 Xianghan Li1,2†
Xianghan Li1,2† Yiran Zou1,2†
Yiran Zou1,2† Teng Li1,2Thomas K. F. Wong1Ryan T. Bushey3
Teng Li1,2Thomas K. F. Wong1Ryan T. Bushey3 Michael J. Campa3
Michael J. Campa3 Elizabeth B. Gottlin3Hongliang Liu4,5
Elizabeth B. Gottlin3Hongliang Liu4,5 Qingyi Wei4,5,6Allen Rodrigo1,2
Qingyi Wei4,5,6Allen Rodrigo1,2 Edward F. Patz Jr.3,4,7*
Edward F. Patz Jr.3,4,7*- 1Research School of Biology, Australian National University, Canberra, ACT, Australia
- 2School of Biological Sciences, University of Auckland, Auckland, New Zealand
- 3Department of Radiology, Duke University Medical Center, Durham, NC, United States
- 4Duke Cancer Institute, Duke University Medical Center, Durham, NC, United States
- 5Department of Population Health Sciences, Duke University School of Medicine, Durham, NC, United States
- 6Department of Medicine, Duke University School of Medicine, Durham, NC, United States
- 7Department of Pharmacology and Cancer Biology, Duke University Medical Center, Durham, NC, United States
Background: Single nucleotide polymorphisms (SNPs) are often associated with distinct phenotypes in cancer. The present study investigated associations of cancer risk and outcomes with SNPs discovered by whole exome sequencing of normal lung tissue DNA of 15 non-small cell lung cancer (NSCLC) patients, 10 early stage and 5 advanced stage.
Methods: DNA extracted from normal lung tissue of the 15 NSCLC patients was subjected to whole genome amplification and sequencing and analyzed for the occurrence of SNPs. The association of SNPs with the risk of lung cancer and survival was surveyed using the OncoArray study dataset of 85,716 patients (29,266 cases and 56,450 cancer-free controls) and the Prostate, Lung, Colorectal and Ovarian study subset of 1,175 lung cancer patients.
Results: We identified 4 SNPs exclusive to the 5 patients with advanced stage NSCLC: rs10420388 and rs10418574 in the CLPP gene, and rs11126435 and rs2021725 in the M1AP gene. The variant alleles G of SNP rs10420388 and A of SNP rs10418574 in the CLPP gene were associated with increased risk of squamous cell carcinoma (OR = 1.07 and 1.07; P = 0.013 and 0.016, respectively). The variant allele T of SNP rs11126435 in the M1AP gene was associated with decreased risk of adenocarcinoma (OR = 0.95; P = 0.027). There was no significant association of these SNPs with the overall survival of lung cancer patients (P > 0.05).
Conclusions: SNPs identified in the CLPP and M1AP genes may be useful in risk prediction models for lung cancer. The previously established association of the CLPP gene with cancer progression lends relevance to our findings.
Introduction
Lung cancer is the leading cause of cancer death in the world with 2 million new cases and 1.7 million deaths in 2018 (1). In the United States, 228,820 new cases of lung cancer and 135,720 deaths were projected to occur in 2020 (2). The majority (~85%) of lung cancer cases are non-small cell lung cancer (NSCLC), and the overall 5-year survival rate is only ~20%. New targeted and immune modulating therapies are expected to improve survival rates, but it often remains unclear which patients will respond and are at high risk for recurrence. Since genetic predisposition plays a role in tumor phenotype, the identification of risk, predictive, and prognostic markers may guide clinical decisions (3).
The current study initially began as an exploration of tumor cell evolution as a method of developing a prognostic biomarker for NSCLC. Tumor growth and metastasis can be viewed in an evolutionary context (4, 5). The clonal heterogeneity of tumors is a reflection of environmental pressures on a genetically diverse population of cells. As part of this study, we examined the most appropriate germ line reference standard against which to search for cancer mutations, and unexpectedly discovered four single nucleotide polymorphisms (SNPs) from resected normal lung tissue in patients with advanced stage NSCLC (n = 5) that were not found in patients with early stage disease (n = 10). We then determined in independent validation datasets (OncoArray and PLCO databases) that two of the four SNPs are associated with a higher risk of developing squamous cell carcinoma and one of the four SNPs was associated with a lower risk of developing adenocarcinoma. Despite these SNPs being identified in advanced stage patients, however, we did not find an association with survival.
Methods
Discovery Patients and Samples
Duke University follows the set of ethical principles outlined in “The Belmont Report: Ethical Principles and Guidelines for the Protection of Human Subjects of Research”. The Duke University Health System Institutional Review Board approved this study and all subjects gave written informed consent for the use of their tissue in this study. Fifteen sequential NSCLC patients were chosen for this study. These patients are described in Table 1. The 10 patients with early stage NSCLC had a post-resection recurrence-free duration ranging from 14-45 months. The duration endpoint for the non-metastatic designation in each of these cases was the last time they were examined in the clinic following initial diagnosis. Five other patients had metastasis at presentation: in four cases, metastasis to a lymph node, in one case, to the lung.
