Sonics Vibra-Cell. A 50 μl sample of the supernatant was retained for analysis. The chromatin was incubated with magnetic beads and antibodies against c-MYC (Abcam, ab32072), MYCN (Abcam, ab16898), BRD4 (Bethyl, A301-985A50), or IgG (Cell Signaling) at room temperature for 6 h. Immunocomplexes were eluted in 1% SDS and 50 mM NaHCO3, and cross-links were reversed for 6 h at 65°C. The samples were digested using proteinase K for 1 h at 50°C, and DNA was extracted using a DNA isolation kit. Eluted DNA was subjected to qRT-PCR to detect enriched genomic DNA regions using the corresponding PCR primers. The primer sequences used in this study are summarized in Supplementary Table 1.
Cell Viability Assay
Small cell lung cancer cells were treated with increasing concentrations of BMN673 (1 μM) or JQ1 (1 μM) alone or in combination. After 3 days of treatment, the cells were analyzed using the CellTiter-Glo luminescent assay according to the manufacturer’s instructions. The combined effect of BMN673 and JQ1 was evaluated by generating a combination index (CI) using Calcusyn software (Biosoft).
Three-Dimensional Sphere Culture
Three-dimensional cell culture experiments were performed as previously described (29). Human SCLC cells were seeded on 96-well plates coated with 50% matrigel and 50% medium without FBS. The cells were grown in media with 2% matrigel and 2% FBS for 3 days. The media were then replaced with media containing drugs, and these media were refreshed every 3 days. Three-dimensional structures were visualized using a Leica microscope, and over 50 spherical structures were scored in the control group and each treatment group.
Western Blot Analysis
Following drug treatment (BMN673: 1 μM, JQ1: 1 μM), the cells were lysed in RIPA buffer supplemented with protease and phosphatase inhibitors. Total proteins were quantified using a BCA Protein Assay Kit, separated using SDS-PAGE, then transferred to PVDF membranes. Antibodies against the following proteins were used for our studies: c-MYC (1:1,000, abcam ab32072), PARP1 (1:1,000, Affinity DF7198), GAPDH (1:10,000, Affinity AF7021), Rad51 (1:10,000, Abcam ab133534), PARP (1:1,000, CST 9542), γH2AX (1:1,000, CST 2577), p-CHK1 (1:1,000, Ser317, CST 12302), BRD2 (1:1,000, Proteintech 22236-1-AP), BRD3 (1:1,000, Proteintech 11859-1-AP), BRD4 (1:500, Bethyl A301-985A50), p-DNA-PKcs (1:1,000, Abcam ab124918), p-RPA32 (1:5,000, Ser4/Ser8, Novus), MYCN (1:1,000, CST 9405), MYCL (1:1,000 R&D, AF4050) and β-actin (1:10,000, Transgen HC201-02). Rabbit IgG (1:10,000, CST 7074) and mouse IgG (1:10,000, CST 7076) were used as secondary antibodies.
Comet Assay
Comet assay was performed as previously described (30). After 48 h of drug treatment (BMN673: 1 μM, JQ1: 1 μM), the cells were harvested and subjected to neutral comet assays. Following electrophoresis, the cells were stained using SYBR gold and visualized using a Zeiss fluorescence microscope.
Immunofluorescence Staining
Immunofluorescence staining was performed as described previously (6). Suspended cells were fixed onto glass using 1% paraformaldehyde and ethanol (70%) at the treatment endpoint. The cells were incubated with primary antibodies against γH2AX (1:500, CST 2577), RAD51 (1:500, Abcam ab133534), RPA (1:200, Abcam ab2175), and p-CHK1 (1:200, Ser317, CST 12302), then incubated with secondary antibody. The cells were visualized using a Zeiss fluorescence microscope.
RNA Isolation and Quantitative RT-PCR Analysis
Total RNA from SCLC cells was extracted using Trizol reagent (Thermo Scientific, Rockford, IL, USA) and reverse transcribed using a cDNA Synthesis Kit (Roche, Mannheim, Germany). Quantitative PCR was performed using FastStart Essential DNA Green Master (Roche, Mannheim, Germany) on a Roche LightCycler 96 Real-Time PCR System. The primer sequences used are summarized in Supplementary Table 2.
Plasmid Constructs and Stable Cell Line Generation
Lentiviral shRNA constructs to knock down BRD2, BRD3, or BRD4 were obtained from the RNAi Consortium (Broad Institute). These shRNA sequences are summarized in Supplementary Table 3. In addition, DMS53 and DMS273 cells were seeded one day prior to transfection with siRNA. Effectene Transfection Reagent (QIAGEN, Hilden, Germany) was used to transfect human c-MYC and negative control siNC into the cells according to the manufacturer’s instructions. Small interfering RNA sequences are listed in Supplementary Table 4. For overexpression, the complementary DNA (cDNA) clone of c-MYC was subcloned into a pWZL-blast vector (Primer sequences are listed in Supplementary Table 5). SHP77 cells that stably expressed c-MYC were generated using a retroviral packaging system, and selected using blasticidin (Solarbio, China).
Histological and Immunohistochemical Analyses
Tumor tissues were fixed in 4% formalin overnight, and then embedded in paraffin. Four-micrometer paraffin-embedded sections were stained with hematoxylin and eosin (H & E). Immunohistochemistry (IHC) was performed as previously described (31). Slides were incubated at 4°C overnight with the following primary antibodies: Ki67 (1:1,000, CST 9449), cleaved-caspase 3 (1:300, CST 9661), RAD51 (1:200, Abcam ab133534), or γH2AX (1:500, Ser139, CST 2577). Three to five random 40X fields were scored for each tumor sample. Staining intensity was quantified as the percentage of nuclear-positive cells. For the quantification of immunohistochemistry staining of human SCLC specimens, the PARP1and c-MYC staining were quantified by integrated optical density (IOD) equaled to optical density by area by using image pro plus software.
Small Cell Lung Cancer Xenograft Mouse Models
Six-week-old athymic nude mice were subcutaneously injected with 5 x 106 DMS273, H526, or H196 cells in 100 μl of PBS and 100 μl of Matrigel (BD Biosciences, Franklin, NJ, USA). Drug treatment was initiated when the tumors reached 100 mm3. Prior to administration, BMN673 was dissolved in 10% hydroxypropyl-β-cyclodextrin, and then administered via oral gavage at a dose of 0.33 mg/kg/day. In addition, JQ1 was dissolved in 0.5% methylcellulose solution, then administered via intraperitoneal injection at a dose of 40 mg/kg/day. Tumor sizes were measured using a caliper, and tumor volumes were determined using the following equation: tumor volume [mm3]=(tumor length X tumor width2)/2.
Patients-Derived Xenograft (PDX) Mouse Model of Small Cell Lung Cancer and Ex Vivo Organ Culture of PDX Tumor Specimens
Primary tumor tissues were transplanted into the subcutaneous fat pad space of mice. Ex vivo organ cultures were generated by dissecting fresh tumor tissues into 1-mm3 blocks, which were then cultured on absorbable gelatin sponges, as previously described (32). At least three pieces of tumor tissue per sponge were examined for each treatment condition. After being allowed to recover for 24 h, the tissue blocks were treated with BMN673 or JQ1 alone or in combination for 48 h. Following treatment, the tissue blocks were fixed using paraformaldehyde and processed for histological analysis.
Statistical Analysis
All in vitro analyses were repeated at least in triplicate. P-values < 0.05 were considered statistically significant. Quantitative results were analyzed using two-tailed unpaired Student’s t tests using GraphPad Prism software. For the prognostic significance study, patients were classified into two groups based on median expression levels of PARP1 in the cohort of patients with SCLC. The Kaplan–Meier product-limit method and log-rank tests were used to estimate the differences in probabilities in OS and PFS between the different groups.
Results
MYC Paralogs Positively Correlate With PARP1 Expression, and Transcriptionally Regulate PARP1 in SCLC Cells
We investigated whether PARP1 could serve as a prognostic marker of SCLC by analyzing RNA-seq data from human patients with tumors (16). Patients with high levels of PARP1 had better OS (median survival of 42 months of high PARP1 group vs 23 months of low PARP1 group, P = 0.027, log-rank test; Figure 1A) and progression-free survival (PFS) (median survival of 27 months of high PARP1 group vs 12 months of low PARP1 group; P = 0.061, log-rank test; Figure 1B) than those with low levels of PARP1. To gain insight into the mechanisms leading to high PARP1 expression in SCLC, we investigated MYC paralog expression in multiple SCLC RNA-seq data sets. An SCLC data set generated by George et al. (16) (77 human SCLC samples) showed that PARP1 expression was higher in the group with high MYC paralog expression than that in the group with low MYC paralog expression (Figure 1C). In the RNA-seq data sets generated by Peifer et al. (17) (15 human SCLC samples), we noticed a strong positive correlation between PARP1 expression and MYC paralog expression (data not shown). Similar results were observed in the CCLE SCLC RNA-seq data set (50 human SCLC cell lines, Figure 1D) and the GSE89660 data set (14 murine SCLC tumors, Figure 1E). To further characterize the correlation between PARP1 and MYC paralog at the protein level in human SCLC samples, 17 human SCLC specimens were collected and subjected to immunohistochemistry analysis for c-MYC expression (Figure 1F). The results showed that PARP1 expression was positively associated with c-MYC expression (Figure 1G). These findings suggest that PARP1 expression may be a novel prognostic marker of SCLC, and MYC paralog expression positively correlates with PARP1 expression.
