 Li-Xin Wu1†Hao Jiang1†Ying-Jun Chang1Ya-Lan Zhou1Jing Wang1Zi-Long Wang1Lei-Ming Cao1Jin-Lan Li1Qiu-Yu Sun1Shan-Bo Cao2Feng Lou2Tao Zhou2
Li-Xin Wu1†Hao Jiang1†Ying-Jun Chang1Ya-Lan Zhou1Jing Wang1Zi-Long Wang1Lei-Ming Cao1Jin-Lan Li1Qiu-Yu Sun1Shan-Bo Cao2Feng Lou2Tao Zhou2 Li-Xia Liu2
Li-Xia Liu2 Cheng-Cheng Wang2
Cheng-Cheng Wang2 Yu Wang1
Yu Wang1 Qian Jiang1
Qian Jiang1 Lan-Ping Xu1Xiao-Hui Zhang1Kai-Yan Liu1
Lan-Ping Xu1Xiao-Hui Zhang1Kai-Yan Liu1 Xiao-Jun Huang1,3,4*
Xiao-Jun Huang1,3,4* Guo-Rui Ruan1*
Guo-Rui Ruan1*- 1Beijing Key Laboratory of Hematopoietic Stem Cell Transplantation, Peking University People’s Hospital, Peking University Institute of Hematology, National Clinical Research Center for Hematologic Disease, Beijing, China
- 2Department of Bioinformatics, AcornMed Biotechnology Co., Ltd., Beijing, China
- 3Peking-Tsinghua Center for Life Sciences, Academy for Advanced Interdisciplinary Studies, Peking University, Beijing, China
- 4Research Unit of Key Technique for Diagnosis and Treatments of Hematologic Malignancies, Chinese Academy of Medical Sciences, Beijing, China
Background: Approximately 30% of Chinese individuals with cytogenetically normal acute myeloid leukemia (CN-AML) have biallelic CEBPA (biCEBPA) mutations. The prognosis and optimal therapy for these patients are controversial in clinical practice.
Methods: In this study, we performed targeted region sequencing of 236 genes in 158 individuals with this genotype and constructed a nomogram model based on leukemia-free survival (LFS). Patients were randomly assigned to a training cohort (N =111) and a validation cohort (N =47) at a ratio of 7:3. Risk stratification was performed by the prognostic factors to investigate the risk-adapted post-remission therapy by Kaplan–Meier method.
Results: At least 1 mutated gene other than CEBPA was identified in patients and mutation number was associated with LFS (61.6% vs. 39.0%, P =0.033), survival (85.6% vs. 62.9%, P =0.030) and cumulative incidence of relapse (CIR) (38.4% vs. 59.5%, P =0.0496). White blood cell count, mutations in CFS3R, KMT2A and DNA methylation related genes were weighted to construct a nomogram model and differentiate two risk subgroups. Regarding LFS, low-risk patients were superior to the high-risk (89.3% vs. 33.8%, P <0.001 in training cohort; 87.5% vs. 18.2%, P =0.009 in validation cohort). Compared with chemotherapy, allogenic hematopoietic stem cell transplantation (allo-HSCT) improved 5-year LFS (89.6% vs. 32.6%, P <0.001), survival (96.9% vs. 63.6%, P =0.001) and CIR (7.2% vs. 65.8%, P <0.001) in high-risk patients but not low-risk patients (LFS, 77.4% vs. 88.9%, P =0.424; survival, 83.9% vs. 95.5%, P =0.173; CIR, 11.7% vs. 11.1%, P =0.901).
Conclusions: Our study indicated that biCEBPA mutant-positive CN-AML patients could be further classified into two risk subgroups by four factors and allo-HSCT should be recommended for high-risk patients as post-remission therapy. These data will help physicians refine treatment decision-making in biCEBPA mutant-positive CN-AML patients.
Introduction
Acute myeloid leukemia (AML) is one of the adult malignancies bearing the fewest mutations (1, 2). However, this disorder still comprises heterogeneous subgroups with variable responses to therapy stratified by identified leukemia driver events such as abnormalities in FLT3-ITD, NPM1, and BCR-ABL1 fusion. Patients without adverse or favorable genetic alterations were classified into the intermediate-risk subgroup and allogenic hematopoietic stem cell transplantation (allo-HSCT) was recommended to improve survival (3). Some of the intermediate-risk patients with normal karyotype were refined as the favorable risk ones in the revised 2016 WHO classification of AML because they had the prognostically favorable alteration, biallelic CEBPA (biCEBPA) mutations, compared with patients with wild-type or monoallelically mutated CEBPA (4, 5). However, this subgroup is still not homogeneous with relapse rate reaching approximately 40% (4, 6) and thus the best post-remission therapy remains controversial. Elucidation of cooperating events in this subgroup is urgently required.
Approximately 86% of AML patients have two or more driver mutations and such gene-gene interactions significantly alter the prognosis (5). To clarify the potential risk factors in biCEBPA mutated AML patients, next-generation sequencing has been adopted in many studies for the detection of co-mutated genes with sensitivity reaching 1 in 107 cells (7). GATA2, CSF3R and other tyrosine kinase genes (KIT, JAK3 and FLT3-ITD), WT1 and genes involved in chromatin/DNA modification, cohesin complex, and splicing were identified as hotspots in recent studies to decipher prognostic stratification in biCEBPA mutated AML (6, 8–12). Despite promising results, the true status of these concomitant mutations and their prognostic impact on biCEBPA mutated AML remain to be fully defined (13). This discordance may be attributed to two reasons. First, the sample size of biCEBPA mutated AML patients was small (<100 in most studies), thus limiting the statistical significance of the conclusions to some extent. Second, dozens of genes, or just the hotspot genes, were detected, hindering analysis of the relationships among different mutations.
In addition to mutational information, clinical data are also of significance. In our previous study, we established the prognostic value of pretreatment parameter, such as higher white blood cell (WBC) count, and posttreatment parameter, such as minimal residual disease detected by multiparameter flow cytometry (MFC-MRD) in biCEBPA mutated AML (14, 15). Patients with positive MFC-MRD after consolidation therapy showed a high risk of relapse and benefited from transplantation (15). Therefore, chemotherapy would no longer be appropriate as the first-line treatment for some biCEBPA mutated AML patients and identification of additional risk factors is required to refine treatment decision-making. However, a comprehensive and risk-adapted estimation of the most appropriate post-remission therapy based on clinical and molecular data at diagnosis (pretreatment parameters) in this population remains to be established.
In this study, we conducted high-depth (≥1 000×) targeted region sequencing (TRS) in a large panel with 236 known and potential driver genes to investigate the mutational context in 158 newly diagnosed patients with cytogenetically normal AML (CN-AML) and biCEBPA mutations. Mutational and clinical data at diagnosis were combined and weighted in a nomogram model for refined risk stratification. This study will provide practical prognosis information for biCEBPA mutated CN-AML patients and pave the way for precision treatment.
