 Mahnaz Norouzi1
Mahnaz Norouzi1 Mohammad Shafiei1*†
Mohammad Shafiei1*† Zeinab Abdollahi2
Zeinab Abdollahi2 Paniz Miar2Hamid Galehdari1Mohammad Hasan Emami3
Paniz Miar2Hamid Galehdari1Mohammad Hasan Emami3 Mehrdad Zeinalian2
Mehrdad Zeinalian2 Mohammad Amin Tabatabaiefar2,4*†
Mohammad Amin Tabatabaiefar2,4*†- 1Department of Biology, Faculty of Sciences, Shahid Chamran University of Ahvaz, Ahvaz, Iran
- 2Department of Genetics and Molecular Biology, School of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran
- 3Department of Gastroenterology, Poursina Hakim Digestive Disease Research Center, Isfahan University of Medical Sciences, Isfahan, Iran
- 4Pediatric Inherited Diseases Research Center, Research Institute for Primordial Prevention of Noncommunicable Disease, Isfahan University of Medical Sciences, Isfahan, Iran
Background: Familial cancers comprise a considerable distribution of colorectal cancers (CRCs), of which only about 5% occurs through well-established hereditary syndromes. It has been demonstrated that deleterious variants at the newly identified cancer-predisposing genes could describe the etiology of undefined familial cancers.
Methods: The present study aimed to identify the genetic etiology in a 32-year-old man with early onset familial CRC employing several molecular diagnostic techniques. DNA was extracted from tumoral and normal formalin-fixed-paraffin-embedded (FFPE) blocks, and microsatellite instability (MSI) was evaluated. Immunohistochemistry staining of MMR proteins was performed on tumoral FFPE blocks. Next-generation sequencing (NGS), multiplex ligation-dependent amplification (MLPA) assay, and Sanger sequencing were applied on the genomic DNA extracted from peripheral blood. Data analysis was performed using bioinformatics tools. Genetic variants interpretation was based on ACMG.
Results: MSI analysis indicated MSI-H phenotype, and IHC staining proved no expressions of MSH2 and MSH6 proteins. MLPA and NGS data showed no pathogenic variants in MMR genes. Further analysis of NGS data revealed a candidate WRN frameshift variant (p.R389Efs*3), which was validated with Sanger sequencing. The variant was interpreted as pathogenic since it met the criteria based on the ACMG guideline including very strong (PVS1), strong (PS3), and moderate (PM2).
Conclusion: WRN is a DNA helicase participating in DNA repair pathways to sustain genomic stability. WRN deficient function may contribute to CRC development that is valuable for further investigation as a candidate gene in hereditary cancer syndrome diagnosis.
Introduction
Hereditary cancer syndromes account for a small percentage of all cancers diagnosed each year, although they are common in some cancer forms, such as colorectal and breast cancers (1, 2). Colorectal cancer (CRC) is considered one of the most frequent malignancies worldwide, and a family history of the condition is a major risk factor (1, 3). Notably, the relative risk of developing CRC increases in individuals with a family history of CRC. Alongside screening and surveillance, another advantage of determining the molecular basis of familial CRC is its therapeutic relevance (4, 5). Meanwhile, chromosomal instability arises in a remarkable portion of CRC tumors. Microsatellite instability (MSI), which is caused by the aberrant function of the DNA mismatch repair (MMR) system, accounts for nearly 15% of all CRC patients. In fact, insertion or deletion of a repetitive unit in a short tandemly repeated sequence (STR), designated as MSI, is a robust diagnostic marker for identifying Lynch syndrome (LS) suspicious individuals (6–8).
LS is an autosomal dominant hereditary cancer syndrome caused by germline defects in the DNA mismatch repair genes, including MLH1, MSH2, MSH6, PMS2, and EPCAM (9, 10). Most of the pathogenic variants of MMR genes include small insertions/deletions or large genetic rearrangements, missense, and nonsense variants (11–13). The majority of these gene defects were found in MLH1 and MSH2 (60 to 80%) genes. Other detrimental variants in MSH6 or PMS2 and, rarely, to EpCAM genes have been identified associated with Lynch phenotype. Nevertheless, recent population studies have found a higher frequency for germline pathogenic variants in MSH6 and PMS2 genes (14–16). It was demonstrated that these genes encompass lower penetrance pathogenic variants, and MSH6 deleterious variant carriers represent late-onset cancer compared to MLH1 or MSH2 carriers (13). Moreover, an increased risk of extracolonic tumors, particularly endometrial cancer, has been shown in MSH2 pathogenic variant carriers (13). Endometrial and ovarian cancers are the most prevalent Lynch-associated tumors. Other associated cancers include pancreatic, biliary tract, ureter, renal pelvis, gastric, small intestine, brain, sebaceous adenomas, sebaceous carcinomas, and keratoacanthomas (9).
LS is also known as hereditary non-polyposis colorectal cancer (HNPCC), with an approximate prevalence of one in 600 to one in 3,000 individuals (17, 18). HNPCC is a heterogeneous group of disorders with overlapping features that are clinically evaluated using Amsterdam (I or II) or Bethesda guidelines (19–22). Clinical guidelines are based on family and personal history of cancer, and their use as diagnostic tools for LS is restricted due to their low sensitivity. Patients with HNPPCC are at increased risk for metachronous or synchronous colon and extracolonic Lynch-related cancers. Since many patients with Lynch syndrome (UP TO 50%) do not even meet the revised Bethesda guidelines, an alternative approach referred to as “universal tumor screening” is recommended. Both MMR immunohistochemistry (IHC) staining and MSI analyses (either by themselves or in combination) are used to test MMR system proteins in all newly diagnosed CRC patients (9, 10). MSI is not exclusive to the Lynch-related CRCs. It may be observed in other CRC tumors. Somatic MLH1 gene inactivation due to promoter hypermethylation characterizes sporadic MMR deficient (dMMR) colorectal tumors, which typically arise in patients with late-onset cancer (12, 13). Unexplained dMMR CRC, termed “Lynch-like syndrome”, is another MSI colorectal tumor for which neither the deleterious germline variant of MMR genes nor the hypermethylation of the MLH1 gene promoter can be identified (12, 15, 23). Somatic bi-allelic mutations in the MMR genes may explain a large number of these Lynch-like dMMR tumors (nearly 70%). However, the etiology of a large number of these unclassified MSI/dMMR tumors is unclear (12, 23, 24). Moreover, dMMR CRCs encompass around 60% of HNPCC cases. About 40% of HNPCC patients do not represent MMR defects, and they are accordingly introduced as familial colorectal cancer type X (FCCTX) with undefined etiology. FCCTX is a group of heterogeneous diseases characterized by microsatellite stable (MSS) and late-onset clinically different from LS (21, 25, 26). Despite the recent advances in gene identification technologies, the genetic etiology of many hereditary cancers remains undefined, especially FCCTX groups (27, 28).
By the poor clinical diagnostic approaches, it is difficult to differentiate Lynch CRC tumors from non-hereditary ones due to the high frequency of MSI occurrence in sporadic tumors. This situation becomes more complicated when there is a positive family history of developing CRC or different related cancer types (19). In the present study, a CRC-affected individual from a fulfilled Amsterdam II criteria pedigree, showing Lynch-associated MSI and IHC phenotypes, underwent whole-exome sequencing (WES). The analysis of WES data revealed no pathogenic variants in any of the MMR genes. Further evaluation unveiled a frameshift variant in WRN (RECQL2; HGNC ID: 12791) gene as a potential candidate gene deserving subsequent considerations.
