 Yasser Tabana
Yasser Tabana Isobel S. Okoye
Isobel S. Okoye Arno Siraki1
Arno Siraki1 Khaled H. Barakat
Khaled H. Barakat- 1Faculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, AB, Canada
- 2School of Dentistry, University of Alberta, Edmonton, AB, Canada
- 3Department of Oncology, University of Alberta, Edmonton, AB, Canada
- 4Department of Medical Microbiology and Immunology, University of Alberta, Edmonton, AB, Canada
The burden of breast cancer is imposing a huge global problem. Drug discovery research and novel approaches to treat breast cancer have been carried out extensively over the last decades. Although immune checkpoint inhibitors are showing promising preclinical and clinical results in treating breast cancer, they are facing multiple limitations. From an immunological perspective, a recent report highlighted breast cancer as an “inflamed tumor” with an immunosuppressive microenvironment. Consequently, researchers have been focusing on identifying novel immunological targets that can tune up the tumor immune microenvironment. In this context, several novel non-classical immune targets have been targeted to determine their ability to uncouple immunoregulatory pathways at play in the tumor microenvironment. This article will highlight strategies designed to increase the immunogenicity of the breast tumor microenvironment. It also addresses the latest studies on targets which can enhance immune responses to breast cancer and discusses examples of preclinical and clinical trial landscapes that utilize these targets.
Introduction
The Global Cancer Statistics (GLOBOCAN 2018) report of 2018 flags breast cancer as the second most diagnosed cancer, with a prevalence of ~11.6% of all cancer cases (1). Breast cancer is the first diagnosed cancer and the leading cause of death among women, with over 450,000 mortalities annually (2). Based on the status of the tumor receptors, three types of breast cancers have been reported: estrogen/progesterone receptor-positive (ER+), human epidermal growth factor receptor 2-positive (HER2+), and triple-negative (TNBC) breast cancer (3). ER+ breast cancer is the most diagnosed breast cancer, with an incidence rate of ~80% (4, 5). Recently, the reactivation of the immune system has emerged as a strategy for cancer treatment other than traditional methods (6). Due to the immunological quiescent nature of breast tumors, immunotherapy has not been considered as a strategy for breast cancer treatment. However, this strategy has been reconsidered following the identification of tumor immune infiltrates. Since tumor-infiltrating lymphocytes (TILs: CD8+ cytotoxic T cells and helper CD4+ cells, regulatory T cells, B cells, NK cells), tumor-associated macrophages and myeloid-derived suppressor cells (MDSCs) are observed in some breast tumors (7, 8). Hence, the alteration and manipulation of the immune responses are now the focus of breast cancer therapeutic strategies (9). The discovery of inhibitory immune checkpoints has revolutionized cancer treatment (10). Understanding their role in promoting immunosuppression in the tumor microenvironment (TME) has resulted in the use of checkpoint inhibitors (generally monoclonal antibodies), which can reactivate immune cells (11, 12). Checkpoint inhibitors that target PD-1 or CTLA-4 have been used for treating metastatic breast cancer (13). However, the response rates were lower than other types of cancers; the overall response rate to anti-PD-1 (Pembrolizumab) was only 18.5% when used as monotherapy for patients with advanced triple-negative breast cancer (TNBC) (14). However, the KEYNOTE 355 study was initiated in 2016 to compare the effectiveness of using pembrolizumab in combination with chemotherapy with placebo plus chemotherapy for treating patients with unresectable locally advanced or metastatic PD-L1-positive TNBC (ClinicalTrials.gov Identifier: NCT02819518). Reports from this study indicated that pembrolizumab combined with several chemotherapy agents showed a statistically significant and clinically meaningful improvement in progression-free survival with 9.7 months vs. only 5.6 months with using chemotherapy alone in these patients. Pembrolizumab combined with chemotherapy showed adverse event rates 68% while 67% with chemotherapy. This combination was generally well-tolerated, with no safety concerns (15, 16). Based on the results of this trial, the FDA approved the use of pembrolizumab (anti-PD1) in combination with chemotherapy for the treatment of unresectable locally advanced or metastatic PD-L1-positive TNBC, in November 2020.
Nevertheless, identifying novel targets and developing new therapeutic agents are needed for breast cancer treatment. Other therapeutic targets that can modulate immune responses against breast tumors are currently under investigation. Co-stimulatory receptors are promising targets, which can improve anti-tumor immunity in breast cancer (13). Purinergic ectoenzymes attenuate the immune response by increasing the level of extracellular adenosine, which has immunosuppressive properties (17, 18). Inhibiting purinergic ectoenzymes will increase the anti-tumor immune responses (19). Similarly, targeting the immunosuppressive enzyme arginase 1 (ARG1), could also improve anti-tumor immune responses (20, 21). Studies have shown that various cytokines, chemokines, growth factors, and their receptors such as vascular endothelial growth factor (VEGF), VEGF Receptor (22), CXC receptor 1(CXCR1), CCL2 receptor (CCR2) (23), colony-stimulating factor-1 (CSF-1) (24) and toll-like receptors (TLRs) (25) are essential for breast tumor proliferation and metastasis. Furthermore, studies on targeting tryptophan catabolism enzymes, such as indoleamine-2,3-dioxygenase (IDO1/IDO2), and tryptophan-2,3-dioxygenase (TDO/TDO2), which are expressed by many immune cells and solid tumors, including breast cancer are underway (26). Moreover, the development of agents, which can modulate the COX2/PGE2 (27) and STING (28) signaling pathways, are ongoing.
The effects of blocking different immune checkpoints in breast cancer have been recently reviewed by Swoboda A, and Nanda R (29). Furthermore, the effectiveness of combining PD1/PD-L1 blockade with chemotherapy, targeted therapies and radiotherapy for the treatment of metastatic breast cancer has been reviewed by Page et al. (30). In this review, we will discuss the pathways that modulate immune responses to breast cancer (Figure 1). We will also discuss novel therapies and clinical trials designed to target these pathways (Table 1).
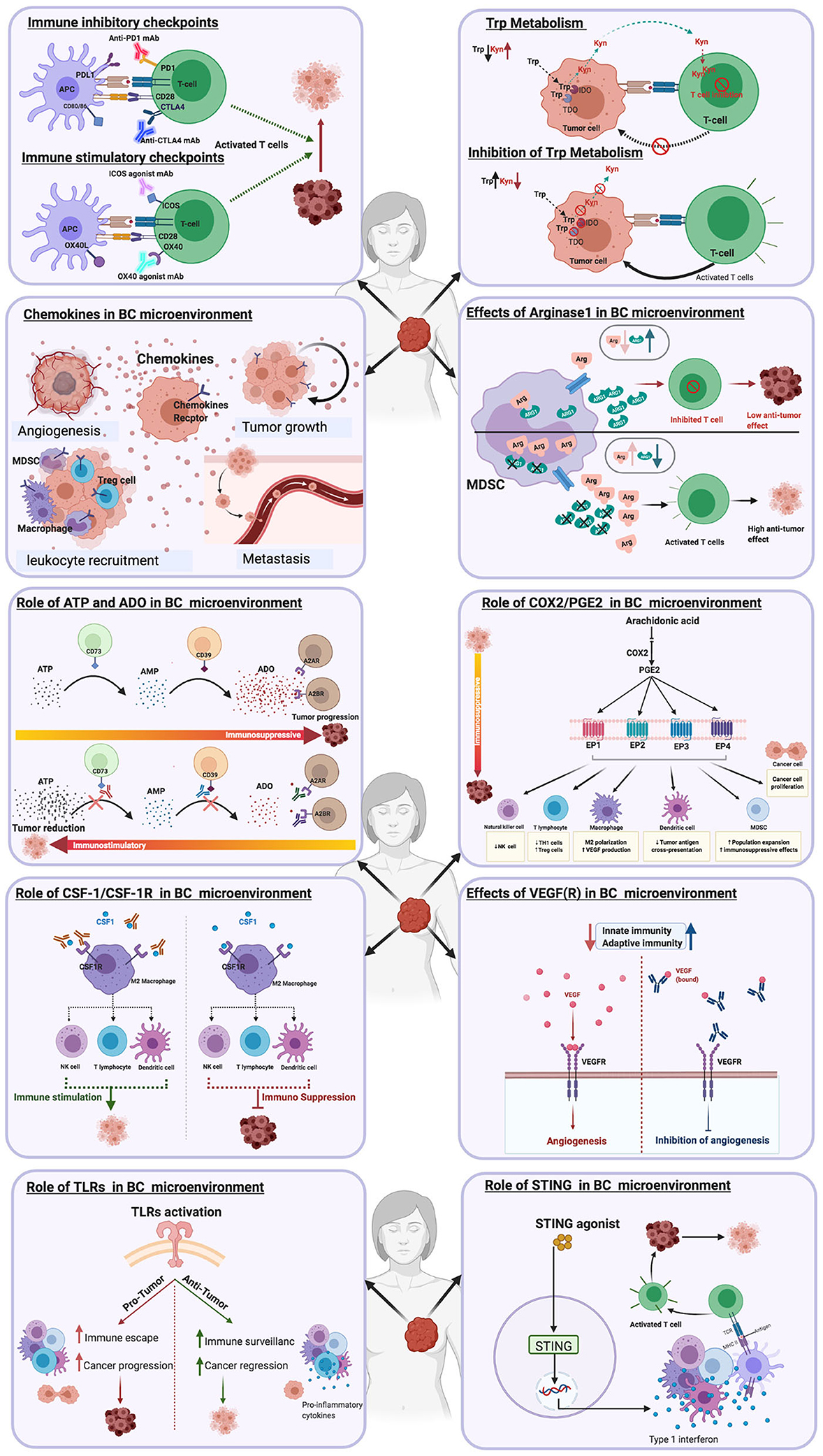
Figure 1. Immune targets in breast cancer immunotherapy.
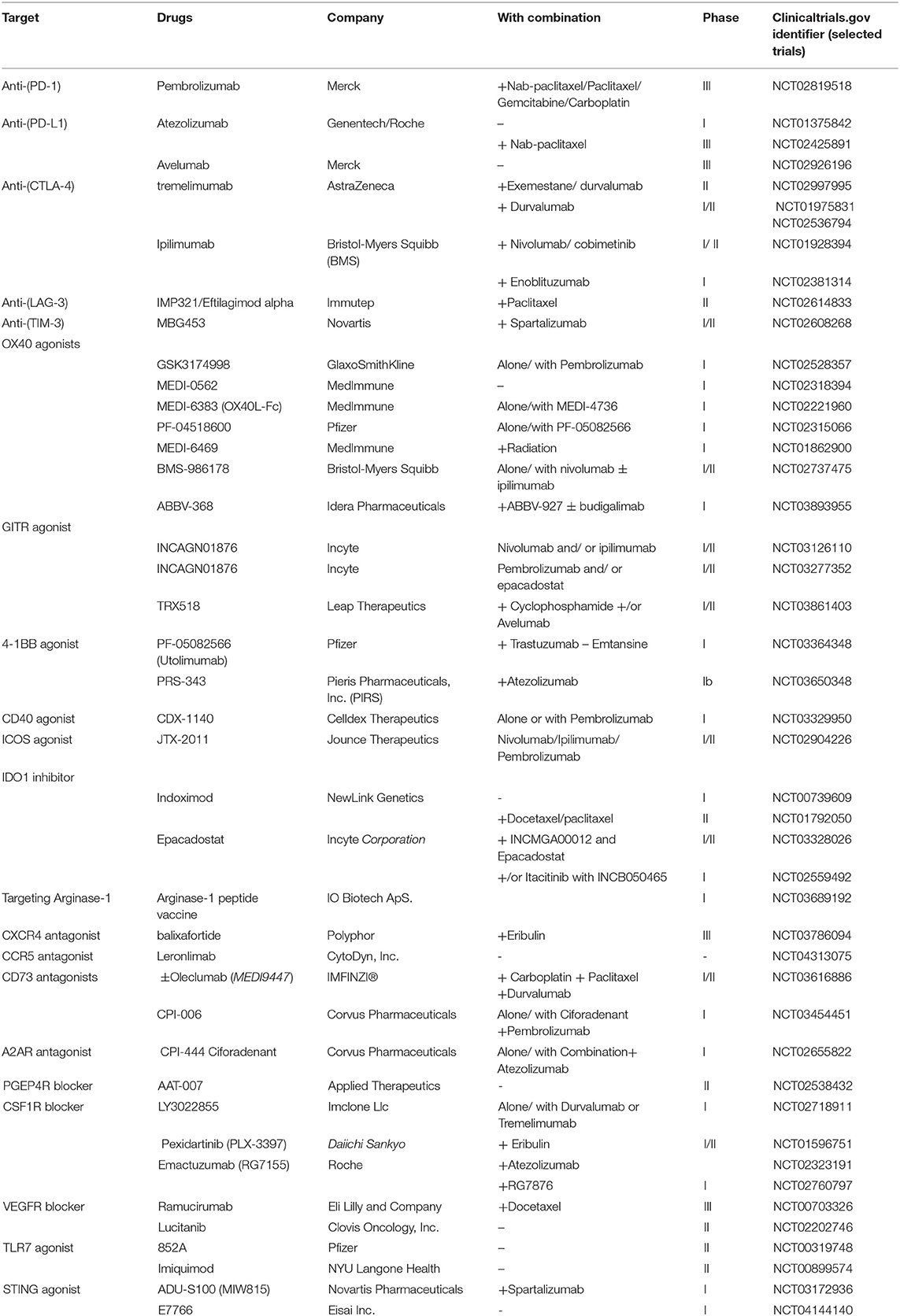
Table 1. Examples of clinical trials of Immune targets in breast cancer immunotherapy.
Stimulatory Checkpoints
A major characteristic of tumors is the paucity of, or ability to downregulate the expression of co-stimulatory molecules and upregulate co-inhibitory receptor expression (31, 32). The ligation of co-stimulatory molecules expressed by antigen-presenting cells (APCs) with their receptors on T cells provides the second signal necessary for T cell activation and differentiation. Hence, the use of co-stimulatory molecule agonist antibodies, is a strategy which may enhance T cell function in the TME (31, 32) (Figure 2A). Targeting co-stimulatory molecules that belong to the tumor necrosis factor receptor (TNFR) family such as OX40, ICOS, GITR, CD40L, and 4-1BB with agonist antibodies have been found to improve T cell function, with favorable outcomes in some cancer patients [reviewed in Moran et al. (33)].
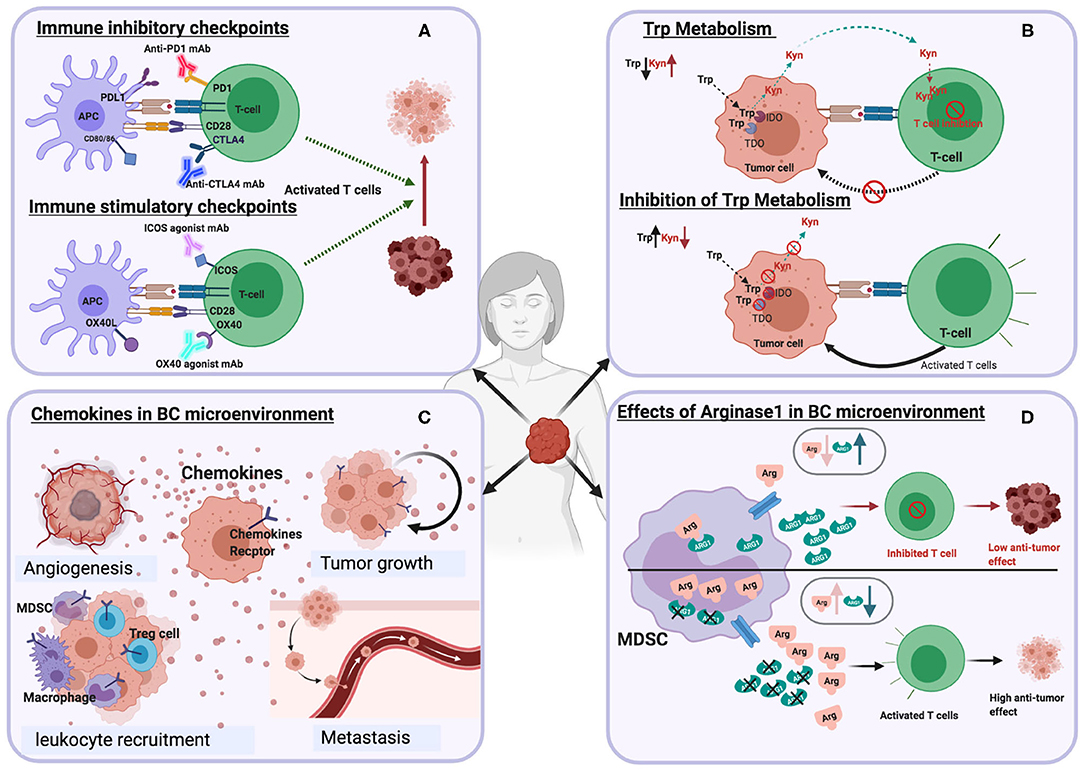
Figure 2. Schematic illustrations depicting the effects of different immune targets on breast cancer (A) Immune checkpoints (B) Tryptophan metabolism (C) Chemokines (D) Arginase enzyme.
