 Nathalie Faumont1
Nathalie Faumont1 Oussama Taoui1
Oussama Taoui1 Davi Collares2
Davi Collares2 Jean-Philippe Jais3Karen Leroy4
Jean-Philippe Jais3Karen Leroy4 Léa Prévaud1
Léa Prévaud1 Fabrice Jardin5Thierry J. Molina2,6Christiane Copie-Bergman7Barbara Petit8Marie-Pierre Gourin9Dominique Bordessoule1,9Danielle Troutaud10
Fabrice Jardin5Thierry J. Molina2,6Christiane Copie-Bergman7Barbara Petit8Marie-Pierre Gourin9Dominique Bordessoule1,9Danielle Troutaud10 Véronique Baud2*
Véronique Baud2* Jean Feuillard1*
Jean Feuillard1*- 1CNRS UMR-7276, INSERM U1262, CRIBL, University of Limoges, and Hematology Laboratory of Dupuytren Hospital University Center (CHU) of Limoges, Limoges, France
- 2Université de Paris, NF-κappaB, Differentiation and Cancer, Paris, France
- 3Biostatistics Department, Imagine Institute, Paris, France
- 4UMRS1138, Centre de Recherche des Cordeliers, Paris Descartes University, CARPEM, Department of Genetics and Molecular Biology, Cochin Hospital, Assistance Publique Hôpitaux de Paris, Paris, France
- 5Inserm U1245 and Department of Henri-Becquerel Hematology Center and Normandie Univ UNIROUEN, Rouen, France
- 6Pathology Department, Necker Hospital, Assistance Publique Hôpitaux de Paris (AP-HP), Paris, France
- 7IMRB-Inserm U955, AP-HP Henri-Mondor Hospital, Créteil, France
- 8Pathology Department, CHU of Limoges, Limoges, France
- 9Regional Reference Structure of Limousin Lymphomas, Clinical Hematology Department, CHU of Limoges, Limoges, France
- 10EA3842, CAPTuR, University of Limoges, Limoges, France
Relationships between c-Rel and GCB-DLBCLs remain unclear. We found that strong c-Rel DNA-binding activity was mostly found in GCBs on two independent series of 48 DLBCLs and 66 DLBCLs, the latter issued from the GHEDI series. c-Rel DNA-binding activity was associated with increased REL mRNA expression. Extending the study to the whole GHEDI and Lenz DLBCL published series of 202 and 233 cases, it was found that the c-Rel gene expression profile (GEP) overlapped partially (12%) but only with the GCB GEP and not with the GEP of ABC-DLBCLs. Cases with both overexpression of REL mRNA and c-Rel GEP were defined as those having a c-Rel signature. These cases were GCBs in 88 and 83% of the GHEDI or Lenz’s DLBCL series respectively. The c-Rel signature was also associated with various recurrent GCB-DLBCL genetic events, including REL gains, BCL2 translocation, MEF2B, EZH2, CREBBP, and TNFRSF14 mutations and with the EZB GCB genetic subtype. By CGH array, the c-Rel signature was specifically correlated with 2p15-16.1 amplification that includes XPO1, BCL11A, and USP34 and with the 22q11.22 deletion that covers IGLL5 and PRAME. The total number of gene copy number aberrations, so-called genomic imbalance complexity, was decreased in cases with the c-Rel signature. These cases exhibited a better overall survival. Functionally, overexpression of c-Rel induced its constitutive nuclear localization and protected cells against apoptosis while its repression tended to increase cell death. These results show that, clinically and biologically, c-Rel is the pivotal NF-κB subunit in the GCB-DLBCL subgroup. Functionally, c-Rel overexpression could directly promote DLBCL tumorigenesis without need for further activation signals.
Introduction
Diffuse large B-cell lymphomas (DLBCL) are the most frequent among aggressive non-Hodgkin’s lymphomas (NHLs). DLBCLs are clinically, molecularly, and genetically heterogeneous, suggesting different oncogenic mechanisms. Gene-expression profiling (GEP) studies led to the proposal of two cell-of-origin (COO) molecular subtypes known as germinal center B-cell-like (GCB) DLBCLs, and activated B-cell like (ABC) DLBCLs, with a subset of cases showing an intermediate, unclassifiable phenotype (1). GCB-DLBCLs seem to arise from normal germinal center B-cells, whereas ABC-DLBCLs would originate from post-germinal center activated B-cells arrested before terminal plasma cell differentiation.
When compared to GCB-DLBCLs, several studies have shown that ABC-DLBCLs are the more aggressive subtype with the worst patient outcome (2, 3). ABC-DLBCLs exhibit constitutive activation of the NF-κB pathway that drives tumor proliferation and survival and confers chemotherapy resistance (4). Constitutive activation of NF-κB in ABC-DLBCLs is due to a variety of mutations in NF-κB regulator coding genes, such as MYD88, TNFAIP3 (A20), CD79A/B, CARD11, TRAF2, TRAF5, MAP3K7 (TAK1), or TNFRSF11A (RANK) (5).
In contrast to ABC-DLBCLs, GCB-DLBCLs do not exhibit a NF-κB transcriptomic signature (1). GCB-DLBCL is the most frequent DLBCL subtype and, with primary mediastinal B-cell lymphomas (PMBLs), represents almost all DLBCL cases diagnosed in children, adolescents, and young adults (6). Genetic abnormalities strongly associated with GCB-DLBCLs result in epigenetic modifications such as histone methylation or acetylation (EZH2, EP300, CREBBP, KMT2D), B-cell migration (GNA13, GNAI2, SIPR2), PI3K/AKT/mTOR pathway activation (FOXO1, SGK1, PTEN), and immune-regulation (TNFRSF14) (5). Additionally, the t(14;18) translocation that brings BCL2 under the control of IGH locus regulatory regions is detected in about 30% of GCB-DLBCLs but is rarely found in ABC-DLBCLs (7). Despite being reported at variable frequencies ranging 15 to 37%, the REL locus (2p16 region) gains including amplification (≥four copies) are among the most frequently observed alterations in the GCB subtype (8). REL gains are also recurrent gene abnormalities in other hematopoietic cancers such as classical Hodgkin’s lymphoma, follicular lymphoma, MALT lymphoma, Burkitt’s lymphoma, and PMBL.
The REL gene encodes the c-Rel subunit of NF-κB. c-Rel was first identified as a cellular homolog of the avian retroviral oncoprotein v-Rel, which rapidly causes fatal lymphoid cell tumors in young birds (9). Like the other members of the NF-κB family, c-Rel is characterized by the presence of a highly conserved amino-terminal domain so called RHD (Rel Homology Domain) responsible for dimerization, DNA-binding, and IκB inhibitory protein binding. c-Rel is the only member of the NF-κB family that can transform avian lymphoid cells in vitro (10). Artificial mutants of c-Rel indicate that the transactivation domain in the carboxy-terminal part is responsible for this transforming potential (11).
The role of c-Rel is important in many aspects of lymphoid cell function. Expression of c-Rel is ubiquitous in B-cells and increases during B-cell development, particularly in germinal center B-cells (12). Experimental ectopic c-Rel expression blocks plasma cell differentiation by inhibiting the expression of the transcription factor Blimp1 (13). Reciprocally, c-Rel is repressed by Blimp1 through its binding to the REL locus when the plasma cell differentiation process is engaged (13). Transgenic mice lacking c-Rel develop normally without effects on hematopoietic bone marrow development, but exhibit numerous peripheral immunological defects, including reduced proliferation and activation of mature B-cells in response to mitogenic stimuli, impaired germinal center formation, and reduced numbers of B-cells in the marginal area (14). Furthermore, inhibition of c-Rel by small hairpin RNA results in reduced cell survival and cell cycle progression in murine lymphoma B-cell lines (15). Recently, establishment of a B-cell specific c-Rel overexpressing mouse model demonstrated that c-Rel led to expansion of germinal center B-cells (16). All these data underscore a direct link between c-Rel and B-cells especially those of the germinal center.
Despite its association with REL amplification, it is notorious that GCB-DLBCLs do not exhibit NF-κB signature. This paradox led to question the functional relationship between c-Rel and GCB-DLBCLs. To shed light on the role of c-Rel in these tumors, we analyzed c-Rel DNA-binding activity in DLBCLs together with the gene expression profile of tumors, patient survival, genetic abnormalities, and REL imbalances. The role of c-Rel on apoptosis was functionally evaluated in vitro.
Materials and Methods
Tumors and Patient Cohorts for EMSA Studies, Transcriptomic and Overall Survival Analyses
Clinical data of the patients studied are presented in Supplementary Tables S1 and S2 and in Supplementary Materials and Methods. Tumors were diagnosed according to the World Health Organization classification (17). The study was performed with approval of an Institutional review board and written informed consent was obtained from all participants at the time of enrollment. All tumor samples were reviewed for tumor infiltration, and selected cases were those for which tumor infiltration was over 90%. EMSA and transcriptomic analysis (HGU133 +2.0 Affymetrix Gene Chip microarray; accession number GSE87371; Supplementary Table S3) were performed on frozen material (see Supplementary Materials and Methods for details).
Survival and Cox multivariate analysis was done with the Survival package (URL: https://github.com/therneau/survival). Overall survival (OS) was evaluated from the date of enrollment to the date of death from any cause. C-Harrel concordance index was calculated as described (18). Details are given in the Supplementary Materials and Methods.
Real-Time Quantitative Reverse-Transcription PCR
Quantification of REL mRNA was done after extraction of total RNA from frozen biopsies using the TaqMan® Universal PCR Master Mix and TaqMan® Gene Expression Assays: REL, Hs00968436_m1; and HPRT1, Hs02800695_m1. The HPRT1 gene was used as a reference gene for the control of amplification (details are in Supplementary Materials and Methods).
Analysis of Gene Copy Number Aberrations; BCL2, BCL6, and MYC Translocations; and Oncogenic-Related Mutations
Gene copy number aberrations were identified by performing Comparative Genomic Hybridization (CGH) on 180 patients after whole-genome amplification, using Agilent SurePrint G3 4 × 180 K microarrays, and analysis was done with GISTIC version 2.0.22 as previously described (19). Genomic imbalance complexity was calculated as the number of all gene loci for which the GISTIC score was −2 (homozygous deletion) or +2 (copy number gain of 2+ copies).
Fluorescence in situ hybridization (FISH) for BCL2, BCL6, and MYC translocations was performed as previously detailed (19). Probes are listed in the Supplementary Materials and Methods.
The Lymphopanel was designed to identify mutations in 34 important lymphomagenesis genes as detailed previously (19). NGS data was available for 213 patients. Ion Torrent Personal Genome Machine (PGM) Sequencing and PGM data analysis were performed as previously described (19).
