Abstract
Lack of demonstrable mutations affecting JAK2, CALR, or MPL driver genes within the spectrum of BCR-ABL1-negative myeloproliferative neoplasms (MPNs) is currently referred to as a triple-negative genotype, which is found in about 10% of patients with essential thrombocythemia (ET) and 5–10% of those with primary myelofibrosis (PMF). Very few papers are presently available on triple-negative ET, which is basically described as an indolent disease, differently from triple-negative PMF, which is an aggressive myeloid neoplasm, with a significantly higher risk of leukemic evolution. The aim of the present study was to evaluate the bone marrow morphology and the clinical-laboratory parameters of triple-negative ET patients, as well as to determine their molecular profile using next-generation sequencing (NGS) to identify any potential clonal biomarkers. We evaluated a single-center series of 40 triple-negative ET patients, diagnosed according to the 2017 WHO classification criteria and regularly followed up at the Hematology Unit of our Institution, between January 1983 and January 2019. In all patients, NGS was performed using the Illumina Ampliseq Myeloid Panel; morphological and immunohistochemical features of the bone marrow trephine biopsies were also thoroughly reviewed. Nucleotide variants were detected in 35 out of 40 patients. In detail, 29 subjects harbored one or two variants and six cases showed three or more concomitant nucleotide changes. The most frequent sequence variants involved the TET2 gene (55.0%), followed by KIT (27.5%). Histologically, most of the cases displayed a classical ET morphology. Interestingly, prevalent megakaryocytes morphology was more frequently polymorphic with a mixture of giant megakaryocytes with hyperlobulated nuclei, normal and small sized maturing elements, and naked nuclei. Finally, in five cases a mild degree of reticulin fibrosis (MF-1) was evident together with an increase in the micro-vessel density. By means of NGS we were able to identify nucleotide variants in most cases, thus we suggest that a sizeable proportion of triple-negative ET patients do have a clonal disease. In analogy with driver genes-mutated MPNs, these observations may prevent issues arising concerning triple-negative ET treatment, especially when a cytoreductive therapy may be warranted.
Introduction
Essential thrombocythemia (ET), together with polycythemia vera (PV) and primary myelofibrosis (PMF), belongs to the so-called “classic” BCR-ABL1-negative myeloproliferative neoplasms (MPN) category. It is mainly characterized by an increased platelet production and sustained thrombocytosis which leads to an increased risk of thrombo-hemorrhagic complications (1, 2). It is the most common BCR-ABL1-negative MPNs, with an annual incidence estimated at 0.38–1.7 ×105 persons (3), a reported median age at presentation of 68 years and a female-to-male ratio of 2:1 (4).
Essential thrombocythemia diagnosis is currently made according to the 2017 WHO criteria and is based on a composite assessment of clinical-laboratory features, including also molecular analyses (5). Indeed, in 2005 the JAK2V617F mutation has been reported to play a crucial role in the pathogenesis of these disorders, being identifiable in about 50–60% of ET patients (6–9). Furthermore, this mutation seems to be associated with peculiar phenotypes in the different BCR-ABL1-negative MPNs: specifically, in ET its presence is consistently associated with older age at diagnosis, higher hemoglobin levels, higher white blood cells counts, and lower platelet counts; it also clusters with a lower risk of fibrotic evolution and a higher risk of thrombosis (10–14).
Subsequently, mutations in genes other than JAK2 have been described: MPL mutations, with a frequency ranging from 3 to 8% (15, 16), and, more recently, somatically acquired mutations in the calreticulin (CALR) gene, reported in about 25–35% of all ET cases (17, 18).
The clinical course of CALR-mutated ET patients was reported to be better than in the presence of the JAK2V617F mutation, as subjects with CALR mutations seem to suffer from a lower risk of thrombosis when compared with both JAK2- and MPL-mutated cases, with a favorable impact on thrombosis-free survival (TFS) (19–23).
Lack of demonstrable mutations affecting the above-mentioned driver genes is currently referred to as a triple-negative genotype, which is found in about 10% of patients with ET and 5–10% of those with PMF. A few triple-negative ET cases have been shown to carry activating mutations of MPL gene outside exon 10, and these non-canonical mutations may be either somatically acquired or inherited (24, 25). Therefore, these subjects may have a true BCR-ABL1-negative MPN associated with non-canonical MPL mutations, or, alternatively, hereditary thrombocytosis (26).
Very few papers are presently available on triple-negative ET, which is basically described as an indolent disease with a low incidence of vascular events, differently from triple-negative PMF, which is an aggressive myeloid neoplasm, with a significantly higher risk of leukemic evolution (27, 28). More specifically, in WHO-defined ET, survival is near-normal (15-year survival of about 80%) and similar to that of the sex- and age-standardized European population (29), and the 10-year risk of MPN – blast phase (MPN-BP) or post-ET myelofibrosis (PET-MF) evolution is <1% (29–32).
Accordingly, triple-negative ET differential diagnosis is of crucial relevance, particularly vs. reactive thrombocytosis and, most notably, pre-fibrotic PMF, as the latter condition significantly impacts on patients' management due to its increased risk of fibrotic progression/leukemic evolution and reduced survival (29).
The advent of next-generation sequencing (NGS) technologies has greatly improved the understanding of MPN pathogenesis. Molecular profiling of triple-negative ET, using targeted gene panels or whole-exome sequencing, revealed the presence of so-called “non-driver” mutations. These variants may chronologically precede or follow the acquisition of driver mutations, thus conferring a selective advantage to neoplastic cells, and affect genes involved in epigenetic regulation (TET2, DNMT3A, ASXL1, EZH2, and IDH1/2), RNA splicing (SF3B1, SRSF2, and U2AF1) and regulation of cytokine signaling (CBL) (25, 33). Although mutations in these genes are not restricted to MPNs, their presence can be of diagnostic value in the triple-negative ET cohort, thus providing evidence of clonality (25, 33).
The aim of the present study was to evaluate the bone marrow (BM) morphology and the clinical-laboratory parameters of ET patients, defined upon strict adherence to the 2017 WHO criteria and featuring a triple-negative genotype, and to determine their molecular profile by means of a targeted NGS panel of current clinical use, in order to identify potential clonal biomarkers.