OX40 (i.e., CD134) is expressed by TILs in various types of cancers, including breast cancer (34), while its receptor OX40L, is upregulated on monocytes, neutrophils, macrophages and dendritic cells. Studies have shown that OX40–OX40L signaling reduces immunosuppression mediated by regulatory T cells (Tregs) and enhances the expansion and proliferation of T cells (34). A study to assess the safety and tolerability of the OX40 agonist (PF-04518600) alone, or in combination with the 4-1BB agonist, PF-05082566, in patients with metastatic carcinoma, including TNBC was concluded in December 2020 (ClinicalTrials.gov Identifier: NCT02315066), (35). However, a clinical study that had planned to test the agonistic anti-OX40 antibody, MEDI6469, in combination with immune checkpoint inhibitors in patients diagnosed with advanced solid tumors, was terminated (32, 35, 36). Another phase I/II study, which investigated the use of MEDI6469 in combination with radiation for the treatment of metastatic breast cancer has been completed (ClinicalTrials.gov Identifier: NCT01862900). An additional phase I study has been initiated to investigate the effectiveness of using a CD40 agonist, ABBV-927 plus OX40 agonist ABBV-368 in combination or without the PD1 inhibitor, budigalimab in patients with advanced solid tumors, including TNBC (ClinicalTrials.gov Identifier: NCT03893955). Observations from a recent study indicated that OX40 agonists enhanced the production of IL-2 by conventional TILs, which increases the proliferation of both tumor-infiltrating Tregs and conventional T cells. Hence, in contrast to what has been postulated by previous studies, Tregs retain their immunosuppressive abilities in response to OX40 agonist treatment. However, results from this study also indicate that Tregs acquire a Th1 phenotype (IFN-g and granzyme B production) in response to OX40 agonist treatment (37). These observations imply that OX40 agonist treatment may be more suitable for combination therapies for cancer treatment. The importance of investigating the sequence of administering monoclonal antibodies in combination treatments that include anti-PD1 and OX40 agonists has been highlighted by Messenheimer et al. (38). They showed that using a preclinical model of oncogene-driven mammary cancer that concurrent administration of anti-PD1 antibody and an OX40 agonist compromised tumor regression. In contrast, sequential administration of the OX40 agonist and anti-PD1 facilitated tumor elimination, which was dependent on CD4+ and CD8+ T cell responses (38). These results indicate that sequential, rather than simultaneous administration of OX40 agonists and anti–PD-1 can revert PD-1 resistance and improve responses to combination therapy. Consequently, one of the approaches in a Bristol-Myers Squibb (BMS) clinical study (39) involves exploring the effectiveness of sequentially administering an OX40 agonist, BMS-986178, anti-PD1 (Nivolumab), an allogeneic autophagosome-enriched vaccine, DPV-001 and cyclophosphamide in TNBC patients (ClinicalTrials.gov Identifier: NCT02737475).
Another co-stimulatory molecule, the inducible co-stimulator (ICOS), is mainly expressed by activated CD4+ and CD8+ T cells and constitutively by Tregs. ICOS binds to its ligand, ICOS-L (B7RP1), expressed by APCs, epithelial cells, endothelial cells and tumor cells (40). ICOS-mediated co-stimulation does not induce IL-2 production, hence it is regarded as less potent relative to co-stimulation elicited by CD28 (41, 42). However, various clinical studies have shown that high expression of ICOS by T cells in patients treated with PD-1 and CTLA-4 checkpoint inhibitors correlates with positive treatment responses (43, 44). Hence, current immunotherapy strategies include the administration of ICOS or ICOS-L agonists with CTLA-4 checkpoint inhibitors (43, 45). A Phase 1/2 first in-human clinical trial has been set up to evaluate JTX-2011, an agonist monoclonal antibody that binds to ICOS, alone or in combination with checkpoint inhibitors for the treatment of advanced solid tumors, including TNBC breast cancer (46). A recently completed phase 1 clinical trial, which involved the use of another ICOS agonist, GSK3359609, in combination with anti-PD-1 shows promising anti-tumor activity in anti-PD-1/L1 naive patients with head and neck squamous cell carcinoma (HNSCC) (32, 36). Furthermore, the findings from this study indicate that GSK3359609 is also suitable for monotherapy of HNSCC in patients with anti-PD-1/L1 -experienced HNSCC (GSK Press Release September 28, 2019).
The glucocorticoid-induced TNFR related protein (GITR) is preferentially expressed on NK cells and T cells, particularly Tregs. GITR interaction with its ligand, GITRL, on dendritic cells, boosts effector T cell differentiation and IL-2 production (11, 13). Importantly, GITR has been detected on lymphocytes and carcinoma cells from a subset of breast cancer tumor specimens (47). Furthermore, observations from a study by Krausz et al. indicated that Tregs from tumor-positive lymph nodes from advanced breast cancer patients express increased levels of GITR, compared to tumor-negative lymph nodes (48). The potential for GITR-mediated co-stimulation to promote high effector CD8+ T cell to Treg ratios, is now harnessed as an immunotherapy strategy (49, 50). In fact, the first in-human phase 1 trial of GITR agonism with the anti-GITR antibody TRX518, has been initiated and a report indicates reduction in circulating and intratumoral Tregs at similar levels (51). However, a combination of GITR agonism with PD-1 blockade has been postponed due to sub-optimal clinical responses induced by TRX518 (51, 52). A clinical trial using another anti-GITR agonistic mAb, INCAGN01876, in combination with pembrolizumab and epacadostat for the treatment of advanced or metastatic malignancies is underway (ClinicalTrials.gov Identifier: NCT03277352).
CD40 is upregulated on the surface of activated APCs and its interaction with its ligand (CD40-L), expressed on activated B cells and T cells, leads to the initiation and progression of cellular and humoral adaptive immunity (53, 54). CD40 is also expressed in breast and lung carcinomas and carcinomas of the urinary bladder, nasopharynx, and colon, in contrast to normal non-proliferating tissues, which are CD40-negative (55, 56). Observations from a study approximately two decades ago by Tong et al., indicated that the interaction of soluble recombinant CD40L with CD40+ human breast cancer cell lines directly inhibits breast cancer cell growth. By examining primary tumor biopsies, they also found that infiltrating ductal, lobular carcinomas and carcinomas expressed CD40 while benign epithelial tissues of these biopsies exhibited weaker expression of CD40 (57). Interestingly, tumor infiltrating lymphocytes from most of the breast cancers examined expressed very low levels of CD40L (57). Other studies have suggested that CD40 may induce apoptosis in breast carcinoma cells by upregulating Fas expression induced by CD40 ligation (58).
A clinical study of CDX-1140, a CD40 agonist, for use as a monotherapy or in combination with the anti-PD-1 mAB, pembrolizumab, has been initiated in patients with advanced malignancies, including breast cancer (ClinicalTrials.gov Identifier: NCT03329950). Furthermore, results from a recent orthotopic breast cancer study suggest that combination treatment using anti-PD-1 and a CD40 agonist promote tumor immunogenicity (59).
4-1BB (CD137) is another member of the TNFR family of co-stimulatory molecules. It is expressed on many hematopoietic cells, including T cells and NK cells. Its ligand, 4-1BBL (CD137L), is predominantly expressed on APCs. 4-1BB:4-1BBL ligation potentiates CTL responses, induces antibody-dependent cell-mediated cytotoxicity in NK cells and modulates the activity of CD4+ T cells, B cells, DCs, monocytes and macrophages (60). For instance, CD8+ TILs from TNBC tumors were successfully propagated with a 4-1BB agonistic antibody (urelumab) (61). Based on these properties, harnessing the 4-1BB signaling pathway through the use of agonistic monoclonal antibodies can serve as a cancer immunotherapy strategy.
Significant breast tumor reduction in xenograft models has been achieved by targeting 4-1BB, combined with trastuzumab (anti-HER2) and rituximab (anti-CD20) treatment (32, 62, 63). In 2017, a clinical trial to investigate the optimal dosage and side effects of the 4-1BB agonist, utomilumab with trastuzumab emtansine or trastuzumab in patients with metastatic HER2-positive breast cancer was initiated (ClinicalTrials.gov Identifier: NCT03364348). However, a dependency of 4-1BB agonists on the Fcγ receptor–mediated hyperclustering and liver toxicity in patients, have been reported (64). Consequently, strategies that will restrict 4-1BB agonism to the TME, thereby minimizing off-target toxicities, have been proposed. A recent study has adopted a protein engineering approach to develop proteins that simultaneously target 4-1BB and tumor stroma or tumor antigens (65).
Amino Acid Catabolism
Amino acid metabolism is an immune regulatory mechanism (52). The breakdown of amino acids, particularly tryptophan and arginine by immunoregulatory myeloid cells, is one mechanism whereby T cell proliferation and activation are suppressed (29). Furthermore, these catabolic pathways are harnessed by solid tumors to induce the development of immunosuppressive tumor microenvironments and poor anti-tumor T cell responses. Hence, the use of inhibitors of arginase-1 and indolamine- 2, 3- dioxygenase-1 enzymes, which catabolise L-arginine and tryptophan, respectively, are now exploited as new cancer immunotherapy strategies.
Catabolism of Tryptophan
Tryptophan is the rarest essential amino acid found in food. It is a precursor to the synthesis of niacin (vitamin B3), neurotransmitter serotonin, and the hormone melatonin. Tryptophan metabolism is associated with immune regulation and tumor progression (66). Tryptophan catabolism occurs through the kynurenine pathway with the aid of two enzymes, indoleamine-2,3-dioxygenase (IDO1) and tryptophan-2,3-dioxygenase (TDO), which catalyze the first rate-limiting step by facilitating the oxidative breakdown of the tryptophan indole group. The generation of kynurenine (Kyn) and the concomitant release of kynurenine metabolites by myeloid cells, suppresses T cell and NK cell activity. The activities of IDO and TDO have been investigated due to their link with various diseases, including diabetes, mental disorders, inflammatory, and cancer (67, 68) (Figure 2B).
Indoleamine-Pyrrole 2,3-Dioxygenase (IDO1)
The upregulation and sustained expression of IDO by tumor cells is a well-characterized immunosuppressive strategy, orchestrated in conjunction with MDSCs and Tregs (69). IDO1 and TDO, through their catalytic activity, function as tryptophan sinks, leading to the suppression of T cell proliferation, apoptosis and Tregs differentiation. Indeed, T cell activation and function are highly dependent on the levels of tryptophan in their microenvironment, as the zeta chain of TCR complex is downregulated upon tryptophan withdrawal. IDO1 also suppresses anti-tumor responses through the generation of L-kynurenine, an endogenous agonist of the arylhydrocarbon receptor (AhR). AhR activation promotes the differentiation of Tregs and the concomitant upregulation of IDO1 by DCs (70). Furthermore, long-term expression of IDO1 by DCs is facilitated when IDO functions as a signal-transducing molecule (70).
The expression of IDO has been observed in breast carcinomas, particularly among triple negative (TNBC) basal-like breast cancers (71, 72). In a study by Dill et al., the authors assessed 281 primary and metastatic breast cancers and identified a correlation between IDO1 and PD-L1 expression, particularly in high-grade TNBC (73). Their observations imply that IDO1 expression contributes to the resistance of breast cancer to anti-PD-1/PD-L1 treatment.
A positive correlation between the high expression of PD-1 by T cells and high levels of kynurenine in the plasma and the TME of breast cancer patients has also been reported (74). IFN-γ produced by CD8+ T cells induces the production of IDO and kynurenine by CD45 negative tumor cells. Kynurenine promotes the translocation of AhR from cytosol to the nucleus of in vitro-treated and tumor-infiltrating CD8+ T cells and subsequently upregulates PD-1 (60).
IDO1 also induces cancer progression in a non-immune manner by regulating angiogenesis (59). The expression of IDO and levels of CD105+ micro vessel density by breast cancer specimens were found to be associated with metastasis and poor prognosis (75). Furthermore, MCF-7 cells which produce high levels of IDO significantly induced the proliferation of human umbilical vein endothelial (HUVEC) cells (75). Thus, the pharmacological modulation of IDO1 and other enzymes that target amino acids have been included in cancer therapy strategies (20). Preclinical and clinical studies to test the efficacy of IDO inhibitors for cancer treatment are discussed extensively in a recent review (76).
A number of studies in which IDO1 is targeted alone or in combination with immune checkpoint inhibitors have been proposed. In 2017, a phase II clinical study investigated the effect of the combined use of chemotherapy and the IDO1 inhibitor, 1-Methyl-D-tryptophan (Indoximod) in metastatic breast cancer patients (ClinicalTrials.gov Identifier: NCT01792050). Results from the phase I study indicated no drug-drug interactions and partial responses in breast cancer and patients with other metastatic tumors (77). Four of the breast cancer patients achieved a reduction in tumor burden; a patient that had hitherto only received only adjuvant endocrine therapy achieved the best response (77). Results from another phase I study on the use of a small molecule inhibitor of IDO1 (Navoximod) alone, or in combination with a PD-L1 inhibitor (Atezolizumab) to treat TNBC and other solid tumors indicated tolerability, partial responses and complete responses in some patients (78). However, there were no clear benefits associated with the use of atezolizumab with navoximod (78). Results from another phase I/II study of another IDO inhibitor, Epacadostat, used in combination with anti-PD-1 (pembrolizumab) for the treatment of TNBC and ovarian cancer indicated tolerability, safety and anti-tumor activity (79). However, in another study, there was no difference in progression-free or overall survival in patients with unresectable stage III or IV melanoma administered with Epacadostat in combination with anti-PD1 (pembrolizumab), compared to placebo plus pembrolizumab (80). Hence, the usefulness of IDO1 inhibition as a strategy to enhance anti-PD-1 therapy activity in cancer yet to be clarified.
Other approaches which utilized nanodelivery systems designed to use Indoximod in conjunction with a-PD-L1 or the induction of immunogenic cell death using doxorubicin for breast cancer treatment, have also been investigated (81). Taken together, the outcomes of these studies suggest that IDO1 inhibitors can be used as standard-of-care treatment for breast cancer and other solid tumors, alone or in combination with other cancer therapeutic strategies.
Tryptophan-2,3-Dioxygenase (TDO)
Unlike IDO1, which is induced in immune cells such as DCs, TDO is constitutively expressed in the liver, where it regulates tryptophan homoeostasis in the blood (82–84). Similar to IDO1, TDO suppresses T cell activation by tryptophan depletion and is also overexpressed in the microenvironment of various tumors, including breast cancer (26). Preclinical studies have demonstrated that TDO expression by breast cancer cells is associated with increased cancer cell migration and invasion (66, 85). In a study by Greene et al., the authors demonstrated that triple-negative breast cancer (TNBC) cells use TDO to suppress CD8+ T-cell viability (86). Furthermore, in an earlier preclinical study, D'Amato et al., showed that NF-kB-dependent upregulation of TDO and AhR is linked to anchorage-independent cell survival and anoikis resistance of TNBC cells (85). These observations imply that the overexpression of TDO by tumors such as TNBC is associated with disease metastatic.
