 Lisa Mayr1
Lisa Mayr1 Johannes Gojo1
Johannes Gojo1 Andreas Peyrl1
Andreas Peyrl1 Amedeo A. Azizi1
Amedeo A. Azizi1 Natalia M. Stepien1Thomas Pletschko1Thomas Czech2
Natalia M. Stepien1Thomas Pletschko1Thomas Czech2 Christian Dorfer2Sander Lambo3,4Karin Dieckmann5Christine Haberler6Marcel Kool3,4,7
Christian Dorfer2Sander Lambo3,4Karin Dieckmann5Christine Haberler6Marcel Kool3,4,7 Irene Slavc1*
Irene Slavc1*- 1Department of Pediatrics and Adolescent Medicine and Comprehensive Center for Pediatrics, Medical University of Vienna, Vienna, Austria
- 2Department of Neurosurgery, Medical University of Vienna, Vienna, Austria
- 3Division of Pediatric Neurooncology, Hopp Children’s Cancer Center Heidelberg (KiTZ), Heidelberg, Germany
- 4Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany
- 5Department of Radiotherapy, Medical University of Vienna, Vienna, Austria
- 6Division of Neuropathology and Neurochemistry, Department of Neurology, Medical University of Vienna, Vienna, Austria
- 7Research Department, Princess Máxima Center for Pediatric Oncology, Utrecht, Netherlands
Embryonal tumor with multilayered rosettes (ETMR) is a rare, aggressive embryonal central nervous system tumor characterized by LIN28A expression and alterations in the C19MC locus. ETMRs predominantly occur in young children, have a dismal prognosis, and no definitive treatment guidelines have been established. We report on nine consecutive patients and review the role of initiation/timing of radiotherapy on survival. Between 2006 and 2018, nine patients were diagnosed with ETMR. Diagnosis was confirmed histopathologically, immunohistochemically and molecularly. Median age was 25 months (5–38). Location was supratentorial in five, pineal in three, and brainstem in one. Seven patients had a gross total resection, one a partial resection and one a biopsy at initial diagnosis. Chemotherapy augmented with intrathecal therapy started a median of 10 days (7–20) after surgery. Only two patients who after gross total resection received radiotherapy very early on (six weeks after diagnosis) are alive and in complete remission 56 and 50 months after diagnosis. All remaining patients for whom radiotherapy was deferred until the end of chemotherapy recurred, albeit none with leptomeningeal disease. A literature research identified 228 patients with ETMR. Including our patients only 26 (11%) of 237 patients survived >36 months with no evidence of disease at last follow-up. All but two long-term (>36 months) survivors received radiotherapy, ten of whom early on following gross total resection (GTR). GTR followed by early focal radiotherapy and intrathecal therapy to prevent leptomeningeal disease are potentially important to improve survival of ETMR in the absence of effective targeted therapies.
Introduction
Embryonal tumors with multilayered rosettes (ETMR) are rare and highly aggressive embryonal brain tumors primarily affecting infants and young children under the age of four years. ETMRs encompass three histological variants: embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma (1, 2), all of which were previously regarded as subtypes of central nervous system (CNS) primitive neuroectodermal tumors (PNETs). Histologically, the presence of undifferentiated neuroepithelial cells forming multi-layered rosettes is characteristic of this tumor entity. Broad bands of well-differentiated neuropil islands, ependymoblastic rosettes, or medulloepitheliomatous rosettes may be present (3). Furthermore, they are typically characterized by a focal amplification spanning the C19MC microRNA cluster at 19q13.42 and high expression of the stem cell marker LIN28A (4). Based on this sensitive and specific marker irrespective of other features, Paulus and Kleihues proposed the term “Embryonal tumors with multilayered rosettes” as unifying entity for these tumors (5). In the updated version of the World Health Organisation (WHO) Classification of Tumours of the Central Nervous System, ETMRs are therefore defined as aggressive CNS embryonal tumors with multi-layered rosettes and alterations in the C19MC locus at 19q13.42, i.e. ETMR, C19MC-altered, grade IV. Cases of embryonal tumors with multi-layered rosettes without alteration in the C19MC locus or that were not tested for alteration are classified as ETMR, not otherwise specified (NOS) (6). ETMR may originate anywhere in the brain with approximately two thirds occurring in the cerebral hemispheres and one third in the brain stem and cerebellum (7). Neuroimaging usually reveals large, solid masses featuring irregular- or no-contrast enhancement, often with significant mass effect. Some cases show cystic components and microcalcifications (8, 9). ETMRs may present with or develop leptomeningeal dissemination as well as extracranial invasive growth in the soft tissues and extracranial metastases. Patients with ETMR have a dismal prognosis with reported survival times averaging 12 months despite maximal safe resection followed by intensive multimodal therapy often including high-dose chemotherapy (HDCT) and delayed radiotherapy (8). Due to the rarity of the disease fewer than 300 cases have been reported so far. Given the young age of the patients, radiotherapy was often deferred and no definitive guidelines for optimal treatment have been established.
We report on the importance of an early focal radiotherapy on the outcome in a series of nine consecutive patients with ETMR, treated at the Medical University of Vienna (MUV) since 2006.
Methods
Reassessment of all highly malignant embryonal brain tumors formerly classified as ETANTR, medulloepithelioma, ependymoblastoma, or CNS PNET with regard to LIN28A expression and amplification of the C19MC microRNA cluster disclosed nine patients treated at the MUV since 2006.
Seven patients previously diagnosed as ETANTR or ETMR were reclassified as ETMR. Two patients (cases 1 and 3) originally diagnosed as CNS PNET were reclassified. Male to female ratio was 2:7. Median age at diagnosis was 25 months (range 5–38). Five tumors were located supratentorially, three in the pineal region and one in the pons. At presentation no patient showed signs of leptomeningeal dissemination. The extent of surgical resection was defined on postoperative magnetic resonance imaging (MRI) performed within 48 hours as gross total resection (GTR, no obvious residual tumor), subtotal resection (STR with <10% residual tumor), partial resection (PR with 10–50% residual tumor) and biopsy (>50% residual tumor). Determination of progression or response was based on response assessment in neuro-oncology (RANO) criteria (10). Clinical characteristics are depicted in Table 1. Informed consent was obtained for all patients.