WRN encodes a DNA helicase enzyme from the highly conserved RECQ gene family and has a pivotal role in DNA repair and maintenance of genomic stability (29, 30). The WRN has both helicase and exonuclease activity and has been considered a genomic caretaker (31). Homozygous truncating variants in the WRN gene cause Werner syndrome (WS; OMIM 277700) manifests, which are characterized by early onset age-associated diseases such as cancer (32, 33). In addition to the elevated risk of developing cancer in WS patients, several studies have indicated a tumor suppressor-like function for WRN, while making it a prominent cancer-predisposing gene (34, 35). More importantly, various evaluations have suggested a crucial role for WRN associated with different types of familial cancers, specifically breast and ovarian cancers (36–38). However, the association of germline WRN gene variants with familial CRC has not been established well. Herein, we aimed to introduce a truncating variant in WRN (NM_000553:exon9:p.R389Efs*3) as a pathogenic candidate based on ACMG standard guideline (39) in a patient affected by early onset familial CRC.
Subject and Methods
Ethics Statement
The Ethics Committee of Shahid Chamran University of Ahvaz approved the present study (Ref. EE/97.24.3.70385/scu.ac.ir). After genetic counseling and obtaining a comprehensive family history, we received informed written consent.
Clinical Evaluation
A CRC affected patient in a family with different types of Lynch-associated cancer (n = 6), CRC, breast, and brain, was recruited from our other study on MSI optimizing in Lynch suspected patients (not published yet). An expert clinical pathologist assessed clinical and pathological data of surgically resected tumors.
DNA Extraction and MSI Analysis
Whole blood and the formalin-fixed-paraffin-embedded (FFPE) blocks were obtained, and DNA extraction was performed based on standard methods. The evaluation of the quality and quantity of the extracted DNA was performed by agarose gel electrophoresis and nanodrop (Thermo Scientific, NanoDrop One C). Promega MSI Analysis version 1.2, (Promega, USA) was utilized containing fluorescently labeled five mononucleotide and two pentanucleotide markers to compare MSI status on tumoral and matched normal tissues according to the protocol. Fragment analysis was performed using the ABI3500 genetic analyzer (Applied Biosystems) and gene marker software V 1.85. The cutoff of MSI-H classification was two or more unstable markers (≥30%) (40).
Immunohistochemistry
Immunohistochemistry analysis was carried out to evaluate MMR protein expression employing mouse monoclonal antibodies against MSH2 (Leica Biosystems: Novocastra, UK, Lyophilized, Product Code (PC): NCL-MSH2; at 1/80 dilution), MLH1 (Leica Biosystems: Novocastra, UK, Liquid, PC: NCL-L-MLH1; at 1/100 dilution), MSH6 (Leica Biosystems: Novocastra, UK, Liquid, PC: NCL-L-MSH6; at 1/100 dilution), and PMS2 (Leica Biosystems: Novocastra, UK, Liquid, PC: NCL-L-PMS2; at 1/100 dilution). In brief, 4 µm-thick FFPE sections were initially de-waxed and rehydrated by using xylene and graded ethanol, respectively. The sections were placed in citrate buffer (10 mmol/L, pH 6) and preheated at 99°C for 30 min. Endogenous peroxidase activity was quenched by exposure to 3% hydrogen peroxidase for 5 min. Subsequently, the sections were incubated for 20 min at room temperature with monoclonal antibodies. The detection was performed by applying Ultra-vision streptavidin-biotin peroxidase kit (Dako, Carpinteria, CA) according to the manufacturer’s instructions. The complete absence of each MMR protein expression in neoplastic cells was considered MMR deficient (7).
Whole Exome Sequencing and Data Analysis
The DNA extracted from peripheral blood was fragmented with the hydrodynamic shearing System (Covaris, Massachusetts, USA). Afterward, Agilent SureSelect Human All Exon kit (Agilent Technologies, CA, USA) was used for enriching the exonic library. The uncaptured DNA was washed, and the trapped DNA was subjected to sequencing on Illumina NovaSeq 6000 with over 100× coverage. The generated fastq file was mapped to the reference genome (hg19, NCBI Build 37) employing the BWA aligner tool. The identified single nucleotide (SNV) and Indel variants, detected by GATK- v3.1, were annotated by ANNOVAR.
Primarily, based on the ACMG guidelines for genetic testing of inherited CRC (41), and the ACMG standard and guidelines for the interpretation of sequence variants (39) we evaluated the pathogenicity of the identified variants in highly associated genes, MSH2, MSH6, EpCAM, MLH1, and PMS2. We also searched the InSIGHT database for the known MMR gene pathogenic variants. Furthermore, the coverage of sequencing in the mentioned gene regions was assessed using IGVD software. Afterward, public databases were employed, including dbSNP version 147, 1000 Genome Project phase 3, and Exome Aggregation Consortium (ExAC), to filter the variants based on minor allele frequency (MAF < 1%). Further filtration was performed based on the functional consequence and inheritance pattern. The Human Gene Card Database (GeneCards) was utilized to prioritize the remaining gene variants according to the clinical phenotype. Catalog of Somatic Mutation Database (COSMIC), Human Gene Mutation Database (HGMD), and ClinVar were investigated, and literature was exhaustively reviewed to figure out deleterious variants associated with the disease.
MLPA
Multiplex ligation-dependent amplification assay (MLPA) was performed using MLH1/MSH2 MLPA probe mix (MRC-Holland Amsterdam, the Netherlands, Lot N: D1-0815). According to the manufacturer’s instruction, the thermal cycles were performed at 98°C to denature 5 µl of genomic DNA and prepare it for hybridization at 60°C with MLPA probes. Following overnight hybridization, the ligation process was done at 54°C for 15 min. After the inactivation of ligase enzyme at 98°C Polymerase mastermix and SALSA PCR primer mix were added, followed by 35 cycles of thermocycler program; including 30 s at 95°C; 30 s at 60°C; 30 s at 72°C; and 20 min at 72°C. PCR products were separated via capillary electrophoresis on ABI 3500 genetic analyzer (Applied Biosystems, USA). The data were collected and analyzed through Coffalyser software.
Sanger Sequencing
Primer 3 software was used to design appropriate primers for WRN exon 9 forward (F-AAAAGCTTCACAGTTTGTCCTTG) and reverse (R-GGGAATAAAGTCTGCCAGAACC) primers. To design the relevant forward and reverse primers of MSH2 (F-TGGGACATAGCAGGCCATAT and R-GCGGCAACCAATCATAAGCA) and MSH6 (F-CTGGGTCAAAGGGCTCAGAT and R-TCGGACGGAGCTCCTAAAAG) gene promoter regions (42–44) Primer 3 software was used. PCR reaction was carried out using specific primers. The PCR products were subjected to Sanger sequencing on an ABI3500 genetic analyzer (Applied Biosystems, USA) instrument according to the standard protocols. Obtained sequences were aligned against the human reference genome (hg19, NCBI Build 37) using MEGA-X software.
Results
Clinical Features and Molecular Profiling
The patient was a 32-year-old man from an inbreed family, subjected to partial colectomy due to CRC adenocarcinoma. Amsterdam II criteria were fulfilled considering maternal and paternal family history (Figure 1A). CRC was diagnosed at the age of 32 for the proband (III:1). The affected individuals existed in two generations; two of them were first-degree relatives. The proband’s younger brother (III:2) died because of neuroblastoma at the age of 6. Six individuals were affected with different types of cancers in this family, and five of whom were diagnosed with malignant lesions under the age of 50. The spectrum of extracolonic cancers included neuroblastoma, breast, and brain tumors in this family. A 42-year-old female was diagnosed with breast carcinoma at the age of 38 (II:8). Two individuals, II:6 and II:1, passed away because of CRC at the age of 60 and 49 years, respectively. II:7 died due to brain cancer at the age of 47.
