 Filipa Lopes-Coelho1,2
Filipa Lopes-Coelho1,2 Filipa Martins1,2
Filipa Martins1,2 Ana Hipólito1,2
Ana Hipólito1,2 Cindy Mendes1,2
Cindy Mendes1,2 Catarina O. Sequeira2Rita F. Pires3
Catarina O. Sequeira2Rita F. Pires3 António M. Almeida1,4
António M. Almeida1,4 Vasco D. B. Bonifácio3
Vasco D. B. Bonifácio3 Sofia A. Pereira2
Sofia A. Pereira2 Jacinta Serpa1,2*
Jacinta Serpa1,2*- 1Unidade de Investigação em Patobiologia Molecular (UIPM), Instituto Português de Oncologia de Lisboa Francisco Gentil (IPOLFG), Lisboa, Portugal
- 2CEDOC, Chronic Diseases Research Centre, NOVA Medical School, Faculdade de Ciências Médicas, Universidade NOVA de Lisboa, Lisboa, Portugal
- 3iBB-Institute for Bioengineering and Biosciences, Instituto Superior Técnico, Universidade de Lisboa, Lisboa, Portugal
- 4Hematology, Hospital da Luz, Lisboa, Portugal
The activation of endothelial cells (ECs) is a crucial step on the road map of tumor angiogenesis and expanding evidence indicates that a pro-oxidant tumor microenvironment, conditioned by cancer metabolic rewiring, is a relevant controller of this process. Herein, we investigated the contribution of oxidative stress-induced ferroptosis to ECs activation. Moreover, we also addressed the anti-angiogenic effect of Propranolol. We observed that a ferroptosis-like mechanism, induced by xCT inhibition with Erastin, at a non-lethal level, promoted features of ECs activation, such as proliferation, migration and vessel-like structures formation, concomitantly with the depletion of reduced glutathione (GSH) and increased levels of oxidative stress and lipid peroxides. Additionally, this ferroptosis-like mechanism promoted vascular endothelial cadherin (VE-cadherin) junctional gaps and potentiated cancer cell adhesion to ECs and transendothelial migration. Propranolol was able to revert Erastin-dependent activation of ECs and increased levels of hydrogen sulfide (H2S) underlie the mechanism of action of Propranolol. Furthermore, we tested a dual-effect therapy by promoting ECs stability with Propranolol and boosting oxidative stress to induce cancer cell death with a nanoformulation comprising selenium-containing chrysin (SeChry) encapsulated in a fourth generation polyurea dendrimer (SeChry@PUREG4). Our data showed that novel developments in cancer treatment may rely on multi-targeting strategies focusing on nanoformulations for a safer induction of cancer cell death, taking advantage of tumor vasculature stabilization.
Introduction
Tumor blood vessels are essential to provide nutrients and oxygen to cancer cells and for the elimination of waste products. Besides the promotion of tumor growth, the neovasculature acts as a gatekeeper for tumor cell invasion and metastasis (1). In an ideal scenario, the tackling of tumor angiogenesis would be an efficient anti-cancer approach, yet, so far, the anti-angiogenic therapies have shown a lack of efficacy and drug resistance (2, 3).
During angiogenesis, the balance between pro-angiogenic factors (e.g., vascular endothelial growth factor - VEGF, fibroblast growth factors - FGFs, and angiopoietins - ANGPTs) and anti-angiogenic factors (e.g., endostatin, thrombospondin, and angiostatin) plays a vital role in the regulation of the angiogenic switch, a process characterized by the activation of the quiescent endothelial cells (ECs) to form new blood vessels (4). However, contrarily to physiological angiogenesis, cancer neovessels are unorganized and leakier, suggesting that an imbalance in pro- and anti-angiogenic factors or the activation of unknown signaling pathways triggers a hyperactivation of the angiogenic switch and further unstable cancer neovessel formation (5).
Still, for both physiological and pathological angiogenesis the oxidative stress represents a pro-angiogenic stimulus (6, 7). Cancer neoangiogenesis seems to be more responsive to oxidative stress than physiological angiogenesis because the metabolic remodeling of malignant cells and tumor-associated stromal cells contributes to the generation of a pro-oxidative tumor microenvironment (8). At a molecular level, reactive oxygen species (ROS) inhibit PHDs (prolyl hydroxylases) leading to hypoxia-inducible factor 1α (HIF1α) stabilization, and consequently to the transcription of VEGF and other pro-angiogenic factors (6, 7).
The oxidative stress-dependent generation of lipid peroxides underlies ferroptosis, a recently discovered process of programmed cell death. In this process, iron ions (Fe2+) promote lipid oxidation in a Fenton-like reaction, increasing ROS levels alongside with intracellular glutathione (GSH) depletion, leading to the impairment of the activity of glutathione peroxidase 4 (GPX4), a GSH-dependent hydroperoxidase responsible for the scavenging of lipid peroxides (9, 10). The ROS-induced lipid peroxidation damages membranar phospholipids directly and can also act as cell death signal. Recent observations have shown that ferroptosis is not strictly a cell death type; it can also be associated with the regulation of biological and pathophysiological processes, including carcinogenesis (9, 11). Ferroptosis can be induced by angiopoietin-like 4 (ANGPTL4), a potent angiogenic mediator that activates the TAZ-ANGPTL4-NOX2 (transcriptional coactivator with PDZ-binding motif-ANGPTL4-NADPH oxidase 2) axis. This axis is responsible for the activation of NADPH oxidase 2 (NOX2), that induces the superoxide radical generation, which in turn acts as an activator of ferroptosis (12), suggesting a correlation between ferroptosis and angiogenesis induction.
In the last years, Propranolol, a liposoluble non-selective β-blocker, without an intrinsic activity (13) and with a well-known membrane stabilizing effect (14), firstly indicated as an anti-hypertensive drug was repurposed as a first-line therapy for vascular tumors, such as infantile hemangiomas and cavernomas (15, 16). Furthermore, breast cancer patients exposed to β-blockers prior or after diagnosis had a better disease prognosis and less metastases (17–19). So far, it is recognized that the impairment of angiogenesis by Propranolol involves the downregulation of VEGF and FGF expression, and consequently the inhibition of mitogen-activated protein kinase (MAPK) signaling pathway (20, 21). Although it seems to be independent of its β-blocker action (22), the specific mechanism(s) of action by which Propranolol affects ECs and angiogenesis remains to be clarified. A paper from Sasaki’s team demonstrated that Propranolol downregulates the expression of ANGPTL4 in hemangioma cells (22). ANGPTL4 regulates angiogenesis in a context-dependent manner, acting as a pro- or as an anti-angiogenic factor (23, 24).
Apart from the inhibition of ANGPTL4 expression and consequent abrogation of NOX2 activation, Propranolol could also interfere with ferroptosis through the inhibition of cytochrome P450 (CYP450) enzymes, whose activity promotes ROS and lipid peroxidation (25, 26). Propranolol is a substrate of CYP450s (27–29), acting as an inhibitor of CYP2D6 (30), CYP2C19 (31) and CYP1A2 (27, 32) isoenzymes. Recently, Mishima et al. identified the anti-ferroptotic properties of Propranolol, which promoted lipid peroxyl radicals scavenging in a β1 activity-independent manner (29). Although the mechanism underlying the anti-angiogenic effect of Propranolol is not clear, it was shown that it does not involve apoptosis (33).
Here, we investigated if ECs activation needed for angiogenesis might be triggered by a ferroptosis-like mechanism. In addition, we disclosed if this mechanism contributes to the anti-angiogenic effect of Propranolol. Finally, we made a pilot proof of concept approach to address the hypothesis that a promising chemotherapeutic strategy could combine inducers of cell death and ECs stabilizers.
Material and Methods
Cell Culture
Three different batches corresponding to three different-original donors of Human umbilical vein endothelial cells (HUVECs: CRL-1730, ATCC) were used. HUVECs were cultured in Endothelial Cell Growth Basal Medium-2 (EBM-2: CC-3156, Lonza, Bioscience) supplemented with EGM-2 SingleQuots Supplements (CC-4176, Lonza, Bioscience). All experiments were performed until the passage 10. Triple-negative breast cancer (MDA-MB-231: HTB-26™, ATCC) were used as tumor models, being cultured in Dulbecco’s Modified Eagles Medium (DMEM) (41965-039, Gibco, Life Technologies), supplemented with 10% fetal bovine serum (FBS) (S 0615, Merck), 1% Antibiotic-Antimycotic (AA; P06-07300, PAN Biotech) and 50 µg/mL Gentamicin (15750-060, Gibco, Life Technologies). Cell cultures were maintained at 37°C in a humidified environment of 5% CO2. Cells were detached with 0.05% Trypsin-EDTA 1 × (25300-054, Invitrogen, Thermo Fisher Scientific) at 37°C for approximately 5 min and split to new plates according to the experimental procedures.
Regarding experimental conditions, cells were cultured with 15 μM hydrogen peroxide (H2O2; 1.07210.0250, Merck), as a ROS generator, 1.5 μM Erastin (E7781, Sigma) as a ferroptosis inducer, 100 μM Propranolol (P8688, Sigma Aldrich) and 160 and 200 μM SeChry@PUREG4, for 6 and 16 h.
Cell Death Analysis by Flow Cytometry
To analyze the effects of Propranolol, Erastin and SeChry on cell death, HUVECs (5×104 cells/well) and MDA-MB-231 (5×104 cells/well) were seeded in 24-well plates. After exposure to the experimental conditions, supernatants and cells were collected and centrifuged at 155 × g for 5 min. Cell pellets were incubated with 0.5 μL FITC-labeled Annexin V (640906, BioLegend) in Annexin V binding buffer 1× (10 mM Hepes (pH 7.4) (391333, Millipore), 0.14 M sodium chloride (NaCl; 106404, Merck), 2.5 mM calcium chloride (CaCl2; 449709, Sigma Aldrich) and incubated at room temperature for 15 min, in the dark. After incubation, cells were rinsed in 200 μL PBS 1×/0.1% (v/w) bovine serum albumin (BSA) and centrifuged at 155 × g for 2 min. The remaining pellet was resuspended in 200 μL of annexin V binding buffer 1× and 2.5 μL of 50 μg/mL propidium iodide (PI; P4170, Sigma Aldrich Aldrich) and analyzed by flow cytometry (FACScalibur – Becton Dickinson). FlowJo X v10.0.7 software (https://www.flowjo.com/) was used to analyze data.
Wound Healing Assay
Cells were plated in 24-well plates (1×105 cells/well) until the formation of a confluent monolayer. Once confluent, cells were incubated for 3 h with 5 μg/mL mitomycin-C (M4287, Sigma Aldrich) and a linear scratch in each monolayer was made with a P200 pipette tip, creating a wound across the well diameter. The media was replaced to remove debris and cells in suspension and the experimental conditions were added. Bright-field images of each well were acquired on the Olympus IX53 Inverted Microscope at the following timepoints: 0, 2, 4, 6, 8, 10 and 24 h. The wound closure was quantified using the ImageJ software (imagej.nih.gov/ij/).
Tube-Forming Assay
A 48-well plate was coated with 100 µL matrigel (354230, Corning) and incubated at 37°C for 30 min until solidification. HUVECs were incubated with 2 μg/mL calcein (C1430, Invitrogen), a fluorescent cell permeable dye, for 30 min at 37°C and 5% CO2 and seeded (3×104 cells/well) on the top of matrigel. Cells were exposed to the experimental conditions for 6h and representative images of the formed tube-like structures were acquired on an Olympus IX53 Inverted Microscope and analyzed with ImageJ software (imagej.nih.gov/ij/). The density of vessel-like structures formation (branch points number/μm2) was calculated as representative of vascular density.
Reactive Oxygen Species (ROS) Quantification by Flow Cytometry
HUVECs (5×104 cells/well) and MDA-MB-231 cells (5×104 cells/well) were plated in 24-well plates. The intracellular ROS were detected in cells incubated with 10 μM DCF-DA probe (D6883, Sigma Aldrich) and mitochondrial ROS were detected in cells incubated with 5 μM MitoSox Red probe (M36008, Invitrogen), both at 37°C for 30 min. The acquisition was performed with FACScalibur (Becton Dickinson) and data were analyzed with FlowJo X v10.0.7 software (https://www.flowjo.com/).