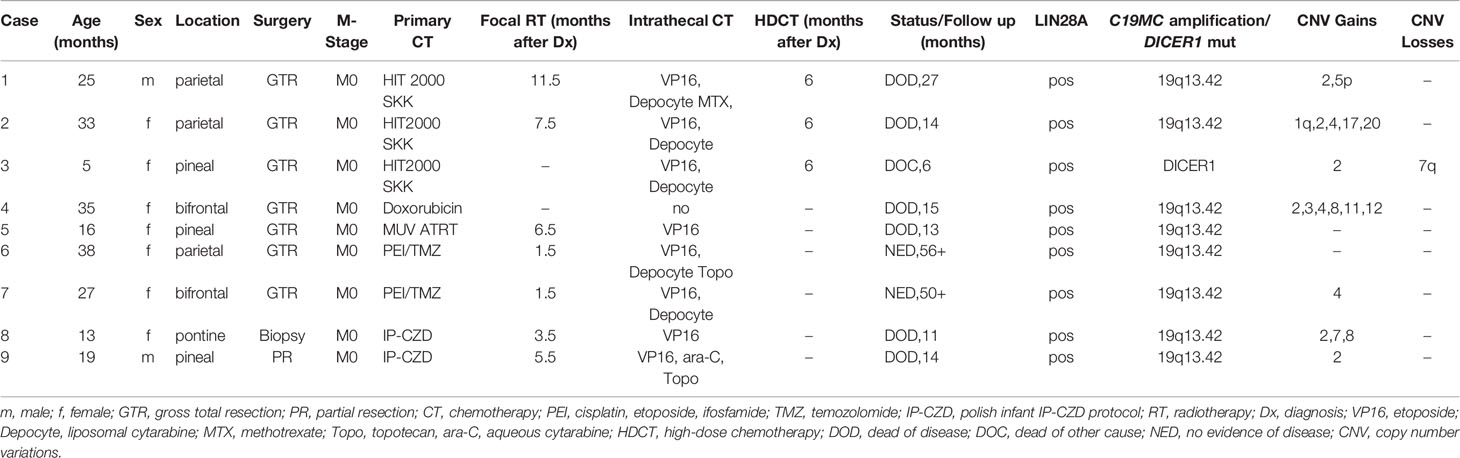
Table 1 Patient characteristics, treatment and outcome.
Histopathology, Interphase Fluorescence In Situ Hybridization
For diagnostic purposes routine histopathological examination on formalin-fixed and paraffin embedded (FFPE) tissue was performed including immunohistochemical (IHC) analysis with an anti-LIN28A polyclonal antibody (antibody (A177 #3978, 1:800, Cell Signaling Inc; Danvers, MA 01923, USA). Interphase Flourescence In Situ Hybridization (iFISH) was carried out on FFPE tissue sections as previously described (11) using a commercially available 19q13.42 (C19MC, spectrum orange) and 19.13.12 (spectrum green) probe (CytoTest C19MC/TPM4), Diagnostic Technology Pty Ltd; Belrose, Australia).
Methylation Array and Whole Genome Sequencing Analyses
To analyze methylation-specific subgrouping, copy number aberrations and somatic/germline mutations in more detail DNA was extracted from fresh frozen tumor tissue in all nine cases. Matched blood samples for germline sequencing were available in six cases. Methylation array (Illumina 850K platform) and whole genome sequencing (40×) were performed via the DKFZ core facility. All analyses were performed according to national and international biosafety guidelines and as previously published (12, 13).
Systemic Chemotherapy
All patients started chemotherapy a median of 10 days (range 7–20 days) after surgery. Intensive chemotherapy followed various brain tumor protocols (HIT SKK (14), MUV ATRT (15), and the Polish infant protocol IP-CZD consisting of VCR 1.5 mg/m2, etoposide 300 mg/m2, cisplatin 100 mg/m2 alternating with cyclophosphamide 1.5 g/m2 per course) and included cisplatin or carboplatin and etoposide in all except one patient (hypoxic brain injury) who received doxorubicin based treatment. Three patients who were in complete remission after GTR and conventional chemotherapy received HDCT consisting of a modified “Head Start” protocol (carboplatin, etoposide and Thiotepa) (16) (Table 1). Chemotherapy was intended to be given for approximately six months and to be followed by HDCT in patients still in remission in order to delay radiotherapy in this vulnerable population.
Two patients aged 38 and 27 months at diagnosis (cases 6 and 7) were treated differently. Both patients started focal radiotherapy exactly six weeks after GTR. The time for planning radiotherapy (as well as second look surgery in one of the patients) was bridged by two courses of PEI (cisplatin 20 mg/m2 d1-5, ifosfamide 1.500 mg/m2 d1-5, and etoposide 100 mg/m2 d1-3) in both patients. Temozolomide was given concomitant to focal radiotherapy and continued thereafter for a total of 12 courses (17).
Intrathecal Therapy
Therapy was augmented with intrathecal chemotherapy in all except one patient (hypoxic brain injury) and consisted of alternating etoposide (0.25 mg <1-year-old; 0.5 mg >-year-old on five consecutive days), and/or methotrexate (2 mg/day for four days), and/or liposomal cytarabine (Depocyte) (25 mg <3-year-old and 35 mg >3 and <9-year-old and 35–50 mg >9-year-old) in combination with oral dexamethason to prevent chemical arachnoiditis and/or topotecan (0.25 mg >1 and <2–year-old, 0.32 mg >2 and <3-year-old, and 0.4 mg >3-year-old) twice a week and/or aqueous cytarabin 30 mg twice a week (16 mg <1-year-old, 20 mg >1 and <2-year-old, 26 mg >2 and <3-year-old) (18–23).
Radiotherapy
All except two patients (case 3 who died after bone marrow transplant and case 4 with hypoxic brain injury) received focal irradiation. Six were treated with photons and one with protons. Three dimensional (3D) treatment planning was performed in all patients. Target definitions were based on pre- and postoperative MRIs.
Gross tumor volume (GTV) included the postoperative tumor bed. Clinical target volume (CTV) was defined as GTV plus 1 cm margin adapted to the anatomical structures. Planning target volume (PTV) was CTV plus 3 mm margin. All but one patient were treated with conformal volume modulated radiotherapy (VMAT) and photons. One patient was treated with protons. Target definition was identical for protons and photons based on MRI. Fraction sizes were 1.8 Gy for all target volumes with a total dose of 54 Gy in all patients. None of the patients was reirradiated.
Treatment at Recurrence
Besides additional resections and radiotherapy three patients received temozolomide and three patients received a metronomic antiangiogenic therapy according to MEMMAT (ClinicalTrials.gov Identifier: NCT01356290), one also topotecan and olaparib. Olaparib was given continuously at a dose of 50 mg BID (100 mg total dose per day) in this 2.4-year-old male (body weight 13 kg) and combined with topotecan given at a dose of 1.5 mg/m2 for five consecutive days every three weeks (Supplementary Table S1).
