 Tashbib Khan1
Tashbib Khan1 Mitchell A. Sullivan1
Mitchell A. Sullivan1 Jennifer H. Gunter2
Jennifer H. Gunter2 Thomas Kryza1Nicholas Lyons1Yaowu He1
Thomas Kryza1Nicholas Lyons1Yaowu He1 John D. Hooper1*
John D. Hooper1*- 1Mater Research Institute—The University of Queensland, Translational Research Institute, Woolloongabba, QLD, Australia
- 2Faculty of Health, Australian Prostate Cancer Research Centre-Queensland, School of Biomedical Sciences, Institute of Health and Biomedical Innovation, Translational Research Institute, Queensland University of Technology, Woolloongabba, QLD, Australia
Once thought to be exclusively a storage hub for glucose, glycogen is now known to be essential in a range of physiological processes and pathological conditions. Glycogen lies at the nexus of diverse processes that promote malignancy, including proliferation, migration, invasion, and chemoresistance of cancer cells. It is also implicated in processes associated with the tumor microenvironment such as immune cell effector function and crosstalk with cancer-associated fibroblasts to promote metastasis. The enzymes of glycogen metabolism are dysregulated in a wide variety of malignancies, including cancers of the kidney, ovary, lung, bladder, liver, blood, and breast. Understanding and targeting glycogen metabolism in cancer presents a promising but under-explored therapeutic avenue. In this review, we summarize the current literature on the role of glycogen in cancer progression and discuss its potential as a therapeutic target for cancer treatment.
Introduction
Glycogen is a highly branched polymer of glucose that is used for the efficient storage and release of energy (1). This function is highlighted by the importance of glycogen in organs such as the brain, liver, heart, and muscle (2–5). With critical roles as a feedstock for respiration and biomolecule biosynthesis, it is unsurprising that glycogen also plays an active role in human pathology, driving certain epilepsies in the brain, and contributing to diabetes-induced kidney damage and severe muscle wasting disorders (2, 6, 7). Glycogen is also implicated in malignancy, and glycogen-based metabolic reprogramming has recently received renewed attention as a targetable phenotype in the tumor microenvironment (8, 9).
Figure 1 summarizes key aspects of glycogen metabolism and overlays inhibitors that have been employed to disrupt it in disease processes. The first step of glycogen synthesis involves glucose entry into cells via glucose transporters. Upon entry, glucose is phosphorylated to glucose-6-phosphate (G-6-P) by hexokinase (HK), followed by transfer of this phosphate group to carbon 1 by phosphoglucomutase (PGM). The resultant glucose-1-phosphate (G-1-P) is converted to uridine diphosphate glucose (UDP)-glucose by UDP-glucose pyrophosphorylase (UPP). Then, dimeric glycogenin (GYG), a specialized primer of glycogen synthesis, auto-glucosylates UDP-glucose to generate an initial chain of (1 → 4)-α linked glucose units on each of the two glycogenin monomers. After auto-glucosylation, glycogen synthase (GS) elongates these initial chains and creates further (1 → 4)-α glyosidic linkages, recruiting more UDP-glucose as the substrate (9, 10). The creation of branchpoints [(1 → 6)-α glycosidic linkages] is catalyzed by glycogen branching enzyme (GBE).
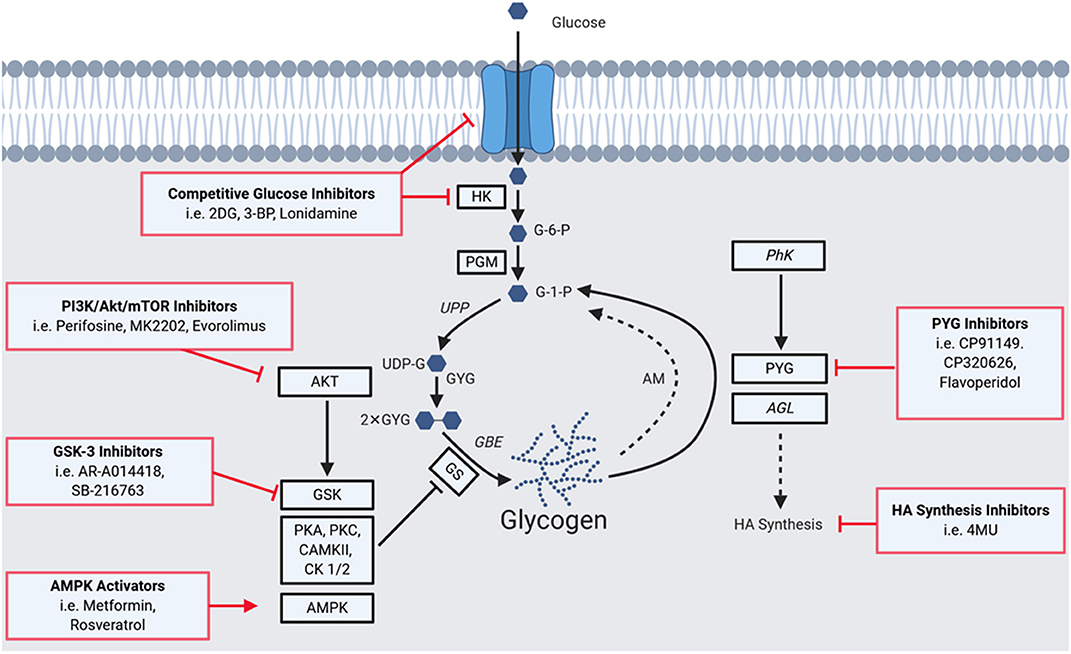
Figure 1. A Summary of the glycogen metabolism axis, including agents that pharmacologically modulate the axis. Glucose enters cells via glucose transporters (GLUTs), where it is phosphorylated to glucose-6-phosphate (G-6-P) by hexokinase (HK), followed by a transfer of the phosphate group from carbon 6 to 1 by phosphoglucomutase (PGM) to yieldglucose-1-phosphate (G-1-P). The resultant G-1-P is converted to UDP-glucose (UDP-G) by UDP-glucose pyrophosphorylase (UPP). This is then used by glycogenin (GYG; in dimerized form), a specialized primer of glycogen synthesis, to auto-glucosylate and extend its chain. After auto-glucosylation, glycogen synthase (GS) elongates these initial chains and creates further α-1,4-glycosidic linkages. Branchpoints of α-1,6-glycosidic linkages are mediated by the glycogen branching enzyme (GBE). GS is heavily regulated allosterically and by a network of kinases. Phosphorylation of GS decreases its activity and hence the rate of glycogen synthesis. GS has phosphorylation sites for Glycogen Synthase Kinase 3 (GSK3), protein kinase A (PKA), protein kinase C (PKC), calmodulin-dependent protein kinase II (CaMKII), AMP-activated protein kinase (AMPK), casein kinase 1 (CK1), and casein kinase 2 (CK2). GS is also positively regulated: allosterically by glucose-6-phosphate, and via dephosphorylation by protein phosphatase 1, regulatory subunit 3 (PPP1R3C). Phosphorylation can also mediate activity with phosphorylase kinase (PhK) activating the enzyme and driving glycogenolysis. A proportion of glycogen is also degraded via the lysosomal autophagy route by the enzyme acid maltase (AM). Enzymes of the pathway implicated in tumorigenesis are highlighted by black squares, those implicated in tumor suppressive roles (PhK and AGL) are italicized. Red boxes indicate means of pharmacologically modulating the axis, highlighting glycogen-targeted drugs that have already been used for clear cell cancers. Glucose entry and processing can be inhibited by 2-deoxy-D-glucose (2DG), 3-bromopyruvate (3BP), and lonidamine. AKT and GSK which inactivate GS by phosphorylation can be targeted with GSK3B inhibitors (e.g., AR-A014418 and SB-2216763), as well as PI3K (Perifosine), AKT (MK2202) and mTOR (Everolimus) inhibitors. AMPK can be activated to promote glycogen synthesis, as has been performed with metformin and resveratrol. PYG has been targeted using inhibitors such as CP91149, CP320626, and flavoperidol, to prevent glycogen mobilization. Furthermore, the downstream effects of AGL on hyaluronic acid synthesis can be disrupted using 4-methylumbelliferone (4MU).
Glycogen can be degraded to release glucose via two distinct pathways. The first occurs in the cytoplasm and involves the enzymes glycogen phosphorylase (PYG) and glycogen debranching enzyme (AGL). The second is an autophagic lysosomal route via the enzyme acid maltase that recycles ~10% of glycogen, the dysfunction of which gives rise to glycogen storage disease type II, known as Pompe's disease (9, 10).