Lipid Peroxides Quantification by Flow Cytometry
HUVECs (5×104 cells/well) were plated in 24-well plates. After experimental conditions, cells were incubated with 2 μM C11-Bodipy 581/591 (D3861, Invitrogen), for 30 min at 37°C in the dark. The excess dye was removed by washing with 2% FBS-1X PBS and cell pellets were resuspended in 2% FBS-1× PBS for the acquisition by flow cytometry (FACScalibur – Becton Dickinson). FlowJo X v10.0.7 software (https://www.flowjo.com/) was used to analyze data.
High-Performance Liquid Chromatography (HPLC)
HUVECs were plated in 6-well plates (2×105 cells/well) and after collection, cell pellets were lysed with 0.01% triton-PBS and centrifuged at 20 000 g, for 10 min at 4 °C. The assessment of pools of cysteine and GSH (total availability, total free fraction, reduced free, protein bound and oxidized pools) of lysed cells and supernatants was performed according to (34) and adapted to cell culture. The cysteine (Cys) and glutathione (GSH) metabolites were separated on a reversed-Phase C18 LiChroCART 250-4 column (LiChrospher 100 RP-18, 5 µm, VWR, USA) on isocratic elution mode for 22 min, at a flow rate of 0.6 mL/min by HPLC system (Shimadzu Scientific Instruments Inc) with a fluorescence detector operating at excitation and emission wavelengths of 385 and 515 nm, respectively. The mobile phase consisted of 100 mM acetate buffer (pH 4.5) and methanol (98:2 (v/v)). The concentrations of these thiols were normalized to the protein assessed with Bradford method (500-0006, Bio Rad). Results are presented as: Total GSH: levels of free form of oxidized or reduced GSH and GSH bound to proteins, it is the total pool of GSH in the cell; Free-total GSH: levels of free form of oxidized or reduced GSH, the total pool of GSH that is not bound to proteins; Free-reduced GSH: levels of reduced GSH free in the cell, and GSSG: levels of oxidized GSH free in the cell.
Reverse Transcription and Quantifying PCR (RT-qPCR)
HUVECs (2×105 cells/well) were plated in 6-well plates and after exposure to the experimental conditions, the RNA was extracted using RNeasy Mini Extraction kit (74104, Qiagen) and the cDNA synthesized from 1 µg RNA and reversely-transcribed by SuperScript II Reverse Transcriptase (18080e44, Invitrogen), both according to the manufacturer’s protocol. Quantitative Real-Time PCR was performed using Power SYBR Green PCR Master Mix (4367659, Applied Biosystems), according to manufacturer’s protocol and carried out in a LightCycler 480 instrument (Roche). The transcriptional expression of genes encoding prostaglandin-endoperoxide synthase 2 (PTGS2), glutathione peroxidase 4 (GPX4) and glutathione synthetase (GSS) was evaluated, using the primers: PTGS2 (Fwd: CTGGCAGGGTTGCTGGTG; Rev: CATCTGCCTGCTCTGGTC); GPX4 (Fwd: GCAGGAGCCAGGGAGTAAC; Rev: CCTTGGGTTGGATCTTCATCC), and GSS (Fwd: GAGAGAGGGTGGAGGTAAC; Rev: CCATGAGGATGTAGGAGGCC). Hypoxanthine-guanine phosphoribosyltransferase (HPRT; Fwd: TGACACTGGCAAAACAATGCA; Rev: GGTCCTTTTCACCAGCAAGCT) was used as housekeeping gene.
Quantification of H2S in Cell Homogenates
HUVECs (2×105 cells/well) were seeded in 6-well plates and cultured under the experimental conditions, for 16 h. After, cells were scrapped in PBS 1× and centrifuged at 210 × g for 5 min. The cell pellet was homogenized in NP40 lysis buffer (1% NP40, 150 mM NaCl, 50 mM Tris-Cl, pH 8.0) on ice for 30 min and centrifuged for 5 min at 20,000 × g 4°C. Cell homogenates (20 µL) were incubated in black 96-well plates with 80 µL of 10 µM 7-Azido-4-Methylcoumarin probe (AzMC, L511455, Sigma Aldrich). The protein concentration was determined with Bradford method using protein assay dye reagent concentrate (500-0006, Bio Rad). The H2S measurements were posteriorly normalized to the total protein concentration and to a blank sample (cellular lysates without probe). H2S production was monitored following fluorescent signal of AzMC probe (355 nm/460 nm) every 30 min for 2 h, in a VIKTOR3 instrument from PerkinElmer/Wallac 1420 v3.0 software.
Immunofluorescence
For Ki67, ICAM and VCAM immunodetection, HUVECs (5×104 cells/well) were cultured on glass slides with 0.2% gelatin coating and fixed in 2% paraformaldehyde, for 15 min at 4°C. After blocking with 1% BSA-1× PBS, cells were incubated with primary antibodies (anti-Ki67, 1:100 in 1% BSA-0.1% triton X-100- 1× PBS (w/v/v); sc-15402, Santa Cruz; anti-ICAM and anti-VCAM, 1:500 in 0.1% BSA-0.1% triton X-100-PBS (w/v/v); SRC023, Millipore), overnight at 4°C, followed by an incubation with secondary antibodies (Alexa Fluor 488 goat anti-rabbit, 1:1000 in 1% BSA-0.1% triton X-100-PBS; A-11078, Invitrogen - Thermo Fisher Scientific; Alexa Fluor 488 goat anti-mouse;115-545-003, Thermo Fisher Scientific and Alexa Fluor 594 donkey anti-mouse; A21203, Thermo Fisher Scientific both at 1:1000 in 0.1% BSA-0.1% triton X-100-PBS), for 2 h at room temperature.
For the cystine/glutamate antiporter system xc- (xCT) immunodetection, after fixation cell were incubated with 50 mM ammonium chloride (NH4Cl) for 10 min, followed by blocking and incubation with anti-xCT (1:100 in 0.5% BSA-0.1% saponin-PBS (w/v/v); ab1756, Millipore), for 30 min at room temperature.
For VE-Cadherin (VE-Cad) immunodetection, HUVECs (1×105 cells/well) were cultured in 24-well plate with glass slides coated with 0.2% gelatin, until the formation of a confluent monolayer. After fixation and blocking, cells were incubated anti-VE-Cad (1:50 in 3% BSA-0.1% triton X-100-PBS (w/v/v); AF938, R&D), for 2 h at room temperature, followed by an incubation with the secondary antibody (Alexa Fluor 488 donkey anti-goat, 1:500 in 3% BSA-0.1% triton X-100-PBS; A11055, Thermo Fisher Scientific), for 2 h at room temperature.
All slides were mounted in VECTASHIELD media with DAPI (4′-6-diamidino-2-phenylindole; H-1200, Vector Labs) and examined by standard fluorescence microscopy, using an Axio Imager.Z1 microscope (Zeiss) with a CytoVision® software.
The determination of cell proliferation rate was based on the ratio of total and Ki67+ nuclei and the quantification of ICAM and VCAM expression per cell was calculated according to the formula CTCF (corrected total cell fluorescence) = integrated density – (area of selected cell × mean fluorescence of background reading), both using ImageJ software (imagej.nih.gov/ij/).
Cancer Cells Endothelial Adhesion
In a 24-well plate, calcein labelled-MDA-MB-231 (5×104 cells/well) were seeded on the top of a HUVECs (1×105 cells/well) monolayer pretreated with 100 ng/mL TNFα (H8916, Sigma), for 24 h. MDA-MB-231 were incubated with HUVECs (exposed previously to TNFα and experimental conditions) for 40 min, at 37°C in a humidified environment of 5% CO2. The non-adherent cells were removed by washing with PBS1X and images were acquired on an Olympus IX53 Inverted Microscope and analyzed using ImageJ software (imagej.nih.gov/ij/). Three fields in each well were evaluated (10× magnification).
Transendothelial Cancer Cells Migration
HUVECs (5×104 cells/well) were plated in 8µm pore transwells (upper wells) (3422, Corning) and exposed to 100 ng/mL TNFα for 24 h, and to experimental conditions for 16 h. MDA-MB-231 previously plated under starvation using serum-free DMEM, for 24 h, were incubated with calcein (2 μg/mL) and seeded (1.5×104 cells/well) in serum free DMEM on the top of the HUVECs monolayer, for 5 h. Complete media was added to the lower well and used as chemoattractant. Cells on the upper Transwell® surface were removed with a cotton swab and the invading MDA-MB-231- calcein labeled cells were photographed on an Olympus IX53 Inverted Microscope. Three fields in each well were counted (10× magnification) using the ImageJ software (imagej.nih.gov/ij/).
Monocytes Isolation, Culture and Characterization
Monocytes isolation from peripheral blood of healthy blood donors (IPOLFG-Ethical committee UIC-1137) and further cell characterization was performed as described by Lopes-Coelho et al. (35). Briefly, monocytes cultured for 4 days in colony-forming unit (CFU) medium (130-091-277, MACS Technology) and for 1 day in complete EBM-2 were incubated with von Willebrand factor (vWF; 1:500 in 0.5% BSA-0.1% saponin-PBS; A0082, Dako), for 60 min at 4°C with gentle shaking, followed by the incubation with Alexa Fluor 488 anti-rabbit, for 30 min at 4°C in the dark, with gentle shaking. H2O2 (15 μM) was used as a promoter of monocytes differentiation into ECs (35). vWF expression was detected by flow cytometry in a FACScalibur–Becton and data were analyzed using the FlowJo X v10.0.7 software (https://www.flowjo.com/).
SeChry@PUREG4 Synthesis
Selenium-containing chrysin (SeChry) was synthesized following a reported protocol (36). After purification, the formation of the product was confirmed by 1H NMR. 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.96 (2H, d, J= 8.0 Hz), 7.76 (1H, s), 7.61 (1H, t, J= 8.0 Hz), 7.52 (2H, t, J= 8.0 Hz), 6.51 (1H, d, J= 4.0 Hz), 6.46 (1H, d, J= 4.0 Hz). Polyurea dendrimer generation four (PUREG4) was obtained using our supercritical-assisted polymerization protocol (37). SeChry was encapsulated in PUREG4 nanoparticles following our protocol (38). Briefly, SeChry (6.5 mg) was added to an aqueous solution (10 mL) of PUREG4 (125 mg) and stirred overnight. Then, the aqueous solution was extracted with CHCl3 to remove non-encapsulated or surface bound SeChry. No SeChry was found in the CHCl3 extracts (control by thin layer chromatography, TLC), thus confirming a full encapsulation. The release profile follows the usual profile reported for this nanodelivery system (39, 40).
Statistical Analysis
All data were analyzed using student’s t-test, one-way ANOVA or two-way ANOVA in GraphPad Prism v7 software (www.graphpad.com/). The assays were performed with at least 3 biological replicates per condition and the differences were determined statistically significant at p value < 0.05.
Results
A Ferroptosis-Like Mechanism Induced by Erastin Promotes ECs Activation, Which Is Impaired by Propranolol Through the Increase of H2S Levels
Erastin was used as a ferroptosis activator since it inhibits xCT (encoded by SLC7A11), which is responsible for the import of cystine (41, 42), the main source of cysteine to sustain GSH synthesis (43). xCT inhibition, impairs GPX4 and consequently leads to an increase in lipid peroxidation that is mediated by free active iron (44), and further ferroptosis (45).
