 Borja Guerra*
Borja Guerra* Carlota Recio
Carlota Recio Haidée Aranda-TavíoMiguel Guerra-Rodríguez
Haidée Aranda-TavíoMiguel Guerra-Rodríguez José M. García-Castellano
José M. García-Castellano Leandro Fernández-Pérez*
Leandro Fernández-Pérez*- Molecular and Translational Pharmacology Lab, Institute for Biomedical and Health Research (IUIBS), University of Las Palmas de Gran Canaria, Las Palmas de Gran Canaria, Spain
A hallmark of cancer cells includes a metabolic reprograming that provides energy, the essential building blocks, and signaling required to maintain survival, rapid growth, metastasis, and drug resistance of many cancers. The influence of tumor microenviroment on cancer cells also results an essential driving force for cancer progression and drug resistance. Lipid-related enzymes, lipid-derived metabolites and/or signaling pathways linked to critical regulators of lipid metabolism can influence gene expression and chromatin remodeling, cellular differentiation, stress response pathways, or tumor microenviroment, and, collectively, drive tumor development. Reprograming of lipid metabolism includes a deregulated activity of mevalonate (MVA)/cholesterol biosynthetic pathway in specific cancer cells which, in comparison with normal cell counterparts, are dependent of the continuous availability of MVA/cholesterol-derived metabolites (i.e., sterols and non-sterol intermediates) for tumor development. Accordingly, there are increasing amount of data, from preclinical and epidemiological studies, that support an inverse association between the use of statins, potent inhibitors of MVA biosynthetic pathway, and mortality rate in specific cancers (e.g., colon, prostate, liver, breast, hematological malignances). In contrast, despite the tolerance and therapeutic efficacy shown by statins in cardiovascular disease, cancer treatment demands the use of relatively high doses of single statins for a prolonged period, thereby limiting this therapeutic strategy due to adverse effects. Clinically relevant, synergistic effects of tolerable doses of statins with conventional chemotherapy might enhance efficacy with lower doses of each drug and, probably, reduce adverse effects and resistance. In spite of that, clinical trials to identify combinatory therapies that improve therapeutic window are still a challenge. In the present review, we revisit molecular evidences showing that deregulated activity of MVA biosynthetic pathway has an essential role in oncogenesis and drug resistance, and the potential use of MVA pathway inhibitors to improve therapeutic window in cancer.
Introduction
Adaptive metabolic reprogramming is often observed in cancer cells. It is widely accepted that metabolic disruptions of carbohydrates, proteins, and lipids are one of the hallmarks of cancer (1–3). Metabolic adaptations provide energy and the crucial building blocks needed to maintain abnormal survival, rapid growth, metastasis, and drug resistance in many tumors. In addition to tumor microenviroment, they are main driving forces for cancer progression (4, 5). Lipid metabolism reprograming involves lipid-related enzymes, metabolites, and signaling pathways linked to key regulators that can directly influence gene expression and chromatin remodeling, cellular differentiation, stress response pathways, or tumor microenviroment that collectively drive tumor development (6). An elevated or deregulated activity of mevalonate (MVA)/cholesterol biosynthetic pathway in specific cancer cells suggests that they are dependent of the continuous availability of MVA-derived metabolites (7–10). Furthermore, the aberrant activity of 3-hydroxy-3-methylglutaryl coenzyme A (HMGCoA) reductase (HMGCR), the rate-limiting enzyme of MVA pathway, can promote malignant transformation (7) and provides essential metabolites (i.e., sterols and non-sterol intermediates) that collectively drive tumor growth and development. Despite clinical evidences supporting the use of MVA pathway inhibitors (i.e., statins) for limiting cancer morbimortality are relatively low, increasing preclinical (11–19) and epidemiological (20–28) studies sustain the inverse association between statins and cancer-specific mortality rate. This beneficial effects of statins have been described in several types of cancer, including osteosarsocoma/chondrosarcoma (16–18), prostate (24, 26), colon (29, 30), breast (19, 31), liver (32, 33), pancreas (34), ovarian (35, 36), esophageal (37, 38), lung (39), and hematological malignances (40). Interestingly, statins may suppress epithelial-mesenchymal transition (EMT) program together with the inhibition of cancer stem cell generation, maintenance, and expansion (6, 41). Unluckily, the use of statins in cancer is currently limited by the requirement of using high doses for prolonged periods, thus generating adverse effects. Therefore, studies focused on elucidating new strategies targeting the MVA signaling pathway to improve the therapeutic window in cancer are urgently needed (42). Clinically relevant, synergistic effects of tolerable doses of statins with conventional chemotherapy could enhance treatment efficacy, by reducing doses of each drug and, probably, adverse effects. To date, clinical trials that identify combinatory therapies (statins-chemotherapy) that improve therapeutic window in different cancer types are still a challenge. In this review, we revisit preclinical and molecular evidences showing that aberrant MVA biosynthetic pathway may has an essential role in oncogenesis and we discuss how potent inhibitors of MVA pathway may best be applied to improve cancer therapy.
The MVA Biosynthetic Pathway
In normal cells, cellular cholesterol can arise from receptor-mediated uptake of LDL-cholesterol from circulation, or be de novo synthesized from acetyl-CoA by the MVA biosynthetic pathway. The precise regulation of MVA pathway is essential to guarantee continuous production of MVA-derived products, and to guard cells from accumulation of toxic end products, including cholesterol (43, 44). MVA pathway produces lipoproteins, dolichol, ubiquinone or cholesterol derived products (i.e., steroid hormones, oxysterols, vitamin D, bile acids) which are essential regulators of cellular metabolism. Cholesterol is essential for the buildup and maintenance of the structure and function of cellular membranes, cholesterol-rich microdomains or membrane rafts (lipid rafts). These structures constitute a core of organization for several signaling pathways and intracellular transport systems where cholesterol acts as a signaling molecule. The MVA biosynthetic pathway (Figure 1) starts with the formation of HMGCoA from three molecules of acetyl-CoA (43, 45), the end product of glycolysis. This reaction is catalyzed by the enzyme HMGCoA synthase. Subsequently, HMGCR converts HMGCoA to MVA which is the rate-limiting step of whole MVA pathway. The MVA is phosphorylated by the MVA kinase and converted to isopentenyl pyrophosphate (IPP). This step is decisive for the biosynthesis of farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP) and is regulated by a cascade of different synthases including the farnesyl diphosphate synthase (FDPS) and the GGPP synthase (GGPS). Then, FPP can be converted to squalene and, subsequently, by further enzymes such as squalene synthase and squalene epoxidase, to cholesterol. Further lipid products FPP downstream include dolichol and ubiquinone, both with antioxidant properties, and crucial for glycosylation and mitochondrial electron transport processes. The synthesis of FPP and GGPP is essential for protein prenylation, a key posttranslational modification for localization, membrane anchoring and function of many signaling proteins. Protein prenylation is mediated by the enzymes farnesyltransferase (FTase) I and geranylgeranyl transferases (GGTase) I and II. The MVA pathway also participates in other biological mechanisms including long-term memory of innate immune cells, survival, and polarization of effector immune cells (i.e., macrophages) or metabolic reprograming in cancer cells (46–48).
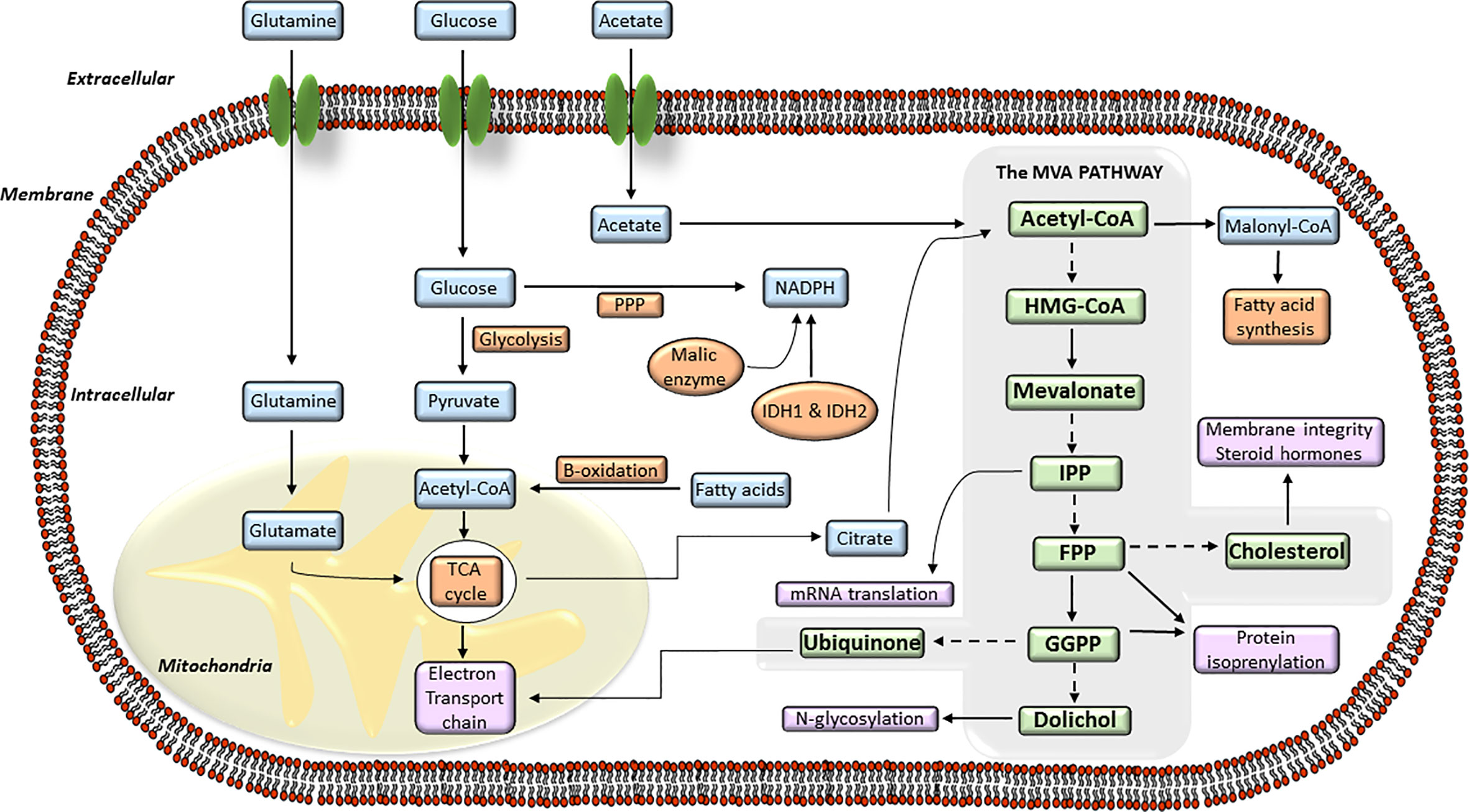
Figure 1 The mevalonate (MVA) pathway and its connection with the intracellular energy metabolism signaling. Diagram of the different steps of the intracellular MVA anabolic pathway, from the entry of acetyl-coenzyme A (CoA) to the production of isoprenoid metabolites. Acetyl-CoA is transformed into hydroxy-methylglutaryl-CoA (HMG-CoA) which is used by the enzyme hydroxy-methyl-glutaryl-CoA reductase (HMGCR) to synthesize MVA. MVA is further metabolized to farnesyl pyrophosphate (FPP), a precursor of cholesterol and sterols. FPP is also converted to geranylgeranyl pyrophosphate (GGPP), and these lipids are used for post-translational modification of proteins, including N-glycosylation and protein prenylation.
Regulation of the MVA Biosynthetic Pathway
The MVA biosynthetic pathway is regulated by transcriptional and post-transcriptional mechanisms including modulation of gene transcription, mRNA translation, protein degradation, and enzymatic activity (44, 49). The HMGCR enzyme, which regulates the rates of cholesterol synthesis, is in turn controlled by very fine-tuned regulatory mechanisms. Transcriptional regulation of HMGCR is mediated by two members of the sterol regulatory element binding proteins (SREBP) family called SREBP1 and SREBP2 (44, 49). SREBP proteins are encoded by two separate genes, SREBP-1 and -2. An alternative splicing of SREBP-1 can be produced, driving the synthesis of two isoforms, SREBP-1a and -1c. Whereas SREBP-1 has been clearly associated with homeostasis of cholesterol and fatty acids, SREBP-2 is mainly involved in synthesis and uptake of cholesterol. Thus, in response to intracellular sterol levels, SREBPs regulate the MVA biosynthetic pathway. Briefly, when the amount of intracellular sterol increases, SREBPs are held in an inactive form at the endoplasmic reticulum (ER) by their binding partner SREBP cleavage-activating protein (SCAP) and the insulin-induced genes (INSIG)-1 and -2. However, in response to sterol deprivation (e.g., when HMGCR activity is inhibited), intracellular end products of the MVA biosynthetic pathway are depleted. As the number of sterols diminishes, they no longer bind SCAP, thus producing a conformational change that triggers the SCAP-SREBP complex dissociation from the INSIGs and translocation from the ER to the Golgi. The SREBPs are successively cleaved by Golgi-resident proteases and released on their activated form, so they can translocate to the nucleus where they bind to sterol regulatory elements (SRE). This initiates the transcription of target genes that translate into key proteins involved in the biosynthesis of MVA-derived metabolites (i.e., HMGCoA synthase, HMGCR, FPP synthase, Insig-1) and cholesterol uptake (i.e., LDLR) to restore intracellular isoprenoid and sterol levels. Intracellular sterol levels are also regulated by oxysterols, metabolites derived from cholesterol oxidation. The 7α- and 27-hydroxycholesterols are synthesized in the liver by CYP7A1 and CYP27A1, the genes encoding the rate-limiting enzymes of neutral and acid bile synthetic pathways, respectively, which contribute to eliminate cholesterol. Oxysterols contribute to cholesterol homeostasis through activation of Liver X receptors (LXR) (50). LXR were originally characterized by their role in the positive regulation of the gene CYP7A. This relevant physiological role was further confirmed by the phenotype of LXRα null mice, which appear healthy when fed on a standard mouse diet but, when fed with a cholesterol-enriched diet, failed to induce CYP7A (51). Consequently, LXRα null mice suffered from a dramatic accumulation of cholesteryl ester in the liver and a reduction in bile acid production. Upon binding to LXR, oxysterols induce the transcription of specific ATP-binding cassette (ABC) transporters A1 and G1, that increase cholesterol efflux from enterocytes and macrophages, respectively (52). In addition to LXR activation, oxysterols (e.g., 25-hydroxysterol (25HC)), and high sterol concentrations, lanosterol, or Insig, can provoke ubiquitination and proteasomal degradation of HMGCR (53). Furthermore, the sterol-accelerated degradation of HMGCR is strengthen by non-sterol isoprenoids, including derivatives from FPP and GGPP. Notably, lanosterol does not interact with the sterol-sensing domain of SCAP and, therefore, does not suppress the processing of SREBP. Thereby, oxysterols downregulate HMGCR by increasing its ubiquitination-mediated degradation as well as suppress HMGCR gene transcription by inhibiting the delivery of SREBP-SCAP complex from ER. In contrast, lanosterol enhances the HMGCR degradation rate, and cholesterol limits the translocation of SREBP-SCAP (54). Furthermore, negative feedback responses of IPP, FPP, and GGPP suppress the activity of the MVA kinase. Expression of HMGCR is further modulated at the translational level, where the translation rate of HMGCR mRNA is controlled by the demand of the cell for non-steroid isoprenoids (e.g., MVA). When HMGCR dependent-MVA production is inhibited by statins, HMGCR mRNA is efficiently translated, even in the presence of sterols, being in contrast reduced when MVA is added. Finally, as mentioned below, the catalytic activity of HMGCR can be inhibited via phosphorylation by AMPK, a sensor of cellular energy state (55).
The MVA Pathway in Cancer
The Warburg phenomenon (56) is the best studied metabolic adaptation program developed by cancer cells. It was described as the preference of cancer cells to use aerobic glycolysis to obtain most of their energy, even in the presence of abundant oxygen supply, when normal cells would typically use the aerobic cellular respiration. Therefore, tumor cells will be highly dependent on glucose to produce large quantities of energy and provide other cells with intermediates necessary for the biosynthesis of amino acids, nucleic acids, and lipids (5, 57). This metabolic reprograming provides energy, the crucial building blocks, and signaling required to keep survival, rapid growth, and drug resistance of many cancers (1–3). Glycolysis generates acetyl-CoA (45), a molecule that is derived from acetate and/or glutamine metabolism. Acetyl-CoA can be incorporated into the MVA biosynthetic pathway, into lipids by fatty acid synthase (FAS) or into phospholipids by the action of different enzymes, including the pro-oncogene choline kinase (CK) (58). Acetyl-CoA feeds the MVA biosynthetic pathway to generate metabolites that are essential to maintain survival and rapid growth of multiple tumors. Accordingly, transcriptional profiling studies support the hypothesis that genes involved in cholesterol and fatty acid metabolism are upregulated in cancer cells and play an essential role in transformation (59). Several studies suggest that an elevated requirement for cholesterol is an innate metabolic hallmark in cancer cells, which could be used in a prophylactic and therapeutic manner. However, the complexities of how lipid metabolism interconnects with oncogenesis and tumor progression are not yet well understood. The list of molecules functionally connected with the MVA biosynthetic pathway in cancer is wide and diverse. It includes: 1) enzymes [e.g., HMGCR (8, 60–62), small GTPases (63, 64), ATP citrate lyase (ACL) (5), AMPK (65–67), FAS (68), pyruvate kinase M2 (PKM2) (69)]; 2) CD36, a fatty acid transporter (68); 3) signaling pathways [e.g., PI3K-AKT-mTOR (70, 71), Hippo (72, 73), Hedgehog (74, 75)]; 4) transcriptional regulators [e.g., SREBPs (68, 76), HIF-1 (77), STAT3 (78–80)], c-MYC (6, 81), YAP/TAZ (72, 73); and 5) nuclear receptors such as LXRs (82), ERα (62, 83), and Estrogen-Related Receptor (ERRα) (84, 85). Additionally, the loss-of-function of tumor suppressor proteins such as p53 (86, 87) and pRb (88–90) can also contribute to adapt lipid metabolism to tumor growth, metastasis, and drug resistance (Figure 2).