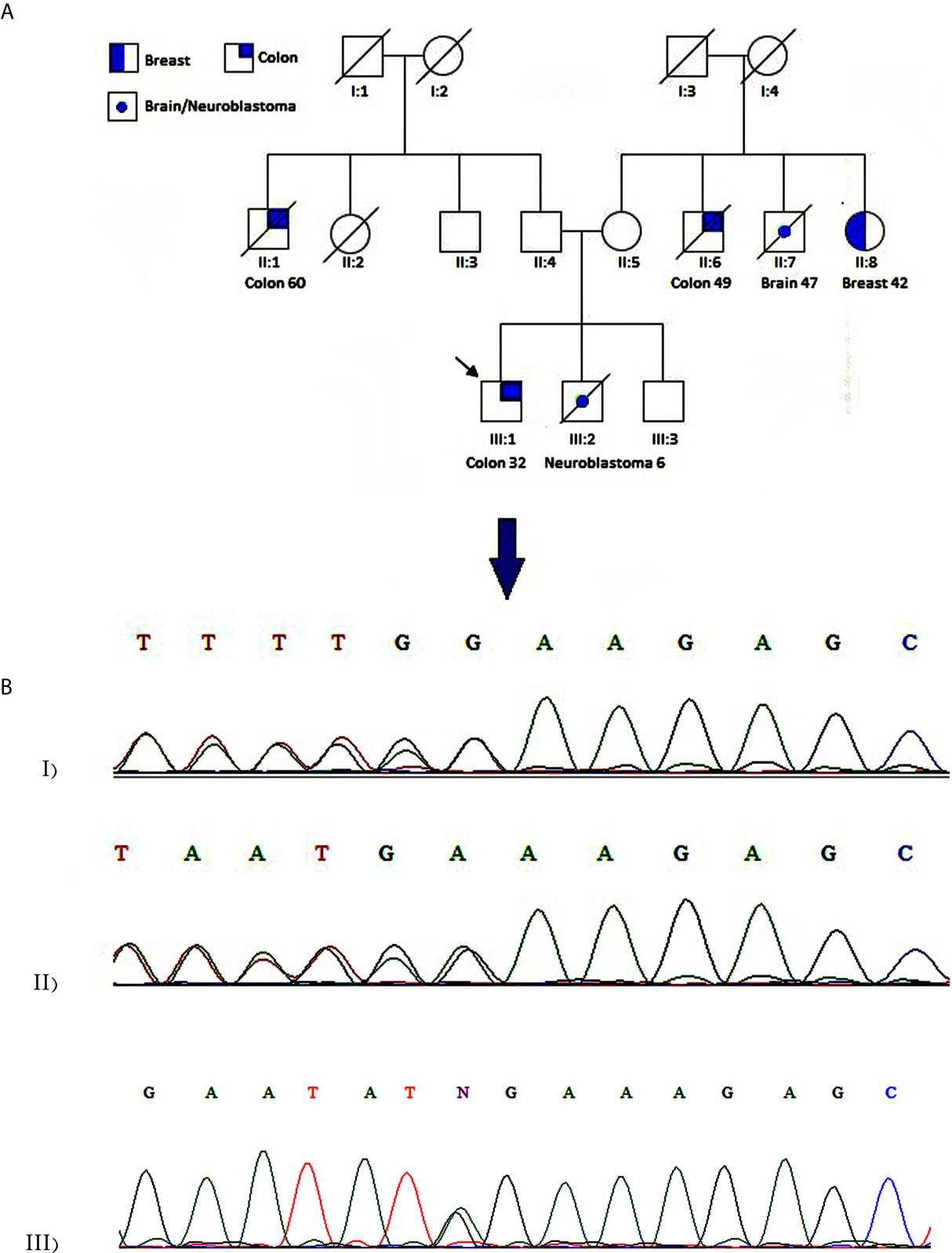
Figure 1 Case presentation (A) Pedigree of the family. The panel shows a family affected with different types of cancer. An arrow designates the studied patient. Colon and breast cancers are indicated by half and quarter filled colors, respectively. A central spot demonstrates neuroblastoma or brain cancer. (B) Chromatogram showing heterozygous c.1163delA mutation in WRN gene for proband (I) and patient II:8 (II). A homozygous wild-type variant was detected in the proband’s unaffected brother (III).
MSI analysis was performed on the DNA samples extracted from FFPE blocks of the proband (III:1). The result implied that instability existed in five Promega mononucleotide markers, BAT-25, BAT-26, NR-21, NR-24, and MONO-27, in the tumoral DNA compared to the normal tissue (Figure 2A) indicating MSI-H profile. IHC analysis using MLH1, MSH2, MSH6, and PMS2 antibodies showed loss of MSH2 and MSH6 proteins in CRC tumor tissue. MLH1 and PMS2 antibodies represented the intact expression of these two proteins (Figure 2B).
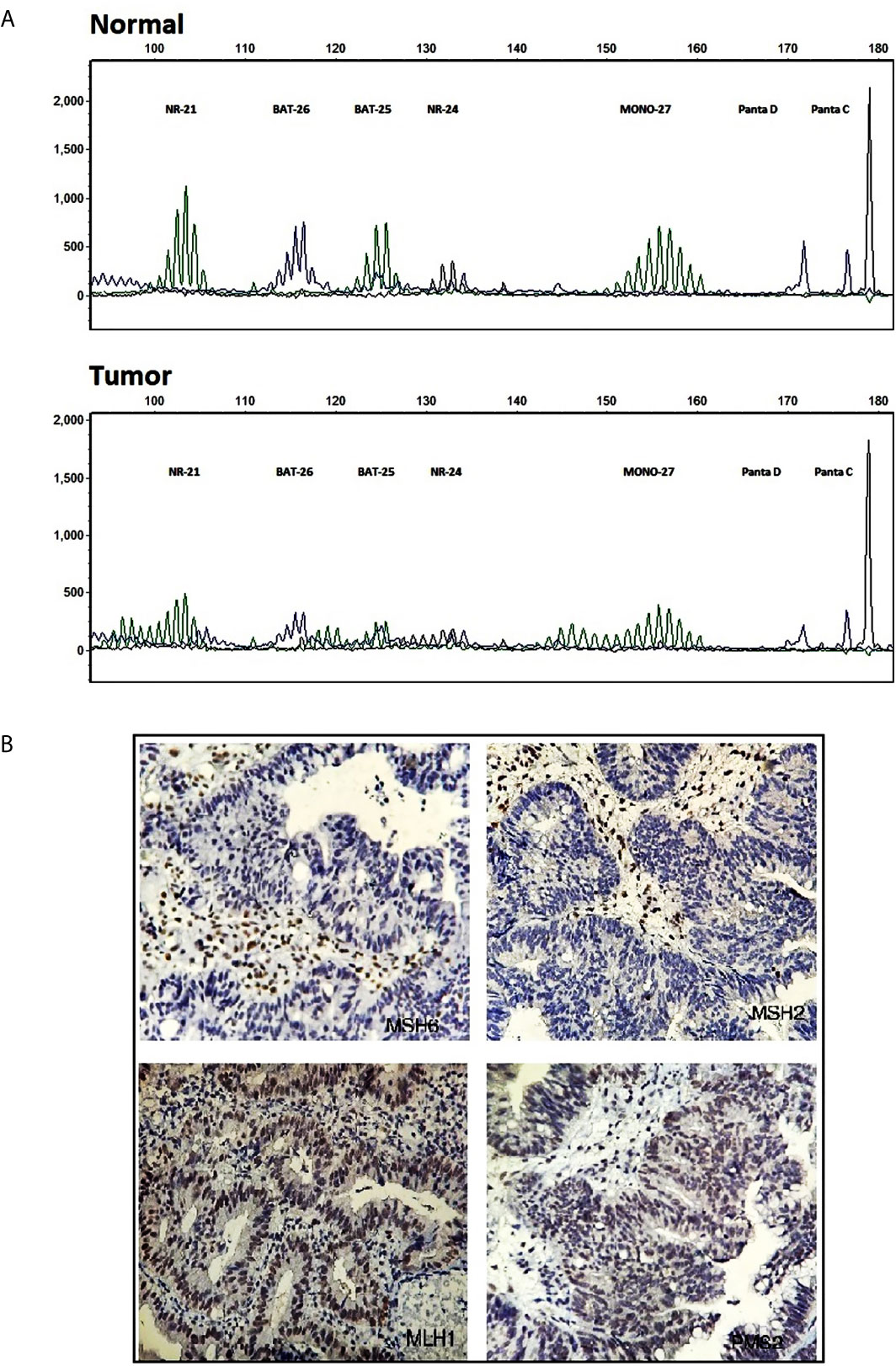
Figure 2 Molecular testing results. (A) Fragment profile of the Promega MSI analysis system (GeneMarker v1.85 Software). Instability is detectable at tumoral tissue (bottom) compared to normal at BAT-25, BAT-26, NR-21, NR-24, and MONO27 markers. (B) Immunohistochemistry staining for MMR system proteins, MSH2, MSH6 (mutant) MLH1, PMS2 (intact), in tumoral tissue.