After confirming the expression of xCT in HUVECs (Figure S1A) and upon Erastin administration the ferroptosis-related features were analyzed. HUVECs exposed to Erastin presented an increase in intracellular levels of ROS (Figure 1A) and lipid peroxide levels (Figure 1C), without affecting mitochondrial ROS (Figure 1B) but decreasing the GSH levels (Figure 1D). Moreover, the inhibitory effect of Erastin in cyst(e)ine uptake was confirmed, since cells exposed to Erastin presented high levels of total cysteine (free oxidized+free reduced+protein bound) in culture medium (Figure 1E). Interestingly, after Erastin exposure the increased generation of ROS (Figure 1A) and lipid peroxides (Figure 1C), the depletion of GSH (Figure 1D) and the downregulation of genes involved in the scavenging of lipid peroxides (Figures 1F, G) and GSH synthesis (Figure 1H), did not promote ferroptosis-induced ECs death (Figure 1I). Moreover, we observed that Propranolol decreased intracellular ROS levels (Figure 1A) and also reverted the levels of ROS (Figure 1A) and lipid peroxides induced by Erastin (Figure 1C). Accordingly, Propranolol, alone or in combination with Erastin, decreased the levels of free (oxidized + reduced) and total GSH, maintaining the oxidized GSH levels similar to the control condition (Figure 1D).
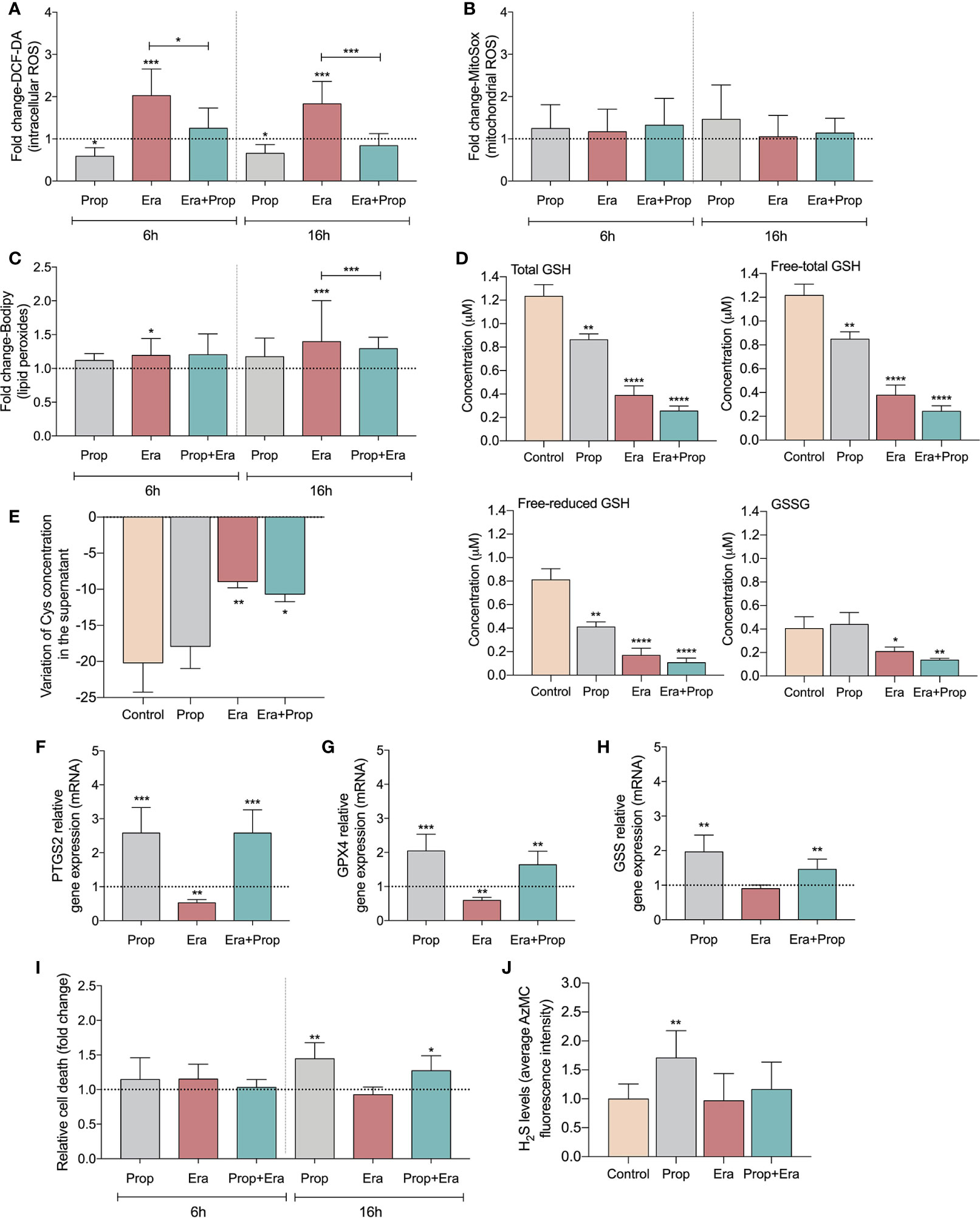
Figure 1 Erastin (Era) promotes increased levels of ROS-induced lipid peroxides and Propranolol (Prop), through the generation of hydrogen sulfide (H2S), is able to revert it. (A) The levels of intracellular ROS (DCF-DA) decrease upon Prop exposure, and Era, although increasing the ROS levels, when co-administrated with Prop the levels are similar to the control, at 6 and 16h. (B) The levels of mitochondrial ROS, assessed by MitoSox, are not affected by the presence of Era and/or Prop, at 6 and 16h. (C) Era induces lipid peroxides (C11-Bodipy) generation and although Prop alone did not affect the lipid peroxides content, its combination with Era reverts the levels generated by Era, being this effect more prominent at 16h. (D) The levels of GSH (total and LT:free total) decrease upon exposure to Era and/or Prop for 16h. (E) The variation of the extracellular levels of cysteine (Cys) indicate that Era inhibits the uptake of Cys by HUVECs, while Prop does not interfere with this process. (F–H) Show the regulation of transcriptional expression of genes encoding, respectively, prostaglandin-endoperoxide synthase 2 (PTGS2), glutathione peroxidase 4 (GPX4) and glutathione synthetase (GSS). Erastin decreases significantly GPX4 and PTGS2 expression and tend to decrease GSS expression, being this effect rescued by propranolol. (I) Era does not affect HUVECs death (annexin V plus PI positive cells) while 16h of Prop exposure, with and without Era, increases the ratio of HUVECs death. (J) Era does not affect H2S levels of HUVECs while Prop increases, at 16h. In graphs the dashed line represents the control condition. All data are normalized to the control condition and represented as mean ± SD. *p<0.5, **p<0.01, ***p<0.001, ****p<0.0001.
Since GSH levels were not increased, we hypothesized that hydrogen sulfide (H2S) generation might underlie the antioxidant properties of Propranolol. H2S, a product of cysteine degradation (46, 47), is a powerful antioxidant (48) with a reduction potential similar to the couple glutathione disulfide/glutathione (GSSG/GSH) (49). There was an increase in H2S production upon Propranolol administration, while Erastin did not affect H2S levels (Figure 1J). This suggests that Propranolol favors cysteine flux for catabolism and not for GSH synthesis. Moreover, when Propranolol was combined with Erastin, the levels of H2S were similar to the control condition. Together, these results suggest that Propranolol’s antioxidant effect is mediated by H2S.
Afterwards, we investigated if the generation of ROS-induced lipid peroxides promoted ECs activation, through this ferroptosis-like mechanism. It was observed that Erastin exposure increased HUVECs proliferation (Figure 2A) and migration (Figure 2B), suggesting that the ferroptosis-like mechanism has a role in the promotion of ECs activation, without affecting cell viability (Figure 1I).
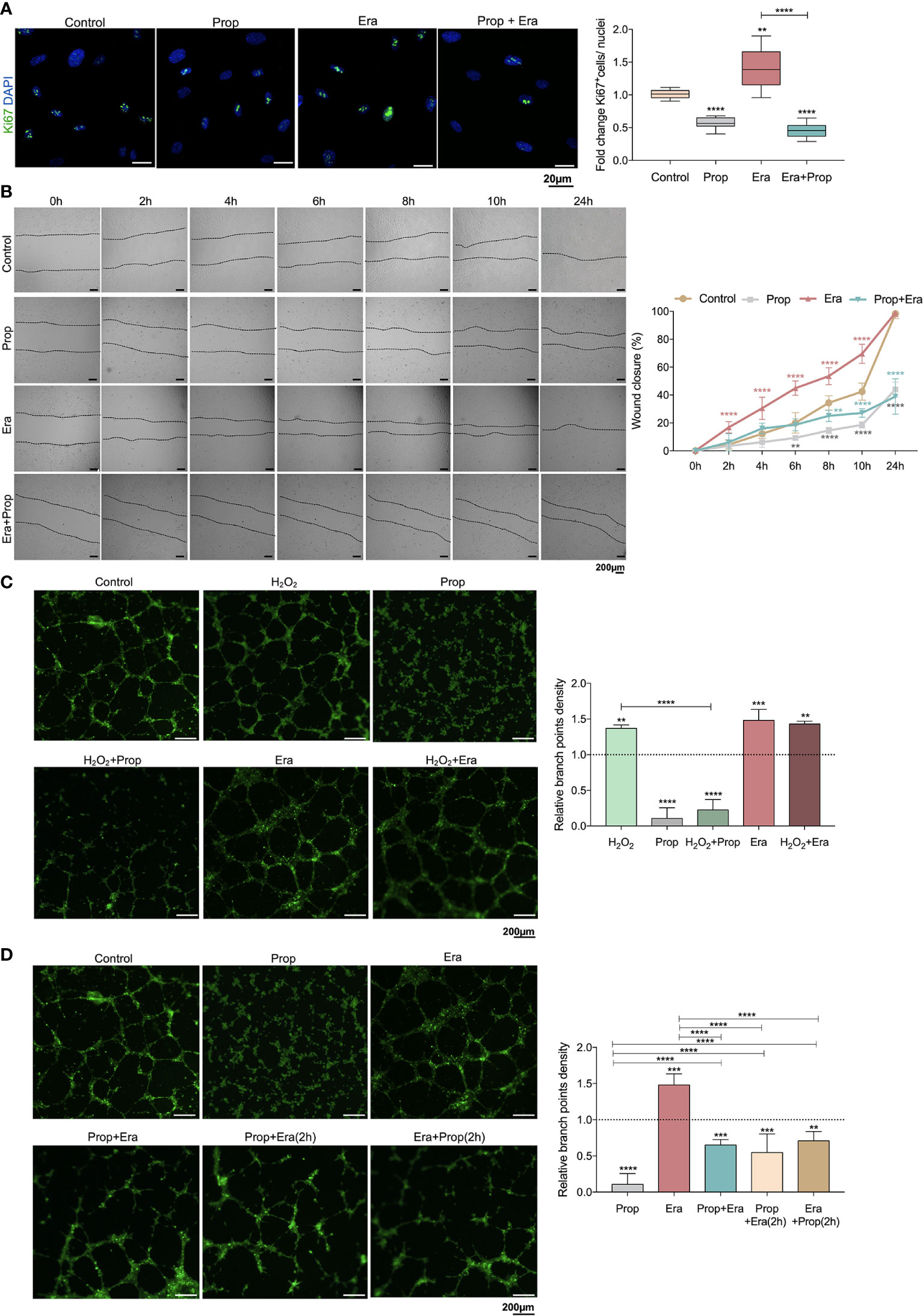
Figure 2 The ferroptosis-like mechanism driven by Erastin (Era) promotes endothelial cell (ECs) activation and Propranolol (Prop) impairs the phenotype induced by Era exposure. (A) The ferroptosis-like mechanism, generated by Era exposure, promotes HUVECs proliferation (increased ratio of Ki67+ (green) nuclei/total nuclei), while Prop decreases the rate of HUVECs proliferation and impairs the phenotype induced by Era. The panel shows representative microscope images of the Ki67 staining. (B) Era fosters HUVECs migration (increased % wound closure) and Prop inhibits and reverts the phenotype induced by Era. (C, D) Era increases the branch point density of vessel-like structures (proxy for vascular density) at the same range of H2O2 (ROS; positive control), with no additive effect. Prop, besides the impairment of vessel-like structures formation (decreased branch point density), inhibits the phenotype induced by Era, even when Prop is added to the vessel-like structures already formed in the presence of Era (Era+Prop (2h)) and contrariwise (Prop+Era (2h)). In graphs the dashed line represents the control condition. All data are normalized to the control condition and represented as mean ± SD. **p<0.01, ***p<0.001, ****p<0.0001.