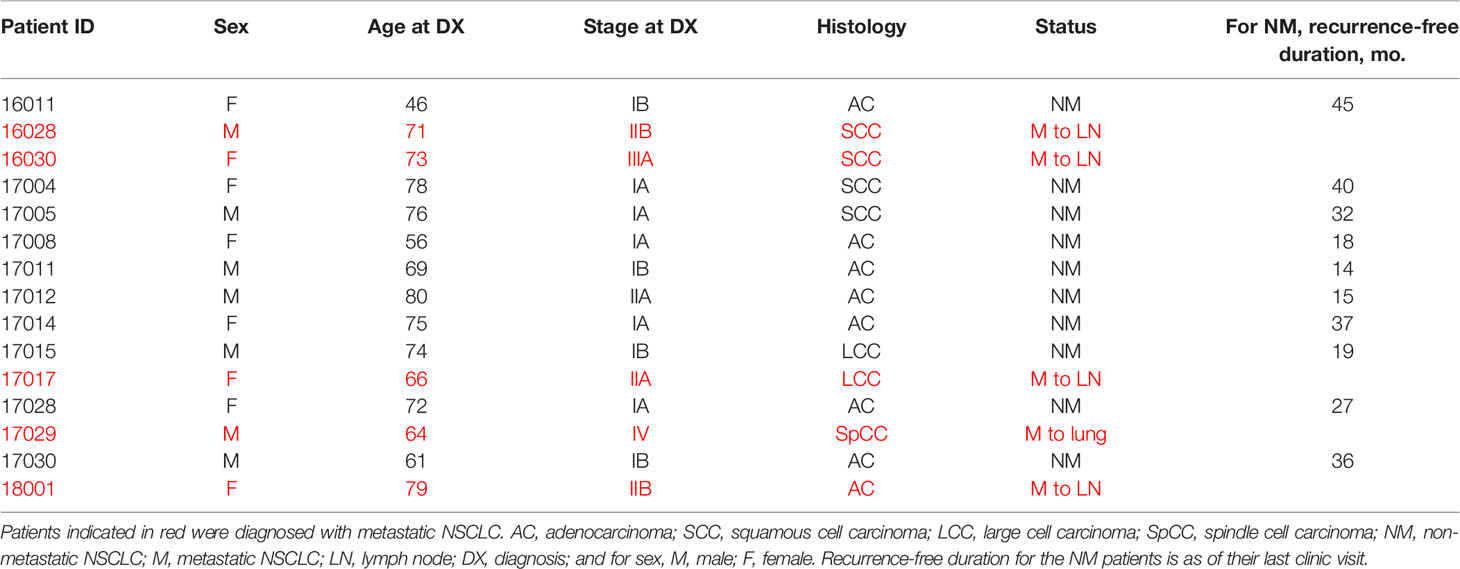
Table 1 Study patients.
Isolation of DNA From Normal Lung Tissue
Normal lung DNA was isolated from lung tissue that had been obtained at surgery and stored at −80°C or from FFPE sections using either a QIAamp Fast DNA Tissue kit or a QIAamp DNA FFPE Tissue kit (Qiagen; Hilden, Germany), respectively, according to the manufacturer’s recommendations.
Whole Genome Amplification
Whole genome amplification (WGA) was carried out using a REPLI-g Single Cell kit (Qiagen). This kit, which was used for bulk DNA amplification from normal lung tissue, uses multiple displacement amplification (MDA) technology (6). Prior to exome sequencing, WGA was verified by PCR amplification of a region of the kinesin light chain 2 gene (KCL2) followed by agarose gel electrophoresis.
Whole Exome Sequencing
Whole genome amplified lung DNA was first treated with KAPA Beads (Roche; Basel, Switzerland) to remove any residual buffer or EDTA prior to library preparation. Sequencing libraries were then created using KAPA HyperPLUS reagents (Roche) on the Hamilton NGS Star liquid handler (Hamilton Robotics; Cary, NC, USA) according to the manufacturer’s instructions. Briefly, samples were enzymatically fragmented, end repaired, A-tailed, and ligated to sample barcodes (Integrated DNA Technologies [IDT]; Coralville, IA, USA). Following library preparation and amplification, libraries were normalized and pooled for exome capture. Sequence capture was performed on the Hamilton NGS Star liquid handler using the IDT xGen Exome Research panel according to the rapid capture protocol. Captured libraries were visualized on the AATI Fragment Analyzer (Agilent Technologies; Santa Clara, CA, USA) for quality. Finally, samples were quantitated by qPCR with KAPA Quant (Roche) and normalized prior to pooling and sequencing on the Illumina NovaSeq 6000 sequencer (Illumina, Inc.; San Diego, CA, USA).