The enzymes that mediate the rate-limiting steps of glycogen metabolism are GS, for synthesis, and PYG, for degradation (9, 10). GS is negatively regulated by an extensive network of kinases including GS kinase 3 (GSK3), protein kinase A (PKA), protein kinase C (PKC), calmodulin-dependent protein kinase II (CaMKII), AMP-activated protein kinase (AMPK), casein kinase 1 (CK1), and casein kinase 2 (CK2) (2, 9). Conversely, GS is positively regulated by allosteric mechanisms including by the allosteric stimulator G-6-P and via its dephosphorylation by protein phosphatase 1 regulatory subunit 3C (2, 9).
PYGs catalyse the first step of glycogen degradation, glycogenolysis, converting glycogen to G-1-P, which is transformed to G-6-P for energy production. There are three PYG isoforms, that display restricted expression patterns: PYGL (liver), PYGM (muscle) and PYGB (brain). PYG exists as a heterodimer with multiple activity states, and activity can be regulated by both negative and positive allosteric effectors at regulatory sites on each monomer. Phosphorylation can also modulate activity with phosphorylase kinase (PhK) activating the enzyme and driving glycogenolysis (9, 10).
The present work details the mechanisms by which glycogen metabolism is hijacked during cancer progression and summarizes the latest data on visualizing, quantifying, and targeting aberrant glycogen metabolism in cancer.
Reprogrammed Glycogen Metabolism in Cancer
The contributions of glycogen metabolism to cancer progression are summarized in Figure 2. The importance of glycogen in carcinogenesis was suggested by a 1981 study demonstrating that it accumulates in a range of carcinomas at levels higher than in surrounding tissues (11). PYG, the key enzyme in glycogenolysis, and upstream and downstream mediators of glycogen degradation, are associated with poor prognosis and diverse malignant phenotypes in a range of cancers (9, 12–17). Interestingly, other enzymes involved in glycogenolysis are thought to have tumor-suppressive roles. This includes the glycogen debranching enzyme AGL which had a suppressing function in models of bladder and lung cancers, and the kinase PhK β-subunit (PHKB) which suppressed models of hepatocellular carcinoma (18–21). Interestingly, these tumor suppressive roles are potentially independent of glycogen metabolism (19, 21). In the case of AGL, this involves the maintenance of amino acid homeostasis and the nucleotide precursor pool, whilst PHKB negatively regulates AKT and STAT3 signaling (18, 19).
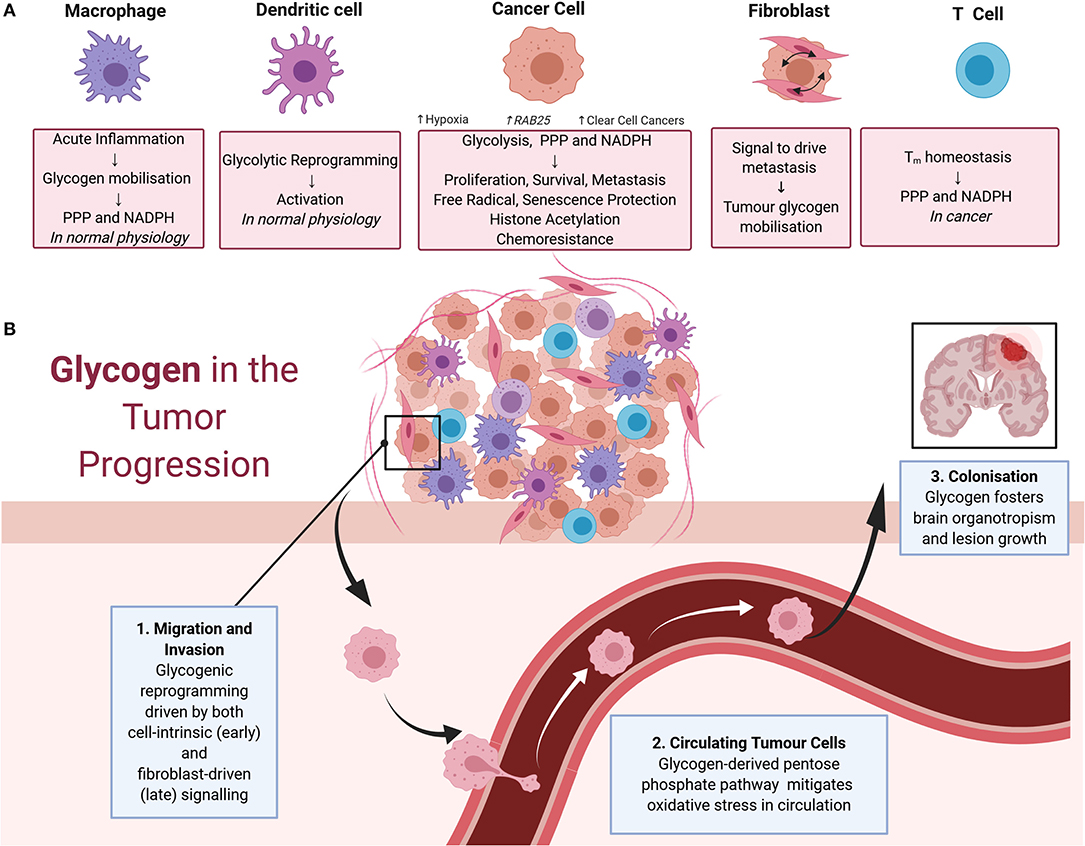
Figure 2. (A) Impacts of glycogen on cells of the tumor microenvironment. In macrophages, physiologically acute inflammation causes glycogen mobilization as a feedstock for the pentose phosphate pathway and NADPH production. Glycogen also drives the early stages of dendritic cell activation via glycolytic reprogramming. These physiological processes are also thought to contribute to cancer progression. In cancer cells, glycogen fuels glycolysis, the pentose phosphate pathway, and NADPH production to support cell proliferation, survival, metastasis, free radical protection, cell senescence protection, histone acetylation, and chemoresistance. Tumor glycogen metabolism increases in a variety of scenarios, including in hypoxic conditions and via the induction of the RAB25 oncogene. Tumor glycogen metabolism is also ubiquitously high in clear cell cancers. In cancer associated fibroblasts, bi-directional signaling between fibroblasts, and neighboring cancer cells drives tumor glycogen mobilization to fuel metastasis. For T cells, glycogen stores consumed via the pentose phosphate pathway for NADPH production are essential for CD8+ memory T cell survival. (B) Glycogen in tumor progression. Glycogen mobilization is implicated in metastasis including as fuel for: (1) migration and invasion of cancer cells in the early stages of metastasis with crosstalk between fibroblasts and malignant cells also eliciting glycogenolysis to fuel migration and invasion; (2) the pentose phosphate pathway in circulating malignant cells facilitating detoxification of reactive oxygen species and promotion of survival; and (3) colonization of distant sites including in the survival of brain metastases.
The following paragraphs detail examples of the causes and phenotypic consequences of altered glycogen metabolism in cancer.
Common Mediators of Tumor Glycogenesis
Even before molecular studies on the roles of glycogen in cancer, it was apparent from histological analysis that a sub-set of carcinomas are enriched in glycogen. The malignant cells of these glycogen-enriched tumors display clear cytoplasm that is vacuolated due to the loss of glycogen that results from histological processing (10, 22). These “clear cell” malignancies include cancers of the kidney, ovary, breast, endometrium, and colorectum (23–26). The two most common clear cell cancers—renal and ovarian—have been well-characterized molecularly and histologically and provide useful systems to interrogate the roles of glycogen in tumorigenesis (26–28).
Glycogenesis occurs in response to diverse physiological and pathological stimuli including oxygen and nutrient deprivation (1, 3, 6, 29, 30). In the normal brain, both glial and neuronal glycogen are protective against hypoxia-induced death (29–31). In cancer, the enzymes of both glycogen synthesis and catabolism rise sharply in response to oxygen and nutrient deficiency, suggesting that both glycogenesis and glycogenolysis are important as tumors evolve in response to local and systemic challenges (32, 33). Key mediators of these responses are hypoxia-inducible factors (HIFs) which regulate tumor survival, metastasis, and therapy resistance (34, 35). For clear cell renal cell carcinoma aberrant hypoxic signaling is initiated by loss of the Von Hippel Lindau (VHL) gene, a negative regulator of HIFs. For ovarian clear cell carcinoma, which arises in the hypoxic environment of endometriotic cysts, the molecular aberrations initiating the hypoxic response remain to be elucidated (22, 36–38). Interestingly, oncogene amplification can also be an important mediator in altered tumor glycogenesis. An example is amplification of the RAB25 gene, which occurs in subtypes of ovarian, renal, prostate, liver, and breast cancer (39, 40). The RAB25 protein is a small GTPase involved in endosomal recycling which interacts with AKT to drive glycogenesis, generating tumor glycogen that can be mobilized in settings of nutrient stress (39). At least in ovarian cancer, RAB25 regulates HIF-1α activity in an oxygen-independent manner, suggesting that RAB25 lies upstream to hypoxia-based induction of glycogen metabolism (41). Despite differences in the molecular initiators and drivers of HIF activity, clear cell cancers employ common mediators of the glycogenesis pathway during malignant transformation, including RAB25, GLUT1, AKT, GSK3, and AMPK, that represent a “glycogen addicted” signature (22, 38, 42, 43).