Considering that Propranolol acts as an inhibitor of angiogenesis and ferroptosis, its interference with ferroptosis-mediated angiogenesis was also evaluated. In fact, HUVECs exposed to Propranolol showed decreased proliferation (Figure 2A) and migration (Figure 2B) and increased cell death, at 16h (Figure 1I). Moreover, Propranolol was able to revert the Erastin effect at 16h, leading to the inhibition of HUVECs proliferation (Figure 2A) and migration (Figure 2B), as well as disturbing cell viability (Figure 1I).
Since the pro-oxidative microenvironment promotes angiogenesis (6, 7), the HUVECs capacity to form vessels-like structures was tested under oxidative conditions, in an in vitro tube-forming assay, upon H2O2 exposure. Erastin exposure increased the branch points density at the same range of H2O2, and no cumulative effects between Erastin and H2O2 were observed (Figure 2C). Moreover, Propranolol decreased the vessel-like structure formation (Figure 2C) and reverted the stimulation by H2O2 and Erastin (Figure 2C). This effect of Propranolol was observed even when Propranolol was added to the vessel-like structures already formed in the presence of Erastin (Figure 2D). These results indicate that the Propranolol anti-angiogenic effect is related, at least in part, to the abrogation of the ferroptosis-like mechanism induced by Erastin.
A Ferroptosis-Like Mechanism Induced by Erastin Promotes the Generation of Leakier ECs Structures Which Are Normalized by Propranolol, Blocking Cancer Cell Adhesion and Transendothelial Migration
The generation of a pro-oxidative microenvironment is correlated with the formation of disorganized and leakier vessel networks (5). Thus, the adhesion structures in HUVECs were evaluated, through VE-Cad immunodetection. VE-Cad is a component of ECs adherents junctions, crucial for the stability and function of the mature vessels (50). Erastin affected the HUVECs monolayer stability, by increasing VE-Cad intercellular junctional gaps, and Propranolol reverted this effect (Figures 3A, B). This indicates that the proliferation of ECs is mediated by a ferroptosis like-mechanism (Figure 2A), although they form more instable structures due to increased intercellular junctional gaps between ECs (Figures 3A, B). Interestingly, Propranolol reverted this phenotype (Figures 3A, B), inducing the stabilization of the ECs monolayer.
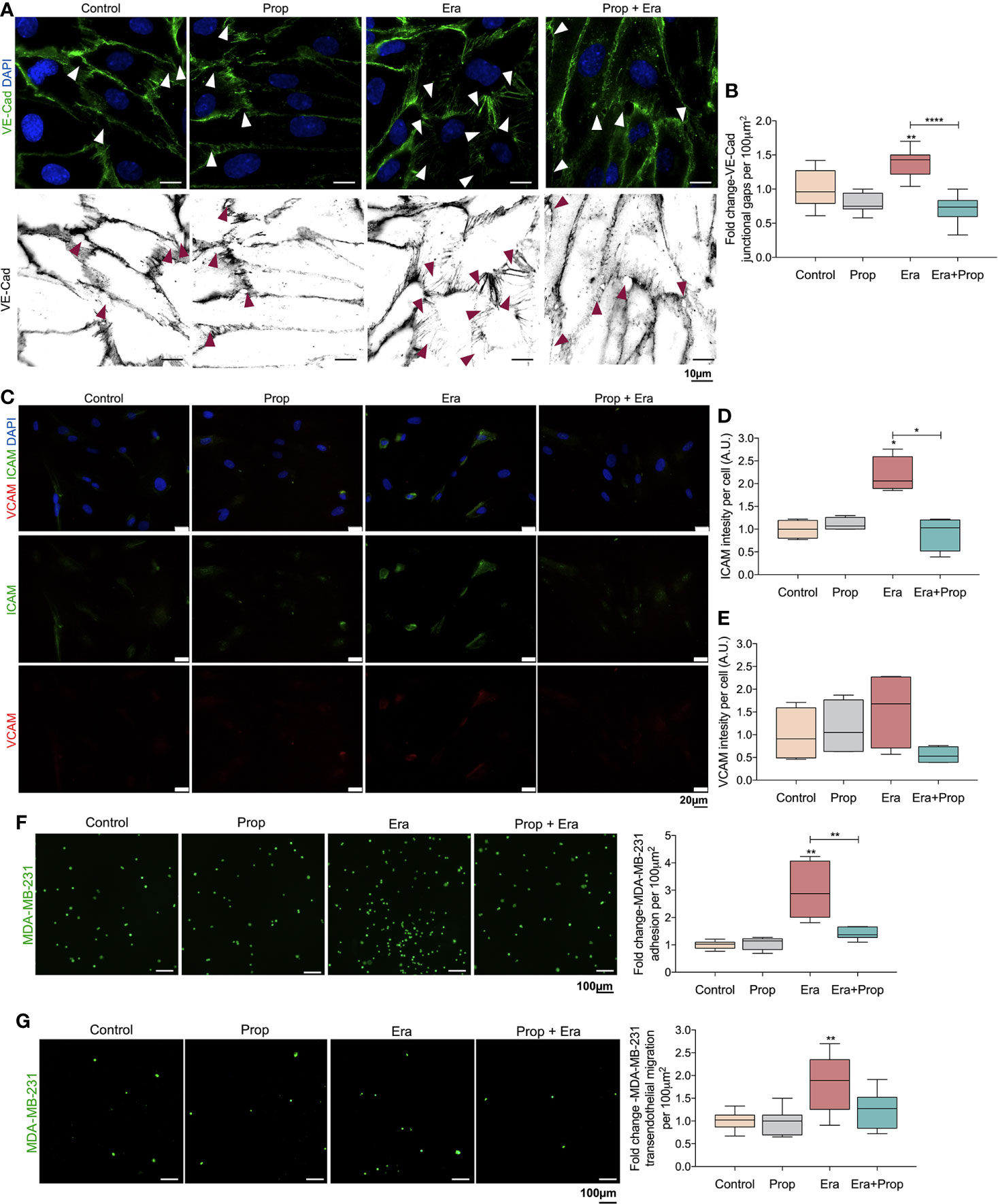
Figure 3 Erastin (Era) promotes the generation of a leakier EC monolayer while increases cancer cell-EC interaction and transendothelial migration. (A) The ferroptosis-like mechanism driven by Era promotes an increased generation of intercellular VE-Cadherin (VE-Cad) gaps per 100µm2, while Propranolol (Prop) is able to revert this phenotype. The panel shows representative images (scale: 10 µm) of VE-Cadherin (green) intercellular junctional gaps (arrows) in HUVECs exposed to Era and/or Prop for 16h. (B) Quantification of VE-Cad junctional gaps. (C) Immunofluorescence for ICAM and VCAM detection. (D) ICAM intensity per cell (HUVECs; A.U.: arbitrary units) increases upon Era exposure and although Prop alone does not affect ICAM expression, it is able to abrogate the expression induced by Era, at 16h. (E) VCAM intensity per cell (A.U.: arbitrary units) shows a tendency to increase under Era exposure. The ferroptosis-like mechanism, induced by Era, promotes cancer cell (MDA-MB-231-calcein labelled cells) adhesion to HUVECs (F) and transendothelial migration (G). Prop alone has no effect but impairs cancer cell adhesion and transendothelial migration induced by Era. The panels show representative microscope images (scale: 100 µm; MDA-MB-231-calcein labelled cells (green)). All data are normalized to the control condition and represented as mean ± SD. *p<0.05, **p<0.01, ***p<0.001.
ICAM and VCAM adhesion molecules are important for cancer cell-EC interaction during the metastatic cascade (51–53), thus, we evaluated their expression. HUVECs exposed to Erastin significantly increased ICAM expression, as well as VCAM expression, although not in a statistically significant level (Figures 3C–E). On the contrary, Propranolol alone did not alter ICAM and VCAM expression, but it reverted the effect of Erastin by decreasing ICAM and VCAM levels (Figures 3C–E).
Considering the increased expression of ICAM (Figures 3C, D), we have also explored the effect of Erastin and Propranolol in the adhesion of cancer cells to ECs and in transendothelial migration. The triple negative breast cancer cell line MDA-MB-231 was co-cultured on the top of a previously established HUVECs monolayer exposed to Erastin and/or Propranolol. Erastin increased the number of cancer cells adherent to the ECs monolayer and stimulated transendothelial migration, while Propranolol reverted both Erastin effects (Figures 3F, G). Our results indicate that a ferroptosis-like mechanism has also a role in the promotion of vessel permeability and in cancer cell adhesion and extravasation. Summing up, Propranolol was able to revert the phenotype induced by Erastin, decreasing the intercellular junctional gaps (Figures 3A, B), the cancer cell adhesion to ECs (Figure 3F) and transendothelial migration (Figure 3G), therefore suggesting that Propranolol could eventually impair, or at least retard, the metastatic process (17).
Neither Erastin nor Propranolol Affect Monocytes (EPCs) Differentiation in ECs
Recently our group disclosed that monocytes act as endothelial progenitor cells and their differentiation into ECs and incorporation in blood vessels depend on a ROS-enriched microenvironment (35). Considering the effect of a ferroptosis-like mechanism in ECs activation, we explored its impact on the differentiation route of monocytes into ECs, assessed by the gain of von Willebrand factor (vWF). Erastin and/or Propranolol exposure had no effect in vWF expression and their concomitant exposure with a H2O2 stimulation (positive control of the differentiation pattern of monocytes) did not affect the vWF expression stimulated by H2O2 (Figure 4A). Moreover, ROS and lipid peroxide (Figures 4B, C) levels did not alter with Erastin exposure after H2O2 stimulation. In contrast to ECs, Propranolol did not affect ROS and lipid peroxide levels in monocytes (Figures 4B, C), suggesting that the antioxidant role of Propranolol verified during ECs activation did not interfere with the differentiation process of monocytes towards ECs.
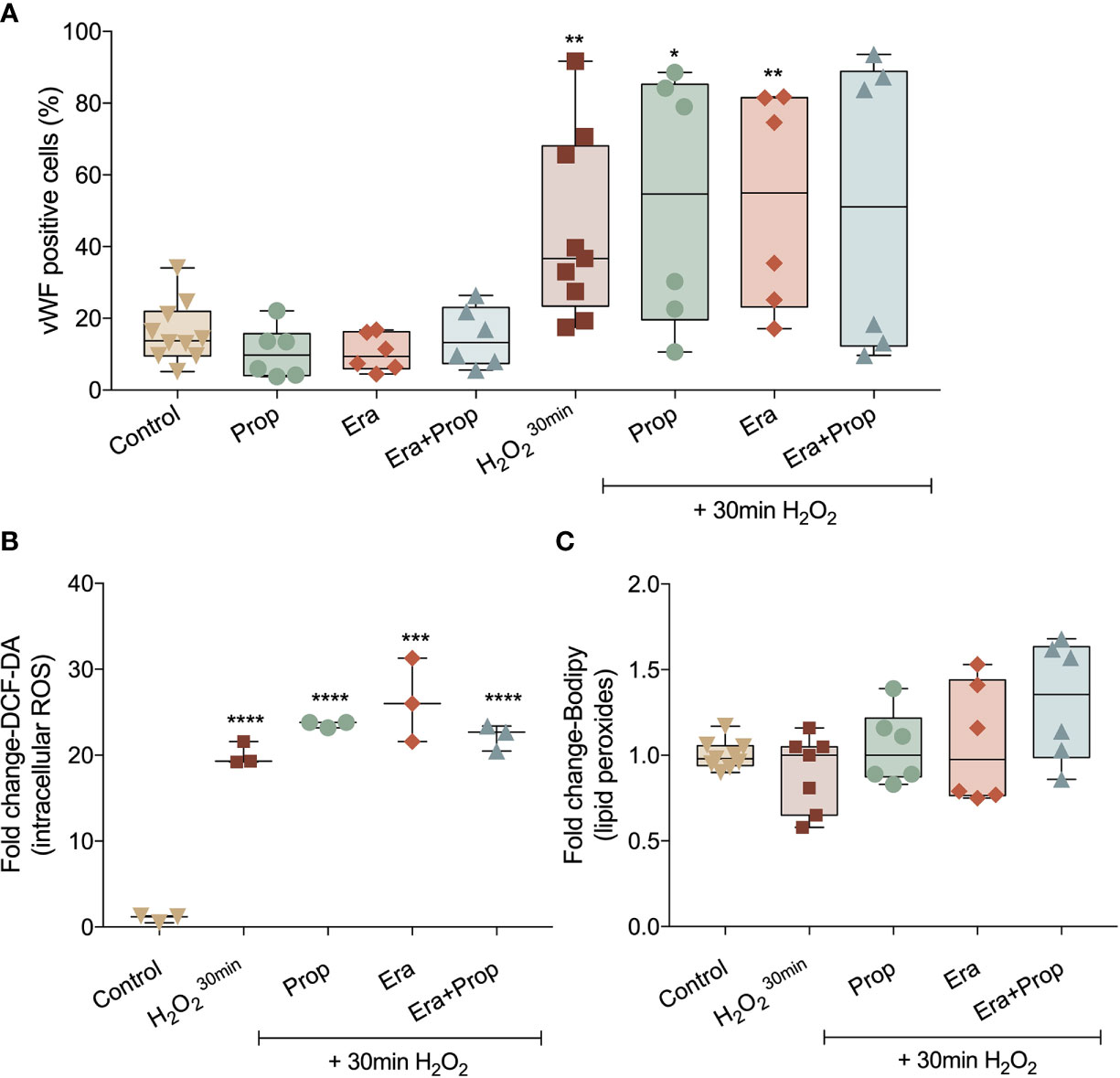
Figure 4 Erastin (Era) and Propranolol (Prop) do not affect the differentiation route of monocytes into ECs. (A) Neither Era and/or Prop affect the expression of vWF, even when co-exposed with a short H2O2 stimulation (positive control of the differentiation pattern of monocytes), indicating that Era and/or Prop have no impact in the differentiation process of monocytes-derived cells into ECs. Era and/or Prop exposure before the short H2O2 stimulation does not influence the intracellular ROS (DCF-DA; B) and lipid peroxide (11C-Bodipy; C) levels. In (B, C) data are normalized to the control condition and represented as mean ± SD. *p<0.5, **p<0.01, ***p<0.001, ****p<0.0001.
