 Laurence C. Cheung1,2
Laurence C. Cheung1,2 Rebecca de Kraa3
Rebecca de Kraa3 Joyce Oommen1
Joyce Oommen1 Grace-Alyssa Chua1Sajla Singh1Anastasia M. Hughes1
Grace-Alyssa Chua1Sajla Singh1Anastasia M. Hughes1 Emanuela Ferrari1Jette Ford1Sung K. Chiu1
Emanuela Ferrari1Jette Ford1Sung K. Chiu1 Ronald W. Stam4Ursula R. Kees1
Ronald W. Stam4Ursula R. Kees1 Sébastien Malinge1
Sébastien Malinge1 Rishi S. Kotecha1,2,5*
Rishi S. Kotecha1,2,5*- 1Division of Children’s Leukaemia and Cancer Research, Telethon Kids Cancer Centre, Telethon Kids Institute, University of Western Australia, Perth, WA, Australia
- 2Curtin Medical School, Curtin University, Perth, WA, Australia
- 3PathWest Laboratory Medicine WA, Perth, WA, Australia
- 4Princess Máxima Center for Pediatric Oncology, Utrecht, Netherlands
- 5Department of Clinical Haematology, Oncology, Blood and Marrow Transplantation, Perth Children’s Hospital, Perth, WA, Australia
Background: Infants with KMT2A-rearranged B-cell precursor acute lymphoblastic leukemia (ALL) have poor outcomes. There is an urgent need to identify novel agents to improve survival. Proteasome inhibition has emerged as a promising therapeutic strategy for several hematological malignancies. The aim of this study was to determine the preclinical efficacy of the selective proteasome inhibitor carfilzomib, for infants with KMT2A-rearranged ALL.
Methods: Eight infant ALL cell lines were extensively characterized for immunophenotypic and cytogenetic features. In vitro cytotoxicity to carfilzomib was assessed using a modified Alamar Blue assay with cells in logarithmic growth. The Bliss Independence model was applied to determine synergy between carfilzomib and the nine conventional chemotherapeutic agents used to treat infants with ALL. Established xenograft models were used to identify the maximal tolerated dose of carfilzomib and determine in vivo efficacy.
Results: Carfilzomib demonstrated low IC50 concentrations within the nanomolar range (6.0–15.8 nm) across the panel of cell lines. Combination drug testing indicated in vitro synergy between carfilzomib and several conventional chemotherapeutic agents including vincristine, daunorubicin, dexamethasone, L-asparaginase, and 4-hydroperoxycyclophosphamide. In vivo assessment did not lead to a survival advantage for either carfilzomib monotherapy, when used to treat both low or high disease burden, or for carfilzomib in combination with multi-agent induction chemotherapy comprising of vincristine, dexamethasone, and L-asparaginase.
Conclusions: Our study highlights that in vitro efficacy does not necessarily translate to benefit in vivo and emphasizes the importance of in vivo validation prior to suggesting an agent for clinical use. Whilst proteasome inhibitors have an important role to play in several hematological malignancies, our findings guard against prioritization of carfilzomib for treatment of KMT2A-rearranged infant ALL in the clinical setting.
Introduction
The last 70 years have seen significant progress in the treatment of children with acute lymphoblastic leukemia (ALL). Initially considered an incurable disease, 5-year overall survival rates are now over 90%. This has largely been achieved through successive randomized clinical trials facilitated by international co-operative study groups, with risk stratification of patients and treatment adaptation based on presenting features, blast genetics, and early response to therapy (1). However, there remain several subgroups who continue to have significantly inferior outcomes. Infants, defined as less than 1 year of age at diagnosis, remain notoriously difficult to treat. Infants with B-cell precursor ALL harbor a KMT2A-rearrangement in up to 80% of cases, which is an aggressive driver mutation associated with chemo-resistance, high rates of relapse, and a 5-year event-free survival (EFS) of less than 40% (2–4). In addition, infants are vulnerable to the toxic effects of conventional chemotherapeutic agents, necessitating dose-limitations during therapy (2). As such, there is an urgent need to identify novel agents to improve outcome of infants with KMT2A-rearranged ALL without further increasing toxicity. In this study, we aimed to investigate the potential of the selective proteasome inhibitor carfilzomib, for the treatment of infants with KMT2A-rearranged ALL.
Materials and Methods
Cell Culture and Differentiation
A panel of eight human cell lines were established as previously described from bone marrow or peripheral blood leukocytes of infants with KMT2A-rearranged ALL (5). Following informed consent from a parent/guardian, primary patient specimens were sourced from infants enrolled on legacy Children’s Oncology Group (COG) trials in Perth, Western Australia. Briefly, cells obtained at the time of diagnosis or relapse were collected in preservative-free heparin and separated on Ficoll-Hypaque (Pharmacia Fine Chemicals AB, Uppsala, Sweden) at 841 × g for 25 min. Washed cells at a concentration of 1 × 106/ml were placed in round bottom 96-well culture plates (Falcon, Corning, NY, USA) seeding 100 ul per well. The cells were cultured in RPMI 1640 medium (Life Technologies, CA, USA) supplemented with L-glutamine (2 mM final concentration) (Sigma-Aldrich, MO, USA), 2-mercaptoethanol (50 µM final concentration) (Sigma-Aldrich, MO, USA), sodium pyruvate (1 mM) (MP Biomedicals, CA, USA), 1% non-essential amino acids (MP Biomedicals, CA, USA), and 20% heat-inactivated fetal calf serum (FCS) (Flow Laboratories, Irvine, Scotland). The cells were cultured in a humidified 37°C incubator containing 5% CO2 in air. Fifty percent of the medium was replaced by fresh medium every third day. Cultures were passaged provided they had reached cell concentrations of 2 × 106/ml. Cells were maintained in RPMI 1640 containing 20 or 10% FCS, 1% non-essential amino acid mix (100×), and 1% 100 mM sodium pyruvate solution. Cell viability was assessed through live cell counting by exclusion of 0.2% Trypan blue (Sigma-Aldrich, MO, USA). For all experiments, cells were seeded at pre-determined densities and incubated at 37°C and 5% CO2.
The cell lines are presently growing in 24-well cell culture plates (Nunclon, Thermo Fisher Scientific, MA, USA) and are passaged twice a week. The cell lines are repeatedly shown to be free of mycoplasma using the MycoAlert™ PLUS Mycoplasma Detection Kit (Lonza, Basel, Switzerland). DNA Fingerprinting was performed on the primary patient samples and on an annual basis for cell line authentication by the Australian Genome Research Facility (Melbourne, Australia), using the GenePrint 10 kit (Promega, WI, USA).
Cell Line Characterization
The AIEOP-BFM consensus antibody panel for pediatric ALL was used to assess the immunophenotype of the cell lines by flow cytometry (6). The cells were stained with manufacturer recommended quantities of fluorochrome-conjugated antigen-specific monoclonal antibodies in 5 ml polystyrene round bottom tubes (Falcon, Corning, NY, USA). The cells were incubated with the antibodies for 20 min at room temperature shielded from light. Phosphate-buffered saline (PBS) containing 2% FCS was added to the tubes and then centrifuged at 473 × g for 5 min. The supernatant was discarded and the cells were resuspended in 300 µl of PBS-2% FCS and used for flow cytometry analysis. For intracytoplasmic antigens, cells were fixed and permeabilized using the Fixation/Permeabilization Solution Kit (BD Cytofix/Cytoperm, BD Biosciences, CA, USA) as per manufacturer instructions. A comprehensive antibody panel with 6–10 color direct immunofluorescent labeling was used, according to manufacturer recommended volumes, for immunophenotyping with a BD LSRFortessa™ X-20 flow cytometer (BD Biosciences, CA, USA) (Supplementary Table 1). Cell populations of interest were gated on the basis of light scattering properties. Where appropriate, unstained cells, isotype controls, or cells known to lack certain cell surface markers were used as appropriate negative controls to set quadrant gates. Conversely, cells known to express particular markers (e.g. Jurkat E6-1) were used as positive controls where required. The percentage of antigen-positive cells was decided using a combination of quadrant statistics and ranged histogram gates.
The cell lines were cultured using unsynchronized cell cultures and G-band conventional analysis was performed on metaphase chromosomes according to standard methods. Fluorescent in situ hybridization (FISH) testing using a commercially available KMT2A (MLL) break apart probe to screen interphase cells was carried out according to the manufacturer’s instructions (MetaSystems, Altlussheim, Germany). Directed metaphase and sequential FISH studies using whole chromosome paints and other FISH probes were performed to further characterize complex rearrangements. Chromosomal abnormalities were defined and reported according to the International System for Human Cytogenetic Nomenclature (7).
