 Li Liu1†
Li Liu1† Jingjing Qu2†Jianfu Heng1,3Chunhua Zhou1Yi Xiong1,3Haiyan Yang1Wenjuan Jiang1Liang Zeng1Songlin Zhu3
Jingjing Qu2†Jianfu Heng1,3Chunhua Zhou1Yi Xiong1,3Haiyan Yang1Wenjuan Jiang1Liang Zeng1Songlin Zhu3 Yongchang Zhang1
Yongchang Zhang1 Jiarong Tan4Chengping Hu4
Jiarong Tan4Chengping Hu4 Pengbo Deng4*Nong Yang1*
Pengbo Deng4*Nong Yang1*- 1Department of Lung Cancer and Gastroenterology, Hunan Cancer Hospital, Affiliated Tumor Hospital of Xiangya Medical School of Central South University, Changsha, China
- 2Department of Respiratory Disease, Thoracic Disease Centre, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, China
- 3Department of Clinical Pharmaceutical Research Institution, Hunan Cancer Hospital, Affiliated Tumor Hospital of Xiangya Medical School of Central South University, Changsha, China
- 4Department of Respiratory Medicine, Xiangya Hospital of Central South University, Changsha, China
Background: MET proto-oncogene amplification (amp) is an important mechanism underlying acquired resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs). However, the optimal treatment strategy after acquiring MET-amp-mediated EGFR-TKI resistance remains controversial. Our study compared three treatment strategies for patients with EGFR-mutant non-small-cell lung cancer (NSCLC) who were detected with MET-amp at EGFR-TKI progression using next-generation sequencing.
Methods: Of the 70 patients included in the study, 38 received EGFR-TKI + crizotinib, 10 received crizotinib monotherapy, and 22 received chemotherapy. Clinical outcomes and molecular profiles were analyzed.
Results: The objective response rate was 48.6% for EGFR-TKI + crizotinib group, 40.0% for crizotinib monotherapy group, and 18.2% for chemotherapy group. Patients who received EGFR-TKI + crizotinib had significantly longer progression-free survival than those who received crizotinib or chemotherapy (5.0 vs. 2.3 vs. 2.9 months, p = 0.010), but overall survival was comparable (10.0 vs. 4.1 vs. 8.5 months, p = 0.088). TP53 mutation (58.5%) and EGFR-amp (42.9%) were frequent concurrent mutations of the cohort. Progression-free survival was significantly longer for patients with either concurrent TP53 mutation (n = 17) (6.0 vs. 2.3 vs. 2.9 months, p = 0.009) or EGFR-amp (n = 13) (5.0 vs. 1.2 vs. 2.4 months, p = 0.016) in the EGFR-TKI + crizotinib group than the other two regimen. Potential acquired resistance mechanisms to EGFR-TKI + crizotinib included EGFR-T790M (n = 2), EGFR-L718Q (n = 1), EGFR-S645C (n = 1), MET-D1228H (n = 1), BRAF-V600E (n = 1), NRAS-Q61H (n = 1), KRAS-amp (n = 1), ERBB2-amp (n = 1), CDK4-amp (n = 1), and MYC-amp (n = 1).
Conclusion: Our study provides real-world clinical evidence from a large cohort that simultaneous inhibition of EGFR and MET could be a more effective therapeutic strategy for patients with MET-amp acquired from EGFR-TKI therapy.
Introduction
The discovery of oncogenic drivers has transformed the management of advanced non-small-cell lung cancer (NSCLC) into a more personalized approach by tailoring the treatment regimen of a patient based on the presence or absence of molecular biomarkers (1). Sensitizing mutations in epidermal growth factor receptor (EGFR) are the most common predictive biomarkers and are identified in approximately 60% of Chinese patients with advanced NSCLC (2). In patients with EGFR-mutant advanced NSCLC, frontline therapy with EGFR tyrosine kinase inhibitors (TKIs) provides superior clinical outcomes as compared to chemotherapy (3–6). However, resistance to EGFR-TKI inevitably develops in most patients within 10–12 months of therapy through the acquisition of either EGFR-dependent or EGFR-independent mechanisms (7–10). EGFR T790M is the most frequent mechanism of acquired resistance to first- and second-generation EGFR-TKIs and is successfully targeted by third-generation EGFR-TKIs (11). EGFR-independent pathways that bypass EGFR inhibition, including aberrations in MET proto-oncogene (MET), also mediate EGFR-TKI resistance (7–10). MET, a receptor tyrosine kinase, is activated by the binding of hepatocyte growth factor (HGF), leading to the activation of various downstream signaling pathways including the phosphatidylinositol-4-biphosphate 3-kinase (PI3K) pathway (12–14). Dysregulation of MET signaling is implicated as one of the oncogenic drivers in lung cancer development and progression (12–14). MET gene amplification has been reported in 2–8% of EGFR-TKI-naive advanced NSCLCs and in approximately 20% of EGFR-TKI-treated relapsed NSCLCs (8, 9, 15, 16). Moreover, patients who harbor concurrent EGFR mutation with MET amplification after EGFR-TKI progression demonstrated short progression-free and overall survival outcomes with MET-TKI monotherapy (16), indicating the urgent need for identifying effective therapeutic strategies that could overcome the MET amplification-mediated EGFR-TKI resistance.