Patients and Methods
Patients
A total of 1 255 patients with newly diagnosed AML were enrolled from February 2010 to December 2019 at Peking University People’s Hospital. All participants included in our study met the following criteria: (1) age ≥15 years; (2) normal cytogenetics; (3) achieved complete remission (CR); (4) biCEBPA mutant-positive (Figure 1). In total, 158 participants qualified for subsequent analyses. The protocols for induction therapy and post-remission therapy are described in our previous study (14, 16–18). Induction treatment included 1–2 cycles of IA10 (idarubicin 10 mg/m2 for 3 days and cytarabine 100 mg/m2 for 7 days), HAA (homoharringtonine 2 mg/m2 for 7 days, aclarubicin 20 mg/day for 7 days and cytarabine 100 mg/m2 for 7 days) or CAG (cytarabine 10 mg/m2 every 12 hours for 14 days, aclarubicin 20 mg/day for 4 days and granulocyte-colony stimulating factor 300μg/day for 14 days). When CR was achieved, patients were recommended to receive at least 6 cycles of consolidation chemotherapy, including 4 cycles of intermediate-dose cytarabine (2 g/m2 every 12 hours for 3 days) and 2 or more cycles of anthracycline (daunorubicin 45 mg/m2 or idarubicin 10 mg/m2 for 3 days or mitoxantrone 8 mg/m2 for 3 days) in combination with cytarabine (100 mg/m2 for 7 days). Patients proceeded to undergo an allo-HSCT received at least 2 cycles of consolidation chemotherapy. Donors were selected from human leukocyte antigen (HLA) matched siblings, HLA matched unrelated donors or HLA haploidentical related donors. MFC-MRD monitoring was described as previously reported (15). The sensitivity was 0.01% and any measurable level of MRD was considered positive (19). For patients with positive MRD after allo-HSCT, preemptive antileukemic chemotherapy in combination with donor lymphocyte infusion (DLI) or interferon-α was given (20). For patients with hematologic relapse, chemotherapy followed by DLI was given as the first-line strategy. And for relapse prophylaxis, only DLI was used. Details of DLI were described previously (21, 22).
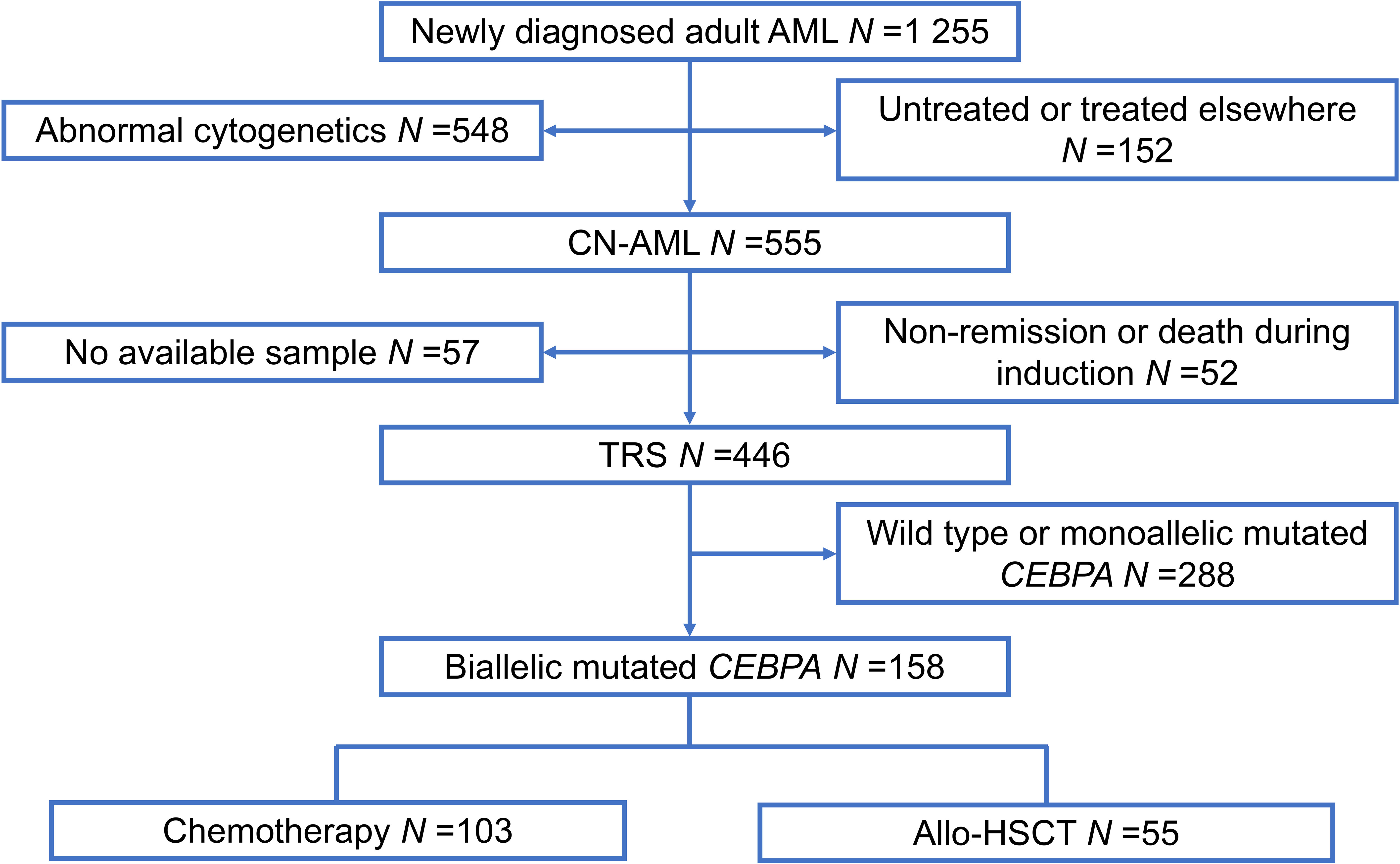
Figure 1 Patient recruitment and cohort assignment. AML, acute myeloid leukemia; CN, cytogenetically normal; TRS, targeted region sequencing; Allo-HSCT, allogenic hematopoietic stem cell transplantation.
High-Depth TRS and Analysis
We designed a panel of 236 known and potential driver genes for TRS (Supplementary Table 1). DNA was extracted from bone marrow samples using DNAzol® kits (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The sequencing process was performed according to our previous report (23). The average sequencing depth on target per sample was ≥1 000×. Typical mutations in NPM1 (type A/B/D) were validated by real-time quantitative polymerase chain reaction and atypical mutations were validated by Sanger sequencing (24). Mutations in FLT3-ITD were validated by Sanger sequencing.
Nomogram Model and Risk Stratification
Participants were assigned to a training cohort (N =111) and a validation cohort (N =47) at a ratio of 7:3 randomly. A nomogram was constructed based on the variables selected from the Cox regression model. The discrimination ability of the prediction model was measured by the concordance index (C-index) and the calibration was evaluated graphically by the calibration plots. Risk stratification was performed based on the nomogram model.
Endpoints and Statistical Analyses
The primary endpoint in this study was leukemia-free survival (LFS), which was calculated from the date of CR to relapse, death from any cause, last contact, or June 30th, 2020. The secondary endpoints included survival, cumulative incidence of relapse (CIR) and non-relapse mortality (NRM). Survival was calculated from the date of diagnosis to death from any cause, last contact, or June 30th, 2020. CIR and NRM were used in a competing risk setting and death without disease progression or relapse was treated as a competing event. Continuous variables were analyzed by Mann-Whitney U test. Categorized variables were analyzed by Pearson Chi-square test. Survival functions were estimated using the Kaplan-Meier method and compared by the log-rank test. Variables were selected by univariate Cox regression model and those with P <0.15 were subsequently enrolled in the multivariate Cox regression model. Receiving an allo-HSCT was recorded as a censored event to identify the prognostic factors before an allo-HSCT. Landmark analysis was performed to revise bias from early relapse or death when comparing the outcomes of post-remission therapies. Analyses were performed using SPSS software version 22.0 (Chicago, IL, USA), GraphPad Prism 7.04® (San Diego, CA, USA) and R software version 4.0.2 (http://www.Rproject.org). P <0.05 was considered to indicate statistical significance.