Data Pre-Processing
For every sample, sequencing reads were aligned to the human reference genome (hs37d5) using BWA MEM (7) as implemented in the Sentieon Genomics bioinformatics suite (8). Reads with a mapping quality score less than 40 were discarded. Duplicated reads in the alignment were removed using SAMtools (7). The alignment was further indel-realigned based on known indels from the 1000 Genomes Project (9), while the base quality scores were then recalibrated based on known indels from the 1000 Genomes Project (9) and SNPs from dbSNP (10), both using GATK (11) in Sentieon Genomics (8). To evaluate the quality of sequencing data, the mean coverage of each sequence on the targeted exome regions was measured using BED tools (12). The target region was downloaded from the xGen Exome Research Panel website (https://sg.idtdna.com/pages/products/next-generation-sequencing/hybridization-capture/lockdown-panels/xgen-exome-research-panel).
SNP Detection From Normal Bulk Sequences
Varscan2 (13) and Haplotype Caller of GATK (11) with default parameters were used to detect single nucleotide polymorphisms (SNPs) from normal bulk sequences. The following command line setting was used with VarScan2: samtools mpileup -B -f reference.fasta myData.bam | java -jar VarScan.v2.2.jar mpileup2snp.
For GATK, the recommended pipeline “Germline SNVs + Indels” given in the link https://software.broadinstitute.org/gatk/best-practices/workflow?id=11145 was used.
Only SNPs detected by both Varscan2 and Haplotype Caller were retained to avoid false-positive variant calls. Each SNP inside the VCF file was subjected to further filtering to require having at least 10 supported reads and had coverage across all bulk sequences. The filtered VCF file was annotated using ANNOVAR (14). After annotation, the SNPs that differentiated the metastatic patients from non-metastatic patients were selected.
Independent Validation of SNPs
We investigated the associations of the identified SNPs with the risk of lung cancer as well as the survival of lung cancer patients by using the available data from OncoArray and PLCO, respectively (15–18).
The GWAS data of OncoArray were requested from dbGAP (dbGAP accession #: phs001273.v3.p2), in which genotyping was performed with Illumina Infinium OncoArray-500k and imputation was performed with IMPUTE2 with the reference dataset of 1000 Genomes Project Phase 3 (Haplotype release date October 2014) (18). The OncoArray dataset contained data derived from the germline DNA sequences of 85,716 patients (29,266 cases and 56,450 cancer-free controls). Logistic regression was used to assess the associations between SNPs and lung cancer risk in each study with adjustment for the top significant principal components. Fixed-effects meta-analysis was used to combine the results of different studies if the Cochran’s Q-test P > 0.100 and the heterogeneity statistic (I2) < 50%. Otherwise, a random-effects model was applied.
For survival analysis, there were 1,185 NSCLC patients from PLCO with both survival and GWAS data available. The GWAS dataset was requested from dbGaP (the approval project number: PLCO-95), for which genomic DNA samples extracted from the whole blood were genotyped with Illumina HumanHap240Sv1.0, HumanHap300v1.1 and HumanHap550v3.0 (dbGaP accession #: phs000093.v2.p2 and phs000336.v1.p1). Imputation was performed with ±500kb flanking buffer regions around the targeted SNPs using miniMac4 and the reference panel of the European data from the 1,000 Genomes Project (phase 3). We then performed a Cox proportional hazards regression analysis to evaluate associations between each of the SNPs and overall survival (in an additive model) with adjustment for age, sex, smoking status, histology, tumor stage, chemotherapy, radiotherapy, surgery and the first four principal components of the population structures in the genotyping data. The distribution of the applied clinical variables can be found in the previous publication (19).
Functional Annotation and Predictive Analysis
To further explore the functional associations of the four SNPs examined in this study (rs11126435, rs2021725, rs10420388 and rs10418574), we annotated non-coding variants using ANNOVAR (14). To analyze the predicted functional effects of these variants, we used several web tools. PredictSNP2 (https://loschmidt.chemi.muni.cz/predictsnp2/) was used to identify potential functional effects of the SNPs (20). HaploReg (http://pubs.broadinstitute.org/mammals/haploreg/haploreg.php) was used to identify SNPs that are in linkage disequilibrium (LD) with the SNPs of interest in order to shed light on their potential regulatory effects (21). The Genotype-Tissue Expression (GTEx) project portal (https://gtexportal.org/home/) was used to investigate relevant expression quantitative trait loci (eQTLs) for the four SNPs in lung tissue (22). RegulomeDB 2.0 (https://regulomedb.org/regulome-search/) was used to estimate the functional relevance of the SNPs. RegulomeDB 2.0 ranks the possibility of the SNP to be a regulatory element from 1a (most likely) to 6 (minimal binding evidence) (23).