Eukaryote Expression Vectors and Cell Lines
Complementary DNAs (cDNAs) for REL, super-repressor IκBαS32,36A,12, IκBε, and Luciferase were cloned into the previously described pRT-1 doxycycline-inducible episomal vector (20). The inducible bidirectional promoter allows concomitant expression of the cDNA of interest and a membrane marker, truncated NGFR (NGFRt). The GCB SUDHL-4 cell line is from the L Staudt group (4). The cell cycle arrested EREB2–5 cell line is described in the Supplementary Materials and Methods and has been published (21). Cytoplasm and nuclear extracts and immunoblot were performed as previously described (20). Antibodies and apoptosis experiments are described in the Supplementary Materials and Methods.
Results
DNA-Binding Activity of c-Rel Was Related to GCB-DLBCLs
We studied the REL mRNA expression and DNA-binding activity on a test series of 48 DLBCLs classified as GCB (30/48) or non-GCB (18/48) according to the Hans’s algorithm (see Supplementary Figure S1 for the analysis pipeline). As shown in Figure 1A, REL mRNA levels tended to be higher in GCB-DLBCLs (Mann–Whitney Test, p = 0.0016). EMSA with super-shift was performed to assess the RelA and c-Rel DNA-binding activities for 32 frozen tumor samples (19 GCBs and 13 non-GCBs; Figure 1B as a typical result). A high c-Rel DNA-binding activity was defined as a c-Rel DNA-binding higher than that of RelA in gel shift assays. Using this criterion, patients were divided into those with a high and a low or negative c-Rel DNA-binding activity. High c-Rel DNA-binding activity was more frequently found in GCB-DLBCLs when compared to non-GCB subgroup (Figure 1C, Fisher’s Exact Test, p = 0.0497). Cases with strong c-Rel DNA-binding were those with increased REL mRNA expression (Figure 1D, Mann–Whitney Test, p < 0.0001).
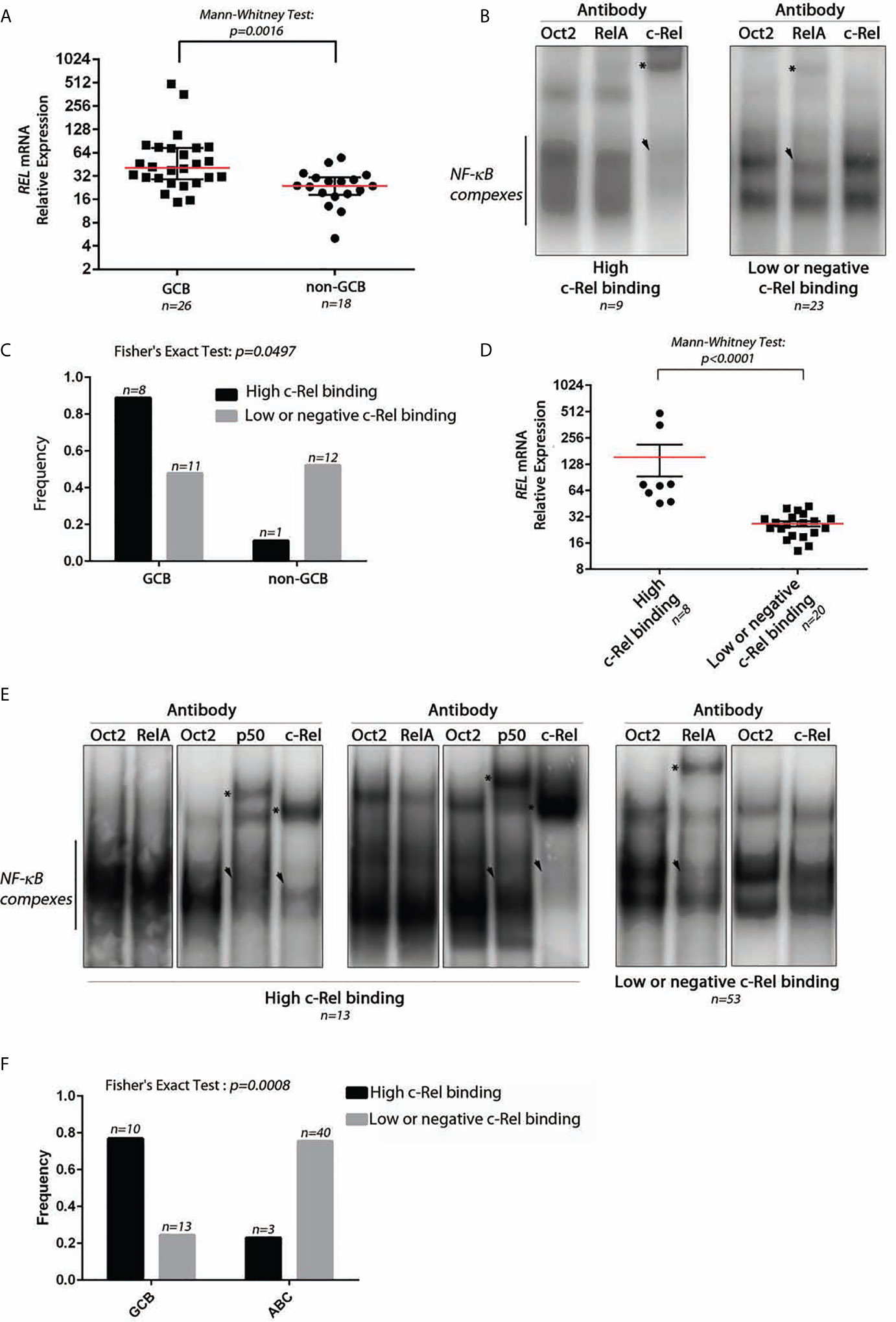
Figure 1 Expression and DNA-binding activity of c-Rel in DLBCLs. (A) Relative expression level of REL mRNA using TaqMan quantitative PCR from 26 GCB-DLBCL and 18 non-GCB-DLBCL samples from a test series. The red line indicates the median with interquartile range. The approximate p-value of a Mann–Whitney test is indicated. (B) Two representative EMSAs showing NF-κB complex DNA-binding on a κB binding site containing probe from DLBCL cases with high c-Rel binding (left) and low or negative c-Rel binding (right). n indicates the number of cases with the same DNA-binding profile. The use of anti-RelA, anti-c-Rel, or anti-Oct2 (irrelevant control) antibodies is noted at the top. Asterisks point on the super-shifted EMSA signal. Arrowhead indicates decreased NF-κB complex DNA-binding. (C) Frequencies of high c-Rel binding and low or negative c-Rel binding among GCB-DLBCLs and non-GCB-DLBCLs. (D) Relative expression levels of REL mRNA using TaqMan quantitative PCR from eight high c-Rel binding DLBCLs and 20 low or negative c-Rel binding DLBCLs. The red line indicates the median with interquartile range. The approximate p-value of a Mann–Whitney test is indicated. (E) DNA-binding activity of c-Rel from the training cohort of the GHEDI (Dubois) DLBCL series. EMSAs with supershift experiments (anti-Oct2, RelA, p50, or c-Rel antibodies) were done with proteins extracted from 66 frozen biopsies from 43 ABC-DLBCLs and 23 GCB-DLBCLs. Representative EMSAs showing NF-κB complex DNA-binding on a κB binding site containing probe from DLBCL cases with high c-Rel binding (left) and low or negative c-Rel binding (right). n indicates the number of cases with the same DNA-binding profile. Antibodies used for supershift are indicated at the top. Asterisks point on the super-shifted EMSA signal. Arrowhead indicates decreased NF-κB complex DNA-binding. (F) Frequencies of high c-Rel binding and low or negative c-Rel binding among GCB-DLBCLs and ABC-DLBCLs.
To confirm these results, an independent series of 66 DLBCLs, 43 ABCs, and 23 GCBs classified according the cell of origin and issued from the GHEDI cohort (22) was analyzed. A typical EMSA result with c-Rel DNA-binding-high, low, or negative is shown in Figure 1E. Consistently with the test series, the 10/13 (77%) DLBCL cases with high c-Rel binding were GCBs, while most low or negative c-Rel binding cases were ABCs (Figure 1F, Fisher’s Exact Test, p = 0.0008).
The c-Rel Signature Is a Hallmark of the Majority of GCB-DLBCLs
To identify c-Rel DNA-binding activity associated genes, the Affimetrix gene expression profiles of the 13 DLBCL cases of the GHEDI series with a high c-Rel DNA-binding activity were compared to the 53 other cases with a low or negative c-Rel DNA-binding activity (see Supplementary Figure S2 for the experimental design and the analysis pipeline). With a fold change of two and a Benjamini–Hochberg adjusted p-value below 0.05 between high and low or negative c-Rel binding cases, a set of 343 probesets/237 genes was selected after LIMMA analysis (Supplementary Table S4). This c-Rel gene expression profile (GEP) was used to clusterize the entire GHEDI series (83 ABCs, 85 GCBs, and 34 Others). PMBLs were excluded from the analysis. As shown in Figure 2A, 74/202 (37%) cases exhibited a c-Rel GEP: 63/85 (74%) GCBs, 3/83 (4%) ABCs, and 8/34 (24%) Others. Among the 128 patients with a non-c-Rel GEP 80 (63%), 22 (17%), and 26 (20%) were ABCs, GCBs, and Others respectively (Chi2 Test, p << 10−6).