Neuropsychological and Quality Of Life Testing
All patients were tested with age-appropriate methods. Domains tested in the two long-term survivors included fluid intelligence, verbal comprehension, processing speed, memory, working memory, attention, fine motor functions, executive functions and emotion recognition (24–33). The test-battery was accompanied by a brief behavioral screening (26, 34, 35). Additionally, patients’ health-related quality of life (HRQoL) was assessed using proxy-reports (parents) (36).
Statistical Analysis
For clustering of the DNA methylation profiles of ETMR cases treated at MUV t-Stochastic neighborhood embedding with 193 ETMRs (12), 534 other CNS tumors (37) and 44 pineoblastomas (13) were used.
Ethics Approval
Ethics approval and consent to participate: The ethics committee of the MUV granted their permission in 2016 with the number 1244/2016.
Results
Surgery
Seven patients had primary GTRs as evidenced by MRI performed within 48 h after surgery. Two patients who had an initial biopsy and PR, respectively, at another hospital and were progressive under adjuvant intensive chemotherapy had a second PR and STR, respectively, three and four months after original diagnosis (cases 8 and 9).
Histopathology, iFISH
All tumors showed characteristic morphological features of ETMR including multi-layered rosettes and neuropil islands as well as immunohistochemical expression of LIN28A. Gain/amplification at the C19MC locus was detected in eight of nine tumors by iFISH.
Methylation Array and Whole Genome Sequencing
Methylation-based classification was performed in all nine cases (Figure 1A, Supplementary Figures S1–S9). In one case (case 6) tumor cell content was too low to allow classification by methylation array despite a clear amplification of C19MC confirming the diagnosis of ETMR (Supplementary Figure S6). In accordance to iFISH, all except one case harbored the typical C19MC-amplification. All tumors were further analyzed by whole genome sequencing which revealed DICER1-mutation in the single case without C19MC-amplifcation (Supplementary Figure S3). For this case no germline material was available to test for germline DICER1-mutation, but the presence of two different mutations being mutually exclusive in the reads suggested that both alleles were affected (Figure 1B). Further detected somatic mutations are summarized in the supplementary material (Supplementary Figures S1–S9). With respect to other copy number variations (CNVs) gain of chromosome 2 was most common (6/9) as previously reported by Sin-Chan et al. (38) followed by gain of chromosomes 4 (3/9) and 8 (2/9). Interestingly, we also found gain of chromosome 5p in one case (Table 1, Supplementary Figure S1).
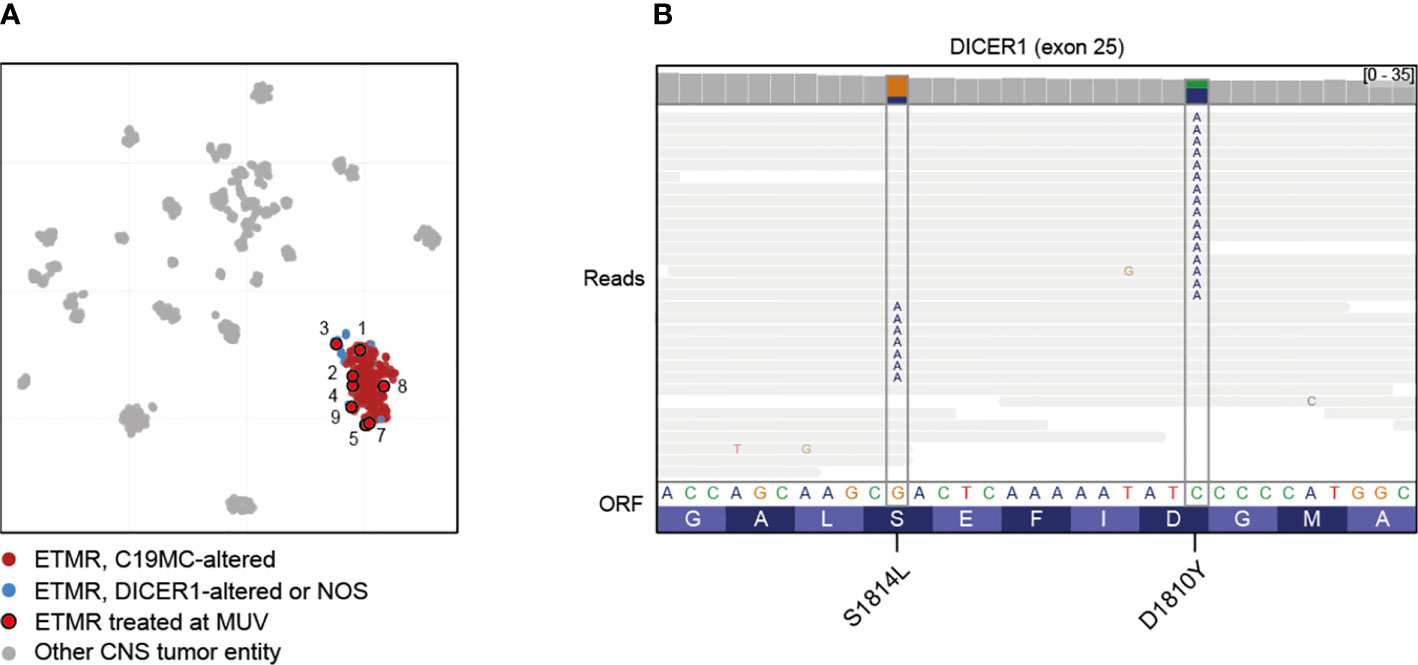
Figure 1 Molecular characterization of treated cases. (A) Clustering of ETMRs treated at MUV using t-Stochastic neighborhood embedding with 193 ETMRs (12), 534 other CNS tumors (37) and 44 pineoblastomas (13). Case 6 was not included due to low tumor content. (B) Mutual exclusivity of mutations affecting DICER1 exon 25 in case 3, suggesting that the mutations are biallelic. Lines represent reads; substitutions are annotated in the reads at the appropriate position.
Response to Systemic Treatment
Only two patients who after GTR received radiotherapy very early on (six weeks after diagnosis) following two courses of PEI are alive and in complete remission 56 and 50 months after diagnosis. All remaining patients in whom radiotherapy was deferred until the end of chemotherapy recurred. In cases with an initial GTR relapse occurred after a median of six months (range 9 weeks–11 months) and the patients succumbed to their disease after a median of 14 months (Table 1). In the two patients with an initial biopsy and PR, respectively, the tumor progressed steadily as evidenced in six-weekly MRIs despite intensive therapy. Both received a very good PR and STR before focal radiotherapy concomitant and followed by temozolomide and both also died of tumor recurrence/progression 11 and 14 months after initial diagnosis.