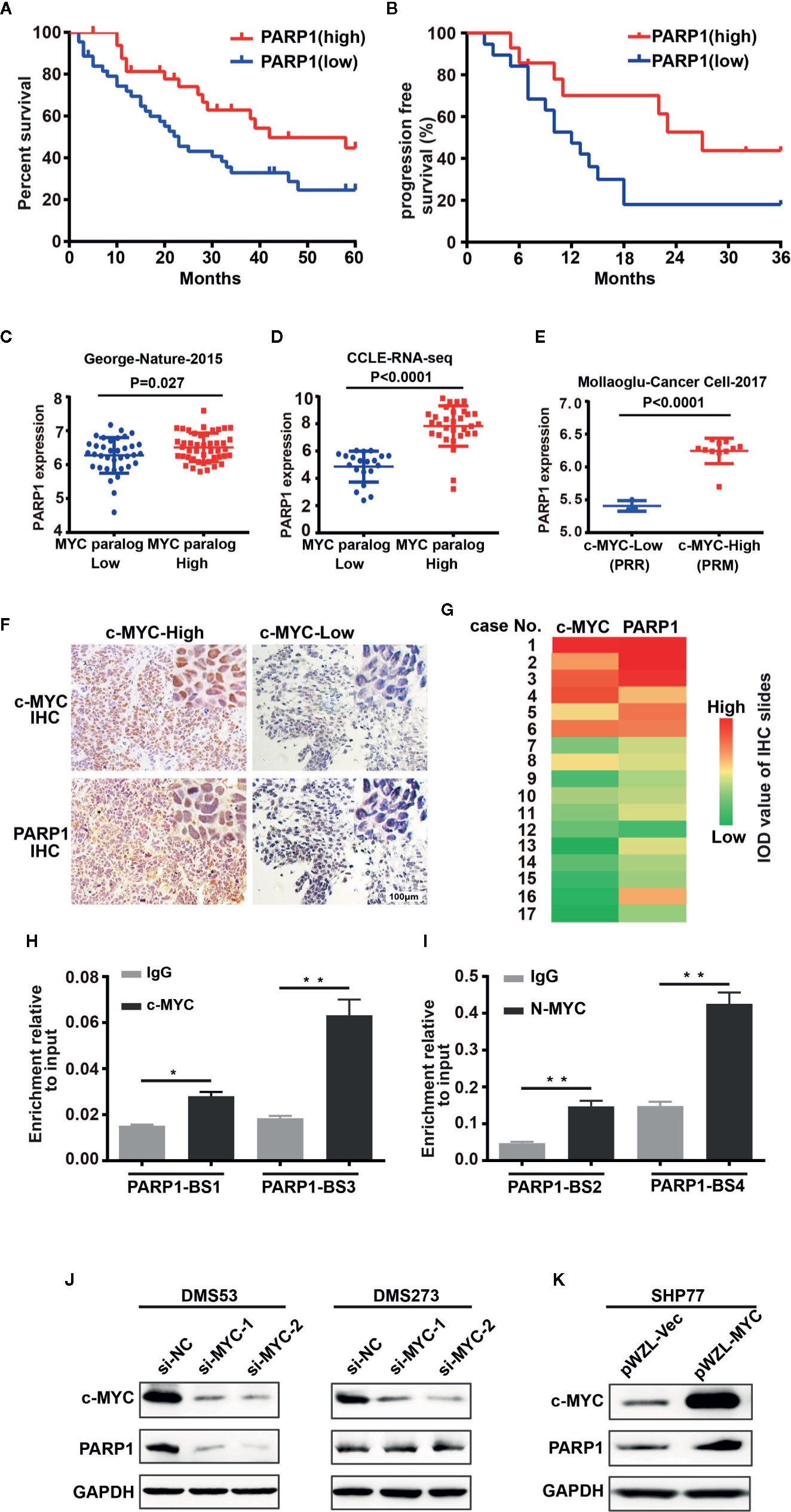
Figure 1 PARP1 mRNA tightly correlates with MYC paralog expression and is an independent prognostic marker of survival in patients with SCLC. (A) Kaplan–Meier analysis of the correlation between PARP1 expression and overall survival (OS, n = 77) and (B) progression-free survival (PFS, n=33). (C–E) Scatter plots of PARP1 mRNA relative to expression of MYC paralogs in SCLC primary tumors (n=81) (C), CCLE cell lines (n=50) (D), murine SCLC tumors (n=14) (E). (F) Representative images of IHC analysis of PARP1 and c-MYC in two independent cases. Scale bar, 100 μm. (G) Heat map showing the correlation between PARP1 and c-MYC in 17 paraffin-embedded SCLC tumor tissues. The heat map was depicted according to the IOD value of each IHC slides (red indicates c-MYC and PARP1 positive staining, green indicates negative staining). The significance analysis was performed by Student’s t test. (H–I) ChIP-qRT-PCR experiment indicating the direct binding of c-MYC and MYCN to the PARP1 promoter in DMS273 (H) and H526 (I) cells. (J) Western blot analysis showing the downregulated proteins in DMS53 and DMS273 cells upon c-MYC knockdown. (K) Western blot analysis showing the upregulated proteins in SHP77 cells with ectopic c-MYC overexpression. GAPDH was used as a loading control. BS, binding site.
To provide mechanistic evidence of the potential regulation of PARP1 by MYC paralog, we performed ChIP assay followed by qPCR analysis to evaluate whether MYC paralog transcriptionally activated PARP1. Four potential MYC paralog-binding sites in the PARP1 promoter were analyzed. The results showed that c-MYC and MYCN directly bound to the promoter region of PARP1 (Figures 1H, I). To further determine whether c-MYC regulated PARP1 expression, the effects of knockdown and overexpression of c-MYC were evaluated in SCLC cell culture models. DMS53 and DMS273 cells were chose for the siRNA-based knockdown assay since both cell lines grow adherently and have high constitutive c-MYC expression and low c-MYC expressing SHP77 cells was used for the overexpression assay. Western blot analysis showed that knockdown of c-MYC using siRNA significantly suppressed the expression of PARP1 in DMS53 cells while knockdown of c-MYC did not significantly suppress PARP1 in DMS273 cells (Figure 1J). Furthermore, ectopic overexpression of c-MYC using a retrovirus packaging system in SHP77 cells markedly induced PARP1 expression (Figure 1K). These results show that c-MYC and MYCN transcriptionally regulate PARP1 in SCLC cells.
DNA Damage Response-Related Genes Were Enriched in MYC Paralog-Dependent SCLC
The above observation prompted us to further investigate the role of MYC paralog in the DDR pathway in SCLC. We first evaluated the CCLE RNA-seq data set for the expression of MYC paralogs and DDR genes. Unsupervised clustering analysis of MYC paralogs, PARP1, and other common DDR genes showed that a number of genes involved in the replication stress response and the homologous recombination repair pathways were positively correlated with the expression levels of MYC paralog and PARP1 (Figure 2A). In contrast, genes related to non-homologous end joining (NHEJ) repair were negatively associated with MYC paralog expression (Figure 2A). Furthermore, integrative analysis of the CCLE SCLC RNA-seq data set and the RPPA data set showed that several DDR genes were highly expressed at the protein level in SCLC cells that expressed high levels of either MYC paralogs or PARP1 (Figure 2B). In addition, several genes involved in the RSR and HR repair pathways tend to be highly expressed in SCLC specimens that exhibited high expression of MYC paralogs in the SCLC data set generated by George et al. (16) (Supplementary Figure 1). Interestingly, ATR, BRCA2, and RAD51 were the genes most significantly associated with PARP1 expression (Figure 2C), and CHEK1, NBN, and RAD51 were the genes most significantly associated with MYC paralog expression (Figure 2D). We then wondered whether MYC paralogs transcriptionally regulated other DDR-related genes in addition to PARP1 using ChIP-PCR analysis. The results showed that c-MYC and MYCN directly bound to the promoter region of RAD51 in DMS273 (Figure 2E) and H526 cells (Figure 2F). In addition, knockdown of c-MYC using siRNA led to downregulation of RAD51 in DMS53, but not in DMS273 cells (Figure 2G). Furthermore, c-MYC overexpression significantly induced RAD51 expression in SHP77 cells (Figure 2H), which supported the observation that c-MYC transcriptionally regulated RAD51 in SCLC cells. Our results indicate that genes involved in the RSR and HR repair pathways were necessary for maintenance of MYC paralog-dependent SCLC tumor growth.
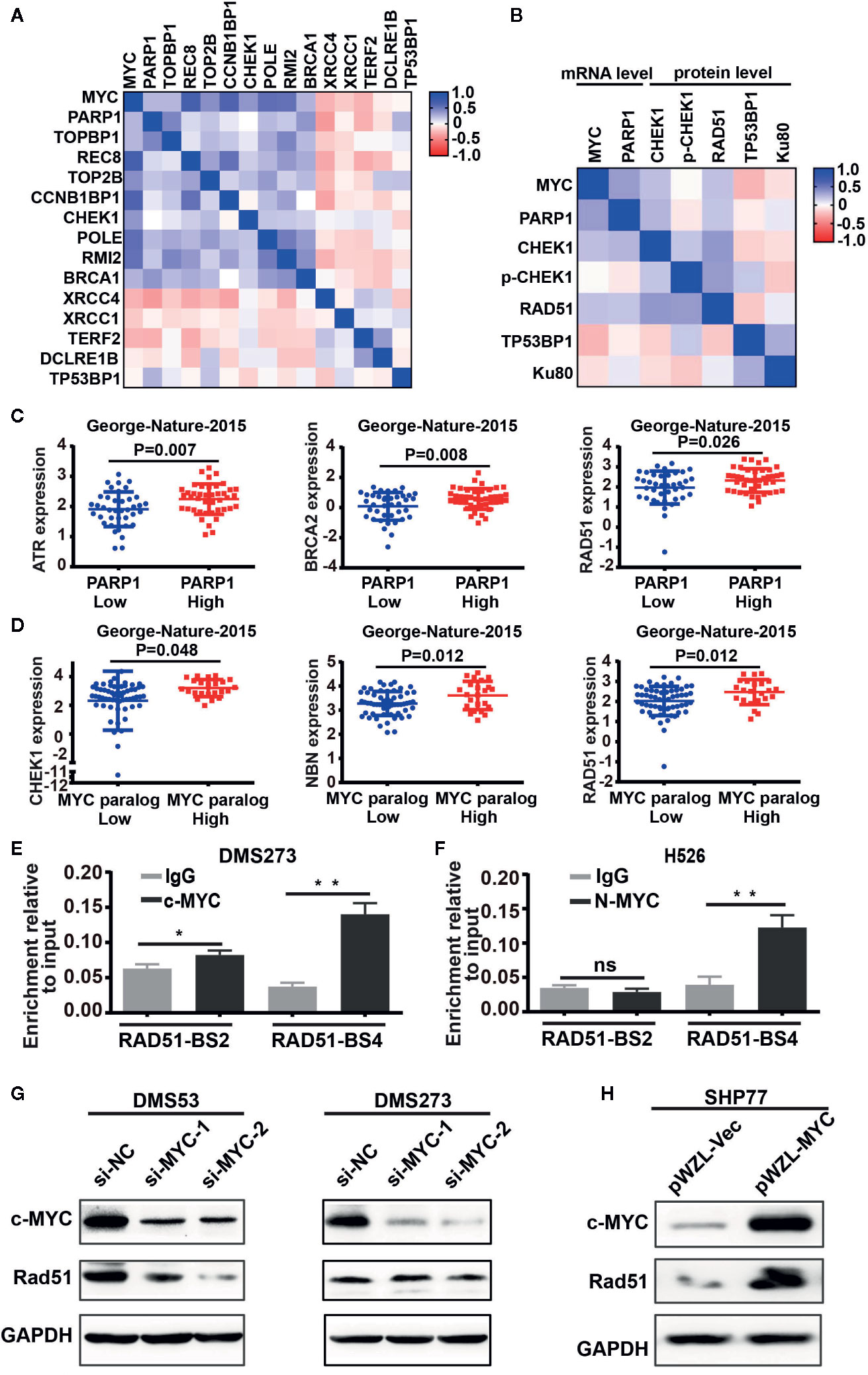
Figure 2 DNA damage response genes were highly enriched in SCLC cells with high expression of MYC paralogs or PARP1 at mRNA levels and protein levels. (A, B) Unsupervised clustering of DDR genes and MYC paralogs in CCLE RNA-seq data set (n = 50) and reverse-phase protein array (RPPA) data set (B, n=50). (C) Scatter plots of PARP1 expression relative to the expression of ATR, BRCA2, and RAD51 in SCLC primary tumors (n=81). (D) Scatter plots of the expression of MYC paralogs relative to the expression of CHEK1, NBN, and RAD51 in SCLC primary tumors (n=81). The significance analysis was performed by Student’s t test. (E, F) ChIP -PCR experiment indicating the binding of c-MYC and MYCN to the RAD51 promoter in DMS273 (E) and H526 cells (F) *P < 0.05; **P < 0.01; ns, no significance. (G) Western blot analysis of Rad51 expression in DMS53 and DMS273 cells upon c-MYC knockdown. (H) Western blot analysis of Rad51 expression in SHP77 cells with c-MYC overexpression. GAPDH was used as a loading control. BS, binding site.