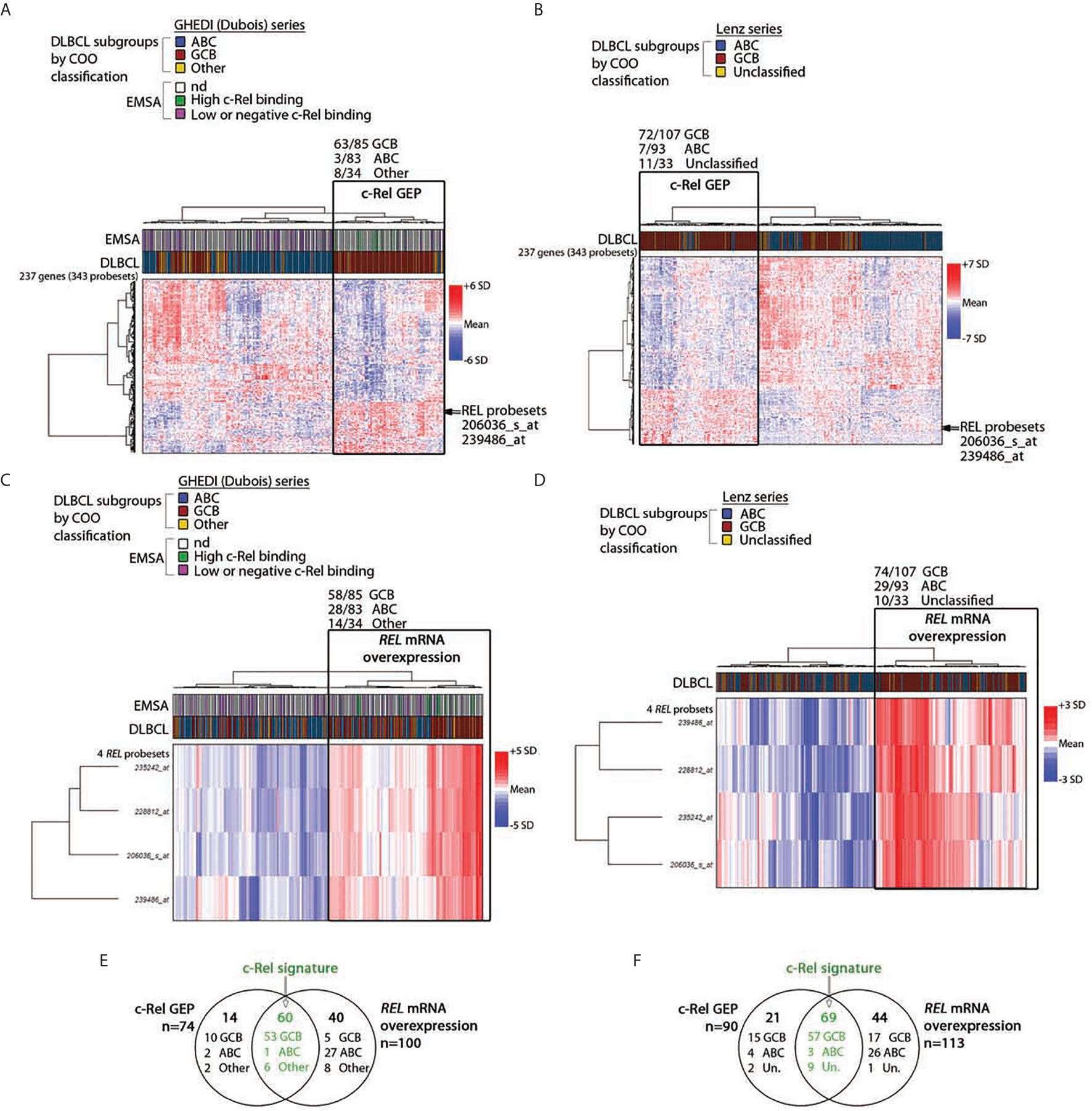
Figure 2 c-Rel related transcriptomic signature of DLBCL patients. By comparing cases with high and low or negative c-Rel DNA-binding, a set of differentially expressed 343 probesets/237 genes with a fold change of two, and a p-value <0.05 was selected after LIMMA analysis of 66 DLBCLs from the GHEDI cohort for which EMSAs with super-shift were performed. (A, B) Unsupervised hierarchical Ward clustering of the whole GHEDI cohort and the Lenz series respectively with the 343 selected probesets. Lines and columns of the heatmap correspond to probesets and patients respectively. Up- and down-regulated genes are color coded according to the color scale shown on the right of the heatmap. The blue, red, and yellow color codes above the heatmap give the original COO ABC/GCB/Other and the ABC/GC/Unclassified diagnosis of the GHEDI and Lenz series respectively. Position of the c-Rel probsesets on gene clusters is indicated by an arrow. For the GHEDI series, high and low c-Rel binding cases by EMSA are color coded in green and purple respectively (nd: EMSA not done). The branch of patients with the c-Rel gene expression profile (c-Rel GEP) is highlighted by a black rectangle on each heatmap. Number of COO cases within this specific branch is indicated at the top of the black rectangle. (C, D) Unsupervised hierarchical Ward clustering of the entire GHEDI cohort and the Lenz series respectively with c-Rel probesets. Orientation of the heatmap and color codes are the same as in panel (A) The branch of patients overexpressing REL mRNA is highlighted by a black rectangle on each heatmap. Number of COO cases within this specific branch is indicated at the top of the black rectangle. (E, F) Venn diagram showing the overlap of patients between c-Rel GEP and REL mRNA overexpression corresponding to c-Rel signature (in green). Number of cases in each group is indicated. Un., unclassified.
Of note, the 10 GCB-DLBCLs with a high c-Rel DNA-binding activity by EMSA were all coclusterized with patients exhibiting the c-Rel GEP. In contrast, 47/53 (89%) EMSA cases with low or negative c-Rel DNA-binding activity were coclusterized with patients that did not exhibit the c-Rel GEP (Fisher’s exact test, p = 8.10−6). Therefore, GEP and EMSA results were highly concordant, strongly suggesting that c-Rel was transcriptionally active and showing the c-Rel GEP was indeed associated with a majority of GCB-DLBCLs.
To validate this result, the DLBCL series published by Lenz et al. (23) (93 ABCs, 107 GCBs and 33 Unclassified) was also analyzed with these 343 probesets. As shown in Figure 2B, this analysis gave similar results since 90/233 (39%) cases were clusterized together with the c-Rel GEP, among them 72/107 (67%) GCBs, 7/93 (8%) ABCs, and 11/33 (33%) Unclassified (Chi2 Test, p << 10−6). Overlaps between genes from clustering of these two independent series were almost perfect, indicating the reproducibility of clustering between both series (Supplementary Table S5).
Among the up-regulated genes in GCB-DLBCLs with the c-Rel GEP was the REL gene. Restricting the analysis to the four REL probesets led to clusterize 100/202 (50%) cases of the GHEDI series, among them 58/85 (68%) GCBs, 28/83 (34%) ABCs, and 14/34 (41%) Others (Figure 2C, Chi2 Test, p = 2.5.10−5). Similarly, clustering of the Lenz series with the REL probesets identified 113/233 (48%) cases with increased REL mRNA expression among them 74/107 (69%) GCBs, 29/93 (31%) ABCs, and 10/33 (30%) Unclassified (Figure 2D, Chi2 test, p = 4.5.10−8). These cases were those with REL mRNA overexpression. This was in contrast with RELA or RELB because no association was established between mRNA expression of these two NF-κB subunits and the three COO subsets (not shown). To be stringent and because we have experimental evidence showing that over-expression of REL mRNA is associated with nuclear translocation of c-Rel protein with increased transcriptional activity (see below), we defined the c-Rel signature by both c-Rel GEP and REL mRNA overexpression. Among cases with the c-Rel signature, 53/60 (88%) and 57/69 (83%) were GCBs for the GHEDI and Lenz series respectively (Figures 2E, F).
Relationships Between c-Rel and ABC-GCB Gene Expression Profiles
In the next step, we investigated the relationship between c-Rel and ABC-GCB gene expression profiles (GEPs). On one hand, we first selected a set of 424 genes (622 probesets) that were differentially expressed with a two-fold change and an adjusted p-value below 0.05 between COO classified ABC and GCB-DLBCLs from the same series of 66 cases tested by EMSA. This “re-established” ABC-GCB GEP consisted of 177 (279 probesets) and 247 (343 probesets) up-regulated genes in ABC (ABC-up) and GCB (GCB-up) DLBCLs respectively (Supplementary Table S6). It included 18/44 (41%) genes of the ABC/GCB Wright’s predictor (2) such as FOXP1, CCND2, IRF4, TCF4, or IL16 for ABCs and LMO2, MYBL1, or VCL for GCBs.
On the other hand and as mentioned above, the c-Rel GEP consisted of 237 genes: 65 up-regulated genes (101 probesets) and 172 down-regulated genes (242 probesets). The intersection between c-Rel and ABC-GCB GEP contained 55 genes (88 probesets) (Figure 3, and Supplementary Table S7). As shown in Figure 3, Venn diagrams indicate that c-Rel up-regulated genes overlapped partially, but only, with the GCB-up genes (26 genes; 42 probesets). Oppositely, c-Rel down-regulated genes overlapped only with ABC-up genes (28 genes; 46 probesets).
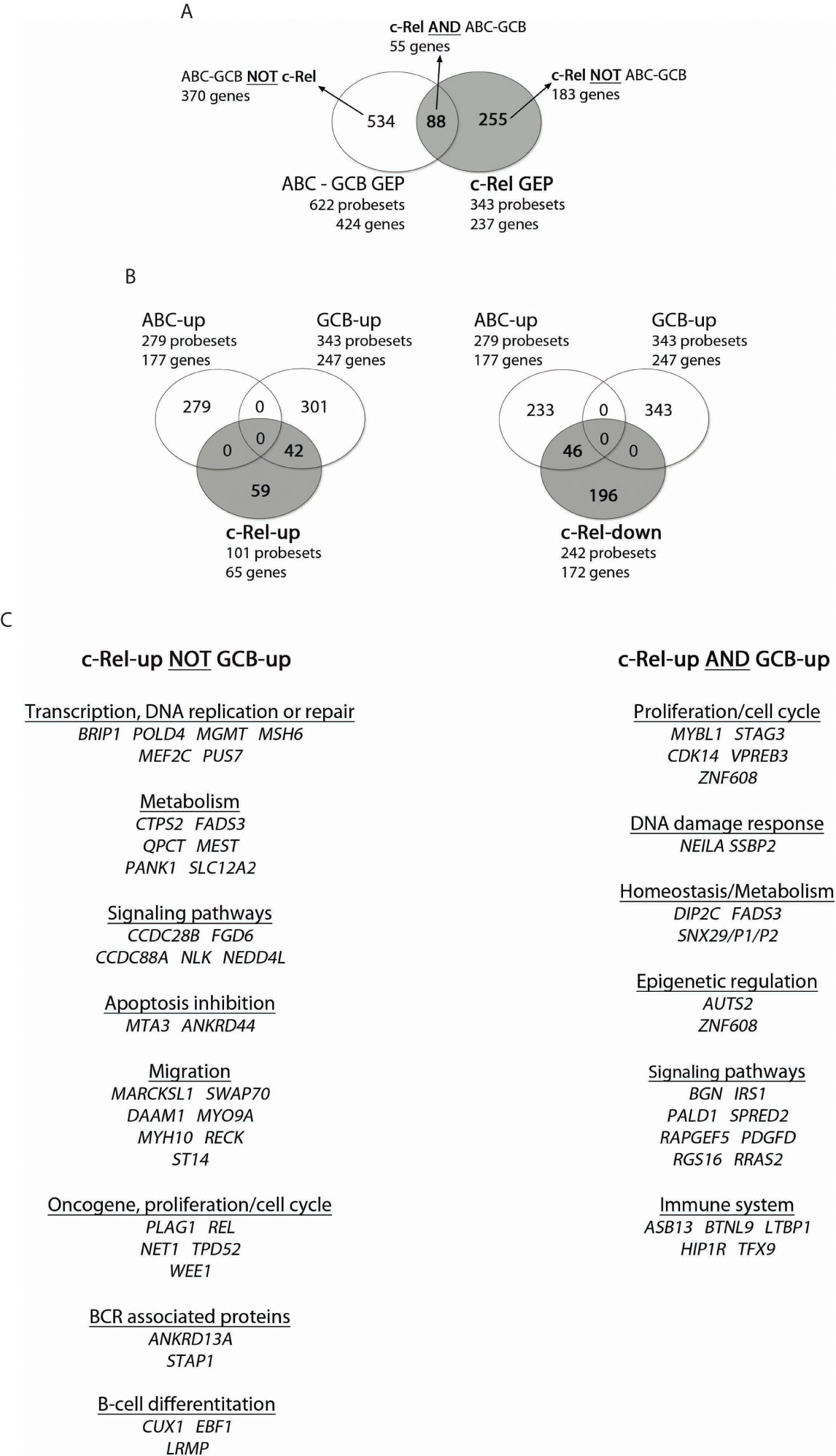
Figure 3 Overlap between c-Rel and ABC-GCB gene expression profiles (GEPs). c-Rel GEP (237 genes, 65 up-regulated genes with 101 probesets, and 172 down-regulated genes with 242 probesets) and ABC-GCB GEP (424 genes, 177 up-regulated genes with 279 probesets, 247 down-regulated with 343 probesets) were established (fold change ≥2) from the 66 DLBCL cases of the GHEDI cohort for which the c-Rel DNA-binding profile was established. (A) Venn diagram showing overlaps of probesets and genes between c-Rel GEP and ABC-GCB GEP. Set of genes named “c-Rel AND ABC-GCB” corresponds to the intersection between c-Rel and ABC-GCB GEP with 55 genes and 88 probesets (list in Supplementary Table S9). “ABC-GCB NOT c-Rel” are genes found in ABC-GCB GEP but not in c-Rel GEP. “c-Rel NOT ABC-GCB” are genes differentially expressed in c-Rel GEP but not in ABC-GCB GEP. This nomenclature is used in Figure 5. (B) Venn diagrams showing overlap between c-Rel up-regulated genes (at left) or c-Rel down-regulated genes (at right) and ABC or GCB up genes. Indicated number corresponds to probesets that overlap or not. (C) c-Rel-up NOT GCB-up and c-Rel-up AND GCB-up gene lists. Genes are ordered related to their function.