Results from preclinical studies investigating the impact of TDO inhibition using knockout mice or compounds have shown that deletion of the TDO gene (TDO2) in mice results in tryptophan accumulation in the blood and neurologic changes, which may be associated with serotonin production (84) Consequently, the utilization of TDO inhibitors may have safety implications with respect to liver and CNS complications. Dose-dependent reduction of the 4T1 breast or CT26 colon tumor growth was achieved by dual inhibition of IDO and TDO using a lead compound, CB548, in a mouse preclinical model (87). Also, the administration of CMG017, another dual inhibitor of IDO and TDO, to tumor-bearing mice resulted in reduced kynurenine concentration, differential expression of immune-related genes and the infiltration of effector CD8+ T cells in the TME (87). Furthermore, co-administration of CMG017 with checkpoint inhibitors (a-PDL1 and a-CTLA-4) to tumor-challenged mice resulted in tumor regression and the establishment of memory CD8+ T cell responses (87).
In 2017, a phase I study was initiated to investigate the safety, pharmacokinetics, pharmacodynamics and efficacy of HTI-1090, a small molecule dual inhibitor of IDO1 and TDO, in patients with advanced solid tumors (ClinicalTrials.gov Identifier: NCT03208959). Although this study was completed in 2019, the outcomes are yet to be disclosed. The utilization of other TDO and IDO1 inhibitors such as 680C9, LM101 are still under preclinical investigation.
Catabolism of Arginine
Arginase
L-arginine is a non-essential amino acid that plays a vital role in cellular activity such as metabolic programming and maintenance of T cell fitness (88, 89). The administration of L-arginine to breast tumor-bearing BALB/c mice suppressed tumor growth significantly and prolonged the survival time of treated mice. L-arginine supplementation also enhanced the levels of IL-10, TNF-α, IFN-γ; macrophage and T cell numbers and suppressed the activity of MDSCs. The activity of arginase enzymes (ARG1 and ARG2), which catalyze L-arginine into ornithine and urea, is increased in the TME of multiple cancers including breast cancer. Arginase enzymes facilitate localized immune suppression mediated by cancer-associated fibroblasts (ARG2), MDSCs, DCs, tumor-associated macrophages (TAMs) and tumor-infiltrating macrophages (ARG1) (90, 91). These cells in turn, produce ARG1 in response to a milieu of tumor cues, such as HIF-1α, M-CSF, GM-CSF, IL-4, IL-13 and IL-6 (89). Another key enzyme associated with L-arginine metabolism, nitric oxide synthase (NOS), produces nitric oxide (NO) from L-arginine and oxygen. In low L-arginine conditions, characteristic of tumor sites, NOS can induce the production of superoxide anion, which can combine with NO to generate various reactive nitrogen species that can also hamper T cell activity at tumor sites (89).
The reduction of extracellular arginine by ARG1 leads to suppression of T cell function (Figure 2D) by the activation of GCN2 kinase, which blocks the expression of several cell cycle genes such as cdk4, cyclin D3, and CD3 (21). High levels of ARG1 have been identified in the serum of preoperative breast cancer patients compared to healthy controls (92). In addition, elevated ARG1 is expressed by MDSCs from patients diagnosed with early-stage breast cancer, which is reduced upon surgical tumor resection (2).
A number of preclinical strategies that target ARG1 have been implemented with promising results. The cell viability and arginase activity of a TNBC cell line with high levels of arginase (MDA-MB-468), were decreased in response to L-lysine induced arginase inhibition, in comparison to a cell line with less arginase levels (MDA-MB-231) (93). The treatment of tumor bearing mice (CT26, 4T1, B16, and LLC) with CB-1158, a small molecule inhibitor of ARG1, elicited increased cytotoxic T cell infiltration and decreased myeloid cell numbers (71). This correlated with increased activation markers, cytokine production and expression of interferon genes. Furthermore, CB-1158 efficacy was enhanced when combined with checkpoint blockade, chemotherapy and adoptive cell therapy (94).
Treatment with the arginase inhibitor (INCB001158) alone inhibited plasma arginase activity with concomitant increase in the plasma arginine in a colorectal carcinoma patient cohort. INCB001158 used in combination with a-PD-1 (pembrolizumab) for the treatment of advanced/metastatic solid tumors. INCB001158-pembrolizumab combination treatment elicited increased frequencies of intratumoural CD8+ T cells and a 7% partial response (ClinicalTrials.gov Identifier: NCT02903914). A clinical study has been initiated to evaluate the safety, toxicity and immune correlates of administering an Arginase-1 peptide vaccine (ARG1-18,19,20) to patients with breast cancer and other solid tumors (ClinicalTrials.gov Identifier: NCT03689192).
Chemokines and Chemokine Receptors
Chemokines and their receptors play a pivotal role in various biological and pathological processes, including chronic inflammation, tissue development, hematopoiesis, and immune modulation (95). Many studies revealed chemokines' role as essential mediators of immunity, angiogenesis (96), metastasis (97), drug resistance (98), breast cancer occurrence and progression (Figure 2C) (23, 99, 100). Chemokines have been classified into four main groups, CXC, CC, XC, and CX3C. The CXC family consists of 17 subfamily members (CXCL1-CXCL16), while CC family is the largest subgroup (CCL1-CCL28). The XC family has two subgroups (XCL1 and XCL2), while there is only one CX3C chemokine (CX3CL1) (95, 101).
Tumor cell migration and the ensuing invasion into specific organs occur in response to receptor-ligand interactions, the rearrangement of the actin cytoskeleton and multiple environmental cues which favor trafficking. Mueller et al., in investigating the role of chemokine receptors in promoting breast cancer metastasis almost two decades ago, found that breast cancer cells express CXCR4 and CCR7 (90). Consequently, targeting chemokines and their receptors has been evaluated in preclinical and clinical cancer immunotherapy studies. The detailed roles of chemokines in cancer biology have been reviewed elsewhere (23, 95, 102). We will highlight a few examples of the roles of chemokine–chemokine receptor interactions in the breast cancer microenvironment.
CXCR Family
CXCL8 (IL-8) is a chemokine whose physiological effects are mediated by two receptors, namely CXCR1 and CXCR2 (103). CXCR2 (IL-8 receptor) is expressed on MDSCs, neutrophils, lymphocytes, and breast cancer cells. CXCR2 and CXCL8 regulate breast cancer progression in the TME by modulating several related pathological processes, including promoting breast cancer growth, angiogenesis, invasion, metastasis, and reducing cancer cell sensitivity to chemotherapy (99, 104, 105). CXCR2 modulates the trafficking of neutrophils from the bone marrow to breast cancer sites, leading to increased tumor growth (106). CXCR2 also induces the migration of MDSCs, thus, promoting local immunosuppression (107). Studies show that cancer patients with high levels of CXCR2 have low overall survival and poor prognosis (108). The CXCL8-CXCR2 axis can also stimulate the transcription of VEGF and activate its receptor,VEGFR2, in endothelial cells by the NF-κB pathway (109). Like CXCR2, CXCR1 is expressed significantly in breast cancer stem cells, which increases the growth of breast cancer when stimulated by inflammation or tissue damage (110). Consequently, targeting the CXCL8-CXCR1/CXCR2 axis has been adopted as a breast cancer therapy strategy (111). The utilization of reparixin, a small molecular weight antagonist of CXCR1/2 as a breast cancer therapeutic agent has been investigated in preclinical and clinical studies (99, 112). Results from a phase Ib trial on the co-administration of reparixin and paclitaxel to patients with HER-2- negative metastatic breast cancer yielded a 30% response rate (88). In another study on the treatment of women with HER-2- negative operative breast cancer with reparixin only, the frequency of cancer stem cells, indicated by aldehyde dehydrogenase, CD44+/CD24- expression, was reduced (113).
Several studies have assessed the impact of CXCR4 in breast cancer cell survival, proliferation, angiogenesis, migration, and metastasis (114, 115). CXCR4 induces breast cancer metastases by binding to its ligand stromal cell-derived factor-1α (SDF-1), which is overexpressed in the bone marrow, liver, lung, and breast tumors sites (100, 116). CXCR4 promotes cancer cell proliferation by activating several signaling pathways, including Src/ERK1-2, PI3K/AKT, STAT3, and NF-κB. The cross-link between CXCR4 and other pathways such as Notch, Wnt, and SHH is also associated with increased breast cancer growth (117). Injecting immunocompromised mice subcutaneously with a CXCR4-low-expressing breast cancer cell line (MCF-7), resulted in reduced tumor growth compared to mice inoculated with the MDA-MB-231 cell line, which expresses high levels of CXCR4 (118). Also, results from a human study in which surgically resected ductal carcinomas were evaluated, indicate that high CXCR4 expression correlates with extensive nodal metastasis (119). Preclinical studies of CXCR4 inhibitors have demonstrated its ability to attenuate the proliferation and metastasis of breast tumors; AMD3100 is a CXCR4 antagonist that decreases lung metastases in breast cancer (120). However, Lefort et al., have shown that AMD3100 and TN14003, another CXCR4 inhibitor, impair only the growth and metastasis of HER2 breast cancers, but not TNBC (121).
In contrast to the preclinical outcomes, the efficacy of CXCR4 blockade in clinical trials has not shown clear success with respect to dosage and the manifestation of undesirable side effects. In a clinical study by Pernas et al., the safety, tolerability, pharmacokinetics, and preliminary phase 1 dose-escalation activity of the CXCR4 antagonist, balixafortide, in combination with eribulin (antineoplastic) chemotherapy, was assessed in patients with relapsed metastatic breast cancer who had hitherto received chemotherapy (96). Partial responses were observed and serious side effects occurred in 30 and 38% of the study patients, respectively. Furthermore, two patients died from septic shock and pneumonia, respectively (96). Based on the observations of the Phase 1 trial, a phase 3 study has been set up to investigate the safety, efficacy and tolerability of intravenous balixafortide administered with eribulin compared to eribulin monotherapy for the treatment of HER2 negative, locally recurrent or metastatic breast cancer patients (ClinicalTrials.gov Identifier: NCT03786094).
The CCR Family
CCL2 is overexpressed in tumor cells, including breast, ovarian, and lung cancer. CCL2 stimulates the migration of macrophages that express the chemokine CCL2 receptor (CCR2), into the TME. It also induces cancer proliferation and invasion (122). CCL2 can induce the migration of various breast cancer cell lines, including T47D, MCF-7, and ZR-75-1 (123). Studies using breast tumor xenografts show that blocking CCL2-CCR2 axis suppresses the recruitment process of inflammatory monocytes, increases tumor growth, and promotes metastasis and invasion (124). These studies suggest that CCL2-CCR2 signaling promotes breast cancer progression, and targeting this pathway might be adopted as a breast cancer therapy strategy.
CCL5/CCR5 pathway also plays a critical role in promoting breast cancer progression. CCL5 ligand is overexpressed in breast cancer cells, mesenchymal stem cells (MSCs), and infiltrating leukocytes. Results from a clinical study indicate that levels of CCL5 in breast cancer patients are higher than that of healthy controls (125). CCL5 can maintain the immunosuppressive activity of human MDSCs (126). The CCL5 receptor (CCR5) is also upregulated on breast cancer cells (127). A study conducted on breast cancer patients showed that 50% of breast tumors express CCR5, with >95% TNBC tumors being CCR5+ (128). The blockade of CCR5 suppresses breast cancer proliferation, migration, colony formation, and metastasis (129). Therefore, targeting CCR5 could be promising strategy for metastatic breast cancer. Met-CCL5, a competitive CCR5 inhibitor, reduces breast cancer proliferation and infiltrating macrophages in animal preclinical models (130). Treatment with maraviroc, CCR5 antagonist, significantly suppresses bone metastasis in a xenograft rat model implanted with breast cancer cells (MDA-MB-231) (131). Leronlimab (PRO 140) is another CCR5 antagonist under investigation in breast cancer clinical trials (129, 132, 133).
Purinergic Signaling
Purinergic signaling plays a prominent role in inflammation and cancer. It modulates cell growth, migration, and cell death (134). In this pathway, two potent molecules (ATP and Adenosine) involved in the immune response are released into the TME (Figure 3A) (135). Intracellular ATP levels are sustained at millimolar concentrations under physiological conditions, while extracellular levels are regulated in nanomolar concentrations. However, in the TME, ATP concentrations arise due to release from necrotic or apoptotic cells (136). Adenosine concentrations in solid tumors are also higher than that of healthy tissues (137, 138). It is well-reported that ATP and Adenosine have opposite effects. ATP is immunostimulatory as it enhances the activation of dendritic cells (DC), macrophages, IL-1β secretion, and cytotoxicity of CD8+ T cells. Hence, ATP activity can mediate the suppression of proliferating cancer cells. Adenosine, on the other hand, has immunosuppressive properties. It inhibits immune effector cells, DC maturation, cytokine production and stabilizes immunosuppressive Tregs (139). Purinergic cell surfaces ectoenzymes (P2Xs, P2Ys, CD73, CD39, and CD38), mediate the biological activities of ATP and Adenosine, and adenosine receptors (A1R, A2AR, A2BR, A3R), are overexpressed by breast cancer cells and tumor-infiltrating immune cells (19). Several therapeutic agents are developed to target these receptors to enhance anti-tumor immune responses against breast cancer.
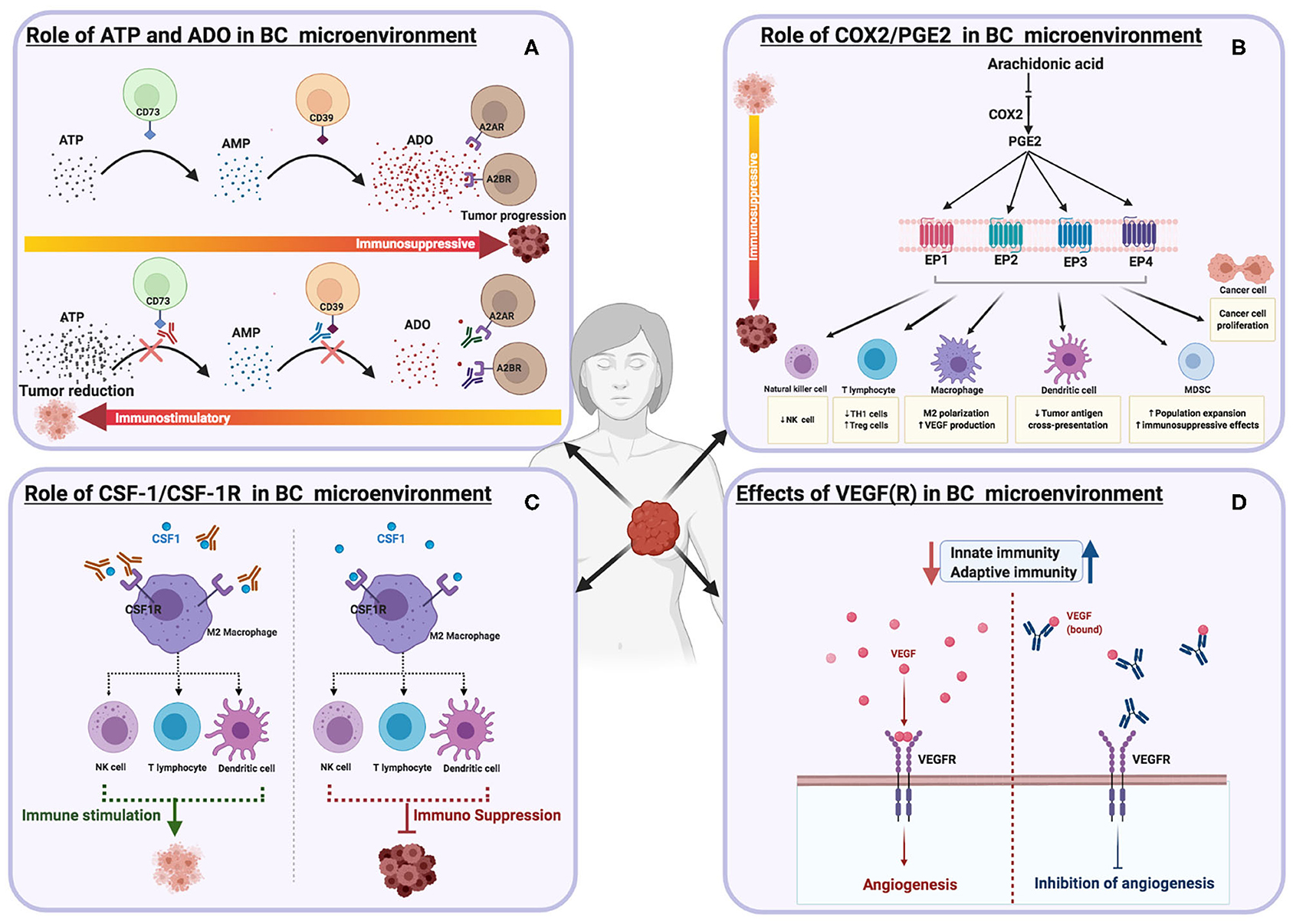
Figure 3. Schematic illustrations depicting the effects of different immune targets on breast cancer (A) ATP and Adenosine signaling (B) COX2/PGE2 pathways (C) CSF-1/CSF-1R (D) VEGF(R).