Sechry@PUREG4 Plus Propranolol: A Putative Anti-Cancer Strategy, Acting on Both Cancer Cell Death and ECs Hyperactivation Prevention
The anti-angiogenic therapy to treat cancer has an efficiency far from the expected. Thus, a promising therapeutic approach would be a dual-effect therapy, which is cytotoxic to cancer cells and stabilizes the vessels, in order to improve the delivery of chemotherapy to the tumor.
Recently, our team showed that SeChry@PUREG4 nanoformulation had an anti-tumoral effect thereby depleting GSH and inhibiting the H2S producing enzyme, cystathionine beta synthase (CBS) (38). Thus, SeChry@PUREG4 is a good candidate to be tested with Propranolol, in a dual-effect therapy, inducing cancer cell death and stabilizing tumor vessels. Hence, we investigated the SeChry@PUREG4 effect with and without Propranolol on MDA-MB-231 and HUVECs. Interestingly, MDA-MB-231 were more sensitive to Propranolol exposure than ECs (Figures 5A and 1I), increasing MDA-MB-231 cell death by 2-fold (Figure 5A). The two tested concentrations of SeChry@PUREG4 (160 and 200 μM) promoted cancer cell death, mainly when combined with Propranolol (Figure 5B). Interestingly, HUVECs cell death was not altered by 160 μM SeChry@PUREG4 and a slight increase was observed with 200 μM SeChry@PUREG4 (Figure 5B). These results confirmed the anti-tumoral function of SeChry and indicate that Propranolol has distinct effects on the viability of cancer cells and ECs.
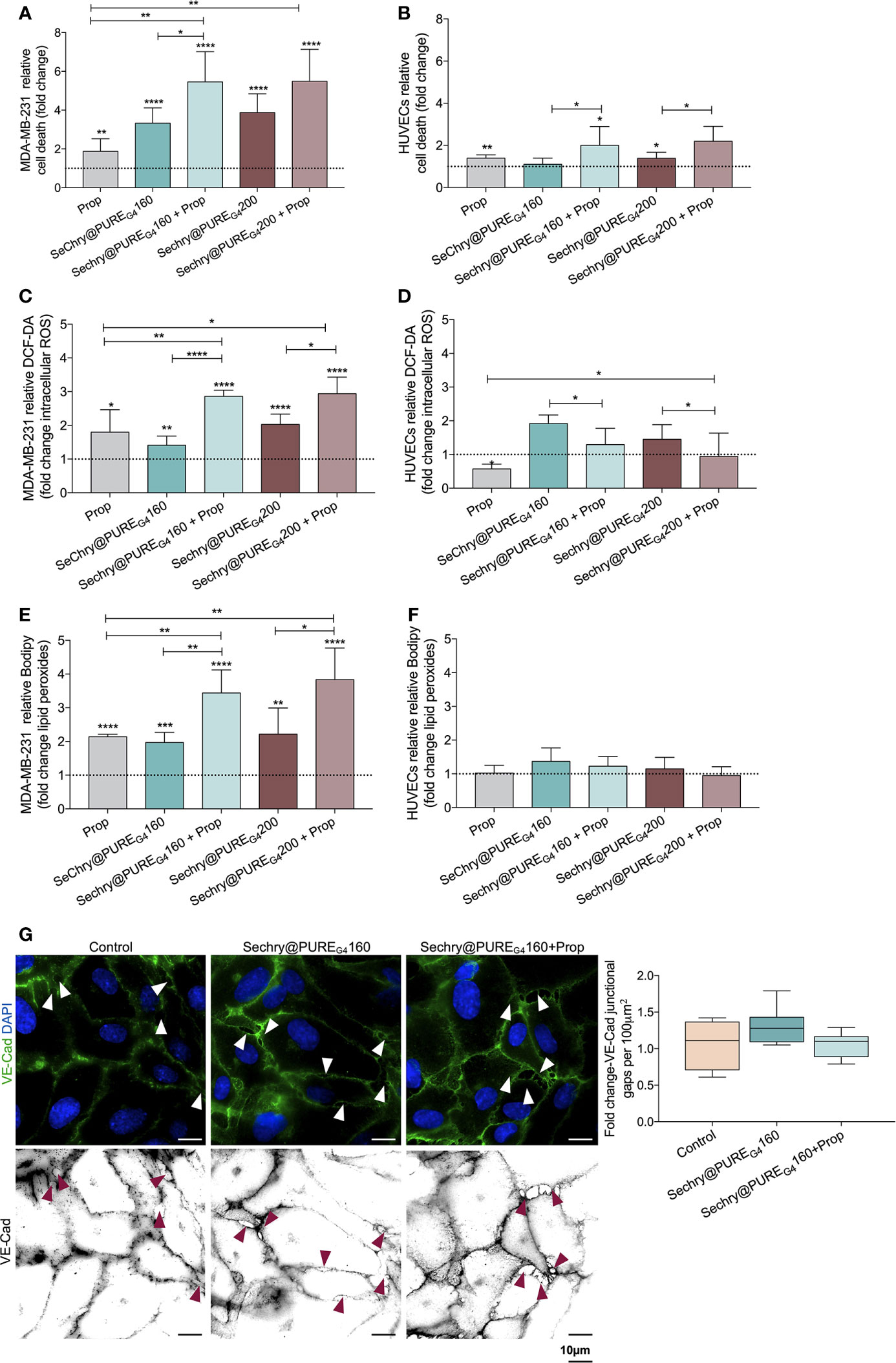
Figure 5 SeChry@PUREG4 plus Propranolol (Prop) increases cancer cell death through the generation of oxidative stress, while in ECs Prop acts as an antioxidant, reverting ROS levels induced by SeChry@PUREG4. (A) SeChry@PUREG4 (160 µM and 200 µM) exposure promotes cancer cell death (MDA-MB-231), being this effect boosted by Prop. (B) ECs (HUVECs) are more resistant to SeChry@PUREG4 -induced cell death, even under Prop exposure. (C–F) Contrarily to HUVECs, in MDA-MB-231 Prop alone increases (C, D) intracellular ROS (DCF-DA) and (E, F) lipid peroxide (11C-Bodipy) levels and it does not revert the generation of ROS-induced lipid peroxidation induced by SeChry@PUREG4. (G) SeChry@PUREG4 does not impact the generation of VE-Cad intercellular junctional gaps. The panel shows representative images (scale: 10 µm) of VE-Cadherin (green) intercellular junctional gaps (arrows). In graphs the dashed line represents the control condition. All data are normalized to the control condition and represented as mean ± SD. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Regarding the effect SeChry@PUREG4 exposure on the intracellular ROS levels, while in cancer cells the combination of SeChry@PUREG4 and Propranolol induced an additive increase in ROS levels, in HUVECs the addition of Propranolol had the opposite effect, reinforcing the previously observed antioxidant role of Propranolol in ECs (Figures 5C, D). Accordingly, the ROS-induced lipid peroxide levels increased upon SeChry@PUREG4 with or without Propranolol exposure, in MDA-MB-231 (Figure 5E) but not in HUVECs (Figure 5F). Moreover, HUVECs exposed to SeChry@PUREG4 presented a more stable monolayer with reduced VE-Cad intercellular junctional gaps (Figure 5G).
Together, these results suggest that SeChry@PUREG4 plus Propranolol could be an interesting strategy for cancer treatment, targeting both cancer cells and ECs. In cancer cells, SeChry@PUREG4 and Propranolol would have anti-tumor effects through the promotion of cell death mediated by ferroptosis, whereas in ECs, Propranolol would impair oxidative stress-induced mechanisms, blunting ECs hyperactivation and promoting stability, which in turn might decrease transendothelial cancer cell migration and impair metastasis.
Discussion
During tumorigenesis, the increased metabolic rate of cancer cells drives the generation of a pro-oxidative tumor microenvironment, responsible for the production and release of pro-angiogenic factors by cancer and tumor-associated stromal cells (54, 55). The ROS production within the tumor microenvironment is also stimulated by the main players of ECs activation, VEGF and hypoxia (56–59). The major production of ROS depends on NOX, which generate O2.- through the transfer of electrons from NADPH to oxygen (56–59). The generation of NOX-dependent ROS increases VEGF secretion and angiogenesis, in a HIF1α-dependent manner (60). Therefore, the generation of a pro-oxidative and pro-angiogenic tumor microenvironment seems to work synergistically in the promotion of tumor angiogenesis.
Ferroptosis has been firstly described as an iron-dependent programed cell death characterized by the accumulation of lipid peroxides (61), although new evidences have shown that ferroptosis is not a strict cell death mechanism, being relevant in the regulation of biological and pathophysiological processes (9–11, 62–64). Ferroptosis-inducing compounds, as Erastin, affect the antioxidant capacity of cells through the inhibition of xCT, also expressed in ECs (HUVECs) (Figure S1A). The impairment of xCT activity disturbs cystine import, the main source of cysteine for GSH synthesis. GSH acts as an electron donor to reduce lipid hydroperoxides upon GPX4 action (9, 10, 65). Besides the role of GSH as a scavenger, the proteins S-glutathionylation should be explored in the future, since it seems to be a mechanism of redox switch in ECs accounting for vascular homeostasis (66). So far, there are no specific markers of ferroptosis as a cell death mechanism and because we were exploring the non-lethal effect of ferroptosis, in this work we evaluated the levels of ROS-induced lipids peroxides underlying ferroptosis and GSH dynamics (67). The expression of genes pointed as being decreased upon ferroptosis activation, PTGS2, GPX4 and GSS was also assessed (10, 68–70). In our experimental conditions, we observed that Erastin significantly decreases cyst(e)ine uptake (Figure 1E) and promotes the increase of intracellular ROS (Figure 1A) and lipid peroxide levels (Figure 1C), without affecting the mitochondrial ROS content (Figure 1B). In agreement, the GSH depletion (Figure 1D) and the decreased expression of PTGS2, GPX4 and GSS genes (Figures 1F–H) was observed upon Erastin exposure. All these ferroptosis-like features did not account for ECs death (Figure 1I).