The reciprocal crosstalk between EGFR and MET in lung adenocarcinoma suggests that simultaneous inhibition through the combined use of EGFR-TKI and MET-TKI would benefit and potentially improve the survival outcomes of patients with concurrent EGFR and MET aberrations (17, 18). Over the years, a growing number of studies have demonstrated the clinical benefit of different combinations of various generations of EGFR-TKIs and various MET-TKIs including crizotinib (16, 19–23), capmatinib (21, 24), and savolitinib (25–27). Recently, tepotinib plus gefitinib showed promising antitumor activity in NSCLC with concomitant EGFR mutation and MET amplification or high MET overexpression compared with chemotherapy (28). The promising clinical outcomes demonstrated by the combinatorial therapy of EGFR-TKI and MET-TKI highlight the survival benefit of targeted therapy over chemotherapy or possibly MET-TKI monotherapy. However, due to the lack of solid clinical evidence derived from a larger cohort or a controlled clinical trial, there is still no consensus on the standard treatment for patients with advanced EGFR-mutant NSCLC who acquire MET amplification during EGFR-TKI therapy. In current clinical practice, after progression from EGFR-TKI with MET amplification or MET overexpression, patients are managed with any of the three regimens: combination therapy consisting of an EGFR-TKI and a MET-TKI, MET-TKI monotherapy, or chemotherapy. In this real-world observational study, we aimed to identify the treatment regimen that could impart better clinical benefit for patients with concurrent MET amplification and EGFR sensitizing mutation after progression from EGFR-TKI therapy. To achieve this aim, we retrospectively analyzed and compared the clinical outcomes of 70 patients who were detected with MET amplification using next-generation sequencing (NGS) after progression from EGFR-TKI regimen and received any of the three regimens. We also explored the molecular mechanism of acquired resistance to the EGFR-TKI and crizotinib combination therapy.
Patients and Methods
Study Subjects and Inclusion/Exclusion Criteria
Patients diagnosed with advanced EGFR-mutant NSCLC who received treatment from Hunan Cancer Hospital or Xiangya Hospital between March 2015 and March 2020 were included in this study. The baseline EGFR mutation status was assessed from blood samples or tissue samples obtained by needle biopsy of lung lesions or lymph nodes using either NGS or amplification refractory mutation system (ARMS) PCR. At progression from EGFR-TKI therapy, all patients underwent rebiopsy of tissue, blood, or other biological samples for NGS analysis. Patients with confirmed mutation status for EGFR and MET amplification at progression from first- or subsequent-line EGFR-TKI therapy were included in the study. Patients with baseline MET amplification or those who had received prior crizotinib therapy were excluded from the study. The patient data were retrieved from the medical records. This study was approved by the Institutional Ethics Committee of Hunan Cancer Hospital. All the patients provided written informed consent for the use of their data for research purposes.
Treatment Procedures
Crizotinib at 250 mg twice daily was administered with or without continued treatment of EGFR-TKI. Patients without EGFR T790M were administered with erlotinib, gefitinib, or afatinib daily at an oral dose of 150, 250, or 40 mg, respectively. Patients with EGFR T790M received osimertinib at a daily oral dose of 80 mg. Some patients were administered platinum-based chemotherapy combined with pemetrexed (500 mg/cm2, 21 days per cycle). Treatment was continued until one or more of the following conditions occurred: unacceptable toxicity, disease progression or death, patient refusal, or treatment withdrawal for any other reason including pregnancy. Treatment responses were investigator assessed every two cycles (approximately 2 months) using enhanced computed tomography (CT) scanning according to the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 (29). Objective response rate (ORR) was defined as the proportion of patients achieving complete response (CR) or partial response (PR). Disease control rate (DCR) was defined as the proportion of patients achieving CR, PR, and stable disease (SD). Treatment-related toxicity was evaluated according to the Common Terminology Criteria for Adverse Event (CTCAE) version 4.03. Real-world progression-free survival (rwPFS) was defined as the period between the date of initiating the treatment regimen with crizotinib, EGFR-TKI combined with crizotinib, or pemetrexed combined with platinum and the date of treatment discontinuation due to radiologically confirmed disease progression or intolerable side effect or death. Overall survival (OS) was defined as the period between the date of treatment administration after developing MET amplification-mediated EGFR-TKI resistance until death or the day of the last follow-up.