Combined PARP1 and BET Inhibition Induces Synergistic Anti-Tumor Activity in MYC Paralog-Dependent SCLC Cells
Our results highlighted the pivotal role of the MYC paralog-PARP1-DDR axis in SCLC. Based on these findings, we evaluated whether targeting of MYC paralog using JQ1 and PARP1 using BMN673 in combination could be a potential strategy for treatment of SCLC. We used a panel of SCLC cell lines with exclusive MYC paralog amplification and two SCLC cell lines without MYC paralog amplification or overexpression to assess the anti-tumor effects of JQ1 and BMN673 (Supplementary Table 6). The SCLC cell lines were treated with a range of concentrations of JQ1 and BMN673, alone or in combination, for 72 h. Cell viability was determined using the CellTiter-Glo luminescent assay. Combination treatment with BMN673 and JQ1 resulted in synergistic growth inhibition in all MYC paralog-amplified SCLC cells, but not in MYC paralog-non amplified SCLC cells (Figures 3A, B, and Supplementary Figure 2). Interestingly, c-MYC knockdown prevented the synergistic effects induced by combined treatment with BMN673 and JQ1 in DMS53 cells (Figure 3C), and c-MYC overexpression enhanced the sensitivity of SHP77 cells to combined treatment with BMN673 and JQ1 (Figure 3D). Western blot analysis showed that c-MYC expression was reduced by JQ1 in H446 and H82 cells but not in DMS273 cells, expressions of MYCN and MYCL were significantly suppressed by JQ1 in H526, H69 and H1963 cells (Figure 3E). The levels of cleaved PARP protein were significantly increased in the combined treatment group in MYC paralog-dependent SCLC cells compared with those in MYC paralog-independent SCLC cells (H196, Figure 3E). Furthermore, overexpression of c-MYC resulted in significantly increased levels of cleaved PARP protein in response to combined treatment with BMN673 and JQ1 (Figure 3F). It is worth noting, JQ1 treatment did not suppress c-MYC expression in SHP77 cells with c-MYC overexpression (Figure 3F). These results indicate that combined treatment with JQ1 and BMN673 induces synergistic effects, as evidenced by significant MYC paralog-dependent SCLC cell apoptosis.
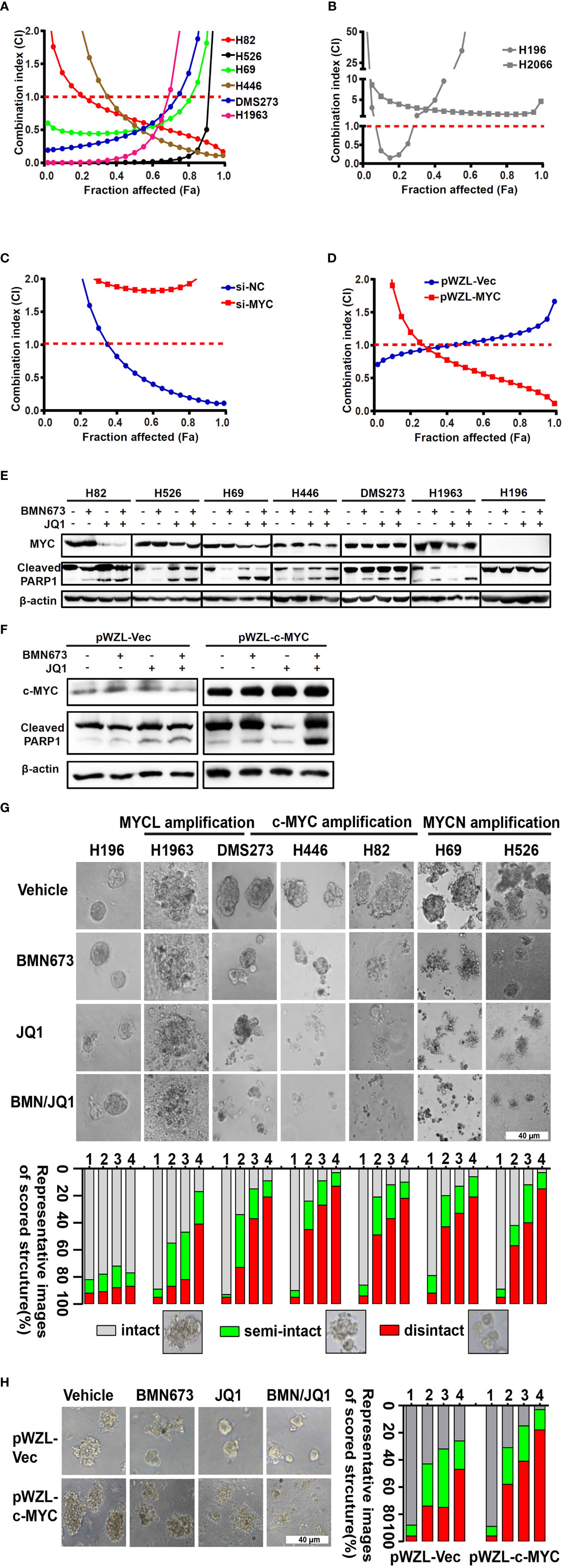
Figure 3 The combination effects of JQ1 and BMN673 in SCLC cells. (A–D) CellTiter-Glo Luminescent assays demonstrating the effects of JQ1 and BMN673 as single agents or in combination in MYC paralog-dependent (A), independent (B) SCLC cells, DMS53 cells with c-MYC knockdown (C), and SHP77 cells with c-MYC overexpression (D). A mathematical model was applied to calculate the combination index using the CalcuSyn software program. (E) Western blot analysis of cleaved PARP and MYC paralogs in SCLC cells treated with BMN673 or JQ1 alone or in combination for 24 h. c-MYC for H82, H446 and DMS273, MYCN for H526 and H69, MYCL for H1963. (F), Western blot analysis of cleaved PARP and c-MYC in SHP77 cells with c-MYC overexpression followed by BMN673 and JQ1 treatment alone or in combination for 24 h. GAPDH was used as a loading control. (G) Tumor sphere structures in 3D matrigel were captured under a phase-contrast microscope upon treatment of JQ1 and BMN673 as single agents or in combination for 10 to 15 days. Representative images of tumor spheres were shown in the top panel. Quantification of scored tumor sphere structures (disintegrated, semi-disintegrated, and intact) was shown in the bottom panel. Scale bar, 40 μm. (H) 3D matrigel assays showing the effect of JQ1 and BMN673 in SHP77 cells with or without c-MYC overexpression. 1, Vehicle; 2, BMN673; 3, JQ1; 4, JQ1+BMN673.
BMN673 and JQ1 Synergistically Inhibit Cell Growth
To evaluate the effects of combined treatment with BMN673 and JQ1 in conditions that mimic the cellular microenvironment, we generated a 3D model of SCLC cells in matrigel. Combined treatment with BMN673 and JQ1 resulted in much greater structural disintegration of 3D spheroids than treatment with JQ1 or BMN673 alone in MYC paralog-dependent SCLC cells (Figure 3G). In contrast, 3D cultures of MYC paralog-independent SCLC cells (H196) did not respond to BMN673 or JQ1, alone or in combination (Figure 3G). Furthermore, ectopic overexpression of c-MYC significantly increased the structural disintegration of 3D spheroids induced by combined treatment with BMN673 and JQ1 in SHP77 cells (Figure 3H). These results indicate that combined administration of JQ1 and BMN673 might be a potential treatment option for MYC paralog-dependent SCLC cells.
BMN673 and JQ1 Cooperate to Induce DNA Damage and Attenuate the Replication Stress Response
To discern the molecular mechanism underlying the synergistic effects of BMN673 and JQ1 observed in SCLC cells, we evaluated the mechanisms by which BET inhibition modified RSR signaling. We first determined the extent of DNA damage using comet assay. Administration of BMN673 or JQ1 as monotherapy induced moderate DNA damage in SCLC cells, and combined treatment resulted in substantially increased DNA damage, as shown by substantial DNA tails (Figure 4A). In contrast, MYC paralog-independent SCLC cells (H196) did not show obvious DNA damage following treatment with BMN673 or JQ1, alone or in combination (Figure 4A). Furthermore, SHP77 cells that overexpressed c-MYC showed much more DNA damage than SHP77 cells that did not overexpress c-MYC upon the dual treatment (Figure 4B). In support of the comet assay results, immunofluorescence further confirmed much more elevated γH2AX foci (a marker for DNA double-strand breaks) in cells treated with the combination of BMN673 and JQ1 than those treated with either drug alone (Figure 4C). Cells without MYC paralogs amplification did not show any γH2AX foci upon BMN673 and JQ1 as monotherapy or in combination treatment (Figure 4C). Furthermore, western blot analysis revealed that c-MYC overexpression resulted in increased protein levels of γH2AX following combined treatment (Figure 4D). These findings indicate that combined treatment with BMN673 and JQ1 results in substantial DNA damage in MYC paralog-dependent SCLC cells.