Results
Detection of SNPs That Differentiate Between Metastatic and Non-Metastatic Groups
We performed whole exome sequencing of bulk normal lung DNA from 15 NSCLC patients, 10 with early stage, non-metastatic disease and 5 with advanced stage disease. Each bulk sequence had >100× mean sequencing depth.
In exome regions, there were no SNPs that differentiated the two groups of patients, while in intron regions, four SNPs differentiated the two groups (Table 2). Two intron variants (rs11126435 and rs2021725) belong to the meiosis 1 associated protein (M1AP) gene. Two intron variants (rs10420388 and rs10418574) belong to the caseinolytic mitochondrial matrix peptidase proteolytic subunit (CLPP) gene. None of the early stage patients had polymorphisms at those four sites, while all the metastatic patients were heterozygous at all the sites. The SNP genotypes for each patient are shown in Table 3.
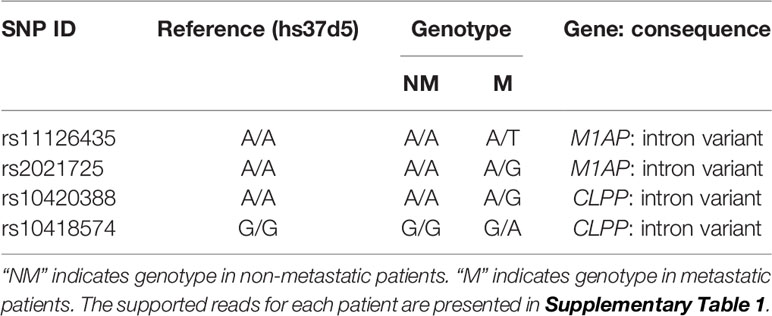
Table 2 SNPs that differentiate between groups.
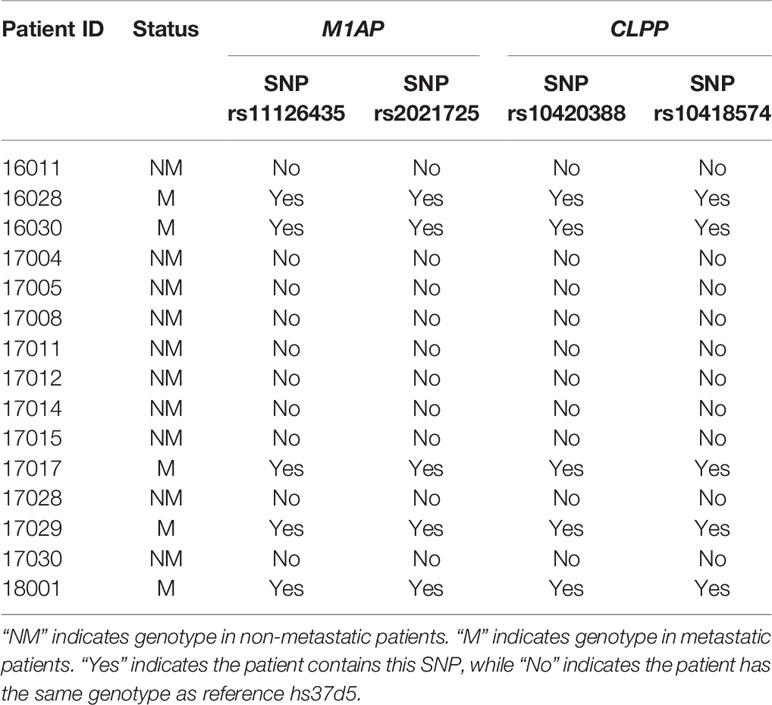
Table 3 SNP genotypes for each NSCLC patient.
Association of SNPs and Survival
Four SNPs were extracted from the imputation data, and their associations with lung cancer risk and survival were assessed in the OncoArray study and the PLCO study. (The rs2021725 SNP was available in the PLCO dataset but not in the OncoArray dataset.) As shown in Supplementary Table 2, we found that the variant alleles G and A of the two SNPs (rs10420388 and rs10418574) in CLPP were associated with an increased risk of squamous cell carcinoma (OR = 1.07 and 1.07; P = 0.013 and 0.016, respectively), while the variant allele T of SNP rs11126435 in M1AP was associated with a decreased risk of adenocarcinoma (OR = 0.95; P = 0.027). However, no significance was found for these SNPs in association with the overall survival of lung cancer patients, either in the overall analysis or stratified analysis by stage and histological type (P > 0.05; Supplementary Table 3).
Functional Annotation and Predictive Analysis
ANNOVAR was used to determine whether non-coding variants rs11126435, rs2021725, rs10420388 and rs10418574 may disrupt regulatory elements in the human genome. The results show that M1AP variants rs11126435 and rs2021725 fall in a weak enhancer unit, while CLPP variants rs10420388 and rs10418574 fall in a transcriptional elongation unit. PredictSNP2 predicts that all four variants have a neutral influence on regulatory function (Supplementary Table 4).