Materials and Methods
Patients
The present single-center cohort study involved a consecutive series of 40 patients, regularly followed up at the Hematology Unit of our Institution between January 1983 and January 2019, who received a diagnosis of triple-negative ET according to the 2017 WHO classification criteria. Inclusion criteria were the availability of demographic, clinical, hematologic, and histologic data at diagnosis, stored BM, and at least one granulocyte DNA sample (collected at the time of JAK2 mutational screening) to perform NGS analysis. All patients lacked mutations in JAK2, CALR, or MPL genes, as routinely assessed by allele-specific PCR or Sanger sequencing and confirmed after deep targeted NGS analysis (see below). Baseline clinical characteristics and outcome measures (thrombosis, hemorrhages, PET-MF and MPN-BP, death, and overall and thrombosis-free survival) were evaluated. We considered as main cardiovascular risk factors the presence of arterial hypertension, diabetes mellitus, hyperlipidemia, and smoking attitude. Events of major thrombosis and bleeding were recorded when objectively documented and according to standard definitions as described in detail by the Cytoreductive Therapy in Polycythemia Vera Collaborative Group (34). Diagnosis of PET-MF was made in accordance with the International Working Group for Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) criteria (35). Diagnosis of MPN-BP was made with a 20% BM or peripheral blood blast threshold (36). The patients were treated with antiplatelet and cytoreductive agents (hydroxycarbamide and anagrelide) according to institutional guidelines which were based on current recommendations [the Italian guidelines after 2004 (37) and the European LeukemiaNet guidelines after 2011 (38, 39)]. Thrombosis-free survival was measured from the date of diagnosis to the date of the first documented major thrombosis. Follow-up information was updated in August 2020.
Methods
Molecular Analyses
The JAK2V617F mutation was detected by allele-specific PCR according to the protocol of Baxter et al. (7) and confirmed by direct Sanger sequencing. Quantitative analysis of the allele burden of the JAK2V617F mutation was performed by RQ-PCR using JAK2 MutaQuant (Ipsogen Inc, New Haven, CT, USA). The cut-off used for defining a case as negative for JAK2V617F mutation was 0.5%.
MPL mutations, in particular W515L, W515K, W515A, S505N, and G509C, were tested by direct sequencing of exon 10. Primer used were: MPL10F 5' TAGCCTGGATCTCCTTGGTG 3'; MPL10R 5' CCTGTTTACAGGCCTTCGGC 3'.
Mutations in exon 9 of the CALR gene were also assessed by the bidirectional sequencing approach as previously described (18). All sequencing analyses were performed on ABI PRISM 310 Genetic Analyzer (Applied Biosystems, Warrington, UK) using the Big Dye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Warrington, UK).
Next-generation sequencing analysis was performed using DNA extracted from peripheral blood granulocytes by the Ampliseq Myeloid Panel (Illumina), a commercially available NGS panel of current use in specialized laboratory for clinical practice, which allows sequencing of 40 genes mostly involved in myeloid malignancies (Supplementary Table 1). Genomic libraries were prepared using 20 ng of gDNA template and sequenced following Illumina's standardized protocol. The final amplicon pool of 14 samples/run was loaded at 9 pM on MiSeq System (v3 chemistry). The sequencing was performed sequentially from both ends each for 151 cycles. Obtained sequences were aligned to the reference genome (GRCh37/hg19) using MiSeq Reporter software (Illumina), which detected discrepancies among samples and reference genomes and determined their type, such as deletions, insertions, and SNPs. We considered data showing the following parameters acceptable for analyses: sequencing coverage >100X (bi-directional true paired-end sequencing), quality of 100, and a variant frequency (VAF) ≥ 3%. Concerning NGS data, a minimum quality score of Q30 (corresponding to a 1:1,000 error rate) was required for a minimum of 90% of bases sequenced and we excluded runs failing these metrics. The cluster density of each run was 600/800 Mean unique depth of coverage across the capture region of 3,000 amplicons.
To annotate sequence variants, Illumina Base Space Sequence Hub software was used. We included in our analysis: (1) low frequency single nucleotide variants, predicted as pathogenic by at least one of the following interpretation tools: FATHMM-MKL (http://fathmm.biocompute.org.uk/), SIFT (https://sift.bii.a-star.edu.sg/), DANN (https://cbcl.ics.uci.edu/public_data/DANN/), Poly-phen 2 (http://genetics.bwh.harvard.edu/pph2/); (2) single nucleotide variants with a minor allele frequency ≥ 0.01 (MAF ≥ 1%) predicted as pathogenic by at least one of the same tools and located in genes already described as associated with MPNs; (3) insertion/deletion variants, reported as pathogenic/likely pathogenic by clinical databases (Supplementary Table 2).
Cytogenetic Analyses
G-banding with trypsin performed on fresh BM aspirates was the standard technique for chromosome analysis with at least 20 metaphases described (40). Normal karyotype was defined as the absence of any chromosomal abnormality in a minimum of 20 metaphases examined. Chromosomal abnormalities were considered clonal if the same structural abnormality or extra chromosome appears in at least two and monosomy in at least three metaphases. A complex karyotype was defined as the presence of three or more distinct structural or numeric abnormalities. Monosomal karyotype was defined as two or more distinct autosomal monosomies or single autosomal monosomy associated with at least one structural abnormality.
Bone Marrow Biopsy
Histologic confirmation of the clinical diagnosis of ET as defined by the 2017 WHO classification was performed by two experienced pathologists (G.A.C and U.G.). Formalin-fixed, paraffin-embedded BM biopsy samples obtained at diagnosis were available for all patients. Sections were stained with hematoxylin-eosin, Giemsa, and Gomori's silver impregnation for evaluation of morphologic features and BM fibrosis. Immunohistochemistry for the evaluation of CD34+ precursors and micro-vessel density was performed via automated Dako OMNIS system, by means of the streptavidin-biotin-peroxidase-conjugated method.
Morphologic parameters were assessed according to the WHO classification, as follows: age-related cellularity, quantitative assessment of granulopoiesis, erythropoiesis and megakaryocytes, left-shifting of granulopoiesis and erythropoiesis (relative increase in immature to intermediate forms or proerythroblasts), presence, size (small vs. large) and density (loose vs. dense) of megakaryocyte clusters, morphologic features of megakaryocytes (hyperlobulated nuclei, cloud-like/bulbous nuclei, naked nuclei, maturation defects), grade of BM fibrosis, presence of osteosclerosis, density of vessels, presence of vascular ectasia and endoluminal hemopoiesis, presence of reactive lymphoid aggregates.
Results
Clinical Characterization
After NGS confirmation, 40 patients with triple-negative ET, diagnosed from January 1983 to January 2019, were included in this study. Their clinical-laboratory features at diagnosis are reported in Table 1. Median age at diagnosis was 50.2 years (range, 20–85) and 60.0% of the patients were females.