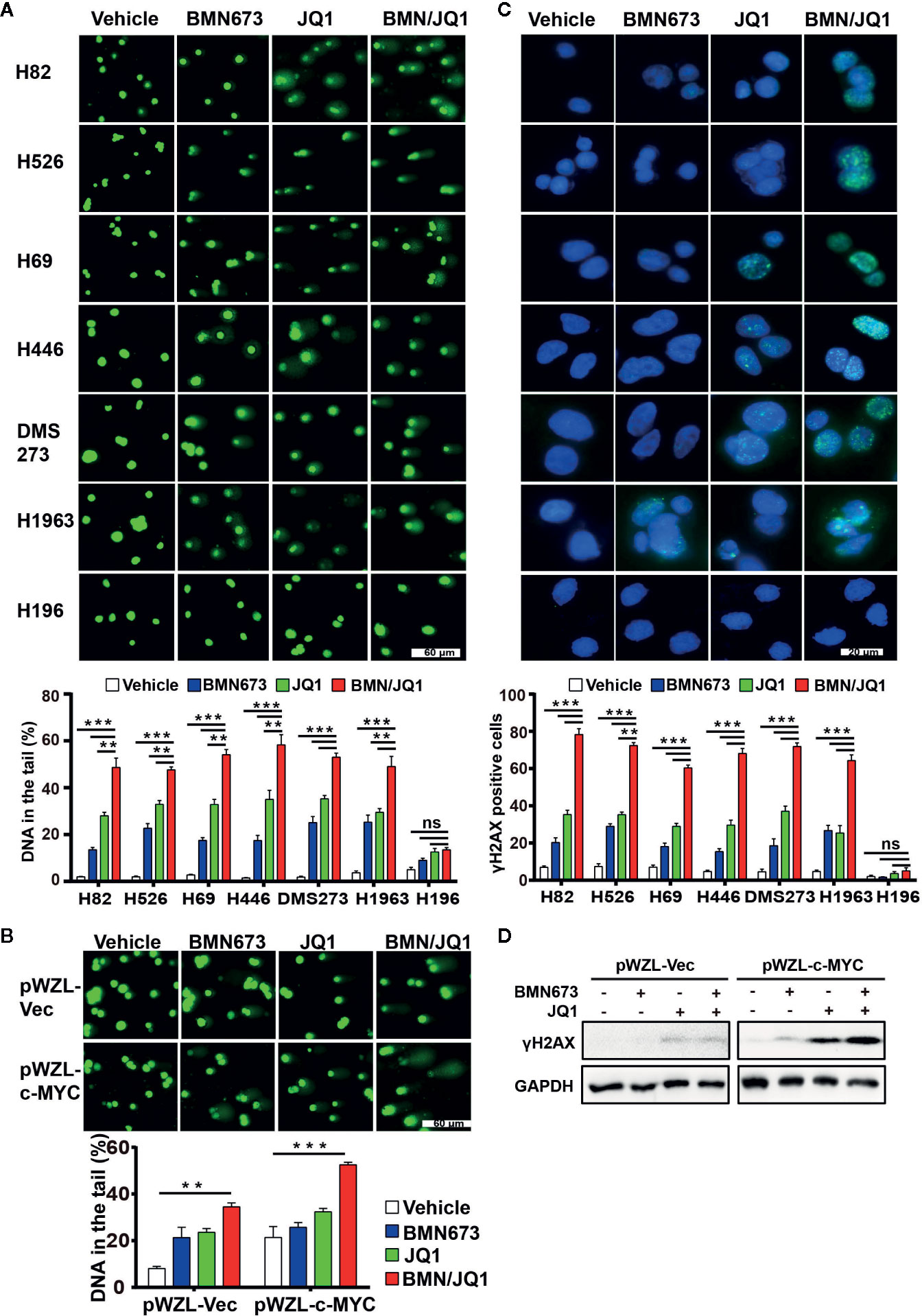
Figure 4 Effects of JQ1 and BMN673 on DNA damage in MYC paralog-dependent SCLC cells. (A) Comet assays detecting DNA damage in SCLC cells upon treatment with BMN673 or JQ1 alone and in combination for 48 h. **P < 0.01; ***P < 0.001. (B) Comet assays detecting DNA damage in SHP77 cells with or without c-MYC overexpression, treated with indicated drugs for 48 h. Scale bar, 60 μm. DNA in the tail was used to measure DNA damage and assessed by CASP software (CaspLab). Quantification of the amount of DNA damage was shown mean ± S.D. **P < 0.01; ***P < 0.001 (Student’s t test). (C) Representative images of immunofluorescent staining for γH2AX in SCLC cells treated with indicated drugs for 24 h, Scale bar, 20 μm. Cells with more than 5 foci were considered as positive. **P < 0.01; ***P < 0.001. (D) Western blot analysis of γH2AX protein levels in SHP77 cells with or without c-MYC overexpression treated with BMN673 or JQ1 alone or in combination for 24 h. GAPDH was used as a loading control.
Recently, PARP inhibition was shown to elicit a strong RSR in multiple cell types (33, 34). Based on this finding, we evaluated whether JQ1 could augment replication-associated DNA damage through blockade of PARPi-induced RSR. Quantitative analysis of immunofluorescence staining for the ssDNA-binding protein RPA showed that JQ1 treatment severely impaired PARPi-induced formation of RPA foci (a marker for DNA replication stress, Figure 5A). The ATR-CHK1 signaling axis is an essential pathway in response to replication stress (35, 36). Treatment with BMN673 resulted in substantially increased CHK1 phosphorylation in S317 and S345 sites, and this effect was reversed by treatment with JQ1, which suggested that JQ1 suppressed RSR in MYC paralog-dependent SCLC cells (Figure 5B). We also checked the phosphorylation of CHK1 in S296 site, we noticed that BMN673 did not induce strong activation of p-CHK1 in S296 site, however, JQ1 as single agent or in combination with BMN673 significantly suppressed the p-CHK1 in S296 site in most of the cell lines examined (Figure 5B). The MYC family of oncogenes plays a substantial role in induction of replication stress (37–39). We further showed that c-MYC-overexpressing SHP77 cells exhibited a strong RSR compared with WT SHP77 cells, as indicated by increased levels of p-CHK1, p-RPA (Figure 5C). Inhibition of PARP further enhanced phosphorylation of CHK1 and RPA, and JQ1 reversed this effect in c-MYC-overexpressing SHP77 cells (Figure 5C and Supplementary Figures 3A, B). These results were consistent with those observed in non-genetically-modified MYC paralog-dependent SCLC cells (Figure 5B). Together, these results show that BETi strongly attenuates the basal and PARPi-induced RSR in MYC paralog-dependent SCLC cells.
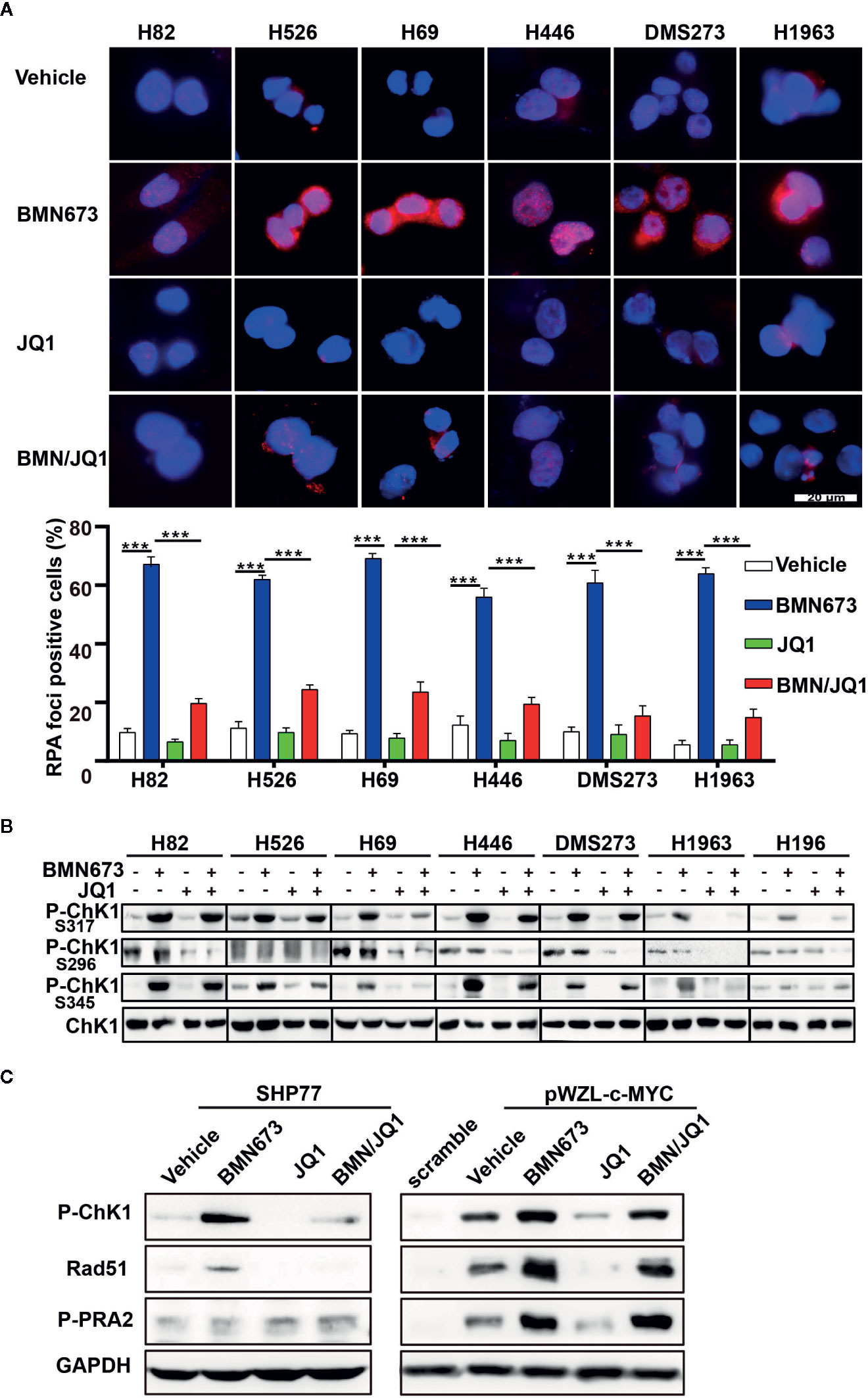
Figure 5 Effects of JQ1 and BMN673 on the DNA replication stress response. (A) Representative images of immunofluorescence staining for RPA2 in SCLC cells treated with drugs as indicated for 24 h. Scale bar, 20 μm. Quantification of RPA2 fluorescence intensities from three independent experiments was shown as mean ± S.D. ***P < 0.001. (B) Western blot analysis of p-CHK1 levels in MYC paralog-dependent and independent-SCLC cells treated with drugs as indicated for 24 h. (C) Western blot analysis of indicated proteins in SHP77 cells with or without c-MYC overexpression followed by BMN673 and JQ1 as single agents or in combination treatment for 24 h. GAPDH was used as a loading control.