Results
Patient Characteristics
Among the 158 patients with biCEBPA mutations, 103 received chemotherapy only, while 55 received an allo-HSCT. Rate of patients receiving an allo-HSCT was significantly decreased after 2016 than before (22.5% vs. 44.8%, P =0.003). The median time from the first CR (CR1) to receiving an allo-HSCT was 4.77 months. According to the landmark analysis, 10 patients with LFS ≤4.77 months would be excluded from the subsequent analyses unless receiving an allo-HSCT was treated as a censored event. As shown in Table 1, there were no significant differences between the consolidation chemotherapy and allo-HSCT cohorts in terms of sex, WBC, hemoglobin, platelets, French-American-British (FAB) type and MRD after induction (MRDint) (all P >0.05). Age and CR rate after first induction were significantly greater in the consolidation chemotherapy cohort than that in the allo-HSCT cohort (age, median, 41 y vs. 33 y, P <0.001; CR rate, 92.6% vs. 79.6%, P =0.021). The allo-HSCT cohort had better 5-year LFS (84.8% vs. 51.2%, P <0.001) and 5-year survival (91.9% vs. 74.1%, P =0.018), lower 5-year CIR (9.1% vs. 47.7%, P <0.001) but comparable NRM (6.1% vs. 1.1%, P =0.125) (Supplementary Figure 1).
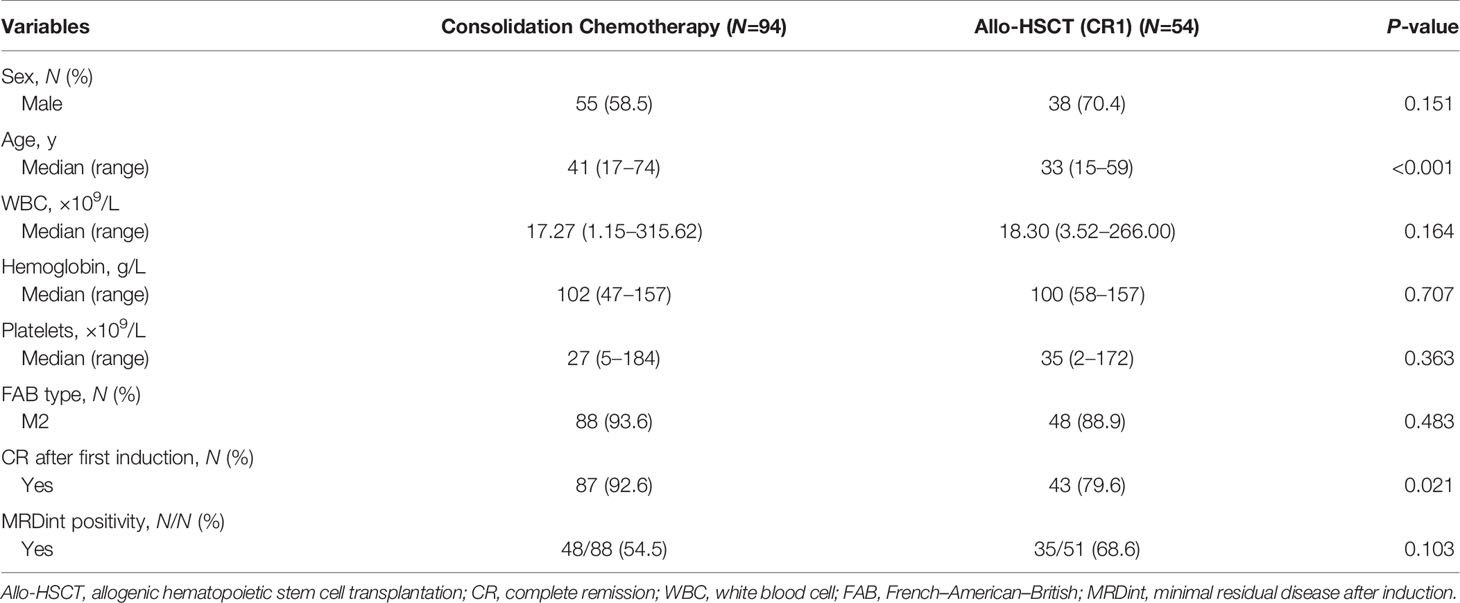
Table 1 Patient characteristics categorized by post-remission therapy.
Genomic Analysis of biCEBPA Mutated CN-AML
Of the 158 biCEBPA mutated ones, two patients carried two frameshift deletion mutations respectively and one carried two frameshift insertion mutations (Figure 2). We identified 1 306 mutations in 203 genes other than CEBPA. The median mutation number was 8 (1–20). Interestingly, additional mutations of ≤5, 6–7, 8, 9–10, >10 were identified uniformly with ~20% of patients (Supplementary Figure 2). Missense mutations were the predominant type (N =1 024; 78.4%), followed by frame-shift (N =103; 7.9%), in-frame (N =98; 7.5%), nonsense (N =55; 4.2%) and splice-site (N =26; 2.0%) mutations. These genes (mutated in ≥10 patients) were classified into 9 genetic subgroups: transcription factors (GATA2 and MYC), tumor suppressors (WT1 and MPL), activated signaling (NRAS, CSF3R, LILRB3, JAK3, MACF1, MST1, FLT3-ITD, KIT, NCOR2 and LAMA5), chromatin modifiers (EP300, SRCAP, DPF2, ASXL2 and ALK), cell metabolism (HERC2), DNA methylation (TET2), cohesin complex (RAD21), histone methylation (KMT2A and KMT2D) and others (AHNAK2, PCLO, EPPK1, POTEG, TRIO, POTEH, LOXHD1, AHNAK, HMCN1 and PLEC). Two genetic subgroups (spliceosome and adhesion) were not listed because of the low frequency of their mutated genes. GATA2 was the most frequently affected gene in 48 patients (30.4%), followed by WT1 (N =42, 26.6%), NRAS (N =31, 19.6%), AHNAK2 (N =31, 19.6%), PCLO (N =28, 17.7%) and CSF3R (N =24, 15.2%). FLT3-ITD represented 10.1% (N =16) in this population and 14 of them were identified by Sanger sequencing. The missed two variants were attributed to the low mutational burden (7.7% and 7.9% respectively). Only 1 patient had NPM1 mutation and this variant was further validated by real-time quantitative polymerase chain reaction.
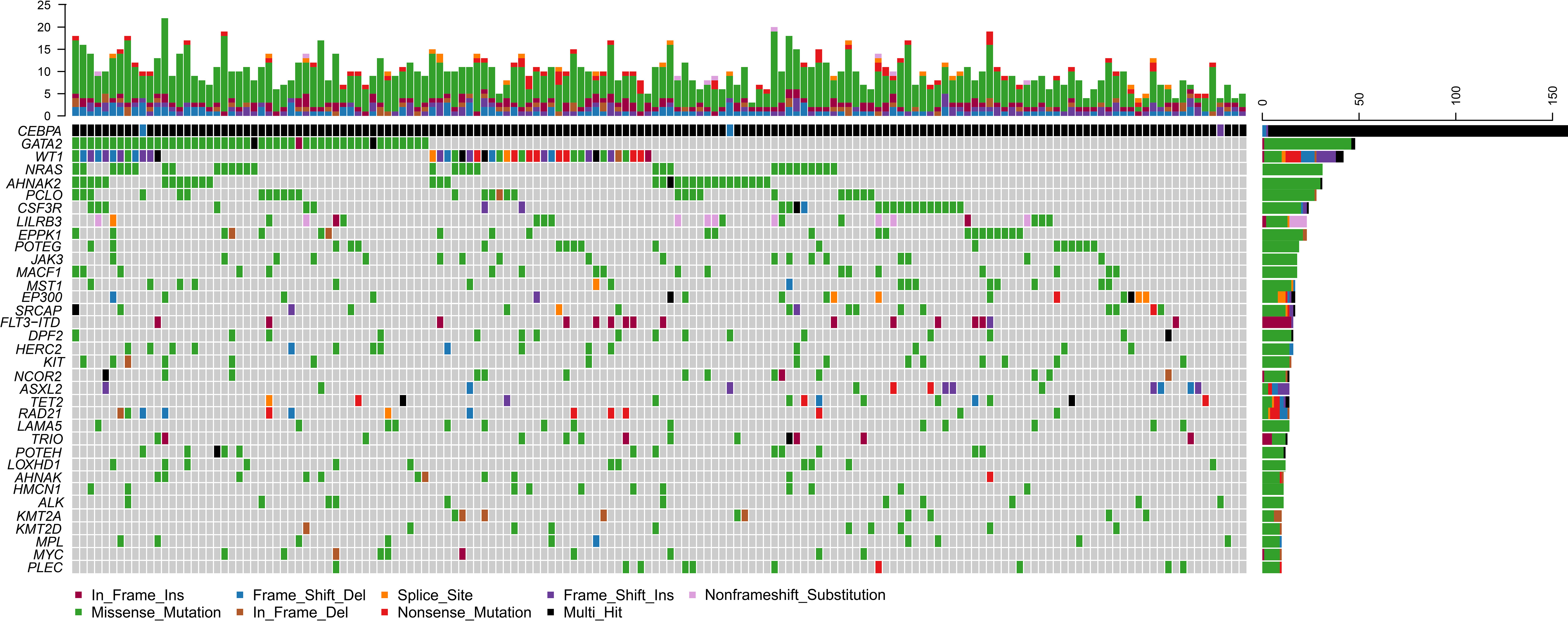
Figure 2 Genomic landscape of 158 CN-AML patients with biCEBPA mutations. Genes mutated in ≥10 patients are shown. Boxes are colored according to the mutation type. Black box indicates multi-hit of mutation type. Non-black box of CEBPA indicates the same mutation type in one patient. The top bar indicates mutation load (mutation/Mb DNA) and the right bar indicates mutation frequency.