MMR Evaluation
The CRC-affected patient (III:1), whose clinical and molecular examination suggested hereditary Lynch phenotype, was subjected to WES. Considering full coverage Lynch-association gene loci, we achieved 99 variants, eight of which were in the exonic regions. Among these MMR exonic variants, there were no deleterious variants except for three non-synonymous variants in MLH1, MSH6, and EpCAM with MAF >1%. Furthermore, we performed Sanger sequencing by using specific primers of the promoter regions of MSH2 and MSH6 genes (44, 45). Sequence alignment against the human reference genome (hg19, NCBI Build 37) detected no deleterious variant in the corresponding regions (Supplementary Figure 1). In general, none of these variants were recognized as pathogenic based on the ACMG guideline and the InSIGHT database (39). To trace down large deletion or duplication MLPA was performed on the patient’s genomic DNA, and no deleterious variants were detected at MLH1 or MSH2 genes (Figure 3).
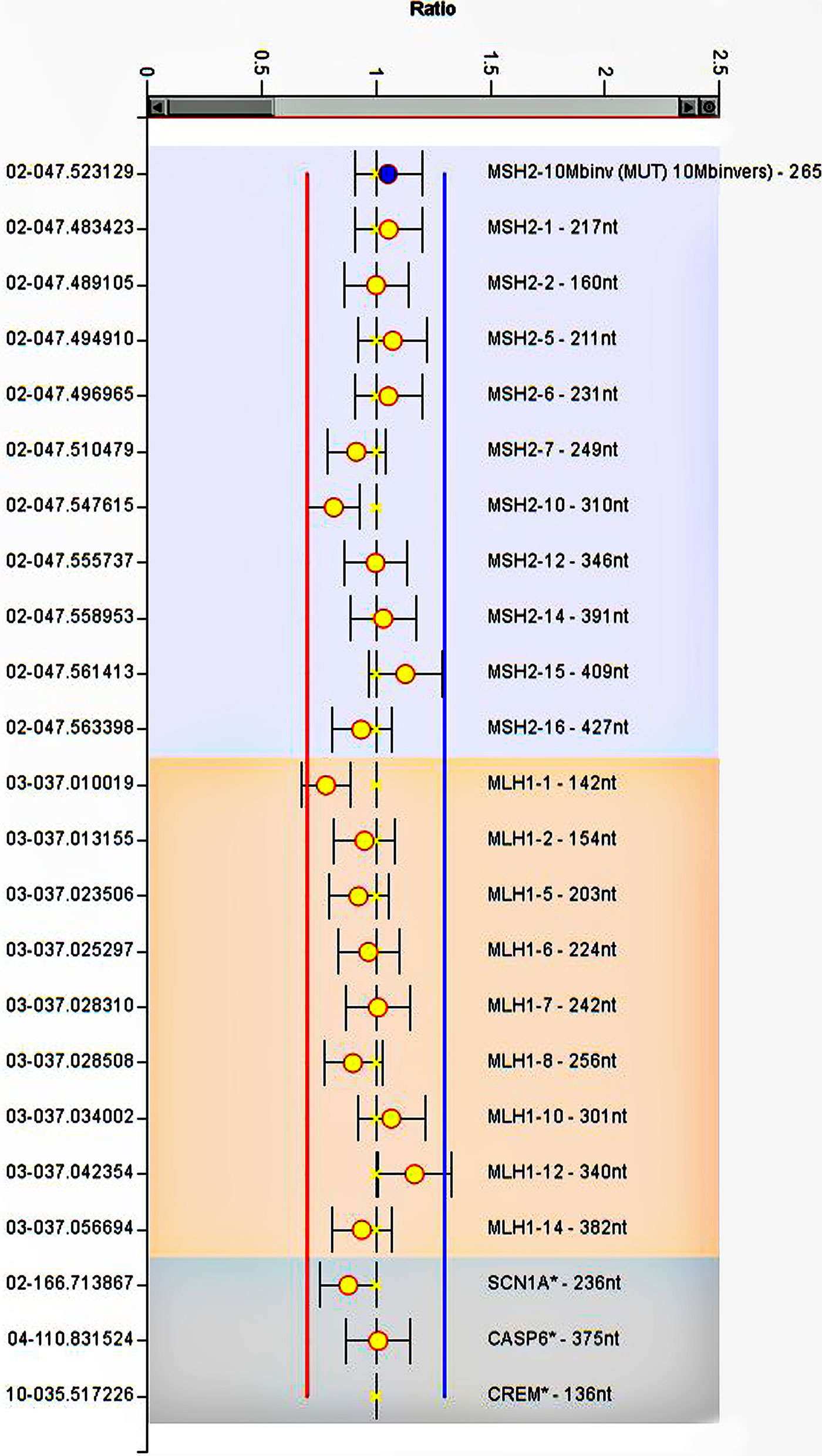
Figure 3 MLPA analysis of MLH1 and MSH2. The image represents no deletion or duplication at gene regions. The error bars show standard deviation. Ratios of the signals normalized to the control reference individual.
Pathogenic Variant Identification and Validation
Screening for prioritized genes provided us a list of candidate exonic variants. Regardless of synonymous and non-synonymous variants, we narrowed down our investigation by incorporating genes harboring truncating variants, including nonsense, start loss, splice site, or frameshift variants (Table 1). After a comprehensive database survey and comprehensive literature review, we reached a frameshift variant in WRN gene exon9:c.1163delA (p.R389Efs*3). It introduced a truncating gene product supported by functional studies to render pathogenicity (34, 35, 39, 46). Population data indicates a frequency lower than 0.0001 in genomeAD-Exome Asian and ExAC Asian databases. Moreover, no frequency was reported in genomeAD-genome East Asian and in-house databases Iranome for instance. The variant was represented as pathogenic or likely-pathogenic at the ClinVar database (Accession number: RCV000232269.6) for WS. Sanger sequencing data using designed primers confirmed exon9:c.1163delA (GRCh37.p13) as diagnosed through WES in a heterozygous pattern in the proband and patient II:8 (Figure 1B). Proband’s unaffected brother (III:3), aged 38 was homozygote for the wild type of the variant. Unfortunately, we could not have access to the DNA samples of other family members to complete the cosegregation analysis.