Glycogen Driving Carcinogenesis
Several recent molecular studies have contributed to elucidating the functions of glycogen in cancer. A seminal loss of function genetic study targeting PYG in models of glioblastoma, breast, and colon cancer demonstrated that glycogen catabolism is a key driver of cancer cell proliferation, survival, and protection from hypoxia-induced free radicals (12). Depletion of the PYG liver isoform PYGL induced premature cell senescence via a ROS-dependent mechanism, and reduced glucose input into the pentose phosphate pathway (PPP) (10, 12). Cell accumulation of glycogen granules is a hallmark of several physiological processes associated with senescence, including the replicative senescence of primary human fibroblasts and neuronal and hepatic aging (12, 44, 45). In the context of cancer, senescence is often viewed as a double-edged sword, on one hand driving tumor stasis, but on the other providing a means of therapy resistance, and fostering a microenvironment favoring inflammation, invasion, and angiogenesis (44–48). Further understanding the role of glycogen in cancer senescence may provide a window into modulating this fine balance (49, 50).
Interestingly, PYG via a mechanism involving the E3 ubiquitin ligase malin, can also translocate to the nucleus to drive nuclear glycogenolysis non-small cell in lung cancer, fueling compartmentalized pyruvate production and histone acetylation (51). Aberrant histone acetylation resulting in an altered epigenetic landscape is a hallmark of multiple cancers including non-small cell lung cancer (52, 53). Depletion of malin impairs nuclear glycogenolysis by blocking nuclear translocation of PYG. The resulting accumulation of nuclear glycogen is reversed by re-introduction of malin in lung cancer cells which restores nuclear catabolism of glycogen, increases histone acetylation, and decreases growth of non-small-cell lung cancer xenografts in mice (51). This highlights the importance of glycogen beyond a cytoplasmic glucose reservoir, revealing this macromolecule as a key driver of compartmentalized tumor metabolism.
Glycogen and the Immune Compartment: Potential Pro-tumoral and Anti-tumoral Roles?
Aberrant immune system responses, including those mediated by metabolic reprogramming, are integral to cancer progression (54, 55). As summarized in Figure 2A, dendritic cells (DCs), macrophages, and T cells have been implicated in metabolic reprogramming in cancer. Mobilization of cell-intrinsic glycogen fuels glycolytic reprogramming and subsequent immune activation of DCs (56–58). For macrophages, glycogen metabolism regulates M1-mediated acute inflammatory responses by fueling the pentose phosphate pathway to generate NADPH for survival and also potentially facilitating the breakdown of immunological tolerance in cancer settings (59). Similarly, maintenance of CD8+ memory T (Tm) cells requires glycogenolysis to fuel the pentose phosphate pathway, generate NADPH, and promote oxidative homeostasis (60). Consistent with this role of glycogen metabolism in immune system responses, disruption of glycogenolysis in the Tm cell compartment caused growth of subcutaneous melanoma cell xenografts and reduced survival of tumor-bearing mice (60). Disruption of the glycogen axis is thought to impair nascent anti-tumor immunity to drive cancer progression and also inhibit immunotherapeutic responses mediated by T-cells (61, 62). Here, it is important to note that immune system responses can elicit both pro- and anti-tumoral actions, and metabolic reprogramming underlies this fine balance (54, 63, 64). Thus, it is possible that global disruption of glycogen metabolism as a therapeutic approach will have adverse consequences for cancer patients because of unwanted effects on the immune compartment. However, it appears that this is not an unassailable problem because metabolic targeting has progressed in the past despite effects on both malignant cells and the immune system. For example, inhibitors of the monocarboxylate transporter MCT1 have progressed to clinical trials despite early concern about deleterious effects on the T cell division that is essential for effective immune response (65, 66). In fact, MCT1 inhibitors have since been shown to improve the infiltration of anti-tumor immune cells (67). Similar progress with glycogen-directed therapies will require a greater understanding of the distinguishing metabolic features of immune and malignant cells, with the key perhaps lying in developing rational combinations of therapeutic agents that selectively synergize in cancer cells (54).
Glycogen as a Metastatic Fuel
Death from cancer is due largely to metastatic rather than primary tumor burden (68, 69). As summarized in Figure 2B, the multistep processes required for metastasis are facilitated to varying degrees by cancer-induced reprogramming of glycogen metabolism. Vascular and lymphatic metastasis involve release of malignant cells from the primary tumor, intravasation, dissemination via the vasculature or lymph, extravasation at secondary sites, establishment of a blood supply, and evasion of internal and external cues to undergo cell death, then thriving in the foreign environment (68–70). The early stages of this metastatic cascade, involving degradation of extracellular matrix (ECM) and local invasion involves HIF-mediated metabolic reprograming that drives acidification of the extracellular space and glycolysis including from glycogen stores (69). In models of breast cancer and osteosarcoma mobilization of glycogen stores by brain glycogen phosphorylase PYGB promotes migration and invasion (71, 72). Also, in pro-metastatic hepatocellular carcinoma, metabolic reprogramming toward glycogenesis, in particular involving UPP, is a key mediator of metastasis (73).
Metastasis also requires the interaction of cancer cells with the cells of the tumor microenvironment including fibroblasts (69). Cancer-associated fibroblasts (CAFs) can mobilize cancer cell glycogen to drive metastasis in models of ovarian cancer (74). It is mediated by the p38α MAPK pathway in CAFs releasing soluble factors to act on malignant cells. This reprogramming event is particularly important in stages of metastasis involving growth of cells in suspension. Cells exposed to death-inducing levels of oxidative stress are protected by ROS-detoxifying metabolites generated via glycogen catabolism feeding the pentose phosphate and one-carbon metabolism pathways, which release NADH, NADPH, and glutathione (74, 75). Glycogen is an important fuel to overcome oxidative stress, allowing cancer cells to successfully survive in suspension during this step of metastasis.
The final stage of metastasis, successful colonization of other organs (68, 69), presents a considerable challenge to malignant cells as the microenvironment at the distant site can be hostile. As a result, metastasizing cancer cells display a propensity to disseminate to particular organs, a relationship called organotropism, that display favorable environments including metabolic requirements (68, 69). There are indications of such an organotrophic relationship between glycogen rich cancers and brain metastases. Notably, glycogenesis sustains glucose-independent growth and survival of breast cancer cells metastasized to the brain (76). In this regard, it was interesting to note that glycogen-rich clear cell carcinoma of the breast displays a higher proportion of brain metastases than other subtypes of breast cancer (23). These findings are perhaps unsurprising, given the roles of glycogen in brain physiology, including neuronal and glial growth, and pathology, including certain epilepsies (31, 77, 78). However, based on the difficulty associated with effective treatment of brain metastasis, targeting of glycogen could perhaps provide a new treatment avenue. There are also suggestions that glycogen mobilization facilitates omental metastases of certain peritoneal tumors, such as ovarian, endometrial serous, colon, gastric, and pancreatic lesions (74), thus inhibition of glycogen catabolism could benefit these patients (79).
Glycogen and Chemoresistance
The importance of glycogen to chemoresistance is highlighted by the observation that glycogen-rich clear cell carcinomas are distinguished by higher chemoresistance than non-clear cell malignancies (43, 80, 81). Clear cell ovarian carcinomas are generally more resistant to conventional platinum and taxol-based chemotherapies compared to other ovarian cancer histotypes, and clear cell renal cell carcinomas rapidly acquire resistance to standard of care tyrosine kinase inhibitors as well as other targeted therapies (43, 81–83). Consistent with this, increased activity of key mediators of glycogen metabolism (glucose, AKT, GSK, and AMPK) is strongly associated with chemoresistance (84–86). Mechanistically, it appears that glycogen-derived metabolites contribute to chemotherapy resistance via a variety of mechanisms, including via ATP-based drug efflux and ROS protection, however more work is required to understand these processes (87).