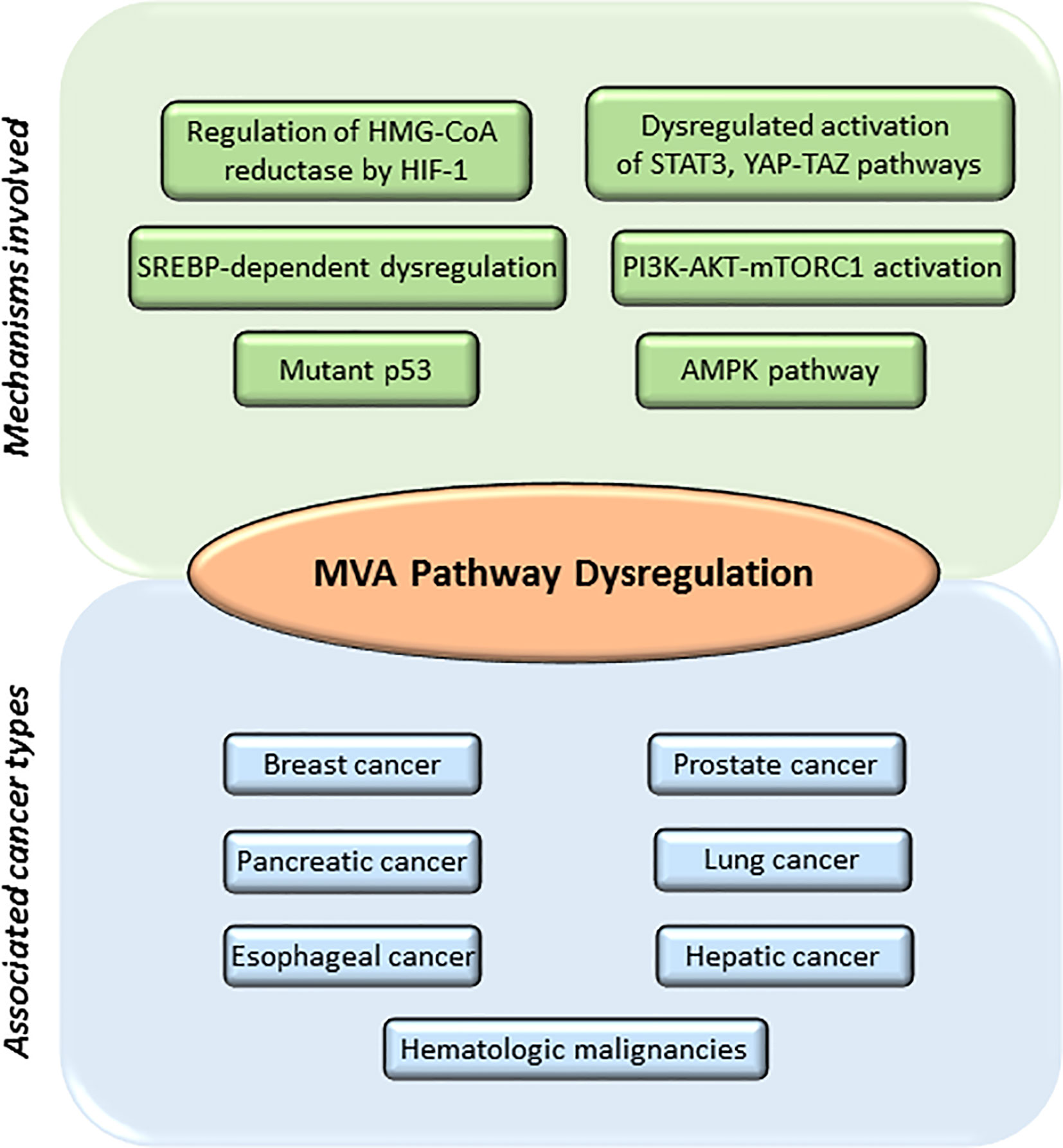
Figure 2 Main mechanisms involved in mevalonate (MVA) pathway dysregulation and different cancers associated. MVA pathway is upregulated in several cancers including breast, prostate, pancreatic, lung, esophageal, hepatic, and leukemia. Main mechanisms involved in the dysregulation of MVA pathway include: abnormal regulation of the enzyme hydroxy-methyl-glutaryl-CoA reductase (HMGCR) by different transcription factors such as hypoxia-inducible factor 1 (HIF-1); mutations or abnormal activation of sterol regulatory element-binding proteins (SREBPs); mutations on tumor suppressor proteins such as tumor protein (p53); decreased AMP-activated protein kinase (AMPK) activation; increased activation of signaling pathways such as phosphoinositide 3-kinase (PI3K)—protein kinase B (AKT)—mammalian target of rapamycin complex 1 (mTORC1), Janus Kinase (JAK)/Signal Transducer and Activator of Transcription 3 (STAT3) or Hippo signaling pathway (YAP-TAZ).
HMGCR
Originally, the hypothesis that MVA-derived metabolites have a role in cancer cell biology was suggested by studying liver cancer (91) and primary chronic lymphocytic leukemia cells (92). Further gene expression profiling and inmunohystochemical analysis identified that HMGCR expression can be associated with a molecular gene signature of certain subtypes of breast cancer (93). The proto-oncogenic role of HMGCR was functionally shown by overexpression of constitutively active HMGCR, which potentiated both anchorage-independent cellular growth in soft agar as well as the development of xenografts (7). Furthermore, dysregulated HMGCR was shown to induce anchorage-independent growth of an immortalized, non-transformed cell line, and support the formation of myeloid colonies from normal hematopoietic progenitors. A link between MVA pathway and oncogenic signaling was also reported with the cooperation between HMGCR and the small GTPase Ras to promote cell transformation (7). Clinically relevant, increased levels of HMGCR were shown to correlate with poor prognosis in breast (93) and prostate cancer (94) patients. Several epidemiological studies have also evidenced that hypercholesterolemia and increased oxysterol production are associated with higher cancer risk (e.g., postmenopausal breast cancer, colon cancer, lung cancer, non-Hodking lymphoma, acute mieloide leukemia) (95). Accordingly, high levels of cholesterol could provide cancer cells with immune surveillance and/or resistance to drug therapy (9, 60). Thus, cholesterol is recognized as an inherent metabolic demand in cancer cells and increased rates of cholesterol synthesis can potentiate the progression of numerous types of cancer (7, 8). This is explained by the fact that highly proliferative cancer cells need to rapidly produce membranes, so requiring higher cholesterol availability than normal cells (8). Besides, cholesterol is an integral component of lipid rafts, which constitute a core of organization for several signaling pathways and intracellular transport systems (96), and is also a precursor of downstream products such as oxysterols and steroid hormones which can drive activation of nuclear receptors in several cancers (97). Thus, decreasing intracellular cholesterol biosynthesis could be a promising strategy to restrain cancer progression. Indeed, it was reported that acute myeloid leukemia (AML) cells exposed to high-cholesterol media in vitro, increased their cholesterol synthesis and influx compared to their normal, non-tumorigenic counterparts. Moreover, AML cells did not usually display efficient feedback repression of cholesterol synthesis and influx, and this appeared to be associated with increased survival of leukemic cells. Interestingly, synthetic LXR ligands can block tumor cell proliferation, tumorigenesis, and metastasis in multiple cancer models, which emphasizes the potential role of LXRs in cancer therapy (82, 98). Cholesterol is also the precursor of steroid hormones, responsible for driving the initiation and progression of hormone-dependent breast and prostate cancers. Recently, it has been shown that long-term E2 withdrawal of ERα-positive breast cancers triggers to the stable epigenetic activation of the MVA pathway and cholesterol synthesis (61). The resulting augmented level of 27-hidroxycholesterol was enough to induce ERα signaling in the absence of exogenous E2, promoting the activation of genes that give rise to an invasive phenotype (62). Likewise, in prostate cancer, the de novo biosynthesis of androgens from cholesterol activates androgen receptor (AR) activity in castration resistant disease (99), thus suggesting a role for the MVA pathway in prostate cancer progression, also considering the observations that SREBP expression is enhanced in advanced stages of prostate cancer. However, these findings require further research into the utility of inhibitors of the MVA pathway and/or SREBPs in the treatment of hormone-driven cancers. All these evidences indicate that increased cellular cholesterol and/or oxysterols, represent another hallmark in many cancers, and suggest that limiting cellular cholesterol levels, or LXR activity, should be considered to improve therapeutic window and sensitivity of cancer cells to chemotherapy. Unluckily, the mechanisms by which HMGCR and the MVA pathway become dysregulated, or how precisely this deregulation promotes carcinogenesis, are still poorly understood, so further studies would be needed in order to elucidate these key questions.
Small GTPases
Rho GTPases belong to the Ras superfamily which comprises more than 20 members classified into eight subfamilies (Rho, Rac, Cdc42, RhoD/RhoF, RhoH, RhoU/RhoV, Rnd, and RhoBTB) according to their structure and function (63, 64). Most Rho family proteins act as molecular switches cycling between an inactive guanosine diphosphate GDP-bound state in the cytoplasm, and an active guanosine triphosphate (GTP)-bound state in the cell membrane. The activation state of Rho GTPases is tightly regulated and occurs in a cell-type and pathway-dependent manner. Although Rho GTPases are mostly known as master regulators of the actin cytoskeleton, they are also involved in cell proliferation, apoptosis, cell cycle progression, and genomic stability, and they are dysregulated in several human cancers (100). Notably, some Rho GTPases have been related to tumor metabolism through activation of glutaminase, which catalyzes the conversion of glutamine to glutamate and ammonia, a crucial step in glutamine metabolism and contributor to the Warburg phenomenon. As Rho GTPases need isoprenylation to properly function, their activity essentially depends on the HMGCR enzyme, thus providing a critical link between the MVA pathway and tumor cell metabolism. Specifically, the isoprenoids FPP and GGPP post-translationally modify proteins with C-terminal CAAX, CXC or CC motifs, such as small GTPases, with very well established roles in carcinogenesis (100). Rho can only be geranylgeranylated, whereas H-Ras is purely farnesylated, and K-Ras and N-Ras can be both farnesylated and geranylgeranylated. Accordingly, inhibition of the MVA pathway can reduce the isoprenylation of these GTPasas and promote apoptosis of cancer cells (100–102). This antitumoral effect can be prevented by the addition of GGPP, and sometimes FPP, suggesting that these MVA-derived metabolites are vital for cancer cell viability. Isoprenoids are also involved in the production of the ubiquinone (quinone coenzyme Q). In this case, the hydrophobic isoprenoid chain places the ubiquinone to the inner membrane of the mitochondria, where the quinone group transfers electrons from complex I or II to complex III of the electron transport chain (ETC) (103). Therefore, ubiquinone is essential for ATP production in cancer cells that rely on oxidative phosphorylation to generate energy. It seems that the depletion of isoprenoid pools, which potentially affect the many proteins that are isoprenylated, mediates the anticancer activity of HMGCR inhibitors (i.e., statins). However, despite dependency of isoprenoids, inhibitors that directly inhibit isoprenylation of small GTPases have not been a successful anticancer strategy to date because, in part, their narrow therapeutic window.
CD36
The fatty acid transporter CD36 is considered as a novel connection between lipids and cancer. It may contribute to regulate cholesterol synthesis (88) and phenotypic changes linked to tumor growth and metastasis (68, 104, 105). In hepatocytes, activation of CD36 increases phosphorylation of Ser872 in HMGCR, and the recruitment of the Insig 1/2 contribute to degradation of HMGCR by the ubiquitin-proteasome pathway. In addition, genes encoding key enzymes involved in MVA pathway, and under the control of SREBP2, remained unresponsive to sterol depletion, due to retention of Scap by Insig-1/2. Interestingly, some fatty acids (e.g., palmitic acid), or a high-fat diet, enhance the metastatic potential of cells in a CD36-dependent manner, whereas blocking CD36 causes inhibition of metastasis in mouse models of human oral cancer, with no side effects (104). Relevant to the connection between oncogenic STAT3 and aberrant lipid metabolism, it has been shown that STAT3-activated CD36 contributes to fatty acid uptake in chronic lymphocytic leukemia cells (106), which supports a critical role of STAT3 in the regulation of CD36-dependent leukemia.
AMPK
The AMP-activated protein kinase (AMPK) was originally described as a protein to lessen anabolic pathways activity when intracellular ATP levels are low (66). AMPK acts as an energy sensor and central regulator of glucose, lipid, and cholesterol metabolism in specialized tissues (e.g., liver, muscle, adipose). This function has placed AMPK as a key therapeutic target in cancer. Decreased AMPK activation can promote carcinogenesis, and the pharmacological induction of AMPK has been reported to be cytotoxic to cancer cells (65, 67). This is in part, because AMPK can regulate the MVA pathway through phosphorylation and thereby inhibition of HMGCR (55) and SREBPs (107) activities. AMPK can phosphorylate the Ser872 within the catalytic domain of HMGCR, suppressing its enzymatic activity, independently of its feedback regulation by MVA-derived metabolites. Moreover, the transcription factors SREBPs are direct targets of AMPK phosphorylation, as AMPK inhibits the proteolytic processing, nuclear translocation, and transactivation activity of SREBPs, after their activation (e.g., under hyperglycemic and hyperinsulinemic conditions). Interestingly, activation of AMPK in the liver of insulin-resistant mice was shown to inhibit the transcription of enzymes that participate in lipid and cholesterol biosynthesis, including HMGCS1 and HMGCR, thereby reducing hepatic triglyceride and cholesterol levels. Thus, AMPK can inhibit the activity of MVA pathway both, directly, via HMGCR phosphorylation and, indirectly, through SREBPs inhibition. However, the relevance of this regulation in the context of cancer is still scarcely regarded. The MVA pathway may besides modulate AMPK activity, thereby forming a feedback loop (108). The discovery that the serine-threonine kinase Liver Kinase B1 (LKB1), a known tumor suppressor, was a crucial upstream activator of the AMPK, added a relevant piece of information to our understanding about the connection between cell metabolism and cancer (109). It is therefore feasible that the anticancer effects of AMPK activation and the tumor suppressor role of its upstream kinase LKB, are in part due to the inhibition of HMGCR and the MVA pathway. LKB1 is modified by protein farnesylation and it phosphorylates and activates AMPK. This suggests a negative feedback loop, where AMPK activation, in response to reduced cellular energy, results in the suppression of the MVA pathway via the phosphorylation of HMGCR and SREBPs. This reduces in turn the FPP pool inside the cell, thereby hampering LKB1 farnesylation and blocking activation of AMPK. Remarkably, AMPK activation has also been reported to suppress cell proliferation in normal and cancer cells by regulating cell cycle progression or inhibiting protein synthesis (110, 111). In line with this, recent studies have shown that simvastatin, a potent HMGCR inhibitor, induces apoptosis and cell cycle arrest by activating AMPK and inhibiting the Signal Transducer and Activator of Transcription 3 (STAT3) axis, both in liver cancer cells and tumor xenografts (112, 113). However, restoration of MVA reversed the activation of AMPK and the suppression of STAT3 caused by statin treatment. These findings contributed to demonstrate that AMPK induction and STAT3 inhibition in liver cancer cells are dependent on HMGCR activity. Thereby, MVA signaling pathway, AMPK and STAT3 activities may represent potential therapeutic targets in liver cancer.