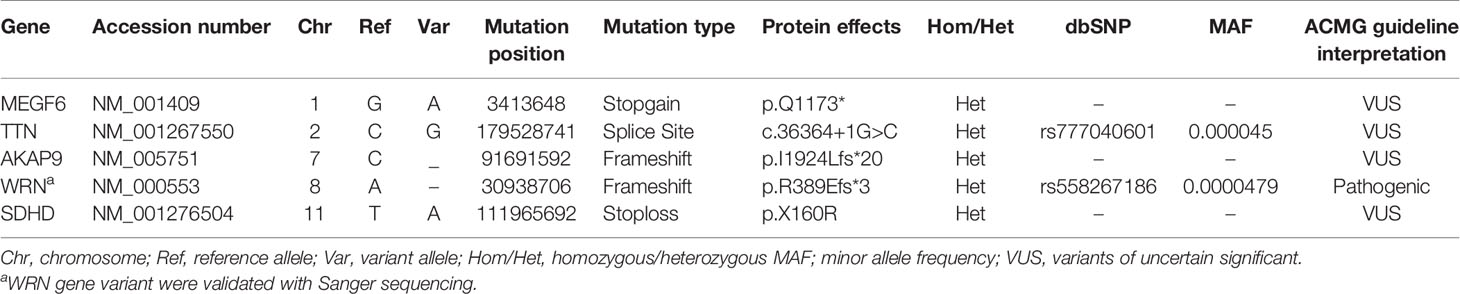
Table 1 Truncating heterozygous variants (i.e., putative frameshifts, stopgain, nonsense mutations) in candidate CRC predisposing genes.
Discussion
While many cancers have a positive family history, pathogenic variants in the well-known cancer-predisposition genes are responsible for up to 10% of hereditary cancers. The number of genes involved in cancer susceptibility is growing and further clinical and molecular evidence is needed to explain the physiopathology of these variants in cancer (47).
In the present study, we investigated the genetic etiology of the familial CRC in a pedigree meeting the Amsterdam criteria (48, 49). Since the clinical and pathological data and molecular analysis strongly implicated the LS (41), we originally focused on the evaluation of MMR system gene variants. Furthermore, we performed Sanger sequencing to assess the presence of any candidate pathogenic variant in the MSH2 and MSH6 gene promoters (43, 45). According to the InSIGHT database and the ACMG guideline, no pathogenic variant was identified in the exonic and regulatory elements of MMR genes. Moreover, MLPA testing was done. The data ruled out the presence of deleterious duplication or deletion in MLH1 or MSH2 genes (50). Accordingly, reanalysis of exome data through GeneCard and COSMIC dataset resulted in a list of relevant gene variants (Table 1). The subsequent assessment suggested that a WRN frameshift pathogenic variant (p.R389Efs*3) could be related to the etiology. Interestingly, the proband’s maternal aunt, II:8, suffering from breast cancer, was found to carry this variant.
Although WRN has been introduced as a candidate gene for hereditary breast cancer (37), the inheritance of the variant from the maternal side has sparked a debate on its role in developing colorectal cancer. The observation of dMMR phenotype might suggest the possibility of developing sporadic CRC due to bi-allelic somatic inactivation of MSH2 and MSH6 genes (12, 24). However, the positive history of cancer in this family and the patient’s young age weaken this assumption in the proband. Zimmer et al. showed that 56% of CRC tumors with bi-allelic mutant WRN gene presented MSI-H/dMMR status (51). The wide spectrum of reported cancers related to different tumor suppressor genes (BRCA1, BRCA2, MSH2, MLH1, etc.) would support the hypothesis that WRN, which has been previously confirmed as a candidate gene for hereditary breast cancer, may also lead to CRC (52, 53).
However, there remains a slight chance for the inheritance of an MMR gene pathogenic variant from the paternal side possibly not detected through the applied methods in the present study. According to several studies, MSI has a multi-pathway mechanism, and non-mutational events and implications of novel genes may explain MSI/dMMR tumors with no detectable germline mutation (32, 54, 55). For example, it was demonstrated that BCL2 gene overexpression, commonly expressed in various tumor types, can downregulate the MSH2 gene and subsequently reduce MMR protein cellular activity via the hypophosphorylation of pRb protein (56). Likewise, it was revealed that WRN gene silencing is linked to the MSI in CRC tumors and in vitro MSI models (54, 57). In this regard, WRN plays an essential role in the survival of MSI/dMMR cancer cell lines (not in MMR-proficient and MSS cells). It has been proposed that WRN is involved in an EXO1 independent pathway for mismatch repair in MSI-H cells (58, 59). Besides, MSI-H/dMMR high frequency in the WRN mutant CRC tumors would support this hypothesis that WRN may be involved in the MSI events as an alternative pathway in undefined dMMR CRCs (51). Our finding in the present study will put forth WRN as a novel gene possibly leading to CRC associated with MSI, although its exact mechanism remains unknown.
The identified variant was interpreted as pathogenic based on the ACMG guideline. Since this premature stop codon variant disrupted the helicase domain, the protein function is lost (PVS1) (39, 60). The frequency of this variant was notably low in genomeAD-Exome Asian and ExAC Asian databases and even absent in genomeAD-genome East Asian population data (PM2). This variant was represented in ClinVar (Variation ID: 238117) associated with WS (PS3). The functional assessments indicated that the damaging effects on WRN gene products were attributed to genomic instability in colorectal tumor cells (35). Furthermore, aberrant WRN function due to hemizygous gene loss was demonstrated in advanced clinical CRC (61). In addition, the genetic instability revealed in cell lines from WS patients indicates haploinsufficiency for the WRN gene (32).
The defective WRN, a member of the RecQ helicase family, implicates WS (29). RecQ helicases are known as genome caretakers and play a critical role in genomic stability (62). The involvement of WRN, BLM, and RecQL4 family members has been determined in the pathogenesis of well-defined genetic disorders with an elevated risk of cancer (63). The WRN gene is located at 8p12 and contains 35 coding exons. The protein comprises ATPase, RQC, and HRDC domains, as well as an N-terminal exonuclease domain. It was shown that WRN participates in several DNA metabolic pathways, including replication, recombination, and repair to preserve the genome (30, 31, 64). To date, 140 pathogenic and 31 likely pathogenic variants have been identified in the WRN gene based on the ClinVar database. These variants include deletion, duplication, indel, insertion, and single nucleotide substitution leading to the destabilizing or protein failure to localization to the nucleus. The recessive truncating variants are described in association with WS well, suggesting that complete loss of WRN is necessary to develop the disease (32).
It was determined that truncating variants, which destroy the helicase activity of WRN, participate in WS pathogenesis (32, 60). The complete loss of WRN in WS patients born with a germline homozygous deleterious variant will lead to premature senescence and neoplastic alteration (65). WRN inactivation can induce DNA double-strand breaks and subsequently activate DNA damage responses such as apoptosis and cell cycle arrest (57). WS patients are susceptible to a variety of cancer types, including colorectal, ovarian, pancreatic, skin, and breast cancers, as well as soft tissue sarcomas, osteosarcomas, and leukemia (65, 66). A common pathogenic variant was shown to play a pivotal role in the genesis of thyroid carcinoma in Japanese WS patients. Furthermore, the association of the follicular and papillary carcinomas with the WRN c.3139-1G>C (exon 26 skip) and c.1105C>T (p.R369*) variants was found in WS patients, respectively (67, 68). Considering the significant frequency of different neoplasms in WS patients, WS was considered a heritable cancer predisposition syndrome. However, limited studies have investigated the role of WRN in developing cancer in non-WS individuals. Experiments revealed a degree of genetic instability for carriers of the WRN heterozygous pathogenic variants. These studies demonstrated that WRN inactivation promotes genetic instability in different human somatic cell lineages (32, 54, 68). However, it should be noted that WRN has a tumor suppressor function, and the loss of its second wild-type allele is necessary for carcinogenesis events to fulfill Knudson’s two-hit hypothesis of tumor suppressor genes (11).