The anti-ferroptotic property of Propranolol has been recently described, showing that the peroxyl radicals scavenging property is independent of β1-blockade activity (29). Here, we observed that Propranolol decreased reduced and total GSH levels, but it kept the GSSG (oxidized GSH) levels similar to control conditions (Figure 1D), as well as it decreased the intracellular ROS levels (Figure 1A) and reverted the accumulation of ROS-induced lipid peroxides induced by Erastin (Figures 1A, C). The expression of PTGS2, GPX4 and GSS was rescued by Propranolol (Figures 1F–H). In fact, the increased H2S levels upon Propranolol exposure (Figure 1J), indicated that the antioxidant Propranolol property might be mediated by H2S, since it is a gasotransmitter capable of regulating oxidative stress by directly scavenging ROS (48, 71).
Considering that the generation of a pro-oxidative microenvironment is implicated in the promotion of the angiogenic switch and further angiogenesis (6, 7), we unraveled, for the first time, the role of the ferroptosis-like mechanism in ECs activation. We showed that the ferroptosis-like mechanism induces ECs hyperactivation, by increasing cell proliferation and migration (Figures 2A, B) and also promoting the formation of vessel-like structures (Figure 2C), mimicking the in vivo capacity of ECs to form blood vessels. Interestingly, Erastin stimulates the formation of vessel-like structures at the same range of H2O2 (ROS) (Figure 2C), supporting the involvement of the ferroptosis-like mechanism on angiogenic processes.
Propranolol acts as an inhibitor of angiogenesis and it was recently described to suppress proliferation, migration and tube formation of hemangioma cells through the HIF-1α-VEGF-A axis (72, 73) and to decrease the expression of angiogenic growth factors, as VEGF and FGF (20). However, the precise cellular mechanism underlying blood vessels disruption and angiogenesis impairment is still unknown. In this study, we observed that Propranolol, besides its anti-angiogenic effect under basal culture conditions, is able to abrogate the stimulation of ECs activation induced by Erastin and to disrupt already established vessel-like structures (Figures 2A–D), suggesting that the Propranolol anti-angiogenic effect is related, at least in part, to the abrogation of the ferroptosis-like mechanism.
The cancer-associated vasculature is characterized by an increased permeability and interstitial fluid pressure due to the disruption of ECs junctions, which reveals to be pivotal in cancer cell adhesion, intravasion and metastasis (74, 75). The increased VEGF levels, in the tumor microenvironment, promote VE-Cad adherens junction phosphorylation and internalization in ECs, leading to an impaired adhesiveness and non-functional vessels (74, 76). Besides the role of the ferroptosis-like mechanism on ECs activation, Erastin exposure promotes the formation of VE-Cad intercellular junctional gaps (Figures 3A, B), which results in the generation of a leakier ECs structure. This is accompanied by an increase in ICAM adhesion molecule expression (Figures 3C, D), a crucial protein for cancer cell and ECs interaction during the metastatic cascade (51–53). Increased ICAM expression (Figures 3C, D) promotes cancer cell adhesion to ECs and transendothelial migration (Figures 3F, G). Our results support that Propranolol contributes to vessel stabilization and putatively disturbs metastasis, since Propranolol did not alter VE-Cad intercellular junctional gaps (Figures 3A, B), ICAM expression and further cancer cell: ECs adhesion (Figure 3F) and transendothelial cancer cell migration (Figure 3G). Accordingly, Propranolol reverted the pro-metastatic ECs phenotype induced by Erastin, by decreasing VE-Cad intercellular junctional gaps (Figures 3A, B) and ICAM expression (Figure 3C); and by disturbing cancer cell adhesion to ECs and transendothelial cancer cell migration (Figures 3F, G). Therefore, the effect of the ferroptosis-like mechanism, at a non-lethal level, mimics the pathophysiological angiogenic process in cancer, characterized by an ECs hyperactivation that leads to the formation of a leakier vascular network (74, 75, 77, 78). On the other hand, Propranolol administration blocks the angiogenic switch, decelerating angiogenesis and further tumor growth, while preventing the generation of a leakier vasculature, decreasing metastasis and putatively increasing the delivery of cytotoxic drugs to the cancer cells.
Recently our group showed that monocytes act as endothelial progenitor cells that differentiate into ECs upon oxidative stress and are capable of incorporating into the tumor neo-vasculature, contributing to cancer progression (35). In this context, we explored if the pro-angiogenic stimuli pushing monocytes towards ECs differentiation, also benefited from a ferroptosis-like mechanism. In fact, we observed that neither Erastin nor Propranolol interfered with monocytes differentiation into ECs (Figure 4).
In cancer cells, selenium compounds interfere with the selenium uptake, selenocysteine biosynthesis and the production of selenoproteins, such as GPX4, consequently abrogating cell protection against ferroptosis (79). Moreover, selenium compounds display antioxidant or pro-oxidant properties, depending on their concentrations (80, 81); and they can be used as cytotoxic compounds showing anti-cancer properties and overcoming cisplatin resistance and multiple drug resistance (36, 38, 82, 83).
Considering the dual effect of these compounds and the antioxidant role of Propranolol in ECs, we explored if a combination of Propranolol with SeChry encapsulated in PUREG4 nanoparticles (SeChry@PUREG4) could be used as a strategy to simultaneously induce cancer cell death and stabilize ECs, resembling tumor vessels. Interestingly, in breast cancer cells (MDA-MB-231), Propranolol alone increased cell death by 2-fold (Figure 5A), demonstrating that, besides its role in preventing ECs hyperactivation, Propranolol could also promote cancer cells death. Moreover, MDA-MB-231 were more sensitive to SeChry@PUREG4 than HUVECs, showing higher cell death levels upon exposure with or without Propranolol (Figures 5A, B). These results reinforce the anti-tumoral SeChry@PUREG4 effect on ovarian cancer cells, recently described by us (38), and confirmed the differential effect of Propranolol on cancer cells and ECs. Interestingly, SeChry@PUREG4 exposure induced ferroptosis in cancer cells, as it increased the generation of intracellular ROS and the accumulation of lipid peroxides (Figures 5C, E), which ultimately promote cancer cell death (Figure 5A). Contrarily to the observations in HUVECs, in MDA-MB-231, Propranolol did not display antioxidant features, since the levels of ROS and ROS-induced lipid peroxides increased upon Propranolol exposure (Figures 5C, E). In HUVECs, SeChry@PUREG4 had no effect on the generation of lipid peroxides (Figure 5F) and did not affect the formation of VE-Cad junctional gaps (Figure 5G), suggesting that SeChry@PUREG4 does not compromise vessels stability. Together, these results demonstrate that SeChry@PUREG4 plus Propranolol administration is a promising strategy for cancer treatment, since this combination is able to induce cancer cell death through ferroptosis, while avoiding the formation of a leakier vasculature, which ultimately impairs cancer cell intravasation and metastasis.
Conclusion
The ferroptosis-like mechanism, mediated by Erastin, through GSH depletion and ROS-induced lipid peroxide generation is implicated in the regulation of some pathophysiological ECs features, promoting ECs hyperactivation, leakiness and cancer cell migration. Propranolol scavenging activity mediated by H2S impairs the generation of oxidative stress, reverting the ECs phenotype observed under Erastin exposure. Additionally, despite the effects of the ferroptosis-like mechanism on ECs activation, it did not affect the differentiation process of monocytes into ECs.
Moreover, in this paper we disclose the potential use of SeChry@PUREG4, a selenium-containing nanoformulation, in combination with Propranolol, as a good strategy for cancer treatment. The combination of SeChry@PUREG4 with Propranolol induces cancer cell death mediated by pro-oxidant features, while in ECs it prevents the formation of a leakier vasculature (Figure 6), pivotal in cancer cell intravasation and metastasis.
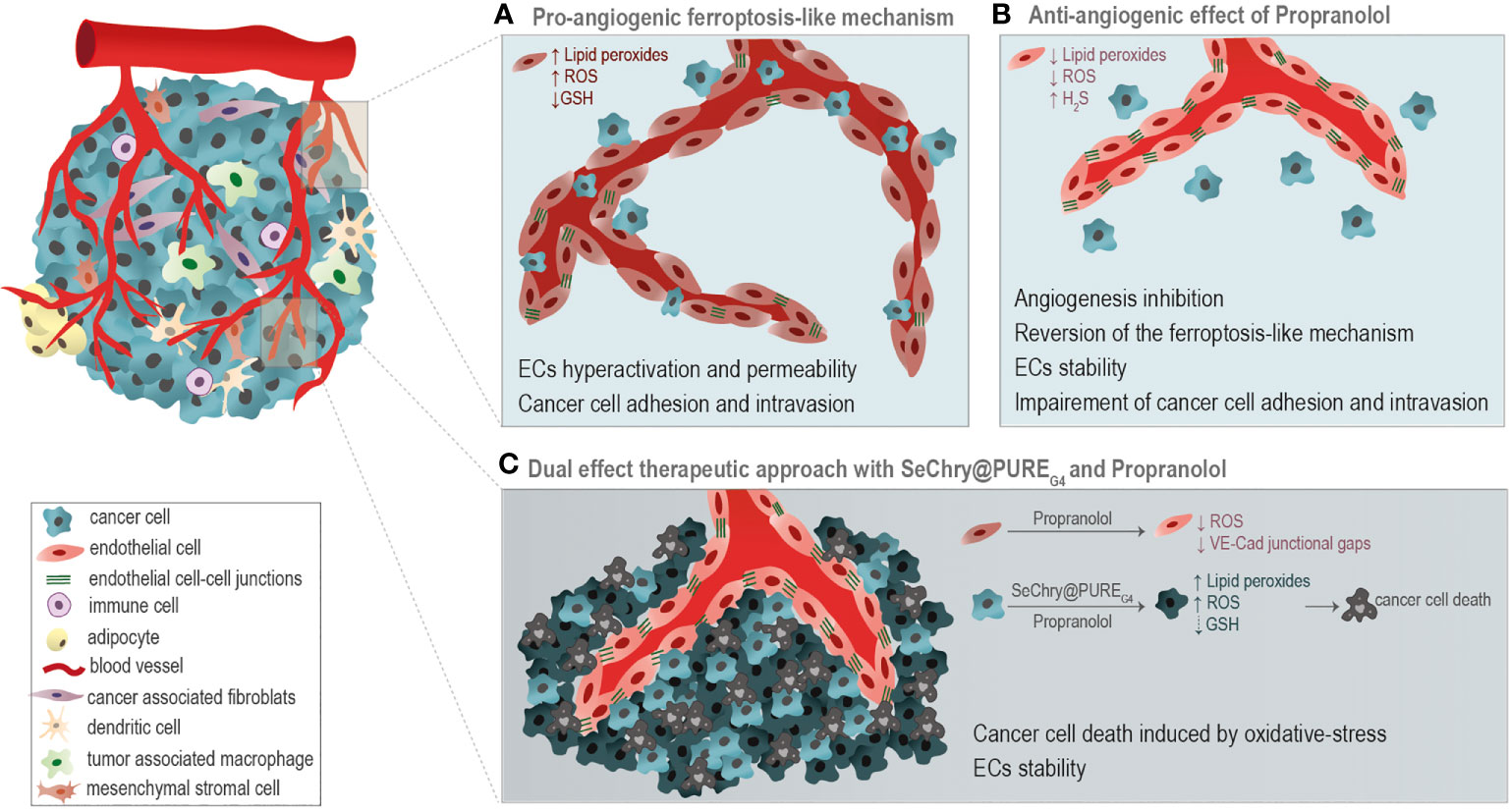
Figure 6 Taking advantage of the differential oxidative stress response of cancer cells and endothelial cells (ECs). (A) A pro-angiogenic ferroptosis-like mechanism, through the generation of ROS, accumulation of lipid peroxides and glutathione (GSH) depletion, is implicated in the promotion of ECs hyperactivation, vessels leakiness and cancer cell adhesion and intravasation. (B) Propranolol (Prop) ROS scavenging activity is anti-angiogenic, impairing ECs activation underlined by the ferroptosis-like mechanism. (C) The combination of SeChry@PUREG4 nanoparticles and Prop was unraveled as a potential cancer therapy. SeChry@PUREG4 induces cancer cell death mediated by pro-oxidative features, while Prop stabilizes ECs and prevents the formation of a leakier vasculature, avoiding metastasis. Prop enhances the pro-oxidative features of Sechry@PUREG4 effect.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
FL-C-Planned, executed the majority of the experimental assays and wrote the first draft of the manuscript. FM, AH, CM and RP-Performed experimental assays and revised the manuscript. AA, VB and SP-Supervised the project and revised and discussed the manuscript. JS-Supervised the project, provided funding, and revised and discussed the manuscript. All authors contributed to the article and approved the submitted version
Funding
The project was funded by IPOLFG EPE and by iNOVA4Health (UID/Multi/04462/2019) a program financially supported by Fundação para a Ciência e Tecnologia (FCT)/Ministério da Educação e Ciência, through national funds. We also acknowledge funding from FCT-MCTES through the project DREAM—PTDC/MEC-ONC/29327/2017. FL-C PhD fellowship was funded by FCT (PD/BD/128337/2017).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Serviço de Imuno-Hemoterapia from Instituto Português de Oncologia de Lisboa Francisco Gentil (IPOLFG EPE) for providing the buffy coats from healthy donors.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.656229/full#supplementary-material
Supplementary Figure 1 | xCT is expressed in endothelial cells (HUVECs) and in comparison to cancer cells, are more sensitive to the generation of lipid peroxides induced by Erastin (Era). (A) HUVECs expresses xCT (green) mainly in mitochondria (TOM20, red), and its expression is not affected by Era and/or Propranolol (Prop), for 16 h (scale: 20μm). (B) HUVECs are more sensitive to the generation of Era-induced lipid peroxides than cancer cells (MDA-MB-231). Two Era concentrations were tested (1,5μM and 5 μM) during different time points (1, 6 and 16h). In graphs the dashed line represents the control condition. All data are normalized to the control condition and represented as mean ± SD. *p<0.05, **p<0.01, ***p<0.001.