The P2 Family
The pyrogenic receptors P2Xs (ion channel receptors) and P2Ys (G protein-coupled receptors) are overexpressed on several immune cells within the TME (140). Among the pyrogenic receptors, P2X7 receptor (P2X7R) has been studied extensively due to its contrasting effects (134). In some studies the role of P2X7 in inducing antitumor immune responses by activating NK cells, CD4+, and CD8+ effector T cells, and promoting Treg apoptosis, has been shown (141, 142). Two P2X7 receptor agonists ATPγS and BzATP, reduce tumor growth and metastasis (143, 144). Other pieces of evidence propose the P2X7 receptor as promoters of tumor progression, mediated by inducing tumor growth, metastasis, and survival (145). P2X7R is upregulated in various tumors, including malignant breast cancers, and its expression is higher in tumors compared to the healthy tissue. This indicates that P2X7 can be used as an effective early cancer biomarker (40, 146). Many inhibitors that target P2X7R have been developed, such as Anthraquinone Emodin, which can potently suppress invasive breast cancer cells in vitro (147). AZ10606120 is another P2X7R antagonist reported to be a potent inhibitor of tumor growth (91).
CD39 and CD73
ATP and ADP are converted into AMP by the catalytic activity of CD39, while AMP is irreversibly converted to adenosine by CD37 (148). CD39 and CD73 are expressed significantly by breast cancer and various immune cells, including T cells, NK cells, B cells, MDSC, macrophages, and neutrophils (17). The high expression of CD39 and CD73 results in increasing adenosine levels in the TME, which in turn stimulates the adenosine A2A and A2B receptors. The adenosine A2A and A2B receptors promote tumor progression by triggering angiogenesis, tumor cell survival, and metastasis (149–151). They also increase the immunosuppressive efficacy of Tregs, macrophages, MDSCs and development of effector T cells. Breast cancer patients with positive clinical outcomes exhibited low expression of CD39 and CD73 compared to patients with poorer clinical outcomes, which indicates that CD39 and CD73 can serve as biomarkers of patients' progress (152–154). Blocking CD73 and CD39 promoted anti-tumor responses; anti-CD73 mAbs, enhances the cytotoxicity of CD8+ T cells and inhibits the activity of Tregs and MDSCs (155). Small molecules against CD73 such as LaSOM 63 and APCP, inhibit tumor progression and increase the efficacy of effector T cells (150, 156). Preclinical studies indicate that anti-CD73 mAbs can hinder metastasis in human breast cancer (157). Three CD73 antagonists (MEDI9447, BMS-986179, CPI-006), which target TNBC are currently under clinical investigation (158). Similarly, preclinical studies of anti-CD39 monoclonal antibodies, BY40 and BA54G, have demonstrated anti-tumor efficacy (159). Therapeutic agents that target CD39 are still in the developmental stage (160).
Adenosine A2A Receptor (A2AR) and Adenosine A2B Receptor (A2BR)
Extracellular adenosine stimulates the immunosuppressive pathway through engagement with specific G-protein-coupled adenosine receptors such as (A2a and A2b) (160). A2aR (high affinity receptor) is upregulated on a variety of immune cell subsets, including monocytes, macrophages, DCs, T cells, and natural killer T (NKT) cells. Adenosine signaling pathway through the A2aR suppresses T cell proliferation by increasing the expression of anti-inflammatory cytokines (IL-10, TGF-β) and reducing the expression of pro-inflammatory cytokines (IFN-γ, IL-2) (161). It also triggers increased expression of immune checkpoints such as LAG-3, PD-1, and CTLA-4 (162, 163). A2aR is overexpressed in many cancer cells, including breast cancer cells. Activation of A2aR leads to an increase in the proliferation of MCF-7 breast cancer cells (164). A2aR promotes proliferation and metastasis by stimulating various signaling pathways, including PLC/PKC, ERK-MAPK, PI3K/AKT/mTOR (165). CPI-444, an A2AR antagonist, is used as monotherapy or combined with anti-PD-L1 (Atezolizumab) to treat TNBC (166). A2bR, on the other hand, is a low-affinity receptor which needs more Adenosine to be activated. A2bR is overexpressed by macrophages, DCs, and multiple tumors such as breast tumors (167, 168). Its upregulation is associated with poor survival and worse prognosis in human TNBC (169). In vitro activation of A2bR, increases the growth and migration of breast cancer (MDA-MB-231) cells (170). Results from an in vivo study indicate that blocking A2bR reduces the metastasis of TNBC and enhances the activities of chemotherapy and immune checkpoint inhibitors (169). Several studies indicate that stimulating A2bR promotes tumor growth and metastasis through the activation of the ERK1/2 and angiogenesis pathways; blocking this receptor reverses these effects (19, 171, 172). A selective A2bR blocker (ATL801) promotes the inhibition of bladder and breast cancer growth when injected intratumorally (173).
Targeting the COX2/PGE2 Pathways
Increased levels of COX2 enzymes have been reported in nearly half of breast cancer patients (174), with other studies reporting a range of 17 to 84% (175, 176). The silencing of COX-2 expressed by the human breast cancer cell line, MDA-MB-231, inhibits cell migration in vitro and metastasis in vivo (177). PGE2, an enzymatic product of COX2, functions by signaling through one of the four G-protein coupled receptors (EP1, EP2, EP3, and EP4) (Figure 3B). The COX2/PGE2 axis promotes breast cancer progression by increasing cancer migration, metastasis, and angiogenesis (178–180). In addition, PGE2 regulates different immune cells- it suppresses the proliferation of CD4+ T cells by reducing intracellular calcium release and suppressing the activity of the p59 protein tyrosine kinase (181, 182). PGE2 decreases the production of effector cytokines, such as IL-2 and IFN-γ, and it can also inhibit NK cell function and B cell proliferation (183–185). PGE2 elevates cAMP by the stimulation of its receptors, EP2 and EP4 (186). COX2/PGE2 and its receptors are potential target(s) for breast cancer therapy. Preclinical studies indicate that celecoxib, a selective COX-2 inhibitor, reduces breast cancer metastasis (176, 187). The daily intake of COX-2 inhibitors such as non-steroidal anti-inflammatory drugs (NSAIDs) reduce the risk of breast cancer occurrence significantly (188). The PGEP4 receptor blocker (AAT-007) is currently in phase 2 for the treatment of patients with solid tumors, including breast cancer (179). A newer version of the PGEP4 receptor antagonist called (AAT-008) has shown significant bioavailability and pharmacological profiles in preclinical investigations (189). The PGE2 EP1 antagonist (ONO-8711) suppresses breast cancer progression in rats (190). Using different breast cancer cell lines in vitro, the PGEP3 receptor antagonist (L798,106), demonstrated potency in reducing breast cancer proliferation and migrations (191).
CSF-1/CSF-1R
Activated macrophages are divided, for simplicity, into anti-tumor (M1) macrophages and pro-tumor (M2) macrophages. M1 macrophages are activated by GM-CSF, IFN-γ, LPS, and other cytokines. M1 macrophages, referred to as “fight” macrophages, play a significant role in producing pro-inflammatory cytokines and inducing anti-tumor immune responses (192, 193). The growth factor, GM-CSF, regulates the differentiation of DCs and macrophages (194, 195). Results from in vivo studies indicate that GM-CSF suppresses breast cancer growth and metastasis (196). In contrast, M2 macrophages induce tumor proliferation, therapy resistance, tumor invasion, angiogenesis, and metastasis. M2 macrophages are polarized by colony-stimulating factor 1 (CSF1), IL-13, IL-10, IL-4, TGF-β, and prostaglandin E2 (197, 198). The upregulation of CSF-1 signaling correlates with increased breast cancer progression (Figure 3C) (199). CSF1R is expressed by both M1/M2 TAMs, MDSCs, neutrophils, and DCs (200). CSF1/CSF1R signaling increases angiogenesis, cancer growth, metastases, invasion, CD8+ T cell suppression, tumor macrophage recruitment, and resistance to therapy (201, 202). CSF1 can also stimulate VEGF production (196). Blocking CSF1 in breast cancer-bearing mice reversed these effects and increased mouse survival rate (203). There are currently many therapeutic agents that target CSF1 and its receptor CSF1R, in preclinical or clinical development stages. For example, LY3022855, a CSF1R blocker used as a single agent or in combination with Durvalumab (anti-PDL1 mAb) or Tremelimumab (anti-CTLA4 mAb) for patients with a solid tumors, including breast cancers (24). Pexidartinib is another inhibitor of CSF1R that is used in combination with a microtubule inhibitor (Eribulin) for breast cancer patients (24). Anti- CSF1R (Emactuzumab) combined with Atezolizumab (anti-PDL1 mAb) are used to treat TNBC (24, 204).
Vascular Endothelial Growth Factor A (VEGF-A)
VEGF binding to its receptors promotes the progression, proliferation, and metastasis of breast cancer (Figure 3D) (22, 205, 206). Among the five identified VEGF subfamilies (VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E), VEGFA, also called VEGF, is the dominant and most researched isoform (207). VEGF isoforms bind with varying affinities to VEGFR1, VEGFR2, and VEGFR3, which mediates VEGF downstream signaling (208). VEGFA is overexpressed in several types of cancer, including breast cancer (209), and plays a vital role in angiogenesis (210). VEGF halts the differentiation and activation of DCs and promotes the exhaustion of CD8+ T cells by increasing the expression of inhibitory receptors, such as PD-1, TIM-3, LAG-3, and CTLA-4 (211). High VEGF plasma levels in breast cancer patients is associated with a significant reduction of DCs in the peripheral blood of cancer patients. The appearance of immature DCs in the blood correlates with the duration and disease stage; surgical removal of tumors showed a partial reversal of the noted effects (212). On the other hand, inhibiting VEGF increases tumor-infiltrating effector T-cells and reduces the recruitment of Tregs and MDSCs to the TME (213). Blocking VEGF stops the growth of tumor blood vessels in murine models and promotes cancer cell death and tumor-shrinkage (214). Therefore, targeting VEGF and its receptor VEGFR are key therapeutic targets for breast cancer treatment. Many angiogenesis inhibitors have been approved by the FDA, however, only a few have been tested in breast cancer patients such as bevacizumab, which binds to VEGFA and blocks its efficacy (215). Bevacizumab was the first FDA approved antiangiogenic agent (216, 217). In 2008, it was approved to be used in combination with chemotherapy to treat metastatic HER2-negative breast cancer (218). However, it showed several adverse side effects and poor overall survival, which led the FDA to revoke its approval in 2011 (219, 220). An example of a VEGFR inhibitor is DC101, a monoclonal antibody which binds to VEGFR2, and exhibits potential antiangiogenic efficacy against breast tumors in xenograft models. In another in vivo study, DC101 enhanced tumor-specific CD8+ T cells and accelerated tumor regression. Combining DC101 with neu-specific vaccination also suppressed tumor progression and increased the activity of CD8+ T cells (221). Ramucirumab, a VEGFR2 blocker, has shown preclinical and clinical promise in targeting breast cancer angiogenesis, growth, and metastasis (222). Axitinib is a small molecule that binds selectively to VEGFR-1,−2, and−3, and blocks their activities (223); murine studies indicate its potency in inhibiting breast cancer growth (224). However, clinical studies have only demonstrated its activity in combination with chemotherapy (paclitaxel). Sorafenib is another small molecule VEGFR blocker; reports indicate encouraging clinical trial results from the treatment of breast cancer patients. However, the utilization of sunitinib, a VEGFR inhibitor, has not shown any clinical benefit in breast cancer patients (225).
Overall, the preclinical results obtained from the use of anti-VEGF agents showed a significant decrease in tumor angiogenesis. However, the outcome of clinical trials exhibited an average response (22, 226).
Toll-Like Receptor (TLR)
TLRs are expressed by both cancer and immune cells (227, 228). Among the thirteen TLRs (TLR1-13) that have been characterized, ten (TLR1-10) were identified in humans, six of which are expressed on the cell surface TLR (1, 2, 4, 5, 6, and 10) and four on endosomal membranes (229).
Several TLRs are upregulated in human breast tumors. TLR4 is the most expressed among the TLR family, on breast cancer cells (MDA-MB-231 cells). Deletion of the TLR4 gene resulted in an increase in cell death and suppression of IL-6 and IL-8 expression (230). The overexpression of TLR9 in human breast cancer enhances tumor cell invasion, which is mechanistically linked to the induction of MMP13 and COX-2 secretion (231). Various studies have reported positive correlations between TLR expression and the activation of the immune system. TLR stimulates DCs and macrophages and promotes the secretion of pro-inflammatory cytokines and the facilitation of anti-tumor immune responses (232, 233). The role of TLRs as pro-tumor agents has also been investigated (234). The function of TLRs in cancer can be described as a “double-edged sword” (Figure 4). On the one hand, agonists that bind to TLR(s) on tumor cells can promote cancer progression by promoting immune escape and cancer cell proliferation and survival. The engagement of TLR4 expressed by human breast cancer cells results in increased production immunosuppressive factors such as NO, VEGF, and MMPs, thereby promoting the tumor invasion (230, 235, 236).
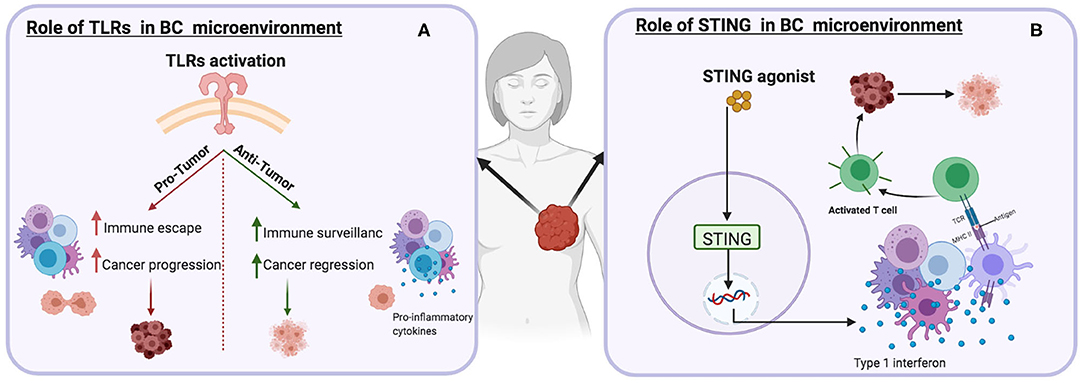
Figure 4. Schematic illustrations depicting the effects of different immune targets on breast cancer (A) Toll-like receptor(s) (B) Stimulator of interferon genes protein.
On the other hand, activating TLR5 in the breast cancer mouse model resulted in anti-proliferative efficacy through the promotion of necrosis, increased neutrophil infiltration and down-regulation of cyclin B1, cyclin D1, and cyclin E2 (237). TLR3 expressed by human and mouse breast cancer cells promotes apoptosis by inducing type I IFN signaling (238). Preclinical studies have demonstrated that TLR agonists, combined with other therapeutic agents, can potentially reduce and suppress tumor progression (239, 240). The different roles of TLRs are linked to the proximal signaling pathways stimulated in cancer cells and immune cells. For example, even though TLR5 is overexpressed in both gastric and breast cancers, it has opposite effects as it suppresses the proliferation of breast cancer and induces the growth of gastric cancer cells (237, 241). Many TLR agonists have been investigated for clinical use. The TLR5 agonist, flagellin, suppresses breast cancer by induction of caspase-1 activation-dependent pyroptosis. It also enhances the expression of granzyme B, TNF-α, and IFN-γ in CD8+ T cells (242, 243). The TLR3 ligand, poly-AU, increases the survival rate in patients with TLR3-positive breast cancer (244). Imiquimod is a well-tolerated TLR7 agonist that can promote the rejection of immune-mediated skin metastasis in breast cancer patients (245) 852A is another TLR7 agonist used for the treatment of metastatic breast cancer patients (240).