Table 1
| Patients (n = 40) | |
|---|---|
| Male/female | 16/24 |
| Age (years), median (range) | 50.2 (20–85) |
| Hb (g/dl), median (range) | 13.8 (10.8–16.1) |
| Hct (%), median (range) | 41.8 (33.2–49.1) |
| WBC count (×109/L), median (range) | 8.01 (5.23–12.5) |
| PLT count (×109/L), median (range) | 595 (443–2456) |
| LDH (IU/L), median (range) | 220 (105–462) |
| Serum erythropoietin (mIU/mL), median (range) | 7.15 (0.6–14.1) |
| Circulating CD34+ cells (/μl), median (range) | 5 (1–9) |
| Cytogenetic abnormalities, n (%) | 7 (17.5) |
| Palpable splenomegaly, n (%) | 5 (12.5) |
| Previous thrombosis, n (%) | 2 (5.0) |
| Previous hemorrhages, n (%) | / |
| Conventional thrombotic score, n(%) | |
| Low risk | 25 (62.5) |
| High risk | 15 (37.5) |
| IPSET-Thrombosis, n(%) | |
| Low risk | 28 (70.0) |
| Intermediate risk | 10 (25.0) |
| High risk | 2 (5.0) |
| Revised IPSET-Thrombosis, n(%) | |
| Very Low risk | 26 (65.0) |
| Low risk | / |
| Intermediate risk | 12 (30.0) |
| High risk | 2 (5.0) |
| Cytoreductive therapy, n (%) | 22 (55.0) |
| Antiplatelet therapy, n (%) | 37 (92.5) |
| Follow-up (years), median (range) | 7.0 (1.2–36.8) |
| Deceased, n (%) | 4 (10.0) |
Baseline clinical and laboratory features of 40 triple-negative ET patients.
ET, essential thrombocythemia; Hb, hemoglobin; Hct, hematocrit; WBC, white blood cells; PLT, platelets; LDH, lactate dehydrogenase.
After a median duration of follow-up of 7.0 years (range, 1.2–36.8), no case of fibrotic transformation or leukemic evolution was recorded. On the contrary, myelodysplastic syndrome (MDS) was diagnosed in one (2.5%) patient after more than 32 years of follow-up.
After a median time from diagnosis of 9.9 years (range, 4.4–15.4), two (5.0%) patients experienced an arterial thrombosis (acute myocardial infarction and ischemic stroke in one case each). Instead, no case of venous thrombosis or significant hemorrhage was reported. Four (10.0%) subjects had died and only in two cases (5.0%) the cause of death was defined as ET-related.
As far as the treatment of these patients is concerned, hydroxycarbamide (20 patients, 50.0%) was the primary cytoreductive agent used, followed by anagrelide (two patients, 5.0%). Sixteen (40.0%) cases were on antiplatelet therapy alone, whereas two (5.0%) patients received no specific therapy.
Cytogenetics
As reported in Table 1, cytogenetic data were available at diagnosis in all patients, with a normal karyotype being reported in most of the cases (33/40, 82.5%). Specifically concerning chromosomal abnormalities, they were more frequently represented by sexual chromosome abnormalities (-Y or -X, 5/7), with +8 detected in the remaining two cases. No complex or monosomal karyotype was reported. Interestingly, the single case who evolved into MDS showed a normal karyotype at diagnosis and maintained it during follow-up.
Molecular Characterization
As detailed in Figure 1 and Supplementary Table 2, no variants were detected in five (12.5%) cases, with the remaining 35 (87.5%) patients harboring at least one likely damaging variant (i.e., VUS excluded). In detail, most of the subjects (29/40, 72.5%) harbored one or two mutations, whereas the remaining six (15.0%) showed three or more concomitant variants. No specific mutational pattern was identified. The most frequent sequence variants involved the TET2 gene (22/40, 55.0%), followed by KIT (11/40, 27.5%). Other variants detected in at least one case also include EZH2, RILP/PRPF8, IDH1, ASXL1, IKZF1, RUNX1, RB1, ETV6, and STAG2. Notably, VAF was observed in a 0.03/0.98 range, as detailed in Supplementary Table 2.
Figure 1
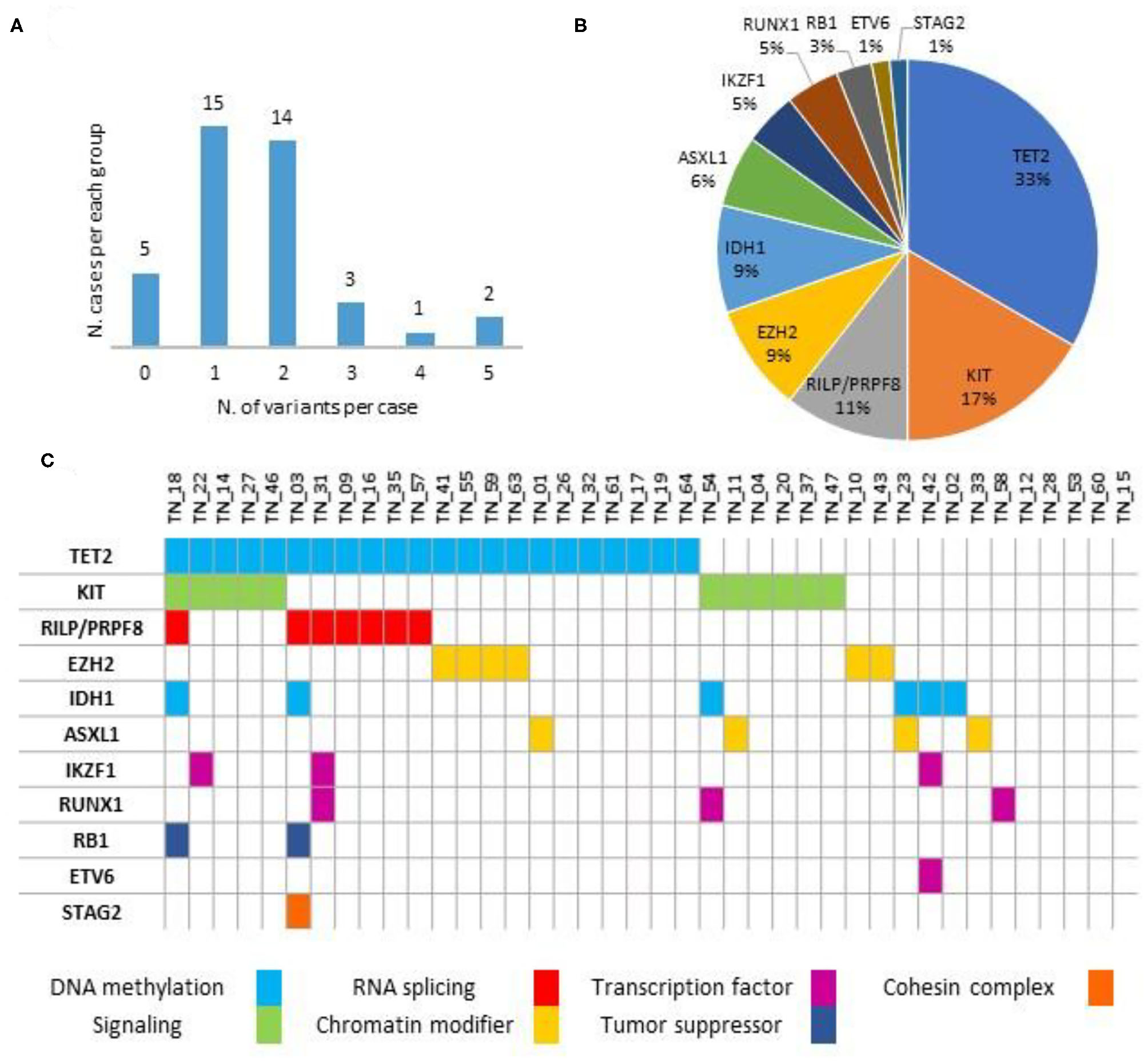
Variants distribution (A), relative frequencies of pathogenic variants (B) and mutational spectrum (C) along the series (n = 40).