The PI3K-AKT-mTORC1 Axis
In normal cells, the mTORC1 activity can be activated by diverse stimuli (i.e., growth factors, nutrients, energy, and stress signals), and key signaling pathways (i.e., PI3K-AKT, MAPK and AMPK), in order to regulate cell growth, proliferation and survival. Upon stimuli, PI3K produces PtdIns(3,4,5)P3 which binds to AKT and 3-phosphoinositide-dependent protein kinase (PDK1). In contrast, the inactivation of AKT is regulated by PTEN that converts PtdIns(3,4,5)P3 into PtdIns(4,5)P2, driving to a lower recruitment of AKT to the cell membrane (114, 115). An increased mTORC1 activity is observed in 40–90% of the most frequent human cancers. The aberrant activation of PI3K-AKT-mTORC1 signaling leads to an increase activity of the MVA biosynthetic pathway and lipogenesis, and the reprograming of lipid metabolism contributes to potentiate tumor growth (70, 71). Several mechanisms are implicated, including the inactivating mutation of PTEN (116, 117), the mutation/amplification of PI3K-AKT (118), the hyperactivation of PI3K-AKT signaling pathway by growth factors (e.g., insulin, PDGF, VEGF, HER-2, IGF-I), the overexpression of mTORC1 targets (i.e., S6K1, 4BP1, eIF4E), or the loss of tumor suppressors (e.g., PTEN, LKB1, or TSC). These are common mechanisms that increase de novo cholesterol synthesis and fatty acid biosynthesis in cancer (119, 120). Upregulated PI3K-AKT activity increases glucose uptake and glycolysis rate in cancer cells, a mechanism that provides NADPH and acetyl-CoA to the MVA pathway. Conversely, inhibition of the MVA pathway can decrease PI3K activity. The PI3K-AKT-mTOR pathway connects with SREBP-mediated lipid biosynthesis by using complex protein-protein interactions and phosphorylation of regulatory elements (121, 122). Interestingly, AKT prevents proteasomal degradation of nuclear SREBPs which increases de novo cholesterol and fatty acid biosynthesis. This role of AKT on lipogenesis, and tumorogenesis, is blocked after gene silencing of SREBPs. Furthermore, the connection of mTOR with SREBPs was evidenced by enhanced lipogenesis in response to mTORC1 activation whereas inhibition of mTORC1 with rapamycin blocked both active SREBP and expression of SREBP target genes. In addition to a positive regulation of SREBP, mTORC1 has a main role in regulation of protein synthesis through phosphorylation of downstream effectors such as 4EBP1 and S6K1. Targets of S6K1 include 40S ribosomal protein S6, protein elongation factors, and IGF-II. Clinically relevant, human primary breast cancer samples with high levels of pS6K1, as a marker of mTORC1 activity, had high expression of SREBP target genes (e.g. FASN, LDLR, MVA kinase). In contrast, breast cancer cell lines with silenced SREBPs (1 and/or 2) showed reduced proliferation and increased cell death despite activation of mTORC1. Finally, mTORC1 activation has also been linked to proteins such as STAT3, STAT5 and PPARγ, in a rapamycin sensitive manner. Thereby, aberrant activation of PI3K-AKT-mTOR axis can reprogram protein and lipid biosynthesis in an orchestrated manner to provide efficient tumor growth.
The Hippo Pathway
The Yes-associated protein (YAP) and the transcriptional co-activator with PDZ-binding motif (TAZ) are key downstream terminal effectors of the Hippo signaling pathway (123). In normal tissues, YAP-TAZ proteins are phosphorylated at specific serine residues in order to confine their subsequent degradation in the cytoplasm (124). However, in cancer, YAP-TAZ proteins are translocated into the nucleus where they bind to TEA domain (TEAD) proteins which drive the transcriptional activation of proliferative genes, the repression of pro-apoptotic genes and the amplification of stem/progenitor cells. Increasing evidences have shown that deregulated Hippo pathway is significantly associated with cancer development (72, 73). Remarkably, YAP and TAZ require the MVA biosynthetic pathway to translocate into the nucleus and be fully functional (72). In fact, it has been reported that the concurrent knockdown of SREBPs (1 and 2) reduces nuclear localization of YAP-TAZ, suggesting the importance of SREBP-mediated induction of the MVA for YAP and TAZ nuclear localization (72). Interestingly, activation of both the MVA pathway and YAP-TAZ is correlated with mutant p53 expression in primary tumors, suggesting a dysfunctional mutant p53-SREBP-YAP-TAZ axis in cancer (72). Relevant to this review, the MVA pathway is an essential intermediate in the oncogenic activation of YAP and TAZ by mutant p53 (72). When statins are used to inhibit the HMGCR activity in the MVA pathway, the nuclear localization and transcriptional activity of YAP-TAZ are also inhibited. GGPP may be involved in this process, as it is known to promote YAP-TAZ nuclear translocation and increase their transcriptional activity via activation of Rho GTPases. Thus, when MVA pathway is inhibited, also GGPP is, thereby reducing YAP-TAZ activity. Additionally, it has been shown that YAP-TAZ can be activated by SREBPs, main regulators of MVA pathway, in a breast cancer cell line. Interestingly, mutant p53 promoted YAP-TAZ transcriptional activity and contributed to cancer cell malignancy by maintaining SREBP expression in MVA pathway. Taken together, these data clearly show that MVA participates in the regulation of YAP-TAZ expression and transcriptional activity and reveal an original process through which statins have anticancer effects.
The Hedgehog Pathway
Members of the Hedgehog (Hh) family of secreted signaling proteins have an essential role in the regulation of vertebrate development and adult tissue homeostasis, including regulation of stem cell physiology (74, 75). Reduced Hh pathway activity can cause development defects in mice and humans, and aberrant increased activity of this pathway is linked to tumorigenesis. The core components of the Hh pathway include: the secreted signaling ligand Hh, the twelve-pass transmembrane receptor Patched (PTCH), the seven-pass transmembrane co-receptor G-protein-coupled receptor (GPCR)-like transducer Smoothened (SMO), and the glioma associated-oncogene (GLI) (74, 75). After secreted from the producing cells, Hh binds to PTCH on the cell surface, and subsequently release suppression of PTCH on SMO. Then, activation of SMO triggers GLI-dependent expression of downstream target genes through a complex network of post-translational modifications and translocations. There are positive and negative feedback loops that ensure a homeostatic regulation of Hh signaling pathway, which include an increment of GLI levels or the potentiation of the activity of negative regulators such as PTCH1, respectively. Relevant to this review, the Hh signaling pathway is regulated by cholesterol and oxysterols, main products of MVA biosynthetic pathway (74). It has been established that cellular cholesterol is an endogenous ligand of SMO. Thereby, cholesterol levels can modulate the Hh signaling pathway by direct binding to GPCR-SMO (50). Thus, cholesterol itself can be used as a substrate for the post-translational modification of Hh ligands, required for biological activities of Hh, as well as a molecule for long-distance and local Hh signal communication. Thereby, inhibitors of MVA pathway (e.g., statins) that modulate Hh pathway activity could represent potential drugs in Hh pathway-related cancers.
Hypoxia-Inducible Factors (HIF)
Under hypoxic conditions, cells respond by suppressing energy-consuming processes to preserve energy, including mitochondrial respiration (125, 126). These conditions promote the activation of the Hypoxia-Inducible Factors (HIF). The HIF protein family consists of three α members (i.e., HIF-1α, HIF-2α, and HIF-3α) and two β members (i.e., HIF-β and ARNT2), which have a similar domain structure (127). Under hypoxic conditions, HIF-1α is stabilized, binds DNA, and regulates the transcription of glycolytic target genes in cancer cells (125, 128). Several observations have shown that the MVA pathway can be directly or indirectly modulated under hypoxic conditions, in part, because HMGCR expression is regulated through the transcriptional activity of HIF-1α (129, 130). It has been reported that HIF-1α connects pathways for oxygen sensing and feedback regulation of cholesterol synthesis in human fibroblasts by directly inducing the transcription of the INSIG-2 gene. INSIG-2 is an ER membrane protein that inhibits cholesterol synthesis by mediating sterol-induced ubiquitination and subsequent degradation of the HMGCR. Furthermore, pharmacologic stabilization of HIF-1α in the liver was shown to trigger accelerated HMGCR degradation by prior ubiquitination (131). Pharmacologically relevant, in other pathologic fields such as Alzheimer´s disease, statins (simvastatin) have been shown to reduce intracellular levels of HIF-1 expression (132). Likewise, fluvastatin was shown to accelerate ubiquitin/proteasome-dependent degradation of HIF-1, effect that was reversed by concomitant treatment with mevalonate, farnesyl pyrophosphate, or geranylgeranyl pyrophosphate (133). While HIF has been broadly studied as an essential protein for modulation of transcriptional program during the hypoxia response, many other transcription factors (e.g., NFkB, Nrf2, Myc, STAT3) and/or tumor suppressors also function under hypoxic conditions to promote the acquisition and maintenance of metabolic reprogramming phenotype in cancer. Further understanding about the connections between these transcription factors and the MVA pathway constitutes an ongoing challenge.
Signal Transducers and Activators of Transcription (STAT)
The STAT family of transcription factors consists of 7 members within STAT3 highlights by its oncogenic activity. STAT3 appears constitutively active in a broad variety of cancers that often become addicted to its activity (78–80). In contrast to normal STAT3 activity, which is transient, constitutively active STAT3 is associated with abnormal cell growth and survival, angiogenesis and metastasis, tumor immune evasion, and aberrant mitochondrial function. While tyrosine phosphorylation by Janus Kinases (JAK), represents the main activation mechanism of STATs, alternative mechanisms, such as the interaction with HIF signaling pathway, phosphorylation of STAT3 on S727 (134), and regulation of activity and nuclear traffic by small GTPases (135), appear to play important roles that connect STAT3 oncogenic activities with deregulated metabolism in cancer cells. An important component of STAT3 oncogenic activity resides in the induction of aerobic glycolysis, making cancer cells highly sensitive to glucose deprivation, whereas they are protected from apoptosis and senescence. Accordingly, inhibition of STAT3 tyrosine phosphorylation in several cancer cells down-regulates glycolysis prior to leading to growth arrest and cell death. STAT3-addicted cancer cells can develop a switch towards aerobic glycolysis program through two mechanisms: a) the up-regulation of HIF-1α, which in turn mediates the induction of several glycolytic genes [e.g., hexokinase 2, LDH-A, pyruvate dehydrogenase kinase 1 (PDH), PKM2] (136); and b) the down-regulation of mitochondrial activity, which is totally or partially independent of HIF-1α. HIF-1α induces PKM2 expression, which maintains STAT3 tyrosine phosphorylation, a mechanism that initiates a positive feedback loop that leads breast cancer cells to adapt and grow into hypoxia conditions (137). Similarly, hypoxia can activate oncogenic STAT3 in prostate cancers cells, and, together with the AKT and HIF-pathways, induces an androgen-independent and invasive phenotype (138). In addition, STAT3 phosphorylation on S727 has emerged as a crucial regulator of metabolic processes in the mitochondria. Indeed, S727-STAT3 was found to enhance Complex I and II activities and reduce ROS production within the mitochondria (139, 140). This function appears to be essential for cellular survival under certain stress conditions such as heart ischemia, where mitochondrial STAT3 protects cardiac cells from apoptosis (140). Furthermore, mitochondrial STAT3 potentiates RAS-mediated oncogenic transformation. This finding supports the role of STAT3 in maintaining cell survival and oncogenesis, linked to a metabolic adaptation of cancer cells. In contrast, mitochondrial expression of an inactive mutant S727A-STAT3 was shown to inhibit growth and metastatic capacity of the breast cancer cell line 4T1, and this inhibition correlated with reduction of Complex I activity under hypoxia (141). The form S727-STAT3 can also be induced by the mTOR pathway to potentiate the expression of STAT3 target genes (e.g., Bcl-xL, VEGF, cyclin D2) (142, 143). Moreover, activated forms of small GTPasas such as Rac1, Cdc42 or RhoA directly or indirectly promote the phosphorylation and activation of STAT3 (134, 144, 145). Particularly, Rac1 specifically induces an increase in Rac1 and Cdc42 protein levels and activities, and stimulates autocrine IL6 secretion, which contributes to an increase of STAT3 activity. Interestingly, the activated form of Rac1, but not its inactive variant, forms a complex with STAT3 to regulate its phosphorylation and activity (146, 147). Interestingly, the Rac (and Cdc42) GTPase activating protein MgcRacGAP plays also a critical role in STAT3 activation (148, 149). When activated, the complex MgcRacGAP-Rac-GTP interacts with STAT3 to promote its binding to the IL6 receptor thus facilitating that JAK phosphorylates and activates STAT3. These observations suggest that MgcRacGAP, a core regulator of cytokinesis, and other Rho proteins, support oncogenic properties of STAT3. All observations indicate that STAT3 can integrate different pro-survival and growth signals in a context of energy and respiratory metabolism, emerging as a key molecule to target within the mitochondrial metabolism.
Tumor immune microenvironment (TIM), including surrounding (niche) and inflammatory cells, plays a key role in the development of angiogenesis, proliferation, immunosuppression, and tumor progression (150). These biological effects depend, at least in part, on the aberrant activation of STAT pathway which is an immuno-inflammatory-carcinogenic pathway. Thus, in addition to their roles in adaptive metabolism of cancer cells, aberrant STAT activity can drive cancer development through the regulation of TIM. This is particularly relevant for STAT3 and STAT5 which are highly expressed in Tumor-Associated Macrophages (TAMs), a critical cellular component of TIM. It is well known that TAMs are recruited into tumor formation by chemo-attractant cytokines and, once inside the tumor, tumor cells secrete cytokines that prolong the survival of TAMs; these cells, in turn, express multiple factors that promote tumor development and immunosuppression. STAT3 and STAT5 have been reported to act by inhibiting the antitumor immune response by activating, at least in part, the production of inflammatory cytokines (IL-1, IL-17, IL-10, TGF-β, or VEGF) and promoting tumor growth and metastasis (150). Moreover, it has been described that TAMs could favor the development of tumor resistance to conventional chemotherapy, highlighting the importance of the microenvironment in tumor development. In addition to TAMs, the influence of niche stem cells on tumor development and drug resistant is also relevant. Interestingly, many studies have shown that STAT activity is essential to localize, maintain, and renew Hematopoietic Stem/Progenitors cells (HSPC) into tumoral niche and that STAT hyperactivation is associated with uncontrolled proliferation of HSPC (151–155). Thereby, dual strategies targeting both tumor cell proliferation and tumor niche and/or regulation of TIM represent a promising therapeutic strategy (156, 157). Interestingly, the effects of MVA biosynthetic pathway inhibitors (i.e., statins, bisphosphonates) on TAMs suggest that TIM can be regulated by MVA biosynthetic pathway (158–160). However, despite TIM is known to be highly dependent of cholesterol biosynthesis (10, 161–164), its interplay with MVA biosynthetic pathway and STAT signaling, remains unexplored.
MYC
MYC belongs to the Myc gene family that is comprised by C-MYC, N-MYC, and L-MYC, and they have been shown to influence almost all aspects of carcinogenesis, including rapid cell growth, inhibition of cell differentiation, genomic instability, metastasis, or angiogenesis (165–167). Aberrant regulation of MYC is observed in more than 50% of cancers, where this oncoprotein is overexpressed, either due to enhanced transcription of the Myc gene or to dysregulated stability of MYC protein. The stability of MYC can be modulated by a) the ubiquitin/26S proteasome pathway, and b) the sequential phosphorylation of MYC at S62 and T58. The phosphorylation of S62 is controlled by the MAPK/ERK pathway and leads to the stabilization of MYC, whereas its phosphorylation on T58 is mediated by GSK3β and promotes ubiquitin-dependent MYC degradation once S62 is dephosphorylated by, for example, the serine/threonine-protein phosphatase 2A (PP2A) (165, 168). Mutations on the phosphorylation sites that stabilize MYC have been identified in human cancers, thus highlighting the relevance of S62 and T58 phosphorylation as regulators of MYC tumorigenic activity (169). MYC is a major driver of metabolic reprogramming in cancer, where this transcription factor regulates the expression of genes involved in anabolic metabolism, cellular bioenergetics and lipid metabolism (167, 170, 171). This oncoprotein can reprogram cancer cell metabolism toward glycolysis and MVA pathway to drive the proliferation and survival of cancer cells. Accordingly, it has been reported that knockdown of c-Myc in gastric cancer cells suppresses glycolysis rates and cell proliferation capacity. MYC can also bind SREBP to drive somatic cell reprogramming into induced pluripotent stem cells (171), or bind to promoters of MVA pathway genes in close proximity to SREBPs (8), suggesting that MYC may contribute to the expression of MVA pathway enzymes. Notably, HMGCR is a positive regulator of phosphorylation, activation, and tumorigenic properties of MYC in a MYC-driven model of hepatocellular carcinoma where exogenous mevalonate deliver can enhance cancer growth (168). In agreement with the positive role of MVA pathway in MYC-induced oncogenic activities, the antitumoral effects of statin were prevented by mevalonate. This effect was associated with a reduction of small GTPase RAC isoprenylation levels and PP2A activation. Moreover, when tumors that expressed active phosphorylated mutants of MYC at S62 or Th58 were studied, there was an increase of tumor resistance to statin treatment which supported the role of serine/threonine phosphatase PP2A as a negative regulator of MYC (168). Recently, studies on MYC null mice showed that mice had improved lifespan, which was linked to the decreased expression of MVA pathway genes, including HMGCR and SREBP2, and most likely to caloric restriction (172). Finally, RAS, whose activity is also regulated by the MVA pathway is thought to modulate MYC activity and enhance levels of HIF-1, independently of hypoxia conditions (173, 174). These findings reinforce the hypotheses that MYC dependent oncogenesis is linked to a deregulated MVA biosynthetic pathway.