HaploReg was used to find SNPs in LD with the SNPs of interest and provide clues to their possible regulatory effects. There are 33 SNPs in LD (r2 > 0.8) with CLPP variants rs10420388 and rs10418574 (Supplementary Table 5). These SNPs affect transcription factor (TF) binding sites, either promoters or enhancers.
There are 9 SNPs in LD (r2 > 0.8) with M1AP variant rs11126435. These SNPs affect TF binding. There are 13 SNPs in LD (r2 > 0.8) with M1AP variant rs2021725. Most of these SNPs are histone marks for promoters and enhancers and thus would also be indicative of sites of TF binding.
In the GTEx portal (https://gtexportal.org/home/), the M1AP variants rs11126435 and rs2021725 are both inferred to be eQTLs for LBX2, LBX2-AS1, INO80B, RP11-523H20.3 and MOGS genes (P-values ranging from 0.000016 to 1.70×10-13), indicating the two SNPs are associated with the expression level of these five genes in lung tissue (Supplementary Table 6). It should be noted that, among these five genes, LBX2 and LBX2-AS1 genes have been found to be related to lung cancer development (24, 25). The M1AP variants rs11126435 and rs2021725 are not inferred to be eQTLs for the M1AP gene in the GTEx portal.
GTEx predicts that both CLPP variants rs10418574 and rs10420388 are eQTLs for GTF2F1 and CLPP genes in lung tissue (P-values ranging from 0.000001 to 5.00×10-7), suggesting that the two SNPs are relevant to the expression level of GTF2F1 and CLPP genes.
For M1AP variant rs11126435, RegulomeDB 2.0 shows a rank of 1b, indicating a high probability of functional relevance for this SNP. For M1AP variant rs2021725 and CLPP variant rs10420388, RegulomeDB 2.0 scores are 1f and 2b, respectively, indicating a likely probability of functionality for each of these two SNPs. CLPP variant rs10418574 has a rank of 4 in RegulomeDB 2.0, suggesting minimal functionality for this SNP (Supplementary Table 7).
Discussion
Lung cancer remains a significant worldwide public health problem, and improvements in early detection and therapy are clearly needed to improve outcomes. The genome-wide association study (GWAS) is an effective method of detecting SNPs, typically using Illumina genotyping platforms (26). This approach identifies SNPs at the genome level, which may be very useful in identifying biomarkers and relevant therapeutic targets. To determine whether the SNP locus is associated with cancer, the allelic frequency of the SNP marker has to significantly differ between the cancer group and the control group (26).
To date, GWAS have successfully identified susceptibility loci associated with lung cancer (27). Moreover, to investigate further biological functions of the associated SNPs, the analysis strategies have moved from single-marker analyses to pathway-based analyses (27). However, most GWAS studies focus on the statistical and biological significance of individual SNPs. In the present study, for each patient, we traced the genetic changes of four SNP sites in patients with different NSCLC phenotypes: early stage, non-metastatic or metastatic. Note that the SNPs were found in intron regions despite our having used exome sequencing technology. A previous study illustrated that exome sequencing can also produce high-quality sequence outside the target region (28). Hence, those intronic SNPs were considered in our analysis.
In the present study, we discovered 4 SNPs from normal lung tissue of patients with advanced stage lung cancer, and then explored if they were associated with increased risk of developing the disease and survival in independent validation datasets. These types of biomarkers could complement current risk prediction models and potentially help guide therapeutic options. Lung cancer risk was assessed for rs10420388 and rs10418574 in CLPP and rs11126435 in M1AP in the OncoArray study. (M1AP rs2021725 was not available in this dataset.) Notably, patients with rs10420388 and rs10418574 in CLPP showed a significantly increased risk of squamous cell carcinoma, while rs11126435 in M1AP was associated with decreased risk of adenocarcinoma. Thus, in clinical practice these SNPs could be used, most likely in combination with other SNPs or markers, to more efficiently determine which patients may benefit from more intense screening programs. This is currently an area of intense interest. To our knowledge, this is the first report that M1AP rs11126435, CLPP rs10420388 and CLPP rs10418574 variant alleles may be associated with an NSCLC risk phenotype. However, we did not find an association of any of the 4 SNPs with survival, despite the fact that the SNPs were identified in patients with advanced stage disease and presumably worse outcomes.