We further identified 21 pairs of genes with co-occurrence and 1 pair with mutual exclusivity with significance (P<0.05, Figure 3A). Both NRAS and NCOR2 had 4 pairwise associated genes. Mutations in NRAS, JAK3 and KIT showed significant associations with each other. Positive pairwise associations were also found in TET2 and POTEG, GATA2 and AHNAK, and CSF3R and ASXL2. Only GATA2 showed significant mutual exclusivity with KMT2A. KEGG pathway enrichment analysis revealed that the mutated genes, including CEBPA, were mainly involved in cancer (Figure 3B). These genes represent pan-cancer biomarkers not only in myelogenous leukemia (acute and chronic myeloid leukemia) but also in many solid tumors. Apart from several pivotal cancer-related pathways in signal transduction, we also enriched pathways in central carbon metabolism in cancer, microRNAs in cancer and EGFR tyrosine kinase inhibitor resistance.
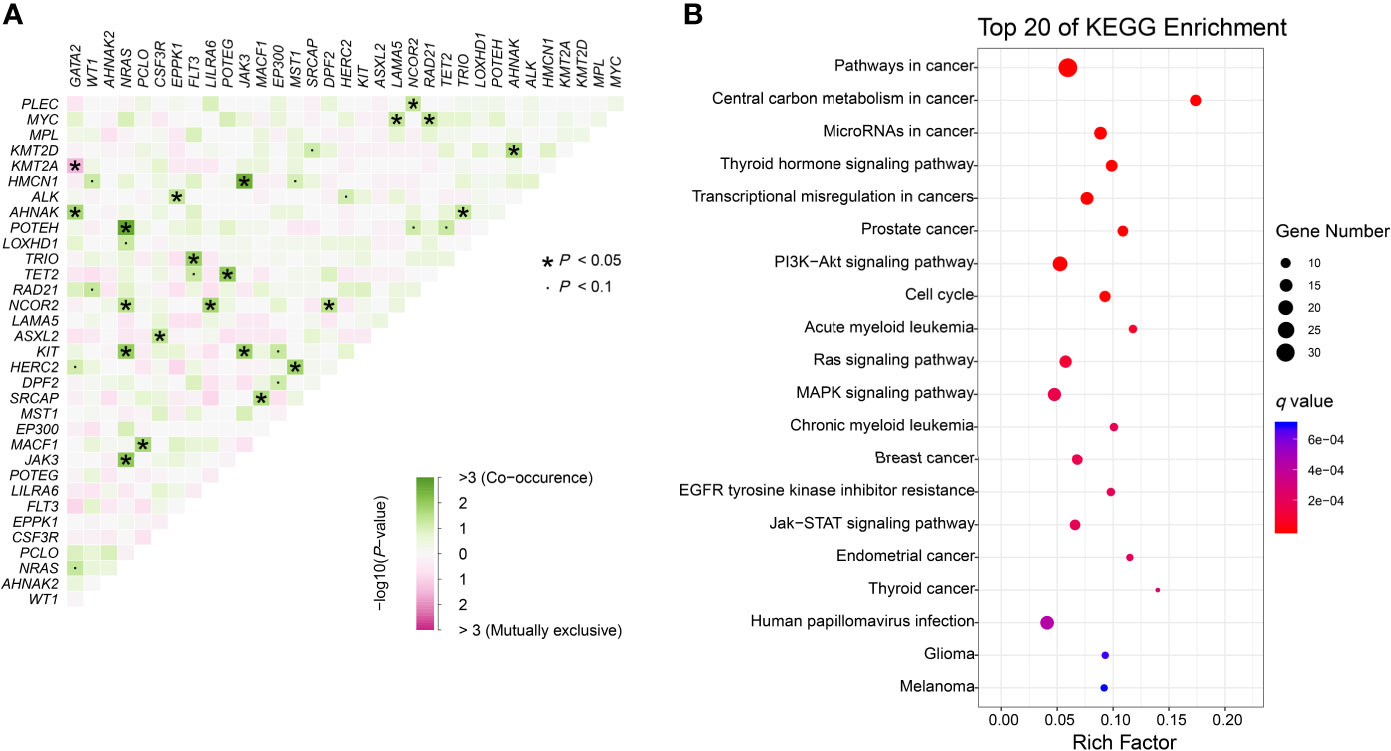
Figure 3 Genomic analyses. (A) Pairwise association between genes mutated in ≥10 patients. Green colors indicate positive association and pink colors indicate negative association. (B) KEGG enrichment analysis. The top 20 pathways are shown. Dot size depends on the mutation number and color depends on the q value (adjust P value).
Mutational Context and Clinical Relevance
The general relapse rate was 32.3% (51/158) in our cohort. We further explored the correlation of mutational complexity with disease progression. A higher median mutation number was seen in ones with events (9 [2–18] vs. 8 [1–20]; P =0.050) for the patients receiving consolidation chemotherapy only. According to the median mutation number (N =8), patients were simply divided into two subgroups: patients with low mutational burden (mutation number <8, N=66) and high mutational burden (mutation number ≥8, N =92). Patients with low mutational burden showed significantly higher 5-year LFS (61.6% vs. 39.0%, P =0.033), higher 5-year survival (85.6% vs. 62.9%, P =0.030), lower 5-year CIR (38.4% vs. 59.5%, P =0.0496) and comparable 5-year NRM (0 vs. 1.5%, P =0.407) compared with those with high (Supplementary Figure 3).
Nomogram for LFS Analysis
Clinical variables, genes, and genetic groups with mutations in ≥10 patients were enrolled for univariate analysis of LFS (allo-HSCT was recorded as a censored event). Eight variables were eligible for subsequent analysis with P <0.15 in the training cohort (Supplementary Table 2) and 4 of them eventually entered the nomogram model after multivariate Cox analysis: WBC (high vs. low, represents >18.30×109/L vs. ≤18.30×109/L), CSF3R mutation (+ vs. -), KMT2A mutation (+ vs. -) and DNA methylation related mutation (mutations in TET2, DNMT3A, BAZ2A, IDH2 and IDH1, mutated in 14, 9, 5, 5 and 3 patients respectively) (+ vs. -) (Figure 4A). All points corresponding to the 4 variables were summed to predict individual probabilities of 1-, 3- and 5-year LFS. The model showed good discrimination with C-index value of 0.750 (95% confidence interval, 0.670–0.830) as well as good calibration (Supplementary Figure 4A). In validation cohort, the model also had good discrimination (C-index, 0.771; 95% confidence interval, 0.661–0.881) and calibration (Supplementary Figure 4B).