References
1. Potente M, Gerhardt H, Carmeliet P. Basic and Therapeutic Aspects of Angiogenesis. Cell (2011) 146:873–87. doi: 10.1016/j.cell.2011.08.039
2. Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, et al. Inhibition of Vascular Endothelial Growth Factor-Induced Angiogenesis Suppresses Tumour Growth In Vivo. Nature (1993) 362:841–4. doi: 10.1038/362841a0
3. Vasudev NS, Reynolds AR. Anti-Angiogenic Therapy for Cancer: Current Progress, Unresolved Questions and Future Directions. Angiogenesis (2014) 17:471–94. doi: 10.1007/s10456-014-9420-y
4. Hanahan D, Folkman J. Patterns and Emerging Mechanisms of the Angiogenic Switch During Tumorigenesis. Cell (1996) 86:353–64. doi: S0092-8674(00)80108-7
5. Lugano R, Ramachandran M, Dimberg A. Tumor Angiogenesis: Causes, Consequences, Challenges and Opportunities. Cell Mol Life Sci (2020) 77:1745–70. doi: 10.1007/s00018-019-03351-7
6. Kim YW, Byzova TV. Oxidative Stress in Angiogenesis and Vascular Disease. Blood (2014) 123:625–31. doi: 10.1182/blood-2013-09-512749
7. Prieto-Bermejo R, Hernández-Hernández A. The Importance of NADPH Oxidases and Redox Signaling in Angiogenesis. Antioxidants (2017) 6:32. doi: 10.3390/antiox6020032
8. Martinez-Outschoorn UE, Lin Z, Trimmer C, Flomenberg N, Wang C, Pavlides S, et al. Cancer Cells Metabolically “Fertilize” the Tumor Microenvironment With Hydrogen Peroxide, Driving the Warburg Effect: Implications for PET Imaging of Human Tumors. Cell Cycle (2011) 10:2504–20. doi: 10.4161/cc.10.15.16585
9. Li J, Cao F, Yin Hl, Huang Zj, Lin Zt, Mao N, et al. Ferroptosis: Past, Present and Future. Cell Death Dis (2020) 11:88. doi: 10.1038/s41419-020-2298-2
10. Yang WS, Sriramaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
11. Lei P, Bai T, Sun Y. Mechanisms of Ferroptosis and Relations With Regulated Cell Death: A Review. Front Physiol (2019) 10:139. doi: 10.3389/fphys.2019.00139
12. Yang WH, Huang Z, Wu J, Ding CKC, Murphy SK, Chi JT. A TAZ-ANGPTL4-NOX2 Axis Regulates Ferroptotic Cell Death and Chemoresistance in Epithelial Ovarian Cancer. Mol Cancer Res (2020) 18:79–90. doi: 10.1158/1541-7786.MCR-19-0691
13. Al-Majed AA, Bakheit AHH, Abdel Aziz HA, Alajmi FM, AlRabiah H. Propranolol. In: Profiles of Drug Substances, Excipients and Related Methodology. Academic Press (2017) p. 287–338. doi: 10.1016/bs.podrm.2017.02.006
14. Anderson R, Ramafi G, Theron AJ. Membrane Stabilizing, Anti-Oxidative Interactions of Propranolol and Dexpropranolol With Neutrophils. Biochem Pharmacol (1996) 52:341–9. doi: 10.1016/0006-2952(96)00212-2
15. Rotter A, de Oliveira ZNP. Infantile Hemangioma: Pathogenesis and Mechanisms of Action of Propranolol. JDDG J der Dtsch Dermatol Gesellschaft (2017) 15:1185–90. doi: 10.1111/ddg.13365
16. Apra C, Dumot C, Bourdillon P, Pelissou-Guyotat I. Could Propranolol be Beneficial in Adult Cerebral Cavernous Malformations? Neurosurg Rev (2019) 42:403–8. doi: 10.1007/s10143-018-01074-0
17. Hiller JG, Cole SW, Crone EM, Byrne DJ, Shackleford DM, Pang JMB, et al. Preoperative β-Blockade With Propranolol Reduces Biomarkers of Metastasis in Breast Cancer: A Phase II Randomized Trial. Clin Cancer Res (2020) 26:1803–11. doi: 10.1158/1078-0432.CCR-19-2641
18. Pantziarka P, Bryan BA, Crispino S, Dickerson EB. Propranolol and Breast Cancer—a Work in Progress. Ecancermedicalscience (2018) 12. doi: 10.3332/ecancer.2018.ed82
19. Cardwell CR, Pottegård A, Vaes E, Garmo H, Murray LJ, Brown C, et al. Propranolol and Survival From Breast Cancer: A Pooled Analysis of European Breast Cancer Cohorts. Breast Cancer Res (2016) 18:119. doi: 10.1186/s13058-016-0782-5
20. Sharifpanah F, Saliu F, Bekhite MM, Wartenberg M, Sauer H. β-Adrenergic Receptor Antagonists Inhibit Vasculogenesis of Embryonic Stem Cells by Downregulation of Nitric Oxide Generation and Interference With VEGF Signalling. Cell Tissue Res (2014) 358:443–52. doi: 10.1007/s00441-014-1976-8
21. Cavalheiro S, do Amaral Campos HG, da Costa MDS. A Case of Giant Fetal Intracranial Capillary Hemangioma Cured With Propranolol. J Neurosurg Pediatr PED (2016) 17:711–6.
22. Sasaki M, North PE, Elsey J, Bubley J, Rao S, Jung Y, et al. Propranolol Exhibits Activity Against Hemangiomas Independent of Beta Blockade. NPJ Precis Oncol (2019) 3:27. doi: 10.1038/s41698-019-0099-9
23. Okochi-Takada E, Hattori N, Tsukamoto T, Miyamoto K, Ando T, Ito S, et al. ANGPTL4 is a Secreted Tumor Suppressor That Inhibits Angiogenesis. Oncogene (2014) 33:2273–8. doi: 10.1038/onc.2013.174
24. Carbone C, Piro G, Merz V, Simionato F, Santoro R, Zecchetto C, et al. Angiopoietin-Like Proteins in Angiogenesis, Inflammation and Cancer. Int J Mol Sci (2018) 19:1–22. doi: 10.3390/ijms19020431
25. Veith A, Moorthy B. Role of Cytochrome P450s in the Generation and Metabolism of Reactive Oxygen Species. Curr Opin Toxicol (2018) 7:44–51. doi: 10.1016/j.cotox.2017.10.003
26. Albertolle ME, Peter Guengerich F. The Relationships Between Cytochromes P450 and H2O2: Production, Reaction, and Inhibition. J Inorg Biochem (2018) 186:228–34. doi: 10.1016/j.jinorgbio.2018.05.014
27. Johnson JA, Herring VL, Wolfe MS, Relling MV, Pharmacy C, Tennessee JAJ, et al. CYP1A2 and CYP2D6 4-Hydroxylate Propranolol and Both Reactions Exhibit Racial Differences 1. J Pharmacol Exp Ther (2000) 294:1099–105.
28. Ayano G. Psychotropic Medications Metabolized by Cytochromes P450 ( CYP ) 2d6 Enzyme and Relevant Drug Interactions. Clin Pharmacol (2016) 5:4–7. doi: 10.4172/2167-065X.1000162
29. Mishima E, Sato E, Ito J, Yamada KI, Suzuki C, Oikawa Y, et al. Drugs Repurposed as Antiferroptosis Agents Suppress Organ Damage, Including Aki, by Functioning as Lipid Peroxyl Radical Scavengers. J Am Soc Nephrol (2020) 31:280–96. doi: 10.1681/ASN.2019060570
30. Rowland K, Yeo WW, Ellis SW, Chadwick IG, Haq I, Lennard MS, et al. Inhibition of CYP2D6 Activity by Treatment With Propranolol and the Role of 4-Hydroxy Propranolol. Br J Clin Pharmacol (1994) 38:9–14.
31. Lewis DFV, Modi S, Dickins M, Limited D, Road P. Structure-Activity Relationship for Human Cytochrome P450 Substrates and Inhibitors. Drug Metab Rev (2002) 34:69–82.
32. Wang B, Zhou S-F. Synthetic and Natural Compounds That Interact With Human Cytochrome P450 1A2 and Implications in Drug Development. Curr Med Chem (2009) 16:4066–218. doi: 10.2174/092986709789378198
33. Kum J. β-Adrenergic Receptor-Dependent and -Independent Effects of Propranolol in Infantile Hemangioma. Electronic Thesis and Dissertation Repository (2015).
34. Grilo NM, João Correia M, Miranda JP, Cipriano M, Serpa J, Matilde Marques M, et al. Unmasking Efavirenz Neurotoxicity: Time Matters to the Underlying Mechanisms. Eur J Pharm Sci (2017) 105:47–54. doi: 10.1016/j.ejps.2017.05.010
35. Lopes-Coelho F, Silva F, Gouveia-Fernandes S, Martins C, Lopes N, Domingues G, et al. Monocytes as Endothelial Progenitor Cells (Epcs), Another Brick in the Wall to Disentangle Tumor Angiogenesis. Cells (2020) 9:107. doi: 10.3390/cells9010107
36. Martins IL, Charneira C, Gandin V, Ferreira Da Silva JL, Justino GC, Telo JP, et al. Selenium-Containing Chrysin and Quercetin Derivatives: Attractive Scaffolds for Cancer Therapy. J Med Chem (2015) 58:4250–65. doi: 10.1021/acs.jmedchem.5b00230
37. Restani RB, Morgado PI, Ribeiro MP, Correia IJ, Aguiar-Ricardo A, Bonifácio VDB. Biocompatible Polyurea Dendrimers With pH-Dependent Fluorescence. Angew Chemie Int Ed (2012) 51:5162–5. doi: 10.1002/anie.201200362
38. Santos I, Ramos C, Mendes C, Sequeira CO, Tomé CS, Fernandes DGH, et al. Targeting Glutathione and Cystathionine β-Synthase in Ovarian Cancer Treatment by Selenium–Chrysin Polyurea Dendrimer Nanoformulation. Nutrients (2019) 11:2523. doi: 10.3390/nu11102523
39. Restani RB, Conde J, Pires RF, Martins P, Fernandes AR, Baptista PV, et al. Poxylated Polyurea Dendrimers: Smart Core-Shell Vectors With IC 50 Lowering Capacity. Macromol Biosci (2015) 15:1045–51. doi: 10.1002/mabi.201500131
40. Kunsang Y, David BM, Milton HJ, Zhihao F, Tacey V. Multifunctional Forms of Polyoxazoline Copolymers and Drug Compositions Comprising the Same (2010). Available at: https://www.surechembl.org/document/US-20100330023-A1.