M1AP, located on chromosome 2, encodes meiosis 1 associated/arrest protein that is likely to function in the progression of meiosis. Previous studies demonstrated that mutations in M1AP were associated with oligozoospermia (29, 30). A retroviral insertion mutagenesis study showed that M1AP synergized with Cbfb (core-binding factor)-MYH11 (myosin, heavy chain 11) translocation during the onset of acute myeloid leukemia (31). However, the role of M1AP in cancer progression remains unclear. The two SNPs detected in the intron region of M1AP are not inferred to be eQTLs for M1AP itself. Although they are not predicted to be associated with expression level of M1AP, these two SNPs are predicted as eQTLs for two lung cancer-related genes: LBX2, encoding a transcription factor causally implicated in lung adenocarcinoma (24) and LBX2-AS1 (25), encoding a regulatory RNA that promotes cell proliferation and metastasis of NSCLC. The two adjacent genes are located approximately 60 and 72 kilobases from rs11126435 and rs2021725, respectively. Therefore, these SNPs may affect the expression of one or both of LBX2 and LBX2-AS and not M1AP.
CLPP is a protein-coding gene located on chromosome 19 (32). The protein encoded by CLPP is a serine protease associated with the inner mitochondrial membrane that degrades misfolded proteins and is required for mitochondrial respiration (33–36). The CLpP protein is overexpressed in many human cancers, including NSCLC regardless of histotype, and is most highly expressed in primary NSCLC lesions that developed metastasis (36). In several types of cancer (including breast, lung, and uveal cancer), overexpressed CLpP is associated with shorter metastasis-free survival (36). It was also shown that CLpP supports tumor cell proliferation and motility (33, 36). Gene sequencing plus predictive functional analyses indicate that the rs10420388 and rs10418574 CLPP variants are associated with intronic elements involved in transcriptional regulation. Therefore, we propose that these variants may cause overexpression of the CLPP gene.
As noted above, this study was designed as a pilot project to examine the evolutionary dynamics of tumor cells in patients with early stage vs. late stage disease. As such, the identification of SNPs reported here is an incidental outcome of a study that was not, in itself, focused on identifying either prognostic markers or causal genetic mechanisms. This means, among other things, that there may be (indeed, are likely to be) more SNPs that have not been identified in our study. However, the small sample size in this study and the variability of coverage also means that we have low statistical power to identify SNPs that may be good markers. Because it is likely that we have not characterized the constellation of SNPs that may separate the two groups of patients, it is difficult to construct a hypothesis about the functional and regulatory pathways that steer individuals with NSCLC into metastatic or non-metastatic progression.
It is also worth noting that the GWAS datasets of OncoArray and PLCO used to validate the markers we identified in this study only include Caucasian populations. Thus, our results may not be generalizable to other ethnic groups, and further studies are needed to determine the true impact on lung cancer risk in the general population.
Because cancer is a disease of altered genes, GWAS and identification of SNPs offers an important strategy to studying cancer risk and biologic features that may be essential for tumor development and progression. In the current study we focused on 4 SNPs discovered from the normal lung of patients with advanced stage NSCLC as compared to early stage disease and showed that three of these markers were associated with lung cancer risk. The exact molecular mechanisms by which they influence cancer cells remain unclear, and although these data may be useful in risk-prediction models, further biochemical studies and functional experiments are warranted.
Data Availability Statement
The datasets presented in this study can be found in the NCBI Bioproject repository under accession number PRJNA750918; link: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA750918.
Ethics Statement
The studies involving human participants were reviewed and approved by Duke University Health System Institutional Review Board. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
AR, QW, and EP conceived of and designed the study and performed supervision. MC and EG selected the patients for the study and followed their cancer statuses. MC and RB performed DNA manipulations. XL, YZ, TL, TW, and HL curated and analyzed the data. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by Department of Defense Idea Award W81XWH-15-1-0243 to EP.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all the participants of the OncoArray studies and PLCO Cancer Screening Trial. We also thank the National Cancer Institute for providing the access to the data collected by the PLCO trial. The statements contained herein are solely those of the authors and do not represent or imply concurrence or endorsement by National Cancer Institute. The authors would also like to acknowledge dbGaP repository for providing cancer genotyping datasets. The accession numbers for the datasets of lung cancer are phs001273.v3.p2, phs000336.v1.p1 and phs000093.v2.p2. A list of contributing investigators and funding agencies for these studies can be found in the supplementary data.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.709829/full#supplementary-material
References
1. Eirew P, Steif A, Khattra J, Ha G, Yap D, Farahani H, et al. Dynamics of Genomic Clones in Breast Cancer Patient Xenografts at Single-Cell Resolution. Nature (2015) 518(7539):422–6. doi: 10.1038/nature13952
2. Itzhak Y. “Multiple Opioid Binding Sites”. In: Pasternak GW, editor. The Opiate Receptors. New Jersey: The Humana Press (1988). p. 95–142.