The ERRα Pathway
Estrogen-Related Receptors (ERRs) are a group of nuclear receptors with three isoforms (α, β, and γ) (84, 85, 175). ERRα is mainly expressed in high-energy demanding tissues where it associates with the co-regulator peroxisome proliferator-activated receptor—γ co-activator 1 (PGC-1). In differentiated cells, ERRα, together with PGC, controls cellular metabolism, assists the growth of rapidly proliferating cells, directs metabolic programs necessary for cell differentiation, and keeps cellular energy homeostasis. In several cancer cells, the expression, and the activity of ERRα, and its cofactor PGC-1, is further influenced by oncogenic signals (e.g., IGF1 receptor pathway, estrogen signaling, mTOR pathway) and induces metabolic programs favoring cell growth and tumor progression. This is particularly relevant when there is a functional relation between augmented cholesterol levels and certain cancer phenotypes, with an overexpression of ERRα [i.e. colorectal cancer (176), prostatic, and breast cancers (177)]. Notably, ERRα activity promotes an inflammatory environment by the production of cytokines that supports a protumoral microenviroment (178). Recently, affinity chromatography and transcriptional assays have identified cholesterol as an endogenous ligand and agonist of ERRα (84). A functional link between cholesterol (or MVA pathway) and ERRα has been described in bone, muscle, and in the immune system (macrophages). Particularly, cholesterol-induced bone loss or bisphosphonate osteoprotection are lost in ERRα knockout mice. In addition, statin induction of muscle toxicity and cholesterol suppression of macrophage cytokine secretion are impaired by loss or inhibition of ERRα. These findings showed that cholesterol is an ERRα agonist and that the MVA biosynthetic pathway impacts biological functions of ERRα (85). Thereby, the use of therapeutic strategies that aim to decrease cholesterol levels (e.g., statins, biphosphonates) could be an encouraging way to counteract metabolic reprograming in cancer cells where ERRα plays a critical role.
ERα
The MVA biosynthesis pathway was recently reported to be up‐regulated in ERα‐positive breast cancer cells lines that are resistant to E2 withdrawal (61, 62, 83). This suggests that dysregulation of cholesterol biosynthesis may be a mechanism of anti-estrogen resistance in ER‐positive breast cancer. Mechanistically relevant, chronic estrogen removal in ERα‐positive breast cancer cells seems to stabilize the epigenetic activation of the MVA pathway and cholesterol biosynthesis (61). This leads to the accumulation of cholesterol-derivative metabolites (i.e., 27HC) which, in the absence of estrogens, acts as ERα agonist, and then potentiates ERα signaling to induce the transcription of genes involved in proliferation and invasion. Therefore, statins might act as anti-breast cancer drug by reducing circulating cholesterol and 27HC, and the availability of these ERα agonists in breast cancer cells. Furthermore, direct suppression of HMGCR by statins depletes intratumoral levels of isoprenoids, which are also key modulators of breast cancer cell proliferation and metastasis.
Tumor Suppressor Proteins
Loss-of-function of tumor suppressor protein p53 (TP53) (86, 87) and cyclin-dependent kinases (cdks)- retinoblastome suppressor protein (Rb)- transcription factor E2F Transcription Factor 1 (E2F1) pathway (88–90), novel regulators of metabolism, promotes the acquisition and maintenance of glucose and/or lipid metabolism reprogramming phenotype in cancer. The mutated forms of the tumor suppressor protein TP53 confer oncogenic properties to p53 in a broad range of cancer types (87). Specific oncogenic mutations lead p53 to functionally interact with nuclear SREBP2 and enhance the transcription of MVA genes (86) (Figure 3). Furthermore, an increased expression of mutant p53 in primary breast cancer tissues has been associated to the augmented expression of MVA pathway genes. In contrast, wild-type p53 can decrease lipid synthesis under glucose starving conditions by inducing the expression of phosphatide phosphatase LPIN1, a protein that can prevent SREBPs-DNA binding. Thus, the interaction between p53 and the MVA axis suggests that this pathway may be a novel therapeutic target for tumors with specific p53 gain-of-function mutations. Another example of mutated tumor suppressor gene that leads to an oncogenic phenotype is the cdks-Rb-E2F1 pathway. Analysis of genetically engineered mice deficient in cdk, E2F1, or Rb protein, showed an adaptive reprogramming to metabolism of glucose and/or lipids, including MVA biosynthetic pathway. This showed that the cdk-Rb-E2F1 pathway acts as a key regulator of cell growth, proliferation, and development by sensing external signals that require a particular adaptive metabolic reprograming. Particularly, this cell cycle regulatory pathway is an essential regulator for decreasing oxidative metabolism and, at the same time, to increase lipid synthesis and glycolytic metabolism. Interestingly, loss of Rb causes abnormal expression of the farnesyl diphosphate synthase (FDPS), many prenyltransferases, and their upstream regulators SREBPs, in an E2F-dependent manner, leading to an increased isoprenylation and activation of N-Ras (89). Additionally, loss of Rb reduces the suppression of E2F (1 and 3), a mechanism that leads to promoter activation of prenyltransferase genes. Conversely, the presence of active Rb prevents the association of SREBPs with the FDPS promoter, suggesting that Rb negatively modulates the MVA pathway at both the transcriptional and the post-translational level.
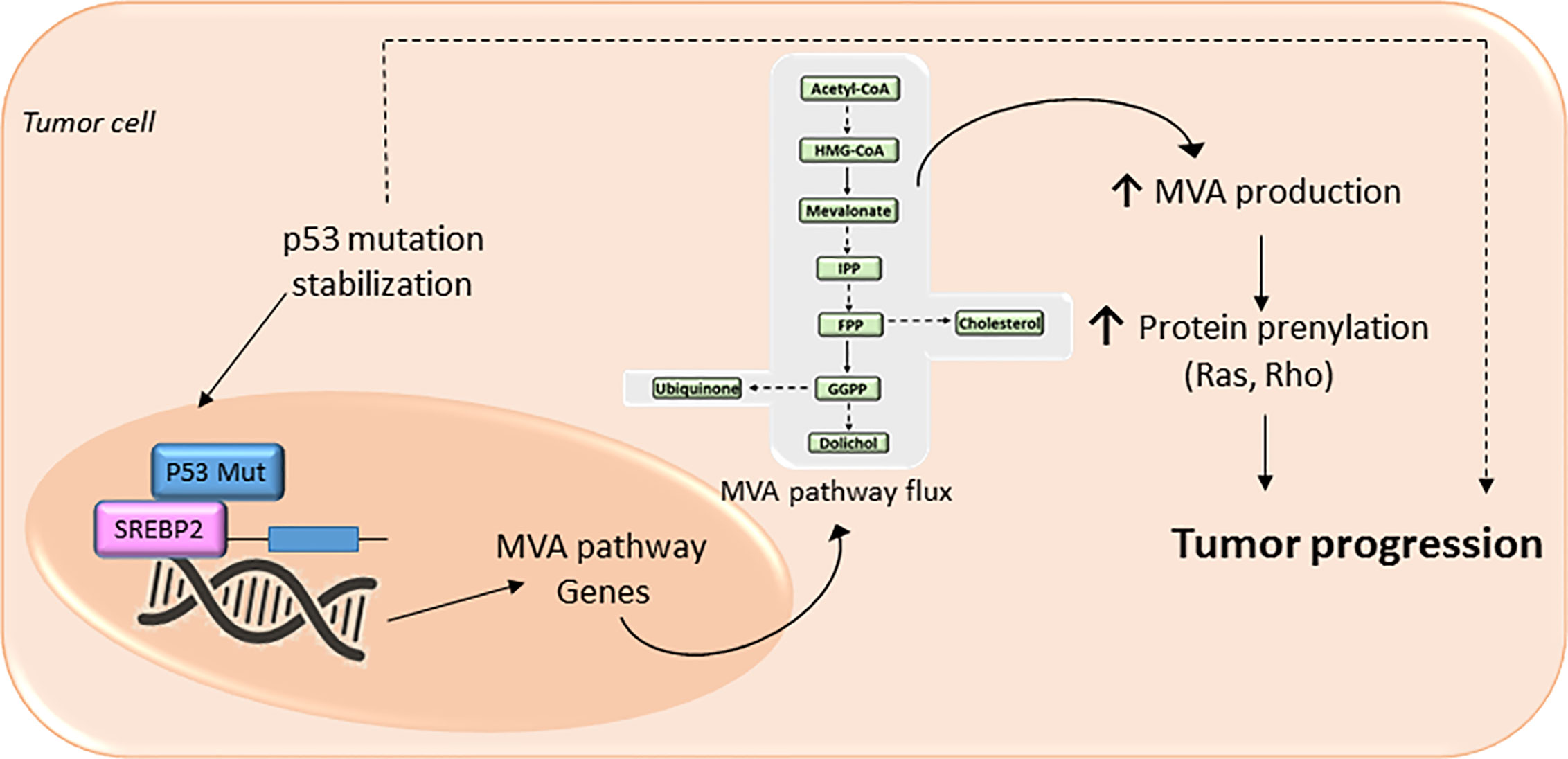
Figure 3 The mevalonate (MVA) pathway in cancer progression. The MVA pathway is dysregulated in several cancer cells due to mutations or abnormal signaling of different proteins/pathways. Upregulation of MVA pathway drives to increased protein prenylation thus promoting a malignant phenotype of cancer cells with an uncontrolled cell invasive growth and survival. In cancer cells expressing a mutation of tumor protein p53, there is a positive-feedback loop where p53 interacts with sterol regulatory element-binding protein (SREBP), leading to increased activation of the MVA pathway activity, and therefore higher levels of MVA. This MVA leads to the stabilization of p53 mutation as well as promotes protein prenylation, thus accelerating cancer progression.
Cholesterol Contributes to Chemotherapy Resistance
Upregulated MVA pathway contributes to chemotherapy resistance by increasing both isoprenoids and cholesterol levels (10), thus generating a serious problem that arises in the treatment of many cancers. It has been shown that, in response to chemotherapy in vitro, some leukemic cells (i.e., AML cells) abnormally increased their cholesterol levels, whereas when this response is blocked with HMGCR inhibitors (i.e., statins), they increased its sensitivity to cytotoxic drugs (179). Interestingly, apoptosis resistance, typically observed in cancer (i.e., hepatocellular carcinoma (HCC), colon cancer and HeLa cells), has been related to cholesterol accumulation in mitochondria, resulting in decreased membrane fluidity (180–182). These data suggest that high mitochondrial cholesterol content contributes to chemotherapeutic resistance, especially to chemotherapeutic agents targeting mitochondria (182). On the other hand, as deeply discussed in a later section of the present review, it has been reported that HMGCR inhibitors (i.e., simvastatin) are able to overcome resistance or to potentiate the antitumoral effects of conventional chemotherapy in several models of cancer (in vitro and in vivo), such as, non-small cell lung cancer (183), resistant colorectal tumors (184) and human gastric cancer (39, 185). Therefore, inhibition of de novo cholesterol synthesis by statins may restore the efficacy and overcome resistance to conventional chemotherapy.
Efficacy and Resistance to MVA Pathway Inhibitors in Human Tumors
MVA biosynthetic pathway is considered a potential drug target to improve therapeutic window in cancer (11) (Figure 4). However, despite mounting body of preclinical and epidemiological evidences suggesting MVA pathway inhibitors (i.e., statins) as anticancer agents, many patients remained non-responsive to drug treatment in some cancer clinical trials (13, 20, 23, 30, 33, 186). This is, in part, because cancer cell selectivity, as well as predictive biomarkers of drug efficacy and drug resistance, is still poorly understood. Therefore, clinical trials are still required to further characterize the subset of cancers that are especially sensitive to MVA pathway inhibitors. The major limitation for the development of MVA pathway-based therapy is the absence of predictive biomarkers of efficacy and chemotherapy resistance, which is due to, at least in part, the lack of routine genotyping of human tumors. Therefore, predictive biomarkers, stratifications of patients, and selection of drug combination-based therapies may lead to a more effective MVA pathway-based therapy in cancer. Nowadays, there are a few completed clinical trials in which statins are used as monotherapy. Some of them have exhibited promising evidence of therapeutic potential and survival benefit mainly in breast cancer (187–189) and multiple myeloma (MM) (190, 191) (Table 1). Breast cancer clinical trials, using atorvastatin and fluvastatin, have shown decreased proliferation index marker in a subset of patients who were treated with cholesterol-management doses of statins between cancer diagnosis and surgery (187, 189) (Table 1). Moreover, a phase II window-of-opportunity trial has shown that high-dose atorvastatin (80 mg/day) induced anti-proliferative effects in breast cancer through cell cycle regulation via cyclin D1 and p27 (188) (Table 1). Although the molecular mechanisms are still unknown, hepatocarcinoma also seems to be particularly responsive to statins (193). Exposition to simvastatin has also been associated to reduced risk of hematological malignancies (194). Although clinical trials with statins show that some tumors may be more sensitive to statins than others, few of them have specifically enriched for subsets of patients whose tumors are preferentially sensitive to statins. As described above, tumors harboring an aberrant MVA pathway may be more sensitive to the antitumoral action of statins. This hypothesis follows the general principles of oncogene addiction and may provide the basis on which patients should be treated with statins. However, follow-up studies are still needed before validating those biomarkers to predict which cancers will be specifically sensitive to statin therapy. For example, certain phases of cancer progression, such as breast cancer recurrence, are particularly sensitive to the antitumoral effects of statins (195, 196). This is in line with the current paradigm of inter-patient cancer biodiversity. This lack of response might also be expected considering the evidence that the MVA pathway is regulated by many critical oncogenic signals. For example, a poor outcome has been reported in clinical breast cancer samples that carry a mutant form of p53 that stimulates the activity of MVA pathway (86). A molecular hallmark of basal transcriptome has been developed to forecast statin response in breast cancer in vitro (197) and aberrant MYC expression has been proposed as an indicator of statin response in specific cancer types (198). Notably, subsets of statin-sensitive and statin-insensitive cells were described in MM cell lines (199, 200). Remarkably, insensitive cells exhibited a robust feedback response, like normal cells, with an immediate up-regulation of different SREBP target genes, including HMGCR. In fact, recently, it has been reported that resistance of breast cancer cells to statins is, at least in part, due to the induction of HMGCR (201). Although the sterol feedback response tried to reinstate the MVA pathway, sensitive cells appear to show, in comparison with statin-insensitive cells, a weaker feedback response. This suggest that statin-sensitive cells have either lost checkpoint controls maintaining the MVA pathway intact, or that the pathway is deregulated and decreased HMGCR activity was not detected by the common intracellular sensors (i.e., SCAP, INSIGs). It appears that the sterol feedback response may serve as a protective mechanism, warranting that normal or statin-insensitive tumor cells are protected from the effect of statins. However, the loss of this sterol feedback response may not be a universal phenomenon across all statin-sensitive cancer types as it has been shown an intact sterol feedback response in AML cells (202). Moreover, tumor cholesterol may also be used a as a biomarker of statin sensitivity in many AML cells exposed to chemotherapy in vitro (202, 203). All these observations suggest that aberrant MVA pathway may be both a promoter of transformation and an indicator of statin sensitivity. Moreover, these data also establish the bases to further developing biomarker tools that could allow to predict which cancers are more sensitive to statins. That may provide a personalized medicine approach in which statins, and/or other inhibitors of MVA pathway, would constitute a successful class of anti-cancer drugs.