Assessment of In Vitro Sensitivity and Synergy
In vitro cytotoxicity assays were performed using a modified Alamar Blue (resazurin reduction) assay with cells in logarithmic growth. All in vitro assays were performed in triplicate in 384-well clear tissue-culture treated plates (Corning, NY, USA). Cells were seeded at pre-determined optimal seeding densities (Supplementary Table 2) in 50 µl media using an automated Multidrop Combi Reagent Dispenser (Thermo Fisher Scientific, MA, USA). Cells were treated with drugs dispensed by the automated Tecan D300e Digital Dispenser (Tecan Group Ltd., Männedorf, Switzerland) at concentration ranges listed in Supplementary Table 2 at fixed ratios. Drugs were dissolved in dimethyl sulfoxide (DMSO) or water for those with poor DMSO solubility or stability (Supplementary Table 3). Final DMSO concentration in assay wells receiving drugs dissolved in DMSO was 0.5%. After 72 h drug exposure at 37°C and 5% CO2, Alamar Blue reagent comprising resazurin, methylene blue, potassium hexacyanoferrate (III), and potassium hexacyanoferrate (II) (Sigma-Aldrich, MO, USA) was added (10% v/v) and cell viability determined by absorbance (570 and 600 nm) 6 h after addition of Alamar Blue using a Synergy™ Mx microplate reader (Biotek, VT, USA). Raw absorbance readings were used to generate dose-response curves. Half maximum inhibitory concentrations (IC50) for each compound were calculated in Microsoft Excel by plotting a log dose-response curve utilizing the slope from the linear trendline to determine the IC50. Cell viability was reported as a percent of the untreated control cells for each cell line.
For the synergy assays, we used a 14 × 7 pairwise checker-board matrix format that assessed each of the nine conventional chemotherapy agents used to treat infants with ALL and carfilzomib, at 13 and 6 concentrations respectively, using a series of twofold dilutions. The fraction of cells affected was calculated for each drug concentration and 72 h percent viability imputed from the growth curves. Excess over Bliss as a measurement for synergy was calculated by the Bliss Independence model using the Chalice Bioinformatics Software (Horizon Discovery, Waterbeach, United Kingdom) (8, 9).
Assessment of In Vivo Efficacy
Female NOD/SCID mice were purchased from the Animal Research Centre, Perth. Animals were housed under pathogen-free conditions and all studies were approved by the Animal Ethics Committee, Telethon Kids Institute, Perth (Ethics number #312). Seven-week-old mice were inoculated with 1 × 106 PER-785, 1 × 106 PER-826, 1 × 106 MLL-5, or 2 × 106 MLL-14 cells. PER-785 and PER-826 xenografts were established from our cell line panel, whereas MLL-5 and MLL-14 are well characterized patient-derived xenograft models (10–12). Carfilzomib (Selleckchem, Houston, USA) was formulated in an aqueous solution of sulfobutylether-beta-cyclodextrin (Captisol®, CyDex Pharmaceuticals, CA, USA) or 2-hydroxypropyl-beta-cyclodextrin (Sigma-Aldrich, MO, USA). The maximum tolerated dose (MTD) of carfilzomib was determined in the PER-785 xenograft. Drug treatment was commenced when the percentage of human CD19+ CD45+ cells reached 1% in the bone marrow, identified from extensive mapping of leukemia cell kinetics (10). Following a 12-day engraftment period, mice were randomized into seven groups of five mice. The delivery schedule was designed to mimic that of a recently completed phase 1b study of carfilzomib in combination with induction therapy for children with relapsed/refractory ALL (13). Carfilzomib was administered intravenously via tail vein injection on days 1, 2, 8, 9, 15, and 16 with treatment groups comprising of 1.5, 2.0, 2.5, 3.0, 3.5, or 4.0 mg/kg dosing of carfilzomib or vehicle control. Mice were sacrificed 3 weeks from the start of therapy and leukemia burden was determined by measuring the percentage of human CD19+ CD45+ cells in the bone marrow, spleen, and peripheral blood by flow cytometry with anti-human CD19-APC and CD45-PE antibodies.
All four xenograft models were used to determine the response to drug treatment by EFS and leukemia burden in lymphoid organs post-treatment. The PER-785 and PER-826 xenograft models were used to evaluate the clinical potential of carfilzomib in the setting of minimal disease. For each xenograft model, mice were randomized into two groups of eight mice, comprising of a carfilzomib treatment group and vehicle control. Carfilzomib monotherapy was administered at the established MTD, commencing on day 12 following injection of cells for the PER-785 xenograft and on day 18 for the PER-826 xenograft, corresponding to 1% human CD19+ CD45+ cells in the bone marrow. MLL-5 and MLL-14 xenografts were used to evaluate the effect of carfilzomib at high leukemic burden and in combination with conventional chemotherapy. For each xenograft model, treatment commenced on day 17 following injection of cells for MLL-5 and on day 21 for MLL-14, corresponding to 1% human CD19+ CD45+ cells in the peripheral blood. Individual mouse EFS was calculated as the time in days from treatment initiation until mice reached a humane end point with evidence of leukemia-related morbidity.
Assessment of Proteasome Activity
For in vitro assessment of proteasome activity, the eight cell lines were seeded in 24-well plates and incubated with carfilzomib at their respective IC50 concentrations for 72 h. Untreated cell lines incubated under the same conditions were used as a control. Cells were subsequently harvested and washed and proteasome activity was measured using the Abcam Proteasome Activity Assay Kit (Abcam, Cambridge, United Kingdom) according to manufacturer’s instructions.
For in vivo assessment of proteasome activity, seven-week-old mice were inoculated with 1 × 106 MLL-5 cells. Drug treatment was commenced when the percentage of human CD19+ CD45+ cells reached 1% in the peripheral blood. Mice were randomized into two groups of five mice, comprising of a carfilzomib treatment group and vehicle control. A 1.5 mg/kg dose of carfilzomib was administered intravenously via tail vein injection on days 1 and 2. Blood was collected from all mice 10 min after the second dose was administered to the carfilzomib treatment group and red cell lysis performed using BD Pharm Lyse™ lysing buffer (BD Biosciences, CA, USA). Proteasome activity was measured using the Abcam Proteasome Activity Assay Kit (Abcam, Cambridge, United Kingdom) according to manufacturer’s instructions.
Statistical Analyses
Statistical analyses and graphics were performed using Prism 8 (GraphPad, La Jolla, CA, USA). Data were analyzed using the two-tailed unpaired Student’s t-test. Survival studies were analyzed using log-rank test. A p value <0.05 was considered statistically significant.
Results
Characteristics of a Panel of Infant KMT2A-Rearranged Cell Lines
Eight cell lines were established from six infants with KMT2A-rearranged ALL (Table 1). Four of the infants were less than 90 days old at diagnosis, with one infant diagnosed with congenital leukemia on day 9 of life. A cell line was established at both diagnosis (PER-494) and relapse (PER-485) from one infant (P272). Two cell lines were derived at diagnosis (PER-784 and PER-826) from P337 and the MLL-14 patient-derived xenograft was also established from this infant at diagnosis (14). Immunophenotyping with the AIEOP-BFM consensus antibody panel revealed B-lineage for PER-494, PER-784, PER-826, PER-785, and PER-910, corresponding with the immunophenotypic classification from the original patient (Table 2). The dominant lineage assignment for PER-490 was B-cell precursor, however there was co-expression of myeloid markers in keeping with the primary patient, P287, from which it was derived. Co-expression of B and myeloid markers was also seen at relapse for patient P272 and at diagnosis for patient P810. Subsequent to in vitro selection pressure, the corresponding cell lines, PER-485 and PER-703, exhibit a myeloid dominant immunophenotype (Table 2). Cytogenetic analysis identified involvement of the same KMT2A-translocation partner between each cell line and corresponding primary patient sample (Table 3). Sideline clones were identified in several cell lines, with structural and numerical changes that were undetected in patient specimens. This could be explained by less advanced techniques for routine standard of care cytogenetic analysis when the infants were originally diagnosed or may reflect clonal evolution in vitro. The former can be exemplified by the initial diagnostic karyotype of P337 revealing 49,XX,+X,+X,+6[5]/46,XX[16] in 1994, with the complex three-way translocation specified in Table 3 identified in the MLL-14 xenograft and on re-analysis of the corresponding primary patient material using more advanced techniques in 2008 (14). The minor cytogenetic clone identified in the patient was detected in the corresponding cell lines, PER-784 and PER-826. DNA fingerprinting revealed identical short tandem repeat profiles between the primary patient sample and corresponding cell line, except for single markers at the CSF1PO locus for PER-784 and PER-785 and at the TH01 locus for PER-703 (Supplementary Table 4). The high match percentage confirmed authentication of the cell lines.