Response to Intrathecal Therapy
All except the patient with hypoxic brain injury received intrathecal therapy. Intrathecal chemotherapy consisted of etoposide in all eight patients alternating with liposomal cytarabine (Depocyte) every two weeks in five patients. Two patients received only etoposide every two weeks and one patient etoposide alternating with aqueous cytarabine on days 1, 4, 8 and 11 because production of the slow release formulation was discontinued. Two patients also received topotecan twice a week on days 1 and 4 and one patient originally diagnosed as PNET intrathecal methotrexate according to HIT 2000 SKK (12) (Table 1). Despite fatal local recurrences none of the patients developed leptomeningeal disease.
Radiotherapy
Radiotherapy started six weeks after GTR in the two long-term survivors and 3.5 to 12 months after diagnosis in the remainder patients (Table 1). At the time of initiation of radiotherapy only three (cases 2,6, and 7) of the seven patients had not progressed under chemotherapy including the two long-term survivors who were irradiated early, six weeks after diagnosis.
Response to Second Line Treatment
Three patients who received temozolomide following second look surgeries for recurrence progressed. Likewise, three patients who received an antiangiogenic metronomic therapy according to the MEMMAT strategy for their progression/relapse succumbed to their disease. One patient (case 9) received topotecan and olaparib for his second recurrence/progression. Olaparib required discontinuation only for a few days per cycle when the white blood cell count dropped below 1,000/mm3. However, he too progressed under this combination and died (Supplementary Table S1).
Neuropsychological Outcome and HRQoL
Both long-term survivors were last tested 3.6 years after diagnosis (case 6 with 7.1 years and case 7 with 5.75 years). Fluid intelligence was within the average range for both patients, as most of the other domains (26, 28, 29, 32, 33, 35). Case 6 had difficulties in fine motor functions (ETMR was located in the right parietal lobe) (30, 31). Case 7 with a bifrontal tumor showed below average results for processing speed, long-term memory, working memory and emotion recognition (Supplementary Figure S10) (24–27).
The behavioral screening (proxy reports by mothers of the patients) resulted in close to average scores in nearly all domains for both patients.
With respect to HRQoL both patients showed average scores in most of the subscales and also in the total score.
Discussion
ETMR is a rare highly malignant embryonal tumor of young children that emerged as a new entity comprising the former entities ETANTR, ependymoblastomas, and medulloepitheliomas based on overlapping molecular features.
We here describe a consecutive series of nine patients with ETMR treated at our institution since 2006. ETMR was confirmed molecularly in all patients with eight of the tumors harboring a C19MC amplification. The single tumor lacking the C19MC amplification displayed a DICER1 mutation and clustered with other ETMRs (C19MC altered or NOS) (11). Seven had a primary GTR and two a very good second look PR and STR, respectively. All patients received immediate postoperative conventional chemotherapy followed by HDCT in three. Similarly, radiotherapy was part of the therapeutic concept and originally planned to be deferred until the end of chemotherapy. To prevent leptomeningeal seeding all but one patient (hypoxic brain injury) also received intrathecal therapy (Table 1). Interestingly, only the two patients who received GTR followed by early focal radiotherapy are long-term survivors and in excellent clinical condition.
Reviewing the literature in the context of their own series several authors described GTR as favourable prognostic factor (8, 17, 39, 40). Alexiou et al. analyzing 41 cases with available survival data concluded that patients receiving GTR or STR had a survival benefit compared to patients in whom only biopsy was performed (14 vs. 6 months) (39). While Alexiou found no statistically significant difference in the survival time between GTR and STR (14 vs 12 months), Mozes et al. in an analysis of seven relapsed cases stressed that there was a trend towards a somewhat faster recurrence among the four patients with STR (10.3 ± 0.85 months) vs. the three patients with GTR (14.0 ± 8.08 months) (17). Horwitz et al. reported on the French experience including 38 patients with ETMR (8). Thirty of these 38 were treated homogenously according to the PNET-HR protocol (41) and progression was observed in all patients with a STR during induction standard therapy (2 courses of etoposide-carboplatin) or sequential high-dose chemotherapy (melphalan/cisplatin/melphalan/cisplatin/thiotepa), highlighting the importance of a GTR. In our series both patients with biopsy and PR, respectively, demonstrated steady tumor progression as evidenced by six-weekly MRIs despite intensive chemotherapy.
Similar to the importance of GTR, multiple authors described a statistically significant overall survival (OS) benefit in ETMR with the addition of radiotherapy in uni- and multivariate analysis (8, 17, 39, 42, 43). Recently, Jaramillo et al. reported on seven patients with ETMR treated with proton therapy (42). Interestingly, three of his seven patients were alive >36 months after diagnosis.
Since only the two patients who received focal radiotherapy early on became long-term survivors in our series we reviewed the literature with regard to the role of initiation/timing and extent of radiotherapy in the treatment of ETMR published since 1990. The search yielded 228 cases published in peer reviewed journals providing clinical information. Including our nine patients 79 patients had a GTR, 61 a STR, 17 a PR or biopsy and in the remainder extent of resection was not known. Metastases were present at diagnosis in 30 (13%) and not reported in 14; 188 (79%) patients received chemotherapy including HDCT in 60, and 93 (39%) received radiotherapy as part of their treatment. Radiotherapy was performed early, delayed, at recurrence or not known in 19, 36, 20 and 18, respectively. Extent of radiotherapy was not known in 15, craniospinal (CSI) irradiation performed in 30 and focal radiotherapy in 48. Only 26 (11%) of 237 patients survived >36 months with no evidence of disease at last follow-up. ETMR was confirmed by LIN28A expression and/or C19MC amplification in 14 of the 26 long-term survivors (Table 2). Nineteen of the 26 had a GTR, four a STR and in three extent of resection was not available. All but two long-term (>36 months) survivors received radiotherapy. Radiotherapy was CSI in 10, focal in 11, and not known in three. Ten of the survivors were irradiated early on following GTR, two of whom received only radiotherapy (Table 2, patient numbers 1, 4, 9, 11, 12, 14, 23–26). Thirteen received radiotherapy after intensive chemotherapy including HDCT in nine and at least four (Table 2, patient numbers 2, 17, 19, and 22) of these 13 patients had progressed before radiotherapy. For one survivor type of therapy is not known.