3. Tang D, Zhao YC, Qian D, Liu H, Luo S, Patz EF, et al. Novel Genetic Variants in HDAC2 and PPARGC1A of the CREB-Binding Protein Pathway Predict Survival of Non-Small-Cell Lung Cancer. Mol Carcinog (2020) 59(1):104–15. doi: 10.1002/mc.23132
4. Ding L, Raphael BJ, Chen F, Wendl MC. Advances for Studying Clonal Evolution in Cancer. Cancer Lett (2013) 340(2):212–9. doi: 10.1016/j.canlet.2012.12.028
5. Greaves M, Maley CC. Clonal Evolution in Cancer. Nature (2012) 481(7381):306–13. doi: 10.1038/nature10762
6. Deleye L, Tilleman L, Vander Plaetsen A-S, Cornelis S, Deforce D, Van Nieuwerburgh F. Performance of Four Modern Whole Genome Amplification Methods for Copy Number Variant Detection in Single Cells. Sci Rep (2017) 7(1):3422. doi: 10.1038/s41598-017-03711-y
7. Li H, Durbin R. Fast and Accurate Short Read Alignment With Burrows-Wheeler Transform. Bioinformatics (2009) 25(14):1754–60. doi: 10.1093/bioinformatics/btp324
8. Kendig KI, Baheti S, Bockol MA, Drucker TM, Hart SN, Heldenbrand JR, et al. Sentieon DNASeq Variant Calling Workflow Demonstrates Strong Computational Performance and Accuracy. Front Genet (2019) 10:736. doi: 10.3389/fgene.2019.00736
9. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, et al. (Genomes Project Consortium). A Global Reference for Human Genetic Variation. Nature (2015) 526(7571):68–74. doi: 10.1038/nature15393
10. Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: The NCBI Database of Genetic Variation. Nucleic Acids Res (2001) 29(1):308–11. doi: 10.1093/nar/29.1.308
11. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ Data to High Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr Protoc Bioinf (2013) 43:11 0 1– 0 33. doi: 10.1002/0471250953.bi1110s43
12. Quinlan AR, Hall IM. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics (2010) 26(6):841–2. doi: 10.1093/bioinformatics/btq033
13. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: Somatic Mutation and Copy Number Alteration Discovery in Cancer by Exome Sequencing. Genome Res (2012) 22(3):568–76. doi: 10.1101/gr.129684.111
14. Wang K, Li M, Hakonarson H. ANNOVAR: Functional Annotation of Genetic Variants From High-Throughput Sequencing Data. Nucleic Acids Res (2010) 38(16):e164–e. doi: 10.1093/nar/gkq603
15. Hasson MA, Fagerstrom RM, Kahane DC, Walsh JH, Myers MH, Caughman C, et al. Design and Evolution of the Data Management Systems in the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial. Controlled Clin Trials (2000) 21(6 Suppl):329S–48S. doi: 10.1016/s0197-2456(00)00100-8
16. Landi MT, Consonni D, Rotunno M, Bergen AW, Goldstein AM, Lubin JH, et al. Environment And Genetics in Lung Cancer Etiology (EAGLE) Study: An Integrative Population-Based Case-Control Study of Lung Cancer. BMC Public Health (2008) 8:203. doi: 10.1186/1471-2458-8-203
17. Landi MT, Chatterjee N, Yu K, Goldin LR, Goldstein AM, Rotunno M, et al. A Genome-Wide Association Study of Lung Cancer Identifies a Region of Chromosome 5p15 Associated With Risk for Adenocarcinoma. Am J Hum Genet (2009) 85(5):679–91. doi: 10.1016/j.ajhg.2009.09.012
18. McKay JD, Hung RJ, Han Y, Zong X, Carreras-Torres R, Christiani DC, et al. Large-Scale Association Analysis Identifies New Lung Cancer Susceptibility Loci and Heterogeneity in Genetic Susceptibility Across Histological Subtypes. Nat Genet (2017) 49(7):1126–32. doi: 10.1038/ng.3892
19. Wang Y, Liu H, Ready NE, Su L, Wei Y, Christiani DC, et al. Genetic Variants in ABCG1 are Associated With Survival of Nonsmall-Cell Lung Cancer Patients. Int J Cancer (2016) 138(11):2592–601. doi: 10.1002/ijc.29991
20. Bendl J, Musil M, Stourac J, Zendulka J, Damborsky J, Brezovsky J. PredictSNP2: A Unified Platform for Accurately Evaluating SNP Effects by Exploiting the Different Characteristics of Variants in Distinct Genomic Regions. PloS Comput Biol (2016) 12(5):e1004962. doi: 10.1371/journal.pcbi.1004962