Inhibition of BET Attenuates HR Repair in MYC Paralog-Dependent SCLC Cells
PARP inhibitor activity can be modulated by HR status (40). However, previous sequencing results showed that DNA damage repair genes are rarely mutated in SCLC (16, 17). We hypothesized that JQ1 might regulate the expression of DNA damage repair genes. Real time qPCR results displayed decreased expression of BRCA1 and RAD51 but not BRCA2 following JQ1 treatment (Supplementary Figure 4). Western blot analysis showed that JQ1 treatment resulted in dose-dependent down-regulation of Rad51 (Supplementary Figure 5). To further evaluate HR following drug treatment, we used immunofluorescence staining to quantify Rad51 foci (a surrogate marker of HR). Treatment with BMN673 for 24 h resulted in significantly increased formation of Rad51 foci. In contrast, combined treatment with BMN673 and JQ1 inhibited formation of Rad51 foci (Figure 6A), which resulted in accumulation of γH2AX foci in MYC paralog-dependent SCLC cells (Figure 4C). Furthermore, western blot analysis showed that treatment with BMN673 increased Rad51 levels, and this effect was inhibited by treatment with JQ1 (Figure 6B). We then investigated the mechanisms by which JQ1 inhibited formation of Rad51 foci. Messenger RNA stability assays using actinomycin D in DMS273 and H526 cells showed that JQ1 did not affect RAD51 mRNA stability (Supplementary Figure 6). A recent study reported that BET proteins regulated RAD51 in ovarian cancer cells (41), and we showed that knockdown of each BET family member (BRD2, BRD3, and BRD4) in DMS273 cells led to downregulation of Rad51 expression (Figure 6C). These results indicated that the BET family of proteins might regulate RAD51 in SCLC cells. As BRD4 was highly expressed in SCLC cells, we wondered whether BRD4 transcriptionally regulated RAD51 expression, we performed ChIP assay using a BRD4 antibody followed by RT-qPCR in JQ1-treated SCLC cells and control cells. The results showed that JQ1 treatment decreased the interaction between BRD4 and the promoter regions of RAD51 (Figure 6D). Together, these results indicate that JQ1 reduces RAD51 expression through suppression of the transcription of RAD51, but not through the destabilization of RAD51 mRNA.
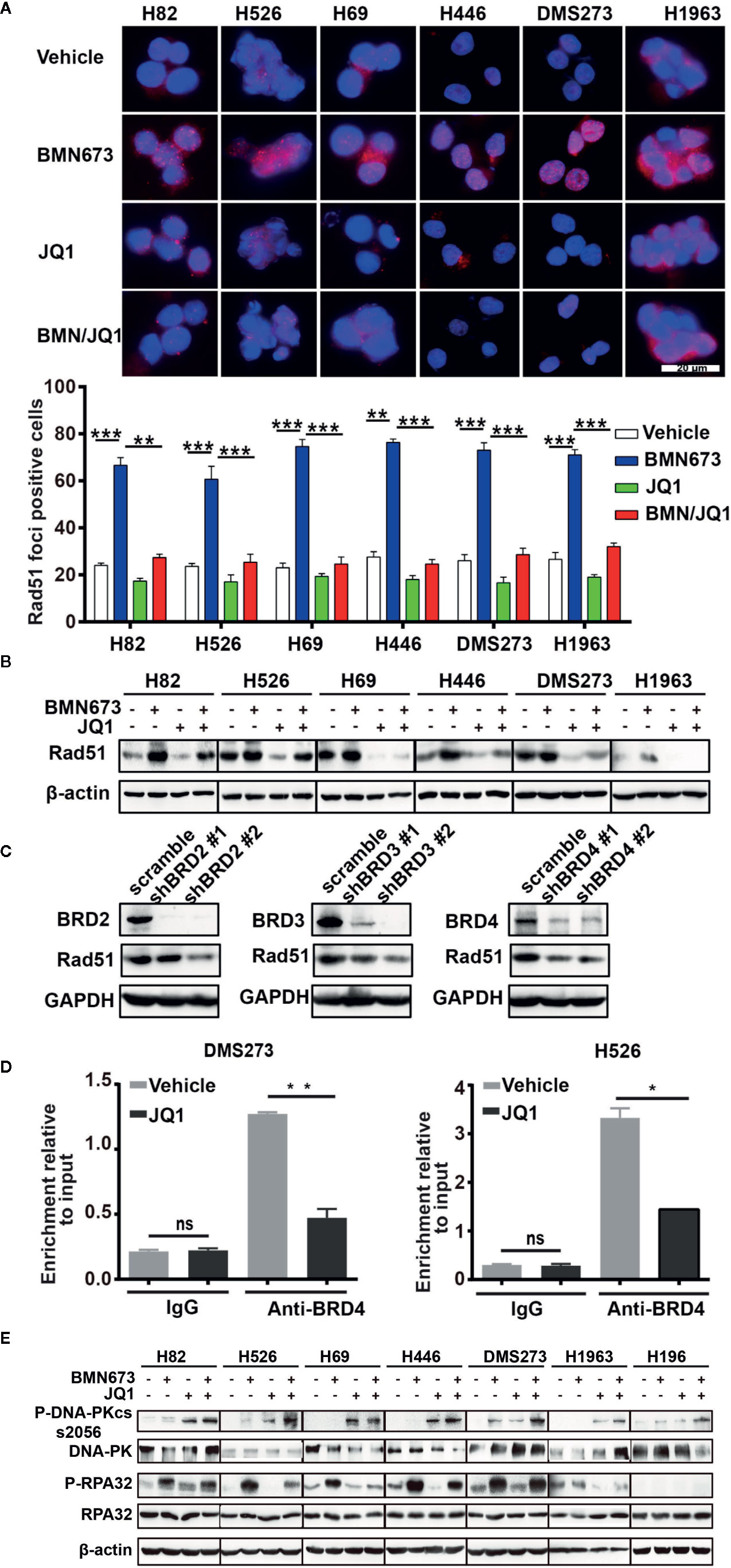
Figure 6 Effects of JQ1 and BMN673 on the HR repair pathway. (A) Representative images of Rad51 immunofluorescence staining in SCLC cells treated with drugs as indicated for 24 h. Scale bar, 20 μm. Quantification of Rad51 fluorescence intensities from three independent experiments was shown as mean ± S.D. **P < 0.01; ***P < 0.001. (B) Western blot analysis of Rad51 expression in SCLC cells treated with drugs as indicated for 24 h. (C) Western blot analysis of indicated proteins in DMS273 cell upon knockdown of BRD2 or BRD3 or BRD4. (D) ChIP-PCR analysis displays the decreased association of BRD4 in the promoter of RAD51 in DMS273 and H526 cells treatment with JQ1 for 24 h. *P < 0.05; **P < 0.01; ns, no significance. (E) Western blot analysis of phosphorylated-DNA-PKcs and phosphorylated-RPA32 in MYC paralog-dependent and independent-SCLC cells treated with indicated drugs for 24 h. β-actin was used as a loading control.
We then investigated whether knockdown of BET family proteins induced synergistic effects with BMN673 in SCLC cells. We first performed clonogenic assays, and showed that knockdown of BET proteins significantly accelerated BMN673-induced cell growth inhibition, with the most pronounced effect in BRD4 knockdown cells (Supplementary Figures 7A, B). Knockdown of BRD2 or BRD3 resulted in modest DNA damage in BMN673-treated cells, whereas knockdown of BRD4 resulted in substantial DNA damage following BMN673 treatment, as determined using comet assay (Supplementary Figures 7C, E). Furthermore, BMN673 induced significantly greater accumulation of γH2AX foci in BRD4 knockdown cells than in BRD2 or BRD3 knockdown cells (Supplementary Figures 7D, F), which suggested that depletion of BRD4 was sufficient to induce synergy with PARP inhibition.
Autophosphorylation of DNA-PK plays a pivotal role in NHEJ. Phosphorylation at S2056 of DNA-PKcs limits end processing during NHEJ (42, 43). Meanwhile, autophosphorylation of DNA-PKcs at S2056 site is cell cycle regulated, occurring primarily in G0/G1 cell cycle phase (44). Western blot analysis showed that JQ1 treatment alone and in combination with BMN673 led to marked increases in phosphorylation of DNA-PK at S2056 site in MYC paralog-dependent SCLC cells (Figure 6E), which suggested that JQ1 might inhibit DNA end processing and induce G1 cell cycle arrest. In accordance with these findings, JQ1 treatment markedly decreased PARPi-induced phosphorylation of RPA32 which is a DNA end-resection marker, in MYC paralog-dependent SCLC cells (45). However, in MYC paralog-independent H196 cells, we did not notice significant expression change of phosphorylated-RPA32 upon drug treatment. (Figure 6E) Furthermore, we showed that c-MYC-overexpressing SHP77 cells exhibited a strong HR, as indicated by increased levels of Rad51 (Figure 5C). BMN673 treatment further enhanced Rad51 expression, which was counteracted by JQ1 (Figure 5C), suggesting JQ1 inhibits HR in SCLC cells. Together, these results show that JQ1 treatment or knockdown of BET family members attenuates HR repair through modulation of RAD51 expression, Rad51 foci formation, and inhibition of DNA end resection.
Dual Inhibition of BET and PARP Is Synergistic In Vivo
The demonstrated synergistic effects induced by BET inhibition and PARP inhibition in vitro led to the evaluation of the therapeutic efficacy of BMN673 and JQ1 as monotherapies or in combination in SCLC xenograft models. SCLC cell lines, including DMS273 (c-MYC and MYCL amplification), H526 (MYCN amplification), and H196 (no MYC paralog amplification or overexpression) were selected for evaluation in vivo. SCLC cells were injected into the subcutaneous tissue of nude mice, and tumors were allowed to grow to about 100 mm3 prior to drug administration. After three weeks treatment, BMN673 or JQ1 alone attenuated tumor growth (Figure 7A, tumor volume reduced by 1.5-fold and 1.36-fold, respectively, for DMS273, tumor volume reduced by 1.86-fold and 1.52-fold, respectively, for H526) and reduced tumor weight (Figure 7B). However, combination treatment with BMN673 and JQ1 inhibited tumor growth and reduced tumor weight to a much greater extent than either drug alone (Figures 7A, B, tumor volumes reduced by 3.53-fold and 2.85-fold, respectively). In contrast, neither BMN673 nor JQ1, alone or in combination, inhibited tumor growth in H196 xenografts, which indicated that combined BMN673-JQ1 treatment was not effective against MYC paralog-independent SCLC in vivo (Figures 7A, B). Notably, all treatments were well tolerated, and did not induce significant body weight loss (Supplementary Figures 8A–C). To evaluate the effects of BMN673 and JQ1 on proliferation, apoptosis, and DDR, tumor tissues were analyzed using immunohistochemical staining. The results showed substantially decreased numbers of Ki67-positive tumor cells and significantly increased numbers of cleaved-caspase 3-positive cells in the group treated with BMN673 and JQ1 compared to those in the vehicle group and the monotherapy groups (Supplementary Figures 9A, B). Tumors in the group administered the combination of BMN673 and JQ1 showed the greatest upregulation of γH2AX expression and downregulation of Rad51 expression (Supplementary Figures 9A, B), which further demonstrated that this combination therapy compromised HR repair activity and enhanced DNA damage.