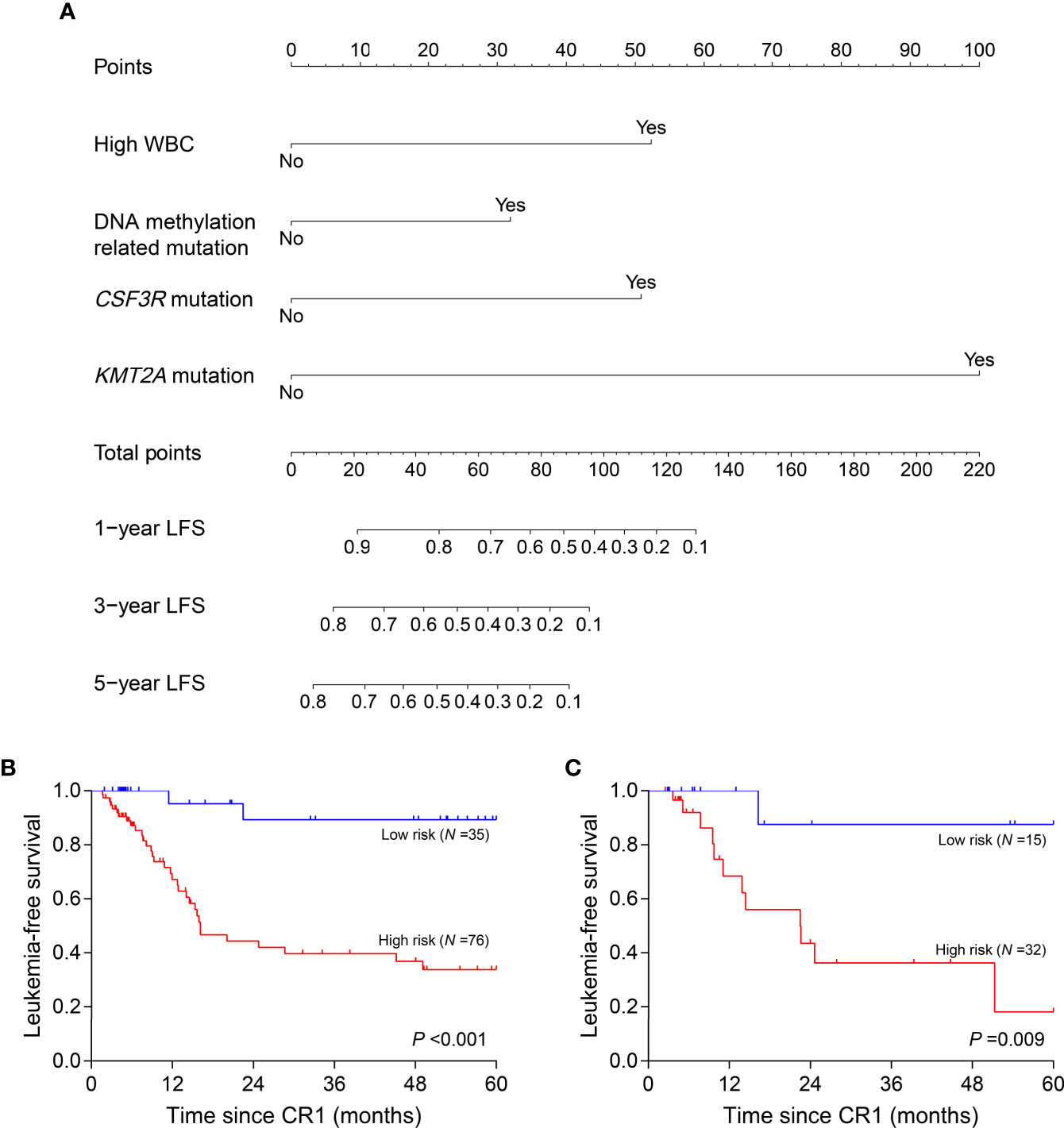
Figure 4 Nomogram model and risk stratification. (A) For WBC, “Yes” represents WBC >18.30×109/L at diagnosis; for genes or genetic group, “Yes” represents mutation. (B, C) Leukemia-free survival analyses by risk stratification in training (B) and validation (C) cohort. WBC, white blood cell.
Risk Stratification Based on Nomogram Model
According to the variables in nomogram model, patients with no identified risk factor were assigned to the low-risk subgroup (N =50) and the remaining to the high-risk (N =108). In training cohort, low-risk patients (N =35) showed better 5-year LFS compared with the high-risk (N =76, 89.3% vs. 33.8%, P <0.001) (Figure 4B). In the validation cohort, there were 15 patients assigned to low-risk subgroup and 32 to high-risk. The validation cohort also differentiated the two risk subgroups (low risk vs. high risk, 87.5% vs. 18.2%, P =0.009) (Figure 4C). MRDint was available in 148 patients and 59 (39.9%) ones were positive. The positive rate was significantly lower in low-risk subgroup (9/45, 20.0%) compared with high-risk (50/103, 48.5%) (P =0.001).
Allo-HSCT Was Superior to Chemotherapy in High-Risk Subgroup
We then interrogated the effect of consolidation chemotherapy and allo-HSCT as post-remission therapies in the two risk subgroups. In the low-risk subgroup (N =49), there was no significant difference in the 5-year LFS (allo-HSCT vs. consolidation chemotherapy, 77.4% vs. 88.9%, P =0.424), 5-year survival (83.9% vs. 95.5%, P =0.173), 5-year CIR (11.7% vs. 11.1%, P =0.901) and 5-year NRM (10.9% vs. 0, P =0.099) (Figures 5A–D). However, in the high-risk subgroup (N =99), allo-HSCT was superior to consolidation chemotherapy alone (5-year LFS, 89.6% vs. 32.6%, P <0.001; 5-year survival, 96.9% vs. 63.6%, P =0.001; 5-year CIR, 7.2% vs. 65.8%, P <0.001) with comparable 5-year NRM (3.1% vs. 1.6%, P =0.685) (Figures 5E–H).