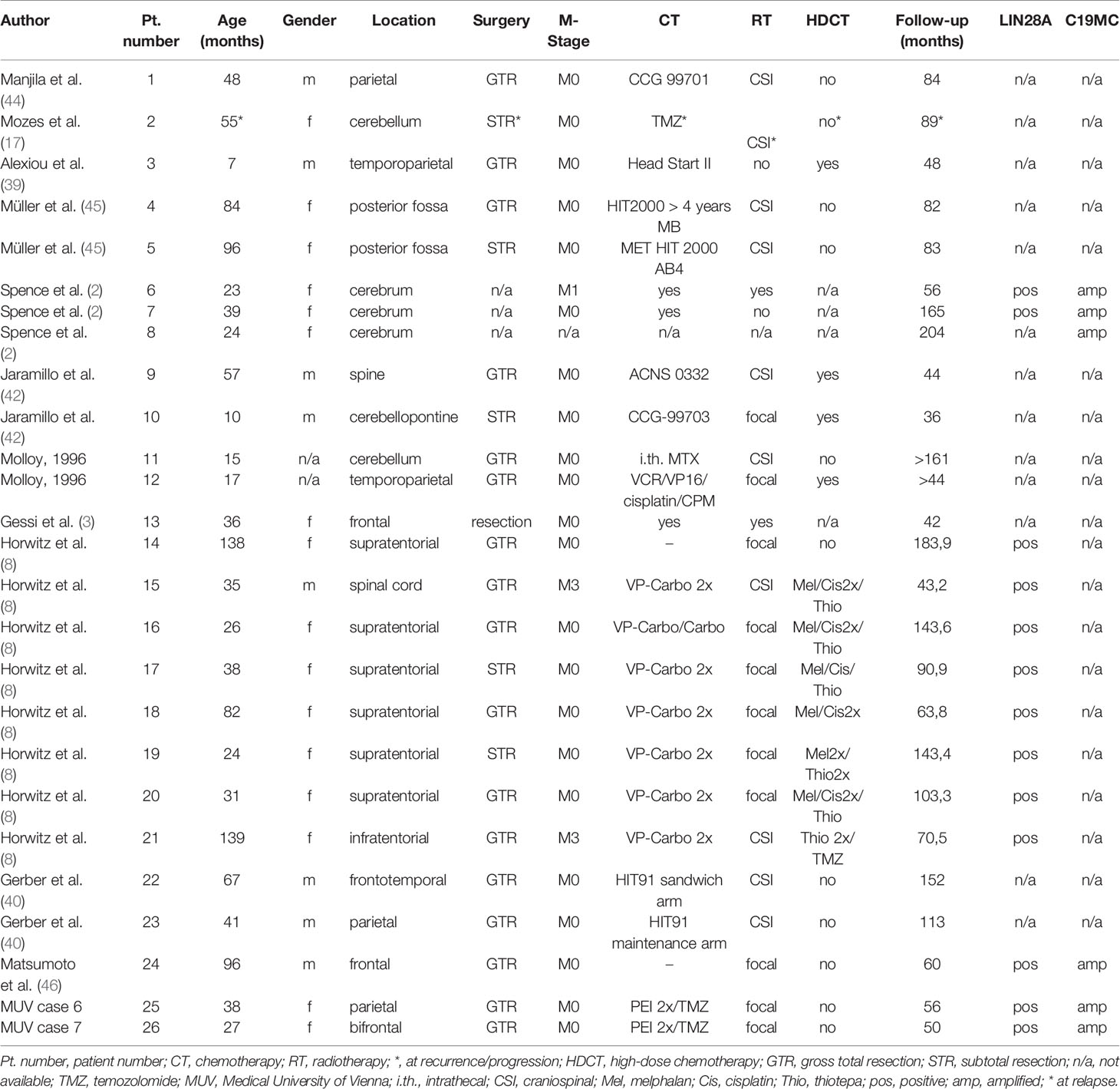
Table 2 Characteristics of all patients with no evidence of disease >36 months.
While GTR and radiation therapy were described as favourable prognostic factors by multiple authors, the impact of HDCT on survival is less clear. Alexiou et al. described a trend towards a better survival in patients receiving HDCT (39). Horowitz et al. reporting on the French series of 38 patients with ETMR found that with decreasing significance GTR, radiotherapy and HDCT, respectively, were associated with a better overall survival (8). Of the two only ones surviving patients who did not receive radiotherapy one received HDCT and in the other information was not available (Table 2).
Except for one patient who died after HDCT all patients in our series in whom radiotherapy was deferred recurred despite intensive chemotherapy and succumbed to their disease. Recently, Lambo et al. reported on the genomic landscape of ETMRs and found that relapsed tumors have a large increase in somatic single nucleotide variants (SNVs) compared to primary tumors (12). Many of these newly acquired SNVs were found to be associated with a mutational signature related to platinum-based agents, suggesting that they were induced by treatment. These findings may explain the fast evolution of resistance to chemotherapy in these patients and emphasize the importance for early use of radiotherapy. The same authors also described a prevalent genomic instability in ETMR caused by widespread occurrence of R-loop structures and demonstrated that topoisomerase 1 (TOP1) inhibitors and Poly ADP-ribose polymerase (PARP) inhibitors act synergistically in increasing the amount of DNA damage by increasing the R-loops in ETMR cells. Based on these findings we used a combination of topotecan and olaparib in one of our patients (case 9) for his second recurrence/progression. Treatment at the chosen dose was well tolerated and could be repeated every three weeks. Unfortunately, the combination treatment was not effective in this recurrent tumor. It is possible that the effect of topotecan could not be enhanced by olaparib, the only PARP inhibitor available to us at the time, because of its poor CNS penetration.