This analysis shows that c-Rel GEP only overlapped with GCB GEP, a very strong indication that c-Rel is linked to GCBs and not to ABCs. It also shows that the c-Rel GEP, including REL itself, was mainly based on an original set of genes.
Association of the c-Rel Signature With Genomic Alterations Related to GCB-DLBCLs
The c-Rel signature was associated with increased frequencies of REL locus gains (Figure 4A, Fisher’s Exact Test, p = 10−4). Consistently, cases with REL gains had higher levels of REL mRNA (Figure 4B). Frequencies of BCL2 gene translocation were increased in cases with the c-Rel signature (Figure 3C, Fisher’s Exact Test, p = 1.2.10−6). Conversely, BCL6 translocation was associated with cases with the non-c-Rel signature, while MYC translocation was equally rare in both cases (Figure 4C, Fisher’s Exact Test, p = 0.023 and p = 0.24 respectively).
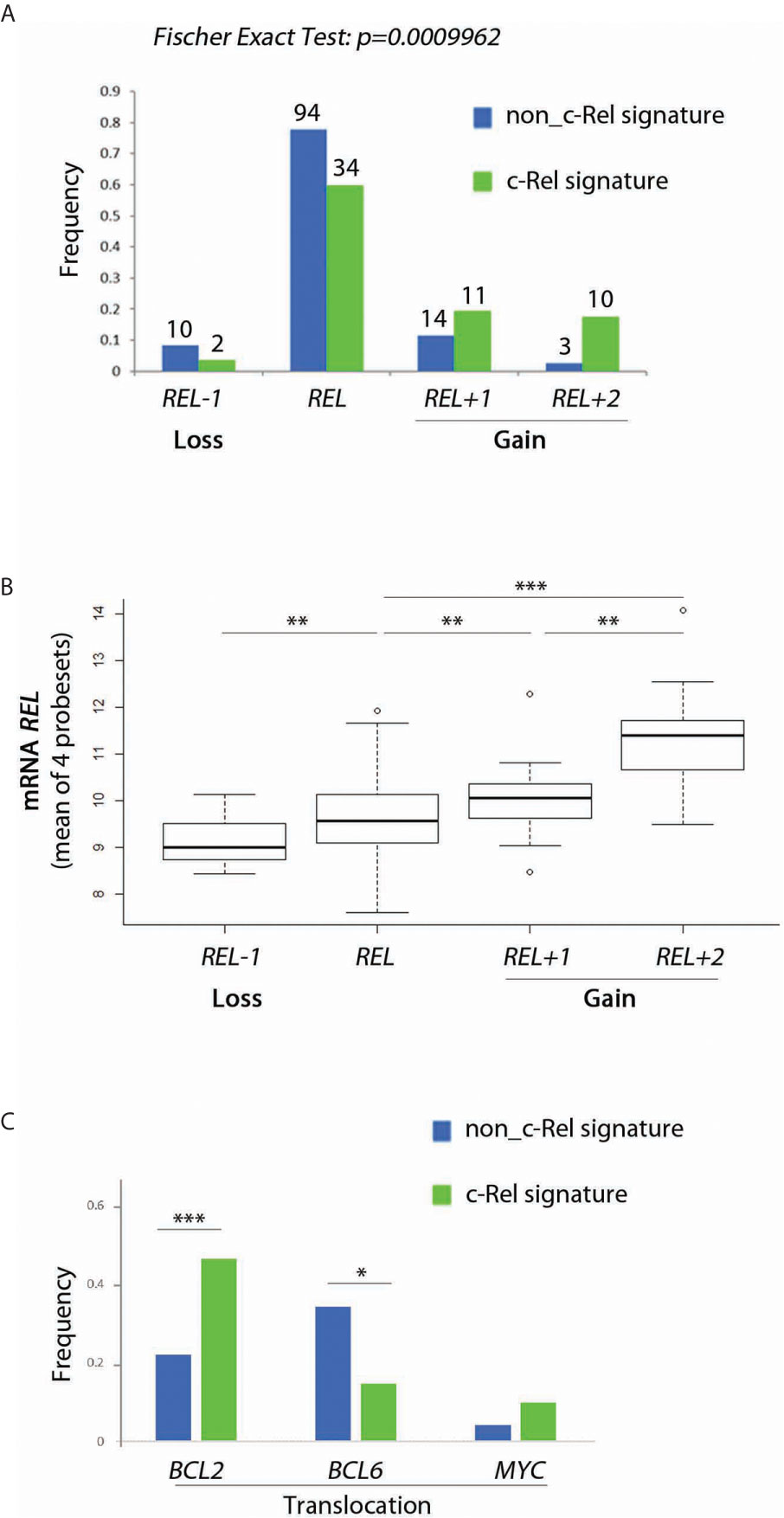
Figure 4 Relationship between c-Rel signature, REL gain, REL mRNA expression and BCL2, BCL6 and MYC translocations for patients from the GHEDI cohort. (A) Frequency of REL deletion or gain according to c-Rel signature. (B) REL mRNA expression levels according to REL loss or gains. (C) Frequency of BCL2, BCL6, and MYC translocations according to the c-Rel signature. In Figures 3A, C, green and blue bars indicate tumors respectively expressing or not the c-Rel signature. In Figures 3A, B, REL deletion, no REL number aberrancy, gain of one REL copy, and of two or more REL copies are encoded by REL−1, REL, REL+1 and REL+2 respectively. Statistical significance was determined by t-test (***p<0.001; **p<0.01; *p<0.05).
Mutational status of 34 genes recurrently altered in DLBCLs was explored in 183/202 cases from the GHEDI cohort, including 59/66 (89%) cases for which c-Rel DNA-binding activity was assessed. As shown in Figures 5A, B, mutational profiles of cases with high c-Rel DNA-binding or with the c-Rel signature were closed, being characterized by increased frequencies of BCL2, CREBBP, EZH2, MEF2B, and TNFRSF14 mutations and decreased frequency of CD79B and PIM1 mutations when compared to cases with the non-c-Rel signature. FISH cytogenetics and mutational status of MYD88, CD79B, NOTCH1, NOTCH2, BCL2, and EZH2 classified 137/202 (67%) DLBCL cases from the GHEDI cohort according to genetic subtypes defined by Schmitz et al. (5). We found 35 BN2 (based on BCL6 fusions and NOTCH2 mutations), 26 EZB (based on EZH2 mutations and BCL2 translocations), 10 MCD (based on the co-occurrence of MYD88L265P and CD79B mutations), six N1 (based on NOTCH1 mutations), 30 “other ABC”, 25 “other GCB”, and five “other unclassified” cases (Supplementary Table S8). As shown in Figure 5C, the c-Rel signature was mostly associated with the GCB EZB subtype, while this signature was absent in MCD cases and almost absent from the “other ABC” subtype. Of note, survival analysis indicated that, even if numbers were small, c-Rel signature tended to be associated with good prognosis among EZB cases (Supplementary Figure S3).