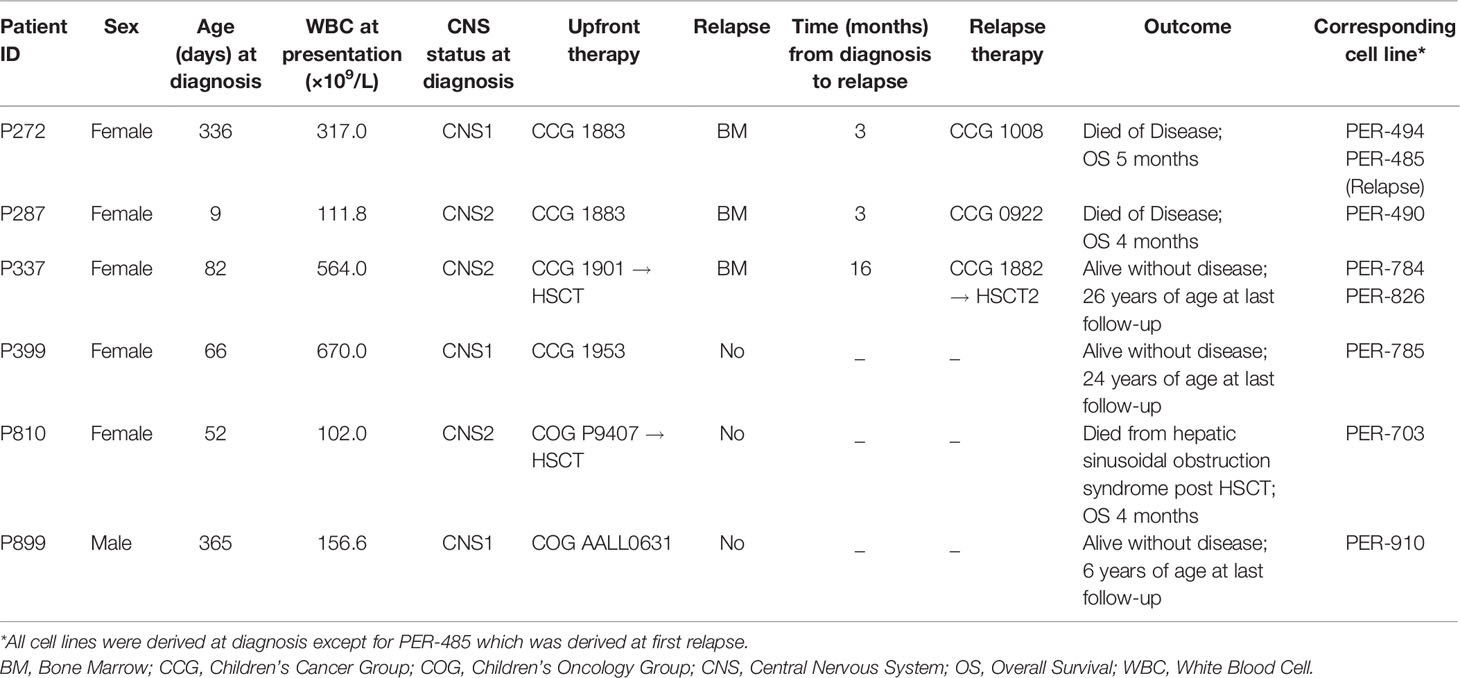
Table 1 Clinical characteristics of six infants with KMT2A-rearranged acute lymphoblastic leukemia and corresponding cell lines.
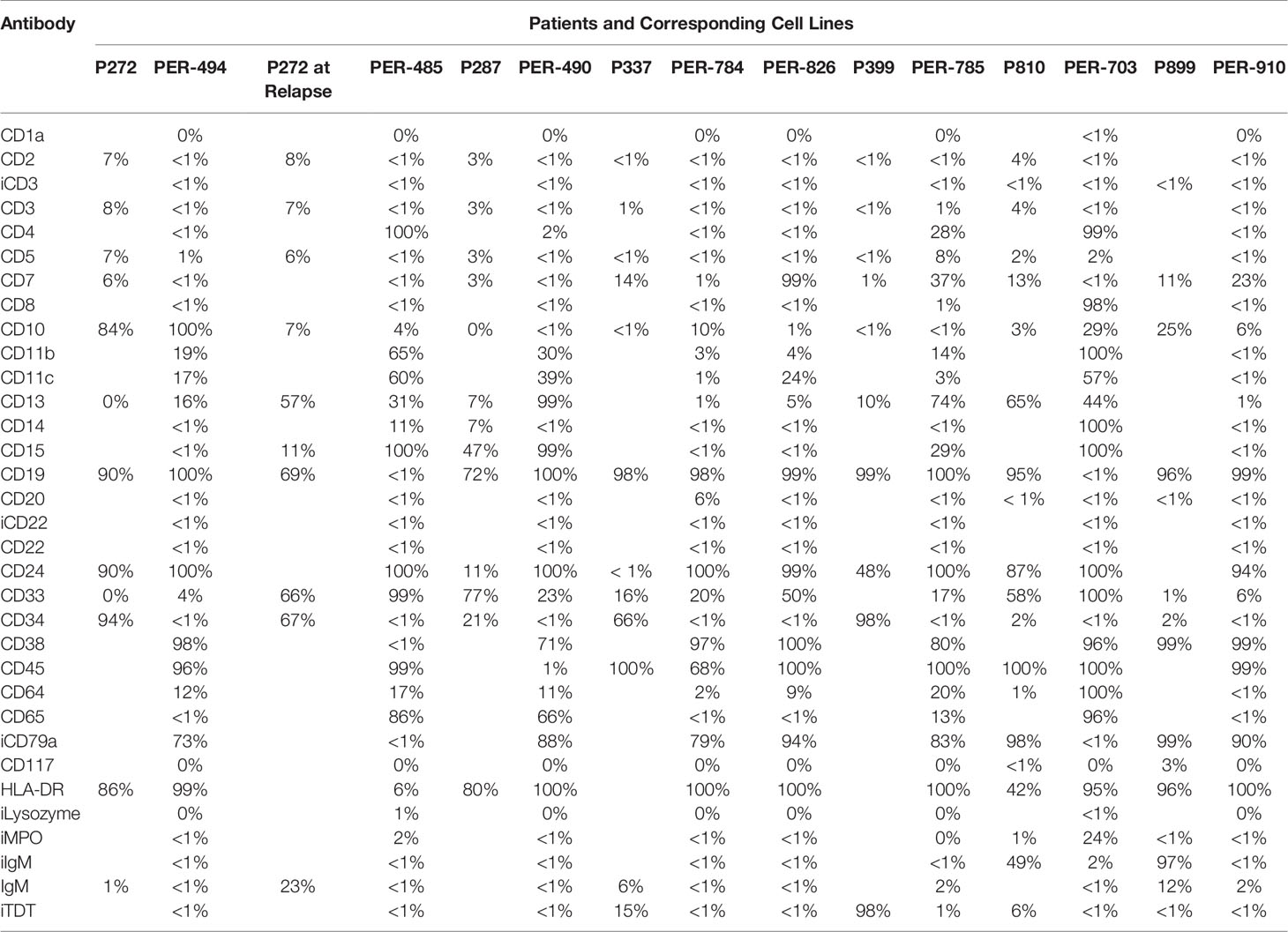
Table 2 Immunophenotype of primary patient samples and corresponding cell lines.