Visualizing and Quantifying Tumor Glycogen
The size and abundance of glycogen facilitate its visualization and use to delineate glycogen-dependent tumors. Hematoxylin and eosin, and periodic acid-Schiff stains are used in routine histopathology assessments to delineate clear cell tumors (9) and antibodies are available to immunohistochemically distinguish glycogen from lipids and glycoproteins (51, 88). Glycogen granules can also be readily detected by electron microscopy, and this technique has been used in studies to detect carcinogenesis-associated changes in glycogen content (10, 12, 32, 60, 74).
The ability to label glycogen with glucose and glucose analogs has facilitated labeling strategies to measure glycogenic flux. Compounds such as 2-[N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose (2NBDG) that readily incorporate into glycogen have provided a relatively cost-effective way to determine broad cellular glycogen content (89). Similarly, glycogen content can be assessed in cell and tissue homogenates spectrophotometrically by taking advantage of degradation of glycogen to glucose which is detected and quantified using luminescent or fluorescent substrates (90, 91). Glycogen content can also be quantified by incorporation of 13C-labeled glucose (6, 8, 25, 51, 71). Similarly, radiolabeling with 18F-N-(methyl-(2-fluoroethyl)-1H-[1,2,3]triazole-4-yl)glucosamine (18F-NFTG) is being used to specifically detect oncogene-driven changes in glycogenesis in in vivo preclinical models (92). 18F-NFTG overcomes certain limitations of FDG PET imaging including high cardiac uptake and low differentiation between neoplastic and inflammatory tissue (92). Clinical use of this tracer could be informative in defining distant metastases, although high background signal associated with glycogen-rich organs such as the kidney and liver presents a challenge that requires mitigation. 18F-NFTG may also provide opportunities for a theranostic-like approach to patient care involving delineation of glycogen-containing tumor burden followed by treatment with glycogen-targeted pharmacological agents. The marriage of metabolic imaging and targeting is beginning to bear fruit for glutamine-addicted (18F-FGln), fat-addicted (11C-acetate), OxPhos-addicted (18F-BnTP), and glycolytic (18FDG) tumors, although it is important to note that radiotracer uptake may not consistently correlate with the activity of the targeted metabolic process in all settings due to the interconnectedness of metabolic pathways (68, 93–95).
Pharmacological Disruption of Glycogen Metabolism in Cancer
The importance of glycogen in a range of processes required for cancer progression suggests that disrupting glycogen metabolism is a means to therapeutically target cancer, in particular tumors displaying glycogen dependence such as clear cell malignancies. Figure 1 highlights several points at which the glycogen axis has potential to be pharmacologically targeted, including the entry of glucose into cells, and during various catalytic processes required for glycogenesis and glycogenolysis. An example of a pharmacological disruptor of glycogen metabolism is the glucose analog 2-deoxy-D-glucose (2DG) which competes with glucose for cell entry via GLUT transporters and as a phosphorylation substrate for hexokinase (96–98). 2DG incorporation into glycogen antagonizes glycogenolysis, and glycogen mediates 2DG resistance via the activity of brain glycogen phosphorylase isoform PYGB (98–101). It has recently been shown that 2DG synergizes with carboplatin against patient-derived mouse models of glycogen-rich ovarian clear cell carcinoma (95). This strategy to increase the efficacy of a key standard-of-care chemotherapy was achieved at a 2DG dose far lower than employed in other studies against non-glycogen dependent cancers (97). This raises the possibility that combining 2DG and chemotherapy could be effective for clear cell cancers generally, including the much more common clear cell renal cell carcinoma (96, 102). The data justify further clinical trials to evaluate the efficacy of these combinations in these glycogen-dependent cancers.
Disruption of glycogen metabolism is also achieved by targeting mediators of glycogen biosynthesis including the PI3K/AKT/mTOR pathway. Modulators of this pathway, such as perifosine, MK-2202, and evorolimus, have progressed into clinical trials, however, it is not yet clear whether anti-cancer effects are mediated by impacts on tumor glycogen metabolism (103, 104). Hyperactivation of the pathway is a prominent molecular feature of both clear cell ovarian and renal cell carcinomas (37, 42, 43, 105). PI3K, AKT, and mTOR inhibitors have shown promise in these malignancies as standalone therapies and in combination with standard-of-care chemotherapies, displaying synergistic anti-proliferative effects in vitro and in vivo (17, 106–109). Furthermore, inhibitors of GSK3β have been pursued as standalone treatments and in combination with multi-kinase inhibition in clear cell renal cell carcinoma, where it potentiates the effects of sorafenib both in vitro and in vivo (110–113).
In certain cancer contexts, it also appears to be useful to promote rapid glycogenolysis to exhaust glycogen stores. For example, myeloid leukemia cells display pathological glycogenesis via a mechanism involving suppression of AMPK activity, with the accumulated glycogen available for mobilization as a glycolytic fuel during times of stress (17). These processes can be targeted with AMPK activators such as metformin and resveratrol, anti-diabetic agents that are becoming more widely studied in cancer (9, 114, 115). Metformin functions by inducing glycogenolysis via insulin signaling, downstream of GSK activity, as well as by AMPK activation (9, 17, 116–118). Pharmacological disruption of processes downstream the glycogen debranching enzyme AGL has also been attempted. This involved via modulation of the hyaluronic acid synthesis axis using drugs such as 4-methylumbelliferone (4MU) which reduced progression of bladder and lung cancers in model systems (19, 21, 119).
Inhibition of glycogenolysis has been most commonly attempted in cancer settings via disruption of PYG (9, 120). PYG inhibitors are often repurposed anti-diabetic therapies which target different allosteric sites of PYG (9) and produce similar phenotypes to genetic depletion of PYG, including impaired glycogenolysis resulting in cell cycle arrest, apoptosis, and ROS-dependent cell senescence (12, 121, 122). CP-320626, an inhibitor of AMP allosteric regulatory and catalytic sites of PYG induces cell cycle arrest and apoptosis in cultured MIA pancreatic cancer cells via MAPK/ERK and TNF-α/NF-κB pathways (123). Interestingly, flavoperidol, a potent inhibitor of CDK activity that progressed to clinical trials, has recently been shown to produce similar effects to genetic disruption of PYG and to be a bona-fide PYG inhibitor (21, 124, 125). The most widely used indole carboxamide PYG inhibitor CP-91149 induces growth inhibition in a range of tumors including hepatocellular carcinoma and pancreatic and prostate cancer (121, 126, 127). It acts synergistically with standard-of-care multi-kinase inhibitors sorafenib and regorafenib in models of hepatocellular carcinoma (121). It will be important to assess the efficacy of a similar regime in models of clear cell renal cancer, for which multi-kinase inhibitors are also standard-of-care. Despite promising in vitro results, PYG inhibitors are yet to be evaluated in in vivo tumor models.
Interestingly, disruption of enzymes involved in glycogen metabolism induces cell death by apoptotic and non-apoptotic processes including necroptosis and ferroptosis (128). PYGL inhibition induces necroptosis by modulating ROS production and aerobic respiration (129, 130). Similarly, an upstream regulator of PYG, phosphorylase kinase G2, is critical to the lipid peroxidation underlying ferroptosis (128, 131). To optimize the efficacy of glycogen metabolism disruptors against cancer, including reducing therapy resistance, further research is required to better understand the mechanisms by which these agents induce cell death. Additional research is also required to minimize adverse events that can occur when targeting metabolic processes which are important in both normal and malignant cells. This is highlighted by toxicities that have been reported from trials of 2DG against a range of advanced solid tumors, where the clinically tolerable dose of this agent was accompanied by significant adverse events including cardiac and gastrointestinal toxicities and neutropenia (28, 29), with one patient experiencing a fatal cardiac arrest (96, 102).
Conclusion
Despite the key role of glycogen in processes that underpin malignant transformation, there are still many gaps in our understanding of the molecular mechanisms that regulate metabolism of this macromolecule in cancer. The importance of glycogen metabolism to tumor cell senescence, metastasis, both in driving the metastatic cascade and organotropism, and in resistance to chemotherapies and targeted therapies, are aspects that warrant particular attention. New information in these areas is expected to lead to further attempts to harness glycogen as an effective therapeutic target (8). It seems likely that the repurposing of glycogen metabolism targeting agents that are currently used for non-cancer pathologies including diabetes, will benefit patients with glycogen-dependent clear cell cancers and potentially other non-clear cell malignancies.