Stimulator of Interferon Genes Protein (STING)
Various studies have suggested that STING (stimulator of interferon (IFN) genes) expression is not only confined to innate and adaptive immune cells (246–248), but is also expressed in various tumors, including breast cancer (249). STING stimulators have shown great potential for activating immune cells, enhancing anti-tumor immunity by inducing a variety of pro-inflammatory cytokines and chemokines (246, 247, 250), priming and activation of T cells (251), enhancement of antigen presentation, promotion of cancer cell death, inducing the recognition and apoptosis of cancer cells by T cells (249, 252, 253). A previous study has revealed the role of STING in promoting death in 4T1 breast cancer cells by increasing the caspase-3 pathway cascade (249). Similarly, the overexpression of STING in two breast cancer cell lines, T47D or MCF-7 has been shown to increase caspase 3 and/or 7 activity (252). The deletion of STING expressed by melanoma cell lines results in the suppression of cytokines (IFN-γ) and chemokines (CCL5 and CXCL10) production (254). Furthermore, STING knockout mice exhibit reduced NK cell responses by mediating the downregulation of perforin, granzyme B, and IFN-γ (253, 254). Numerous STING stimulators are now under clinical investigation for the treatment of various types of cancers. The utilization of ADU-S100 (MIW815), a STING agonist, is currently being tested in combination with anti-PD-1 (spartalizumab) for the treatment of patients with solid tumors, including PD-1-naïve TNBC (255).
Conclusions
Following several years of preclinical and clinical research, our understanding of how the immune system responds to cancer has increased. The limited success of immune checkpoints, like CTLA-4 or PD-1, in clinical trials for breast cancer patients, has prompted research to find alternative targets. Many new emerging data reported novel pathways that stimulate immune responses against breast tumors. These newly discovered pathways are likely to be the future targets of breast cancer immunotherapy.
Author Contributions
YT and IO drafted the manuscript. KB, AS, and SE edited and modified the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the Alberta Cancer Foundation (ACF) (KB). This work was also supported by a grant from the Canadian Institutes of Health Research (CIHR) and Cancer Research Society (SE).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2018) 68:394–424. doi: 10.3322/caac.21492
2. de Boniface J, Mao Y, Schmidt-Mende J, Kiessling R, Poschke I. Expression patterns of the immunomodulatory enzyme arginase 1 in blood, lymph nodes and tumor tissue of early-stage breast cancer patients. Oncoimmunology. (2012) 1:1305–12. doi: 10.4161/onci.21678
3. Tong CW, Wu M, Cho W, To KK. Recent advances in the treatment of breast cancer. Front Oncol. (2018) 8:227. doi: 10.3389/fonc.2018.00227
4. Anderson WF, Katki HA, Rosenberg PS. Incidence of breast cancer in the United States: current and future trends. J Natl Cancer Inst. (2011) 103:1397–402. doi: 10.1093/jnci/djr257
5. Hu J, Hu B, Wang M, Xu F, Miao B, Yang CY, et al. Discovery of ERD-308 as a highly potent proteolysis targeting chimera (PROTAC) degrader of estrogen receptor (ER). J Med Chem. (2019) 62:1420–42. doi: 10.1021/acs.jmedchem.8b01572
6. García-Aranda M, Redondo M. Immunotherapy: a challenge of breast cancer treatment. Cancers. (2019) 11:1822. doi: 10.3390/cancers11121822
7. Cimino-Mathews A, Thompson E, Taube JM, Ye X, Lu Y, Meeker A, et al. PD-L1 (B7-H1) expression and the immune tumor microenvironment in primary and metastatic breast carcinomas. Hum Pathol. (2016) 47:52–63. doi: 10.1016/j.humpath.2015.09.003
8. Del Alcazar CRG, Huh SJ, Ekram MB, Trinh A, Liu LL, Beca F, et al. Immune escape in breast cancer during in situ to invasive carcinoma transition. Cancer Discov. (2017) 7:1098–115. doi: 10.1158/2159-8290.CD-17-0222
9. Nakasone ES, Hurvitz SA, McCann KE. Harnessing the immune system in the battle against breast cancer. Drugs Context. (2018) 7:212520. doi: 10.7573/dic.212520
10. Chen R, Ganesan A, Okoye I, Arutyunova E, Elahi S, Lemieux MJ, et al. Targeting B7-1 in immunotherapy. Med Res Rev. (2020) 40:654–82. doi: 10.1002/med.21632
11. Okoye IS, Houghton M, Tyrrell L, Barakat K, Elahi S. Coinhibitory receptor expression and immune checkpoint blockade: maintaining a balance in CD8+ T cell responses to chronic viral infections and cancer. Front Immunol. (2017) 8:1215. doi: 10.3389/fimmu.2017.01215
12. Ganesan A, Ahmed M, Okoye I, Arutyunova E, Babu D, Turnbull WL, et al. Comprehensive in vitro characterization of PD-L1 small molecule inhibitors. Sci Rep. (2019) 9:1–9.
13. Chrétien S, Zerdes I, Bergh J, Matikas A, Foukakis T. Beyond PD-1/PD-L1 inhibition: what the future holds for breast cancer immunotherapy. Cancers. (2019) 11:628. doi: 10.3390/cancers11050628
14. Nanda R, Chow LQM, Dees EC, Berger R, Gupta S, Geva R, et al. Pembrolizumab in patients with advanced triple-negative breast cancer: phase Ib KEYNOTE-012 study. J Clin Oncol Off J Am Soc Clin Oncol. (2016) 34:2460–7. doi: 10.1200/JCO.2015.64.8931
15. Cortés J, Guo Z, Karantza V, Aktan G. KEYNOTE-355: randomized, double-blind, phase III study of pembrolizumab (pembro)+ chemotherapy (chemo) vs placebo (PBO)+ chemo for previously untreated, locally recurrent, inoperable or metastatic triple-negative breast cancer (mTNBC). J Clin Oncol. (2018) 38:1. doi: 10.1158/1538-7445.AM2017-CT069
16. Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im S-A, Yusof MM, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet. (2020) 396:1817–28. doi: 10.1016/S0140-6736(20)32531-9
17. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD 39 and CD 73: novel checkpoint inhibitor targets. Immunol Rev. (2017) 276:121–44. doi: 10.1111/imr.12528
18. Perrot I, Michaud H-A, Giraudon-Paoli M, Augier S, Docquier A, Gros L, et al. Blocking antibodies targeting the CD39/CD73 immunosuppressive pathway unleash immune responses in combination cancer therapies. Cell Rep. (2019) 27:2411–25. doi: 10.1016/j.celrep.2019.04.091
19. Sek K, Mølck C, Stewart GD, Kats L, Darcy PK, Beavis PA. Targeting adenosine receptor signaling in cancer immunotherapy. Int J Mol Sci. (2018) 19:3837. doi: 10.3390/ijms19123837
20. Mondanelli G, Iacono A, Allegrucci M, Puccetti P, Grohmann U. Immunoregulatory interplay between arginine and tryptophan metabolism in health and disease. Front Immunol. (2019) 10:1565. doi: 10.3389/fimmu.2019.01565
21. Raber P, Ochoa AC, Rodríguez PC. Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: mechanisms of T cell suppression and therapeutic perspectives. Immunol Invest. (2012) 41:614–34. doi: 10.3109/08820139.2012.680634
22. Madu CO, Wang S, Madu CO, Lu Y. Angiogenesis in Breast Cancer Progression, Diagnosis, and Treatment. J Cancer. (2020) 11:4474. doi: 10.7150/jca.44313
23. Ali S, Lazennec G. Chemokines: novel targets for breast cancer metastasis. Cancer Metastasis Rev. (2007) 26:401–20. doi: 10.1007/s10555-007-9073-z
24. Cannarile MA, Weisser M, Jacob W, Jegg A-M, Ries CH, Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. (2017) 5:53. doi: 10.1186/s40425-017-0257-y
25. Yusuf N. Toll-like receptor mediated regulation of breast cancer: a case of mixed blessings. Front Immunol. (2014) 5:224. doi: 10.3389/fimmu.2014.00224
26. Opitz CA, Patterson LFS, Mohapatra SR, Dewi DL, Sadik A, Platten M, et al. The therapeutic potential of targeting tryptophan catabolism in cancer. Br J Cancer. (2019) 122:30–44. doi: 10.1038/s41416-019-0664-6
27. Chen EP, Smyth EM. COX-2 and PGE2-dependent immunomodulation in breast cancer. Prostaglandins Other Lipid Mediat. (2011) 96:14–20. doi: 10.1016/j.prostaglandins.2011.08.005
28. Su T, Zhang Y, Valerie K, Wang XY, Lin S, Zhu G. STING activation in cancer immunotherapy. Theranostics. (2019) 9:7759–71. doi: 10.7150/thno.37574
29. Swoboda A, Nanda R. Immune checkpoint blockade for breast cancer. Cancer Treat Res. (2018) 173:155–65. doi: 10.1007/978-3-319-70197-4_10
30. Page DB, Bear H, Prabhakaran S, Gatti-Mays ME, Thomas A, Cobain E, et al. Two may be better than one: PD-1/PD-L1 blockade combination approaches in metastatic breast cancer. NPJ Breast Cancer. (2019) 5:1–9. doi: 10.1038/s41523-019-0130-x
31. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. (2013) 13:227. doi: 10.1038/nri3405
32. Gaynor N, Crown J, Collins DM. Immune checkpoint inhibitors: key trials and an emerging role in breast cancer. Semin Cancer Biol. (2020) 33:3–15. doi: 10.1016/j.semcancer.2020.06.016
33. Moran AE, Kovacsovics-Bankowski M, Weinberg AD. The TNFRs OX40, 4-1BB, and CD40 as targets for cancer immunotherapy. Curr Opin Immunol. (2013) 25:230–7. doi: 10.1016/j.coi.2013.01.004
34. Deng J, Zhao S, Zhang X, Jia K, Wang H, Zhou C, et al. OX40 (CD134) and OX40 ligand, important immune checkpoints in cancer. OncoTargets Ther. (2019) 12:7347. doi: 10.2147/OTT.S214211
35. Hamid O, Thompson JA, Diab A, Ros W, Eskens F, Bermingham C, et al. First in human (FIH) study of an OX40 agonist monoclonal antibody (mAb) PF-04518600 (PF-8600) in adult patients (pts) with select advanced solid tumors: preliminary safety and pharmacokinetic (PK)/pharmacodynamic results. Am Soc Clin Oncol. (2016) 34:3079. doi: 10.1200/JCO.2016.34.15_suppl.3079
36. Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol OncolJ Hematol Oncol. (2018) 11:39. doi: 10.1186/s13045-018-0582-8
37. Polesso F, Sarker M, Weinberg AD, Murray SE, Moran AE. OX40 agonist tumor immunotherapy does not impact regulatory T cell suppressive function. J Immunol. (2019) 203:2011–9. doi: 10.4049/jimmunol.1900696
38. Messenheimer DJ, Jensen SM, Afentoulis ME, Wegmann KW, Feng Z, Friedman DJ, et al. Timing of PD-1 blockade is critical to effective combination immunotherapy with anti-OX40. Clin Cancer Res. (2017) 23:6165–77. doi: 10.1158/1078-0432.CCR-16-2677
39. Gutierrez M, Moreno V, Heinhuis KM, Olszanski AJ, Spreafico A, Ong M, et al. OX40 agonist BMS-986178 alone or in combination with nivolumab and/or ipilimumab in patients with advanced solid tumors. Clin Cancer Res. (2020) 27:460–72. doi: 10.1158/1078-0432.CCR-20-1830
40. Solinas C, Gu-Trantien C, Willard-Gallo K. The rationale behind targeting the ICOS-ICOS ligand costimulatory pathway in cancer immunotherapy. ESMO Open. (2020) 5:1–7. doi: 10.1136/esmoopen-2019-000544
41. Riley JL, Blair PJ, Musser JT, Abe R, Tezuka K, Tsuji T, et al. ICOS costimulation requires IL-2 and can be prevented by CTLA-4 engagement. J Immunol. (2001) 166:4943–8. doi: 10.4049/jimmunol.166.8.4943
42. Wikenheiser DJ, Stumhofer JS. ICOS co-stimulation: friend or foe? Front Immunol. (2016) 7:304. doi: 10.3389/fimmu.2016.00304
43. Gariepy J, Prodeus A, Sparkes A, Fischer N, Saha S. A powerful ICOS agonist that enhances anti-tumor immune responses restored by immune checkpoint inhibitors. J Immunol. (2019) 202:1–20.