Notably, epigenetic inactivation of the WRN gene was introduced as a common event in sporadic colorectal tumorigenesis as described for other DNA repair tumor suppressor genes, hMLH1 and BRCA1, involved in hereditary cancers (34). Furthermore, the aberrant function of the WRN gene in MSI-H CRC tumors, as a result of frameshift variants, suggested the probability of WRN contribution at MSI-H tumorigenesis as a putative tumor suppressor (35). Additionally, WRN’s contribution to breast cancer susceptibility has been demonstrated by Ding et al. They reported that a WRN specific variant (rs9649886), situated in the C-terminal of WRN, significantly affected breast cancer risk (36). Likewise, Zins et al. examined the significance of a WRN non-synonymous variant (rs1346044) with an elevated risk of breast cancer. They indicated that the risk of breast cancer increased in homozygous individuals due to the reduced helicase activity (38). Even though the potential role of WRN in promoting breast cancer was undetermined, more recently, a novel germline frameshift variant (p.N1370Tfs*23) has been identified in a Chinese family. The truncated variant was confirmed in three affected individuals and an unaffected first-degree relative. The identified variant was interpreted as pathogenic according to the ACMG guidelines, thereby introducing WRN as a potential candidate gene for hereditary breast cancer (37, 39).
In conclusion, hereditary cancer syndromes have provided a remarkable venue in cancer investigation, particularly in diagnostic and therapeutic strategies. Furthermore, the identification of at-risk individuals through germline mutation testing is a key to success in the prevention, early detection, and management of patients (21, 26). However, WRN has not been addressed as a hereditary CRC predisposing gene so far. We noticed a WRN pathogenic variant as a disease-causing variant in a familial CRC patient. Our data could be further authenticated by following up the family, performing additional functional studies, and precisely detecting the loss of heterozygosity feature in colorectal tumor cells. Nevertheless, the potential role of WRN in CRCs remains elucidated, and further studies are needed to reveal its role in hereditary cancers, HNPCC subtypes in particular.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author/s.
Ethics Statement
The study involving human participants was reviewed and approved by the Ethics Committee of Shahid Chamran University of Ahvaz (Ref. EE/97.24.3.70385/scu.ac.ir). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
MS and MT conceived and designed the study, obtained funding, developed methodology, reviewed, and revised the manuscript. MZ and ME managed patients and provided facilities. MN drafted the manuscript. HG revised the manuscript. MN, ZA, and PM performed molecular techniques and interpretation of data. All authors contributed to the article and approved the submitted version.
Funding
Shahid Chamran University of Ahvaz (grant number 97/3/02/26247) and Isfahan University of Medical Science (grant number 195216) financially supported this work.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all individuals who cooperated in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.648649/full#supplementary-material
References
1. Jass JR. Familial Colorectal Cancer: Pathology and Molecular Characteristics. Lancet Oncol (2000) 1(4):220–6. doi: 10.1016/S1470-2045(00)00152-2
2. Vahteristo P, Bartkova J, Eerola H, Syrjäkoski K, Ojala S, Kilpivaara O, et al. A CHEK2 Genetic Variant Contributing to a Substantial Fraction of Familial Breast Cancer. Am J Hum Genet (2002) 71(2):432–8. doi: 10.1086/341943
3. Roncucci L, Mariani F. Prevention of Colorectal Cancer: How Many Tools Do We Have in Our Basket? Eur J Intern Med (2015) 26(10):752–6. doi: 10.1016/j.ejim.2015.08.019
4. Aziz MA, Yousef Z, Saleh AM, Mohammad S, Al Knawy B. Towards Personalized Medicine of Colorectal Cancer. Crit Rev Oncol Hematol (2017) 118:70–8. doi: 10.1016/j.critrevonc.2017.08.007
5. Courtney RJ, Paul CL, Carey ML, Sanson-Fisher RW, Macrae FA, D’Este C, et al. A Population-Based Cross-Sectional Study of Colorectal Cancer Screening Practices of First-Degree Relatives of Colorectal Cancer Patients. BMC Cancer (2013) 13:13. doi: 10.1186/1471-2407-13-13
6. Heinen CD. Mismatch Repair Defects and Lynch Syndrome: The Role of the Basic Scientist in the Battle Against Cancer. DNA Repair (2016) 38:127–34. doi: 10.1016/j.dnarep.2015.11.025
7. Kawakami H, Zaanan A, Sinicrope FA. Microsatellite Instability Testing and Its Role in the Management of Colorectal Cancer. Curr Treat Options Oncol (2015) 16:30. doi: 10.1007/s11864-015-0348-2
8. Svrcek M, Lascols O, Cohen R, Collura A, Jonchère V, Fléjou J-F, et al. MSI/MMR-Deficient Tumor Diagnosis: Which Standard for Screening and for Diagnosis? Diagnostic Modalities for the Colon and Other Sites: Differences Between Tumors. Bull Cancer (2019) 106(2):119–28. doi: 10.1016/j.bulcan.2018.12.008
9. Gupta S, Provenzale D, Llor X, Halverson AL, Grady W, Chung DC, et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Colorectal, Version 2.2019: Featured Updates to the NCCN Guidelines. J Natl Compr Canc Netw (2019) 17:1032–41. doi: 10.6004/jnccn.2019.0044
10. Gupta S, Provenzale D, Regenbogen SE, Hampel H, Slavin TP, Hall MJ, et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Colorectal, Version 3.2017. J Natl Compr Canc Netw (2017) 15:1465–75. doi: 10.6004/jnccn.2017.0176
11. Duraturo F, Liccardo R, De Rosa M, Izzo P. Genetics, Diagnosis and Treatment of Lynch Syndrome: Old Lessons and Current Challenges. Oncol Lett (2019) 17:3048–54. doi: 10.3892/ol.2019.9945
12. Liccardo R, De Rosa M, Izzo P, Duraturo F. Novel Implications in Molecular Diagnosis of Lynch Syndrome. Gastroenterol Res Pract (2017) 2017:2595098. doi: 10.1155/2017/2595098
13. Sinicrope FA. Lynch Syndrome–Associated Colorectal Cancer. N Engl J Med (2018) 379(8):764–73. doi: 10.1056/NEJMcp1714533
14. Biller LH, Syngal S, Yurgelun MB. Recent Advances in Lynch Syndrome. Fam Cancer (2019) 18(2):211–9. doi: 10.1007/s10689-018-00117-1
15. Cohen SA, Pritchard CC, Jarvik GP. Lynch Syndrome: From Screening to Diagnosis to Treatment in the Era of Modern Molecular Oncology. Annual Review of Genomics and Human Genetics. Annu Rev Genomics Hum Genet (2019) 20:293–307. doi: 10.1146/annurev-genom-083118-015406