Author Contributions
TKh wrote the manuscript. MS, JG, TKr, NL, YH, and JH edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by a grant from the Australia New Zealand Gynecological Oncology Group and philanthropic support from the Mater Foundation (JH) and a higher degree scholarship awarded by The University of Queensland Graduate School (TKh). MS is supported by a Mater Foundation McGuckin Early Career Fellowship and the University of Queensland Amplify Initiative.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Roach PJ, Depaoli-Roach AA, Hurley TD, Tagliabracci VS. Glycogen and its metabolism: some new developments and old themes. Biochem J. (2012) 441:763–87. doi: 10.1042/BJ20111416
2. Prats C, Graham TE, Shearer J. The dynamic life of the glycogen granule. J Biol Chem. (2018) 293:7089–98. doi: 10.1074/jbc.R117.802843
3. Adeva-Andany MM, González-Lucán M, Donapetry-García C, Fernández-Fernández C, Ameneiros-Rodríguez E. Glycogen metabolism in humans. BBA Clin. (2016) 5:85–100. doi: 10.1016/j.bbacli.2016.02.001
4. Jensen J, Rustad P, Kolnes A, Lai Y-C. The role of skeletal muscle glycogen breakdown for regulation of insulin sensitivity by exercise. Front Physiol. (2011) 2:112. doi: 10.3389/fphys.2011.00112
5. Calì C, Tauffenberger A, Magistretti P. The strategic location of glycogen and lactate: from body energy reserve to brain plasticity. Front Cell Neurosci. (2019) 13:82. doi: 10.3389/fncel.2019.00082
6. Ashcroft FM, Rohm M, Clark A, Brereton MF. Is type 2 diabetes a glycogen storage disease of pancreatic β cells? Cell Metab. (2017) 26:17–23. doi: 10.1016/j.cmet.2017.05.014
7. Sullivan MA, Forbes JM. Glucose and glycogen in the diabetic kidney: heroes or villains? EBioMedicine. (2019) 47:590–7. doi: 10.1016/j.ebiom.2019.07.067
8. Dauer P, Lengyel E. New roles for glycogen in tumor progression. Trends Cancer. (2019) 5:396–9. doi: 10.1016/j.trecan.2019.05.003
9. Zois CE, Harris AL. Glycogen metabolism has a key role in the cancer microenvironment and provides new targets for cancer therapy. J Mol Med (Berl). (2016) 94:137–54. doi: 10.1007/s00109-015-1377-9
10. Zois CE, Favaro E, Harris AL. Glycogen metabolism in cancer. Biochem Pharmacol. (2014) 92:3–11. doi: 10.1016/j.bcp.2014.09.001
11. Rousset M, Zweibaum A, Fogh J. Presence of glycogen and growth-related variations in 58 cultured human tumor cell lines of various tissue origins. Cancer Res. (1981) 41:1165–70.
12. Favaro E, Bensaad K, Chong MG, Tennant DA, Ferguson DJ, Snell C, et al. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab. (2012) 16:751–64. doi: 10.1016/j.cmet.2012.10.017
13. Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. (2020) 20:74–88. doi: 10.1038/s41568-019-0216-7
14. Song M, Bode AM, Dong Z, Lee M-H. AKT as a therapeutic target for cancer. Cancer Res. (2019) 79:1019–31. doi: 10.1158/0008-5472.CAN-18-2738
15. Yu M, Yongzhi H, Chen S, Luo X, Lin Y, Zhou Y, et al. The prognostic value of GLUT1 in cancers: a systematic review and meta-analysis. Oncotarget. (2017) 8:43356–67. doi: 10.18632/oncotarget.17445
16. Zhou Y, Jin Z, Wang C. Glycogen phosphorylase B promotes ovarian cancer progression via Wnt/β-catenin signaling and is regulated by miR-133a-3p. Biomed Pharmacother. (2019) 120:109449. doi: 10.1016/j.biopha.2019.109449
17. Bhanot H, Reddy MM, Nonami A, Weisberg EL, Bonal D, Kirschmeier PT, et al. Pathological glycogenesis through glycogen synthase 1 and suppression of excessive AMP kinase activity in myeloid leukemia cells. Leukemia. (2015) 29:1555–63. doi: 10.1038/leu.2015.46
18. Yang W, Zhang C, Li Y, Jin A, Sun Y, Yang X, et al. Phosphorylase kinase β represents a novel prognostic biomarker and inhibits malignant phenotypes of liver cancer cell. Int J Biol Sci. (2019) 15:2596–606. doi: 10.7150/ijbs.33278
19. Guin S, Ru Y, Agarwal N, Lew CR, Owens C, Comi GP, et al. Loss of glycogen debranching enzyme AGL drives bladder tumor growth via induction of hyaluronic acid synthesis. Clin Cancer Res. (2016) 22:1274–83. doi: 10.1158/1078-0432.CCR-15-1706
20. Richmond CS, Oldenburg D, Dancik G, Meier DR, Weinhaus B, Theodorescu D, et al. Glycogen debranching enzyme (AGL) is a novel regulator of non-small cell lung cancer growth. Oncotarget. (2018) 9:16718–30. doi: 10.18632/oncotarget.24676
21. Ritterson Lew C, Guin S, Theodorescu D. Targeting glycogen metabolism in bladder cancer. Nat Rev Urol. (2015) 12:383–91. doi: 10.1038/nrurol.2015.111
22. van der Mijn JC, Fu L, Khani F, Zhang T, Molina AM, Barbieri CE, et al. Combined metabolomics and genome-wide transcriptomics analyses show multiple HIF1α-induced changes in lipid metabolism in early stage clear cell renal cell carcinoma. Transl Oncol. (2020) 13:177–85. doi: 10.1016/j.tranon.2019.10.015
23. Zhou Z, Kinslow CJ, Hibshoosh H, Guo H, Cheng SK, He C, et al. Clinical features, survival and prognostic factors of glycogen-rich clear cell carcinoma (GRCC) of the breast in the U.S. Population. J Clin Med. (2019) 8:246. doi: 10.3390/jcm8020246
24. Remo A, Fassan M, Vanoli A, Bonetti LR, Barresi V, Tatangelo F, et al. Morphology and molecular features of rare colorectal carcinoma histotypes. Cancers. (2019) 11:1036. doi: 10.3390/cancers11071036
25. Abdulfatah E, Sakr S, Thomas S, Al-Wahab Z, Mutch DG, Dowdy S, et al. Clear cell carcinoma of the endometrium: evaluation of prognostic parameters in a multi-institutional cohort of 165 cases. Int J Gynecol Cancer. (2017) 27:1714–21. doi: 10.1097/IGC.0000000000001050
26. Ji JX, Wang YK, Cochrane DR, Huntsman DG. Clear cell carcinomas of the ovary and kidney: clarity through genomics. J Pathol. (2018) 244:550–64. doi: 10.1002/path.5037
27. Glasspool RM, McNeish IA. Clear cell carcinoma of ovary and uterus. Curr Oncol Rep. (2013) 15:566–72. doi: 10.1007/s11912-013-0346-0
28. Kato N. Pathology of clear cell carcinoma of the ovary: a basic view based on cultured cells and modern view from comprehensive approaches. Pathol Int. (2020) 70:591–601. doi: 10.1111/pin.12954
29. Saez I, Duran J, Sinadinos C, Beltran A, Yanes O, Tevy MF, et al. Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J Cereb Blood Flow Metab. (2014) 34:945–55. doi: 10.1038/jcbfm.2014.33
30. Schulz A, Sekine Y, Oyeyemi MJ, Abrams AJ, Basavaraju M, Han SM, et al. The stress-responsive gene GDPGP1/mcp-1 regulates neuronal glycogen metabolism and survival. J Cell Biol. (2020) 219:e202001006. doi: 10.1083/jcb.201807127
31. Czech-Damal NU, Geiseler SJ, Hoff ML, Schliep R, Ramirez JM, Folkow LP, et al. The role of glycogen, glucose and lactate in neuronal activity during hypoxia in the hooded seal (Cystophora cristata) brain. Neuroscience. (2014) 275:374–83. doi: 10.1016/j.neuroscience.2014.06.024
32. Pelletier J, Bellot G, Gounon P, Lacas-Gervais S, Pouyssegur J, Mazure NM. Glycogen synthesis is induced in hypoxia by the hypoxia-inducible factor and promotes cancer cell survival. Front Oncol. (2012) 2:18. doi: 10.3389/fonc.2012.00018
33. Pescador N, Villar D, Cifuentes D, Garcia-Rocha M, Ortiz-Barahona A, Vazquez S, et al. Hypoxia promotes glycogen accumulation through hypoxia inducible factor (HIF)-mediated induction of glycogen synthase 1. PLoS One. (2010) 5:e9644. doi: 10.1371/journal.pone.0009644
34. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. (2003) 3:721–32. doi: 10.1038/nrc1187
35. Pezzuto A, Carico E. Role of HIF-1 in cancer progression: novel insights. A review. Curr Mol Med. (2018) 18:343–51. doi: 10.2174/1566524018666181109121849
36. Lee S, Garner EIO, Welch WR, Berkowitz RS, Mok SC. Over-expression of hypoxia-inducible factor 1 alpha in ovarian clear cell carcinoma. Gynecol Oncol. (2007) 106:311–7. doi: 10.1016/j.ygyno.2007.03.041
37. Mandai M, Amano Y, Yamaguchi K, Matsumura N, Baba T, Konishi I. Ovarian clear cell carcinoma meets metabolism; HNF-1β confers survival benefits through the Warburg effect and ROS reduction. Oncotarget. (2015) 6:30704–14. doi: 10.18632/oncotarget.5228
38. Tan TZ, Ye J, Yee CV, Lim D, Ngoi NYL, Tan DSP, et al. Analysis of gene expression signatures identifies prognostic and functionally distinct ovarian clear cell carcinoma subtypes. EBioMedicine. (2019) 50:203–10. doi: 10.1016/j.ebiom.2019.11.017
39. Cheng KW, Agarwal R, Mitra S, Lee J-S, Carey M, Gray JW, et al. Rab25 increases cellular ATP and glycogen stores protecting cancer cells from bioenergetic stress. EMBO Mol Med. (2012) 4:125–41. doi: 10.1002/emmm.201100193
40. Agarwal R, Jurisica I, Mills GB, Cheng KW. The emerging role of the RAB25 small GTPase in cancer. Traffic. (2009) 10:1561–8. doi: 10.1111/j.1600-0854.2009.00969.x
41. Gomez-Roman N, Sahasrabudhe NM, McGregor F, Chalmers AJ, Cassidy J, Plumb J. Hypoxia-inducible factor 1 alpha is required for the tumourigenic and aggressive phenotype associated with Rab25 expression in ovarian cancer. Oncotarget. (2016) 7:22650–64. doi: 10.18632/oncotarget.7998
42. Ricketts CJ, De Cubas AA, Fan H, Smith CC, Lang M, Reznik E, et al. The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. (2018) 23:313–26.e5.