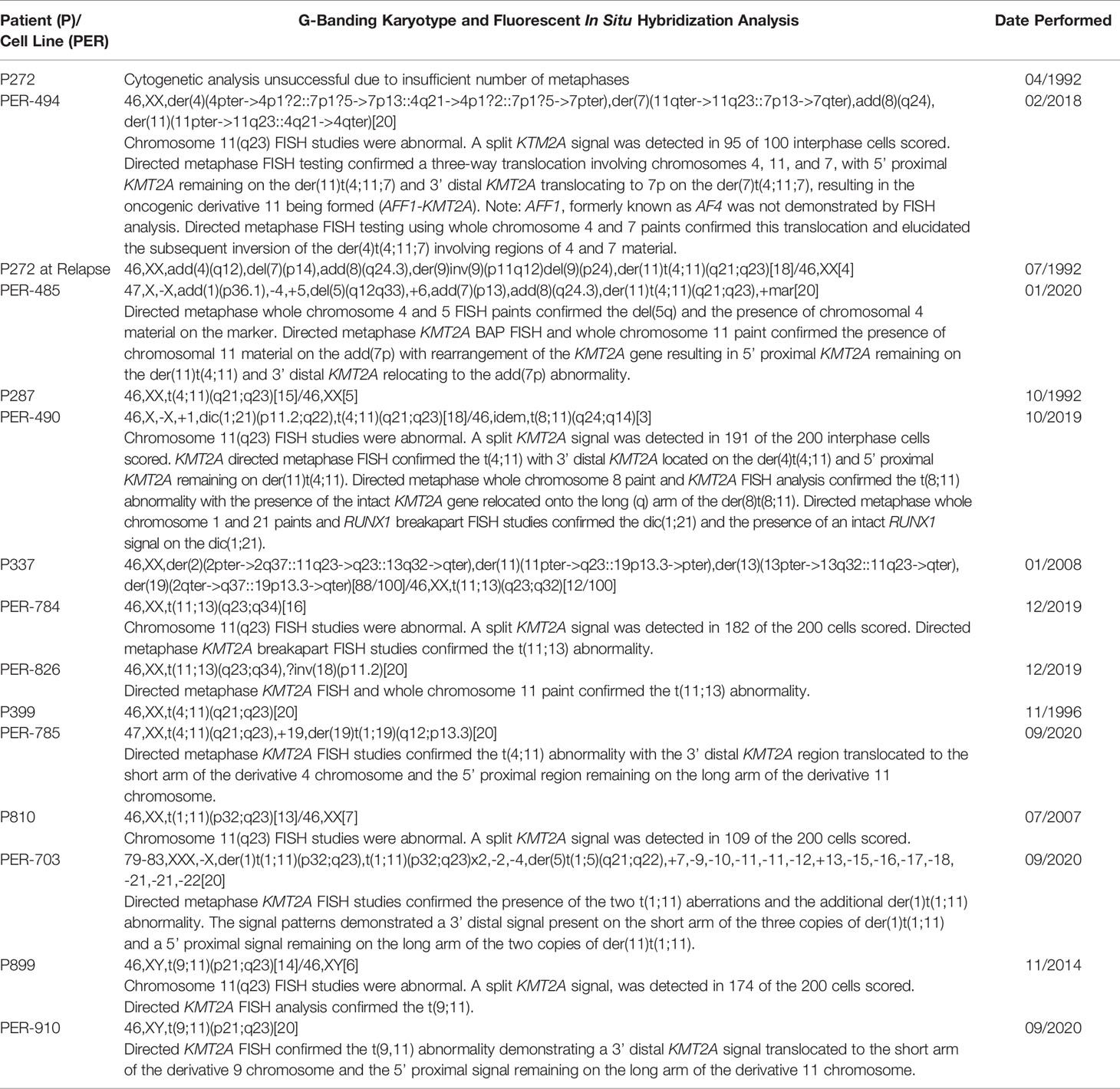
Table 3 Cytogenetic analysis of primary patient samples and corresponding cell lines.
Carfilzomib Is Cytotoxic In Vitro and Exhibits Synergy With Conventional Chemotherapeutic Agents Used in Induction
Carfilzomib demonstrated low IC50 concentrations within the nanomolar range across all cell lines (PER-494: 6.0 nM; PER-485: 15.8 nM; PER-490: 9.1 nM; PER-784: 7.3 nM; PER-826: 10.6 nM; PER-785: 14.8 nM; PER-703: 12.9 nM; PER-910: 12.8 nM) (Figure 1). Carfilzomib was shown to inhibit proteasome activity at the IC50 concentration in all cell lines compared to untreated control (Figure 2A). Combination drug testing indicated in vitro synergy between carfilzomib and several conventional chemotherapeutic agents including vincristine, daunorubicin, dexamethasone, L-asparaginase, and 4-hydroperoxycyclophosphamide, with six or more cell lines demonstrating positive sum of synergy scores according to Excess over Bliss (Table 4). Notably, the highest total synergy scores in the two cell lines with a myeloid dominant immunophenotype (PER-485 and PER-703) occurred in combination with cytarabine (Table 4). As a first step towards assessing the potential of such drug combinations, we examined carfilzomib as single agent in vivo.
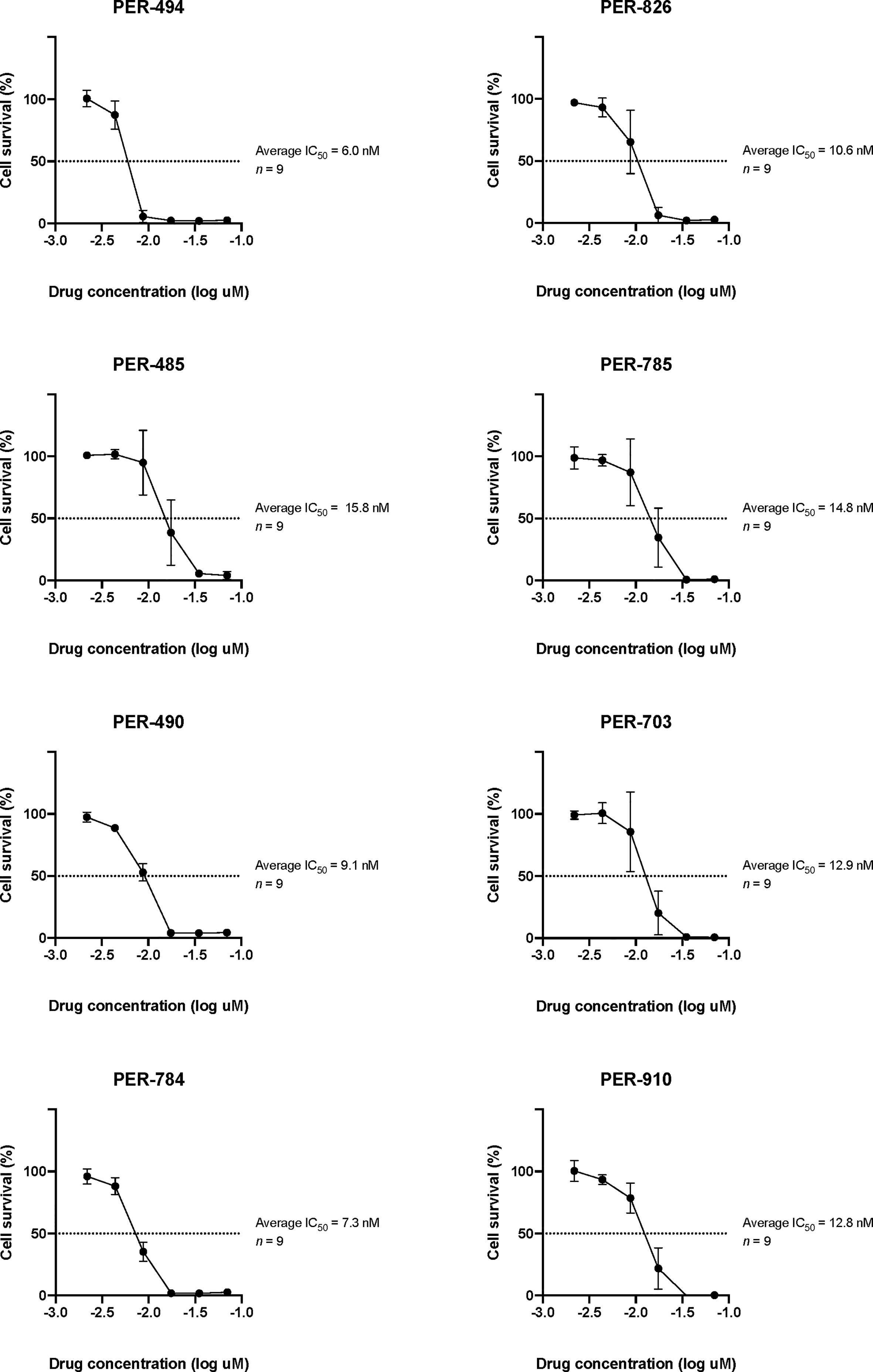
Figure 1 Log dose-response analyses of carfilzomib in PER cell lines derived from infants with KMT2A-rearranged acute lymphoblastic leukemia. The half maximum inhibitory concentrations (IC50) are indicated. Data points are represented as the mean ± standard deviation of n = 9 replicates.