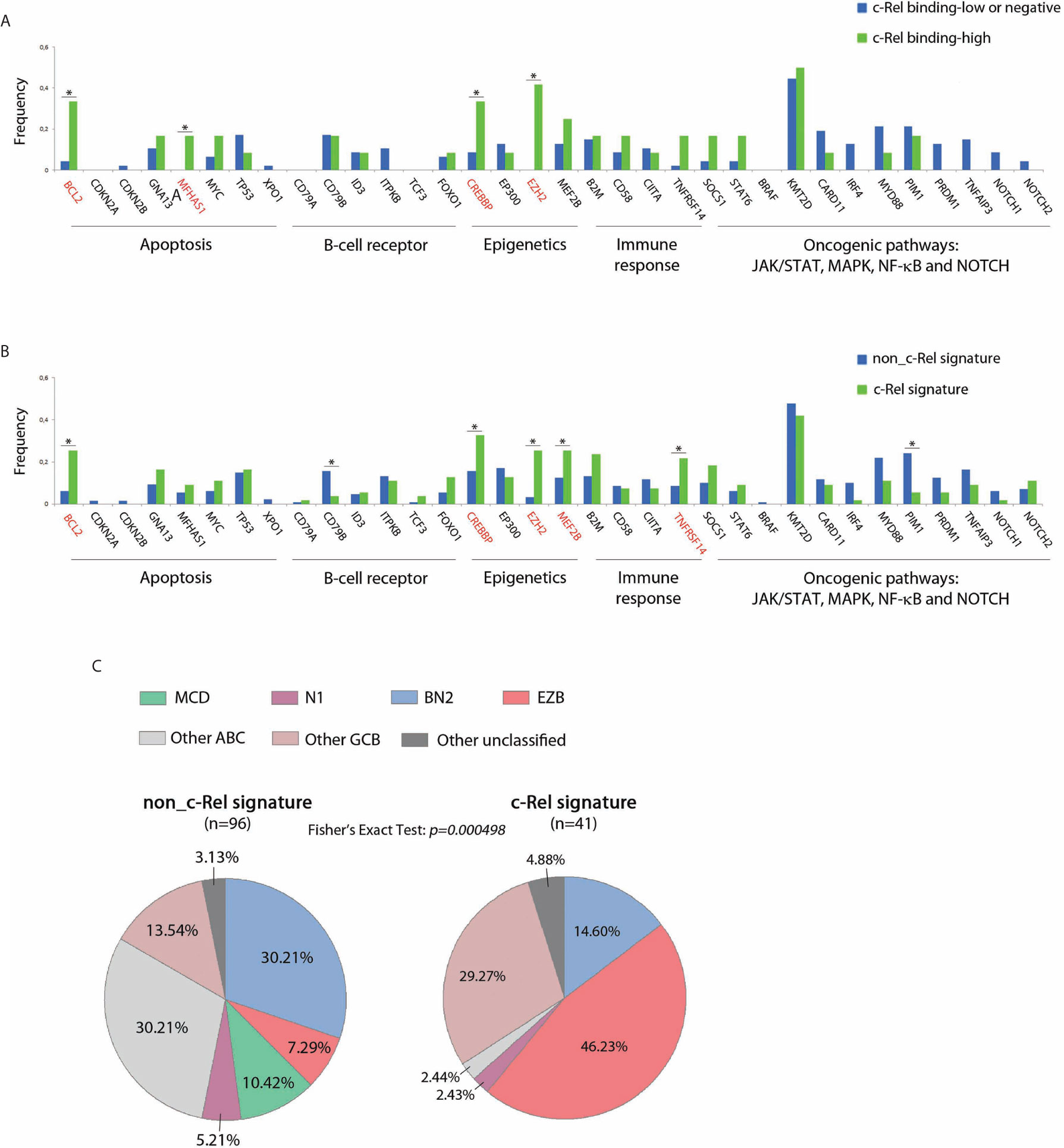
Figure 5 c-Rel related oncogenic mutations in DLBCL patients from the GHEDI cohort. (A, B) Mutation frequencies of 66 cases analyzed by EMSA according to c-Rel DNA-binding activity and the whole GHEDI series according to c-Rel expression signature. Genes known to be mutated at high frequencies in GCB DLBCLs are highlighted in red. The functional pathways are indicated at the bottom of the graph. *: Fisher exact test p-value < 0.05. (C) Percentage of 134 DLBCL cases that could be reclassified in BN2 (based on BCL6 fusions and NOTCH2 mutations), EZB (based on EZH2 mutations and BCL2 translocations), MCD (based on MYD88L265P and CD79B), N1 (based on NOTCH1 mutations), “other ABC”, “other GCB”, and “other unclassified” subtypes according to the c-Rel expression signature.
Thus, the c-Rel signature was related to REL locus gains and to other mutations known to be associated with GCB-DLBCL cases such as BCL2 translocation and EZH2 mutations. Consequently, the c-Rel signature subtype was part of the EZB DLBCL subtype and had a favorable impact on patient survival.
The c-Rel Signature Is Associated With Decreased Genomic Complexity, Specific Gene Imbalances and Better Overall Survival
We next analyzed the whole genome imbalances of the GHEDI series by CGH-array. With the GISTIC pipeline, each of the 23,636 gene loci was scored between −2, −1, 0, 1, 2 according to the level of gene loss (−2 meaning loss of both alleles and +2 meaning gain of two copies or more). Because of the uncertainties of CGH-array signal quantification, only gene scores of −2 or +2 were taken into account. With these criteria, the genomic imbalance complexity was estimated as the number of all genes for which loci were deleted or amplified. Histogram frequencies of the number of gene loci imbalances revealed a bi-modal distribution with a threshold at 1,000 (Figure 6A). The proportion of cases with a genomic imbalance complexity over this threshold was increased in ABC-DLBCLs when compared to GCB or “Other” DLBCLs but without reaching statistical significance (Figure 6B). By contrast, the c-Rel signature was associated with a marked decrease in cases with a genomic imbalance complexity above 1,000 (Figure 6B, Wilcoxon test, p = 0.017). This was still true and even more striking when this analysis was restricted to GCB cases only (Figure 6B, Wilcoxon test, p = 0.003). Indeed while being not significantly different between ABC and GCB-DLBCLs, the genomic imbalance complexity of cases with the c-Rel signature was significantly decreased when compared to non-c-Rel cases (Figures 6C, D).
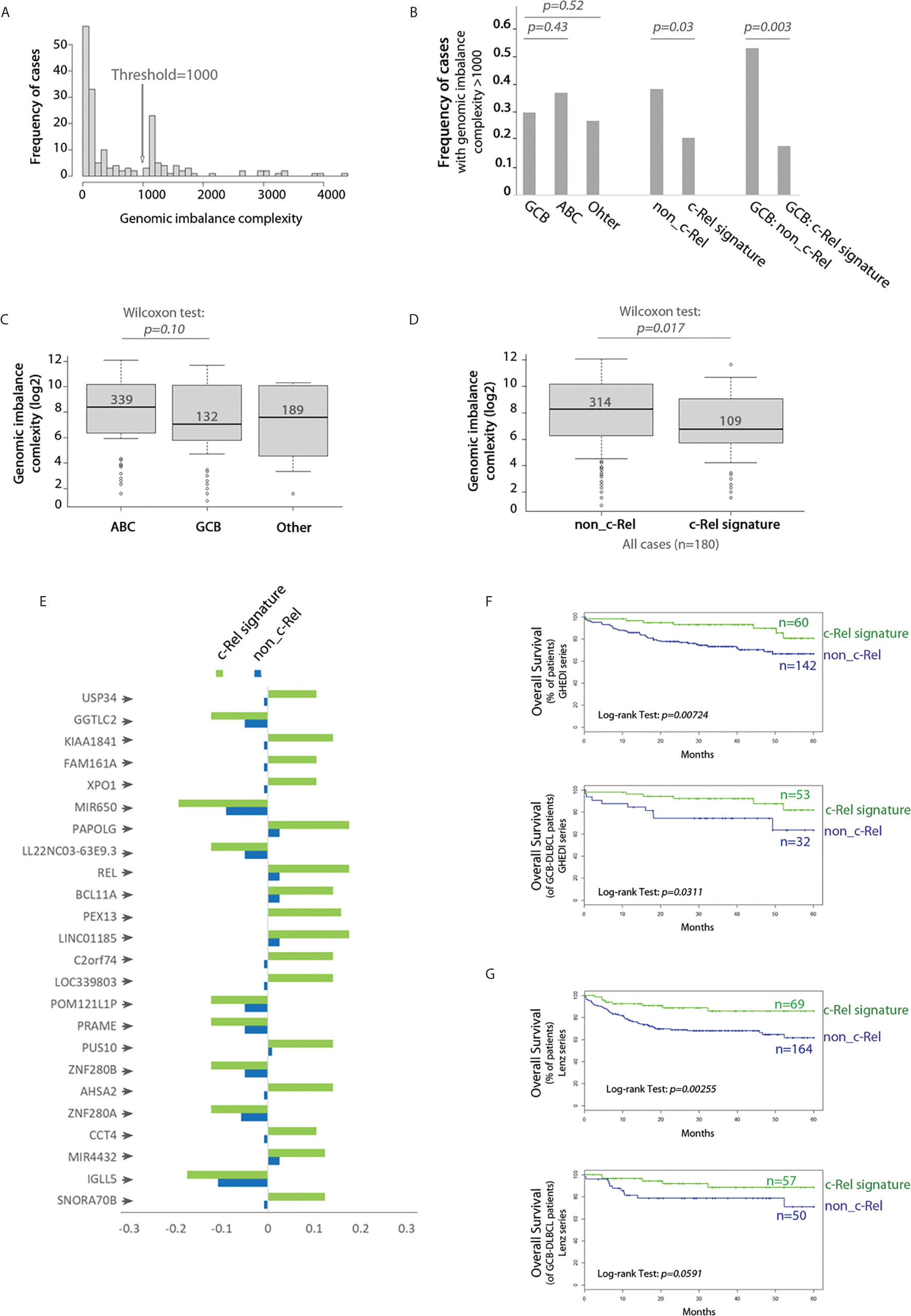
Figure 6 c-Rel related gene copy number aberrations and overall survival. (A) Frequency histogram of the genomic imbalance burden reveals a bimodal distribution with a threshold at 1,000. (B) Frequency of DBCL cases with a genomic imbalance burden >1,000 according to the COO classification (left), absence (non_c-Rel) or presence (c-Rel signature) of the c-Rel signature (middle and right) among the entire GHEDI series (left and middle) or GCB-DLBCLs only (right). Chi2 test p-values are indicated on the top. (C) Boxplot of the genomic imbalance burden according to the COO classification. (D) Boxplot of the genomic imbalance burden according to the absence (non-c-Rel) or presence (c-Rel signature) of the c-Rel signature. (C, D) Median number of gene imbalances is indicated by the black solid line. Wilcoxon test p-values are shown on the top. (E) Frequency of cases with gene copy number aberrations for the 24 gene loci specifically imbalanced in DLBCL cases with the c-Rel signature. Negative values mean deletion while positive values mean amplification. (F, G) Kaplan–Meier overall survival curves of all (upper panel) or GCB (lower panel) patients from GHEDI and Lenz series respectively according to expression of the c-Rel signature (c-Rel signature, green curve) or not (non-c-Rel, blue curve). Number of cases is indicated on the right. The log-rank p-value is given within each graph.
To search for specifically altered gene loci in c-Rel cases, we used as a criterion that a gene copy number aberration had to be retrieved in at least 10% of c-Rel cases with a 50% frequency increase when compared to non-c-Rel cases. With this condition a list of 24 genes was selected (Figure 6E and Supplementary Table S9). All these genes, including REL, were located in the 2p15-16.1 chromosome region when amplified and in the 22q11.22 chromosome region when deleted (Supplementary Table S9). Amplification of 2p15-16.1 chromosome region could be expected (24) and is associated with transformation from follicular lymphoma to DLBCL (25). Deletion of the 22q region was reported once and could be associated in DLBCLs with a more advanced clinical stage (26). Among these genes, some are already known in DLBCLs. For example, we found that USP34 (Ubiquitin Specific Peptidase 34) and BCL11A (BAF Chromatin Remodeling Complex Subunit BCL11A) were co-amplified with REL itself. Co-amplification of these three gene loci is found in transformed DLBCLs and indicates a gain of chromosome 2p15-16.1 (25). Amplification of the XPO1 gene (Exportin 1, also located on chromosome 2p15) was found in 10.5% c-Rel cases of the GHEDI series. XPO1 gene mutations have been associated with PMBLs (22). High XPO1 expression has been reported in DLBCL and its specific inhibition is targetable by the selinexor agent (27). PRAME (Preferentially Expressed Antigen In Melanoma, located in 22q11.22) gene, which overexpression is associated with a worse prognosis in DLBCLs (28), was deleted in 12.3% c-Rel cases, suggesting these cases were oppositely associated with expression of this gene. Deletion of IGLL5 (Immunoglobulin Lambda Like Polypeptide 5, also located in 22q11.22) locus, which is one of the most mutated gene in DLBCLs, was found in 17.5% c-Rel cases.