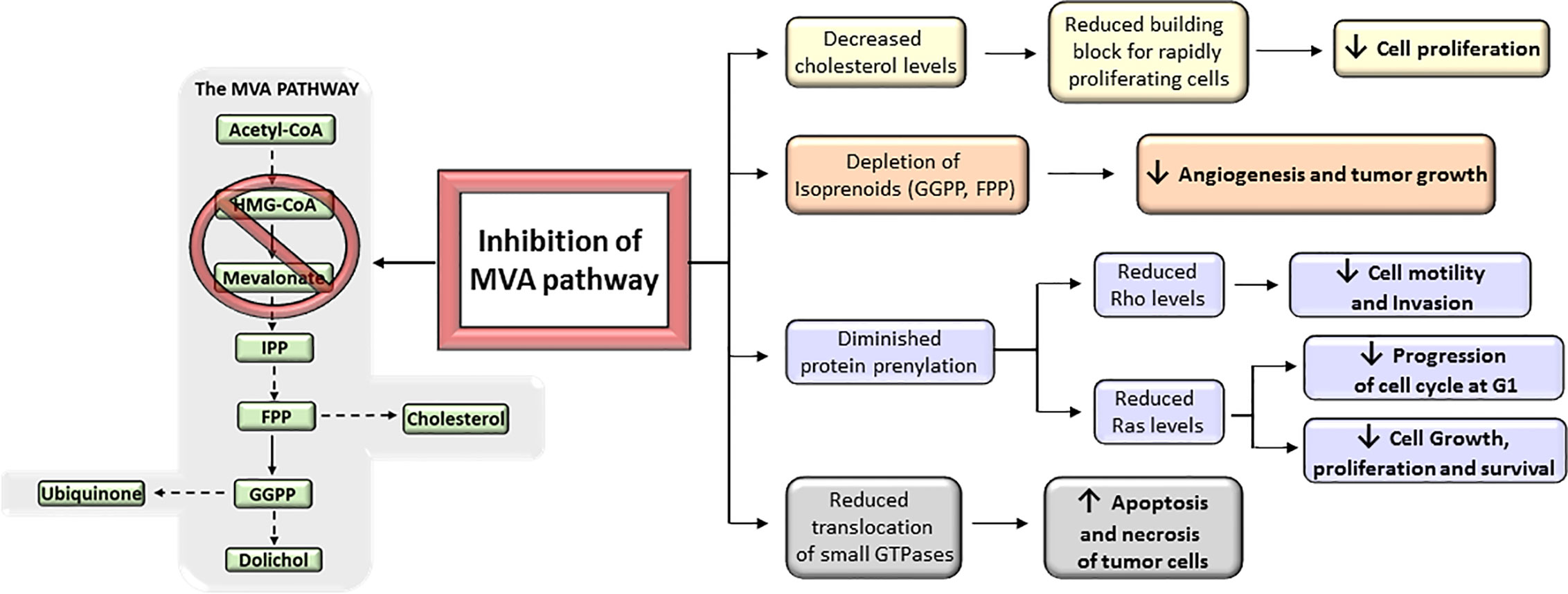
Figure 4 Antitumoral effects of mevalonate (MVA) pathway inhibition. The inhibition of MVA pathway triggers a series of anticancer events that get to inhibit tumor growth and progression. These include the reduction of MVA synthesis, which in turn decreases the levels of downstream products (isoprenoids) and therefore prevents protein prenylation; the reduction in the translocation of small GTPases such as Rho and Ras to the cell membrane; and the inhibition of cholesterol synthesis. All these inhibitory actions suggest that targeting the MVA pathway could represent a key mechanism to prevent cancer progression.

Table 1 Completed clinical trials with statins used in mono- or in combination therapy for cancer treatment.
SREBP, a Drug Target to Increase Statin Efficacy and Overcome Drug Resistance
Inhibition of the MVA pathway leads to the activation of the SREBPs and the increased expression of MVA pathway genes, an effect that may be intensified in cancer cells and be responsible of statin resistance (201, 204). This SREBP-mediated feedback mechanism also increases the expression of the LDLR, and LDL-cholesterol uptake, which has been shown relevant in cancer cells (205, 206). Thus, the SREBPs work to replenish MVA-derived metabolites, which can depress the apoptotic response following statin treatment. Recent studies targeting the maturation or transcriptional activities of SREBPs supply proof of concept for the efficacy of SREBP inhibition in cancer therapy (204). Inhibiting the SREBP-regulated feedback response together with statin therapy could prevent drug resistance and increase the antitumoral efficacy of statins. In addition to HMGCR, the MVA pathway genes HMGCoAS1, GGPS1, SCAP, and SREBP2 are also good candidates to either suppressing other enzymes in the MVA pathway or blocking the SREBP-mediated feedback response in combination with statin therapy (207). Particularly, the clinically approved agent dipyridamole may be repurposed as an inhibitor of statin (fluvastatin)-induced SREBP processing and blocks the SREBP-regulated feedback response. This mechanism can potentiate antitumoral efficacy of statins, at least in prostate cancer, and most likely prevent drug resistance (208). However, preclinical and clinical investigations performed in order to investigate the utility of this combinatory drug strategy in cancer (i.e., HMGCR inhibitors plus SREBP inhibitors), are still a pharmacological challenge. Hopefully, other molecules can be repurposed as potentially antitumoral candidates in combination with statins. Thereby, fatostatin, a nonsterol diarylthiazole derivative originally developed to inhibit insulin-induced adipogenesis, suppresses (in vitro and in vivo) prostate cancer cell proliferation and induces apoptosis through inhibition of SREBP-regulated pathways, such as MVA pathway (209). Moreover, the combination of the anti-chronic myelogenous leukemia (CML) drug imatinib and simvastatin resulted in a synergistic killing effect on imatinib-resistant CML cells (210).
Combinatory Therapy, a Strategy to Improve Therapeutic Windows of MVA Pathway Inhibitors in Cancer
Monotherapy with statins (e.g., simvastatin) displays anticancer activity in vitro (11). However, it is undefined whether lipophilic statins accumulate in tumor tissues at concentrations in which they are cytotoxic to cancer cells and efforts are still underway to determine tolerable and therapeutic dose of statins that could potentially be used in cancer (211). This is particularly jumbling as statins are also known to exert effects on certain normal cells. For example, myopathy is a rare but potentially dangerous side effect of statin treatment that is thought to be consequence of the induction of apoptosis in skeletal muscle cells (212). Interestingly, many studies have shown that statins can directly and specifically trigger the apoptosis of cancer cells (213, 214). Noteworthy, statins trigger apoptosis of cells derived from AML, whereas normal myeloid progenitors do not suffer apoptosis and keep a proliferative phenotype (213). This optimal therapeutic index may be result of the altered metabolic reprogramming of AML cells leading to an increased dependence on MVA-derived metabolites for survival and proliferation. These findings and the widespread use of statins for hypercholesterolemia control strongly suggest that these drugs might have a high therapeutic window to target tumors in vivo, despite the MVA pathway is active in both normal and cancer cells. Therefore, the therapeutic window of statins in cancer patients is being explored in several clinical trials that have been conducted to study the tolerability of high dose statins in cancer patients. Phase I–II clinical trials have shown that statins can be given to cancer patients in relatively high dosages (i.e., 15 mg/kg/day for simvastatin; 25 mg/kg/day for lovastatin). In these studies, the maximum tolerated dose of simvastatin was defined to be 15 mg/kg/day, 25-fold higher compared to a typical dose. However, response may not be satisfactory because to treat human cancer high doses of statins (10–100 μM) need to be used. Moreover, statins can cause anorexia and death in some individuals when serum concentrations reached levels higher than 20–25 μM (215, 216). An efficient strategy that might increase therapeutic window of statins in cancer patients is its combination with conventional chemotherapy in those cancers where altered aberrant cholesterol metabolism is linked to oncogenic signaling. This strategy can improve antitumor efficacy, by taking advantage of the synergistic effects of these drugs, and, potentially, reduces therapy-associated toxicity (8, 13, 217). For example and related to hematological cancers, cholesterol levels are abnormally elevated in many AML cells exposed to chemotherapy in vitro (202, 203). Suppressing this cholesterol response was further shown to sensitize AML cells to drug treatment, suggesting that MVA pathway inhibition by statins may improve the efficacy of conventional chemotherapy (203, 218). Thus, when pravastatin was combined with conventional treatment in AML resulted in complete or partial response in 60% of patients with AML (218). Furthermore, simvastatin also has potential application in oncohematology as it is able to potentiate the effects of imatinib in CML cells, inducing cell cycle arrest and apoptosis through the inactivation of STAT3 and STAT5 (210). Interestingly, lovastatin can enhance the antitumor effects of the antiretroviral drug saquinavir against human lymphoma cells, decreasing some of its side effects while potentiating the antitumor effectiveness (219). In another study, the combination of lovastatin with thalidomide and dexamethasone in patients with relapsed or refractory multiple myeloma prolonged overall survival and progression-free survival (220). Recently, it has been reported that combination of statins (atorvastatin, fluvastatin and simvastatin) and conventional chemotherapy (topotecan, paclitaxel and doxorubicin) acted synergistically to inhibit cell proliferation and to induce cytotoxicity in an aggressive natural killer cell leukemia (221). On the other hand, and related to solid tumors, a combinatory strategy has also been safely used to increase statin efficacy and security in HCC. Thus, pravastatin was combined with conventional treatment in HCC, resulting in significantly longer median survival (193). Moreover, promising results from both epidemiological studies (222, 223) and clinical trials (187, 224) suggest that patients with hormone dependent breast and prostate cancers, may benefit from the addition of statins to their conventional treatment regimens. Accordingly, it has been reported that simvastatin has additive effect with the antiandrogen enzalutamide promoting a greater inhibition of prostate cancer cells (225, 226). Moreover, simvastatin also enhances ex vivo the tumor cell inhibition effects of cisplatin or docetaxel in head and neck squamous carcinoma (HNSCC) (227) and sensitized human osteosarcoma cells to doxorubicin and cisplatin (228). Preclinical data have also shown that simvastatin in combination with cetuximab/irinotecan allows overcoming the resistance to irinotecan and oxaliplatin in KRAS mutant colorectal cancer (184). Moreover, simvastatin can potentiate the antitumor effect of capecitabine by suppressing proliferation and tumor invasion mediated by NFκB in a xenograft mouse model of human gastric cancer (185). In addition, it has been observed that lovastatin increases in vitro TNF-α -induced cell death in two gefitinib-resistant cholangiocarcinoma cell lines (229). Finally, statins can overcome the resistance to EGFR tyrosine kinase inhibitors in a non-small cell lung cancer cells (183) and to gefitinib in KRAS-mutant human non-small cell lung cancer cells (230). Paradoxically, several clinical trials have shown that combinatory therapy with statins does not add any benefit in comparison with conventional therapy. Clinical trials where simvastatin was combined with capecitabine–cisplatin (XP) in patients with previously untreated advanced gastric cancer (AGC) showed that addition of low dose (40 mg) of simvastatin to XP does not increased the median progression free survival (PFS) (192) (Table 1). Moreover, it has been reported that using a combination of pravastatin, a hydrophilic statin, with etoposide plus cisplatin or carboplatin in order to treat small-cell lung cancer does not provide additional benefit for patients (39) (Table 1). Alike, a randomized double-blind phase II clinical trial in which patients with locally advanced and metastatic pancreatic cancer participated, reported no clinical benefits when low doses of simvastatin were added to gemcitabine (34). The limited effectiveness of statins in these and previous studies (231, 232), and in clinical trials mentioned above (34, 39, 192) (Table 1) might be linked to low statin biodisponibility in cancer cells. Pharmacokinetic studies in chronic lymphocytic leukemia patients have shown that when simvastatin is administered at the maximum tolerated dose of 7.5 mg/kg, twice daily, plasma concentrations were dose proportional relative to the hypolipidemic doses, but lower than those required for in vitro cytotoxicity on cancer cells (231). This lower drug bioavailability in cancer cells might explain, at least in part, the absent or low efficacy of statins in cancer patients.
Conclusions and Future Perspectives
Nowadays, an increasing amount of data, from preclinical and epidemiological studies, support an inverse association between the use of potent inhibitors of MVA pathway and the mortality rate in specific cancers (e.g., breast, colon, prostate, liver, hematological malignances). Furthermore, inhibitors of MVA pathway seem to have potential features that overcame main limitations of current chemotherapy: drug resistance and toxicity. Cancer treatment demands the use of relatively high doses of single inhibitors of MVA pathway for a prolonged period, thereby limiting this therapeutic strategy due to adverse effects. Clinically relevant, synergistic effects of tolerable doses of MVA inhibitors with conventional chemotherapy might enhance efficacy with lower doses of each drug and, probably, reduce adverse effects and resistance. In spite of that, clinical trials to identify combinatory therapies that improve therapeutic window are still a challenge. Dual strategies targeting both tumor cell proliferation and tumor niche and/or regulation of TIM represent a promising therapeutic strategy. However, despite TIM is known to be highly dependent of cholesterol biosynthesis, interplay of MVA biosynthetic pathway and TIM remains unexplored. Therefore, research needs to be performed in order to identify an effective MVA pathway inhibitor that may be clinically used, individually or in combination with conventional chemotherapy, in the treatment of cancers with addiction to cholesterol biosynthetic pathway.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
The Ministry of Science, Innovation and Universities (MCIU¸ SAF2015-65113-C2-2) with the funding of European Regional Development Fund-European Social Fund, SODECAN (Canary Islands Government; ET/15020), and FIISC (FUNCANIS2017; FIISC-DISA2018) supported research to Molecular and Translational Pharmacology Lab at University Institute for Biomedical and Health Research (IUIBS). Molecular and Translational Pharmacology Lab was also supported by grant-in-aid from Alfredo Martin-Reyes Foundation (Arehucas S.A.). MCIU-FPU16/00233 and ULPGC-2016 supported pre-doctoral programs to MG-R. and HA-T, respectively. The post-doctoral program Juan de la Cierva (MCIU2017; MCIU2019) supported to CR.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all the authors that have made a contribution to the understanding of the crosstalk between MVA pathway and cancer. We apologize to those whose work deserves to be cited but unfortunately are not quoted because of space limitations.