Histopathologic Characterization
Morphologic features are detailed in Table 2 and Figure 2. In brief, most of the cases displayed a normocellular BM with increased number of megakaryocytes with the classical giant forms with hyperlobulated (“staghorn”) nuclei.
Table 2
| Histologic features | N. cases (%) (n = 40) | Frequency of features in ET according to WHO 2017 |
|---|---|---|
| Cellularity | ||
| Age related increase | 5/40 (12.5%) | 10–19% |
| Granulopoiesis | ||
| Increased in quantity | 3/40 (7.5%) | <10% |
| Left shifted | 5/40 (12.5%) | <10% |
| Erythropoiesis | ||
| Increased in quantity | 10/40 (25%) | <10% |
| Left shifted | 16/40 (40%) | <10% |
| Myeloid erythroid ratio | ||
| <3:1 | 13/40 (32.5%) | NA |
| =3:1 | 26/40 (65%) | |
| >3:1 | 1/40 (2.5%) | |
| Megakaryopoiesis | ||
| Increased in quantity | 40/40 (100%) | >80% |
| Clusters | ||
| Small cluster (>3 cells) | 40/40 (100%) | 10–19% |
| Large cluster (>7 cells) | 0/40 (0%) | 0% |
| Loose cluster | 40/40 (100%) | 20–49% |
| Dense cluster | 11/40 (27.5%) | <5% |
| Size of cells | ||
| Normal/small | 37/40 (92.5%) | <5% |
| Giant | 40/40 (100%) | 20–49% |
| Morphology of cells | ||
| Hyperlobulated nuclei | 40/40 (100%) | 50–80% |
| Cloud-like/bulbous nuclei | 2/40 (5%) | <10% |
| Naked nuclei | 17/40 (42.5%) | 20–49% |
| Maturation defects | 4/40 (10%) | 0% |
| Prevalent morphology | ||
| Giant, hyperlobulated | 17/40 (42.5%) | NA |
| Polymorphic | 23/40 (57.5%) | NA |
| Fibrosis (WHO) | ||
| MF 0 | 34/40 (85%) | <5% |
| MF 1 | 5/40 (12.5%) | 0% |
| MF 2-3 | 0/40 (0%) | 0% |
| Osteosclerosis | 0/40 (0%) | |
| CD34+ vessels | ||
| Increased density | 5/40 (12.5%) | NA |
| Ectasic vessels | 16/40 (40%) | NA |
| Endoluminal hemopoiesis | 1/40 (2.5%) | NA |
| Lymphoid aggregates | ||
| Present | 8/40 (20%) | <5% |
Histologic features of 40 triple-negative ET patients.
ET, essential thrombocythemia; NA, not assessed.
Figure 2
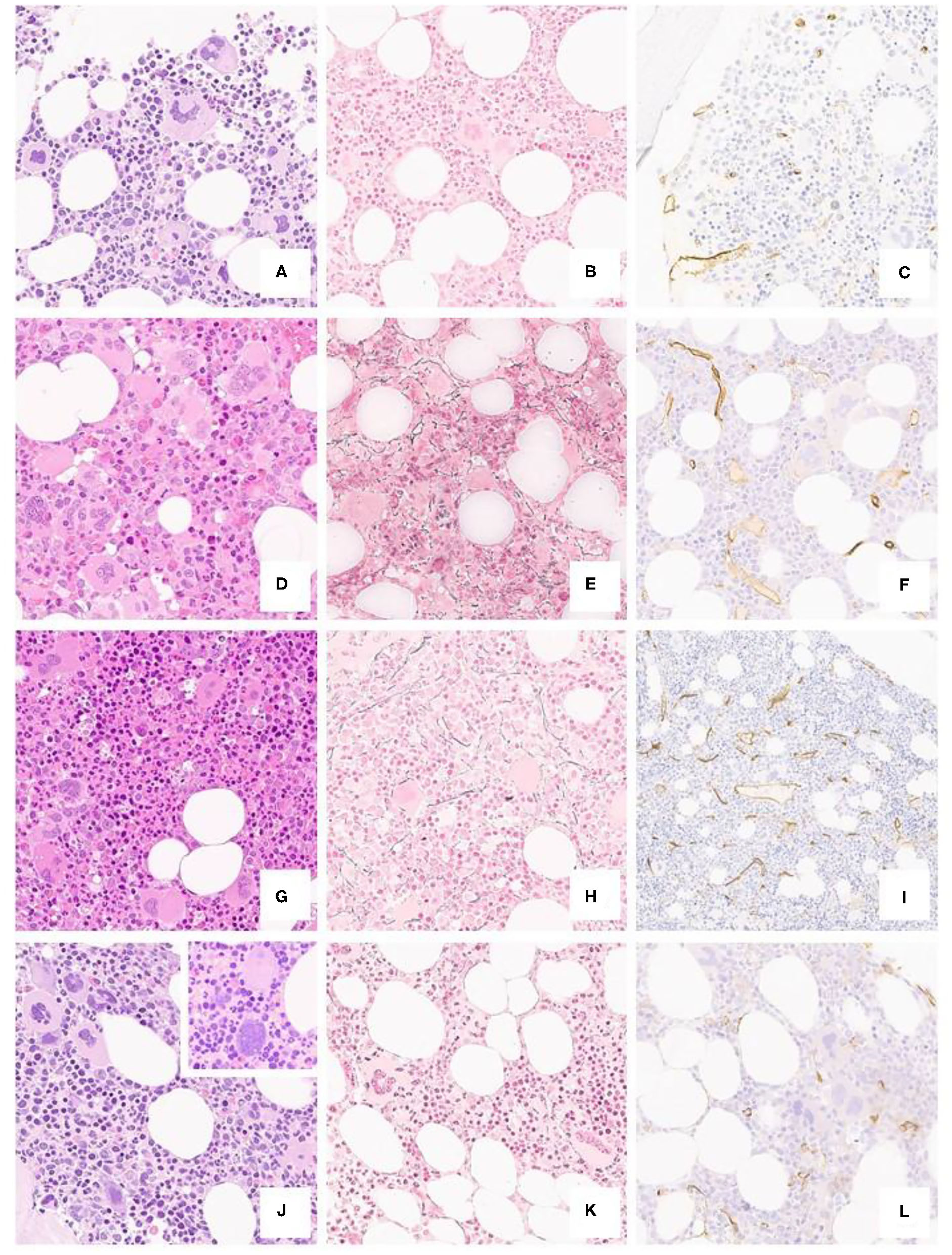
Typical ET case from a 40-year-old patient (a H/E, 20x), featuring a polymorphic spectrum of megakaryocytes, but with giant, hyperlobated forms and scant naked nuclei, MF-0 fibrosis (b Gomori, 20x) and unremarkable CD34+ blasts and vessels (c CD34, 20x). BM biopsy from a 35-year-old subject (d H/E, 20x) featuring mild hypercellularity, increased, left-shifted erythropoiesis with proerythroblasts and absolute predominance of giant, hyperlobated megakaryocytes; there is only a minor increase in reticulin fibers (e Gomori, 20x) and vessel density (f CD34, 20x). A further case from a 31-year-old patient shows mild panmyelosis (g H/E, 20x) with polymorphic megakaryocyte clusters, but featuring ET-like giant forms, along with dim reticulin fibers increase (h Gomori, 20x) and mild micro-vessel proliferation (i CD34, 20x). The last panel from a 52-year-old individual depicts a normocellular bone marrow (j H/E, 20x) with clustering of predominantly hyperlobulated megakaryocytes, with scant bulbous forms (inset) or with maturation defects, with unremarkable fibrosis and CD34+ positive cells (k Gomori, 20x; l CD34, 20x).
In 10 (25%) out of 40 patients the erythropoietic series resulted variably increased with some degree of left-shifting (16/40, 40%) and a reduction of the myeloid/erythroid ratio (13/40, 32.5%). On the contrary, a slight increase of the granulopoietic series was found in three cases (3/40, 7.5%) with left-shifting (5/40, 12.5%).
Megakaryocytes prevalent morphology was polymorphic in more than half of the subjects (23/40, 57.5%) with a mixture of giant forms with hyperlobulated nuclei (40/40, 100%), normal and small sized maturing elements (37/40, 92.5%) and naked nuclei (17/40, 42.5%). In two cases (2/40, 5%) megakaryocytes displayed bulbous (“cloud-like”) nuclei.