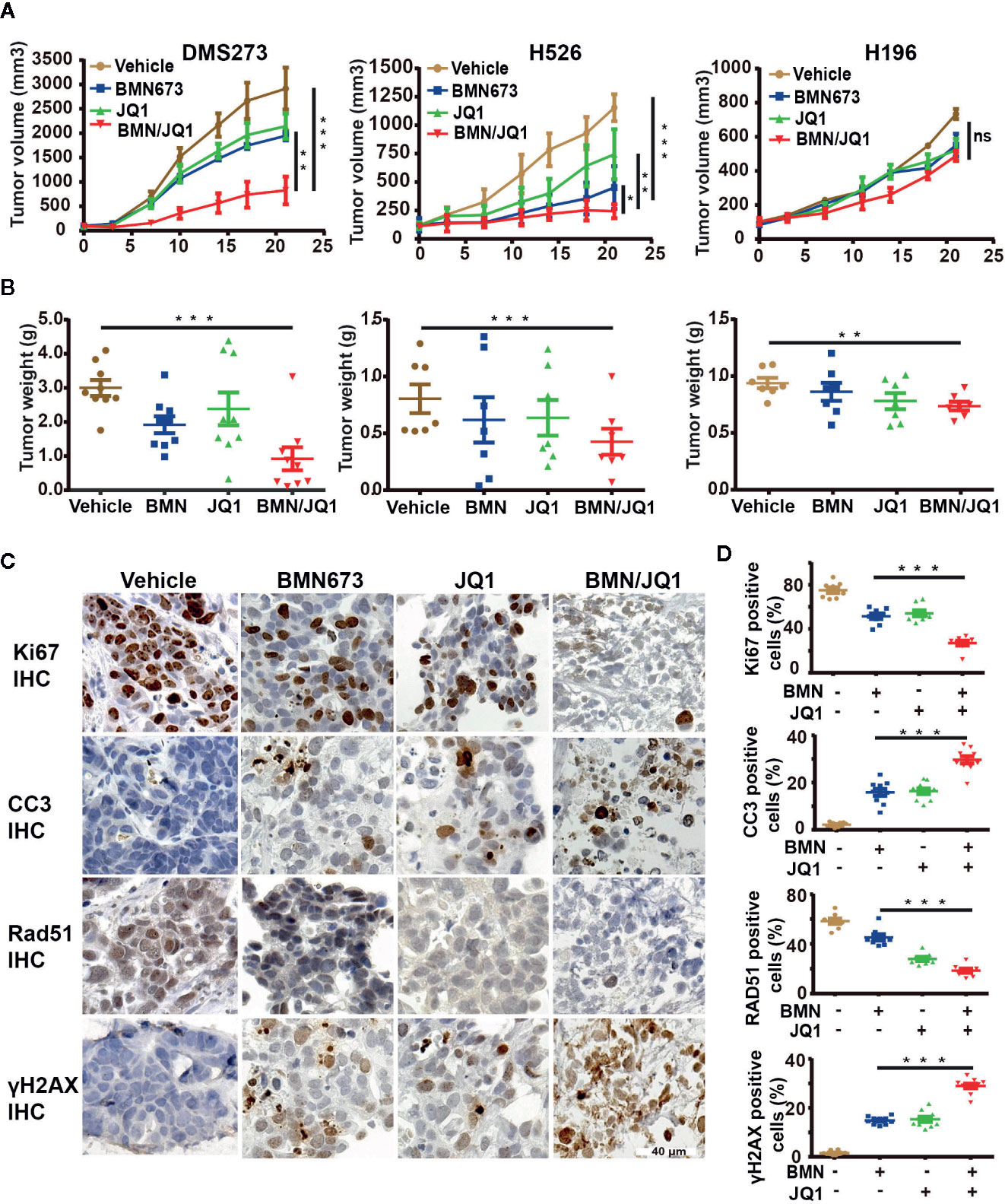
Figure 7 Therapeutic efficacy of JQ1 and BMN673 as single agents or in combination in vivo. (A) Tumor volume curves of DMS273 (left panel) and H526 (middle panel) and H196 (right panel) xenograft mice treated with BMN673, JQ1, or a combination of BMN673 and JQ1. Data are shown as mean ± SD (n = 6). (B) Tumor weights of DMS273 (left panel) and H526 (middle panel) and H196 (right panel) xenograft mice were measured after the 21 days of drug treatment. The statistic analysis was shown as mean ± S.E.M. *P < 0.05; **P < 0.01; ***P < 0.001 (Student’s t test). (C) Representative immunohistochemistry images of Ki67, cleaved-caspase3, Rad51, and γH2AX on PDX tumor explants treated with BMN673 and JQ1 alone or in combination. Scale bar, 40 μm. The statistic analysis was shown at the right panel (D) and indicated as mean ± S.E.M. *P < 0.05; **P < 0.01; ***P < 0.001 (Student’s t test).
To further investigate the treatment effects of BMN673 and JQ1 in vivo, a PDX model of SCLC (c-MYC-overexpression) that maintained the pathological characteristics of the human primary tumor sample was established in our laboratory (Supplementary Figure 10A). We then prepared ex vivo organ cultures to evaluate the therapeutic effects of combination treatment using BMN673 and JQ1. Histological analysis demonstrated that the architecture and cellularity of the explants were very similar to those in the primary tumor (Supplementary Figure 10B). Immunohistochemistry analysis showed that combination treatment resulted in substantially reduced ki67 expression and enhanced cleaved-caspase 3 expression compared with those observed in groups treated with BMN673 or JQ1 monotherapy (Figures 7C, D). Furthermore, the expression of the HR repair protein Rad51 was markedly attenuated, and γH2AX expression levels were significantly increased, in explants treated with the combination of BMN673 and JQ1 (Figures 7C, D). Collectively, our in vivo study suggested that combined administration of BMN673 and JQ1 induced synergistic anti-tumor effects against MYC paralog-dependent SCLC xenografts and PDX explants.
Discussion
Small cell lung cancer is among the most aggressive types of cancer, and therapeutic options are limited. In the current study, we showed that PARP1, a promising therapeutic target in SCLC, might be a potential prognostic marker. Integrative analysis of several gene expression data sets revealed that genes involved in the RSR and HR repair pathways, including PARP1, were strongly associated with the expression of MYC paralogs (Figure 8). We further showed that c-MYC and MYCN transcriptionally activated PARP1 in SCLC cells. These findings highlighted a novel mechanism of induction of high expression of PARP1 in a subset of SCLC. We also showed that the RSR and HR repair pathways might be potential therapeutic targets for the treatment of MYC paralog-dependent SCLC. Furthermore, targeting the MYC paralog-PARP1-DDR axis via concurrent inhibition of BET and PARP1 resulted in synergistic anti-tumor activity in vitro and in vivo against MYC paralog-driven SCLC cells (Figure 8), but not against MYC paralog-independent SCLC cells. Mechanistic investigations confirmed that BETi contributed to PARPi sensitivity through multiple mechanisms. Inhibition of BET resulted in (1) defects in the cellular response to PARPi-induced DNA replication stress, (2) impaired HR repair capacity via interference with DNA end resection and RAD51 function, and (3) limited end processing during NHEJ repair through induction of phosphorylation of DNA-PKcs. Finally, we showed that MYC paralog might be an indicator of treatment response to combination therapy with JQ1 and BMN673 in patients with SCLC.
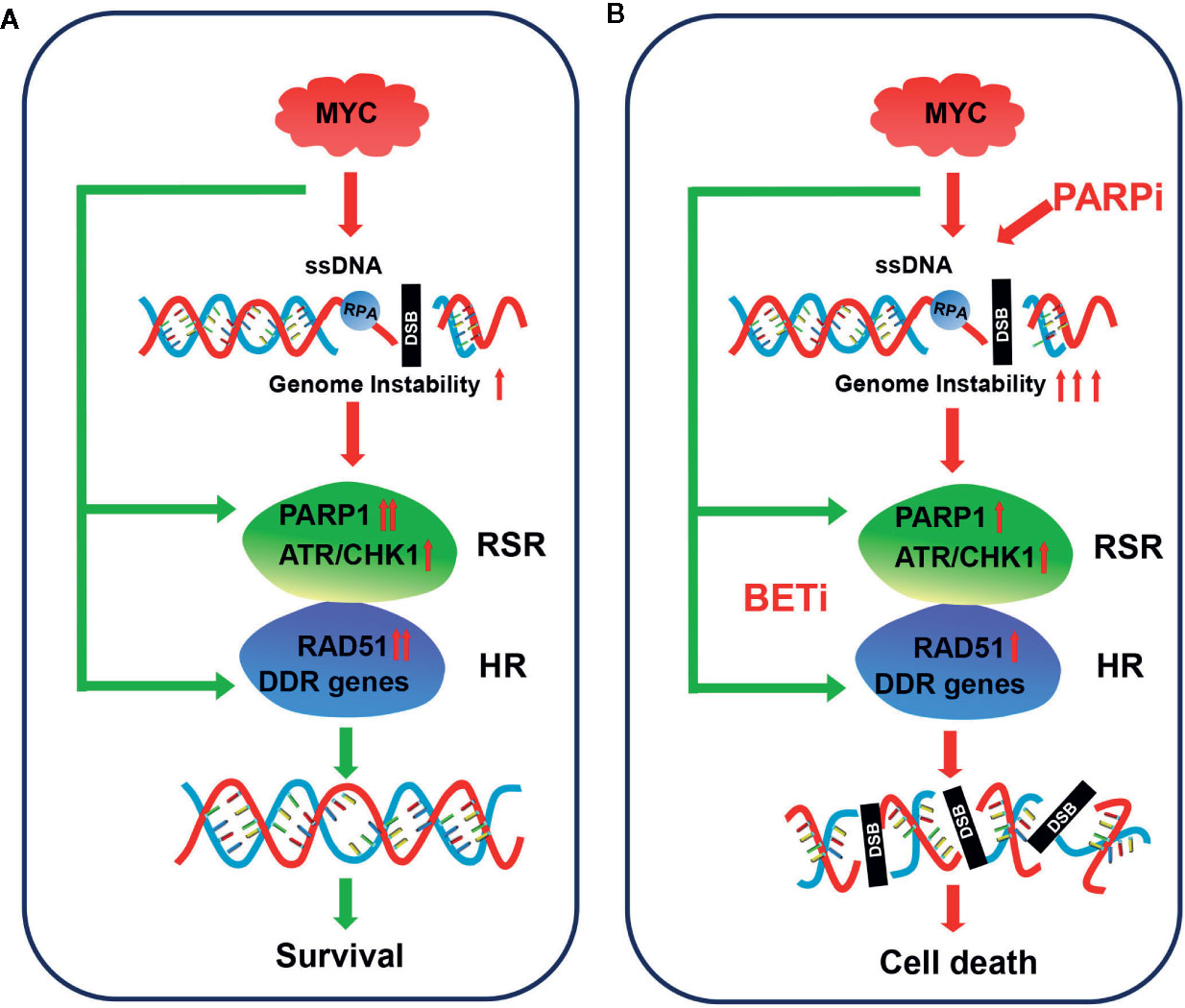
Figure 8 Model for targeting MYC paralog-PARP1 axis in MYC paralog-dependent SCLC cells. (A) Genome instability is common in MYC paralog-activated SCLC cells. DDR genes, including PARP1 and RAD51, were highly expressed to maintain genome stability for cell survival in this context. (B) Combining PARP inhibition (PARPi) with BET inhibition (BETi) yields synergistic anti-tumor effects in MYC paralog-activated SCLC cells. BETi treatment causes defects in the replication stress response (RSR) and homologous recombination (HR) competency. The combination of PARPi and BETi leads to synthetic lethality due to unrepaired DSBs.