44. Xiao Z, Mayer AT, Nobashi TW, Gambhir SS. ICOS is an indicator of T-cell–mediated response to cancer immunotherapy. Cancer Res. (2020) 80:3023–32. doi: 10.1158/0008-5472.CAN-19-3265
45. Soldevilla MM, Villanueva H, Meraviglia-Crivelli D, Menon AP, Ruiz M, Cebollero J, et al. ICOS costimulation at the tumor site in combination with ctla-4 blockade therapy elicits strong tumor immunity. Mol Ther. (2019) 27:1878–91. doi: 10.1016/j.ymthe.2019.07.013
46. Burris HA, Callahan MK, Tolcher AW, Kummar S, Falchook GS, Pachynski RK, et al. Phase 1 safety of ICOS agonist antibody JTX-2011 alone and with nivolumab (nivo) in advanced solid tumors; predicted vs observed pharmacokinetics (PK) in ICONIC. J Clin Oncol. (2017) 35:3033. doi: 10.1200/JCO.2017.35.15_suppl.3033
47. Zhu MM, Burugu S, Gao D, Yu J, Kos Z, Leung S, et al. Evaluation of glucocorticoid-induced TNF receptor (GITR) expression in breast cancer and across multiple tumor types. Mod Pathol. (2020) 33:1753–63. doi: 10.1038/s41379-020-0550-z
48. Krausz L, Fischer-Fodor E, Major ZZ, Fetica B. GITR-expressing regulatory T-cell subsets are increased in tumor-positive lymph nodes from advanced breast cancer patients as compared to tumor-negative lymph nodes. Int J Immunopathol Pharmacol. (2012) 25:59–66. doi: 10.1177/039463201202500108
49. Han S, Toker A, Liu ZQ, Ohashi PS. Turning the tide against regulatory T cells. Front Oncol. (2019) 9:279. doi: 10.3389/fonc.2019.00279
50. Marhelava K, Pilch Z, Bajor M, Graczyk-Jarzynka A, Zagozdzon R. Targeting negative and positive immune checkpoints with monoclonal antibodies in therapy of cancer. Cancers. (2019) 11:1756. doi: 10.3390/cancers11111756
51. Zappasodi R, Sirard C, Li Y, Budhu S, Abu-Akeel M, Liu C, et al. Rational design of anti-GITR-based combination immunotherapy. Nat Med. (2019) 25:759–66. doi: 10.1038/s41591-019-0420-8
52. Koon HB, Shepard DR, Merghoub T, Schaer DA, Sirard CA, Wolchok JD. First-in-human phase 1 single-dose study of TRX-518, an anti-human glucocorticoid-induced tumor necrosis factor receptor (GITR) monoclonal antibody in adults with advanced solid tumors. J Clin Oncol. (2016) 34:3017. doi: 10.1200/JCO.2016.34.15_suppl.3017
53. Elgueta R, Benson MJ, De Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. (2009) 229:152–72. doi: 10.1111/j.1600-065X.2009.00782.x
54. Driessens G, Kline J, Gajewski TF. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol Rev. (2009) 229:126–44. doi: 10.1111/j.1600-065X.2009.00771.x
55. Young L, Dawson C, Brown K, Rickinson A. Identification of a human epithelial cell surface protein sharing an epitope with the C3d/Epstein-Barr virus receptor molecule of B lymphocytes. Int J Cancer. (1989) 43:786–94. doi: 10.1002/ijc.2910430508
56. ÜNVER N, ERMIS DY, Weber BZ, ESENDAGLI G. Transcriptional splice variants of CD40 and its prognostic value in breast cancer. Turk J Biol. (2020) 44:73–81. doi: 10.3906/biy-1912-21
57. Tong AW, Papayoti MH, Netto G, Armstrong DT, Ordonez G, Lawson JM, et al. Growth-inhibitory effects of CD40 ligand (CD154) and its endogenous expression in human breast cancer. Clin Cancer Res. (2001) 7:691–703. Available online at: https://clincancerres.aacrjournals.org/content/7/3/691.long
58. Wingett DG, Vestal RE, Forcier K, Hadjokas N, Nielson CP. CD40 is functionally expressed on human breast carcinomas: variable inducibility by cytokines and enhancement of Fas-mediated apoptosis. Breast Cancer Res Treat. (1998) 50:27–36. doi: 10.1023/A:1006012607452
59. Ma HS, Poudel B, Torres ER, Sidhom J-W, Robinson TM, Christmas B, et al. A CD40 agonist and PD-1 antagonist antibody reprogram the microenvironment of nonimmunogenic tumors to allow T-cell-mediated anticancer activity. Cancer Immunol Res. (2019) 7:428–42. doi: 10.1158/2326-6066.CIR-18-0061
60. Bartkowiak T, Curran MA. 4-1BB agonists: multi-potent potentiators of tumor immunity. Front Oncol. (2015) 5:117. doi: 10.3389/fonc.2015.00117
61. Harao M, Forget M-A, Roszik J, Gao H, Babiera GV, Krishnamurthy S, et al. 4-1BB–enhanced expansion of CD8+ TIL from triple-negative breast cancer unveils mutation-specific CD8+ T cells. Cancer Immunol Res. (2017) 5:439–45. doi: 10.1158/2326-6066.CIR-16-0364
62. Chester C, Ambulkar S, Kohrt HE. 4-1BB agonism: adding the accelerator to cancer immunotherapy. Cancer Immunol Immunother. (2016) 65:1243–8. doi: 10.1007/s00262-016-1829-2
63. Kohrt HE, Houot R, Weiskopf K, Goldstein MJ, Scheeren F, Czerwinski D, et al. Stimulation of natural killer cells with a CD137-specific antibody enhances trastuzumab efficacy in xenotransplant models of breast cancer. J Clin Invest. (2012) 122:1066–75. doi: 10.1172/JCI61226
64. Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood J Am Soc Hematol. (2018) 131:49–57. doi: 10.1182/blood-2017-06-741041
65. Claus C, Ferrara C, Xu W, Sam J, Lang S, Uhlenbrock F, et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci Transl Med. (2019) 11:1–12. doi: 10.1126/scitranslmed.aav5989
66. Platten M, Nollen EA, Röhrig UF, Fallarino F, Opitz CA. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov. (2019) 18:379–401. doi: 10.1038/s41573-019-0016-5
67. Ye Z, Yue L, Shi J, Shao M, Wu T. Role of IDO and TDO in cancers and related diseases and the therapeutic implications. J Cancer. (2019) 10:2771. doi: 10.7150/jca.31727
68. Wu H, Gong J, Liu Y. Indoleamine 2, 3-dioxygenase regulation of immune response. Mol Med Rep. (2018) 17:4867–73. doi: 10.3892/mmr.2018.8537
69. Meireson A, Devos M, Brochez L. IDO expression in cancer: different compartment, different functionality? Front Immunol. (2020) 11:2340. doi: 10.3389/fimmu.2020.531491
70. Mondanelli G, Ugel S, Grohmann U, Bronte V. The immune regulation in cancer by the amino acid metabolizing enzymes ARG and IDO. Curr Opin Pharmacol. (2017) 35:30–9. doi: 10.1016/j.coph.2017.05.002
71. Asghar K, Loya A, Rana IA, Tahseen M, Ishaq M, Farooq A, et al. Indoleamine 2, 3-dioxygenase expression and overall survival in patients diagnosed with breast cancer in Pakistan. Cancer Manag Res. (2019) 11:475. doi: 10.2147/CMAR.S184221
72. Kim S, Park S, Cho MS, Lim W, Moon B-I, Sung SH. Strong Correlation of Indoleamine 2,3-Dioxygenase 1 Expression with Basal-Like Phenotype and Increased Lymphocytic Infiltration in Triple-Negative Breast Cancer. J Cancer. (2017) Jan 1;8:124–30. doi: 10.7150/jca.17437
73. Dill EA, Dillon PM, Bullock TN, Mills AM. IDO expression in breast cancer: an assessment of 281 primary and metastatic cases with comparison to PD-L1. Mod Pathol. (2018) Oct 1;31:1513–22. doi: 10.1038/s41379-018-0061-3
74. Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T, et al. Tumor-repopulating cells induce PD-1 expression in CD8+ T cells by transferring kynurenine and AhR activation. Cancer Cell. (2018) 33:480–94. doi: 10.1016/j.ccell.2018.02.005
75. Wei L, Zhu S, Li M, Li F, Wei F, Liu J, et al. High indoleamine 2, 3-dioxygenase is correlated with microvessel density and worse prognosis in breast cancer. Front Immunol. (2018) 9:724. doi: 10.3389/fimmu.2018.00724
76. Le Naour J, Galluzzi L, Zitvogel L, Kroemer G, Vacchelli E. Trial watch: IDO inhibitors in cancer therapy. OncoImmunology. (2020) 9:1777625. doi: 10.1080/2162402X.2020.1777625
77. Soliman HH, Jackson E, Neuger T, Dees EC, Harvey RD, Han H, et al. A first in man phase I trial of the oral immunomodulator, indoximod, combined with docetaxel in patients with metastatic solid tumors. Oncotarget. (2014) 5:8136–46. doi: 10.18632/oncotarget.2357
78. Jung KH, LoRusso P, Burris H, Gordon M, Bang Y-J, Hellmann MD, et al. Phase I study of the indoleamine 2, 3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) administered with PD-L1 inhibitor (atezolizumab) in advanced solid tumors. Clin Cancer Res. (2019) 25:3220–8. doi: 10.1158/1078-0432.CCR-18-2740
79. Spira AI, Hamid O, Bauer TM, Borges VF, Wasser JS, Smith DC, et al. Efficacy/safety of epacadostat plus pembrolizumab in triple-negative breast cancer and ovarian cancer: phase I/II ECHO-202 study. J Clin Oncol. (2017) 35:1103. doi: 10.1200/JCO.2017.35.15_suppl.1103
80. Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. (2019) 20:1083–97. doi: 10.1016/S1470-2045(19)30274-8
81. Lu J, Liu X, Liao YP, Wang X, Ahmed A, Jiang W, et al. Breast cancer chemo-immunotherapy through liposomal delivery of an immunogenic cell death stimulus plus interference in the IDO-1 pathway. ACS Nano. (2018) 12:11041–61. doi: 10.1021/acsnano.8b05189
82. Zhai L, Spranger S, Binder DC, Gritsina G, Lauing KL, Giles FJ, et al. Molecular pathways: targeting IDO1 and other tryptophan dioxygenases for cancer immunotherapy. Clin Cancer Res. (2015) 21:5427–33. doi: 10.1158/1078-0432.CCR-15-0420
83. Hornyák L, Dobos N, Koncz G, Karányi Z, Páll D, Szabó Z, et al. The role of indoleamine-2, 3-dioxygenase in cancer development, diagnostics, and therapy. Front Immunol. (2018) 9:151. doi: 10.3389/fimmu.2018.00151
84. Prendergast GC, Malachowski WJ, Mondal A, Scherle P, Muller AJ. Indoleamine 2, 3-dioxygenase and its therapeutic inhibition in cancer. Int. Rev. Cell Mol. Biol. (2018) 336:175–203. doi: 10.1016/bs.ircmb.2017.07.004
85. D'Amato NC, Rogers TJ, Gordon MA, Greene LI, Cochrane DR, Spoelstra NS, et al. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res. (2015) 75:4651–64. doi: 10.1158/0008-5472.CAN-15-2011
86. Greene LI, Bruno TC, Christenson JL, D'Alessandro A, Culp-Hill R, Torkko K, et al. A role for tryptophan-2,3-dioxygenase in CD8 T-cell suppression and evidence of tryptophan catabolism in breast cancer patient plasma. Mol Cancer Res. (2019) 17:131–9. doi: 10.1158/1541-7786.MCR-18-0362
87. Kim C, Kim JH, Kim J sung, Chon HJ, Kim JH. A novel dual inhibitor of IDO and TDO, CMG017, potently suppresses the kynurenine pathway and overcomes resistance to immune checkpoint inhibitors. J Clin Oncol. (2019) 37:14228. doi: 10.1200/JCO.2019.37.15_suppl.e14228
88. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. (2016) 167:829–42. doi: 10.1016/j.cell.2016.09.031
89. Mondanelli G, Bianchi R, Pallotta MT, Orabona C, Albini E, Iacono A, et al. A relay pathway between arginine and tryptophan metabolism confers immunosuppressive properties on dendritic cells. Immunity. (2017) 46:233–44. doi: 10.1016/j.immuni.2017.01.005
90. WU G, MORRIS SM Jr. Arginine metabolism: nitric oxide and beyond. Biochem J. (1998) 336:1–17. doi: 10.1042/bj3360001
91. Li J, Van Valkenburgh J, Hong X, Conti PS, Zhang X, Chen K. Small molecules as theranostic agents in cancer immunology. Theranostics. (2019) 9:7849–71. doi: 10.7150/thno.37218
92. Porembska Z, Luboiński G, Chrzanowska A, Mielczarek M, Magnuska J, Barańczyk-Kuzma A. Arginase in patients with breast cancer. Clin Chim Acta. (2003) Feb 1;328:105–11. doi: 10.1016/S0009-8981(02)00391-1
93. Yarani R, Jahani M, Tahmasebi H, Chehri J, Mansouri K. P2-195 L-Arginine metabolism alteration by L-Lysine intervention increased cell death in triple negative breast cancer cell. Ann Oncol. (2019) 30:mdz343-080. doi: 10.1093/annonc/mdz343.080
94. Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer. (2017) 5:101–101. doi: 10.1186/s40425-017-0308-4
95. Liu H, Yang Z, Lu W, Chen Z, Chen L, Han S, et al. Chemokines and chemokine receptors: a new strategy for breast cancer therapy. Cancer Med. (2020) 9:3786–99. doi: 10.1002/cam4.3014
96. Belperio JA, Keane MP, Arenberg DA, Addison CL, Ehlert JE, Burdick MD, et al. CXC chemokines in angiogenesis. J Leukoc Biol. (2000) 68:1–8. doi: 10.1189/jlb.68.1.1
98. Marquette C, Nabell L. Chemotherapy-resistant metastatic breast cancer. Curr Treat Options Oncol. (2012) 13:263–75. doi: 10.1007/s11864-012-0184-6
99. Guo F, Long L, Wang J, Wang Y, Liu Y, Wang L, et al. Insights on CXC chemokine receptor 2 in breast cancer: an emerging target for oncotherapy. Oncol Lett. (2019) 18:5699–708. doi: 10.3892/ol.2019.10957
100. Cortés J, Holgado E, Perez-Garcia J. CXCR4 antagonists for treatment of breast cancer. Oncotarget. (2018) 9:33442. doi: 10.18632/oncotarget.26090
101. Rollins BJ. Chemokines. Blood J Am Soc Hematol. (1997) 90:909–28. doi: 10.1182/blood.V90.3.909.909_909_928
102. Karin N. Chemokines and cancer: new immune checkpoints for cancer therapy. Curr Opin Immunol. (2018) 51:140–5. doi: 10.1016/j.coi.2018.03.004
103. Zlotnik A, Yoshie O. The chemokine superfamily revisited. Immunity. (2012) 36:705–16. doi: 10.1016/j.immuni.2012.05.008
104. Stadtmann A, Zarbock A. CXCR2: from bench to bedside. Front Immunol. (2012) 3:1–10. doi: 10.3389/fimmu.2012.00263
105. Sharma B, Varney ML, Saxena S, Wu L, Singh RK. Induction of CXCR2 ligands, stem cell-like phenotype, and metastasis in chemotherapy-resistant breast cancer cells. Cancer Lett. (2016) 372:192–200. doi: 10.1016/j.canlet.2015.12.011
106. Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. (2010) 120:2423–31. doi: 10.1172/JCI41649
107. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. (2014) 6:237ra67. doi: 10.1126/scitranslmed.3007974
108. Yang Y, Luo B, An Y, Sun H, Cai H, Sun D. Systematic review and meta-analysis of the prognostic value of CXCR2 in solid tumor patients. Oncotarget. (2017) 8:109740. doi: 10.18632/oncotarget.22285
109. Martin D, Galisteo R, Gutkind JS. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFκB through the CBM (Carma3/Bcl10/Malt1) complex. J Biol Chem. (2009) 284:6038–42. doi: 10.1074/jbc.C800207200
110. Xue M-Q, Liu J, Sang J-F, Su L, Yao Y-Z. Expression characteristic of CXCR1 in different breast tissues and the relevance between its expression and efficacy of neo-adjuvant chemotherapy in breast cancer. Oncotarget. (2017) 8:48930. doi: 10.18632/oncotarget.16893
111. Ruffini PA. The CXCL8-CXCR1/2 Axis as a therapeutic target in breast cancer stem-like cells. Front Oncol. (2019) 9:40. doi: 10.3389/fonc.2019.00040
112. Schott AF, Goldstein LJ, Cristofanilli M, Ruffini PA, McCanna S, Reuben JM, et al. Phase Ib pilot study to evaluate reparixin in combination with weekly paclitaxel in patients with HER-2–negative metastatic breast cancer. Clin Cancer Res. (2017) 23:5358–65. doi: 10.1158/1078-0432.CCR-16-2748
113. Goldstein LJ, Perez RP, Yardley D, Han LK, Reuben JM, Gao H, et al. A window-of-opportunity trial of the CXCR1/2 inhibitor reparixin in operable HER-2-negative breast cancer. Breast Cancer Res. (2020) 22:1–9. doi: 10.1186/s13058-020-01294-7
114. Xu T, Shen H, Liu L, Shu Y. The impact of chemokine receptor CXCR4 on breast cancer prognosis: a meta-analysis. Cancer Epidemiol. (2013) 37:725–31. doi: 10.1016/j.canep.2013.04.017
115. Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan M, et al. et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. (2001) 410:6824. doi: 10.1038/35065016
116. Scala S. Molecular pathways: targeting the CXCR4–CXCL12 axis—untapped potential in the tumor microenvironment. Clin Cancer Res. (2015) 21:4278–85. doi: 10.1158/1078-0432.CCR-14-0914
117. Xu C, Zhao H, Chen H, Yao Q. CXCR4 in breast cancer: oncogenic role and therapeutic targeting. Drug Des Devel Ther. (2015) 9:4953. doi: 10.2147/DDDT.S84932
118. Dewan M, Ahmed S, Iwasaki Y, Ohba K, Toi M, Yamamoto N. Stromal cell-derived factor-1 and CXCR4 receptor interaction in tumor growth and metastasis of breast cancer. Biomed Pharmacother. (2006) 60:273–6. doi: 10.1016/j.biopha.2006.06.004
119. Kato M, Kitayama J, Kazama S, Nagawa H. Expression pattern of CXC chemokine receptor-4 is correlated with lymph node metastasis in human invasive ductal carcinoma. Breast Cancer Res. (2003) 5:R144. doi: 10.1186/bcr627
120. Smith MC, Luker KE, Garbow JR, Prior JL, Jackson E, Piwnica-Worms D, et al. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. (2004) 64:8604–12. doi: 10.1158/0008-5472.CAN-04-1844
121. Lefort S, Thuleau A, Kieffer Y, Sirven P, Bieche I, Marangoni E, et al. CXCR4 inhibitors could benefit to HER2 but not to triple-negative breast cancer patients. Oncogene. (2017) 36:1211–22. doi: 10.1038/onc.2016.284
122. Kitamura T, Pollard JW. Therapeutic potential of chemokine signal inhibition for metastatic breast cancer. Pharmacol Res. (2015) 100:266–70. doi: 10.1016/j.phrs.2015.08.004
123. Youngs SJ, Ali SA, Taub DD, Rees RC. Chemokines induce migrational responses in human breast carcinoma cell lines. Int J Cancer. (1997) 71:257–66. doi: 10.1002/(SICI)1097-0215(19970410)71:2<257::AID-IJC22>3.0.CO;2-D
124. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. (2011) 475:222–5. doi: 10.1038/nature10138
125. Dehqanzada ZA, Storrer CE, Hueman MT, Foley RJ, Harris KA, Jama YH, et al. Assessing serum cytokine profiles in breast cancer patients receiving a HER2/neu vaccine using Luminex® technology. Oncol Rep. (2007) 17:687–94. doi: 10.3892/or.17.3.687
126. Zhang Y, Lv D, Kim H-J, Kurt RA, Bu W, Li Y, et al. A novel role of hematopoietic CCL5 in promoting triple-negative mammary tumor progression by regulating generation of myeloid-derived suppressor cells. Cell Res. (2013) 23:394–408. doi: 10.1038/cr.2012.178
127. Aldinucci D, Colombatti A. The inflammatory chemokine CCL5 and cancer progression. Mediators Inflamm. (2014) 2014:292376. doi: 10.1155/2014/292376
128. Velasco-Velázquez M, Jiao X, De La Fuente M, Pestell TG, Ertel A, Lisanti MP, et al. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. (2012) 72:3839. doi: 10.1158/0008-5472.CAN-11-3917
129. Jiao X, Nawab O, Patel T, Kossenkov AV, Halama N, Jaeger D, et al. Recent advances targeting CCR5 for cancer and its role in immuno-oncology. Cancer Res. (2019) 79:4801. doi: 10.1158/0008-5472.CAN-19-1167
130. Robinson SC, Scott KA, Wilson JL, Thompson RG, Proudfoot AEI, Balkwill FR. A chemokine receptor antagonist inhibits experimental breast tumor growth. Cancer Res. (2003) 63:8360. Available online at: https://cancerres.aacrjournals.org/content/63/23/8360.long
131. Pervaiz A, Zepp M, Mahmood S, Ali DM, Berger MR, Adwan H. CCR5 blockage by maraviroc: a potential therapeutic option for metastatic breast cancer. Cell Oncol. (2019) 42:93–106. doi: 10.1007/s13402-018-0415-3
132. Cristofanilli M, Dolezal M, Lalezari J, Rui H, Patterson B, Tang C-M, et al. Abstract CT233: phase Ib/II study of leronlimab (PRO 140) combined with carboplatin in CCR5+ mTNBC patients. Cancer Res. (2020) 80. doi: 10.1158/1538-7445.AM2020-CT233
133. Jiao X, Wang M, Pestell RG. Leronlimab, a humanized monoclonal antibody to CCR5, blocks breast cancer cellular invasion and enhances cell death induced by DNA damaging chemotherapies. Breast Cancer Res. (2019) 23:11. doi: 10.1186/s13058-021-01391-1
134. de Andrade Mello P, Coutinho-Silva R, Savio LEB. Multifaceted effects of extracellular adenosine triphosphate and adenosine in the tumor–host interaction and therapeutic perspectives. Front Immunol. (2017) 8:1526. doi: 10.3389/fimmu.2017.01526
135. Silva-Vilches C, Ring S, Mahnke K. ATP and its metabolite adenosine as regulators of dendritic cell activity. Front Immunol. (2018) 9:2581. doi: 10.3389/fimmu.2018.02581
136. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. (2014) 509:310–7. doi: 10.1038/nature13085
137. Ballarin M, Fredholm B, Ambrosio S, Mahy N. Extracellular levels of adenosine and its metabolites in the striatum of awake rats: inhibition of uptake and metabolism. Acta Physiol Scand. (1991) 142:97–103. doi: 10.1111/j.1748-1716.1991.tb09133.x