16. Seth S, Ager A, Arends MJ, Frayling IM. Lynch Syndrome–Cancer Pathways, Heterogeneity and Immune Escape. J Pathol (2018) 246(2):129–33. doi: 10.1002/path.5139
17. Obstetricians ACo, Gynecologists. Hereditary Cancer Syndromes and Risk Assessment. Committee Opinion No. 634. Obstet Gynecol (2015) 125(6):1538–43. doi: 10.1097/01.AOG.0000466373.71146.51
18. Giardiello FM, Allen JI, Axilbund JE, Boland RC, Burke CA, Burt RW, et al. Guidelines on Genetic Evaluation and Management of Lynch Syndrome: A Consensus Statement by the US Multi-Society Task Force on Colorectal Cancer. Dis Colon Rectum (2014) 57(8):1025–48. doi: 10.1097/DCR.000000000000000
19. Chen E, Xu X, Liu T. Hereditary Nonpolyposis Colorectal Cancer and Cancer Syndromes: Recent Basic and Clinical Discoveries. J Oncol (2018) 2018:3979135. doi: 10.1155/2018/3979135
20. Armelao F, de Pretis G. Familial Colorectal Cancer: A Review. World J Gastroenterol (2014) 20:9292. doi: 10.3748/wjg.v20.i28.9292
21. Da Silva FC, Wernhoff P, Dominguez-Barrera C, Dominguez-Valentin M. Update on Hereditary Colorectal Cancer. Anticancer Res (2016) 36:4399–405. doi: 10.21873/anticanres.10983
22. Takeda T, Tsuji K, Banno K, Yanokura M, Kobayashi Y, Tominaga E, et al. Screening for Lynch Syndrome Using Risk Assessment Criteria in Patients With Ovarian Cancer. Journal of Gynecologic Oncology. J Gynecol Oncol (2018) 29(3):e29. doi: 10.3802/jgo.2018.29.e29
23. Lefol C, Sohier E, Baudet C, Naïbo P, Ruano E, Grand-Masson C, et al. Acquired Somatic MMR Deficiency Is a Major Cause of MSI Tumor in Patients Suspected for “Lynch-Like Syndrome” Including Young Patients. European Journal of Human Genetics. Eur J Hum Genet (2020) 2020:1–7. doi: 10.1038/s41431-020-00778-6
24. Wheeler J, Beck N, Kim H, Tomlinson I, Mortensen NM, Bodmer W. Mechanisms of Inactivation of Mismatch Repair Genes in Human Colorectal Cancer Cell Lines: The Predominant Role of hMLH1. Proc Natl Acad Sci USA (1999) 96(18):10296–301. doi: 10.1073/pnas.96.18.1029696
25. Schulz E, Klampfl P, Holzapfel S, Janecke AR, Ulz P, Renner W, et al. Germline Variants in the SEMA4A Gene Predispose to Familial Colorectal Cancer Type X. Nat Commun (2014) 5:1–11. doi: 10.1038/ncomms6191
26. Valle L. Genetic Predisposition to Colorectal Cancer: Where We Stand and Future Perspectives. World J Gastroenterol (2014) 20:9828. doi: 10.3748/wjg.v20.i29.9828
27. DeRycke MS, Gunawardena SR, Middha S, Asmann YW, Schaid DJ, McDonnell SK, et al. Identification of Novel Variants in Colorectal Cancer Families by High-Throughput Exome Sequencing. Cancer Epidemiol Biomarkers Prev (2013) 22:1239–51. doi: 10.1158/1055-9965.EPI-12-1226
28. Powis Z, Espenschied CR, LaDuca H, Hagman KD, Paudyal T, Li S, et al. Clinical Germline Diagnostic Exome Sequencing for Hereditary Cancer: Findings Within Novel Candidate Genes Are Prevalent. Cancer Genet (2018) 224:12–20. doi: 10.1016/j.cancergen.2018.04.002
29. Futami K, Ishikawa Y, Goto M, Furuichi Y, Sugimoto M. Role of Werner Syndrome Gene Product Helicase in Carcinogenesis and in Resistance to Genotoxins by Cancer Cells. Cancer Sci (2008) 99:843–8. doi: 10.1111/j.1349-7006.2008.00778.x
30. Lebel M, Monnat RJ Jr. Werner Syndrome (WRN) Gene Variants and Their Association With Altered Function and Age-Associated Diseases. Ageing Res Rev (2018) 41:82–97. doi: 10.1016/j.arr.2017.11.003
31. Opresko PL, Calvo JP, von Kobbe C. Role for the Werner Syndrome Protein in the Promotion of Tumor Cell Growth. Mech Ageing Dev (2007) 128:423–36. doi: 10.1016/j.mad.2007.05.009
32. Moser MJ, Kamath-Loeb AS, Jacob JE, Bennett SE, Oshima J, Monnat RJ. WRN Helicase Expression in Werner Syndrome Cell Lines. Nucleic Acids Res (2000) 28:648–54. doi: 10.1093/nar/28.2.648
33. Tavera-Tapia A, de la Hoya M, Calvete O, Martin-Gimeno P, Fernández V, Macías JA, et al. Recql5: Another DNA Helicase Potentially Involved in Hereditary Breast Cancer Susceptibility. Human Mutation. Hum Mutat (2019) 40:566–77. doi: 10.1002/humu.23732
34. Agrelo R, Cheng W-H, Setien F, Ropero S, Espada J, Fraga MF, et al. Epigenetic Inactivation of the Premature Aging Werner Syndrome Gene in Human Cancer. Proc Natl Acad Sci USA (2006) 103:8822–7. doi: 10.1073/pnas.0600645103
35. Lee JH, Kim SS, Kim MS, Yoo NJ, Lee SH. WRN, the Werner Syndrome Gene, Exhibits Frameshift Mutations in Gastric and Colorectal Cancers. Pathol Oncol Res (2017) 23:451–2. doi: 10.1007/s12253-016-0173-3
36. Ding S-l, Yu J-C, Chen S-T, Hsu G-C, Shen C-Y. Genetic Variation in the Premature Aging Gene WRN: A Case-Control Study on Breast Cancer Susceptibility. Cancer Epidemiol Biomarkers Prev (2007) 16:263–9. doi: 10.1158/1055-9965.EPI-06-0678
37. Yang L, Wang G, Zhao X, Ye S, Shen P, Wang W, et al. A Novel WRN Frameshift Mutation Identified by Multiplex Genetic Testing in a Family With Multiple Cases of Cancer. PloS One (2015) 10(8):e0133020. doi: 10.1371/journal.pone.0133020
38. Zins K, Frech B, Taubenschuss E, Schneeberger C, Abraham D, Schreiber M. Association of the rs1346044 Polymorphism of the Werner Syndrome Gene RECQL2 With Increased Risk and Premature Onset of Breast Cancer. Int J Mol Sci (2015) 16:29643–53. doi: 10.3390/ijms161226192
39. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17:405–23. doi: 10.1038/gim.2015.30
40. Ishida H, Yamaguchi T, Tanakaya K, Akagi K, Inoue Y, Kumamoto K, et al. Japanese Society for Cancer of the Colon and Rectum (JSCCR) Guidelines 2016 for the Clinical Practice of Hereditary Colorectal Cancer (Translated Version). J Anus Rectum Colon (2018) 2:S1–S51. doi: 10.23922/jarc.2017-028
41. Hegde M, Ferber M, Mao R, Samowitz W, Ganguly A. ACMG Technical Standards and Guidelines for Genetic Testing for Inherited Colorectal Cancer (Lynch Syndrome, Familial Adenomatous Polyposis, and MYH-Associated Polyposis). Genet Med (2014) 16:101–16. doi: 10.1038/gim.2013.166
42. Gazzoli I, Kolodner RD. Regulation of the Human MSH6 Gene by the Sp1 Transcription Factor and Alteration of Promoter Activity and Expression by Polymorphisms. Mol Cell Biol (2003) 23(22):7992–8007. doi: 10.1128/mcb.23.22.7992-8007.2003
43. Shin K-H, Shin J-H, Kim J-H, Park J-G. Mutational Analysis of Promoters of Mismatch Repair Genes hMSH2 and hMLH1 in Hereditary Nonpolyposis Colorectal Cancer and Early Onset Colorectal Cancer Patients: Identification of Three Novel Germ-Line Mutations in Promoter of the hMSH2 Gene. Cancer Res (2002) 62(1):38–42.