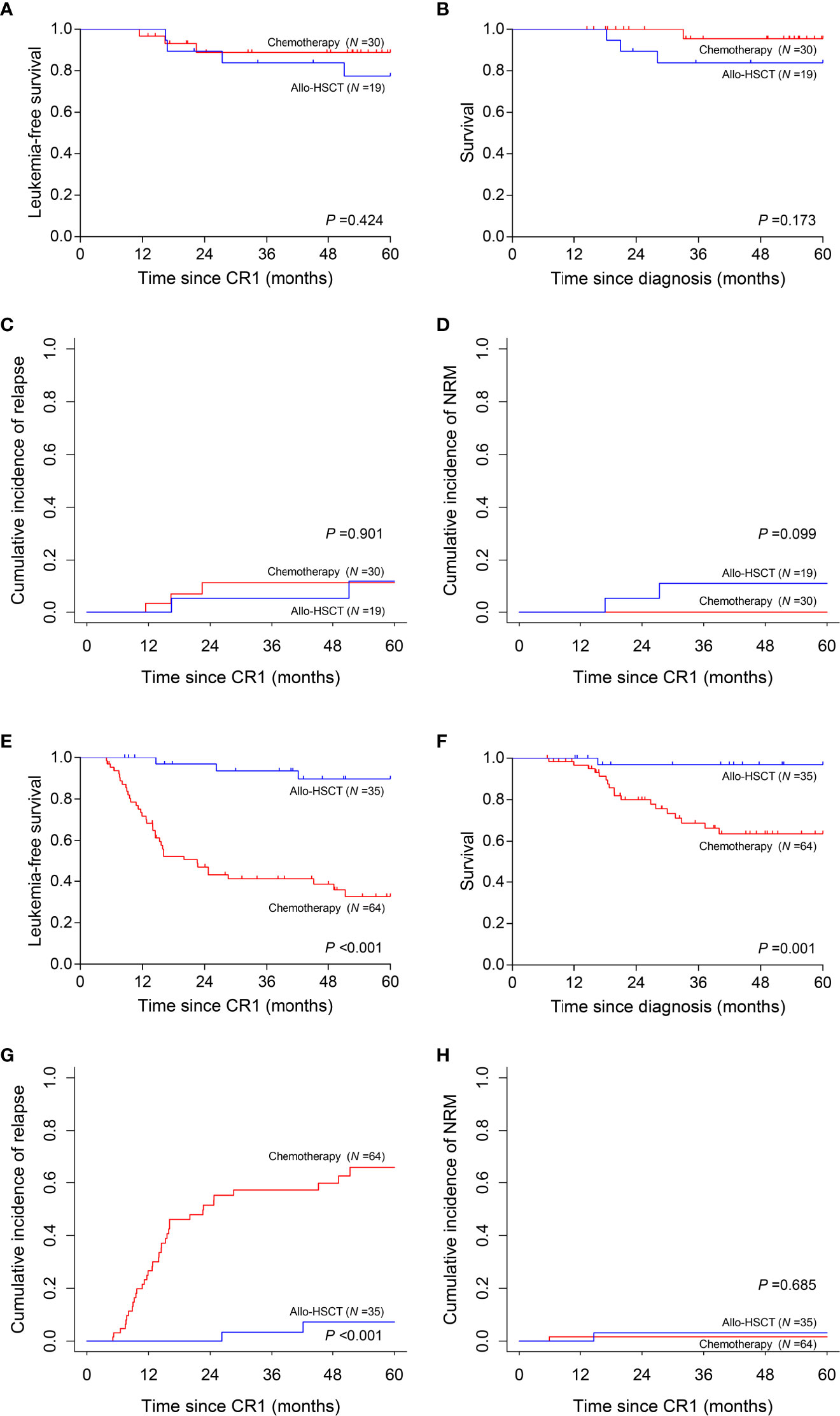
Figure 5 Prognosis of two post-remission therapies by risk stratification. (A–D) Leukemia-free survival, survival, cumulative incidence of relapse and non-relapse mortality analyses in low-risk subgroup. (E–H) Leukemia-free survival, survival, cumulative incidence of relapse and non-relapse mortality analyses in high-risk subgroup.
Discussion
Our study presents comprehensive information on mutational context and detailed risk stratification of biCEBPA mutated CN-AML patients. A significant reduction in the rate of transplantation in recent years was seen in our study. This was attributed to the important insights into biCEBPA mutations in AML and recommendation for consolidation chemotherapy as the first-line post-remission therapy (25). However, in accordance with other studies (4, 6), we observed a considerable relapse rate in the CN-AML patients with biCEBPA mutations. Furthermore, although not limited to CN-AML, our previous study with 36 patients identical to the current study, also supported the heterogeneity of biCEBPA mutations in patients with similar relapse rate (15). It has been found that HSCT reduced the relapse rate in this population; however, the survival benefit is still controversial (26, 27). Our study indicated that allo-HSCT improved the prognosis (Supplementary Figure 1), demonstrating that the first-line post-remission treatment should be tailored according to an individualized risk assessment. Risk factors alone cannot represent the actual status of a patient and a comprehensive and quantitative method such as a nomogram model may provide a refined stratification. We thus sought to elucidate the heterogeneity by a large panel and develop a new prognostic model based on clinical and molecular data in this population.
As expected, higher WBC (median as the cutoff) was identified as the clinical prognostic factor. We found that at least 1 mutation cooccurred with mutated biCEBPA and mutation complexity did confer higher relapse risk, which further verified the heterogeneity of biCEBPA mutated CN-AML. GATA2 mutation was the most frequent co-activated event with biCEBPA mutations (6, 8, 11). Study showed that GATA2 activity affected the mutational dynamics of leukemia in Cbfb-MYH11 knockin mice (28). The prognostic value of this gene is not well established. Several studies have revealed a trend of improvement in GATA2 mutated CN-AML patients with biCEBPA mutations (8, 29), especially when mutations disrupted the zinc finger 1 domain. In our study, GATA2 mutation showed no correlation with prognosis (data not shown). We further identified two mutated genes (CSF3R and KMT2A) and a genetic group (DNA methylation) which conferred prognostic significance in our cohort. Braun et al. (30) confirmed that CEBPA mutations must be the initial event prior to mutant CSF3R since otherwise, AML did not develop and CSF3R and CEBPA mutations cooperated to promote leukemogenesis. CSF3R, which is involved in the JAK-STAT signaling pathway, is a common tyrosine kinase mutated gene in biCEBPA mutated AML patients who were sensitive to JAK inhibition (9, 11, 31). The EGFR tyrosine kinase inhibitor resistance is also a pathway related to tyrosine kinase. Reports of the role of EGFR and its inhibitors (gefitinib and erlotinib) in the origination, progression and treatment of AML were discordant (32–34). Mahmud et al. (35) reported elevated protein levels of EGFR and its activation in a subset of AML and attributed the discordance in other studies to patient selection because the EGFR levels in more than 80% of AML patients did not differ from those in normal individuals. Although EGFR mutations were not identified in this study and its expression was not evaluated, the downstream mutated genes which were enriched in the EGFR tyrosine kinase inhibitor resistance pathway may confer drug resistance in biCEBPA mutated CN-AML patients. Genes involved in DNA methylation (such as TET2 and DNMT3A) were frequently mutated in biCEBPA mutated AML, especially in the older participants and mutated TET2 was not significantly different from wild type in relapse/event-free survival (6, 36). We further studied these genes as a genetic group and found that mutations in this group conferred a worse outcome. However, reports of other epigenetic modifiers involved in histone methylation are rare (13). We identified that KMT2A, as well as KMT2D and EP300 mutations, were mutually exclusive with the most frequent GATA2 mutation (Figure 3A). The infrequent mutation in KMT2 gene family members represents an obstacle to interpretation. In our study, we revealed that mutated KMT2A was also an independent risk factor in biCEBPA mutated CN-AML patients.
Combined with sequencing data, we developed a nomogram model and further stratified the patients by the risk factors. According to our stratification, approximately one third of the patients were categorized into the low-risk subgroup, which had only biCEBPA mutations and no other detrimental clinical or genetic factors. Low-risk patients were more sensitive to induction chemotherapy with lower MRD level after induction therapy. The 5-year LFS and CIR in this subgroup were not significantly improved by allo-HSCT and chemotherapy alone seemed to have better 5-year survival. That was because of the high rate of transplant-related mortality counterbalancing the graft-versus-leukemia effect in allo-HSCT. These data strongly indicated that this subgroup represented the patients with a real favorable prognosis in those with biCEBPA mutated CN-AML. However, allo-HSCT was shown to be a powerful therapy to reverse the high mortality resulting from relapse in the high-risk subgroup.
One limitation of our study was the analysis of FLT3-ITD. The prognostic impact of FLT3-ITD in biCEBPA mutated AML patients was controversial. Grossmann et al. (36) indicated that FLT3-ITD had no impact, while Zhang et al. (11) revealed that FLT3-ITD had worse outcome in biCEBPA mutated CN-AML patients. In our study, 13 FLT3-ITD patients with biCEBPA mutations received allo-HSCT during the CR1 (median time from CR1 to allo-HSCT, 4.53 months). The prognostic value could not be estimated because these patients were censored at the date of allo-HSCT. Although FLT3-ITD was more frequently observed in non-biCEBPA mutated AML patients (6), the contribution of FLT3-ITD to risk stratification warrants further investigation because two of the FLT3-ITD patients receiving the consolidation chemotherapy relapsed (LFS, 20.0 months and 16.1 months respectively) eventually. Other prognostically associated genes in our study, like CSF3R and KMT2A, still need a larger and prospective study to validate.