Finally, in five cases (5/40, 12.5%) a mild degree of reticulin fibrosis (MF-1) was evident together with an increase of the micro-vessel density (6/40, 16%).
Discussion
In this real-life, single-center cohort study, we investigated the mutational profiles and reviewed the clinical data and BM biopsies of 40 consecutive triple-negative ET patients with a median follow-up of 7 years.
From a strictly clinical point-of-view, we first confirmed that triple-negative ET is a very indolent disease, with a low incidence of vascular events as only two patients suffered from thrombosis. Furthermore, no case of fibrotic progression or leukemic evolution was registered, thus confirming the different behavior of this condition when compared with triple-negative PMF. Interestingly, MDS evolution was diagnosed in one (2.5%) patient after more than 32 years of follow-up, underlying therefore the need of a prompt recognition of different types of disease progression in MPNs (41).
It is worth noting that at least one non-driver nucleotide variant predicted as damaging by in silico tools was found in most subjects, the most frequently detected variants involving the TET2 gene (22/40 patients, 55.0%). This finding is in line with a recent report on the occurrence and prognostic relevance of DNA sequence variants/mutations other than JAK2/CALR/MPL in both PV and ET (42, 43). Indeed, NGS revealed that 53% of 183 Mayo Clinic ET patients harbored one or more sequence variants/mutations other than JAK2/CALR/MPL, being TET2 and ASXL1 the most frequent ones.
Interestingly, some of these variants are reported at high VAF in the literature, in a non-negligible fraction of cases within large cohort studies focusing on the topic of triple-negative MPNs (44) and clonal hematopoiesis of indeterminate potential (CHIP) (45, 46). In accordance with these data, in our cohort we also found a high VAF, with a mean value of 43%. Interestingly, with the notable differences of thrombocytosis and a lower median age, our patients share with CHIP individuals an unremarkable clinical history, with a very indolent disease, and variants affecting genes regarded as initiator of clonal expansion (e.g., TET2 or ASXL1), as well as genes with higher impact on disease progression (e.g., RUNX1). On the contrary, CHIP-associated DNMT3A variants were not recorded in our series. From a speculative point of view, in analogy with CHIP, triple-negative ET patients may constitute a very specific subset of individuals featuring low-impact laboratory abnormalities. Furthermore, it may be argued that at least some of the variants identified by our analysis may represent mere polymorphisms, without a phenotypic effect. However, we report an enhanced frequency of variants such as TET2 c.5162T>G (p.L1721W), which features a MAF = 0.11 but it is observed in 27% of our patients, suggesting a non-casual association, though basing on low numbers. The same TET2 variant is also predicted as damaging according to most predictive tools. Furthermore, we report a few variants, such as RB1 c.929G>A (p.G310E), which does not represent a polymorphism (MAF = 0.0003) but features high VAF. As to the interpretation of molecular findings, we deal with the limitations of an in silico approach for assessment of pathogenicity or protein function damage, as we relied on databases for clinical prediction of current use in molecular diagnostics. In absence of functional studies to prove the real biological impact of such variants, their finding may be helpful to determine the clonal nature of disease in triple-negative MPNs and complement the morphological criteria (47). This stands also in accordance with the revised 2017 WHO criteria.