21. Ward LD, Kellis M. HaploReg V4: Systematic Mining of Putative Causal Variants, Cell Types, Regulators and Target Genes for Human Complex Traits and Disease. Nucleic Acids Res (2016) 44(D1):D877–81. doi: 10.1093/nar/gkv1340
22. Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. The Genotype-Tissue Expression (GTEx) Project. Nat Genet (2013) 45(6):580–5. doi: 10.1038/ng.2653
23. Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of Functional Variation in Personal Genomes Using RegulomeDB. Genome Res (2012) 22(9):1790–7. doi: 10.1101/gr.137323.112
24. Hu J, Bai Y, Zhang Q, Li M, Yin R, Xu L. Identification of LBX2 as a Novel Causal Gene of Lung Adenocarcinoma. Thorac Cancer (2020) 11(8):2137–45. doi: 10.1111/1759-7714.13506
25. Tang LX, Su SF, Wan Q, He P, Xhang Y, Cheng XM. Novel Long Non-Coding RNA LBX2-AS1 Indicates Poor Prognosis and Promotes Cell Proliferation and Metastasis Through Notch Signaling in Non-Small Cell Lung Cancer. Eur Rev Med Pharmacol Sci (2019) 23(17):7419–29. doi: 10.26355/eurrev_201909_18851
26. Liang B, Ding H, Huang L, Luo H, Zhu X. GWAS in Cancer: Progress and Challenges. Mol Genet Genomics (2020) 295(3):537–61. doi: 10.1007/s00438-020-01647-z
27. Bosse Y, Amos CI. A Decade of GWAS Results in Lung Cancer. Cancer Epidemiol Biomarkers Prev (2018) 27(4):363–79. doi: 10.1158/1055-9965.EPI-16-0794
28. Guo Y, Long J, He J, Li C-I, Cai Q, Shu X-O, et al. Exome Sequencing Generates High Quality Data in Non-Target Regions. BMC Genomics (2012) 13(1):194. doi: 10.1186/1471-2164-13-194
29. Tu C, Wang Y, Nie H, Meng L, Wang W, Li Y, et al. An M1AP Homozygous Splice-Site Mutation Associated With Severe Oligozoospermia in a Consanguineous Family. Clin Genet (2020) 97(5):741–6. doi: 10.1111/cge.13712
30. Arango NA, Li L, Dabir D, Nicolau F, Pieretti-Vanmarcke R, Koehler C, et al. Meiosis I Arrest Abnormalities Lead to Severe Oligozoospermia in Meiosis 1 Arresting Protein (M1ap)-Deficient Mice. Biol Reprod (2013) 88(3):76. doi: 10.1095/biolreprod.111.098673
31. Castilla LH, Perrat P, Martinez NJ, Landrette SF, Keys R, Oikemus S, et al. Identification of Genes That Synergize With Cbfb-MYH11 in the Pathogenesis of Acute Myeloid Leukemia. Proc Natl Acad Sci USA (2004) 101(14):4924–9. doi: 10.1073/pnas.0400930101
32. Zeiler E, Korotkov VS, Lorenz-Baath K, Bottcher T, Sieber SA. Development and Characterization of Improved Beta-Lactone-Based Anti-Virulence Drugs Targeting ClpP. Bioorg Med Chem (2012) 20(2):583–91. doi: 10.1016/j.bmc.2011.07.047
33. Luo J, Zeng B, Tao C, Lu M, Ren G. ClpP Regulates Breast Cancer Cell Proliferation, Invasion and Apoptosis by Modulating the Src/PI3K/Akt Signaling Pathway. PeerJ (2020) 8:e8754. doi: 10.7717/peerj.8754
34. Ishizawa J, Zarabi SF, Davis RE, Halgas O, Nii T, Jitkova Y, et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell (2019) 35(5):721–37 e9. doi: 10.1016/j.ccell.2019.03.014
35. Graves PR, Aponte-Collazo LJ, Fennell EMJ, Graves AC, Hale AE, Dicheva N, et al. Mitochondrial Protease ClpP is a Target for the Anticancer Compounds ONC201 and Related Analogues. ACS Chem Biol (2019) 14(5):1020–9. doi: 10.1021/acschembio.9b00222
Keywords: non-small cell lung cancer, single nucleotide polymorphism, lung cancer risk, CLPP, M1AP
Citation: Li X, Zou Y, Li T, Wong TKF, Bushey RT, Campa MJ, Gottlin EB, Liu H, Wei Q, Rodrigo A and Patz EF Jr. (2021) Genetic Variants of CLPP and M1AP Are Associated With Risk of Non-Small Cell Lung Cancer. Front. Oncol. 11:709829. doi: 10.3389/fonc.2021.709829
Received: 14 May 2021; Accepted: 20 August 2021;
Published: 15 September 2021.
Edited by:
Mehmet Artac, Meram Faculty of Medicine, TurkeyCopyright © 2021 Li, Zou, Li, Wong, Bushey, Campa, Gottlin, Liu, Wei, Rodrigo and Patz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edward F. Patz, Jr., cGF0ejAwMDJAbWMuZHVrZS5lZHU=
†These authors have contributed equally to this work and share first authorship