The underlying mechanisms that cause a high expression of PARP1 in SCLC have not been characterized. We, for the first time, showed that c-MYC and MYCN directly targeted PARP1, and transcriptionally regulated its expression in SCLC cells. Whether MYCL directly targets PARP1 in SCLC cells requires further study. We also demonstrated that c-MYC directly regulated RAD51 expression using c-MYC overexpression or knockdown models and ChIP-PCR assays. These results agreed with those from studies that reported an association between c-MYC and the RAD51 promoter in other cell types (46, 47). Given that a number of genes required for the RSR and HR repair pathways are highly expressed in MYC paralog-activated cells, we speculated that MYC paralogs might also regulate other DDR genes in SCLC cells. Although PARP1 expression was recently shown to be associated with poor prognosis in patients with neuroblastoma (33), we found that high PARP1 expression was associated with high OS and PFS in patients with SCLC. This discrepancy may be clarified as SCLC is usually detected in a late disease stage and PARP1 mostly acts at later points of cancer development. Meanwhile, a previous related study also indicated the favorable role of PARP1 in cancer development and the fact is that strong PARP activation may result in depletion of NAD+ and ATP which in turn cause cell death (48). These results agreed with those in a previous study that reported high nuclear PARP1 expression resulted in improved survival in patients with PDA (49). Therefore, the data present in this study can only serve as a prognosis outlook, further mechanistic investigation of PARP1 biology in SCLC is required.
PARPi induces synthetic lethality in tumors with BRCA1/2 deficiency. However, the BRCA1/2 mutations are rarely reported in SCLC (17, 50), which might limit PARPi efficacy. In our investigations, BMN673 treatment strongly induced DDR activation and caused modest DNA damage in MYC paralog-dependent SCLC (Figures 4A, C), which might be due to a high competency of RSR and HR in these cells. A recent study identified c-MYC expression as a predictive biomarker of CHK1 inhibitor activity in SCLC (51). Another study showed that defective HR repair or MYC family oncogene amplification predisposed a SCLC CDX model to a combination treatment of PARP inhibitor and WEE1 inhibitor (52). Also, c-MYC and MYCN amplification is a predictive biomarker for PARP inhibitor sensitivity in glioblastoma (53). Most importantly, we and others recently demonstrated that inhibiting RSR pathway is effective in treatment of SCLC (54, 55). Together, these studies demonstrated that targeting one or more DDR proteins is a desirable strategy for the treatment of a subset of SCLC. We further showed that MYC paralog-activated SCLC cells might depend on DDR signaling for cell survival. Targeting the MYC paralog-PARP1-DDR signaling pathway could allow for increased efficacy of PARPi.
Although we showed that c-MYC strongly induced PARP1 and RAD51 expression, in DMS273 cells which harbor both c-MYC and MYCL amplification, knockdown of c-MYC did not significantly repress PARP1 and Rad51 expression, we speculate this discrepancy may result from the transcription regulation of PARP1 and RAD51 by MYCL in DMS273 cells. Therefore, the effects of MYCN or MYCL on SCLC cells require further investigations. In our study, the expression of c-MYC in DMS273 cells and SHP77 cells with c-MYC overexpression was not altered following treatment with JQ1 (Supplementary Figure 5). This finding was consistent with those from a previous study that showed that JQ1 treatment did not reduce c-MYC activity (23, 56). In contrast, MYCN expression was significantly reduced by JQ1 in a dose-dependent manner in SCLC (Supplementary Figure 5). Whether MYC paralogs are required for the anti-proliferative effect of JQ1 in SCLC requires further study.
A recent finding identified a novel role for the BET family member BRD4 in the regulation of the RSR pathway through interaction with the pre-replication factor CDC6 (57). Furthermore, recent studies have shown that BRD4 inhibitors reduced HR competence through the downregulation of DDR-related gene expression (41, 58). Indeed, previous studies have shown that JQ1 can be used effectively to treat SCLC, but the underlying mechanisms of this anti-tumor activity are not well understood. Our study showed that JQ1 induced DNA damage and reduced HR repair and RSR efficiency in MYC paralog-dependent SCLC cells. These findings were consistent with those from a recent study that showed that BRD4 inhibition impaired HR via interfering with DNA end resection (58). In contrast to their proposed mechanism, we found that BRD4 expression was not positively correlated with RBBP8 (CtIP, a key component of DNA end resection) expression at the mRNA level (Supplementary Figures 11A, B). Instead, we found that JQ1 induced a marked increase in phosphorylation of DNA-PKcs at S2056. We hypothesized that JQ1-induced HR deficiency might have been due to DNA-PKcs-mediated attenuation of DNA end resection. A recent study indicated that BET inhibition hampered NHEJ activity through inhibition of ku80 (59). Given that phosphorylation of DNA-PKcs at S2056 enhances NHEJ, it would be interesting to address whether JQ1 promotes NHEJ in SCLC.
Although the FDA recently approved chemotherapy plus immunotherapy as a first-line treatment for patients with SCLC, the results from clinical trials showed only a 2-month increase in overall survival in patients treated with chemotherapy in combination with anti-PD-L1 therapy (5). Therefore, identification of new combination treatment strategies to improve clinical outcomes of patients with SCLC is an urgent need. We showed that targeting epigenetic factors via BET inhibition potentiated sensitivity of MYC paralog-dependent SCLC to the PARP inhibitor BMN673 in vitro and in vivo. Further clinical trials are needed to evaluate the efficacy of combination therapies that inhibit BET and PARP, as this strategy has great potential to improve the prognoses of patients with SCLC with MYC paralog amplification or overexpression.
Data Availability Statement
Publicly available datasets were analyzed in this study, these data can be found here: https://portals.broadinstitute.org/ccle/data, George et al, 2015 (16) and NCBI Gene Expression Omnibus (GSE89660).
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Hefei Institutes of Physical Science, Chinese Academy of Sciences. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
WL and XB conceived and designed the study, and wrote the manuscript. XB and XW performed the experiments and acquired the data. QZ, LM, and JHH performed flow cytometry analysis. AX and GC analyzed and interpreted the data. JH provided plasmids. All authors contributed to the article and approved the submitted version.
Funding
This study is supported by National Natural Science Foundation of China (Grant Numbers: 81972191 and 81672647), Science and Technology Service Network Initiative of Chinese Academy of Sciences (Grant Number: KFJ-STS-SCYD-010), Science and Technology Major Project of Anhui Province (Grant Number: 18030801140), Key program of 13th five-year plan of CASHIPS (Grant Number: KP-2017-26), and the 100-Talent Program of Chinese Academy of Sciences.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Ge Shan (University of Science and Technology of China, China) for kindly providing the Zeiss fluorescence microscope.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.565820/full#supplementary-material
References
1. Gazdar AF, Bunn PA, Minna JD. Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer (2017) 17:725–37. doi: 10.1038/nrc.2017.87
2. Semenova EA, Nagel R, Berns A. Origins, genetic landscape, and emerging therapies of small cell lung cancer. Genes Dev (2015) 29:1447–62. doi: 10.1101/gad.263145.115
3. William WN Jr., Glisson BS. Novel strategies for the treatment of small-cell lung carcinoma. Nat Rev Clin Oncol (2011) 8:611–9. doi: 10.1038/nrclinonc.2011.90
4. Pietanza MC, Byers LA, Minna JD, Rudin CM. Small cell lung cancer: will recent progress lead to improved outcomes? Clin Cancer Res (2015) 21:2244–55. doi: 10.1158/1078-0432.CCR-14-2958
5. Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med (2018) 379:2220–9. doi: 10.1056/NEJMoa1809064
6. Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J, et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov (2012) 2:798–811. doi: 10.1158/2159-8290.CD-12-0112
7. Cardnell RJ, Feng Y, Diao L, Fan YH, Masrorpour F, Wang J, et al. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin Cancer Res (2013) 19:6322–8. doi: 10.1158/1078-0432.CCR-13-1975
8. Lok BH, Gardner EE, Schneeberger VE, Ni A, Desmeules P, Rekhtman N, et al. PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin Cancer Res (2017) 23:523–35. doi: 10.1158/1078-0432.CCR-16-1040
9. Pietanza MC, Waqar SN, Krug LM, Dowlati A, Hann CL, Chiappori A, et al. Randomized, Double-Blind, Phase II Study of Temozolomide in Combination With Either Veliparib or Placebo in Patients With Relapsed-Sensitive or Refractory Small-Cell Lung Cancer. J Clin Oncol (2018) 36:2386–94. doi: 10.1200/JCO.2018.77.7672
10. Owonikoko TK, Dahlberg SE, Sica GL, Wagner LI, Wade JL,3, Srkalovic G, et al. Randomized Phase II Trial of Cisplatin and Etoposide in Combination With Veliparib or Placebo for Extensive-Stage Small-Cell Lung Cancer: ECOG-ACRIN 2511 Study. J Clin Oncol (2019) 37:222–9. doi: 10.1200/JCO.18.00264
11. de Bono J, Ramanathan RK, Mina L, Chugh R, Glaspy J, Rafii S, et al. Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov (2017) 7:620–9. doi: 10.1158/2159-8290.CD-16-1250
12. Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, et al. c-Myc regulates transcriptional pause release. Cell (2010) 141:432–45. doi: 10.1016/j.cell.2010.03.030
13. Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell (2016) 29:536–47. doi: 10.1016/j.ccell.2016.03.001