References
1. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer (2011) 11(2):85–95. doi: 10.1038/nrc2981
2. Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol (2015) 17(4):351–9. doi: 10.1038/ncb3124
3. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab (2016) 23(1):27–47. doi: 10.1016/j.cmet.2015.12.006
4. Sullivan LB, Gui DY, Vander Heiden MG. Altered metabolite levels in cancer: implications for tumour biology and cancer therapy. Nat Rev Cancer (2016) 16(11):680–93. doi: 10.1038/nrc.2016.85
5. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer (2016) 16(10):619–34. doi: 10.1038/nrc.2016.71
6. Li Z, Kang Y. Lipid Metabolism Fuels Cancer’s Spread. Cell Metab (2017) 25(2):228–30. doi: 10.1016/j.cmet.2017.01.016
7. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci U S A (2010) 107(34):15051–6. doi: 10.1073/pnas.0910258107
8. Mullen PJ, Yu R, Longo J, Archer MC, Penn LZ. The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer (2016) 16(11):718–31. doi: 10.1038/nrc.2016.76
9. Oguro H. The Roles of Cholesterol and Its Metabolites in Normal and Malignant Hematopoiesis. Front Endocrinol (2019) 10:204. doi: 10.3389/fendo.2019.00204
10. Huang B, Song BL, Xu C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab (2020) 2(2):132–41. doi: 10.1038/s42255-020-0174-0
11. Gazzerro P, Proto MC, Gangemi G, Malfitano AM, Ciaglia E, Pisanti S, et al. Pharmacological actions of statins: a critical appraisal in the management of cancer. Pharmacol Rev (2012) 64(1):102–46. doi: 10.1124/pr.111.004994
12. Clendening JW, Penn LZ. Targeting tumor cell metabolism with statins. Oncogene (2012) 31(48):4967–78. doi: 10.1038/onc.2012.6
13. Chimento A, Casaburi I, Avena P, Trotta F, De Luca A, Rago V, et al. Cholesterol and Its Metabolites in Tumor Growth: Therapeutic Potential of Statins in Cancer Treatment. Front Endocrinol (2018) 9:807. doi: 10.3389/fendo.2018.00807
14. Mo H, Jeter R, Bachmann A, Yount ST, Shen CL, Yeganehjoo H. The Potential of Isoprenoids in Adjuvant Cancer Therapy to Reduce Adverse Effects of Statins. Front Pharmacol (2018) 9:1515. doi: 10.3389/fphar.2018.01515
15. Iannelli F, Lombardi R, Milone MR, Pucci B, De Rienzo S, Budillon A, et al. Targeting Mevalonate Pathway in Cancer Treatment: Repurposing of Statins. Recent Pat Anticancer Drug Discov (2018) 13(2):184–200. doi: 10.2174/1574892812666171129141211
16. Kamel WA, Sugihara E, Nobusue H, Yamaguchi-Iwai S, Onishi N, Maki K, et al. Simvastatin-Induced Apoptosis in Osteosarcoma Cells: A Key Role of RhoA-AMPK/p38 MAPK Signaling in Antitumor Activity. Mol Cancer Ther (2017) 16(1):182–92. doi: 10.1158/1535-7163.MCT-16-0499
17. Kany S, Woschek M, Kneip N, Sturm R, Kalbitz M, Hanschen M, et al. Simvastatin exerts anticancer effects in osteosarcoma cell lines via geranylgeranylation and c-Jun activation. Int J Oncol (2018) 52(4):1285–94. doi: 10.3892/ijo.2018.4288
18. Cecen B, Keles D, Oktay G, Kozaci LD. Effects of simvastatin on matrix metalloproteinase regulation in IL-1beta-induced SW1353 cells. Chem Biol Interact (2019) 310:108730. doi: 10.1016/j.cbi.2019.108730
19. Beckwitt CH, Clark AM, Ma B, Whaley D, Oltvai ZN, Wells A. Statins attenuate outgrowth of breast cancer metastases. Br J Cancer (2018) 119(9):1094–105. doi: 10.1038/s41416-018-0267-7
20. Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM. Statins and cancer prevention. Nat Rev Cancer (2005) 5(12):930–42. doi: 10.1038/nrc1751
21. Kuoppala J, Lamminpaa A, Pukkala E. Statins and cancer: A systematic review and meta-analysis. Eur J Cancer (2008) 44(15):2122–32. doi: 10.1016/j.ejca.2008.06.025
22. Mei Z, Liang M, Li L, Zhang Y, Wang Q, Yang W. Effects of statins on cancer mortality and progression: A systematic review and meta-analysis of 95 cohorts including 1,111,407 individuals. Int J Cancer (2017) 140(5):1068–81. doi: 10.1002/ijc.30526
23. Jeong GH, Lee KH, Kim JY, Eisenhut M, Kronbichler A, van der Vliet HJ, et al. Statin and Cancer Mortality and Survival: An Umbrella Systematic Review and Meta-Analysis. J Clin Med (2020) 9(2):326. doi: 10.3390/jcm9020326
24. Alfaqih MA, Allott EH, Hamilton RJ, Freeman MR, Freedland SJ. The current evidence on statin use and prostate cancer prevention: are we there yet? Nat Rev Urol (2017) 14(2):107–19. doi: 10.1038/nrurol.2016.199
25. Liu B, Yi Z, Guan X, Zeng YX, Ma F. The relationship between statins and breast cancer prognosis varies by statin type and exposure time: a meta-analysis. Breast Cancer Res Treat (2017) 164(1):1–11. doi: 10.1007/s10549-017-4246-0
26. Chen YA, Lin YJ, Lin CL, Lin HJ, Wu HS, Hsu HY, et al. Simvastatin Therapy for Drug Repositioning to Reduce the Risk of Prostate Cancer Mortality in Patients With Hyperlipidemia. Front Pharmacol (2018) 9:225. doi: 10.3389/fphar.2018.00225
27. Borgquist S, Broberg P, Tojjar J, Olsson H. Statin use and breast cancer survival - a Swedish nationwide study. BMC Cancer (2019) 19(1):54. doi: 10.1186/s12885-018-5263-z
28. Feng JL, Qin X. Does adherence to lipid-lowering medications improve cancer survival? A nationwide study of breast and colorectal cancer, and melanoma. Br J Clin Pharmacol (2020). doi: 10.1111/bcp.14573
29. Poynter JN, Gruber SB, Higgins PD, Almog R, Bonner JD, Rennert HS, et al. Statins and the risk of colorectal cancer. N Engl J Med (2005) 352(21):2184–92. doi: 10.1056/NEJMoa043792
30. Ibanez-Sanz G, Guino E, Pontes C, Quijada-Manuitt MA, de la Pena-Negro LC, Aragon M, et al. Statin use and the risk of colorectal cancer in a population-based electronic health records study. Sci Rep (2019) 9(1):13560. doi: 10.1038/s41598-019-49877-5
31. Ehmsen S, Pedersen MH, Wang G, Terp MG, Arslanagic A, Hood BL, et al. Increased Cholesterol Biosynthesis Is a Key Characteristic of Breast Cancer Stem Cells Influencing Patient Outcome. Cell Rep (2019) 27(13):3927–38 e6. doi: 10.1016/j.celrep.2019.05.104
32. Zhong GC, Liu Y, Ye YY, Hao FB, Wang K, Gong JP. Meta-analysis of studies using statins as a reducer for primary liver cancer risk. Sci Rep (2016) 6:26256. doi: 10.1038/srep26256
33. Facciorusso A, Abd El Aziz MA, Singh S, Pusceddu S, Milione M, Giacomelli L, et al. Statin Use Decreases the Incidence of Hepatocellular Carcinoma: An Updated Meta-Analysis. Cancers (Basel) (2020) 12(4):874. doi: 10.3390/cancers12040874
34. Hong JY, Nam EM, Lee J, Park JO, Lee SC, Song SY, et al. Randomized double-blinded, placebo-controlled phase II trial of simvastatin and gemcitabine in advanced pancreatic cancer patients. Cancer Chemother Pharmacol (2014) 73(1):125–30. doi: 10.1007/s00280-013-2328-1
35. Goldman AR, Bitler BG, Schug Z, Conejo-Garcia JR, Zhang R, Speicher DW. The primary effect on the proteome of ARID1A-mutated ovarian clear cell carcinoma is downregulation of the mevalonate pathway at the post-transcriptional level. Mol Cell Proteomics (2016) 15(11):3348–60. doi: 10.1074/mcp.M116.062539
36. Kobayashi Y, Kashima H, Rahmanto YS, Banno K, Yu Y, Matoba Y, et al. Drug repositioning of mevalonate pathway inhibitors as antitumor agents for ovarian cancer. Oncotarget (2017) 8(42):72147–56. doi: 10.18632/oncotarget.20046
37. Alexandre L, Clark AB, Bhutta HY, Chan SS, Lewis MP, Hart AR. Association Between Statin Use After Diagnosis of Esophageal Cancer and Survival: A Population-Based Cohort Study. Gastroenterology (2016) 150(4):854–65 e1; quiz e16-7. doi: 10.1053/j.gastro.2015.12.039
38. Alexandre L, Clark AB, Bhutta HY, Holt S, Lewis MP, Hart AR. Statin use is associated with reduced risk of histologic subtypes of esophageal cancer: a nested case-control analysis. Gastroenterology (2014) 146(3):661–8. doi: 10.1053/j.gastro.2013.11.046
39. Seckl MJ, Ottensmeier CH, Cullen M, Schmid P, Ngai Y, Muthukumar D, et al. Multicenter, Phase III, Randomized, Double-Blind, Placebo-Controlled Trial of Pravastatin Added to First-Line Standard Chemotherapy in Small-Cell Lung Cancer (LUNGSTAR). J Clin Oncol (2017) 35(14):1506–14. doi: 10.1200/JCO.2016.69.7391
40. Pradelli D, Soranna D, Zambon A, Catapano A, Mancia G, La Vecchia C, et al. Statins use and the risk of all and subtype hematological malignancies: a meta-analysis of observational studies. Cancer Med (2015) 4(5):770–80. doi: 10.1002/cam4.411
41. Warita K, Warita T, Beckwitt CH, Schurdak ME, Vazquez A, Wells A, et al. Statin-induced mevalonate pathway inhibition attenuates the growth of mesenchymal-like cancer cells that lack functional E-cadherin mediated cell cohesion. Sci Rep (2014) 4:7593. doi: 10.1038/srep07593
42. Pandyra AA, Mullen PJ, Goard CA, Ericson E, Sharma P, Kalkat M, et al. Genome-wide RNAi analysis reveals that simultaneous inhibition of specific mevalonate pathway genes potentiates tumor cell death. Oncotarget (2015) 6(29):26909–21. doi: 10.18632/oncotarget.4817
43. Edwards PA, Ericsson J. Sterols and isoprenoids: signaling molecules derived from the cholesterol biosynthetic pathway. Annu Rev Biochem (1999) 68:157–85. doi: 10.1146/annurev.biochem.68.1.157
44. Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol (2017) 13(12):710–30. doi: 10.1038/nrendo.2017.91
45. Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab (2015) 21(6):805–21. doi: 10.1016/j.cmet.2015.05.014
46. Greenwood J, Steinman L, Zamvil SS. Statin therapy and autoimmune disease: from protein prenylation to immunomodulation. Nat Rev Immunol (2006) 6(5):358–70. doi: 10.1038/nri1839
47. Gruenbacher G, Thurnher M. Mevalonate Metabolism in Cancer Stemness and Trained Immunity. Front Oncol (2018) 8:394. doi: 10.3389/fonc.2018.00394
48. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden C, Li Y, et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell (2018) 172(1-2):135–46 e9. doi: 10.1016/j.cell.2017.11.025
49. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest (2002) 109(9):1125–31. doi: 10.1172/JCI0215593
50. Luu W, Sharpe LJ, Capell-Hattam I, Gelissen IC, Brown AJ. Oxysterols: Old Tale, New Twists. Annu Rev Pharmacol Toxicol (2016) 56:447–67. doi: 10.1146/annurev-pharmtox-010715-103233
51. Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, et al. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell (1998) 93(5):693–704. doi: 10.1016/S0092-8674(00)81432-4
52. Sontag TJ, Reardon CA, Getz GS. ABC transporters: lipid transport and inflammation. Curr Opin Lipidol (2010) 21(2):159–60. doi: 10.1097/MOL.0b013e3283376910
53. Leichner GS, Avner R, Harats D, Roitelman J. Dislocation of HMG-CoA reductase and Insig-1, two polytopic endoplasmic reticulum proteins, en route to proteasomal degradation. Mol Biol Cell (2009) 20(14):3330–41. doi: 10.1091/mbc.e08-09-0953
54. Hinson DD, Chambliss KL, Toth MJ, Tanaka RD, Gibson KM. Post-translational regulation of mevalonate kinase by intermediates of the cholesterol and nonsterol isoprene biosynthetic pathways. J Lipid Res (1997) 38(11):2216–23. doi: 10.1016/S0022-2275(20)34935-X
55. Clarke PR, Hardie DG. Regulation of HMG-CoA reductase: identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. EMBO J (1990) 9(8):2439–46. doi: 10.1002/j.1460-2075.1990.tb07420.x
56. Warburg O. On respiratory impairment in cancer cells. Science (1956) 124(3215):269–70. doi: 10.1126/science.124.3215.267
57. Grunt TW. Interacting Cancer Machineries: Cell Signaling, Lipid Metabolism, and Epigenetics. Trends Endocrinol Metab (2018) 29(2):86–98. doi: 10.1016/j.tem.2017.11.003
58. Ramirez de Molina A, Gallego-Ortega D, Sarmentero J, Banez-Coronel M, Martin-Cantalejo Y, Lacal JC. Choline kinase is a novel oncogene that potentiates RhoA-induced carcinogenesis. Cancer Res (2005) 65(13):5647–53. doi: 10.1158/0008-5472.CAN-04-4416
59. Hirsch HA, Iliopoulos D, Joshi A, Zhang Y, Jaeger SA, Bulyk M, et al. A transcriptional signature and common gene networks link cancer with lipid metabolism and diverse human diseases. Cancer Cell (2010) 17(4):348–61. doi: 10.1016/j.ccr.2010.01.022
60. Yang J, Wang L, Jia R. Role of de novo cholesterol synthesis enzymes in cancer. J Cancer (2020) 11(7):1761–7. doi: 10.7150/jca.38598
61. Nguyen VT, Barozzi I, Faronato M, Lombardo Y, Steel JH, Patel N, et al. Differential epigenetic reprogramming in response to specific endocrine therapies promotes cholesterol biosynthesis and cellular invasion. Nat Commun (2015) 6:10044. doi: 10.1038/ncomms10044
62. Silvente-Poirot S, Dalenc F, Poirot M. The Effects of Cholesterol-Derived Oncometabolites on Nuclear Receptor Function in Cancer. Cancer Res (2018) 78(17):4803–8. doi: 10.1158/0008-5472.CAN-18-1487
63. Hafizz A, Zin RRM, Aziz NHA, Kampan NC, Shafiee MN. Beyond lipid-lowering: role of statins in endometrial cancer. Mol Biol Rep (2020) 47(10):8199–207. doi: 10.1007/s11033-020-05760-5
64. Hodge RG, Ridley AJ. Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Biol (2016) 17(8):496–510. doi: 10.1038/nrm.2016.67
65. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer (2009) 9(8):563–75. doi: 10.1038/nrc2676
66. Carling D. The AMP-activated protein kinase cascade–a unifying system for energy control. Trends Biochem Sci (2004) 29(1):18–24. doi: 10.1016/j.tibs.2003.11.005
67. Han F, Li CF, Cai Z, Zhang X, Jin G, Zhang WN, et al. The critical role of AMPK in driving Akt activation under stress, tumorigenesis and drug resistance. Nat Commun (2018) 9(1):4728. doi: 10.1038/s41467-018-07188-9
68. Nickels JTJ. New links between lipid accumulation and cancer progression. J Biol Chem (2018) 293(17):6635–6. doi: 10.1074/jbc.H118.002654
69. Zahra K, Dey T, Ashish, Mishra SP, Pandey U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front Oncol (2020) 10:159. doi: 10.3389/fonc.2020.00159
70. Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol (2013) 10(3):143–53. doi: 10.1038/nrclinonc.2013.10
71. Song M, Bode AM, Dong Z, Lee MH. AKT as a Therapeutic Target for Cancer. Cancer Res (2019) 79(6):1019–31. doi: 10.1158/0008-5472.CAN-18-2738
72. Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol (2014) 16(4):357–66. doi: 10.1038/ncb2936
73. Zhao Y, Yang X. The Hippo pathway in chemotherapeutic drug resistance. Int J Cancer (2015) 137(12):2767–73. doi: 10.1002/ijc.29293
74. Eaton S. Multiple roles for lipids in the Hedgehog signalling pathway. Nat Rev Mol Cell Biol (2008) 9(6):437–45. doi: 10.1038/nrm2414
75. Wu F, Zhang Y, Sun B, McMahon AP, Wang Y. Hedgehog Signaling: From Basic Biology to Cancer Therapy. Cell Chem Biol (2017) 24(3):252–80. doi: 10.1016/j.chembiol.2017.02.010
76. Cheng X, Li J, Guo D. SCAP/SREBPs are Central Players in Lipid Metabolism and Novel Metabolic Targets in Cancer Therapy. Curr Top Med Chem (2018) 18(6):484–93. doi: 10.2174/1568026618666180523104541
77. Cook CC, Kim A, Terao S, Gotoh A, Higuchi M. Consumption of oxygen: a mitochondrial-generated progression signal of advanced cancer. Cell Death Dis (2012) 3:e258. doi: 10.1038/cddis.2011.141
78. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer (2014) 14(11):736–46. doi: 10.1038/nrc3818
79. Poli V, Camporeale A. STAT3-Mediated Metabolic Reprograming in Cellular Transformation and Implications for Drug Resistance. Front Oncol (2015) 5:121. doi: 10.3389/fonc.2015.00121
80. Huynh J, Chand A, Gough D, Ernst M. Therapeutically exploiting STAT3 activity in cancer - using tissue repair as a road map. Nat Rev Cancer (2019) 19(2):82–96. doi: 10.1038/s41568-018-0090-8
81. Li X, Wu JB, Li Q, Shigemura K, Chung LW, Huang WC. SREBP-2 promotes stem cell-like properties and metastasis by transcriptional activation of c-Myc in prostate cancer. Oncotarget (2016) 7(11):12869–84. doi: 10.18632/oncotarget.7331