138. Blay J, White TD, Hoskin DW. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. (1997) 57:2602–5.
139. Azambuja JH, Ludwig N, Braganhol E, Whiteside TL. Inhibition of the adenosinergic pathway in cancer rejuvenates innate and adaptive immunity. Int J Mol Sci. (2019) 20:5698. doi: 10.3390/ijms20225698
140. Abbracchio MP, Burnstock G, Boeynaems J-M, Barnard EA, Boyer JL, Kennedy C, et al. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. (2006) 58:281–341. doi: 10.1124/pr.58.3.3
141. Yip L, Woehrle T, Corriden R, Hirsh M, Chen Y, Inoue Y, et al. Autocrine regulation of T-cell activation by ATP release and P2X7 receptors. FASEB J. (2009) 23:1685–93. doi: 10.1096/fj.08-126458
142. Schenk U, Westendorf AM, Radaelli E, Casati A, Ferro M, Fumagalli M, et al. Purinergic control of T cell activation by ATP released through pannexin-1 hemichannels. Sci Signal. (2008) 1:ra6–ra6. doi: 10.1126/scisignal.1160583
143. Fu W, McCormick T, Qi X, Luo L, Zhou L, Li X, et al. Activation of P2X 7-mediated apoptosis Inhibits DMBA/TPA-induced formation of skin papillomas and cancer in mice. BMC Cancer. (2009) 9:1–20. doi: 10.1186/1471-2407-9-114
144. Zhou JZ, Riquelme MA, Gao X, Ellies LG, Sun L-Z, Jiang JX. Differential impact of adenosine nucleotides released by osteocytes on breast cancer growth and bone metastasis. Oncogene. (2015) 34:1831–42. doi: 10.1038/onc.2014.113
145. Burnstock G, Di Virgilio F. Purinergic signalling and cancer. Purinergic Signal. (2013) 9:491–540. doi: 10.1007/s11302-013-9372-5
146. Slater M, Danieletto S, Barden JA. Expression of the apoptotic calcium channel P2X 7 in the glandular epithelium. J Mol Histol. (2005) 36:159–65. doi: 10.1007/s10735-004-6166-7
147. Jelassi B, Anchelin M, Chamouton J, Cayuela ML, Clarysse L, Li J, et al. Anthraquinone emodin inhibits human cancer cell invasiveness by antagonizing P2X7 receptors. Carcinogenesis. (2013) 34:1487–96. doi: 10.1093/carcin/bgt099
148. Antonioli L, Blandizzi C, Pacher P, Haskó G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer. (2013) 13:842–57. doi: 10.1038/nrc3613
149. Allard B, Turcotte M, Spring K, Pommey S, Royal I, Stagg J. Anti-CD73 therapy impairs tumor angiogenesis. Int J Cancer. (2014) 134:1466–73. doi: 10.1002/ijc.28456
150. Wang L, Tang S, Wang Y, Xu S, Yu J, Zhi X, et al. Ecto-5′-nucleotidase (CD73) promotes tumor angiogenesis. Clin Exp Metastasis. (2013) 30:671–80. doi: 10.1007/s10585-013-9571-z
151. Zhi X, Wang Y, Zhou X, Yu J, Jian R, Tang S, et al. RNAi-mediated CD73 suppression induces apoptosis and cell-cycle arrest in human breast cancer cells. Cancer Sci. (2010) 101:2561–9. doi: 10.1111/j.1349-7006.2010.01733.x
152. Jiang T, Xu X, Qiao M, Li X, Zhao C, Zhou F, et al. Comprehensive evaluation of NT5E/CD73 expression and its prognostic significance in distinct types of cancers. BMC Cancer. (2018) 18:1–10. doi: 10.1186/s12885-018-4073-7
153. Loi S, Pommey S, Haibe-Kains B, Beavis PA, Darcy PK, Smyth MJ, et al. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc Natl Acad Sci. (2013) 110:11091–6. doi: 10.1073/pnas.1222251110
154. Supernat A, Markiewicz A, Welnicka-Jaskiewicz M, Seroczynska B, Skokowski J, Sejda A, et al. CD73 expression as a potential marker of good prognosis in breast carcinoma. Appl Immunohistochem Mol Morphol. (2012) 20:103–7. doi: 10.1097/PAI.0b013e3182311d82
155. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology. (2016) 5:e1208875. doi: 10.1080/2162402X.2016.1208875
156. Figueiro F, Mendes FB, Corbelini PF, Janarelli F, JANDREY EHF, Russowsky D, et al. A monastrol-derived compound, LaSOM 63, inhibits ecto-5'Nucleotidase/CD73 activity and induces apoptotic cell death of glioma cell lines. Anticancer Res. (2014) 34:1837–42. Available online at: https://ar.iiarjournals.org/content/34/4/1837.long
157. Terp MG, Olesen KA, Arnspang EC, Lund RR, Lagerholm BC, Ditzel HJ, et al. Anti-human CD73 monoclonal antibody inhibits metastasis formation in human breast cancer by inducing clustering and internalization of CD73 expressed on the surface of cancer cells. J Immunol. (2013) 191:4165–73. doi: 10.4049/jimmunol.1301274
158. Elia I, Fendt SM. In vivo cancer metabolism is defined by the nutrient microenvironment. Transl Cancer Res. (2016) 5:S1284–7.
159. Bastid J, Cottalorda-Regairaz A, Alberici G, Bonnefoy N, Eliaou J, Bensussan A. ENTPD1/CD39 is a promising therapeutic target in oncology. Oncogene. (2013) 32:1743–51. doi: 10.1038/onc.2012.269
160. Leone RD, Lo Y-C, Powell JD. A2aR antagonists: next generation checkpoint blockade for cancer immunotherapy. Comput Struct Biotechnol J. (2015) 13:265–72. doi: 10.1016/j.csbj.2015.03.008
161. Nowak M, Lynch L, Yue S, Ohta A, Sitkovsky M, Balk SP, et al. The A2aR adenosine receptor controls cytokine production in iNKT cells. Eur J Immunol. (2010) 40:682–7. doi: 10.1002/eji.200939897
162. Sevigny CP, Li L, Awad AS, Huang L, McDuffie M, Linden J, et al. Activation of adenosine 2A receptors attenuates allograft rejection and alloantigen recognition. J Immunol. (2007) 178:4240–9. doi: 10.4049/jimmunol.178.7.4240
163. Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, et al. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood. (2008) 111:251–9. doi: 10.1182/blood-2007-03-081646
164. Di Virgilio F, Adinolfi E. Extracellular purines, purinergic receptors and tumor growth. Oncogene. (2017) 36:293–303. doi: 10.1038/onc.2016.206
165. Gessi S, Bencivenni S, Battistello E, Vincenzi F, Colotta V, Catarzi D, et al. Inhibition of A2A adenosine receptor signaling in cancer cells proliferation by the novel antagonist TP455. Front Pharmacol. (2017) 8:888. doi: 10.3389/fphar.2017.00888
166. Harrison H, Bennett G, Blakeley D, Brown A, Campbell S, Chen L, Lutz RJ, et al. BT1718, a novel bicyclic peptide-maytansinoid conjugate targeting MT1-MMP for the treatment of solid tumours: Design of bicyclic peptide and linker selection. In: Proceedings of the American Association for Cancer Research Annual Conference. Washington, DC (2017). doi: 10.1200/JCO.2017.35.15_suppl.3004
167. Sepúlveda C, Palomo I, Fuentes E. Role of adenosine A2b receptor overexpression in tumor progression. Life Sci. (2016) 166:92–9. doi: 10.1016/j.lfs.2016.10.008
168. Panjehpour M, Castro M, Klotz K. Human breast cancer cell line MDA-MB-231 expresses endogenous A2B adenosine receptors mediating a Ca2+ signal. Br J Pharmacol. (2005) 145:211–8. doi: 10.1038/sj.bjp.0706180
169. Mittal D, Sinha D, Barkauskas D, Young A, Kalimutho M, Stannard K, et al. Adenosine 2B receptor expression on cancer cells promotes metastasis. Cancer Res. (2016) 76:4372–82. doi: 10.1158/0008-5472.CAN-16-0544
170. Fernandez-Gallardo M, González-Ramírez R, Sandoval A, Felix R, Monjaraz E. Adenosine stimulate proliferation and migration in triple negative breast cancer cells. PLoS ONE. (2016) 11:e0167445. doi: 10.1371/journal.pone.0167445
171. Giacomelli C, Daniele S, Romei C, Tavanti L, Neri T, Piano I, et al. The A2B adenosine receptor modulates the epithelial–mesenchymal transition through the balance of cAMP/PKA and MAPK/ERK pathway activation in human epithelial lung cells. Front Pharmacol. (2018) 9:54. doi: 10.3389/fphar.2018.00054
172. Desmet CJ, Gallenne T, Prieur A, Reyal F, Visser NL, Wittner BS, et al. Identification of a pharmacologically tractable Fra-1/ADORA2B axis promoting breast cancer metastasis. Proc Natl Acad Sci USA. (2013) 110:5139–44. doi: 10.1073/pnas.1222085110
173. Cekic C, Sag D, Li Y, Theodorescu D, Strieter RM, Linden J. Adenosine A2B receptor blockade slows growth of bladder and breast tumors. J Immunol. (2012) 188:198. doi: 10.4049/jimmunol.1101845
174. Ristimäki A, Sivula A, Lundin J, Lundin M, Salminen T, Haglund C, et al. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res. (2002) 62:632–5. Available online at: https://cancerres.aacrjournals.org/content/62/3/632.long
175. Boland G, Butt I, Prasad R, Knox W, Bundred N. COX-2 expression is associated with an aggressive phenotype in ductal carcinoma in situ. Br J Cancer. (2004) 90:423–9. doi: 10.1038/sj.bjc.6601534
176. Howe LR. Inflammation and breast cancer. Cyclooxygenase/prostaglandin signaling and breast cancer. Breast Cancer Res. (2007) 9:1–9. doi: 10.1186/bcr1678
177. Stasinopoulos I, O'Brien DR, Wildes F, Glunde K, Bhujwalla ZM. Silencing of cyclooxygenase-2 inhibits metastasis and delays tumor onset of poorly differentiated metastatic breast cancer cells. Mol Cancer Res. (2007) 5:435–42. doi: 10.1158/1541-7786.MCR-07-0010
178. Filipenko I, Schwalm S, Reali L, Pfeilschifter J, Fabbro D, Huwiler A, et al. Upregulation of the S1P3 receptor in metastatic breast cancer cells increases migration and invasion by induction of PGE2 and EP2/EP4 activation. Biochim Biophys Acta BBA-Mol Cell Biol Lipids. (2016) 1861:1840–51. doi: 10.1016/j.bbalip.2016.09.005
179. Majumder M, Nandi P, Omar A, Ugwuagbo KC, Lala PK. EP4 as a therapeutic target for aggressive human breast cancer. Int J Mol Sci. (2018) 19:1019. doi: 10.3390/ijms19041019
180. Singh B, Berry JA, Shoher A, Ramakrishnan V, Lucci A. COX-2 overexpression increases motility and invasion of breast cancer cells. Int J Oncol. (2005) 26:1393–9. doi: 10.3892/ijo.26.5.1393
181. Choudhry MA, Ahmed Z, Sayeed MM. PGE2-mediated inhibition of T cell p59fyn is independent of cAMP. Am J Physiol-Cell Physiol. (1999) 277:C302–9. doi: 10.1152/ajpcell.1999.277.2.C302
182. Reader J, Holt D, Fulton A. Prostaglandin E 2 EP receptors as therapeutic targets in breast cancer. Cancer Metastasis Rev. (2011) 30:449–63. doi: 10.1007/s10555-011-9303-2
183. Garrone P, Galibert L, Rousset F, Fu SM, Banchereau J. Regulatory effects of prostaglandin E2 on the growth and differentiation of human B lymphocytes activated through their CD40 antigen. J Immunol. (1994) 152:4282–90.
184. Porter BO, Malek TR. Prostaglandin E2 inhibits T cell activation-induced apoptosis and Fas-mediated cellular cytotoxicity by blockade of Fas-ligand induction. Eur J Immunol. (1999) 29:2360–5. doi: 10.1002/(SICI)1521-4141(199907)29:07<2360::AID-IMMU2360>3.0.CO;2-A
185. Hilkens C, Snijders A, Snijdewint F, Wierenga E, Kapsenberg M. Modulation of T-cell cytokine secretion by accessory cell-derived products. Eur Respir J Suppl. (1996) 22:90s.