41. Lo M, Wang YZ, Gout PW. The Xc- Cystine/Glutamate Antiporter: A Potential Target for Therapy of Cancer and Other Diseases. J Cell Physiol (2008) 215:593–602. doi: 10.1002/jcp.21366
42. Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, et al. The Cystine / Glutamate Antiporter System X C - in Health and Disease : From Molecular Mechanisms. Antioxid Redox Signal (2013) 18:522–55. doi: 10.1089/ars.2011.4391
43. Sun Y, Zheng Y, Wang C, Liu Y. Glutathione Depletion Induces Ferroptosis , Autophagy , and Premature Cell Senescence in Retinal Pigment Epithelial Cells. Cell Death Dis (2018) 9:753. doi: 10.1038/s41419-018-0794-4
44. Sato M, Kusumi R, Hamashima S, Kobayashi S, Sasaki S, Komiyama Y, et al. The Ferroptosis Inducer Erastin Irreversibly Inhibits System Xc– and Synergizes With Cisplatin to Increase Cisplatin’s Cytotoxicity in Cancer Cells. Sci Rep (2018) 8:968. doi: 10.1038/s41598-018-19213-4
45. Koppula P, Zhang Y, Zhuang L, Gan B. Amino Acid Transporter SLC7A11 / xCT At the Crossroads of Regulating Redox Homeostasis and Nutrient Dependency of Cancer. Cancer Commun (2018) 38:1–13. doi: 10.1186/s40880-018-0288-x
46. Nunes SC, Ramos C, Lopes-Coelho F, Sequeira CO, Silva F, Gouveia-Fernandes S, et al. Cysteine Allows Ovarian Cancer Cells to Adapt to Hypoxia and to Escape From Carboplatin Cytotoxicity. Sci Rep (2018) 8:9513. doi: 10.1038/s41598-018-27753-y
47. Hipólito A, Nunes SC, Vicente JB, Serpa J. Cysteine Aminotransferase (Cat): A Pivotal Sponsor in Metabolic Remodeling and an Ally of 3-Mercaptopyruvate Sulfurtransferase (MST) in Cancer. Molecules (2020) 25:3984. doi: 10.3390/molecules25173984
48. Shefa U, Kim MS, Jeong NY, Jung J. Antioxidant and Cell-Signaling Functions of Hydrogen Sulfide in the Central Nervous System. Oxid Med Cell Longev (2018) 2018:1873962. doi: 10.1155/2018/1873962
49. Giuffrè A, Vicente JB. Hydrogen Sulfide Biochemistry and Interplay With Other Gaseous Mediators in Mammalian Physiology. Oxid Med Cell Longev (2018) 2018:1–31. doi: 10.1155/2018/6290931
50. Harris ES, Nelson WJ. VE-Cadherin: At the Front, Center, and Sides of Endothelial Cell Organization and Function. Curr Opin Cell Biol (2010) 22:651–8. doi: 10.1016/j.ceb.2010.07.006
51. Shenoy AK, Lu J. Cancer Cells Remodel Themselves and Vasculature to Overcome the Endothelial Barrier. Cancer Lett (2016) 380:534–44. doi: 10.1016/j.canlet.2014.10.031
52. Zhang P, Goodrich C, Fu C, Dong C. Melanoma Upregulates ICAM-1 Expression on Endothelial Cells Through Engagement of Tumor CD44 With Endothelial E-selectin and Activation of a Pkcα-P38-SP-1 Pathway. FASEB J (2014) 28:4591–609. doi: 10.1096/fj.11-202747
53. Chen Q, Massagué J. Molecular Pathways: VCAM-1 as a Potential Therapeutic Target in Metastasis. Clin Cancer Res (2012) 18:5520–5. doi: 10.1158/1078-0432.CCR-11-2904
54. De Sá Junior PL, Câmara DAD, Porcacchia AS, Fonseca PMM, Jorge SD, Araldi RP, et al. The Roles of ROS in Cancer Heterogeneity and Therapy. Oxid Med Cell Longev (2017) 2017:2467940. doi: 10.1155/2017/2467940
55. Schito L, Semenza GL. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer (2016) 1136:71–85. doi: 10.1016/j.trecan.2016.10.016
56. Dewhirst MW, Cao Y, Moeller B. Cycling Hypoxia and Free Radicals Regulate Angiogenesis and Radiotherapy Response. Nat Rev Cancer (2008) 8:425–37. doi: 10.1038/nrc2397
57. Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, et al. Mitochondrial Superoxide: Production, Biological Effects, and Activation of Uncoupling Proteins. Free Radic Biol Med (2004) 37:755–67. doi: 10.1016/j.freeradbiomed.2004.05.034
58. Coso S, Harrison I, Harrison CB, Vinh A, Sobey CG, Drummond GR, et al. Nadph Oxidases as Regulators of Tumor Angiogenesis: Current and Emerging Concepts. Antioxid Redox Signal (2012) 16:1229–47. doi: 10.1089/ars.2011.4489
59. Lassègue B, San Martín A, Griendling KK. Biochemistry, Physiology, and Pathophysiology of NADPH Oxidases in the Cardiovascular System. Circ Res (2012) 110:1364–90. doi: 10.1161/CIRCRESAHA.111.243972
60. Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang XR, Jiang BH. Reactive Oxygen Species Regulate Angiogenesis and Tumor Growth Through Vascular Endothelial Growth Factor. Cancer Res (2007) 67:10823–30. doi: 10.1158/0008-5472.CAN-07-0783
61. Hirschhorn T, Stockwell BR. The Development of the Concept of Ferroptosis. Free Radic Biol Med (2019) 133:130–43. doi: 10.1016/j.freeradbiomed.2018.09.043
62. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. Nat Cell Biol (2014) 16:1180–91. doi: 10.1038/ncb3064
63. Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, et al. Synchronized Renal Tubular Cell Death Involves Ferroptosis. Proc Natl Acad Sci U.S.A. (2014) 111:16836–41. doi: 10.1073/pnas.1415518111
64. Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, et al. The Ferroptosis Inducer Erastin Enhances Sensitivity of Acute Myeloid Leukemia Cells to Chemotherapeutic Agents. Mol Cell Oncol (2015) 2:e1054549. doi: 10.1080/23723556.2015.1054549
65. Proneth B, Conrad M. Ferroptosis and Necroinflammation, a Yet Poorly Explored Link. Cell Death Differ (2019) 26:14–24. doi: 10.1038/s41418-018-0173-9
66. Lermant A, Murdoch CE. Cysteine Glutathionylation Acts as a Redox Switch in Endothelial Cells. Antioxidants (2019) 8:315. doi: 10.3390/antiox8080315
67. Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol (2016) 26:165–76. doi: 10.1016/j.tcb.2015.10.014
68. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
69. Chen X, Comish PB, Tang D, Kang R. Characteristics and Biomarkers of Ferroptosis. Front Cell Dev Biol (2021) 9:637162. doi: 10.3389/fcell.2021.637162
70. Yang Y, Lin J, Guo S, Xue X, Wang Y, Qiu S, et al. RRM2 Protects Against Ferroptosis and is a Tumor Biomarker for Liver Cancer. Cancer Cell Int (2020) 20:587. doi: 10.1186/s12935-020-01689-8
71. Olas B. Hydrogen Sulfide as a “Double-Faced” Compound: One With Pro- and Antioxidant Effect. Adv Clin Chem (2017) 78:187–96. doi: 10.1016/bs.acc.2016.07.005
72. Chen YZ, Bai N, Bi JH, Liu XW, Xu GQ, Zhang LF, et al. Propranolol Inhibits the Proliferation, Migration and Tube Formation of Hemangioma Cells Through HIF-1α Dependent Mechanisms. Braz J Med Biol Res (2017) 50:e6138. doi: 10.1590/1414-431x20176138
73. Chim H, Armijo BS, Miller E, Gliniak C, Serret MA, Gosain AK. Propranolol Induces Regression of Hemangioma Cells Through Hif-1α–Mediated Inhibition of VEGF-a. Ann Surg (2012) 256:146–56. doi: 10.1097/SLA.0b013e318254ce7a
74. Rohlenova K, Veys K, Miranda-Santos I, De Bock K, Carmeliet P. Endothelial Cell Metabolism in Health and Disease. Trends Cell Biol (2018) 28:224–36. doi: 10.1016/j.tcb.2017.10.010
75. Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, et al. Openings Between Defective Endothelial Cells Explain Tumor Vessel Leakiness. Am J Pathol (2000) 156:1363–80. doi: 10.1016/S0002-9440(10)65006-7
76. Bentley K, Mariggi G, Gerhardt H, Bates PA. Tipping the Balance: Robustness of Tip Cell Selection, Migration and Fusion in Angiogenesis. PloS Comput Biol (2009) 5:e1000549. doi: 10.1371/journal.pcbi.1000549
77. Trédan O, Galmarini CM, Patel K, Tannock IF. Drug Resistance and the Solid Tumor Microenvironment. J Natl Cancer Inst (2007) 99:1441–54. doi: 10.1093/jnci/djm135
78. Masiero M, Simões FC, Han HD, Snell C, Peterkin T, Bridges E, et al. A Core Human Primary Tumor Angiogenesis Signature Identifies the Endothelial Orphan Receptor ELTD1 as a Key Regulator of Angiogenesis. Cancer Cell (2013) 24:229–41. doi: 10.1016/j.ccr.2013.06.004
79. Carlisle AE, Lee N, Matthew-Onabanjo AN, Spears ME, Park SJ, Youkana D, et al. Selenium Detoxification is Required for Cancer-Cell Survival. Nat Metab (2020) 2:603–11. doi: 10.1038/s42255-020-0224-7
80. Lee KH, Jeong D. Bimodal Actions of Selenium Essential for Antioxidant and Toxic Pro-Oxidant Activities: The Selenium Paradox (Review). Mol Med Rep (2012) 5:299–304. doi: 10.3892/mmr.2011.651
81. Collery P. Strategies for the Development of Selenium-Based Anticancer Drugs. J Trace Elem Med Biol (2018) 50:498–507. doi: 10.1016/j.jtemb.2018.02.024
82. Singh M, Kaur M, Silakari O. Flavones: An Important Scaffold for Medicinal Chemistry. Eur J Med Chem (2014) 84:206–39. doi: 10.1016/j.ejmech.2014.07.013
Keywords: angiogenesis, ferroptosis, oxidative stress, lipid peroxidation, endothelial cell hyperactivation, propranolol, polyurea dendrimers, tumor vasculature stabilizers
Citation: Lopes-Coelho F, Martins F, Hipólito A, Mendes C, Sequeira CO, Pires RF, Almeida AM, Bonifácio VDB, Pereira SA and Serpa J (2021) The Activation of Endothelial Cells Relies on a Ferroptosis-Like Mechanism: Novel Perspectives in Management of Angiogenesis and Cancer Therapy. Front. Oncol. 11:656229. doi: 10.3389/fonc.2021.656229
Received: 20 January 2021; Accepted: 21 April 2021;
Published: 10 May 2021.
Edited by:
Sara Rodriguez-Enriquez, Instituto Nacional de Cardiología, MexicoReviewed by:
Hsueh-Wei Chang, Kaohsiung Medical University, TaiwanAngela Ostuni, University of Basilicata, Italy
Copyright © 2021 Lopes-Coelho, Martins, Hipólito, Mendes, Sequeira, Pires, Almeida, Bonifácio, Pereira and Serpa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jacinta Serpa, amFjaW50YS5zZXJwYUBubXMudW5sLnB0