Thanks to the NGS approach, we could reveal the presence of non-canonical variants. Indeed, KIT mutations were detected in more than one third of patients (27.5%), apparently overrepresented than usually reported in MPNs, although none harbored the canonical D816V variant. Moreover, in four cases we found the c.1621A>C (M541L) variant of KIT, which has been shown to be associated with chronic eosinophilic leukemia, not otherwise specified (48).
Specifically concerning mutations in genes which are already well-known to be associated with poor prognosis in other MPNs (49), particularly in PMF, all ASXL1-mutated cases have a BM biopsy showing histological features typical for ET; with regards to the six IDH1-mutated subjects, one displayed BM fibrosis grade 1. As far as their biologic significance, ASXL1 mutation is recognized within the so-called CHIP, as early initiator of a clonal expansion (50, 51) and as favoring a state of thrombocytosis (52). Furthermore, it should be highlighted that none of our cases harbored variants (affecting TP53, SRSF2 and/or TET2) previously reported to impact on leukemic evolution of MPNs (53).
Interestingly, in all cases a cytogenetic analysis was performed at diagnosis and chromosomal abnormalities were detected in seven (17.5%) patients, most being, however, represented by sexual chromosome abnormalities (-Y or -X, 5/7), which cannot be considered as a marker of disease. Considering instead the two patients with the +8 detected, we can speculate that this chromosomal alteration may help in resolving clonality, even though not representing a chromosomal abnormality typical of ET, as it can be detected also in other myeloid neoplasms, including MPNs (i.e., both PMF and PV), as well as MDS or acute myeloid leukemia (40).
On the contrary, NGS provided a relevant contribution to the diagnosis of MPNs; its use should therefore be implemented in clinical practice, as it also allows the detection of novel variants.
As far as the histologic features of our series are concerned, most of the cases showed a classic ET picture characterized by a normocellular BM with an increased number of large to giant megakaryocytes with hyperlobulated nuclei forming loose clusters while about one fourth of the patients displayed a variable increase of the erythropoietic series with left-shifting and reduced myeloid/erythroid ratio. Furthermore, in about 12% of the cases a mild increment of BM fibrosis with increased micro-vessel density was identified. These histopathologic features, identified singularly, do not inhibit a clinical-pathological diagnosis of ET according to the WHO classification. However, they raise the concern of a more appropriate classification of those patients, questioning whether subjects with triple-negative, persistent thrombocytosis, but lacking the morphological features of PV or pre-fibrotic PMF, should be classified as ET or attributed to the myeloproliferative neoplasms, unclassifiable (MPN, U) category to be followed over time and classified according to the clinical evolution and any progressive BM changes (54, 55).
Finally, it is important to note that in the one single case progressed toward MDS after 32 years of follow-up, mutations of predicted pathogenic significance were found by NGS for ASXL1 and KIT genes, whereas the BM morphology, alongside with the classical ET thrombocytopoiesis, displayed also features of hypercellularity and left-shifted erythropoiesis and presence of megakaryocytes with maturation defects.
We are aware that the main limitations of the present study are represented by its retrospective nature and by the relatively small sample size, preventing us from drawing any definitive conclusion on the prognostic impact of the molecular profile in this specific subgroup of patients suffering from an indolent disease. Further limits include sequencing of JAK2 and MPL hotspot regions, which may certainly impair the capability of detection of novel variants of driver genes (24, 25) and the sensitivity of RQ-PCR and NGS, which may have missed mutations in driver genes at very low VAF. Finally, in absence of matched germline DNA in most of the patients, we cannot reliably rule out the alternative hypothesis of somatic vs. germline variants.
It should be stressed that in the present study a triple-negative definition was initially applied in the context of a molecular characterization performed on a routine basis: when using an extended NGS panel, the clonal nature of hematopoiesis has then been demonstrated in the majority of the cases by identifying sequence variants/mutations affecting other genes involved in myeloid malignancies. In this way, we could demonstrate that a sizeable proportion of triple-negative ET patients do have a clonal disease, thus preventing issues arising concerning their treatment, especially when a cytoreductive therapy may be warranted. Finally, our results stress the pivotal role of strict adherence to histopathologic criteria, with the prime importance given to a thorough assessment of megakaryocyte morphology, to identify those patients that, besides a formal diagnosis of triple-negative ET vs. MPN, U, can be expected to portend an indolent clinical course.
Statements
Data availability statement
The original contributions presented in the study are included in the article as Supplementary Table 1. In addition, COSMIC - Catalogue Of Somatic Mutations in Cancer (RRID:SCR_002260) and dbSNP - Database of Single Nucleotide Variations (RRID:SCR_002338) were used to annotate each sequence variant.
Ethics statement
This study was carried out in accordance with the recommendations of Good Clinical Practice. At their first admission to our Hospital, all subjects gave informed consent to retrospectively collect clinical and laboratory data, in accordance with the Declaration of Helsinki. Accordingly, a new submission to the ethics committee for the present study was not required.
Author contributions
DC, GAC, UG, and AI interpreted the data and prepared the text. GAC and UG reviewed bone marrow biopsies. ST, PB, EF, and SF carried out the mutational analyses. DC, AI, CB, MB, and KB followed the patients and collected the data. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.637116/full#supplementary-material
References
1.
Tefferi A Vardiman JW . Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. (2008) 22:14–22. 10.1038/sj.leu.2404955
2.
Tefferi A . Myeloproliferative neoplasms 2012: the John M. Bennett 80th birthday anniversary lecture. Leuk Res. (2012) 36:1481–9. 10.1016/j.leukres.2012.08.011
3.
Moulard O Mehta J Fryzek J Olivares R Iqbal U Mesa RA . Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur J Haematol. (2014) 92:289–97. 10.1111/ejh.12256
4.
Srour SA Devesa SS Morton LM Check DP Curtis RE Linet MS et al . Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms in the United States, 2001-12. Br J Haematol. (2016) 174:382–96. 10.1111/bjh.14061
5.
Arber DA Orazi A Hasserjian R Thiele J Borowitz MJ Le Beau MM et al . The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–405. 10.1182/blood-2016-03-643544
6.