Based on the literature search including our own patients the median age of patients at presentation was 37 months (range 0 to 276), 53 (22%) were below the age of 18 months and 23 (10%) below 12 months. Only 30 of 237 (13%) were metastatic at diagnosis. This would allow for confining radiotherapy to the tumor bed in the majority of patients if given early on. Recently, encouraging results of long-term HRQoL were reported for 59 pediatric brain tumor survivors receiving proton radiotherapy at a median age of 2.5 years (range 0.3–3.8 years) (47). While the data for HRQoL outcomes were variable and significantly associated with the severity of neurologic injury at the time of diagnosis and treatment, approximately one third of the patients achieved HRQoL scores comparable to those of healthy children, and 90% of children functioned in a regular classroom. This is in accordance with the observation in our two surviving patients whose neuropsychological outcome and HRQol were within the average range in most domains 3.6 years after diagnosis. Case 6 is the best student of her class in elementary school. Concerns over negative sequelae of radiation therapy in young children may therefore be alleviated to a certain extent by an advanced radiotherapy approach such as proton therapy, in particular if only focal therapy with a markedly reduced radiation exposure to the surrounding brain is used. Taking into account area and size of the treatment volume a possible cut-off age of 18 months may be discussed. It has to be noted though that both our patients with a positive neuropsychological outcome were 38 and 27 months old at diagnosis. Nevertheless future protocols or guidelines recommending early focal radiotherapy following for example two cycles of chemotherapy may be suitable for the majority of patients given the median age of presentation for ETMRs of 37 months. Whether the concomitant use of temozolomide to radiotherapy was of additional benefit for the patients cannot be determined independently given the small numbers and the fact that two long-term survivors reported in the literature (8, 44) received focal radiotherapy only. Furthermore, while both patients receiving temozolomide chemoradiotherapy for primary diagnosis are long-term survivors this approach was not effective in two patients following tumor progression under intensive conventional chemotherapy. This is in contrast to the case report by Mozes et al. (17) who reported efficacy of concomitant temozolomide chemoradiotherapy followed by temozolomide cycles in a patient with an ETMR recurrence after intensive chemotherapy including HDCT.
Leptomeningeal spreading in patients receiving focal radiotherapy only might be prevented by intrathecal chemotherapy. In contrast to the French series (8) all our patients recurred locally and none developed leptomeningeal disease suggesting that intrathecal therapy consisting of etoposide alone or etoposide alternating with liposomal or aqueous cytarabine and/or topotecan, is effective although only topotecan is supported by preclinical evidence (48). This is in accordance with an observation by Gessi et al. (3) who described that widespread meningeal metastases resolved transiently in one patient after intrathecal therapy. While early radiotherapy might be an option for the majority of patients, infants <18 months of age, infants with a critical treatment area or volume or primary metastasized tumors will not be eligible for this approach. In these patients early intensive chemotherapy including HDCT possibly followed by maintenance therapy including novel agents might be an option.
Conclusion
ETMR is a very rare, aggressive embryonal tumor with reported survival times averaging 12 months. Because of the median age of 37 months at presentation treatment typically consists of surgery followed by chemotherapy and delayed or no radiotherapy. Our institutional experience and a literature search suggest that GTR and early radiotherapy are potentially pivotal for long-term survival (>36 months) of patients with ETMR in the absence of effective targeted therapies.
Because only 13% of the patients presented with metastatic disease focal radiotherapy may suffice in the majority of patients if intrathecal therapy is used to prevent leptomeningeal dissemination. The use of advanced radiation technology with reduced exposure of uninvolved brain and reports of a reasonably good HRQol also of young children with proton therapy may mitigate the fear of prohibitive late effects of early focal radiotherapy in the majority of patients.
Data Availability Statement
All data generated or analyzed for this study can be found in the article [and its Supplementary Material] or are publicly available within previous publications by Lambo et al. (12) for 193 ETMRs in the manuscript by Sturm et al. (37) for 534 other CNS tumors, the article by Capper et al. (13) for 44 pineoblastomas. The DNA methylation profiles generated during this study have been deposited in GEO with the accession number GSE160958.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of the Medical University of Vienna. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
LM provided clinical data, reviewed the literature, wrote the first draft of the manuscript, and critically reviewed the manuscript for intellectual content. JG provided input on molecular data, designed figures, and critically reviewed the manuscript for intellectual content. AP provided clinical input on interpretation of the results and critically reviewed the manuscript for intellectual content. AA provided clinical input on interpretation of the results and critically reviewed the manuscript for intellectual content. NS provided clinical input and critically reviewed the manuscript for intellectual content. TP provided data on neuropsychological outcome and health-related quality of life. TC provided clinical input on interpretation of the results and critically reviewed the manuscript for intellectual content. CD provided clinical input on interpretation of the results and critically reviewed the manuscript for intellectual content. SL performed molecular analyses and critically reviewed the manuscript for intellectual content. KD provided data on radiotherapy and critically reviewed the manuscript for intellectual content. CH provided neuropathological data and critically reviewed the manuscript for intellectual content. MK provided data on molecular analyses and critically reviewed the manuscript for intellectual content. IS initiated publication of the series, provided input on interpretation of the results, and completed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Austrian Science Fund (I 4164 to J.G.) within the TRANSCAN-2 project “BRCAddict”. SL and MK are supported by the Solving Kid's Cancer foundation and the Bibi Fund for Childhood Cancer Research.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank all patients, their families, and clinical staff for their contributions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.584681/full#supplementary-material
Abbreviations
ETMR, Embryonal tumors with multilayered rosettes; ETANTR, Embryonal tumor with abundant neuropil and true rosettes; CNS, Central nervous system; PNET, Primitive neuroectodermal tumor; WHO, World Health Organisation; NOS, Not otherwise specified; HDCT, High dose chemotherapy; MUV, Medical University of Vienna; GTR, Gross total resection; STR, Subtotal resection; PR, Partial resection; RANO, Response assessment in neuro-oncology; IHC, Immunohistochemical; iFISH, Interphase fluorescence in situ hybridization; FFPE, Formalin-fixed paraffin-embedded tissue; ATRT, Atypical teratoid rhabdoid tumor; IP-CZD, Polish infant protocol; Depocyte, Liposomal cytarabine; 3D, Three dimensional; GTV, Gross tumor volume; CTV, Clinical target volume; PTV, Planning target volume; VMAT, Volume mediated radiotherapy; MEMMAT, (MEMMAT;ClinicalTrials.gov Identifier: NCT01356290); BID, Bis in die (two times per day); PNET-HR protocol, Primitive neuroectodermal tumor high-risk protocol; HRQoL, Health-related quality of life; MRI, Magnetic resonance imaging; CNV, Copy number variations; OS, Overall survival; CSI, Craniospinal irradiation; SNV, Single nucleotide variants; TOP1, Topoisomerase 1; PARP, Poly (ADP-ribose) polymerase.