82. Lin CY, Gustafsson JA. Targeting liver X receptors in cancer therapeutics. Nat Rev Cancer (2015) 15(4):216–24. doi: 10.1038/nrc3912
83. Kulkoyluoglu-Cotul E, Arca A, Madak-Erdogan Z. Crosstalk between Estrogen Signaling and Breast Cancer Metabolism. Trends Endocrinol Metab (2019) 30(1):25–38. doi: 10.1016/j.tem.2018.10.006
84. Wei W, Schwaid AG, Wang X, Chen S, Chu Q, Saghatelian A, et al. Ligand Activation of ERRalpha by Cholesterol Mediates Statin and Bisphosphonate Effects. Cell Metab (2016) 23(3):479–91. doi: 10.1016/j.cmet.2015.12.010
85. Casaburi I, Chimento A, De Luca A, Nocito M, Sculco S, Avena P, et al. Cholesterol as an Endogenous ERRalpha Agonist: A New Perspective to Cancer Treatment. Front Endocrinol (2018) 9:525. doi: 10.3389/fendo.2018.00525
86. Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell (2012) 148(1-2):244–58. doi: 10.1016/j.cell.2011.12.017
87. Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer (2018) 18(2):89–102. doi: 10.1038/nrc.2017.109
88. Kitajima S, Yoshida A, Kohno S, Li F, Suzuki S, Nagatani N, et al. The RB-IL-6 axis controls self-renewal and endocrine therapy resistance by fine-tuning mitochondrial activity. Oncogene (2017) 36(36):5145–57. doi: 10.1038/onc.2017.124
89. Shamma A, Takegami Y, Miki T, Kitajima S, Noda M, Obara T, et al. Rb Regulates DNA damage response and cellular senescence through E2F-dependent suppression of N-ras isoprenylation. Cancer Cell (2009) 15(4):255–69. doi: 10.1016/j.ccr.2009.03.001
90. Wu Q, Ba-Alawi W, Deblois G, Cruickshank J, Duan S, Lima-Fernandes E, et al. GLUT1 inhibition blocks growth of RB1-positive triple negative breast cancer. Nat Commun (2020) 11(1):4205. doi: 10.1038/s41467-020-18020-8
92. Larson RA, Yachnin S. Mevalonic acid induces DNA synthesis in chronic lymphocytic leukemia cells. Blood (1984) 64(1):257–62. doi: 10.1182/blood.V64.1.257.bloodjournal641257
93. Bjarnadottir O, Feldt M, Inasu M, Bendahl PO, Elebro K, Kimbung S, et al. Statin use, HMGCR expression, and breast cancer survival - The Malmo Diet and Cancer Study. Sci Rep (2020) 10(1):558. doi: 10.1038/s41598-019-57323-9
94. Ashida S, Kawada C, Inoue K. Stromal regulation of prostate cancer cell growth by mevalonate pathway enzymes HMGCS1 and HMGCR. Oncol Lett (2017) 14(6):6533–42. doi: 10.3892/ol.2017.7025
95. Hu J, La Vecchia C, de Groh M, Negri E, Morrison H, Mery L. Dietary cholesterol intake and cancer. Ann Oncol (2012) 23(2):491–500. doi: 10.1093/annonc/mdr155
96. Sezgin E, Levental I, Mayor S, Eggeling C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol (2017) 18(6):361–74. doi: 10.1038/nrm.2017.16
97. Zhao L, Zhou S, Gustafsson JA. Nuclear Receptors: Recent Drug Discovery for Cancer Therapies. Endocr Rev (2019) 40(5):1207–49. doi: 10.1210/er.2018-00222
98. Bovenga F, Sabba C, Moschetta A. Uncoupling nuclear receptor LXR and cholesterol metabolism in cancer. Cell Metab (2015) 21(4):517–26. doi: 10.1016/j.cmet.2015.03.002
99. Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res (2008) 68(15):6407–15. doi: 10.1158/0008-5472.CAN-07-5997
100. Jung H, Yoon SR, Lim J, Cho HJ, Lee HG. Dysregulation of Rho GTPases in human cancers. Cancers (Basel) (2020) 12(5):1179. doi: 10.3390/cancers12051179
101. Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer (2011) 11(11):775–91. doi: 10.1038/nrc3151
102. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov (2014) 13(11):828–51. doi: 10.1038/nrd4389
103. Ernster L, Dallner G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim Biophys Acta (1995) 1271(1):195–204. doi: 10.1016/0925-4439(95)00028-3
104. Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature (2017) 541(7635):41–5. doi: 10.1038/nature20791
105. Horton BL, Spranger S. CD36 - the Achilles’ heel of Treg cells. Nat Immunol (2020) 21(3):251–3. doi: 10.1038/s41590-020-0601-0
106. Rozovski U, Harris DM, Li P, Liu Z, Jain P, Ferrajoli A, et al. STAT3-activated CD36 facilitates fatty acid uptake in chronic lymphocytic leukemia cells. Oncotarget (2018) 9(30):21268–80. doi: 10.18632/oncotarget.25066
107. Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab (2011) 13(4):376–88. doi: 10.1016/j.cmet.2011.03.009
108. Collins SP, Reoma JL, Gamm DM, Uhler MD. LKB1, a novel serine/threonine protein kinase and potential tumour suppressor, is phosphorylated by cAMP-dependent protein kinase (PKA) and prenylated in vivo. Biochem J (2000) 345 Pt 3:673–80. doi: 10.1042/bj3450673
109. Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem (2006) 75:137–63. doi: 10.1146/annurev.biochem.75.103004.142702
110. Rao S, Porter DC, Chen X, Herliczek T, Lowe M, Keyomarsi K. Lovastatin-mediated G1 arrest is through inhibition of the proteasome, independent of hydroxymethyl glutaryl-CoA reductase. Proc Natl Acad Sci U S A (1999) 96(14):7797–802. doi: 10.1073/pnas.96.14.7797
111. Motoshima H, Goldstein BJ, Igata M, Araki E. AMPK and cell proliferation–AMPK as a therapeutic target for atherosclerosis and cancer. J Physiol (2006) 574(Pt 1):63–71. doi: 10.1113/jphysiol.2006.108324
112. Kochuparambil ST, Al-Husein B, Goc A, Soliman S, Somanath PR. Anticancer efficacy of simvastatin on prostate cancer cells and tumor xenografts is associated with inhibition of Akt and reduced prostate-specific antigen expression. J Pharmacol Exp Ther (2011) 336(2):496–505. doi: 10.1124/jpet.110.174870
113. Wang ST, Ho HJ, Lin JT, Shieh JJ, Wu CY. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis (2017) 8(2):e2626. doi: 10.1038/cddis.2016.472
114. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol (2002) 4(9):648–57. doi: 10.1038/ncb839
115. Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev (2003) 17(15):1829–34. doi: 10.1101/gad.1110003
116. Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell (1998) 95(1):29–39. doi: 10.1016/S0092-8674(00)81780-8
117. Milella M, Falcone I, Conciatori F, Matteoni S, Sacconi A, De Luca T, et al. PTEN status is a crucial determinant of the functional outcome of combined MEK and mTOR inhibition in cancer. Sci Rep (2017) 7:43013. doi: 10.1038/srep43013
118. Gris-Oliver A, Palafox M, Monserrat L, Braso-Maristany F, Odena A, Sanchez-Guixe M, et al. Genetic alterations in the PI3K/AKT pathway and baseline AKT activity define AKT inhibitor sensitivity in breast cancer patient-derived xenografts. Clin Cancer Res (2020) 26(14):3720–31. doi: 10.1158/1078-0432.CCR-19-3324
119. Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol (2014) 15(3):155–62. doi: 10.1038/nrm3757
120. Ricoult SJ, Yecies JL, Ben-Sahra I, Manning BD. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene (2016) 35(10):1250–60. doi: 10.1038/onc.2015.179
121. Lee G, Zheng Y, Cho S, Jang C, England C, Dempsey JM, et al. Post-transcriptional Regulation of De Novo Lipogenesis by mTORC1-S6K1-SRPK2 Signaling. Cell (2017) 171(7):1545–58 e18. doi: 10.1016/j.cell.2017.10.037
122. Siess KM, Leonard TA. Lipid-dependent Akt-ivity: where, when, and how. Biochem Soc Trans (2019) 47(3):897–908. doi: 10.1042/BST20190013
123. Panciera T, Azzolin L, Cordenonsi M, Piccolo S. Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol (2017) 18(12):758–70. doi: 10.1038/nrm.2017.87
124. Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol (2011) 13(8):877–83. doi: 10.1038/ncb2303
125. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer (2003) 3(10):721–32. doi: 10.1038/nrc1187
126. Pugh CW, Ratcliffe PJ. New horizons in hypoxia signaling pathways. Exp Cell Res (2017) 356(2):116–21. doi: 10.1016/j.yexcr.2017.03.008
127. Bersten DC, Sullivan AE, Peet DJ, Whitelaw ML. bHLH-PAS proteins in cancer. Nat Rev Cancer (2013) 13(12):827–41. doi: 10.1038/nrc3621
128. Koyasu S, Kobayashi M, Goto Y, Hiraoka M, Harada H. Regulatory mechanisms of hypoxia-inducible factor 1 activity: Two decades of knowledge. Cancer Sci (2018) 109(3):560–71. doi: 10.1111/cas.13483
129. Pallottini V, Guantario B, Martini C, Totta P, Filippi I, Carraro F, et al. Regulation of HMG-CoA reductase expression by hypoxia. J Cell Biochem (2008) 104(3):701–9. doi: 10.1002/jcb.21757
130. Thompson JM, Alvarez A, Singha MK, Pavesic MW, Nguyen QH, Nelson LJ, et al. Targeting the Mevalonate Pathway Suppresses VHL-Deficient CC-RCC through an HIF-Dependent Mechanism. Mol Cancer Ther (2018) 17(8):1781–92. doi: 10.1158/1535-7163.MCT-17-1076
131. Hwang S, Nguyen AD, Jo Y, Engelking LJ, Brugarolas J, DeBose-Boyd RA. Hypoxia-inducible factor 1alpha activates insulin-induced gene 2 (Insig-2) transcription for degradation of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase in the liver. J Biol Chem (2017) 292(22):9382–93. doi: 10.1074/jbc.M117.788562
132. Jeong JH, Yum KS, Chang JY, Kim M, Ahn JY, Kim S, et al. Dose-specific effect of simvastatin on hypoxia-induced HIF-1alpha and BACE expression in Alzheimer’s disease cybrid cells. BMC Neurol (2015) 15:127. doi: 10.1186/s12883-015-0390-5
133. Hisada T, Ayaori M, Ohrui N, Nakashima H, Nakaya K, Uto-Kondo H, et al. Statin inhibits hypoxia-induced endothelin-1 via accelerated degradation of HIF-1alpha in vascular smooth muscle cells. Cardiovasc Res (2012) 95(2):251–9. doi: 10.1093/cvr/cvs110
134. Aznar S, Valeron PF, del Rincon SV, Perez LF, Perona R, Lacal JC. Simultaneous tyrosine and serine phosphorylation of STAT3 transcription factor is involved in Rho A GTPase oncogenic transformation. Mol Biol Cell (2001) 12(10):3282–94. doi: 10.1091/mbc.12.10.3282
135. Raptis L, Arulanandam R, Geletu M, Turkson J. The R(h)oads to Stat3: Stat3 activation by the Rho GTPases. Exp Cell Res (2011) 317(13):1787–95. doi: 10.1016/j.yexcr.2011.05.008
136. Jiang S, Zhang LF, Zhang HW, Hu S, Lu MH, Liang S, et al. A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J (2012) 31(8):1985–98. doi: 10.1038/emboj.2012.45
137. Demaria M, Poli V. PKM2, STAT3 and HIF-1alpha: The Warburg’s vicious circle. JAKSTAT (2012) 1(3):194–6. doi: 10.4161/jkst.20662
138. Yamasaki M, Nomura T, Sato F, Mimata H. Chronic hypoxia induces androgen-independent and invasive behavior in LNCaP human prostate cancer cells. Urol Oncol (2013) 31(7):1124–31. doi: 10.1016/j.urolonc.2011.12.007
139. Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, et al. Function of mitochondrial Stat3 in cellular respiration. Science (2009) 323(5915):793–7. doi: 10.1126/science.1164551
140. Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, Szelag M, et al. Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J Biol Chem (2011) 286(34):29610–20. doi: 10.1074/jbc.M111.226209
141. Zhang Q, Raje V, Yakovlev VA, Yacoub A, Szczepanek K, Meier J, et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J Biol Chem (2013) 288(43):31280–8. doi: 10.1074/jbc.M113.505057
142. Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci U S A (2007) 104(41):16158–63. doi: 10.1073/pnas.0702596104
143. Yang F, Zhang W, Li D, Zhan Q. Gadd45a suppresses tumor angiogenesis via inhibition of the mTOR/STAT3 protein pathway. J Biol Chem (2013) 288(9):6552–60. doi: 10.1074/jbc.M112.418335
144. Arulanandam R, Vultur A, Cao J, Carefoot E, Elliott BE, Truesdell PF, et al. Cadherin-cadherin engagement promotes cell survival via Rac1/Cdc42 and signal transducer and activator of transcription-3. Mol Cancer Res (2009) 7(8):1310–27. doi: 10.1158/1541-7786.MCR-08-0469
145. Faruqi TR, Gomez D, Bustelo XR, Bar-Sagi D, Reich NC. Rac1 mediates STAT3 activation by autocrine IL-6. Proc Natl Acad Sci U S A (2001) 98(16):9014–9. doi: 10.1073/pnas.161281298
146. Simon AR, Vikis HG, Stewart S, Fanburg BL, Cochran BH, Guan KL. Regulation of STAT3 by direct binding to the Rac1 GTPase. Science (2000) 290(5489):144–7. doi: 10.1126/science.290.5489.144
147. Tonozuka Y, Minoshima Y, Bao YC, Moon Y, Tsubono Y, Hatori T, et al. A GTPase-activating protein binds STAT3 and is required for IL-6-induced STAT3 activation and for differentiation of a leukemic cell line. Blood (2004) 104(12):3550–7. doi: 10.1182/blood-2004-03-1066
148. Kawashima T, Bao YC, Minoshima Y, Nomura Y, Hatori T, Hori T, et al. A Rac GTPase-activating protein, MgcRacGAP, is a nuclear localizing signal-containing nuclear chaperone in the activation of STAT transcription factors. Mol Cell Biol (2009) 29(7):1796–813. doi: 10.1128/MCB.01423-08
149. van Adrichem AJ, Wennerberg K. MgcRacGAP inhibition stimulates JAK-dependent STAT3 activity. FEBS Lett (2015) 589(24 Pt B):3859–65. doi: 10.1016/j.febslet.2015.11.013
150. Owen KL, Brockwell NK, Parker BS. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers (Basel) (2019) 11(12):2002–27. doi: 10.3390/cancers11122002
151. Krause DS, Scadden DT. A hostel for the hostile: the bone marrow niche in hematologic neoplasms. Haematologica (2015) 100(11):1376–87. doi: 10.3324/haematol.2014.113852
152. Cartledge Wolf DM, Langhans SA. Moving Myeloid Leukemia Drug Discovery Into the Third Dimension. Front Pediatr (2019) 7:314. doi: 10.3389/fped.2019.00314
153. Irey EA, Lassiter CM, Brady NJ, Chuntova P, Wang Y, Knutson TP, et al. JAK/STAT inhibition in macrophages promotes therapeutic resistance by inducing expression of protumorigenic factors. Proc Natl Acad Sci U S A (2019) 116(25):12442–51. doi: 10.1073/pnas.1816410116
154. Petty AJ, Yang Y. Tumor-Associated Macrophages in Hematologic Malignancies: New Insights and Targeted Therapies. Cells (2019) 8(12):1526–40. doi: 10.3390/cells8121526
155. Teodorescu P, Pasca S, Dima D, Tomuleasa C, Ghiaur G. Targeting the Microenvironment in MDS: The Final Frontier. Front Pharmacol (2020) 11:1044. doi: 10.3389/fphar.2020.01044
156. Genard G, Lucas S, Michiels C. Reprogramming of Tumor-Associated Macrophages with Anticancer Therapies: Radiotherapy versus Chemo- and Immunotherapies. Front Immunol (2017) 8:828. doi: 10.3389/fimmu.2017.00828
157. Tabraue C, Lara PC, De Mirecki-Garrido M, De La Rosa JV, Lopez-Blanco F, Fernandez-Perez L, et al. LXR Signaling Regulates Macrophage Survival and Inflammation in Response to Ionizing Radiation. Int J Radiat Oncol Biol Phys (2019) 104(4):913–23. doi: 10.1016/j.ijrobp.2019.03.028
158. Al Dujaily E, Baena J, Das M, Sereno M, Smith C, Kamata T, et al. Reduced Protumorigenic Tumor-Associated Macrophages With Statin Use in Premalignant Human Lung Adenocarcinoma. JNCI Cancer Spectr (2020) 4(2):pkz101. doi: 10.1093/jncics/pkz101
159. Comito G, Pons Segura C, Taddei ML, Lanciotti M, Serni S, Morandi A, et al. Zoledronic acid impairs stromal reactivity by inhibiting M2-macrophages polarization and prostate cancer-associated fibroblasts. Oncotarget (2017) 8(1):118–32. doi: 10.18632/oncotarget.9497
160. Coscia M, Quaglino E, Iezzi M, Curcio C, Pantaleoni F, Riganti C, et al. Zoledronic acid repolarizes tumour-associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. J Cell Mol Med (2010) 14(12):2803–15. doi: 10.1111/j.1582-4934.2009.00926.x
161. Gruenbacher G, Thurnher M. Mevalonate metabolism governs cancer immune surveillance. Oncoimmunology (2017) 6(10):e1342917. doi: 10.1080/2162402X.2017.1342917
162. Baek AE, Yu YA, He S, Wardell SE, Chang CY, Kwon S, et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat Commun (2017) 8(1):864. doi: 10.1038/s41467-017-00910-z
163. Kopecka J, Godel M, Riganti C. Cholesterol metabolism: At the cross road between cancer cells and immune environment. Int J Biochem Cell Biol (2020) 129:105876. doi: 10.1016/j.biocel.2020.105876
164. Mok EHK, Lee TKW. The Pivotal Role of the Dysregulation of Cholesterol Homeostasis in Cancer: Implications for Therapeutic Targets. Cancers (Basel) (2020) 12(6):1410–31. doi: 10.3390/cancers12061410
165. Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol (2004) 6(4):308–18. doi: 10.1038/ncb1110
166. Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol (2005) 6(8):635–45. doi: 10.1038/nrm1703
167. Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, Metabolism, and Cancer. Cancer Discov (2015) 5(10):1024–39. doi: 10.1158/2159-8290.CD-15-0507
168. Cao Z, Fan-Minogue H, Bellovin DI, Yevtodiyenko A, Arzeno J, Yang Q, et al. MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res (2011) 71(6):2286–97. doi: 10.1158/0008-5472.CAN-10-3367
169. Bhatia K, Huppi K, Spangler G, Siwarski D, Iyer R, Magrath I. Point mutations in the c-Myc transactivation domain are common in Burkitt’s lymphoma and mouse plasmacytomas. Nat Genet (1993) 5(1):56–61. doi: 10.1038/ng0993-56
171. Wu Y, Chen K, Liu X, Huang L, Zhao D, Li L, et al. Srebp-1 Interacts with c-Myc to Enhance Somatic Cell Reprogramming. Stem Cells (2016) 34(1):83–92. doi: 10.1002/stem.2209
172. Hofmann JW, Zhao X, De Cecco M, Peterson AL, Pagliaroli L, Manivannan J, et al. Reduced expression of MYC increases longevity and enhances healthspan. Cell (2015) 160(3):477–88. doi: 10.1016/j.cell.2014.12.016
173. Kikuchi H, Pino MS, Zeng M, Shirasawa S, Chung DC. Oncogenic KRAS and BRAF differentially regulate hypoxia-inducible factor-1alpha and -2alpha in colon cancer. Cancer Res (2009) 69(21):8499–506. doi: 10.1158/0008-5472.CAN-09-2213
174. Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Mol Cell (1999) 3(2):169–79. doi: 10.1016/S1097-2765(00)80308-1
175. Giguere V. Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr Rev (2008) 29(6):677–96. doi: 10.1210/er.2008-0017
176. Wang C, Li P, Xuan J, Zhu C, Liu J, Shan L, et al. Cholesterol enhances colorectal cancer progression via ROS elevation and MAPK signaling pathway activation. Cell Physiol Biochem (2017) 42(2):729–42. doi: 10.1159/000477890
177. Gutierrez-Pajares JL, Ben Hassen C, Chevalier S, Frank PG. SR-BI: linking cholesterol and lipoprotein metabolism with breast and prostate cancer. Front Pharmacol (2016) 7:338. doi: 10.3389/fphar.2016.00338
178. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol (2017) 14(7):399–416. doi: 10.1038/nrclinonc.2016.217
179. Gritsman K, Yuzugullu H, Von T, Yan H, Clayton L, Fritsch C, et al. Hematopoiesis and RAS-driven myeloid leukemia differentially require PI3K isoform p110alpha. J Clin Invest (2014) 124(4):1794–809. doi: 10.1172/JCI69927
180. Lucken-Ardjomande S, Montessuit S, Martinou JC. Bax activation and stress-induced apoptosis delayed by the accumulation of cholesterol in mitochondrial membranes. Cell Death Differ (2008) 15(3):484–93. doi: 10.1038/sj.cdd.4402280
181. Montero J, Morales A, Llacuna L, Lluis JM, Terrones O, Basanez G, et al. Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res (2008) 68(13):5246–56. doi: 10.1158/0008-5472.CAN-07-6161
182. Garcia-Ruiz C, de la Rosa LC, Ribas V, Fernandez-Checa JC. Mitochondrial cholesterol and cancer. Semin Cancer Biol (2020). doi: 10.1016/j.semcancer.2020.07.014
183. Hwang KE, Kwon SJ, Kim YS, Park DS, Kim BR, Yoon KH, et al. Effect of simvastatin on the resistance to EGFR tyrosine kinase inhibitors in a non-small cell lung cancer with the T790M mutation of EGFR. Exp Cell Res (2014) 323(2):288–96. doi: 10.1016/j.yexcr.2014.02.026
184. Lee J, Hong YS, Hong JY, Han SW, Kim TW, Kang HJ, et al. Effect of simvastatin plus cetuximab/irinotecan for KRAS mutant colorectal cancer and predictive value of the RAS signature for treatment response to cetuximab. Invest New Drugs (2014) 32(3):535–41. doi: 10.1007/s10637-014-0065-x
185. Manu KA, Shanmugam MK, Li F, Chen L, Siveen KS, Ahn KS, et al. Simvastatin sensitizes human gastric cancer xenograft in nude mice to capecitabine by suppressing nuclear factor-kappa B-regulated gene products. J Mol Med (2014) 92(3):267–76. doi: 10.1007/s00109-013-1095-0
186. Jeong GH, Lee KH, Kim JY, Eisenhut M, Kronbichler A, van der Vliet HJ, et al. Effect of statin on cancer incidence: an umbrella systematic review and meta-analysis. J Clin Med (2019) 8(6):819. doi: 10.3390/jcm8060819
187. Garwood ER, Kumar AS, Baehner FL, Moore DH, Au A, Hylton N, et al. Fluvastatin reduces proliferation and increases apoptosis in women with high grade breast cancer. Breast Cancer Res Treat (2010) 119(1):137–44. doi: 10.1007/s10549-009-0507-x
188. Feldt M, Bjarnadottir O, Kimbung S, Jirstrom K, Bendahl PO, Veerla S, et al. Statin-induced anti-proliferative effects via cyclin D1 and p27 in a window-of-opportunity breast cancer trial. J Transl Med (2015) 13:133. doi: 10.1186/s12967-015-0486-0
189. Arun BK, Gong Y, Liu D, Litton JK, Gutierrez-Barrera AM, Jack Lee J, et al. Phase I biomarker modulation study of atorvastatin in women at increased risk for breast cancer. Breast Cancer Res Treat (2016) 158(1):67–77. doi: 10.1007/s10549-016-3849-1
190. Schmidmaier R, Baumann P, Bumeder I, Meinhardt G, Straka C, Emmerich B. First clinical experience with simvastatin to overcome drug resistance in refractory multiple myeloma. Eur J Haematol (2007) 79(3):240–3. doi: 10.1111/j.1600-0609.2007.00902.x
191. Sondergaard TE, Pedersen PT, Andersen TL, Soe K, Lund T, Ostergaard B, et al. A phase II clinical trial does not show that high dose simvastatin has beneficial effect on markers of bone turnover in multiple myeloma. Hematol Oncol (2009) 27(1):17–22. doi: 10.1002/hon.869
192. Kim ST, Kang JH, Lee J, Park SH, Park JO, Park YS, et al. Simvastatin plus capecitabine-cisplatin versus placebo plus capecitabine-cisplatin in patients with previously untreated advanced gastric cancer: a double-blind randomised phase 3 study. Eur J Cancer (2014) 50(16):2822–30. doi: 10.1016/j.ejca.2014.08.005
193. Graf H, Jungst C, Straub G, Dogan S, Hoffmann RT, Jakobs T, et al. Chemoembolization combined with pravastatin improves survival in patients with hepatocellular carcinoma. Digestion (2008) 78(1):34–8. doi: 10.1159/000156702
194. Yi X, Jia W, Jin Y, Zhen S. Statin use is associated with reduced risk of haematological malignancies: evidence from a meta-analysis. PloS One (2014) 9(1):e87019. doi: 10.1371/journal.pone.0087019
195. Ahern TP, Pedersen L, Tarp M, Cronin-Fenton DP, Garne JP, Silliman RA, et al. Statin prescriptions and breast cancer recurrence risk: a Danish nationwide prospective cohort study. J Natl Cancer Inst (2011) 103(19):1461–8. doi: 10.1093/jnci/djr291
196. Boudreau DM, Yu O, Chubak J, Wirtz HS, Bowles EJ, Fujii M, et al. Comparative safety of cardiovascular medication use and breast cancer outcomes among women with early stage breast cancer. Breast Cancer Res Treat (2014) 144(2):405–16. doi: 10.1007/s10549-014-2870-5
197. Goard CA, Chan-Seng-Yue M, Mullen PJ, Quiroga AD, Wasylishen AR, Clendening JW, et al. Identifying molecular features that distinguish fluvastatin-sensitive breast tumor cells. Breast Cancer Res Treat (2014) 143(2):301–12. doi: 10.1007/s10549-013-2800-y
198. Shachaf CM, Perez OD, Youssef S, Fan AC, Elchuri S, Goldstein MJ, et al. Inhibition of HMGcoA reductase by atorvastatin prevents and reverses MYC-induced lymphomagenesis. Blood (2007) 110(7):2674–84. doi: 10.1182/blood-2006-09-048033
199. Wong WW, Clendening JW, Martirosyan A, Boutros PC, Bros C, Khosravi F, et al. Determinants of sensitivity to lovastatin-induced apoptosis in multiple myeloma. Mol Cancer Ther (2007) 6(6):1886–97. doi: 10.1158/1535-7163.MCT-06-0745
200. Clendening JW, Pandyra A, Li Z, Boutros PC, Martirosyan A, Lehner R, et al. Exploiting the mevalonate pathway to distinguish statin-sensitive multiple myeloma. Blood (2010) 115(23):4787–97. doi: 10.1182/blood-2009-07-230508
201. Gobel A, Breining D, Rauner M, Hofbauer LC, Rachner TD. Induction of 3-hydroxy-3-methylglutaryl-CoA reductase mediates statin resistance in breast cancer cells. Cell Death Dis (2019) 10(2):91. doi: 10.1038/s41419-019-1322-x
202. Banker DE, Mayer SJ, Li HY, Willman CL, Appelbaum FR, Zager RA. Cholesterol synthesis and import contribute to protective cholesterol increments in acute myeloid leukemia cells. Blood (2004) 104(6):1816–24. doi: 10.1182/blood-2004-01-0395
203. Li HY, Appelbaum FR, Willman CL, Zager RA, Banker DE. Cholesterol-modulating agents kill acute myeloid leukemia cells and sensitize them to therapeutics by blocking adaptive cholesterol responses. Blood (2003) 101(9):3628–34. doi: 10.1182/blood-2002-07-2283
204. Xue L, Qi H, Zhang H, Ding L, Huang Q, Zhao D, et al. Targeting SREBP-2-regulated mevalonate metabolism for cancer therapy. Front Oncol (2020) 10:1510. [Review]. doi: 10.3389/fonc.2020.01510
205. Ho YK, Smith RG, Brown MS, Goldstein JL. Low-density lipoprotein (LDL) receptor activity in human acute myelogenous leukemia cells. Blood (1978) 52(6):1099–114. doi: 10.1182/blood.V52.6.1099.1099
206. Guillaumond F, Bidaut G, Ouaissi M, Servais S, Gouirand V, Olivares O, et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc Natl Acad Sci U S A (2015) 112(8):2473–8. doi: 10.1073/pnas.1421601112
207. Gobel A, Rauner M, Hofbauer LC, Rachner TD. Cholesterol and beyond - The role of the mevalonate pathway in cancer biology. Biochim Biophys Acta Rev Cancer (2020) 1873(2):188351. doi: 10.1016/j.bbcan.2020.188351
208. Longo J, Mullen PJ, Yu R, van Leeuwen JE, Masoomian M, Woon DTS, et al. An actionable sterol-regulated feedback loop modulates statin sensitivity in prostate cancer. Mol Metab (2019) 25:119–30. doi: 10.1016/j.molmet.2019.04.003
209. Li X, Chen YT, Hu P, Huang WC. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol Cancer Ther (2014) 13(4):855–66. doi: 10.1158/1535-7163.MCT-13-0797
210. Oh B, Kim TY, Min HJ, Kim M, Kang MS, Huh JY, et al. Synergistic killing effect of imatinib and simvastatin on imatinib-resistant chronic myelogenous leukemia cells. Anticancer Drugs (2013) 24(1):20–31. doi: 10.1097/CAD.0b013e32835a0fbd
211. Moon H, Hill MM, Roberts MJ, Gardiner RA, Brown AJ. Statins: protectors or pretenders in prostate cancer? Trends Endocrinol Metab (2014) 25(4):188–96. doi: 10.1016/j.tem.2013.12.007
212. Dirks AJ, Jones KM. Statin-induced apoptosis and skeletal myopathy. Am J Physiol Cell Physiol (2006) 291(6):C1208. doi: 10.1152/ajpcell.00226.2006
213. Dimitroulakos J, Nohynek D, Backway KL, Hedley DW, Yeger H, Freedman MH, et al. Increased sensitivity of acute myeloid leukemias to lovastatin-induced apoptosis: A potential therapeutic approach. Blood (1999) 93(4):1308–18. doi: 10.1182/blood.V93.4.1308.404k08_1308_1318
214. Wong WW, Dimitroulakos J, Minden MD, Penn LZ. HMG-CoA reductase inhibitors and the malignant cell: the statin family of drugs as triggers of tumor-specific apoptosis. Leukemia (2002) 16(4):508–19. doi: 10.1038/sj.leu.2402476
215. Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet (2005) 366(9493):1267–78. doi: 10.1016/S0140-6736(05)67394-1
216. Adnan M, Mohammad KI, Manik MEH. Anticancer agents in combination with statins. J Bioequiv Availab (2017) 9(4):463–6. doi: 10.4172/jbb.1000345
217. Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov (2004) 3(8):673–83. doi: 10.1038/nrd1468
218. Kornblau SM, Banker DE, Stirewalt D, Shen D, Lemker E, Verstovsek S, et al. Blockade of adaptive defensive changes in cholesterol uptake and synthesis in AML by the addition of pravastatin to idarubicin + high-dose Ara-C: a phase 1 study. Blood (2007) 109(7):2999–3006. doi: 10.1182/blood-2006-08-044446
219. Issat T, Nowis D, Jakobisiak M, Golab J. Lovastatin potentiates antitumor effects of saquinavir against human lymphoma cells. Oncol Rep (2004) 12(6):1371–5. doi: 10.3892/or.12.6.1371
220. Hus M, Grzasko N, Szostek M, Pluta A, Helbig G, Woszczyk D, et al. Thalidomide, dexamethasone and lovastatin with autologous stem cell transplantation as a salvage immunomodulatory therapy in patients with relapsed and refractory multiple myeloma. Ann Hematol (2011) 90(10):1161–6. doi: 10.1007/s00277-011-1276-2
221. Henslee AB, Steele TA. Combination statin and chemotherapy inhibits proliferation and cytotoxicity of an aggressive natural killer cell leukemia. Biomark Res (2018) 6:26. doi: 10.1186/s40364-018-0140-0
222. Hamilton RJ, Banez LL, Aronson WJ, Terris MK, Platz EA, Kane CJ, et al. Statin medication use and the risk of biochemical recurrence after radical prostatectomy: results from the Shared Equal Access Regional Cancer Hospital (SEARCH) Database. Cancer (2010) 116(14):3389–98. doi: 10.1002/cncr.25308
223. Harshman LC, Wang X, Nakabayashi M, Xie W, Valenca L, Werner L, et al. Statin Use at the Time of Initiation of Androgen Deprivation Therapy and Time to Progression in Patients With Hormone-Sensitive Prostate Cancer. JAMA Oncol (2015) 1(4):495–504. doi: 10.1001/jamaoncol.2015.0829
224. Bjarnadottir O, Romero Q, Bendahl PO, Jirstrom K, Ryden L, Loman N, et al. Targeting HMG-CoA reductase with statins in a window-of-opportunity breast cancer trial. Breast Cancer Res Treat (2013) 138(2):499–508. doi: 10.1007/s10549-013-2473-6
225. Syvala H, Pennanen P, Blauer M, Tammela TL, Murtola TJ. Additive inhibitory effects of simvastatin and enzalutamide on androgen-sensitive LNCaP and VCaP prostate cancer cells. Biochem Biophys Res Commun (2016) 481(1-2):46–50. doi: 10.1016/j.bbrc.2016.11.021
226. Pennanen P, Syvala H, Blauer M, Savinainen K, Ylikomi T, Tammela TLJ, et al. The effects of metformin and simvastatin on the growth of LNCaP and RWPE-1 prostate epithelial cell lines. Eur J Pharmacol (2016) 788:160–7. doi: 10.1016/j.ejphar.2016.06.036
227. Stoehr M, Mozet C, Boehm A, Aigner A, Dietz A, Wichmann G. Simvastatin suppresses head and neck squamous cell carcinoma ex vivo and enhances the cytostatic effects of chemotherapeutics. Cancer Chemother Pharmacol (2014) 73(4):827–37. doi: 10.1007/s00280-014-2412-1
228. Fromigue O, Hamidouche Z, Marie PJ. Statin-induced inhibition of 3-hydroxy-3-methyl glutaryl coenzyme a reductase sensitizes human osteosarcoma cells to anticancer drugs. J Pharmacol Exp Ther (2008) 325(2):595–600. doi: 10.1124/jpet.108.136127
229. Yang SH, Lin HY, Chang VH, Chen CC, Liu YR, Wang J, et al. Lovastatin overcomes gefitinib resistance through TNF-alpha signaling in human cholangiocarcinomas with different LKB1 statuses in vitro and in vivo. Oncotarget (2015) 6(27):23857–73. doi: 10.18632/oncotarget.4408
230. Chen J, Bi H, Hou J, Zhang X, Zhang C, Yue L, et al. Atorvastatin overcomes gefitinib resistance in KRAS mutant human non-small cell lung carcinoma cells. Cell Death Dis (2013) 4:e814. doi: 10.1038/cddis.2013.312
231. Ahmed TA, Hayslip J, Leggas M. Pharmacokinetics of high-dose simvastatin in refractory and relapsed chronic lymphocytic leukemia patients. Cancer Chemother Pharmacol (2013) 72(6):1369–74. doi: 10.1007/s00280-013-2326-3
Keywords: mevalonate, cholesterol, oxysterols, isoprenoids, sterol regulatory element binding protein, cancer, statins
Citation: Guerra B, Recio C, Aranda-Tavío H, Guerra-Rodríguez M, García-Castellano JM and Fernández-Pérez L (2021) The Mevalonate Pathway, a Metabolic Target in Cancer Therapy. Front. Oncol. 11:626971. doi: 10.3389/fonc.2021.626971
Received: 07 November 2020; Accepted: 18 January 2021;
Published: 25 February 2021.
Edited by:
Miriam Martini, University of Turin, ItalyReviewed by:
Khalid Omer Alfarouk, Alfarouk Biomedical Research LLC, United StatesParames C. Sil, Bose Institute, India
Copyright © 2021 Guerra, Recio, Aranda-Tavío, Guerra-Rodríguez, García-Castellano and Fernández-Pérez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Borja Guerra, Ym9yamEuZ3VlcnJhQHVscGdjLmVz; Leandro Fernández-Pérez, bGVhbmRyb2Zjby5mZXJuYW5kZXpAdWxwZ2MuZXM=