186. Duan B, Davis R, Sadat EL, Collins J, Sternweis PC, Yuan D, et al. Distinct roles of adenylyl cyclase VII in regulating the immune responses in mice. J Immunol. (2010) 185:335. doi: 10.4049/jimmunol.0903474
187. Liu CH, Chang S-H, Narko K, Trifan OC, Wu M-T, Smith E, et al. Overexpression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. J Biol Chem. (2001) 276:18563–9. doi: 10.1074/jbc.M010787200
188. Harris R. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology. (2009) 17:55–67. doi: 10.1007/s10787-009-8049-8
189. Okumura Y, Yamagishi T, Nukui S, Nakao K. Discovery of AAT-008, a novel, potent, and selective prostaglandin EP4 receptor antagonist. Bioorg Med Chem Lett. (2017) 27:1186–92. doi: 10.1016/j.bmcl.2017.01.067
190. Kawamori T, Uchiya N, Nakatsugi S, Watanabe K, Ohuchida S, Yamamoto H, et al. Chemopreventive effects of ONO-8711, a selective prostaglandin E receptor EP1 antagonist, on breast cancer development. Carcinogenesis. (2001) 22:2001–4. doi: 10.1093/carcin/22.12.2001
191. Hester A, Salzmann B, Rahmeh M, Kolben T, Czogalla B, Ditsch N, et al. EP3 receptor antagonist L798, 106 reduces proliferation and migration of SK-BR-3 breast cancer cells. OncoTargets Ther. (2019) 12:6053. doi: 10.2147/OTT.S204919
192. Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol. (2007) 178:5245–52. doi: 10.4049/jimmunol.178.8.5245
193. Arnold CE, Whyte CS, Gordon P, Barker RN, Rees AJ, Wilson HM. A critical role for suppressor of cytokine signalling 3 in promoting M 1 macrophage activation and function in vitro and in vivo. Immunology. (2014) 141:96–110. doi: 10.1111/imm.12173
194. Zhou J, Tang Z, Gao S, Li C, Feng Y, Zhou X. Tumor-associated macrophages: recent insights and therapies. Front Oncol. (2020) 10:188. doi: 10.3389/fonc.2020.00188
195. Shi Y, Liu CH, Roberts AI, Das J, Xu G, Ren G, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don't know. Cell Res. (2006) 16:126–33. doi: 10.1038/sj.cr.7310017
196. Eubank TD, Roberts RD, Khan M, Curry JM, Nuovo GJ, Kuppusamy P, et al. GM-CSF inhibits breast cancer growth and metastasis by invoking an anti-angiogenic program in tumor-educated macrophages. Cancer Res. (2009) 69:2133. doi: 10.1158/0008-5472.CAN-08-1405
197. Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, et al. Macrophage polarization in tumour progression. Semin Cancer Biol. (2008) 18:349–55. doi: 10.1016/j.semcancer.2008.03.004
198. Jenkins SJ, Ruckerl D, Thomas GD, Hewitson JP, Duncan S, Brombacher F, et al. IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J Exp Med. (2013) 210:2477–91. doi: 10.1084/jem.20121999
199. Richardsen E, Uglehus RD, Johnsen SH, Busund L-T. Macrophage-colony stimulating factor (CSF1) predicts breast cancer progression and mortality. Anticancer Res. (2015) 35:865–74. Available online at: https://ar.iiarjournals.org/content/35/2/865.long
200. Priceman SJ, Sung JL, Shaposhnik Z, Burton JB, Torres-Collado AX, Moughon DL, et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood J Am Soc Hematol. (2010) 115:1461–71. doi: 10.1182/blood-2009-08-237412
201. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
202. Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol. (2015) 36:229–39. doi: 10.1016/j.it.2015.02.004
203. Paulus P, Stanley ER, Schäfer R, Abraham D, Aharinejad S. Colony-stimulating factor-1 antibody reverses chemoresistance in human MCF-7 breast cancer xenografts. Cancer Res. (2006) 66:4349–56. doi: 10.1158/0008-5472.CAN-05-3523
204. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. (2014) 25:846–59. doi: 10.1016/j.ccr.2014.05.016
205. Srabovic N, Mujagic Z, Mujanovic-Mustedanagic J, Softic A, Muminovic Z, Rifatbegovic A, et al. Vascular endothelial growth factor receptor-1 expression in breast cancer and its correlation to vascular endothelial growth factor a. Int J Breast Cancer. (2013) 2013:1–6. doi: 10.1155/2013/746749
206. Linardou H, Kalogeras KT, Kronenwett R, Kouvatseas G, Wirtz RM, Zagouri F, et al. The prognostic and predictive value of mRNA expression of vascular endothelial growth factor family members in breast cancer: a study in primary tumors of high-risk early breast cancer patients participating in a randomized Hellenic Cooperative Oncology Group trial. Breast Cancer Res. (2012) 14:R145. doi: 10.1186/bcr3354
207. Ferrara N, Gerber H-P, LeCouter J. The biology of VEGF and its receptors. Nat Med. (2003) 9:669–76. doi: 10.1038/nm0603-669
208. Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J. (2011) 437:169–83. doi: 10.1042/BJ20110301
209. Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer. (2013) 13:871–82. doi: 10.1038/nrc3627
210. Tabana YM, Hassan LEA, Ahamed MBK, Dahham SS, Iqbal MA, Saeed MA, et al. Scopoletin, an active principle of tree tobacco (Nicotiana glauca) inhibits human tumor vascularization in xenograft models and modulates ERK1, VEGF-A, and FGF-2 in computer model. Microvasc Res. (2016) 107:17–33. doi: 10.1016/j.mvr.2016.04.009
211. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet A-L, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. (2015) 212:139–48. doi: 10.1084/jem.20140559
212. Almand B, Resser JR, Lindman B, Nadaf S, Clark JI, Kwon ED, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. (2000) 6:1755–66. Available online at: https://clincancerres.aacrjournals.org/content/6/5/1755.long
213. Wada J, Suzuki H, Fuchino R, Yamasaki A, Nagai S, Yanai K, et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Res. (2009) 29:881–8. Available online at: https://ar.iiarjournals.org/content/29/3/881.long
214. Marina DD, Carmela R, Alfredo F, Giovanna Maria P. Anti-VEGF therapy in breast and lung mouse models of cancers. BioMed Res Int. (2011) 2011:1–10. doi: 10.1155/2011/947928
215. Ferrara N, Hillan KJ, Novotny W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun. (2005) 333:328–35. doi: 10.1016/j.bbrc.2005.05.132
216. Kabbinavar F, Hurwitz HI, Fehrenbacher L, Meropol NJ, Novotny WF, Lieberman G, et al. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J Clin Oncol. (2003) 21:60–5. doi: 10.1200/JCO.2003.10.066
217. Rini BI. Vascular endothelial growth factor–targeted therapy in renal cell carcinoma: current status and future directions. Clin Cancer Res. (2007) 13:1098–106. doi: 10.1158/1078-0432.CCR-06-1989
219. Miles DW, Chan A, Dirix LY, Cortés J, Pivot X, Tomczak P, et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2–negative metastatic breast cancer. J Clin Oncol. (2010) 28:3239–47. doi: 10.1200/JCO.2008.21.6457
220. Robert NJ, Diéras V, Glaspy J, Brufsky AM, Bondarenko I, Lipatov ON, et al. RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2–negative, locally recurrent or metastatic breast cancer. J Clin Oncol. (2011) 29:1252–60. doi: 10.1200/JCO.2010.28.0982
221. Manning EA, Ullman JG, Leatherman JM, Asquith JM, Hansen TR, Armstrong TD, et al. A vascular endothelial growth factor receptor-2 inhibitor enhances antitumor immunity through an immune-based mechanism. Clin Cancer Res. (2007) 13:3951–9. doi: 10.1158/1078-0432.CCR-07-0374
222. Vahdat LT, Layman R, Yardley DA, Gradishar W, Salkeni MA, Joy AA, et al. Randomized phase II study of ramucirumab or icrucumab in combination with capecitabine in patients with previously treated locally advanced or metastatic breast cancer. The oncologist. (2017) 22:245. doi: 10.1634/theoncologist.2016-0265
223. Escudier B, Gore M. Axitinib for the management of metastatic renal cell carcinoma. Drugs R D. (2011) 11:113–26. doi: 10.2165/11591240-000000000-00000
224. Wilmes LJ, Pallavicini MG, Fleming LM, Gibbs J, Wang D, Li K-L, et al. AG-013736, a novel inhibitor of VEGF receptor tyrosine kinases, inhibits breast cancer growth and decreases vascular permeability as detected by dynamic contrast-enhanced magnetic resonance imaging. Magn Reson Imaging. (2007) 25:319–27. doi: 10.1016/j.mri.2006.09.041
225. Rugo HS, Stopeck AT, Joy AA, Chan S, Verma S, Lluch A, et al. Randomized, placebo-controlled, double-blind, phase II study of axitinib plus docetaxel versus docetaxel plus placebo in patients with metastatic breast cancer. J Clin Oncol. (2011) 29:2459–65. doi: 10.1200/JCO.2010.31.2975
226. Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. (2006) 3:24–40. doi: 10.1038/ncponc0403
227. Iwasaki A. Medzhitov R. Toll- recept control adapt immune responses. Nat Immunol. (2004) 5:987–95. doi: 10.1038/ni1112
228. Shi M, Yao Y, Han F, Li Y, Li Y. MAP1S controls breast cancer cell TLR5 signaling pathway and promotes TLR5 signaling-based tumor suppression. PLoS ONE. (2014) 9:e86839. doi: 10.1371/journal.pone.0086839
229. Botos I, Segal DM, Davies DR. The structural biology of Toll-like receptors. Structure. (2011) 19:447–59. doi: 10.1016/j.str.2011.02.004
230. Yang H, Zhou H, Feng P, Zhou X, Wen H, Xie X, et al. Reduced expression of Toll-like receptor 4 inhibits human breast cancer cells proliferation and inflammatory cytokines secretion. J Exp Clin Cancer Res. (2010) 29:92. doi: 10.1186/1756-9966-29-92
231. Merrell MA, Ilvesaro JM, Lehtonen N, Sorsa T, Gehrs B, Rosenthal E, et al. Toll-like receptor 9 agonists promote cellular invasion by increasing matrix metalloproteinase activity. Mol Cancer Res. (2006) 4:437–47. doi: 10.1158/1541-7786.MCR-06-0007
232. Kalb ML, Glaser A, Stary G, Koszik F, Stingl G. TRAIL+ human plasmacytoid dendritic cells kill tumor cells in vitro: mechanisms of imiquimod-and IFN-α-mediated antitumor reactivity. J Immunol. (2012) 188:1583–91. doi: 10.4049/jimmunol.1102437
233. Li K, Qu S, Chen X, Wu Q, Shi M. Promising targets for cancer immunotherapy: TLRs, RLRs, and STING-mediated innate immune pathways. Int J Mol Sci. (2017) 18:404. doi: 10.3390/ijms18020404
234. Shchebliakov D, Logunov DY, Tukhvatulin A, Shmarov M, Naroditsky B, Ginzburg A. Toll-like receptors (TLRs): the role in tumor progression. Acta Naturae. (2010) 2:21–9. doi: 10.32607/20758251-2010-2-3-21-29
235. Bhattacharya D, Yusuf N. Expression of toll-like receptors on breast tumors: taking a toll on tumor microenvironment. Int J Breast Cancer. (2012) 2012:1–17. doi: 10.1155/2012/716564
236. Javaid N, Choi S. Toll-like receptors from the perspective of cancer treatment. Cancers. (2020) 12:297. doi: 10.3390/cancers12020297
237. Cai Z, Sanchez A, Shi Z, Zhang T, Liu M, Zhang D. Activation of Toll-like receptor 5 on breast cancer cells by flagellin suppresses cell proliferation and tumor growth. Cancer Res. (2011) 71:2466–75. doi: 10.1158/0008-5472.CAN-10-1993
238. Salaun B, Coste I, Rissoan M-C, Lebecque SJ, Renno T. TLR3 can directly trigger apoptosis in human cancer cells. J Immunol. (2006) 176:4894–901. doi: 10.4049/jimmunol.176.8.4894
239. Taura M, Fukuda R, Suico MA, Eguma A, Koga T, Shuto T, et al. TLR3 induction by anticancer drugs potentiates poly I: C-induced tumor cell apoptosis. Cancer Sci. (2010) 101:1610–7. doi: 10.1111/j.1349-7006.2010.01567.x
240. Kaczanowska S, Joseph AM, Davila E. TLR agonists: our best frenemy in cancer immunotherapy. J Leukoc Biol. (2013) 93:847–63. doi: 10.1189/jlb.1012501
241. Song EJ, Kang MJ, Kim YS, Kim SM, Lee SE, Kim CH, et al. Flagellin promotes the proliferation of gastric cancer cells via the Toll-like receptor 5. Int J Mol Med. (2011) 28:115–9. doi: 10.3892/ijmm.2011.656
242. McCarron M, Reen DJ. Activated Human Neonatal CD8+ T Cells Are Subject to Immunomodulation by Direct TLR2 or TLR5 Stimulation. J Immunol. (2009) 182:55. doi: 10.4049/jimmunol.182.1.55
243. Shi M, Chen X, Ye K, Yao Y, Li Y. Application potential of toll-like receptors in cancer immunotherapy: systematic review. Medicine (Baltimore). (2016) 95:e3951. doi: 10.1097/MD.0000000000003951
244. Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. (2007) 13:552–9. doi: 10.1038/nm1589
245. Adams S, Kozhaya L, Martiniuk F, Meng T-C, Chiriboga L, Liebes L, et al. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin Cancer Res Off J Am Assoc Cancer Res. (2012) 18:6748–57. doi: 10.1158/1078-0432.CCR-12-1149
246. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. (2009) 461:788–92. doi: 10.1038/nature08476
247. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. (2008) 455:674–8. doi: 10.1038/nature07317
248. Elahi S, Van Kessel J, Kiros TG, Strom S, Hayakawa Y, Hyodo M, et al. c-di-GMP enhances protective innate immunity in a murine model of pertussis. PloS One. (2014) 9:e109778. doi: 10.1371/journal.pone.0109778
249. Chandra D, Quispe-Tintaya W, Jahangir A, Asafu-Adjei D, Ramos I, Sintim HO, et al. STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer Immunol Res. (2014) 2:901–10. doi: 10.1158/2326-6066.CIR-13-0123
250. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. (2015) 15:760–70. doi: 10.1038/nri3921
251. Pantelidou C, Sonzogni O, Taveira MDO, Mehta AK, Kothari A, Wang D, et al. PARP inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov. (2019) 9:722–37. doi: 10.1158/2159-8290.CD-18-1218
252. Bhatelia K, Singh A, Tomar D, Singh K, Sripada L, Chagtoo M, et al. Antiviral signaling protein MITA acts as a tumor suppressor in breast cancer by regulating NF-κB induced cell death. Biochim Biophys Acta BBA-Mol Basis Dis. (2014) 1842:144–53. doi: 10.1016/j.bbadis.2013.11.006
253. Sokolowska O, Nowis D. STING signaling in cancer cells: important or not? Arch Immunol Ther Exp (Warsz). (2018) 66:125–32. doi: 10.1007/s00005-017-0481-7
254. Takashima K, Takeda Y, Oshiumi H, Shime H, Okabe M, Ikawa M, et al. STING in tumor and host cells cooperatively work for NK cell-mediated tumor growth retardation. Biochem Biophys Res Commun. (2016) 478:1764–71. doi: 10.1016/j.bbrc.2016.09.021
Keywords: breast cancer, immunotherapy, therapeutic agents, immune targets, PD-1, PD-L1
Citation: Tabana Y, Okoye IS, Siraki A, Elahi S and Barakat KH (2021) Tackling Immune Targets for Breast Cancer: Beyond PD-1/PD-L1 Axis. Front. Oncol. 11:628138. doi: 10.3389/fonc.2021.628138
Received: 11 November 2020; Accepted: 08 February 2021;
Published: 05 March 2021.
Edited by:
Sara M. Tolaney, Dana–Farber Cancer Institute, United StatesReviewed by:
David Page, Providence Portland Medical Center, United StatesRomualdo Barroso-Sousa, Hospital Sirio Libanes, Brazil
Copyright © 2021 Tabana, Okoye, Siraki, Elahi and Barakat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Khaled H. Barakat, a2JhcmFrYXRAdWFsYmVydGEuY2E=