43. Creighton CJ, Morgan M, Gunaratne PH, Wheeler DA, Gibbs RA, Gordon Robertson A, et al. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. (2013) 499:43–9. doi: 10.1038/nature12222
44. Kwon SM, Hong SM, Lee YK, Min S, Yoon G. Metabolic features and regulation in cell senescence. BMB Rep. (2019) 52:5–12. doi: 10.5483/BMBRep.2019.52.1.291
45. Seo YH, Jung HJ, Shin HT, Kim YM, Yim H, Chung HY, et al. Enhanced glycogenesis is involved in cellular senescence via GSK3/GS modulation. Aging Cell. (2008) 7:894–907. doi: 10.1111/j.1474-9726.2008.00436.x
46. Zeng S, Shen WH, Liu L. Senescence and cancer. Cancer Transl Med. (2018) 4:70–4. doi: 10.4103/ctm.ctm_22_18
47. Lee S, Schmitt CA. The dynamic nature of senescence in cancer. Nat Cell Biol. (2019) 21:94–101. doi: 10.1038/s41556-018-0249-2
48. Schosserer M, Grillari J, Breitenbach M. The dual role of cellular senescence in developing tumors and their response to cancer therapy. Front Oncol. (2017) 7:278. doi: 10.3389/fonc.2017.00278
49. Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. (2018) 553:96–100. doi: 10.1038/nature25167
50. Ruscetti M, Morris JP, Mezzadra R, Russell J, Leibold J, Romesser PB, et al. Senescence-induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell. (2020) 181:424–41.e21. doi: 10.1016/j.cell.2020.03.008
51. Sun RC, Dukhande VV, Zhou Z, Young LEA, Emanuelle S, Brainson CF, et al. Nuclear glycogenolysis modulates histone acetylation in human non-small cell lung cancers. Cell Metab. (2019) 30:903–16.e7. doi: 10.1016/j.cmet.2019.08.014
52. Cheng Y, He C, Wang M, Ma X, Mo F, Yang S, et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Therapy. (2019) 4:62. doi: 10.1038/s41392-019-0095-0
53. Komatsu N, Kawamata N, Takeuchi S, Yin D, Chien W, Miller CW, et al. SAHA, a HDAC inhibitor, has profound anti-growth activity against non-small cell lung cancer cells. Oncol Rep. (2006) 15:187–91. doi: 10.3892/or.15.1.187
54. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity. (2015) 43:435–49. doi: 10.1016/j.immuni.2015.09.001
55. O'Sullivan D, Sanin DE, Pearce EJ, Pearce EL. Metabolic interventions in the immune response to cancer. Nat Rev Immunol. (2019) 19:324–35. doi: 10.1038/s41577-019-0140-9
56. Thwe PM, Fritz DI, Snyder JP, Smith PR, Curtis KD, O'Donnell A, et al. Syk-dependent glycolytic reprogramming in dendritic cells regulates IL-1β production to β-glucan ligands in a TLR-independent manner. J Leukoc Biol. (2019) 106:1325–35. doi: 10.1002/JLB.3A0819-207RR
57. Thwe PM, Pelgrom LR, Cooper R, Beauchamp S, Reisz JA, D'Alessandro A, et al. Cell-intrinsic glycogen metabolism supports early glycolytic reprogramming required for dendritic cell immune responses. Cell Metab. (2017) 26:558–67.e5. doi: 10.1016/j.cmet.2017.08.012
58. Curtis KD, Smith PR, Despres HW, Snyder JP, Hogan TC, Rodriguez PD, et al. Glycogen metabolism supports early glycolytic reprogramming and activation in dendritic cells in response to both TLR and Syk-dependent CLR agonists. Cells. (2020) 9:715. doi: 10.3390/cells9030715
59. Ma J, Wei K, Liu J, Tang K, Zhang H, Zhu L, et al. Glycogen metabolism regulates macrophage-mediated acute inflammatory responses. Nat Commun. (2020) 11:1769. doi: 10.1038/s41467-020-15636-8
60. Ma R, Ji T, Zhang H, Dong W, Chen X, Xu P, et al. A Pck1-directed glycogen metabolic program regulates formation and maintenance of memory CD8+ T cells. Nat Cell Biol. (2018) 20:21–7. doi: 10.1038/s41556-017-0002-2
61. Le Bourgeois T, Strauss L, Aksoylar H-I, Daneshmandi S, Seth P, Patsoukis N, et al. Targeting T cell metabolism for improvement of cancer immunotherapy. Front Oncol. (2018) 8:237. doi: 10.3389/fonc.2018.00237
62. Xu X, Gnanaprakasam JNR, Sherman J, Wang R. A metabolism toolbox for CAR T therapy. Front Oncol. (2019) 9:322. doi: 10.3389/fonc.2019.00322
63. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. (2017) 14:399–416. doi: 10.1038/nrclinonc.2016.217
64. Mantovani A, Locati M. Macrophage metabolism shapes angiogenesis in tumors. Cell Metab. (2016) 24:653–4. doi: 10.1016/j.cmet.2016.10.016
65. Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Disc. (2011) 10:671. doi: 10.1038/nrd3504
66. Murray CM, Hutchinson R, Bantick JR, Belfield GP, Benjamin AD, Brazma D, et al. Monocarboxylate transporter MCT1 is a target for immunosuppression. Nat Chem Biol. (2005) 1:371–6. doi: 10.1038/nchembio744
67. Beloueche-Babari M, Casals Galobart T, Delgado-Goni T, Wantuch S, Parkes HG, Tandy D, et al. Monocarboxylate transporter 1 blockade with AZD3965 inhibits lipid biosynthesis and increases tumour immune cell infiltration. Br J Cancer. (2020) 122:895–903. doi: 10.1038/s41416-019-0717-x
68. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. (2020) 368:eaaw5473. doi: 10.1126/science.aaw5473
69. Massagué J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. (2016) 529:298–306. doi: 10.1038/nature17038
70. Luengo A, Gui DY, Vander Heiden MG. Targeting metabolism for cancer therapy. Cell Chem Biol. (2017) 24:1161–80. doi: 10.1016/j.chembiol.2017.08.028
71. Altemus MA, Goo LE, Little AC, Yates JA, Cheriyan HG, Wu ZF, et al. Breast cancers utilize hypoxic glycogen stores via PYGB, the brain isoform of glycogen phosphorylase, to promote metastatic phenotypes. PLoS One. (2019) 14:e0220973. doi: 10.1371/journal.pone.0220973
72. Zhang S, Zhou Y, Zha Y, Yang Y, Wang L, Li J, et al. PYGB siRNA inhibits the cell proliferation of human osteosarcoma cell lines. Mol Med Rep. (2018) 18:715–22. doi: 10.3892/mmr.2018.9022
73. Li Y, Zhuang H, Zhang X, Li Y, Liu Y, Yi X, et al. Multiomics integration reveals the landscape of prometastasis metabolism in hepatocellular carcinoma. Mol Cell Proteomics. (2018) 17:607–18. doi: 10.1074/mcp.RA118.000586
74. Curtis M, Kenny HA, Ashcroft B, Mukherjee A, Johnson A, Zhang Y, et al. Fibroblasts mobilize tumor cell glycogen to promote proliferation and metastasis. Cell Metab. (2019) 29:141–55.e9. doi: 10.1016/j.cmet.2018.08.007
75. Rosenzweig A, Blenis J, Gomes AP. Beyond the Warburg effect: how do cancer cells regulate one-carbon metabolism? Front Cell Dev Biol. (2018) 6:90. doi: 10.3389/fcell.2018.00090
76. Chen J, Lee HJ, Wu X, Huo L, Kim SJ, Xu L, et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Cancer Res. (2015) 75:554–65. doi: 10.1158/0008-5472.CAN-14-2268