Kralovics R Passamonti F Buser AS Teo SS Tiedt R Passweg JR et al . Gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. (2005) 352:1779–90. 10.1056/NEJMoa051113
7.
Baxter EJ Scott LM Campbell PJ East C Fourouclas N Swanton S et al . Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. (2005) 365:1054–61. 10.1016/S0140-6736(05)71142-9
8.
Tefferi A . JAK2 mutations and clinical practice in myeloproliferative neoplasms. Cancer J. (2007) 13:366–71. 10.1097/PPO.0b013e318159467b
9.
Levine RL Pardanani A Tefferi A Gilliland DG . Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. (2007) 7:673–83. 10.1038/nrc2210
10.
Vannucchi AM Antonioli E Guglielmelli P Rambaldi A Barosi G Marchioli R et al . Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. (2007) 110:840–6. 10.1182/blood-2006-12-064287
11.
Vannucchi AM Antonioli E Guglielmelli P Pardanani A Tefferi A . Clinical correlates of JAK2V617F presence or allele burden in myeloproliferative neoplasms: a critical reappraisal. Leukemia. (2008) 22:1299–307. 10.1038/leu.2008.113
12.
Passamonti F Rumi E . Clinical relevance of JAK2 (V617F) mutant allele burden. Haematologica. (2009) 94:7–10. 10.3324/haematol.2008.001271
13.
Carobbio A Finazzi G Antonioli E Guglielmelli P Vannucchi AM Dellacasa CM et al . JAK2V617F allele burden and thrombosis: a direct comparison in essential thrombocythemia and polycythemia vera. Exp Hematol. (2009) 37:1016–21. 10.1016/j.exphem.2009.06.006
14.
Tefferi A Barbui T . Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk-stratification and management. Am J Hematol. (2020) 95:1599–613. 10.1002/ajh.26008
15.
Pikman Y Lee BH Mercher T McDowell E Ebert BL Gozo M et al . MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. (2006) 3:e270. 10.1371/journal.pmed.0030270
16.
Pardanani AD Levine RL Lasho T Pikman Y Mesa RA Wadleigh M et al . MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. (2006) 108:3472–76. 10.1182/blood-2006-04-018879
17.
Klampfl T Gisslinger H Harutyunyan AS Nivarthi H Rumi E Milosevic JD et al . Somatic mutations of calreticulin in myeloproliferative neoplasms. N Eng J Med. (2013) 369:2379–90. 10.1056/NEJMoa1311347
18.
Nangalia J Massie CE Baxter EJ Nice FL Gundem G Wedgeet DC et al . Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Eng J Med. (2013) 369:2391–405. 10.1056/NEJMoa1312542
19.
Gangat N Wassie E Lasho T Finke C Ketterling RP Hanson CA et al . Mutations and thrombosis in essential thrombocythemia: prognostic interaction with age and thrombosis history. Eur J Haematol. (2015) 94:31–6. 10.1111/ejh.12389
20.
Rumi E Pietra D Ferretti V Klampfl T Harutyunyan AS Milosevic JD et al . JAK2 or CALR mutation status defines subtypes of essential thrombocytemia with substantially different clinical course and outcomes. Blood. (2014) 123:1544–51. 10.1182/blood-2013-11-539098
21.
Rotunno G Mannarelli C Guglielmelli P Pacilli A Pancrazzi A Pieri L et al . Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood. (2014) 123:1552–5. 10.1182/blood-2013-11-538983
22.
Chen CC Gau JP Chou HJ You JY Huang CE Chen YY et al . Frequencies, clinical characteristics, and outcome of somatic CALR mutations in JAK2-unmutated essential thrombocythemia. Ann Hematol. (2014) 93:2029–36. 10.1007/s00277-014-2151-8
23.
Tefferi A Wassie EA Guglielmelli P Gangat N Belachew AA Lasho TL et al . Type 1 versus Type 2 calreticulin mutations in essential thrombocythemia: a collaborative study of 1027 patients. Am J Hematol. (2014) 89:E121–4. 10.1002/ajh.23743
24.
Milosevic Feenstra JD Nivarthi H Gisslinger H Leroy E Rumi E Chachoua I et al . Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood. (2016) 127:325–32. 10.1182/blood-2015-07-661835
25.
Cabagnols X Favale F Pasquier F Messaoudi K Defour JP Ianotto JC et al . Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood. (2016) 127:333–42. 10.1182/blood-2015-07-661983
26.
Rumi E Cazzola M . Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood. (2017) 129:680–92. 10.1182/blood-2016-10-695957
27.
Tefferi A Lasho TL Finke CM Knudson RA Ketterling R Hanson CH et al . CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia. (2014) 28:1472–7. 10.1038/leu.2014.3
28.
Szuber N Tefferi A . Driver mutations in primary myelofibrosis and their implications. Curr Opin Hematol. (2018) 25:129–35. 10.1097/MOH.0000000000000406
29.
Barbui T Thiele J Passamonti F Rumi E Boveri E Ruggeri M et al . Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: an international study. J Clin Oncol. (2011) 29:3179–84. 10.1200/JCO.2010.34.5298
30.
Cervantes F Alvarez-Larrán A Talarn C Gómez M Montserrat E . Myelofibrosis with myeloid metaplasia following essential thrombocythaemia: actuarial probability, presenting characteristics and evolution in a series of 195 patients. Br J Haematol. (2002) 118:786–90. 10.1046/j.1365-2141.2002.03688.x
31.
Passamonti F Rumi E Arcaini L Boveri E Elena C Pietra D et al . Prognostic factors for thrombosis, myelofibrosis, and leukemia in essential thrombocythemia: a study of 605 patients. Haematologica. (2008) 93:1645–51. 10.3324/haematol.13346
32.
Kiladjian JJ Rain JD Bernard JF Briere J Chomienne C Fenaux P . Long-term incidence of hematological evolution in three French prospective studies of hydroxyurea and pipobroman in polycythemia vera and essential thrombocythemia. Semin Thromb Hemost. (2006) 32:417–21. 10.1055/s-2006-942762
33.
Michail O McCallion P McGimpsey J Hindley A Greenfield G Feerick J et al . Mutational profiling in suspected triple-negative essential thrombocythaemia using targeted next-generation sequencing in a real-world cohort. J Clin Pathol. (2020). 10.1136/jclinpath-2020-206570. [Epub ahead of print].
34.