14. Nau MM, Brooks BJ, Battey J, Sausville E, Gazdar AF, Kirsch IR, et al. L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature (1985) 318:69–73. doi: 10.1038/318069a0
15. Wu R, Lin L, Beer DG, Ellenson LH, Lamb BJ, Rouillard JM, et al. Amplification and overexpression of the L-MYC proto-oncogene in ovarian carcinomas. Am J Pathol (2003) 162:1603–10. doi: 10.1016/S0002-9440(10)64294-0
16. George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature (2015) 524:47–53. doi: 10.1038/nature14664
17. Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet (2012) 44:1104–10. doi: 10.1038/ng.2396
18. Mollaoglu G, Guthrie MR, Bohm S, Bragelmann J, Can I, Ballieu PM, et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell (2017) 31:270–85. doi: 10.1016/j.ccell.2016.12.005
19. Kim DW, Wu N, Kim YC, Cheng PF, Basom R, Kim D, et al. Genetic requirement for Mycl and efficacy of RNA Pol I inhibition in mouse models of small cell lung cancer. Genes Dev (2016) 30:1289–99. doi: 10.1101/gad.279307.116
20. Fletcher S, Prochownik EV. Small-molecule inhibitors of the Myc oncoprotein. Biochim Biophys Acta (2015) 1849:525–43. doi: 10.1016/j.bbagrm.2014.03.005
21. Lam LT, Lin X, Faivre EJ, Yang Z, Huang X, Wilcox DM, et al. Vulnerability of Small-Cell Lung Cancer to Apoptosis Induced by the Combination of BET Bromodomain Proteins and BCL2 Inhibitors. Mol Cancer Ther (2017) 16:1511–20. doi: 10.1158/1535-7163.MCT-16-0459
22. Kato F, Fiorentino FP, Alibes A, Perucho M, Sanchez-Cespedes M, Kohno T, et al. MYCL is a target of a BET bromodomain inhibitor, JQ1, on growth suppression efficacy in small cell lung cancer cells. Oncotarget (2016) 7:77378–88. doi: 10.18632/oncotarget.12671
23. Lenhart R, Kirov S, Desilva H, Cao J, Lei M, Johnston K, et al. Sensitivity of Small Cell Lung Cancer to BET Inhibition Is Mediated by Regulation of ASCL1 Gene Expression. Mol Cancer Ther (2015) 14:2167–74. doi: 10.1158/1535-7163.MCT-15-0037
24. Kaur G, Reinhart RA, Monks A, Evans D, Morris J, Polley E, et al. Bromodomain and hedgehog pathway targets in small cell lung cancer. Cancer Lett (2016) 371:225–39. doi: 10.1016/j.canlet.2015.12.001
25. Bragelmann J, Bohm S, Guthrie MR, Mollaoglu G, Oliver TG, Sos ML. Family matters: How MYC family oncogenes impact small cell lung cancer. Cell Cycle (2017) 16:1489–98. doi: 10.1080/15384101.2017.1339849
26. Mostocotto C, Carbone M, Battistelli C, Ciotti A, Amati P, Maione R. Poly(ADP-ribosyl)ation is required to modulate chromatin changes at c-MYC promoter during emergence from quiescence. PLoS One (2014) 9:e102575. doi: 10.1371/journal.pone.0102575
27. Zhang W, Liu B, Wu W, Li L, Broom BM, Basourakos SP, et al. Targeting the MYCN-PARP-DNA Damage Response Pathway in Neuroendocrine Prostate Cancer. Clin Cancer Res (2018) 24:696–707. doi: 10.1158/1078-0432.CCR-17-1872
28. Hu S, Wang X, Shan G. Insertion of an Alu element in a lncRNA leads to primate-specific modulation of alternative splicing. Nat Struct Mol Biol (2016) 23:1011–9. doi: 10.1038/nsmb.3302
29. Lee GY, Kenny PA, Lee EH, Bissell MJ. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat Methods (2007) 4:359–65. doi: 10.1038/nmeth1015
30. Bian X, Gao J, Luo F, Rui C, Zheng T, Wang D, et al. PTEN deficiency sensitizes endometrioid endometrial cancer to compound PARP-PI3K inhibition but not PARP inhibition as monotherapy. Oncogene (2018) 37:341–51. doi: 10.1038/onc.2017.326
31. Cheng H, Liu P, Zhang F, Xu E, Symonds L, Ohlson CE, et al. A genetic mouse model of invasive endometrial cancer driven by concurrent loss of Pten and Lkb1 Is highly responsive to mTOR inhibition. Cancer Res (2014) 74:15–23. doi: 10.1158/0008-5472.CAN-13-0544
32. Dean JL, McClendon AK, Hickey TE, Butler LM, Tilley WD, Witkiewicz AK, et al. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle (2012) 11:2756–61. doi: 10.4161/cc.21195
33. Colicchia V, Petroni M, Guarguaglini G, Sardina F, Sahun-Roncero M, Carbonari M, et al. PARP inhibitors enhance replication stress and cause mitotic catastrophe in MYCN-dependent neuroblastoma. Oncogene (2017) 36:4682–91. doi: 10.1038/onc.2017.40
34. Parsels LA, Karnak D, Parsels JD, Zhang Q, Velez-Padilla J, Reichert ZR, et al. PARP1 Trapping and DNA Replication Stress Enhance Radiosensitization with Combined WEE1 and PARP Inhibitors. Mol Cancer Res (2018) 16:222–32. doi: 10.1158/1541-7786.MCR-17-0455
35. Liao H, Ji F, Helleday T, Ying S. Mechanisms for stalled replication fork stabilization: new targets for synthetic lethality strategies in cancer treatments. EMBO Rep (2018) 19(9):e46263. doi: 10.15252/embr.201846263
36. Marechal A, Zou L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res (2015) 25:9–23. doi: 10.1038/cr.2014.147
37. Srinivasan SV, Dominguez-Sola D, Wang LC, Hyrien O, Gautier J. Cdc45 is a critical effector of myc-dependent DNA replication stress. Cell Rep (2013) 3:1629–39. doi: 10.1016/j.celrep.2013.04.002
38. Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montana MF, et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol (2011) 18:1331–5. doi: 10.1038/nsmb.2189
39. Petroni M, Sardina F, Heil C, Sahun-Roncero M, Colicchia V, Veschi V, et al. The MRN complex is transcriptionally regulated by MYCN during neural cell proliferation to control replication stress. Cell Death Differ (2016) 23:197–206. doi: 10.1038/cdd.2015.81
40. Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science (2017) 355:1152–8. doi: 10.1126/science.aam7344
41. Yang L, Zhang Y, Shan W, Hu Z, Yuan J, Pi J, et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci Transl Med (2017) 9(400):eaal1645. doi: 10.1126/scitranslmed.aal1645
42. Neal JA, Meek K. Choosing the right path: does DNA-PK help make the decision? Mutat Res (2011) 711:73–86. doi: 10.1016/j.mrfmmm.2011.02.010
43. Cui X, Yu Y, Gupta S, Cho YM, Lees-Miller SP, Meek K. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol Cell Biol (2005) 25:10842–52. doi: 10.1128/MCB.25.24.10842-10852.2005
44. Chen BP, Chan DW, Kobayashi J, Burma S, Asaithamby A, Morotomi-Yano K, et al. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J Biol Chem (2005) 280:14709–15. doi: 10.1074/jbc.M408827200
45. Kousholt AN, Fugger K, Hoffmann S, Larsen BD, Menzel T, Sartori AA, et al. CtIP-dependent DNA resection is required for DNA damage checkpoint maintenance but not initiation. J Cell Biol (2012) 197:869–76. doi: 10.1083/jcb.201111065
46. Luoto KR, Meng AX, Wasylishen AR, Zhao H, Coackley CL, Penn LZ, et al. Tumor cell kill by c-MYC depletion: role of MYC-regulated genes that control DNA double-strand break repair. Cancer Res (2010) 70:8748–59. doi: 10.1158/0008-5472.CAN-10-0944
47. Carey JPW, Karakas C, Bui T, Chen X, Vijayaraghavan S, Zhao Y, et al. Synthetic Lethality of PARP Inhibitors in Combination with MYC Blockade Is Independent of BRCA Status in Triple-Negative Breast Cancer. Cancer Res (2018) 78:742–57. doi: 10.1158/0008-5472.CAN-17-1494
48. Carson DA, Carrera CJ, Wasson DB, Yamanaka H. Programmed cell death and adenine deoxynucleotide metabolism in human lymphocytes. Adv Enzyme Regul (1988) 27:395–404. doi: 10.1016/0065-2571(88)90028-3
49. Klauschen F, von Winterfeld M, Stenzinger A, Sinn BV, Budczies J, Kamphues C, et al. High nuclear poly-(ADP-ribose)-polymerase expression is prognostic of improved survival in pancreatic cancer. Histopathology (2012) 61:409–16. doi: 10.1111/j.1365-2559.2012.04225.x
50. Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet (2012) 44:1111–6. doi: 10.1038/ng.2405
51. Sen T, Tong P, Stewart CA, Cristea S, Valliani A, Shames DS, et al. CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer Res (2017) 77:3870–84. doi: 10.1158/0008-5472.CAN-16-3409
52. Lallo A, Frese KK, Morrow CJ, Sloane R, Gulati S, Schenk MW, et al. The Combination of the PARP Inhibitor Olaparib and the WEE1 Inhibitor AZD1775 as a New Therapeutic Option for Small Cell Lung Cancer. Clin Cancer Res (2018) 24:5153–64. doi: 10.1158/1078-0432.CCR-17-2805
53. Ning JF, Stanciu M, Humphrey MR, Gorham J, Wakimoto H, Nishihara R, et al. Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat Commun (2019) 10:2910. doi: 10.1038/s41467-019-10993-5
54. Nagel R, Avelar AT, Aben N, Proost N, van de Ven M, van der Vliet J, et al. Inhibition of the Replication Stress Response Is a Synthetic Vulnerability in SCLC That Acts Synergistically in Combination with Cisplatin. Mol Cancer Ther (2019) 18:762–70. doi: 10.1158/1535-7163.MCT-18-0972
55. Bian X, Lin W. Targeting DNA Replication Stress and DNA Double-Strand Break Repair for Optimizing SCLC Treatment. Cancers (Basel) (2019) Sep 2; 11:(9):1289. doi: 10.3390/cancers11091289
56. Inoue-Yamauchi A, Jeng PS, Kim K, Chen HC, Han S, Ganesan YT, et al. Targeting the differential addiction to anti-apoptotic BCL-2 family for cancer therapy. Nat Commun (2017) 8:16078. doi: 10.1038/ncomms16078
57. Zhang J, Dulak AM, Hattersley MM, Willis BS, Nikkila J, Wang A, et al. BRD4 facilitates replication stress-induced DNA damage response. Oncogene (2018) 37:3763–77. doi: 10.1038/s41388-018-0194-3
58. Sun C, Yin J, Fang Y, Chen J, Jeong KJ, Chen X, et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell (2018) 33:401–416.e408. doi: 10.1016/j.ccell.2018.01.019
Keywords: small cell lung cancer, MYC paralog, PARP1, BET, DNA damage response
Citation: Bian X, Wang X, Zhang Q, Ma L, Cao G, Xu A, Han J, Huang J and Lin W (2020) The MYC Paralog-PARP1 Axis as a Potential Therapeutic Target in MYC Paralog-Activated Small Cell Lung Cancer. Front. Oncol. 10:565820. doi: 10.3389/fonc.2020.565820
Received: 26 May 2020; Accepted: 22 September 2020;
Published: 08 October 2020.
Edited by:
Shiv K. Gupta, Mayo Clinic, United StatesReviewed by:
Nicola Jane Curtin, Newcastle University, United KingdomLi Ding, Mayo Clinic, United States
Copyright © 2020 Bian, Wang, Zhang, Ma, Cao, Xu, Han, Huang and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenchu Lin, d2VuY2h1QGhtZmwuYWMuY24=