In summary, we validated the heterogeneity of CN-AML patients with biCEBPA mutations and developed a new system of risk stratification based on a nomogram model. Only one third of these patients represented the low-risk subgroup, and consolidation chemotherapy should be the first-line post-remission therapy. While in the high-risk subgroup, allo-HSCT is recommended. These data, if validated, will be greatly beneficial in translating commercial sequencing into clinical testing and directing decision-making during treatment of CN-AML patients with biCEBPA mutations.
Data Availability Statement
The sequencing data presented in the study are deposited in the NCBI Sequence Read Archive (SRA) repository, accession number PRJNA749620.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Peking University People’s Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
G-RR and X-JH designed the project and prepared the manuscript. L-XW, Y-LZ, Z-LW, L-MC, S-BC, FL, TZ, L-XL, and C-CW performed the experiments and statistical analyses. HJ, Y-JC, JW, J-LL, Q-YS, YW, QJ, L-PX, X-HZ, and K-YL provided the clinical data. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from National Key Research and Development Program of China [Grant 2017YFA0104500], National Natural Science Foundation of China [Grant 81770156], Innovative Research Groups of the National Natural Science Foundation of China [Grant 81621001], Beijing Municipal Science and Technology Commission [Grant Z181100009618032] and Beijing Municipal Natural Science Foundation [Grant 7192213].
Conflict of Interest
Authors S-BC, FL, TZ, L-XL, and C-CW are employed by Acornmed Biotechnology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer RL declared a past co-authorship with the authors to the handling editor.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank all our colleagues at Peking University Institute of Hematology for their cooperation in sample collection.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.706935/full#supplementary-material
References
1. Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, et al. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N Engl J Med (2013) 368(22):2059–74. doi: 10.1056/NEJMoa1301689
2. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational Heterogeneity in Cancer and the Search for New Cancer-Associated Genes. Nature (2013) 499(7457):214–8. doi: 10.1038/nature12213
3. Huang XJ, Zhu HH, Chang YJ, Xu LP, Liu DH, Zhang XH, et al. The Superiority of Haploidentical Related Stem Cell Transplantation Over Chemotherapy Alone as Postremission Treatment for Patients With Intermediate- or High-Risk Acute Myeloid Leukemia in First Complete Remission. Blood (2012) 119(23):5584–90. doi: 10.1182/blood-2011-11-389809
4. Dufour A, Schneider F, Metzeler KH, Hoster E, Schneider S, Zellmeier E, et al. Acute Myeloid Leukemia With Biallelic CEBPA Gene Mutations and Normal Karyotype Represents a Distinct Genetic Entity Associated With a Favorable Clinical Outcome. J Clin Oncol Off J Am Soc Clin Oncol (2010) 28(4):570–7. doi: 10.1200/jco.2008.21.6010
5. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med (2016) 374(23):2209–21. doi: 10.1056/NEJMoa1516192
6. Konstandin NP, Pastore F, Herold T, Dufour A, Rothenberg-Thurley M, Hinrichsen T, et al. Genetic Heterogeneity of Cytogenetically Normal AML With Mutations of CEBPA. Blood Adv (2018) 2(20):2724–31. doi: 10.1182/bloodadvances.2018016840
7. Pulsipher MA, Carlson C, Langholz B, Wall DA, Schultz KR, Bunin N, et al. Igh-V(D)J NGS-MRD Measurement Pre- and Early Post-Allotransplant Defines Very Low- and Very High-Risk ALL Patients. Blood (2015) 125(22):3501–8. doi: 10.1182/blood-2014-12-615757
8. Greif PA, Dufour A, Konstandin NP, Ksienzyk B, Zellmeier E, Tizazu B, et al. GATA2 Zinc Finger 1 Mutations Associated With Biallelic CEBPA Mutations Define a Unique Genetic Entity of Acute Myeloid Leukemia. Blood (2012) 120(2):395–403. doi: 10.1182/blood-2012-01-403220
9. Lavallee VP, Krosl J, Lemieux S, Boucher G, Gendron P, Pabst C, et al. Chemo-Genomic Interrogation of CEBPA Mutated AML Reveals Recurrent CSF3R Mutations and Subgroup Sensitivity to JAK Inhibitors. Blood (2016) 127(24):3054–61. doi: 10.1182/blood-2016-03-705053
10. Zhang Y, Wang F, Chen X, Zhang Y, Wang M, Liu H, et al. Csf3r Mutations are Frequently Associated With Abnormalities of RUNX1, Cbfb, CEBPA, and NPM1 Genes in Acute Myeloid Leukemia. Cancer (2018) 124(16):3329–38. doi: 10.1002/cncr.31586
11. Zhang Y, Wang F, Chen X, Zhang Y, Wang M, Liu H, et al. Companion Gene Mutations and Their Clinical Significance in AML With Double Mutant CEBPA. Cancer Gene Ther (2020) 27(7-8):599–606. doi: 10.1038/s41417-019-0133-7
12. Tien FM, Hou HA, Tang JL, Kuo YY, Chen CY, Tsai CH, et al. Concomitant WT1 Mutations Predict Poor Prognosis in Acute Myeloid Leukemia Patients With Double Mutant CEBPA. Haematologica (2018) 103(11):e510–3. doi: 10.3324/haematol.2018.189043
13. Wilhelmson AS, Porse BT. CCAAT Enhancer Binding Protein Alpha (CEBPA) Biallelic Acute Myeloid Leukaemia: Cooperating Lesions, Molecular Mechanisms and Clinical Relevance. Br J Haematol (2020) 190(4):495–507. doi: 10.1111/bjh.16534
14. Deng DX, Zhu HH, Liu YR, Chang YJ, Ruan GR, Jia JS, et al. Minimal Residual Disease Detected by Multiparameter Flow Cytometry Is Complementary to Genetics for Risk Stratification Treatment in Acute Myeloid Leukemia With Biallelic CEBPA Mutations. Leukemia Lymphoma (2019) 60(9):2181–9. doi: 10.1080/10428194.2019.1576868
15. Wang J, Lu R, Wu Y, Jia J, Gong L, Liu X, et al. Detection of Measurable Residual Disease May Better Predict Outcomes Than Mutations Based on Next-Generation Sequencing in Acute Myeloid Leukaemia With Biallelic Mutations of CEBPA. Br J Haematol (2020) 190(4):533–44. doi: 10.1111/bjh.16535
16. Lv M, Wang Y, Chang YJ, Zhang XH, Xu LP, Jiang Q, et al. Myeloablative Haploidentical Transplantation Is Superior to Chemotherapy for Patients With Intermediate-risk Acute Myelogenous Leukemia in First Complete Remission. Clin Cancer Res An Off J Am Assoc Cancer Res (2019) 25(6):1737–48. doi: 10.1158/1078-0432.ccr-18-1637