Since genomic imbalance complexity was decreased in DLBCL cases with the c-Rel signature and that these cases exhibited specific gene copy number aberrations that could be associated with prognostic, we examined the overall survival (OS) impact of the c-Rel signature. DLBCL cases from both GHEDI and Lenz series with the c-Rel signature exhibited an increased overall survival (OS) (upper panel of Figures 6F, G). Interestingly, and most likely related to the lower genomic imbalance burden, this better OS tended to be confirmed on GCB cases (lower panel of Figures 6F, G).
Overexpression of c-Rel Was Associated With Increased Protection Against Apoptosis
To assess the in vitro functional role of c-Rel on proliferation and protection against apoptosis, we used the cell cycle arrested EREB2.5 cell line. This cell line, which proliferation can be arrested by estradiol withdrawal, allows assaying the role of any putative oncogene without interference with its proliferation potential. Cells were stably transfected with a doxycycline regulatable episomal vector allowing for expression of c-Rel. In B-cells, c-Rel is regulated by IκBα and IκBε inhibitors with a predominant role of the latter (29). In consequence and in addition to the luciferase control, we also transfected cells to over-express the IκBαS32,36A super-repressor (20) and the IκBε inhibitor. As shown in Figure 7A, c-Rel overexpression in estradiol starved EREB2.5 B-cells (resting state) was associated with increased expression of RelB, IκBα, TRAF1, and A20 proteins (known to be NF-κB targets) when compared to luciferase-expressing cells, suggesting a constitutive NF-κB transcriptional activity. Indeed, c-Rel was constitutively found in the nucleus of cells when over-expressed (Figure 7B). No effect of c-Rel was seen on cell proliferation in this model (not shown). While induction of IκBα and IκBε inhibitors increased cell apoptosis, c-Rel protected resting EREB2.5 B-cells against cell death when compared to luciferase control cells (Figure 7C). Reciprocally, inhibition of c-Rel expression in the GCB SUDHL-4 cell line tended to increase cell death (Supplementary Figure S4).
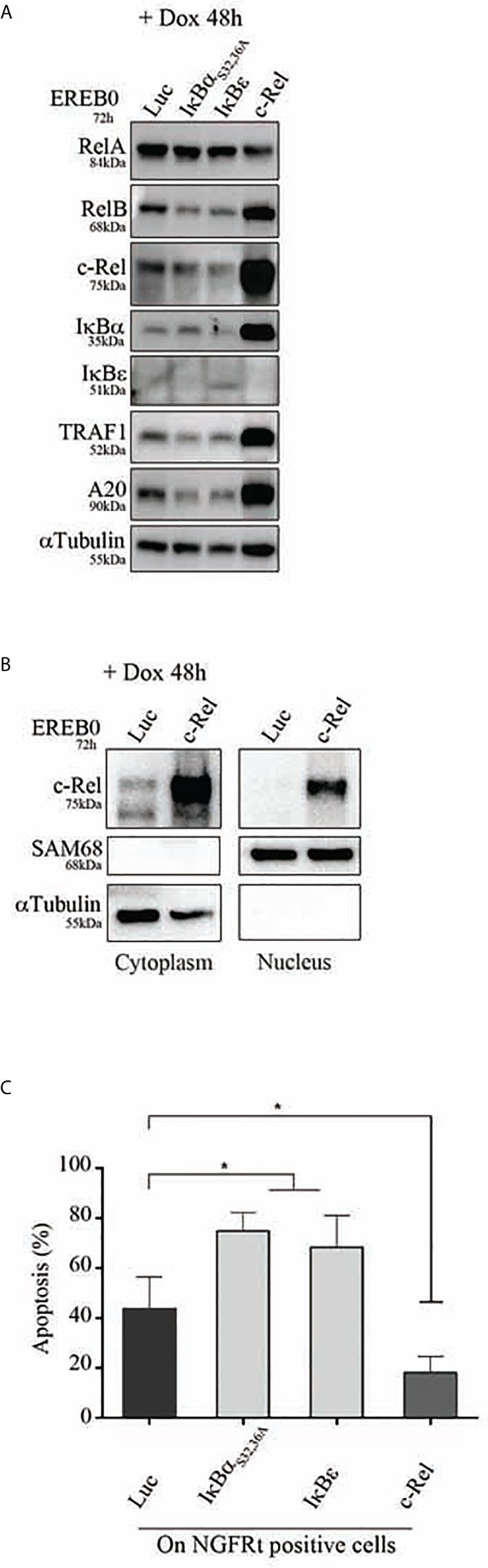
Figure 7 Apoptosis of c-Rel overexpressed human B-cells. EREB2-5 cells were stably transfected with the doxycycline-regulatable pRT-1 vector coding for Luciferase (Luc), IκBαS32,36A, IκBε, or c-Rel. After estradiol starvation for 24 h (EREB0), cells were treated with doxycycline (+ Dox) for 48 h. (A) Immunoblots detecting the expression of RelA, RelB, c-Rel, IκBα, IκBε, TRAF1, A20 as well as αTubulin as loading control. (B) Detection of cytoplasmic and nuclear c-Rel protein by immunoblot. SAM68 and αTubulin revelations are nuclear and cytoplasmic markers, respectively. (C) Percentages of Annexin-V-positive cells gated on NGFRt positive cells. Statistically significant difference (Mann–Whitney test) is indicated by *(p ≤ 0.05).
Therefore, overexpression of c-Rel was associated with its constitutive nuclear translocation, and protection against cell death.
Discussion
Here, we describe a new c-Rel transcriptomic signature related to c-Rel DNA-binding activity in GCB-DLBCLs. Results indicate that this new c-Rel signature defined a good prognosis compared to non-c-Rel cases. Cases with the c-Rel signature were more frequently associated with REL locus gains, BCL2 translocation, and EZH2 mutations. Functionally, overexpression of c-Rel protein was sufficient to induce its nuclear translocation and to protect cells against apoptosis.
The relationship between REL gains and REL mRNA overexpression has been previously reported by others (8). Confirming those published by Li et al. (30), our results show that REL mRNA levels were higher in GCB than in ABC-DLBCLs. Probably due to immunostaining technical limitations, it is not clear from the literature whether a clear relationship between c-Rel protein expression levels, its nuclear localization and REL amplification exists in ABC and GCB-DLBCLs (30, 31). Pham et al. reported significant higher c-Rel DNA-binding activity in GCB compared to ABC-DLBCL subtypes from 14 DLBCL cell lines and 14 DLBCL patient samples using an ELISA-based approach (32). By EMSA with a supershift approach, we show that high c-Rel DNA-binding was more frequently associated with GCB than ABC-DLBCLs in two independent DLBCL series. We also found that REL mRNA expression levels were correlated to its c-Rel DNA-binding activity, to c-Rel GEP, and to REL amplification. This suggests that REL mRNA overexpression is probably a good indicator of c-Rel transcriptional activity. This conducted us to define the c-Rel signature as the association of both REL mRNA over-expression and c-Rel GEP. These DLBCL cases were those with the best overall survival in both the GHEDI and Lenz series.
The c-Rel GEP consisted in 237 differentially expressed genes between high c-Rel binding samples and the others. Among up-regulated genes of the c-Rel GEP were REL and NLK (Nemo-Like Kinase). NLK has been shown to negatively regulate RelA NF-κB activation by targeting CBP300 or IKKβ (33). Increased expression of NLK has been found in various solid cancers such as those of the colon or lung (34). The c-Rel GEP also includes EBF1 or SWAP70 genes. EBF1 coding for a B-cell specific transcription factor is involved in immunoglobulin heavy chain translocations in DLBCLs (35), and SWAP70 is known to be associated with B-cell activation, immunoglobulin class switching, migration and homing processes essential for B-cell entry into lymph nodes, and germinal center formation (36). Genes involved in metabolism such as pyrimidine nucleotide or lipid metabolic processes with CTPS2 (CTP synthesis from UTP) and FADS3 (fatty acid desaturase 3) genes were also among the up-regulated genes of the c-Rel GEP. Some of the c-Rel-up and GCB-up subset of genes are involved in cell proliferation, G2/M transition of the mitotic cell cycle, or in DNA repair such as STAG3, CDK14, and NEIL1. Other genes are also regulators of the Ras signaling pathway such as RAPGEF5, RGS16, and RRAS2 genes. NF-κB activity in cancer can arise from mutations in upstream regulators such as the Ras pathway (37). Oncogenic KRAS driven cancers require NF-κB anti-apoptotic signals involving c-Rel (38). Furthermore, microRNA dysregulation can target the Ras signaling pathway in germinal center derived lymphomas such as DLBCLs and Burkitt’s lymphomas (39). The VPREB3 gene, also belonging to this gene subset, has been shown to be over-expressed in germinal center B-cells as well as in DLBCLs with MYC amplification (40). Consistent with the previously described importance of c-Rel in germinal center B-cells, various genes that are in the c-Rel-down and ABC-up subset are known to be down-regulated in the germinal center B-cells such as BATF, RAB29, CCND2, HCK, CD44, and STAT3.
Interestingly, cases from the GHEDI series with high c-Rel DNA-binding and c-Rel signature more frequently exhibited the genetic events previously ascribed to GCB-DLBCLs and follicular lymphomas (FL) (1), including BCL2 translocation, MEF2B and EZH2 gain of function mutations as well as loss of function of the tumor suppressors CREBBP, and TNFRSF14. MEF2B may control BCL6 which in turn regulates LMO2 and MYBL1 (41), the latter being up-regulated in both GCB and c-Rel signatures. As demonstrated by LM Staudt’s group, the ABC/GCB-DLBCL subgroups can be separated into distinct genetic subtypes, among them EZB, based on EZH2 mutations and BCL2 translocation. This subtype mainly corresponds to the GCB-DLBCL cases (5). EZB is also related to REL amplification (5). Here, EZB DLBCLs frequently exhibited the c-Rel signature that could suggest a relationship between c-Rel transcriptomic activity and EZH2 dysregulation. This could probably, at least partially, be due to the transcriptional regulation of EZH2 by c-Rel as previously shown in activated B and T lymphocytes (42). Patients with EZB tumors have a trend toward increased frequency of MYC translocation and inferior overall survival when compared to “other GCB” patients (5, 43). The number of cases from the GHEDI series with MYC translocation was too low to draw any conclusions. Nevertheless, among all GCB as well as the EZB classified GCB cases, patients with the c-Rel signature had a much more favorable outcome than non-c-Rel patients. This probably illustrates the importance of c-Rel in the biology of GCB cases.