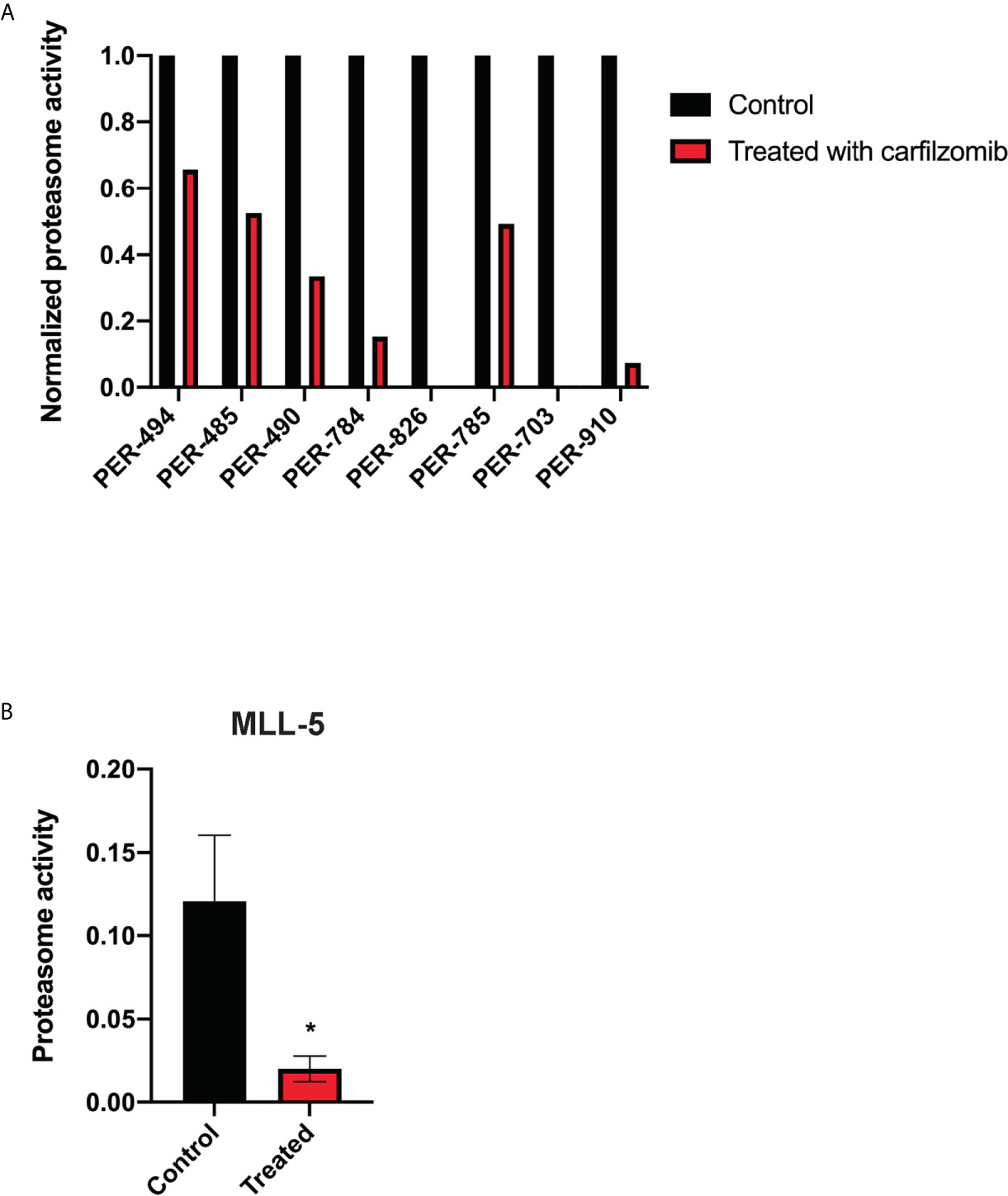
Figure 2 Carfilzomib inhibits proteasome activity. (A) Proteasome activity of PER cell lines derived from infants with KMT2A-rearranged acute lymphoblastic leukemia following incubation with carfilzomib at their respective half maximum inhibitory concentrations (IC50) for 72 h normalized to proteasome activity of untreated cell lines. (B) Proteasome activity in peripheral blood of MLL-5 patient-derived xenografts treated with 1.5 mg/kg of intravenous carfilzomib via tail vein injection on days 1 and 2 compared to untreated mice (n = 5 mice per group). Proteasome activity was measured 10 min after the injection on day 2. Mice were treated at high leukemic burden in the bone marrow corresponding to a mean peripheral blood blast percentage of 5.1% for treated mice and 4.2% for untreated mice. Error bars represent mean ± standard error of mean. *P < 0.05.
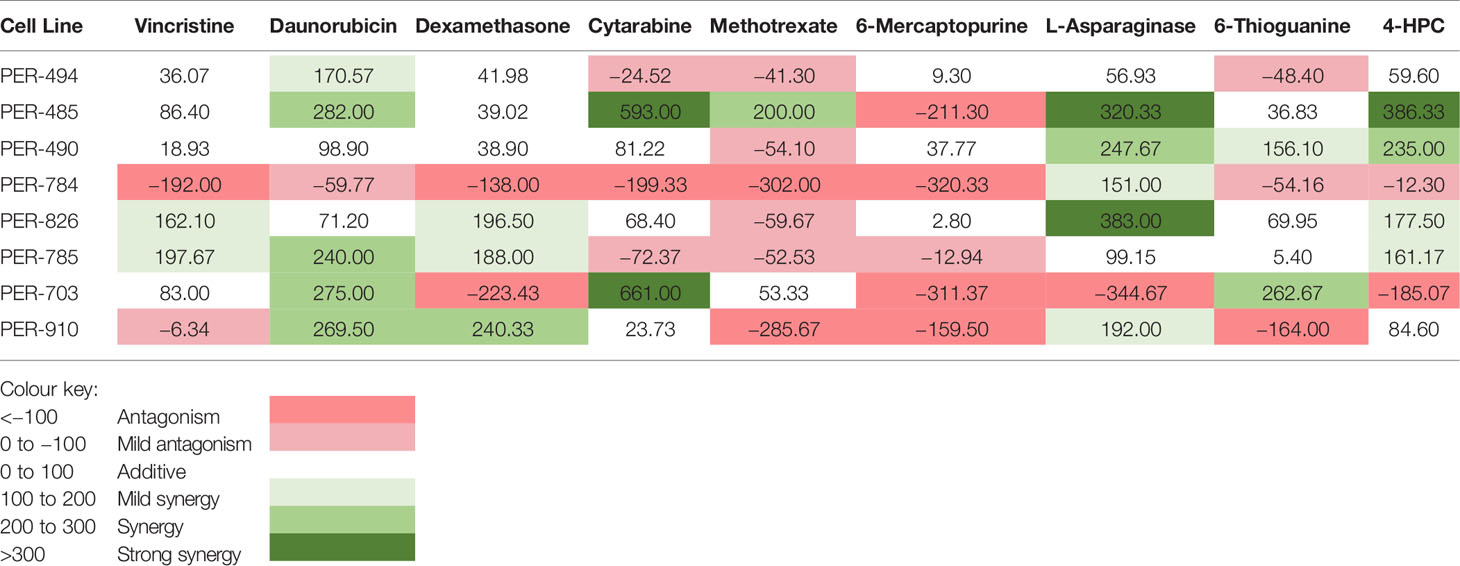
Table 4 Total in vitro synergy scores between carfilzomib and conventional chemotherapy agents.
Carfilzomib Displays Toxicity In Vivo and Does Not Prolong Survival as a Single Agent or in Combination With Induction Chemotherapy
Leukemia-bearing mice receiving 2.5 mg/kg or more of carfilzomib developed weight loss, diarrhea, shortness of breath, slower movement, and swelling at the injection site. Mice that received 4.0 mg/kg were euthanized due to excessive morbidity prior to completion of therapy, thus establishing the MTD of carfilzomib as 2.0 mg/kg. There was no difference in leukemia burden in the bone marrow, spleen, or peripheral blood between any of the doses tested or vehicle control at the defined endpoint (Figure 3). This finding was extended for assessment of EFS, when carfilzomib monotherapy was used to treat minimal disease burden in both the PER-785 and PER-826 xenograft models. Compared to vehicle control, no significant survival benefit was seen for carfilzomib at the MTD of 2.0 mg/kg (Figures 4A, B).