77. Duran J, Gruart A, Varea O, López-Soldado I, Delgado-García JM, Guinovart JJ. Lack of neuronal glycogen impairs memory formation and learning-dependent synaptic plasticity in mice. Front Cell Neurosci. (2019) 13:374. doi: 10.3389/fncel.2019.00374
78. Matsui T, Soya S, Okamoto M, Ichitani Y, Kawanaka K, Soya H. Brain glycogen decreases during prolonged exercise. J Physiol. (2011) 589(Pt 13):3383–93. doi: 10.1113/jphysiol.2011.203570
79. Lengyel E. Ovarian cancer development and metastasis. Am J Pathol. (2010) 177:1053–64. doi: 10.2353/ajpath.2010.100105
80. Yamashita Y. Ovarian cancer: new developments in clear cell carcinoma and hopes for targeted therapy. Jpn J Clin Oncol. (2015) 45:405–7. doi: 10.1093/jjco/hyu221
81. Makhov P, Joshi S, Ghatalia P, Kutikov A, Uzzo RG, Kolenko VM. Resistance to systemic therapies in clear cell renal cell carcinoma: mechanisms and management strategies. Mol Cancer Ther. (2018) 17:1355–64. doi: 10.1158/1535-7163.MCT-17-1299
82. Itamochi H, Kigawa J, Terakawa N. Mechanisms of chemoresistance and poor prognosis in ovarian clear cell carcinoma. Cancer Sci. (2008) 99:653–8. doi: 10.1111/j.1349-7006.2008.00747.x
83. Mabuchi S, Sugiyama T, Kimura T. Clear cell carcinoma of the ovary: molecular insights and future therapeutic perspectives. J Gynecol Oncol. (2016) 27:e31. doi: 10.3802/jgo.2016.27.e31
84. Takehara K, Konishi H, Kojima A, Okame S, Shiroyama Y, Yokoyama T, et al. SUV max of FDG-PET/CT as a prognostic factor in ovarian clear cell adenocarcinoma. J Clin Oncol. (2014) 32:e16509. doi: 10.1200/jco.2014.32.15_suppl.e16509
85. Hopkins BD, Goncalves MD, Cantley LC. Insulin-PI3K signalling: an evolutionarily insulated metabolic driver of cancer. Nat Rev Endocrinol. (2020) 16:276–83. doi: 10.1038/s41574-020-0329-9
86. Mancinelli R, Carpino G, Petrungaro S, Mammola CL, Tomaipitinca L, Filippini A, et al. Multifaceted roles of GSK-3 in cancer and autophagy-related diseases. Oxid Med Cell Longev. (2017) 2017:4629495. doi: 10.1155/2017/4629495
87. Ma L, Zong X. Metabolic symbiosis in chemoresistance: refocusing the role of aerobic glycolysis. Front Oncol. (2020) 10:5. doi: 10.3389/fonc.2020.00005
88. Skurat AV, Segvich DM, DePaoli-Roach AA, Roach PJ. Novel method for detection of glycogen in cells. Glycobiology. (2017) 27:416–24. doi: 10.1093/glycob/cwx005
89. Louzao MC, Espina B, Vieytes MR, Vega FV, Rubiolo JA, Baba O, et al. “Fluorescent glycogen” formation with sensibility for in vivo and in vitro detection. Glycoconj J. (2008) 25:503–10. doi: 10.1007/s10719-007-9075-7
90. Possik E, Pause A. Biochemical measurement of glycogen: method to investigate the AMPK-glycogen relationship. Methods Mol Biol. (2018) 1732:57–67. doi: 10.1007/978-1-4939-7598-3_4
91. Thwe PM, Amiel E. Analysis of glycogen metabolic pathway utilization by dendritic cells and T cells using custom phenotype metabolic assays. J Immunol Methods. (2018) 458:53–7. doi: 10.1016/j.jim.2018.04.006
92. Witney TH, Carroll L, Alam IS, Chandrashekran A, Nguyen Q-D, Sala R. A novel radiotracer to image glycogen metabolism in tumors by positron emission tomography. Cancer Res. (2014) 74:1319–28. doi: 10.1158/0008-5472.CAN-13-2768
93. Venneti S, Dunphy MP, Zhang H, Pitter KL, Zanzonico P, Campos C, et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci Transl Med. (2015) 7:274ra17. doi: 10.1126/scitranslmed.aaa1009
94. Yoshii Y, Furukawa T, Oyama N, Hasegawa Y, Kiyono Y, Nishii R, et al. Fatty acid synthase is a key target in multiple essential tumor functions of prostate cancer: uptake of radiolabeled acetate as a predictor of the targeted therapy outcome. PLoS One. (2013) 8:e64570. doi: 10.1371/journal.pone.0064570
95. Momcilovic M, Jones A, Bailey ST, Waldmann CM, Li R, Lee JT, et al. In vivo imaging of mitochondrial membrane potential in non-small-cell lung cancer. Nature. (2019) 575:380–4. doi: 10.1038/s41586-019-1715-0
96. Stein M, Lin H, Jeyamohan C, Dvorzhinski D, Gounder M, Bray K, et al. Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate. (2010) 70:1388–94. doi: 10.1002/pros.21172
97. Khan T, He Y, Kryza T, Harrington BS, Gunter JH, Sullivan MA, et al. Disruption of glycogen utilization markedly improves the efficacy of carboplatin against preclinical models of clear cell ovarian carcinoma. Cancers. (2020) 12:869. doi: 10.3390/cancers12040869
98. Colwell DR, Higgins JA, Denyer GS. Incorporation of 2-deoxy-D-glucose into glycogen. Implications for measurement of tissue-specific glucose uptake and utilisation. Int J Biochem Cell Biol. (1996) 28:115–21. doi: 10.1016/1357-2725(95)00110-7
99. Ralser M, Wamelink MM, Struys EA, Joppich C, Krobitsch S, Jakobs C, et al. A catabolic block does not sufficiently explain how 2-deoxy-D-glucose inhibits cell growth. Proc Natl Acad Sci U S A. (2008) 105:17807–11. doi: 10.1073/pnas.0803090105
100. Philips KB, Kurtoglu M, Leung HJ, Liu H, Gao N, Lehrman MA, et al. Increased sensitivity to glucose starvation correlates with downregulation of glycogen phosphorylase isoform PYGB in tumor cell lines resistant to 2-deoxy-D-glucose. Cancer Chemother Pharmacol. (2014) 73:349–61. doi: 10.1007/s00280-013-2358-8
101. Abbadi S, Rodarte JJ, Abutaleb A, Lavell E, Smith CL, Ruff W, et al. Glucose-6-phosphatase is a key metabolic regulator of glioblastoma invasion. Mol Cancer Res. (2014) 12:1547–59. doi: 10.1158/1541-7786.MCR-14-0106-T
102. Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. (2013) 71:523–30. doi: 10.1007/s00280-012-2045-1
103. Guo K, Tang W, Zhuo H, Zhao G. Recent advance of Akt inhibitors in clinical trials. ChemistrySelect. (2019) 4:9040–4. doi: 10.1002/slct.201901293
104. Kaley TJ, Panageas KS, Pentsova EI, Mellinghoff IK, Nolan C, Gavrilovic I, et al. Phase I clinical trial of temsirolimus and perifosine for recurrent glioblastoma. Ann Clin Transl Neurol. (2020) 7:429–36. doi: 10.1002/acn3.51009
105. Yamaguchi K, Mandai M, Oura T, Matsumura N, Hamanishi J, Baba T, et al. Identification of an ovarian clear cell carcinoma gene signature that reflects inherent disease biology and the carcinogenic processes. Oncogene. (2010) 29:1741–52. doi: 10.1038/onc.2009.470
106. Sasano T, Mabuchi S, Kuroda H, Kawano M, Matsumoto Y, Takahashi R, et al. Preclinical efficacy for AKT targeting in clear cell carcinoma of the ovary. Mol Cancer Res. (2015) 13:795–806. doi: 10.1158/1541-7786.MCR-14-0314