Under normal conditions, c-Rel is expressed in mature B-cells. Its expression is up-regulated throughout B-cell differentiation upon activation mediated by the B-cell receptor (BCR), and CD40 during T-cell contact. Toll-like receptors 4 and 9 also mediate c-Rel expression in B-cells (44). Germinal center formation and maintenance require c-Rel expression in order for B-cells to shuttle between dark and light zones (45). Furthermore, BCR activated B-cells have elevated levels of apoptosis in the absence of c-Rel (46) suggesting that c-Rel provides an important survival signal to reconfigure the BCR in germinal centers. In pathological conditions, we speculate that aberrant increased c-Rel expression and/or activity corresponding to the c-Rel signature may promote tumorigenesis in DLBCL-precursor cells by disturbing the normal dynamics of germinal center B-cell development. In other words, c-Rel could send a survival signal to a germinal center B-cell normally destined to die (after, for example, loss of antigen recognition ability). Like NF-κB continuous activation in MALT lymphoma that is first due to the B-cell immune environment before acquiring its independence thanks to translocations involving MALT1 and BCL10 genes, it can be hypothesized that the germinal center environment of the initial tumor cell was first responsible for the c-Rel up-regulation. c-Rel up-regulation would become constitutive over time thanks to acquired genetic events such as REL amplification for example or by others not known yet mechanisms including epigenetic, transcriptomic, and/or post-transcriptional oncogenic events. As mentioned above, the dysregulation of the RAS pathway, that we found associated with the c-Rel signature, could be one of this additional oncogenic mechanisms (38). Finally, our in vitro results suggest that REL mRNA overexpression is associated with nuclear translocation of a transcriptionally active c-Rel protein in the absence of any activation signal, and increased protection against apoptosis in a model of resting human B-cells. We previously showed that overexpressed RelA remains stored in the cytoplasm in the absence of adequate activation signals in the EREB2.5 EBV transformed B-cell model [(20) and unpublished results]. In contrast to RelA, c-Rel does not have a nuclear export signal explaining why de novo c-Rel gene transcription and translation can lead to long-term maintenance of nuclear c-Rel-containing heterodimers (47). These different properties of the NF-κB subunits would explain why tumors dependent upon RelA activation must absolutely acquire mutations in the NF-κB activation track while c-Rel overexpression, due for example to REL locus gains, would not require additional activation events to achieve c-Rel effects.
The hypothesis that, to exert its oncogenic effects, c-Rel would not need additional genetic events is fully in agreement with the fact that the genomic imbalance complexity of DLBCL cases with the c-Rel signature was markedly decreased. It has been shown that, rather than the antigenic pressure, oncogenic events are the main driving forces for relapse in DLBCLs (48). Like in chronic lymphocytic leukemia in which the cytogenetic complexity is associated with resistance to therapies, increased imbalance genomic complexity in non-c-Rel cases is very likely to be related to genetic instability that would favor the selection of a therapy resistant DLBCL subclone. Oppositely around, the decreased genomic imbalance complexity of c-Rel DLBCL cases would be associated with a better prognosis.
While ABC-DLBCLs are very likely to be associated with RelA activation, the place of c-Rel in DLBCLs remains rather enigmatic since it is associated with GCB-DLBCLs that are not supposed to harbor any NF-κB activity. An explanation for such a discrepancy between the absence of NF-κB signature and REL amplification may have been provided by TJ de Jesùs and P Ramakrishnan (49). Indeed these authors showed that c-Rel represses TNF-α-induced RelA-mediated transcription. This suggests that long term over-expression of c-Rel could extinguish the RelA signature. In that context, a c-Rel signature would almost never overlap with that of RelA. Consistently, we did not find any overlap in between c-Rel and ABC-DLBCL signatures.
For the first time, we show that a strong c-Rel DNA binding activity was a feature of GCB-DLBCLs. These GCBs specifically exhibited a c-Rel transcriptomic signature, REL gains, BCL2 translocation, EZH2 mutations, and a favorable prognosis. They also exhibited a decreased genomic imbalance burden and some specific gene copy aberrations that are linked to 2p15-16.1 amplification and 22q11.22 deletion. These could likely be associated with the outcome such as PRAME deletion or XPO1 amplification, the latter being also targetable by selinexor. Functionally, c-Rel was responsible for protection against apoptosis. Thus, c-Rel could very likely participate in cell transformation in a significant proportion of GCB-DLBCLs. As a future direction, the better survival advantage for DLBCL patients with a c-Rel signature could be examined with regard to both sensitivity toward apoptosis under classical therapeutic molecules and the ability of tumor cells to be eliminated by the immune microenvironment. Even if this has to be confirmed on prospective studies, R-CHOP regiment is likely to give very good results on DLBCL tumors with the c-Rel signature. This also raises the question whether the c-Rel signature could become a decision criterion to better choose patients who would very likely be able to benefit from R-CHOP regimen or not. In other words, it would be of importance to find alternative therapeutics for patients whose DLBCL tumors do not exhibit the c-Rel signature and have increased genomic imbalance complexity.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The studies involving human participants were reviewed and approved by the LYSA group Institutional Review Board. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
NF, VB, and JF designed the study and supervised the work. NF and JF wrote the manuscript. NF, OT, DC, and LP made the experiments. KL, JF, and TM reviewed and selected the cases from the GHEDI series of patients. NF, JF, BP, M-PG, DB, and DT reviewed and selected the cases issued from the CHU of Limoges. JF and J-PJ made the bioinformatics and the statistics. CC-B made the cytogenetics. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by grants from the Institut National du Cancer (INCA). The group of JF is supported by grants from the Ligue Nationale Contre le Cancer (Equipe labellisée Ligue), the Comité Orientation Recherche Cancer (CORC) and the Haute-Vienne and Corrèze committees of the Ligue Nationale Contre le Cancer. The group of VB is supported by grants from the Institut National du Cancer, Agence Nationale pour la Recherche, the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 766214, Université de Paris, and Fondation ARC pour la Recherche sur le Cancer (Equipe labelisée), and doctoral funding from IDEX doctoral international program and Societé Française d’Hémathologie (to DC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank A Olivrie and A David, Regional Reference Structure of Limousin Lymphomas, Clinical Hematology Department, CHU of Limoges, France, for their helpful assistance in clinical data handling. We thank C Ouk from BISCEm US 42 INSERM/UMS 2015 CNRS platforms for flow cytometry analysis. We thank Dr J Cook Moreau, UMR CNRS 7276/INSERM 1262, Limoges, France, for English editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.638897/full#supplementary-material
References
1. Shaffer AL, Young RM, Staudt LM. Pathogenesis of human B cell lymphomas. Annu Rev Immunol (2012) 30:565–610. doi: 10.1146/annurev-immunol-020711-075027
2. Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci USA (2003) 100:9991–6. doi: 10.1073/pnas.1732008100
3. Scott DW, Mottok A, Ennishi D, Wright GW, Farinha P, Ben-Neriah S, et al. Prognostic Significance of Diffuse Large B-Cell Lymphoma Cell of Origin Determined by Digital Gene Expression in Formalin-Fixed Paraffin-Embedded Tissue Biopsies. J Clin Oncol Off J Am Soc Clin Oncol (2015) 33:2848–56. doi: 10.1200/JCO.2014.60.2383
4. Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med (2001) 194:1861–74. doi: 10.1084/jem.194.12.1861
5. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med (2018) 378:1396–407. doi: 10.1056/NEJMoa1801445
6. Deffenbacher KE, Iqbal J, Sanger W, Shen Y, Lachel C, Liu Z, et al. Molecular distinctions between pediatric and adult mature B-cell non-Hodgkin lymphomas identified through genomic profiling. Blood (2012) 119:3757–66. doi: 10.1182/blood-2011-05-349662
7. Iqbal J, Sanger WG, Horsman DE, Rosenwald A, Pickering DL, Dave B, et al. BCL2 translocation defines a unique tumor subset within the germinal center B-cell-like diffuse large B-cell lymphoma. Am J Pathol (2004) 165:159–66. doi: 10.1016/s0002-9440(10)63284-1
8. Jardin F, Jais J-P, Molina T-J, Parmentier F, Picquenot J-M, Ruminy P, et al. Diffuse large B-cell lymphomas with CDKN2A deletion have a distinct gene expression signature and a poor prognosis under R-CHOP treatment: a GELA study. Blood (2010) 116:1092–104. doi: 10.1182/blood-2009-10-247122
9. Brownell E, O’Brien SJ, Nash WG, Rice N. Genetic characterization of human c-rel sequences. Mol Cell Biol (1985) 5:2826–31. doi: 10.1128/mcb.5.10.2826
10. Gilmore TD, Cormier C, Jean-Jacques J, Gapuzan ME. Malignant transformation of primary chicken spleen cells by human transcription factor c-Rel. Oncogene (2001) 20:7098–103. doi: 10.1038/sj.onc.1204898
11. Fan Y, Gélinas C. An optimal range of transcription potency is necessary for efficient cell transformation by c-Rel to ensure optimal nuclear localization and gene-specific activation. Oncogene (2007) 26:4038–43. doi: 10.1038/sj.onc.1210164