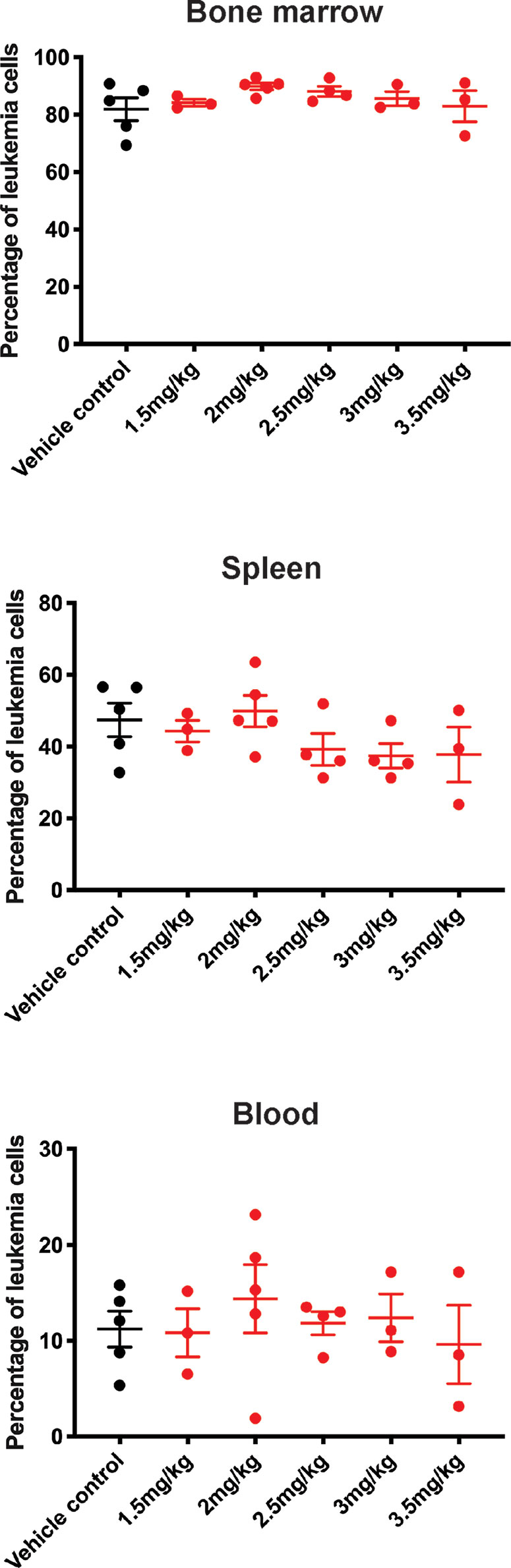
Figure 3 Carfilzomib monotherapy does not reduce leukemic burden in the PER-785 xenograft. Following a 12-day engraftment period, carfilzomib was administered intravenously on days 1, 2, 8, 9, 15, and 16 with treatment groups comprising of vehicle control, 1.5, 2.0, 2.5, 3.0, 3.5, or 4.0 mg/kg dosing of carfilzomib (n = 5 mice per group). Mice were sacrificed 3 weeks from the start of therapy and leukemia burden was determined by measuring the percentage of human CD19+ CD45+ cells in the bone marrow, spleen, and peripheral blood. Error bars represent mean ± standard error of mean.
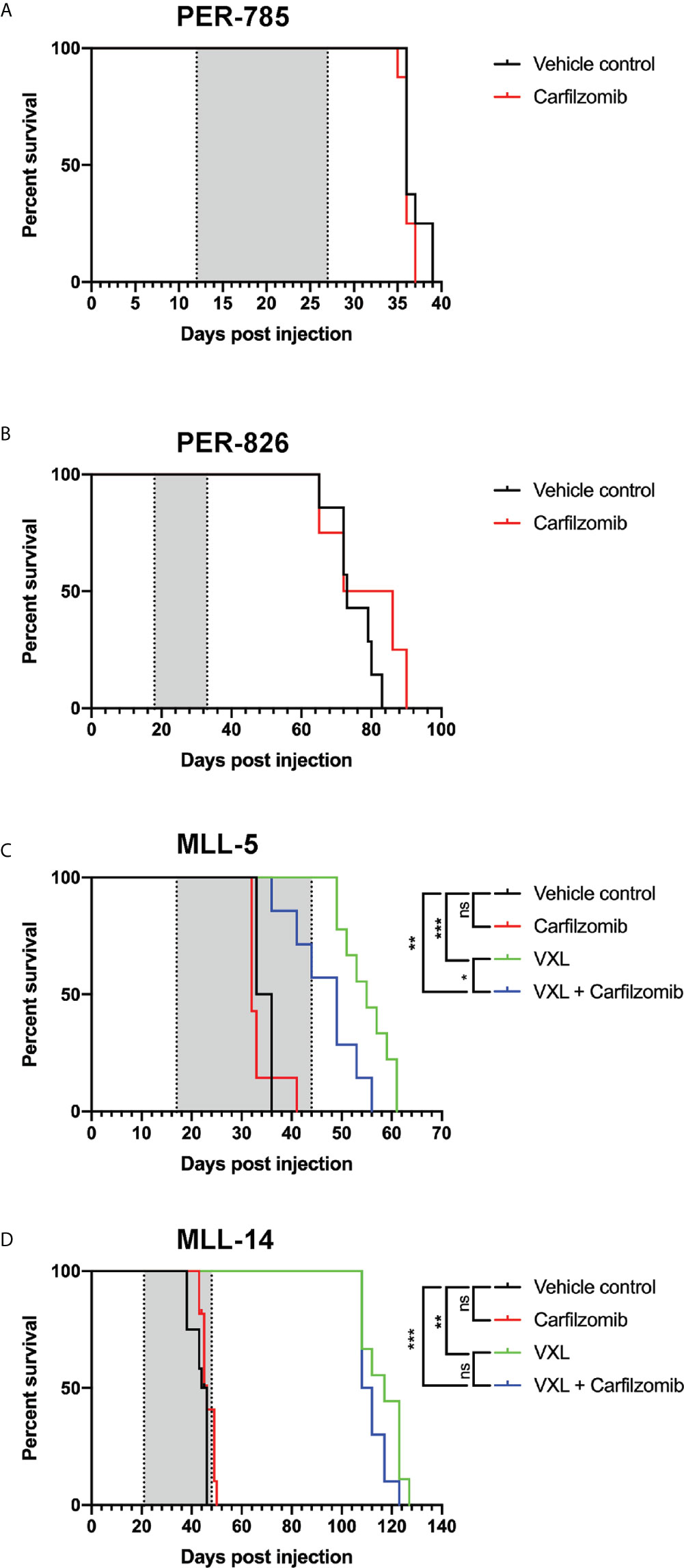
Figure 4 Carfilzomib does not extend survival in infant KMT2A-rearranged acute lymphoblastic leukemia xenografts. (A, B) Kaplan-Meier survival curves of mice injected with PER-785 or PER-826 leukemia cells that were treated at low leukemic burden with vehicle control or 2 mg/kg carfilzomib on days 1, 2, 8, 9, 15, and 16 (n = 8 mice per group). (C, D) Kaplan-Meier survival curves of mice injected with MLL-5 or MLL-14 leukemia cells that were treated at high leukemic burden with carfilzomib, VXL, carfilzomib-VXL combination therapy, or vehicle control (n = 10 mice per group). Carfilzomib was administered intravenously on days 1, 2, 8, 9, 15, and 16 at 1.5 mg/kg. VXL comprised of 0.15 mg/kg of vincristine once a week for 4 weeks, dexamethasone 5 mg/kg 5 days a week for 4 weeks, and L-asparaginase 1,000 U/kg 5 days a week for 4 weeks, administered by intraperitoneal injection. Gray shaded areas indicate treatment duration, with the first dotted line representing the start of therapy and the second dotted line the end of therapy. *P < 0.05, **P < 0.001, ***P < 0.0001.