17. Xu L, Chen H, Chen J, Han M, Huang H, Lai Y, et al. The Consensus on Indications, Conditioning Regimen, and Donor Selection of Allogeneic Hematopoietic Cell Transplantation for Hematological Diseases in China-recommendations From the Chinese Society of Hematology. J Hematol Oncol (2018) 11(1):33. doi: 10.1186/s13045-018-0564-x
18. Wang Y, Chen H, Chen J, Han M, Hu J, Jiong H, et al. The Consensus on the Monitoring, Treatment, and Prevention of Leukemia Relapse After Allogeneic Hematopoietic Stem Cell Transplantation in China. Cancer Lett (2018) 438:63–75. doi: 10.1016/j.canlet.2018.08.030
19. Chang YJ, Wang Y, Liu YR, Xu LP, Zhang XH, Chen H, et al. Haploidentical Allograft Is Superior to Matched Sibling Donor Allograft in Eradicating Pre-Transplantation Minimal Residual Disease of AML Patients as Determined by Multiparameter Flow Cytometry: A Retrospective and Prospective Analysis. J Hematol Oncol (2017) 10(1):134. doi: 10.1186/s13045-017-0502-3
20. Mo XD, Zhang XH, Xu LP, Wang Y, Yan CH, Chen H, et al. Interferon-α: A Potentially Effective Treatment for Minimal Residual Disease in Acute Leukemia/Myelodysplastic Syndrome After Allogeneic Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant J Am Soc Blood Marrow Transplant (2015) 21(11):1939–47. doi: 10.1016/j.bbmt.2015.06.014
21. Chang YJ, Xu LP, Wang Y, Zhang XH, Chen H, Chen YH, et al. Controlled, Randomized, Open-Label Trial of Risk-Stratified Corticosteroid Prevention of Acute Graft-Versus-Host Disease After Haploidentical Transplantation. J Clin Oncol Off J Am Soc Clin Oncol (2016) 34(16):1855–63. doi: 10.1200/jco.2015.63.8817
22. Huang XJ, Liu DH, Liu KY, Xu LP, Chen H, Han W. Donor Lymphocyte Infusion for the Treatment of Leukemia Relapse After HLA-Mismatched/Haploidentical T-Cell-Replete Hematopoietic Stem Cell Transplantation. Haematologica (2007) 92(3):414–7. doi: 10.3324/haematol.10570
23. Zhou YL, Wu LX, Peter Gale R, Wang ZL, Li JL, Jiang H, et al. Mutation Topography and Risk Stratification for De Novo Acute Myeloid Leukaemia With Normal Cytogenetics and No Nucleophosmin 1 (NPM1) Mutation or Fms-like Tyrosine Kinase 3 Internal Tandem Duplication (FLT3-ITD). Br J Haematol (2020) 190(2):274–83. doi: 10.1111/bjh.16526
24. Ruan GR, Li JL, Qin YZ, Li LD, Xie M, Chang Y, et al. Nucleophosmin Mutations in Chinese Adults With Acute Myelogenous Leukemia. Ann Hematol (2009) 88(2):159–66. doi: 10.1007/s00277-008-0591-8
25. Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and Management of AML in Adults: 2017 ELN Recommendations From an International Expert Panel. Blood (2017) 129(4):424–47. doi: 10.1182/blood-2016-08-733196
26. Ahn JS, Kim JY, Kim HJ, Kim YK, Lee SS, Jung SH, et al. Normal Karyotype Acute Myeloid Leukemia Patients With CEBPA Double Mutation Have a Favorable Prognosis But No Survival Benefit From Allogeneic Stem Cell Transplant. Ann Hematol (2016) 95(2):301–10. doi: 10.1007/s00277-015-2540-7
27. Schlenk RF, Taskesen E, van Norden Y, Krauter J, Ganser A, Bullinger L, et al. The Value of Allogeneic and Autologous Hematopoietic Stem Cell Transplantation in Prognostically Favorable Acute Myeloid Leukemia With Double Mutant CEBPA. Blood (2013) 122(9):1576–82. doi: 10.1182/blood-2013-05-503847
28. Saida S, Zhen T, Kim E, Yu K, Lopez G, McReynolds LJ, et al. Gata2 Deficiency Delays Leukemogenesis While Contributing to Aggressive Leukemia Phenotype in Cbfb-MYH11 Knockin Mice. Leukemia (2020) 34(3):759–70. doi: 10.1038/s41375-019-0605-7
29. Tien FM, Hou HA, Tsai CH, Tang JL, Chiu YC, Chen CY, et al. GATA2 Zinc Finger 1 Mutations Are Associated With Distinct Clinico-Biological Features and Outcomes Different From GATA2 Zinc Finger 2 Mutations in Adult Acute Myeloid Leukemia. Blood Cancer J (2018) 8(9):87. doi: 10.1038/s41408-018-0123-2
30. Braun TP, Okhovat M, Coblentz C, Carratt SA, Foley A, Schonrock Z, et al. Myeloid Lineage Enhancers Drive Oncogene Synergy in CEBPA/CSF3R Mutant Acute Myeloid Leukemia. Nat Commun (2019) 10(1):5455. doi: 10.1038/s41467-019-13364-2
31. Braun TP, Coblentz C, Smith BM, Coleman DJ, Schonrock Z, Carratt SA, et al. Combined Inhibition of JAK/STAT Pathway and Lysine-Specific Demethylase 1 as a Therapeutic Strategy in CSF3R/CEBPA Mutant Acute Myeloid Leukemia. Proc Natl Acad Sci USA (2020) 117(24):13670–9. doi: 10.1073/pnas.1918307117
32. Sun JZ, Lu Y, Xu Y, Liu F, Li FQ, Wang QL, et al. Epidermal Growth Factor Receptor Expression in Acute Myelogenous Leukaemia Is Associated With Clinical Prognosis. Hematol Oncol (2012) 30(2):89–97. doi: 10.1002/hon.1002
33. Deangelo DJ, Neuberg D, Amrein PC, Berchuck J, Wadleigh M, Sirulnik LA, et al. A Phase II Study of the EGFR Inhibitor Gefitinib in Patients With Acute Myeloid Leukemia. Leukemia Res (2014) 38(4):430–4. doi: 10.1016/j.leukres.2013.10.026
34. Boehrer S, Ades L, Braun T, Galluzzi L, Grosjean J, Fabre C, et al. Erlotinib Exhibits Antineoplastic Off-Target Effects in AML and MDS: A Preclinical Study. Blood (2008) 111(4):2170–80. doi: 10.1182/blood-2007-07-100362
35. Mahmud H, Kornblau SM, Ter Elst A, Scherpen FJ, Qiu YH, Coombes KR, et al. Epidermal Growth Factor Receptor Is Expressed and Active in a Subset of Acute Myeloid Leukemia. J Hematol Oncol (2016) 9(1):64. doi: 10.1186/s13045-016-0294-x
Keywords: acute myeloid leukemia, targeted region sequencing (TRS), biCEBPA mutations, risk stratification, therapy
Citation: Wu L-X, Jiang H, Chang Y-J, Zhou Y-L, Wang J, Wang Z-L, Cao L-M, Li J-L, Sun Q-Y, Cao S-B, Lou F, Zhou T, Liu L-X, Wang C-C, Wang Y, Jiang Q, Xu L-P, Zhang X-H, Liu K-Y, Huang X-J and Ruan G-R (2021) Risk Stratification of Cytogenetically Normal Acute Myeloid Leukemia With Biallelic CEBPA Mutations Based on a Multi-Gene Panel and Nomogram Model. Front. Oncol. 11:706935. doi: 10.3389/fonc.2021.706935
Received: 08 May 2021; Accepted: 14 July 2021;
Published: 17 August 2021.
Edited by:
Julia T. Geyer, Weill Cornell Medical Center, United StatesReviewed by:
Eduardo Rego, São Paulo State University, BrazilRunqing Lu, First Affiliated Hospital of Zhengzhou University, China
Copyright © 2021 Wu, Jiang, Chang, Zhou, Wang, Wang, Cao, Li, Sun, Cao, Lou, Zhou, Liu, Wang, Wang, Jiang, Xu, Zhang, Liu, Huang and Ruan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guo-Rui Ruan, cnVhbmd1b3J1aUBwa3VwaC5lZHUuY24=; Xiao-Jun Huang, aHVhbmd4aWFvanVuQGJqbXUuZWR1LmNu
†These authors have contributed equally to this work