12. Liou H-C. Regulation of the immune system by NF-kappaB and IkappaB. J Biochem Mol Biol (2002) 35:537–46. doi: 10.5483/bmbrep.2002.35.6.537
13. Roy K, Mitchell S, Liu Y, Ohta S, Lin Y-S, Metzig MO, et al. Hoffmann A. A Regulatory Circuit Controlling the Dynamics of NFκB cRel Transitions B Cells from Proliferation to Plasma Cell Differentiation. Immunity (2019) 50:616–28.e6. doi: 10.1016/j.immuni.2019.02.004
14. Köntgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, et al. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev (1995) 9:1965–77. doi: 10.1101/gad.9.16.1965
15. Tian W, Liou H-C. RNAi-mediated c-Rel silencing leads to apoptosis of B cell tumor cells and suppresses antigenic immune response in vivo. PloS One (2009) 4:e5028. doi: 10.1371/journal.pone.0005028
16. Kober-Hasslacher M, Oh-Strauß H, Kumar D, Soberón V, Diehl C, Lech M, et al. c-Rel gain in B cells drives germinal center reactions and autoantibody production. J Clin Invest (2020) 130:3270–86. doi: 10.1172/JCI124382
17. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood (2016) 127:2375–90. doi: 10.1182/blood-2016-01-643569
18. Chauzeix J, Pastoret C, Donaty L, Gachard N, Fest T, Feuillard J, et al. A reduced panel of eight genes (ATM, SF3B1, NOTCH1, BIRC3, XPO1, MYD88, TNFAIP3, and TP53) as an estimator of the tumor mutational burden in chronic lymphocytic leukemia. Int J Lab Hematol (2020). doi: 10.1111/ijlh.13435
19. Dubois S, Tesson B, Mareschal S, Viailly P-J, Bohers E, Ruminy P, et al. Refining diffuse large B-cell lymphoma subgroups using integrated analysis of molecular profiles. EBioMedicine (2019) 48:58–69. doi: 10.1016/j.ebiom.2019.09.034
20. Chanut A, Duguet F, Marfak A, David A, Petit B, Parrens M, et al. RelA and RelB cross-talk and function in epstein-barr virus transformed B-cells. Leukemia (2013) 28:871–9. doi: 10.1038/leu.2013.274
21. Kempkes B, Zimber-Strobl U, Eissner G, Pawlita M, Falk M, Hammerschmidt W, et al. Epstein-Barr virus nuclear antigen 2 (EBNA2)-oestrogen receptor fusion proteins complement the EBNA2-deficient Epstein-Barr virus strain P3HR1 in transformation of primary B cells but suppress growth of human B cell lymphoma lines. J Gen Virol (1996) 77( Pt 2 ):227–37. doi: 10.1099/0022-1317-77-2-227
22. Dubois S, Viailly P-J, Mareschal S, Bohers E, Bertrand P, Ruminy P, et al. Next-Generation Sequencing in Diffuse Large B-Cell Lymphoma Highlights Molecular Divergence and Therapeutic Opportunities: a LYSA Study. Clin Cancer Res Off J Am Assoc Cancer Res (2016) 22:2919–28. doi: 10.1158/1078-0432.CCR-15-2305
23. Lenz G, Wright GW, Emre NCT, Kohlhammer H, Dave SS, Davis RE, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci USA (2008) 105:13520–5. doi: 10.1073/pnas.0804295105
24. Kober-Hasslacher M, Schmidt-Supprian M. The Unsolved Puzzle of c-Rel in B Cell Lymphoma. Cancers (2019) 11:941–66. doi: 10.3390/cancers11070941
25. Kwiecinska A, Ichimura K, Berglund M, Dinets A, Sulaiman L, Collins VP, et al. Amplification of 2p as a genomic marker for transformation in lymphoma. Genes Chromosomes Cancer (2014) 53:750–68. doi: 10.1002/gcc.22184
26. Berglund M, Enblad G, Flordal E, Lui W-O, Backlin C, Thunberg U, et al. Chromosomal imbalances in diffuse large B-cell lymphoma detected by comparative genomic hybridization. Mod Pathol Off J U S Can Acad Pathol Inc (2002) 15:807–16. doi: 10.1097/01.MP.0000024375.04135.2B
27. Deng M, Zhang M, Xu-Monette ZY, Pham LV, Tzankov A, Visco C, et al. XPO1 expression worsens the prognosis of unfavorable DLBCL that can be effectively targeted by selinexor in the absence of mutant p53. J Hematol OncolJ Hematol Oncol (2020) 13:148. doi: 10.1186/s13045-020-00982-3
28. Mitsuhashi K, Masuda A, Wang Y-H, Shiseki M, Motoji T. Prognostic significance of PRAME expression based on immunohistochemistry for diffuse large B-cell lymphoma patients treated with R-CHOP therapy. Int J Hematol (2014) 100:88–95. doi: 10.1007/s12185-014-1593-z
29. Clark JM, Aleksiyadis K, Martin A, McNamee K, Tharmalingam T, Williams RO, et al. Inhibitor of kappa B epsilon (IκBϵ) is a non-redundant regulator of c-Rel-dependent gene expression in murine T and B cells. PloS One (2011) 6:e24504. doi: 10.1371/journal.pone.0024504
30. Li L, Xu-Monette ZY, Ok CY, Tzankov A, Manyam GC, Sun R, et al. Prognostic impact of c-Rel nuclear expression and REL amplification and crosstalk between c-Rel and the p53 pathway in diffuse large B-cell lymphoma. Oncotarget (2015) 6:23157–80. doi: 10.18632/oncotarget.4319
31. Odqvist L, Montes-Moreno S, Sánchez-Pacheco RE, Young KH, Martín-Sánchez E, Cereceda L, et al. NFκB expression is a feature of both activated B-cell-like and germinal center B-cell-like subtypes of diffuse large B-cell lymphoma. Mod Pathol Off J U S Can Acad Pathol Inc (2014) 27:1331–7. doi: 10.1038/modpathol.2014.34
32. Pham LV, Fu L, Tamayo AT, Bueso-Ramos C, Drakos E, Vega F, et al. Constitutive BR3 receptor signaling in diffuse, large B-cell lymphomas stabilizes nuclear factor-κB-inducing kinase while activating both canonical and alternative nuclear factor-κB pathways. Blood (2011) 117:200–10. doi: 10.1182/blood-2010-06-290437
33. Li S-Z, Zhang H-H, Liang J-B, Song Y, Jin B-X, Xing N-N, et al. Nemo-like kinase (NLK) negatively regulates NF-kappa B activity through disrupting the interaction of TAK1 with IKKβ. Biochim Biophys Acta (2014) 1843:1365–72. doi: 10.1016/j.bbamcr.2014.03.028
34. Lei L, Wang Y, Zheng Y-W, Fei L-R, Shen H-Y, Li Z-H, et al. Overexpression of Nemo-like Kinase Promotes the Proliferation and Invasion of Lung Cancer Cells and Indicates Poor Prognosis. Curr Cancer Drug Targets (2019) 19:674–80. doi: 10.2174/1568009618666181119150521
35. Bouamar H, Abbas S, Lin A-P, Wang L, Jiang D, Holder KN, et al. A capture-sequencing strategy identifies IRF8, EBF1, and APRIL as novel IGH fusion partners in B-cell lymphoma. Blood (2013) 122:726–33. doi: 10.1182/blood-2013-04-495804
36. Quemeneur L, Angeli V, Chopin M, Jessberger R. SWAP-70 deficiency causes high-affinity plasma cell generation despite impaired germinal center formation. Blood (2008) 111:2714–24. doi: 10.1182/blood-2007-07-102822
37. Bradford JW, Baldwin AS. IKK/nuclear factor-kappaB and oncogenesis: roles in tumor-initiating cells and in the tumor microenvironment. Adv Cancer Res (2014) 121:125–45. doi: 10.1016/B978-0-12-800249-0.00003-2
38. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature (2009) 462:108–12. doi: 10.1038/nature08460
39. Hezaveh K, Kloetgen A, Bernhart SH, Mahapatra KD, Lenze D, Richter J, et al. Alterations of microRNA and microRNA-regulated messenger RNA expression in germinal center B-cell lymphomas determined by integrative sequencing analysis. Haematologica (2016) 101:1380–9. doi: 10.3324/haematol.2016.143891
40. Rodig SJ, Kutok JL, Paterson JC, Nitta H, Zhang W, Chapuy B, et al. The pre-B-cell receptor associated protein VpreB3 is a useful diagnostic marker for identifying c-MYC translocated lymphomas. Haematologica (2010) 95:2056–62. doi: 10.3324/haematol.2010.025767
41. Ding J, Dirks WG, Ehrentraut S, Geffers R, MacLeod RAF, Nagel S, et al. BCL6–regulated by AhR/ARNT and wild-type MEF2B–drives expression of germinal center markers MYBL1 and LMO2. Haematologica (2015) 100:801–9. doi: 10.3324/haematol.2014.120048
42. Neo WH, Lim JF, Grumont R, Gerondakis S, Su I. c-Rel Regulates Ezh2 Expression in Activated Lymphocytes and Malignant Lymphoid Cells. J Biol Chem (2014) 289:31693–707. doi: 10.1074/jbc.M114.574517
43. Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell (2020) 37:551–568.e14. doi: 10.1016/j.ccell.2020.03.015
44. Kaileh M, Sen R. NF-κB function in B lymphocytes. Immunol Rev (2012) 246:254–71. doi: 10.1111/j.1600-065X.2012.01106.x
45. Heise N, De Silva NS, Silva K, Carette A, Simonetti G, Pasparakis M, et al. Germinal center B cell maintenance and differentiation are controlled by distinct NF-κB transcription factor subunits. J Exp Med (2014) 211:2103–18. doi: 10.1084/jem.20132613
46. Grumont RJ, Rourke IJ, Gerondakis S. Rel-dependent induction of A1 transcription is required to protect B cells from antigen receptor ligation-induced apoptosis. Genes Dev (1999) 13:400–11. doi: 10.1101/gad.13.4.400
47. Damdinsuren B, Zhang Y, Khalil A, Wood WH, Becker KG, Shlomchik MJ, et al. Single round of antigen receptor signaling programs naive B cells to receive T cell help. Immunity (2010) 32:355–66. doi: 10.1016/j.immuni.2010.02.013
48. Rizzo D, Viailly P-J, Mareschal S, Bohers E, Picquenot J-M, Penther D, et al. Oncogenic events rather than antigen selection pressure may be the main driving forces for relapse in diffuse large B-cell lymphomas. Am J Hematol (2017) 92:68–76. doi: 10.1002/ajh.24584
Keywords: NF-kappaB, DNA binding activity, c-Rel, germinal center B-cell–diffuse large B-cell lymphoma, genetic alterations
Citation: Faumont N, Taoui O, Collares D, Jais J-P, Leroy K, Prévaud L, Jardin F, Molina TJ, Copie-Bergman C, Petit B, Gourin M-P, Bordessoule D, Troutaud D, Baud V and Feuillard J (2021) c-Rel Is the Pivotal NF-κB Subunit in Germinal Center Diffuse Large B-Cell Lymphoma: A LYSA Study. Front. Oncol. 11:638897. doi: 10.3389/fonc.2021.638897
Received: 07 December 2020; Accepted: 15 March 2021;
Published: 20 April 2021.
Edited by:
Stefano Luminari, University of Modena and Reggio Emilia, ItalyReviewed by:
Valentina Tabanelli, European Institute of Oncology (IEO), ItalyAlessandro Isidori, AORMN Hospital, Italy
Copyright © 2021 Faumont, Taoui, Collares, Jais, Leroy, Prévaud, Jardin, Molina, Copie-Bergman, Petit, Gourin, Bordessoule, Troutaud, Baud and Feuillard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Véronique Baud, dmVyb25pcXVlLmJhdWRAaW5zZXJtLmZy; Jean Feuillard, amVhbi5mZXVpbGxhcmRAdW5pbGltLmZy