Results from the in vitro synergy screen identified positive Bliss scores in the majority of cell lines for carfilzomib and chemotherapeutic agents used during induction therapy for children with ALL (Table 4). Given this finding, we sought to determine whether carfilzomib could potentiate the effect of multi-agent induction therapy. This was performed using the MLL-5 and MLL-14 xenografts, as they have been extensively characterized for their response to vincristine, dexamethasone, and L-asparaginase (VXL) (12). As induction therapy is administered to children with high leukemic burden and to additionally determine whether carfilzomib could provide an in vivo survival benefit at high leukemic burden, treatment was commenced when the percentage of human CD19+ CD45+ cells reached 1% in the blood. For each xenograft model, mice were randomized into four groups of 10 mice, with groups comprising of carfilzomib monotherapy, VXL, carfilzomib-VXL combination therapy, or vehicle control. VXL treatment comprised of 0.15 mg/kg of vincristine (Pfizer, NY, USA) once a week for 4 weeks, dexamethasone (Mylan, PA, USA) 5 mg/kg 5 days a week for 4 weeks, and L-asparaginase (Sanofi, Paris, France) 1,000 U/kg 5 days a week for 4 weeks administered by intraperitoneal injection (15). To minimize toxicity when combined with VXL therapy, carfilzomib was administered at one dose level below the established MTD, namely 1.5 mg/kg, with in vivo inhibition of proteasome activity demonstrated at this dose level compared to untreated controls (Figure 2B). However, despite the on-target effect, no survival benefit was seen for carfilzomib, either as a single agent when compared to vehicle control, or in combination with VXL therapy compared to VXL alone in MLL-5 or MLL-14 xenografts treated with high leukemic burden (Figures 4C, D).
Discussion
Infant KMT2A-rearranged ALL has a desperately poor outcome. The high rate of relapse combined with the dose-limiting toxicities of conventional chemotherapeutic agents has led to the search for novel agents that can be integrated into the treatment schedule. However, infant KMT2A-rearranged ALL is a rare disease and there is a consequent limitation to the number of trials that can be concurrently active, with prolonged periods required to meet target accrual. Currently there are four main groups conducting clinical trials for infant ALL, namely the COG, Japanese Pediatric Leukemia/Lymphoma Study Group, St Jude Children’s Research Hospital, and the INTERFANT study group. The most recently completed trial from the latter, INTERFANT-06, required 10 years to accrue 651 patients with no improvement in outcome compared to the preceding INTERFANT-99 study (3, 16). The most recent phase 3 COG study, AALL0631, added the FLT3 inhibitor lestaurtinib to post-induction chemotherapy for infants with KMT2A-rearranged ALL, with no improvement to overall outcome (17). Given these limitations and to ensure progress is made it is imperative that novel agents undergo rigorous and robust preclinical assessment to provide the best evidence to enable clinical trial development committees to make informed decisions regarding selection of the best agents for integration into the clinical setting.
Proteasome inhibitors have emerged as a powerful treatment strategy for multiple myeloma (18). The ubiquitin-proteasome system maintains protein homeostasis by controlling the degradation of damaged, abnormally folded, or short-lived regulatory proteins. Consequently, it has the capacity to influence key cellular functions related to progression through the cell cycle, cellular proliferation, cellular differentiation, DNA damage repair, and cell death. Elevated proteasomal activity has been demonstrated in many cancers, notably hematological neoplasms, because malignant cells are often heavily dependent on proteasomal function, particularly for the degradation of proteins that hinder proliferation. Inhibition of proteasome with proteasome inhibitors impairs turnover of multiple proteins resulting in their accumulation in the cell and disruption of multiple signaling pathways within the cell, leading to disruption of cell cycle and apoptosis.
In recent years, proteasome inhibitors have been increasingly investigated for the treatment of pediatric ALL. Bortezomib was the first-in-class reversible proteasome inhibitor to receive FDA approval and has undergone clinical investigation in the relapsed/refractory setting. Although these studies have shown that the addition of bortezomib to induction chemotherapy improves response rates, it is yet to translate into a survival advantage (19). In particular, encouraging response rates have been seen in children with relapsed T-ALL (20), resulting in development of the COG phase 3 AALL1231 study, which randomized the addition of bortezomib to standard induction therapy in patients with newly diagnosed T-ALL (NCT02112916). However, this trial was prematurely closed due to the need to integrate findings from the predecessor COG AALL0434 study which identified benefit of nelarabine for T-ALL (21). The tolerability of incorporating bortezomib and vorinostat into an ALL chemotherapy backbone for newly diagnosed infants with ALL is currently being investigated by St Jude Children’s Research Hospital in a single arm phase 2 study (NCT02553460). The results of this study will be eagerly anticipated as the addition of two novel agents, bortezomib and vorinostat, to an ALL chemotherapy backbone represents a bold strategy that is required to improve the outcome for infants with ALL. However, elucidating the magnitude of any effect specifically attributable to bortezomib may ultimately prove difficult due to concurrent administration with vorinostat and conventional chemotherapeutic agents. The COG phase 3 AAML1031 trial identified that the addition of bortezomib to standard chemotherapy increased toxicity but did not improve survival in children with acute myeloid leukemia (22).
Carfilzomib is a second-generation irreversible proteasome inhibitor and was the second agent in class to receive FDA approval for the treatment of multiple myeloma. A phase 1b study of carfilzomib in combination with induction therapy for children with relapsed/refractory ALL has recently completed accrual, with tolerability identified from preliminary findings (13). However, infants were excluded from this study precluding the ability to draw any conclusions regarding clinical use of carfilzomib in this population. Our study was able to demonstrate in vitro cytotoxicity of the selective proteasome inhibitor carfilzomib, against infant KMT2A-rearranged cell lines. Carfilzomib exhibited in vitro synergy with several chemotherapeutic agents conventionally used to treat infant ALL, including those administered during induction therapy. Similar to our findings, previous studies have demonstrated effective cytotoxicity of carfilzomib in commercially available pediatric leukemia cell lines, which included one commercially available infant ALL cell line (KOPN8), and in primary pediatric leukemia cells (23, 24). However, despite showing an on-target effect, our findings did not translate to an in vivo benefit, with higher carfilzomib doses leading to excessive toxicity and an inability to demonstrate a survival advantage for carfilzomib alone or in combination with induction-style chemotherapy in KMT2A-rearranged infant ALL patient-derived xenograft models. However, a limitation of the study was that proteasome activity was measured in the peripheral blood and thus the inhibitory effect of carfilzomib was representative of an effect on normal cells as well as blast cells. Proteasome activity was not measured in the bone marrow, raising the potential that carfilzomib did not reach blast cells in the bone marrow or sustain relevant therapeutic concentrations, which could also account for the lack of in vivo efficacy.
In conclusion, our study highlights that in vitro efficacy does not necessarily translate into benefit in vivo and emphasizes the importance of in vivo validation prior to suggesting an agent for clinical use. Whilst proteasome inhibitors have an important role to play in several hematological malignancies, our findings guard against prioritization of carfilzomib for treatment of KMT2A-rearranged infant ALL in the clinical setting. Current clinical trial development for KMT2A-rearranged infant ALL will therefore focus on integration of novel agents which have strong preclinical data, such as venetoclax (25) and menin inhibitors (26), and immunotherapeutic approaches with promising preliminary clinical findings, such as blinatumomab (27) and CAR T-cell therapy (28).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by Telethon Kids Institute Animal Ethics Committee.