References
1. Korshunov A, Sturm D, Ryzhova M, Hovestadt V, Gessi M, Jones DTW, et al. Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity. Acta Neuropathol (Berl) (2014) 128:279–89. doi: 10.1007/s00401-013-1228-0
2. Spence T, Sin-Chan P, Picard D, Barszczyk M, Hoss K, Lu M, et al. CNS-PNETs with C19MC amplification and/or LIN28 expression comprise a distinct histogenetic diagnostic and therapeutic entity. Acta Neuropathol (Berl) (2014) 128:291–303. doi: 10.1007/s00401-014-1291-1
3. Gessi M, Giangaspero F, Lauriola L, Gardiman M, Scheithauer BW, Halliday W, et al. Embryonal tumors with abundant neuropil and true rosettes: a distinctive CNS primitive neuroectodermal tumor. Am J Surg Pathol (2009) 33:211–7. doi: 10.1097/PAS.0b013e318186235b
4. Pfister S, Remke M, Castoldi M, Bai AHC, Muckenthaler MU, Kulozik A, et al. Novel genomic amplification targeting the microRNA cluster at 19q13.42 in a pediatric embryonal tumor with abundant neuropil and true rosettes. Acta Neuropathol (Berl) (2009) 117:457–64. doi: 10.1007/s00401-008-0467-y
5. Paulus W, Kleihues P. Genetic profiling of CNS tumors extends histological classification. Acta Neuropathol (Berl) (2010) 120:269–70. doi: 10.1007/s00401-010-0710-1
6. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol (Berl) (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
7. Kram DE, Henderson JJ, Baig M, Chakraborty D, Gardner MA, Biswas S, et al. Embryonal Tumors of the Central Nervous System in Children: The Era of Targeted Therapeutics. Bioengineering (2018) 5(4):78. doi: 10.3390/bioengineering5040078
8. Horwitz M, Dufour C, Leblond P, Bourdeaut F, Faure-Conter C, Bertozzi A-I, et al. Embryonal tumors with multilayered rosettes in children: the SFCE experience. Childs Nerv Syst (2016) 32:299–305. doi: 10.1007/s00381-015-2920-2
9. Wang B, Gogia B, Fuller GN, Ketonen LM. Embryonal Tumor with Multilayered Rosettes, C19MC-Altered: Clinical, Pathological, and Neuroimaging Findings. J Neuroimaging (2018) 28:483–9. doi: 10.1111/jon.12524
10. Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol (2010) 28:1963–72. doi: 10.1200/JCO.2009.26.3541
11. Woehrer A, Slavc I, Peyrl A, Czech T, Dorfer C, Prayer D, et al. Embryonal tumor with abundant neuropil and true rosettes (ETANTR) with loss of morphological but retained genetic key features during progression. Acta Neuropathol (Berl) (2011) 122:787–90. doi: 10.1007/s00401-011-0903-2
12. Lambo S, Gröbner SN, Rausch T, Waszak SM, Schmidt C, Gorthi A, et al. The molecular landscape of ETMR at diagnosis and relapse. Nature (2019) 576:274–80. doi: 10.1038/s41586-019-1815-x
13. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation-based classification of central nervous system tumours. Nature (2018) 555:469–74. doi: 10.1038/nature26000
14. von Bueren AO, von Hoff K, Pietsch T, Gerber NU, Warmuth-Metz M, Deinlein F, et al. Treatment of young children with localized medulloblastoma by chemotherapy alone: results of the prospective, multicenter trial HIT 2000 confirming the prognostic impact of histology. Neuro-Oncol (2011) 13:669–79. doi: 10.1093/neuonc/nor025
15. Slavc I, Chocholous M, Leiss U, Haberler C, Peyrl A, Azizi AA, et al. Atypical teratoid rhabdoid tumor: improved long-term survival with an intensive multimodal therapy and delayed radiotherapy. The Medical University of Vienna Experience 1992-2012. Cancer Med (2014) 3:91–100. doi: 10.1002/cam4.161
16. Dhall G, Grodman H, Ji L, Sands S, Gardner S, Dunkel IJ, et al. Outcome of children less than three years old at diagnosis with non-metastatic medulloblastoma treated with chemotherapy on the “Head Start” I and II protocols. Pediatr Blood Cancer (2008) 50:1169–75. doi: 10.1002/pbc.21525
17. Mozes P, Hauser P, Hortobágyi T, Benyó G, Peták I, Garami M, et al. Evaluation of the good tumor response of embryonal tumor with abundant neuropil and true rosettes (ETANTR). J Neurooncol (2016) 126:99–105. doi: 10.1007/s11060-015-1938-3
18. Slavc I, Schuller E, Falger J, Günes M, Pillwein K, Czech T, et al. Feasibility of long-term intraventricular therapy with mafosfamide (n = 26) and etoposide (n = 11): experience in 26 children with disseminated malignant brain tumors. J Neurooncol (2003) 64:239–47. doi: 10.1023/a:1025633704071
19. Pajtler KW, Tippelt S, Siegler N, Reichling S, Zimmermann M, Mikasch R, et al. Intraventricular etoposide safety and toxicity profile in children and young adults with refractory or recurrent malignant brain tumors. J Neurooncol (2016) 128:463–71. doi: 10.1007/s11060-016-2133-x
20. Peyrl A, Sauermann R, Traunmueller F, Azizi AA, Gruber-Olipitz M, Gupper A, et al. Pharmacokinetics and safety of intrathecal liposomal cytarabine in children aged <3 years. Clin Pharmacokinet (2009) 48:265–71. doi: 10.2165/00003088-200948040-00004
21. Peyrl A, Sauermann R, Chocholous M, Azizi AA, Jäger W, Höferl M, et al. Pharmacokinetics and toxicity of intrathecal liposomal cytarabine in children and adolescents following age-adapted dosing. Clin Pharmacokinet (2014) 53:165–73. doi: 10.1007/s40262-013-0106-1
22. Potter SLP, Berg S, Ingle AM, Krailo M, Adamson PC, Blaney SM. Phase 2 clinical trial of intrathecal topotecan in children with refractory leptomeningeal leukemia: a Children’s Oncology Group trial (P9962). Pediatr Blood Cancer (2012) 58:362–5. doi: 10.1002/pbc.23317
23. Cairo MS, Sposto R, Gerrard M, Auperin A, Goldman SC, Harrison L, et al. Advanced stage, increased lactate dehydrogenase, and primary site, but not adolescent age (≥ 15 years), are associated with an increased risk of treatment failure in children and adolescents with mature B-cell non-Hodgkin’s lymphoma: results of the FAB LMB 96 study. J Clin Oncol (2012) 30:387–93. doi: 10.1200/JCO.2010.33.3369
24. Petermann F, Daseking M. Wechsler Preschool and Primary Scale of Intelligence – Forth Edition (WPPSI-IV) (German version). Frankfurt: Pearson (2018).