Marchioli R Finazzi G Specchia G Cacciola R Cavazzina R Cilloni D et al . Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. (2013) 368:22–33. 10.1056/NEJMoa1208500
35.
Barosi G Mesa RA Thiele J Cervantes F Campbell PJ S Verstovsek S et al . Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia. (2008) 22:437–8. 10.1038/sj.leu.2404914
36.
Swerdlow SH Campo E Harris L et al . WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th edn.Lyon: IARC (2008). p. 127–9.
37.
Barbui T Barosi G Grossi A Gugliotta L Liberato LN Marchetti M et al . Practice guidelines for the therapy of essential thrombocythemia. A statement from the Italian Society of Hematology, the Italian Society of Experimental Hematology and the Italian Group for Bone Marrow Transplantation. Haematologica. (2004) 89:215–32.
38.
Barbui T Barosi G Birgegard G Cervantes F Finazzi G Griesshammer M et al . Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. (2011) 29:761–70. 10.1200/JCO.2010.31.8436
39.
Barbui T Tefferi A Vannucchi AM Passamonti F Silver RT Hoffman R et al . Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet. Leukemia. (2018) 32:1057–69. 10.1038/s41375-018-0077-1
40.
Rack KA van den Berg E Haferlach C Beverloo HB Costa D Espinet B et al . European recommendations and quality assurance for cytogenomic analysis of haematological neoplasms. Leukemia. (2019) 33:1851–67. 10.1038/s41375-019-0378-z
41.
Geyer JT Margolskee E Krichevsky SA Cattaneo D Boiocchi L Ronchi P et al . Disease progression in myeloproliferative neoplasms: comparing patients in accelerated phase with those in chronic phase with increased blasts (<10%) or with other types of disease progression. Haematologica. (2020) 105:e221–4. 10.3324/haematol.2019.230193
42.
Tefferi A Lasho TL Guglielmelli P Finke CM Rotunno G Elala Y et al . Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. (2016) 1:21–30. 10.1182/bloodadvances.2016000216
43.
Tefferi A Guglielmelli P Lasho TL Coltro G Finke CM Loscocco GG et al . Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br J Haematol. (2020) 189:291–302. 10.1111/bjh.16380
44.
Acha P Xandri M Fuster-Tormo F Palomo L Xicoy B Cabezón M et al . Diagnostic and prognostic contribution of targeted NGS in patients with triple-negative myeloproliferative neoplasms. Am J Hematol. (2019) 94:E264–7. 10.1002/ajh.25580
45.
Buscarlet M Provost S Feroz Zada Y Barhdadi A Bourgoin V Lépine G et al . DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood. (2017) 130:753–62. 10.1182/blood-2017-04-777029
46.
Zink F Stacey SN Norddahl GL Frigge ML Magnusson OT Jonsdottir I et al . Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. (2017) 130:742–52. 10.1182/blood-2017-02-769869
47.
Skov V . Next generation sequencing in MPNs. Lessons from the past and prospects for use as predictors of prognosis and treatment responses. Cancers. (2020) 12:2194. 10.3390/cancers12082194
48.
Iurlo A Gianelli U Beghini A Spinelli O Orofino N Lazzaroni F et al . Identification of kit(M541L) somatic mutation in chronic eosinophilic leukemia, not otherwise specified and its implication in low-dose imatinib response. Oncotarget. (2014) 5:4665–70. 10.18632/oncotarget.1941
49.
Iurlo A Cattaneo D Gianelli U . Blast transformation in myeloproliferative neoplasms: risk factors, biological findings, and targeted therapeutic options. Int J Mol Sci. (2019) 20:1839. 10.3390/ijms20081839
50.
Steensma DP Bejar R Jaiswal S Lindsley RC Sekeres MA Hasserjian RP et al . Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. (2015) 126:9–16. 10.1182/blood-2015-03-631747
51.
Fujino T Kitamura T . ASXL1 mutation in clonal hematopoiesis. Exp Hematol. (2020) 83:74–84. 10.1016/j.exphem.2020.01.002
52.
Veninga A De Simone I Heemskerk JWM Cate HT van der Meijden PEJ . Clonal hematopoietic mutations linked to platelet traits and the risk of thrombosis or bleeding. Haematologica. (2020) 105:2020–31. 10.3324/haematol.2019.235994
53.
Venton G Courtier F Charbonnier A D'incan E Saillard C Mohty B et al . Impact of gene mutations on treatment response and prognosis of acute myeloid leukemia secondary to myeloproliferative neoplasms. Am J Hematol. (2018) 93:330–8. 10.1002/ajh.24973
54.
Gianelli U Cattaneo D Bossi A Cortinovis I Boiocchi L Liu YC et al . The myeloproliferative neoplasms, unclassifiable: clinical and pathological considerations. Mod Pathol. (2017) 30:169–79. 10.1038/modpathol.2016.182
55.
Iurlo A Gianelli U Cattaneo D Thiele J Orazi A . Impact of the 2016 revised WHO criteria for myeloproliferative neoplasms, unclassifiable: Comparison with the 2008 version. Am J Hematol. (2017) 92:E48–51. 10.1002/ajh.24657
Summary
Keywords
essential thrombocytemia, triple-negative, bone marrow morphology, next-generation sequencing, prognosis
Citation
Cattaneo D, Croci GA, Bucelli C, Tabano S, Cannone MG, Gaudioso G, Barbanti MC, Barbullushi K, Bianchi P, Fermo E, Fabris S, Baldini L, Gianelli U and Iurlo A (2021) Triple-Negative Essential Thrombocythemia: Clinical-Pathological and Molecular Features. A Single-Center Cohort Study. Front. Oncol. 11:637116. doi: 10.3389/fonc.2021.637116
Received
02 December 2020
Accepted
18 February 2021
Published
12 March 2021
Volume
11 - 2021
Edited by
Bing Xu, Xiamen University, China
Reviewed by
Stephen Langabeer, St. James's Hospital, Ireland; Barbara Mora, ASST Sette Laghi, Italy; Caleb Gonshen Chen, Mackay Memorial Hospital, Taiwan; Emanuela Sant'Antonio, Azienda USL Toscana Nord Ovest, Italy
Updates
Copyright
© 2021 Cattaneo, Croci, Bucelli, Tabano, Cannone, Gaudioso, Barbanti, Barbullushi, Bianchi, Fermo, Fabris, Baldini, Gianelli and Iurlo.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniele Cattaneo daniele.cattaneo@policlinico.mi.it
This article was submitted to Hematologic Malignancies, a section of the journal Frontiers in Oncology
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.