44. Iwahashi Y, Ito E, Yanagisawa Y, Akiyama Y, Yuasa Y, Onodera T, et al. Promoter Analysis of the Human Mismatch Repair Gene Hmsh2. Gene (1998) 213(1-2):141–7. doi: 10.1016/S0378-1119(98)00187-5
45. Liu Q, Thompson BA, Ward RL, Hesson LB, Sloane MA. Understanding the Pathogenicity of Noncoding Mismatch Repair Gene Promoter Variants in Lynch Syndrome. Hum Mutat (2016) 37(5):417–26. doi: 10.1002/humu.22971
46. Dobbins SE, Broderick P, Chubb D, Kinnersley B, Sherborne AL, Houlston RS. Undefined Familial Colorectal Cancer and the Role of Pleiotropism in Cancer Susceptibility Genes. Fam Cancer (2016) 15:593–9. doi: 10.1007/s10689-016-9914-4
47. Wang Q. Cancer Predisposition Genes: Molecular Mechanisms and Clinical Impact on Personalized Cancer Care: Examples of Lynch and HBOC Syndromes. Acta Pharmacol Sin (2016) 37(2):143–9. doi: 10.1038/aps.2015.89
48. Lynch HT, Drescher K, Knezetic J, Lanspa S. Genetics, Biomarkers, Hereditary Cancer Syndrome Diagnosis, Heterogeneity and Treatment: A Review. Curr Treat Options Oncol (2014) 15(3):429–42. doi: 10.1007/s11864-014-0293-5
49. Xicola RM, Llor X, Pons E, Castells A, Alenda C, Piñol V, et al. Performance of Different Microsatellite Marker Panels for Detection of Mismatch Repair–Deficient Colorectal Tumors. J Natl Cancer Inst (2007) 99:244–52. doi: 10.1093/jnci/djk033
50. Momma T, Gonda K, Akama Y, Endo E, Ujiie D, Fujita S, et al. MLH1 Germline Mutation Associated With Lynch Syndrome in a Family Followed for More Than 45 Years. BMC Med Genet (2019) 20:67. doi: 10.1186/s12881-019-0792
51. Zimmer K, Puccini A, Xiu J, Baca Y, Spizzo G, Lenz H-J, et al. WRN-Mutated Colorectal Cancer is Characterized by a Distinct Genetic Phenotype. Cancers (2020) 12(5):1319. doi: 10.3390/cancers12051319
52. Jarhelle E, Stensland HMFR, Hansen GÅM, Skarsfjord S, Jonsrud C, Ingebrigtsen M, et al. Identifying Sequence Variants Contributing to Hereditary Breast and Ovarian Cancer in BRCA1 and BRCA2 Negative Breast and Ovarian Cancer Patients. Sci Rep (2019) 9(1):1–12. doi: 10.1038/s41598-019-55515-x
53. Rahner N, Steinke V. Hereditary Cancer Syndromes. Dtsch Arztebl Int (2008) 105(41):706. doi: 10.3238/arztebl.2008.0706
54. Moser MJ, Bigbee WL, Grant SG, Emond MJ, Langlois RG, Jensen RH, et al. Genetic Instability and Hematologic Disease Risk in Werner Syndrome Patients and Heterozygotes. Cancer Rese (2000) 60(9):2492–6.
55. Li K, Luo H, Huang L, Luo H, Zhu X. Microsatellite Instability: A Review of What the Oncologist Should Know. Cancer Cell Int (2020) 20(1):1–13. doi: 10.1186/s12935-019-1091-8
56. Youn C-K, Cho H-J, Kim S-H, Kim H-B, Kim M-H, Chang I-Y, et al. Bcl-2 Expression Suppresses Mismatch Repair Activity Through Inhibition of E2F Transcriptional Activity. Nat Cell Biol (2005) 7(2):137–47. doi: 10.1038/ncb1215
57. Chan EM, Shibue T, McFarland JM, Gaeta B, Ghandi M, Dumont N, et al. WRN Helicase Is a Synthetic Lethal Target in Microsatellite Unstable Cancers. Nature (2019) 568(7753):551–6. doi: 10.1038/s41586-019-1102-x
58. Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic Function of Mutlα in Human Mismatch Repair. Cell (2006) 126(2):297–308. doi: 10.1016/j.cell.2006.05.039
59. Kategaya L, Perumal SK, Hager JH, Belmont LD. Werner Syndrome Helicase is Required for the Survival of Cancer Cells With Microsatellite Instability. Iscience (2019) 13:488–97. doi: 10.1016/j.isci.2019.02.006
60. Uhrhammer NA, Lafarge L, Santos LD, Domaszewska A, Lange M, Yang Y, et al. Werner Syndrome and Mutations of the WRN and LMNA Genes in France. Hum Mutat (2006) 27:718–9. doi: 10.1002/humu.9435
61. Lee H, Flaherty P, Ji HP. Systematic Genomic Identification of Colorectal Cancer Genes Delineating Advanced From Early Clinical Stage and Metastasis. BMC Med Genomics (2013) 6:54. doi: 10.1186/1755-8794-6-54
62. Chu WK, Hickson ID. Recq Helicases: Multifunctional Genome Caretakers. Nat Rev Cancer (2009) 9:644–54. doi: 10.1038/nrc2682
63. Brosh RM Jr. DNA Helicases Involved in DNA Repair and Their Roles in Cancer. Nat Rev Cancer (2013) 13:542–58. doi: 10.1038/nrc3560
64. Kitano K. Structural Mechanisms of Human RecQ Helicases WRN and BLM. Front Genet (2014) 5:366. doi: 10.3389/fgene.2014.00366
65. Chun SG, Yee NS. Werner Syndrome as a Hereditary Risk Factor for Exocrine Pancreatic Cancer. Cancer Biol Ther (2010) 10(5):430–7. doi: 10.4161/cbt.10.5.12763
66. Shamanna RA, Croteau DL, Lee J-H, Bohr VA. Recent Advances in Understanding Werner Syndrome. F1000Res (2017) 6:1179. doi: 10.12688/f1000research.12110.1
67. Ishikawa Y, Sugano H, Matsumoto T, Furuichi Y, Miller RW, Goto M. Unusual Features of Thyroid Carcinomas in Japanese Patients With Werner Syndrome and Possible Genotype–Phenotype Relations to Cell Type and Race. Cancer (1999) 85(6):1345–52. doi: 10.1002/(SICI)1097-0142(19990315)85:6<1345::AID-CNCR18>3.0.CO;2-#
Keywords: colorectal cancer, microsatellite instability, hereditary non-polyposis colorectal cancer, next-generation sequencing, WRN
Citation: Norouzi M, Shafiei M, Abdollahi Z, Miar P, Galehdari H, Emami MH, Zeinalian M and Tabatabaiefar MA (2021) WRN Germline Mutation Is the Likely Inherited Etiology of Various Cancer Types in One Iranian Family. Front. Oncol. 11:648649. doi: 10.3389/fonc.2021.648649
Received: 01 January 2021; Accepted: 05 May 2021;
Published: 07 June 2021.
Edited by:
Israel Gomy, Dana–Farber Cancer Institute, United StatesReviewed by:
Mev Dominguez-Valentin, Oslo University Hospital, NorwayReza Ghasemi, Washington University School of Medicine in St. Louis, United States
Copyright © 2021 Norouzi, Shafiei, Abdollahi, Miar, Galehdari, Emami, Zeinalian and Tabatabaiefar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohammad Amin Tabatabaiefar, dGFiYXRhYmFpZWZhckBtZWQubXVpLmFjLmly; dGFiYXRhYmFpZWZhckBnbWFpbC5jb20=; Mohammad Shafiei, c2hhZmllaW1vaEB5YWhvby5jb20=
†These authors have contributed equally to this work and share senior authorship