25. Petermann F. Wechsler Intelligence Scale for Children – Fifth Edition (WISC-V) (German version). Frankfurt: Pearson (2017).
27. Helmstaedter C, Lendt M, Lux S. Verbaler Lern- und Merkfähigkeitstest (VLMT). Göttingen: Beltz Test (2001).
28. Gleißner U, Krause MP, Reuner G. Fragebogen zur Erfassung kognitiver Prozesse bei 4 – 6 –jährigen Kindern (KOPKI 4 – 6). Göttingen: Hogrefe (2011).
29. Zimmermann P, Fimm B. Testbatterie zur Aufmerksamkeitsprüfung (TAP 2.3.1). Herzogenrath: Psytest (2017).
30. Beery KE, Beery NA, Buktenica NA. Beery- Buktenica Developmental Test of Visual-Motor Integration. 6th Ed. Frankfurt: Pearson (2010).
32. Daseking M, Petermann F. Verhaltensinventar zur Beurteilung exekutiver Funktionen für das Kindergartenalter (BRIEF – P). Göttingen: Hogrefe (2013).
33. Drechsler R, Steinhausen H-C. Verhaltensinventar zur Beurteilung exekutiver Funktionen (BRIEF). Göttingen: Hogrefe (2013).
34. Goodman R, Ford T, Simmons H, Gatward R, Meltzer H. Using the Strengths and Difficulties Questionnaire (SDQ) to screen for child psychiatric disorders in a community sample. Br J Psychiatry (2000) 177:534–9.
35. Grob A, Hagmann-von Arx P. Intelligence and Development Scales (2nd Ed.) (IDS-2). Göttingen: Hogrefe (2018).
36. Ravens-Sieberer U, Bullinger M. Assessing health-related quality of life in chronically ill children with the German KINDL: first psychometric and content analytical results. Qual Life Res (1998) 7(5)399–407.
37. Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D, et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell (2016) 164:1060–72. doi: 10.1016/j.cell.2016.01.015
38. Sin-Chan P, Mumal I, Suwal T, Ho B, Fan X, Singh I, et al. A C19MC-LIN28A-MYCN Oncogenic Circuit Driven by Hijacked Super-enhancers Is a Distinct Therapeutic Vulnerability in ETMRs: A Lethal Brain Tumor. Cancer Cell (2019) 36:51–67.e7. doi: 10.1016/j.ccell.2019.06.002
39. Alexiou GA, Stefanaki K, Vartholomatos G, Sfakianos G, Prodromou N, Moschovi M. Embryonal tumor with abundant neuropil and true rosettes: a systematic literature review and report of 2 new cases. J Child Neurol (2013) 28:1709–15. doi: 10.1177/0883073812471434
40. Gerber NU, von Hoff K, Resch A, Ottensmeier H, Kwiecien R, Faldum A, et al. Treatment of children with central nervous system primitive neuroectodermal tumors/pinealoblastomas in the prospective multicentric trial HIT 2000 using hyperfractionated radiation therapy followed by maintenance chemotherapy. Int J Radiat Oncol Biol Phys (2014) 89:863–71. doi: 10.1016/j.ijrobp.2014.04.017
41. Dufour C, Kieffer V, Varlet P, Raquin MA, Dhermain F, Puget S, et al. Tandem high-dose chemotherapy and autologous stem cell rescue in children with newly diagnosed high-risk medulloblastoma or supratentorial primitive neuro-ectodermic tumors. Pediatr Blood Cancer (2014) 61:1398–402. doi: 10.1002/pbc.25009
42. Jaramillo S, Grosshans DR, Philip N, Varan A, Akyüz C, McAleer MF, et al. Radiation for ETMR: Literature review and case series of patients treated with proton therapy. Clin Transl Radiat Oncol (2018) 15:31–7. doi: 10.1016/j.ctro.2018.11.002
43. Ding D, Zhao A, Qiu B, Xing D, Guan G, Guo Z. Ependymoblastoma with cystic change in a child. J Neurosurg Pediatr (2014) 13:658–65. doi: 10.3171/2014.3.PEDS13405
44. Manjila S, Ray A, Hu Y, Cai DX, Cohen ML, Cohen AR. Embryonal tumors with abundant neuropil and true rosettes: 2 illustrative cases and a review of the literature. Neurosurg Focus (2011) 30:E2. doi:10.3171/2010.10.FOCUS10226
45. Müller K, Zwiener I, Welker H, Maass E, Bongartz R, Berthold F, et al. Curative treatment for central nervous system medulloepithelioma despite residual disease after resection. Report of two cases treated according to the GPHO Protocol HIT 2000 and review of the literature. Strahlenther Onkol (2011) 187(11):757–62. doi: 10.1007/s00066-011-2256-0
46. Matsumoto M, Horiuchi K, Sato T, Oinuma M, Sakuma J, Suzuki K, et al. Cerebral medulloepithelioma with long survival. Neurol Med Chir (Tokyo) (2007) 47:428–33. doi: 10.2176/nmc.47.428
47. Eaton BR, Goldberg S, Tarbell NJ, Lawell MP, Gallotto SL, Weyman EA, et al. Long-term Health Related Quality of Life in Pediatric Brain Tumor Survivors Treated with Proton Radiotherapy at <4 Years of Age. Neuro-Oncol (2020) 47(9):428–33. doi: 10.1093/neuonc/noaa042
Keywords: embryonal tumor with multilayered rosette, embryonal tumor with abundant neuropil and true rosette, radiotherapy, focal radiotherapy, intrathecal therapy, embryonal brain tumors
Citation: Mayr L, Gojo J, Peyrl A, Azizi AA, Stepien NM, Pletschko T, Czech T, Dorfer C, Lambo S, Dieckmann K, Haberler C, Kool M and Slavc I (2020) Potential Importance of Early Focal Radiotherapy Following Gross Total Resection for Long-Term Survival in Children With Embryonal Tumors With Multilayered Rosettes. Front. Oncol. 10:584681. doi: 10.3389/fonc.2020.584681
Received: 17 July 2020; Accepted: 12 November 2020;
Published: 17 December 2020.
Edited by:
Theodore Nicolaides, New York University, United StatesReviewed by:
Stergios Zacharoulis, Columbia University, United StatesAllison Martin, Albert Einstein College of Medicine, United States
Copyright © 2020 Mayr, Gojo, Peyrl, Azizi, Stepien, Pletschko, Czech, Dorfer, Lambo, Dieckmann, Haberler, Kool and Slavc. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irene Slavc, aXJlbmUuc2xhdmNAbWVkdW5pd2llbi5hYy5hdA==