107. Oishi T, Itamochi H, Kudoh A, Nonaka M, Kato M, Nishimura M, et al. The PI3K/mTOR dual inhibitor NVP-BEZ235 reduces the growth of ovarian clear cell carcinoma. Oncol Rep. (2014) 32:553–8. doi: 10.3892/or.2014.3268
108. Guo H, German P, Bai S, Barnes S, Guo W, Qi X, et al. The PI3K/AKT pathway and renal cell carcinoma. J Genet Genomics. (2015) 42:343–53. doi: 10.1016/j.jgg.2015.03.003
109. Vlachostergios PJ, Molina AM. PI3K/AKT inhibitors in patients with refractory renal cell carcinoma: what have we learnt so far? Ann Oncol. (2017) 28:914–6. doi: 10.1093/annonc/mdx104
110. Bilim V, Ougolkov A, Yuuki K, Naito S, Kawazoe H, Muto A, et al. Glycogen synthase kinase-3: a new therapeutic target in renal cell carcinoma. Br J Cancer. (2009) 101:2005–14. doi: 10.1038/sj.bjc.6605437
111. Cao Q, Lu X, Feng Y-J. Glycogen synthase kinase-3β positively regulates the proliferation of human ovarian cancer cells. Cell Res. (2006) 16:671–7. doi: 10.1038/sj.cr.7310078
112. Kawazoe H, Bilim VN, Ugolkov AV, Yuuki K, Naito S, Nagaoka A, et al. GSK-3 inhibition in vitro and in vivo enhances antitumor effect of sorafenib in renal cell carcinoma (RCC). Biochem Biophys Res Commun. (2012) 423:490–5. doi: 10.1016/j.bbrc.2012.05.147
113. Walz A, Ugolkov A, Chandra S, Kozikowski A, Carneiro BA, O'Halloran TV, et al. Molecular pathways: revisiting glycogen synthase kinase-3β as a target for the treatment of cancer. Clin Cancer Res. (2017) 23:1891–7. doi: 10.1158/1078-0432.CCR-15-2240
114. Zi F, Zi H, Li Y, He J, Shi Q, Cai Z. Metformin and cancer: an existing drug for cancer prevention and therapy. Oncol Lett. (2018) 15:683–90. doi: 10.3892/ol.2017.7412
115. Buckanovich RJ, Brown J, Shank J, Griffith KA, Reynolds RK, Johnston C, et al. A phase II clinical trial of metformin as a cancer stem cell targeting agent in stage IIc/III/IV ovarian, fallopian tube, and primary peritoneal cancer. J Clin Oncol. (2017) 35:5556. doi: 10.1200/JCO.2017.35.15_suppl.5556
116. Elgendy M, Ciro M, Hosseini A, Weiszmann J, Mazzarella L, Ferrari E, et al. Combination of hypoglycemia and metformin impairs tumor metabolic plasticity and growth by modulating the PP2A-GSK3beta-MCL-1 axis. Cancer cell. (2019) 35:798–815.e5. doi: 10.1016/j.ccell.2019.03.007
117. Sarfstein R, Friedman Y, Attias-Geva Z, Fishman A, Bruchim I, Werner H. Metformin downregulates the insulin/IGF-I signaling pathway and inhibits different uterine serous carcinoma (USC) cells proliferation and migration in p53-dependent or -independent manners. PLoS One. (2013) 8:e61537. doi: 10.1371/journal.pone.0061537
118. Ailanen L, Bezborodkina NN, Virtanen L, Ruohonen ST, Malova AV, Okovityi SV, et al. Metformin normalizes the structural changes in glycogen preceding prediabetes in mice overexpressing neuropeptide Y in noradrenergic neurons. Pharmacol Res Perspect. (2018) 6:e00389. doi: 10.1002/prp2.389
119. Guin S, Pollard C, Ru Y, Ritterson Lew C, Duex JE, Dancik G, et al. Role in tumor growth of a glycogen debranching enzyme lost in glycogen storage disease. J Natl Cancer Inst. (2014) 106:dju062. doi: 10.1093/jnci/dju062
120. Donnier-Marechal M, Vidal S. Glycogen phosphorylase inhibitors: a patent review (2013 - 2015). Expert Opin Ther Pat. (2016) 26:199–212. doi: 10.1517/13543776.2016.1131268
121. Barot S, Abo-Ali EM, Zhou DL, Palaguachi C, Dukhande VV. Inhibition of glycogen catabolism induces intrinsic apoptosis and augments multikinase inhibitors in hepatocellular carcinoma cells. Exp Cell Res. (2019) 381:288–300. doi: 10.1016/j.yexcr.2019.05.017
122. Lee WNP, Guo P, Lim S, Bassilian S, Lee ST, Boren J, et al. Metabolic sensitivity of pancreatic tumour cell apoptosis to glycogen phosphorylase inhibitor treatment. Br J Cancer. (2004) 91:2094–100. doi: 10.1038/sj.bjc.6602243
123. Ma D, Wang J, Zhao Y, Lee W-NP, Xiao J, Go VLW, et al. Inhibition of glycogen phosphorylation induces changes in cellular proteome and signaling pathways in MIA pancreatic cancer cells. Pancreas. (2012) 41:397–408. doi: 10.1097/MPA.0b013e318236f022
124. Oikonomakos NG, Schnier JB, Zographos SE, Skamnaki VT, Tsitsanou KE, Johnson LN. Flavopiridol inhibits glycogen phosphorylase by binding at the inhibitor site. J Biol Chem. (2000) 275:34566–73. doi: 10.1074/jbc.M004485200
125. Shapiro GI. Preclinical and clinical development of the cyclin-dependent kinase inhibitor flavopiridol. Clin Cancer Res. (2004) 10(12 Pt 2):4270s–5s. doi: 10.1158/1078-0432.CCR-040020
126. Schnier JB, Nishi K, Gumerlock PH, Gorin FA, Bradbury EM. Glycogen synthesis correlates with androgen-dependent growth arrest in prostate cancer. BMC Urol. (2005) 5:6. doi: 10.1186/1471-2490-5-6
127. Schnier JB, Nishi K, Monks A, Gorin FA, Bradbury EM. Inhibition of glycogen phosphorylase (GP) by CP-91,149 induces growth inhibition correlating with brain GP expression. Biochem Biophys Res Commun. (2003) 309:126–34. doi: 10.1016/S0006-291X(03)01542-0
128. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. (2019) 35:830–49. doi: 10.1016/j.ccell.2019.04.002
129. Zhang D-W, Shao J, Lin J, Zhang N, Lu B-J, Lin S-C, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science (New York, NY). (2009) 325:332–6. doi: 10.1126/science.1172308
130. Qiu X, Zhang Y, Han J. RIP3 is an upregulator of aerobic metabolism and the enhanced respiration by necrosomal RIP3 feeds back on necrosome to promote necroptosis. Cell Death Differ. (2018) 25:821–4. doi: 10.1038/s41418-018-0075-x
Keywords: glycogen, cancer metabolism, cancer therapy, metabolic reprogramming, chemoresistance, immunometabolism
Citation: Khan T, Sullivan MA, Gunter JH, Kryza T, Lyons N, He Y and Hooper JD (2020) Revisiting Glycogen in Cancer: A Conspicuous and Targetable Enabler of Malignant Transformation. Front. Oncol. 10:592455. doi: 10.3389/fonc.2020.592455
Received: 07 August 2020; Accepted: 15 September 2020;
Published: 30 October 2020.
Edited by:
Fatima Baltazar, University of Minho, PortugalReviewed by:
Domenica Scumaci, Magna Græcia University of Catanzaro, ItalyAngela Ostuni, University of Basilicata, Italy
Copyright © 2020 Khan, Sullivan, Gunter, Kryza, Lyons, He and Hooper. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John D. Hooper, am9obi5ob29wZXJAbWF0ZXIudXEuZWR1LmF1