Author Contributions
LC, UK, and RSK conceived and designed the study. RSK provided study materials and patient information. RdK performed and interpreted the cytogenetic analysis. JF developed the cell lines. JO, AH, and EF performed in vitro experiments. G-AC and SS performed in vivo experiments. RS, SC, SM, and UK provided technical insight and analyzed results. LC and RK interpreted data and supervised the project. JO and RSK wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
RSK is supported by a Fellowship from the National Health and Medical Research Council of Australia (NHMRC APP1142627). SM is supported by a Fellowship from the Cancer Council Western Australia. This study was supported by project grants from the Children’s Leukaemia and Cancer Research Foundation and The Kids’ Cancer Project.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.631594/full#supplementary-material
References
1. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med (2015) 373(16):1541–52. doi: 10.1056/NEJMra1400972
2. Kotecha RS, Gottardo NG, Kees UR, Cole CH. The evolution of clinical trials for infant acute lymphoblastic leukemia. Blood Cancer J (2014) 4(4):e200. doi: 10.1038/bcj.2014.17
3. Pieters R, De Lorenzo P, Ancliffe P, Aversa LA, Brethon B, Biondi A, et al. Outcome of infants younger than 1 year with acute lymphoblastic leukemia treated with the Interfant-06 protocol: results from an international phase III randomized study. J Clin Oncol (2019) 37(25):2246–56. doi: 10.1200/JCO.19.00261
4. Winters AC, Bernt KM. MLL-rearranged leukemias - an update on science and clinical approaches. Front Pediatr (2017) 5:4. doi: 10.3389/fped.2017.00004
5. Kees UR, Ford J, Price PJ, Meyer BF, Herrmann RP. PER-117: a new human ALL cell line with an immature thymic phenotype. Leuk Res (1987) 11(5):489–98. doi: 10.1016/0145-2126(87)90082-8
6. Dworzak MN, Buldini B, Gaipa G, Ratei R, Hrusak O, Luria D, et al. AIEOP-BFM consensus guidelines 2016 for flow cytometric immunophenotyping of pediatric acute lymphoblastic leukemia. Cytometry B Clin Cytom (2018) 94(1):82–93. doi: 10.1002/cyto.b.21518
7. McGowan-Jordan J, Simons A, Schmid M. ISCN 2016: an international system for human cytogenetic nomenclature. Basel: Karger (2016). doi: 10.1159/isbn.978-3-318-06861-0
8. Bliss CI. The toxicity of poisons applied jointly. Ann Appl Biol (1939) 26(3):585–615. doi: 10.1111/j.1744-7348.1939.tb06990.x
9. Greco WR, Bravo G, Parsons JC. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev (1995) 47(2):331–85.
10. Cheung LC, Cruickshank MN, Hughes AM, Singh S, Chua GA, Ford J, et al. Romidepsin enhances the efficacy of cytarabine in vivo, revealing histone deacetylase inhibition as a promising therapeutic strategy for KMT2A-rearranged infant acute lymphoblastic leukemia. Haematologica (2019) 104(7):e300–3. doi: 10.3324/haematol.2018.192906
11. Cruickshank MN, Ford J, Cheung LC, Heng J, Singh S, Wells J, et al. Systematic chemical and molecular profiling of MLL-rearranged infant acute lymphoblastic leukemia reveals efficacy of romidepsin. Leukemia (2017) 31(1):40–50. doi: 10.1038/leu.2016.165
12. Richmond J, Carol H, Evans K, High L, Mendomo A, Robbins A, et al. Effective targeting of the P53-MDM2 axis in preclinical models of infant MLL-rearranged acute lymphoblastic leukemia. Clin Cancer Res (2015) 21(6):1395–405. doi: 10.1158/1078-0432.CCR-14-2300
13. Burke MJ, Ziegler DS, Sirvent FJB, Attarbaschi A, Gore L, Locatelli F, et al. Phase 1b study of carfilzomib in combination with induction chemotherapy in children with relapsed or refractory acute lymphoblastic leukemia (ALL). Blood (2019) 134(Supplement_1):3873. doi: 10.1182/blood-2019-127350
14. Henderson MJ, Choi S, Beesley AH, Baker DL, Wright D, Papa RA, et al. A xenograft model of infant leukaemia reveals a complex MLL translocation. Br J Haematol (2008) 140(6):716–9. doi: 10.1111/j.1365-2141.2007.06966.x
15. Szymanska B, Wilczynska-Kalak U, Kang MH, Liem NL, Carol H, Boehm I, et al. Pharmacokinetic modeling of an induction regimen for in vivo combined testing of novel drugs against pediatric acute lymphoblastic leukemia xenografts. PloS One (2012) 7(3):e33894. doi: 10.1371/journal.pone.0033894
16. Stutterheim J, van der Sluis IM, de Lorenzo P, Alten J, Ancliffe P, Attarbaschi A, et al. Clinical implications of minimal residual disease detection in infants with KMT2A-rearranged acute lymphoblastic leukemia treated on the Interfant-06 protocol. J Clin Oncol (2021) 39(6):652–62. doi: 10.1200/JCO.20.02333
17. Brown P, Kairalla J, Wang C, Dreyer Z, Salzer W, Sorenson M, et al. Addition of FLT3 inhibitor lestaurtinib to post-induction chemotherapy does not improve outcomes in MLL-rearranged infant acute lymphoblastic leukemia (ALL): AALL0631, a Children’s Oncology Group study. Pediatr Blood Cancer (2016) 63(S3):S7. doi: 10.1002/pbc.26233
18. Ito S. Proteasome inhibitors for the treatment of multiple myeloma. Cancers (Basel) (2020) 12(2):265. doi: 10.3390/cancers12020265
19. Messinger YH, Bostrom BC. Bortezomib-based four-drug induction does induce a response in advanced relapsed ALL but cure remains elusive. Pediatr Blood Cancer (2020) 67(3):e28115. doi: 10.1002/pbc.28115
20. Horton TM, Whitlock JA, Lu X, O’Brien MM, Borowitz MJ, Devidas M, et al. Bortezomib reinduction chemotherapy in high-risk ALL in first relapse: a report from the Children’s Oncology Group. Br J Haematol (2019) 186(2):274–85. doi: 10.1111/bjh.15919
21. Dunsmore KP, Winter SS, Devidas M, Wood BL, Esiashvili N, Chen Z, et al. Children’s Oncology Group AALL0434: a phase III randomized clinical trial testing nelarabine in newly diagnosed T-Cell acute lymphoblastic leukemia. J Clin Oncol (2020) 38(28):3282–93. doi: 10.1200/JCO.20.00256
22. Aplenc R, Meshinchi S, Sung L, Alonzo T, Choi J, Fisher B, et al. Bortezomib with standard chemotherapy for children with acute myeloid leukemia does not improve treatment outcomes: a report from the Children’s Oncology Group. Haematologica (2020) 105(7):1879–86. doi: 10.3324/haematol.2019.220962
23. Swift L, Jayanthan A, Ruan Y, Anderson R, Boklan J, Trippett T, et al. Targeting the proteasome in refractory pediatric leukemia cells: characterization of effective cytotoxicity of carfilzomib. Target Oncol (2018) 13(6):779–93. doi: 10.1007/s11523-018-0603-0
24. Niewerth D, Franke NE, Jansen G, Assaraf YG, van Meerloo J, Kirk CJ, et al. Higher ratio immune versus constitutive proteasome level as novel indicator of sensitivity of pediatric acute leukemia cells to proteasome inhibitors. Haematologica (2013) 98(12):1896–904. doi: 10.3324/haematol.2013.092411
25. Khaw SL, Suryani S, Evans K, Richmond J, Robbins A, Kurmasheva RT, et al. Venetoclax responses of pediatric ALL xenografts reveal sensitivity of MLL-rearranged leukemia. Blood (2016) 128(10):1382–95. doi: 10.1182/blood-2016-03-707414
26. Krivtsov AV, Evans K, Gadrey JY, Eschle BK, Hatton C, Uckelmann HJ, et al. A menin-MLL inhibitor induces specific chromatin changes and eradicates disease in models of MLL-rearranged leukemia. Cancer Cell (2019) 36(6):660–73.e11. doi: 10.1016/j.ccell.2019.11.001
27. Clesham K, Rao V, Bartram J, Ancliff P, Ghorashian S, O’Connor D, et al. Blinatumomab for infant acute lymphoblastic leukemia. Blood (2020) 135(17):1501–4. doi: 10.1182/blood.2019004008
Keywords: infant, acute lymphoblastic leukemia, KMT2A, MLL, carfilzomib, PER cell lines
Citation: Cheung LC, de Kraa R, Oommen J, Chua G-A, Singh S, Hughes AM, Ferrari E, Ford J, Chiu SK, Stam RW, Kees UR, Malinge S and Kotecha RS (2021) Preclinical Evaluation of Carfilzomib for Infant KMT2A-Rearranged Acute Lymphoblastic Leukemia. Front. Oncol. 11:631594. doi: 10.3389/fonc.2021.631594
Received: 20 November 2020; Accepted: 25 February 2021;
Published: 15 April 2021.
Edited by:
Charles De Bock, Children’s Cancer Institute Australia, AustraliaReviewed by:
Sarah K. Tasian, Children’s Hospital of Philadelphia, United StatesKeith Sia, Children’s Cancer Institute Australia, Australia
Copyright © 2021 Cheung, de Kraa, Oommen, Chua, Singh, Hughes, Ferrari, Ford, Chiu, Stam, Kees, Malinge and Kotecha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rishi S. Kotecha, cmlzaGkua290ZWNoYUBoZWFsdGgud2EuZ292LmF1