Preparation of Circulating Cell-Free DNA and Tissue DNA
Circulating cell-Free DNA (cfDNA) was extracted from plasma samples and other liquid biopsy samples according to the manufacturer’s instructions using a QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany). Tissue DNA was extracted from formalin-fixed paraffin-embedded (FFPE) cell blocks of tumor biopsy or other cytology samples using QIAamp DNA FFPE tissue kit (Qiagen, Hilden, Germany). CfDNA and tissue DNA concentration were quantified using Qubit 2.0 fluorometer with dsDNA HS Assay Kit (Life Technologies, CA, USA).
NGS-Based Analysis of the MET and EGFR Mutation Status
NGS was performed to establish the mutation status of EGFR and MET and to elucidate the molecular mechanism of acquired resistance to EGFR-TKI and crizotinib. NGS library construction, sequencing procedures, and sequencing data analysis were performed using optimized protocols at Burning Rock Biotech, a clinical laboratory accredited and certified by the College of American Pathologists (CAP) and Clinical Laboratory Improvement Amendments (CLIA), respectively (30). Target capture was performed using commercial gene panels consisting of 8–168 cancer-related genes. All gene panels interrogated all exons and critical introns of the classic lung cancer oncogenic genes including EGFR, ALK, ROS, and MET. The quality and size of the fragments were assessed using Agilent high-sensitivity DNA assay kit with the Bioanalyzer 2100 (Agilent, CA, USA). Sequencing of the indexed samples was performed using a NextSeq500 instrument (Illumina, CA, USA) with paired-end reads at a target sequencing depth of 10,000× for plasma samples and 1,000× for tissue samples.
DNA sequence data were analyzed using an optimized bioinformatics pipeline developed by Burning Rock Biotech, which accurately detected various cancer-related mutation types, including single nucleotide, copy number, and structural variations (30). Sequence mapping to the reference human genome (hg19) was performed using Burrows–Wheeler Aligner (version 0.7.10). Genomic analysis toolkit (GATK version 3.2) and VarScan (version 2.4.3) were used for local alignment optimization, variant calling, and annotation. Plasma samples were compared against paired white blood cells to filter out clonal hematopoiesis-related variants and identify true somatic variants. Factera (version 1.4.3) was used to analyze structural variants. Copy number variations (CNVs) were analyzed based on the depth of coverage and were called when the coverage data of the gene region was quantitatively and statistically significant from its reference control. The copy number cutoff for identifying CN deletion was set at 1.5 and CN amplification set at 2.25.
Statistical Analysis
Mann–Whitney U test was implemented for intergroup comparisons of continuous variables. Fisher’s exact test was used to compare the ORR and DCR among the groups. Survival analyses were performed for each group using the Kaplan–Meier method with log-rank statistics. All the statistical analysis was performed using R statistics package (R version 3.5.3; Vienna, Austria). Statistical significance was defined as p < 0.05. p-values (adjP) were adjusted accordingly with clinical factors including age, smoking history, and treatment line. Hazard ratios (HRs) and their corresponding 95% confidence intervals (CIs) were also calculated using Cox proportional hazards regression method for adjusted survival curves.
Results
Patient Characteristics
Seventy patients who were detected with MET amplification using DNA NGS after progression from EGFR-TKI therapy were included in this study. MET amplification was detected after progression from first-generation EGFR-TKI therapy in 57.1% (n = 40) of patients, after progression from second-generation EGFR-TKI therapy in 4.3% (n = 3), and after progression from third-generation EGFR-TKI therapy in 37.1% (n = 26) of patients. MET amplification was not evaluated using fluorescence in situ hybridization due to lack of or insufficient tissue biopsy sample. The cohort was further stratified into three groups based on the treatment they received after EGFR-TKI progression: 38 patients received a combination of crizotinib and any EGFR-TKI, 10 patients received crizotinib monotherapy, and 22 patients received carboplatin and pemetrexed chemotherapy. All the patients had extensive metastasis at EGFR-TKI progression. Most of the patients included in this study progressed from first-line EGFR-TKI and received one of the three regimens described above as second-line therapy (57.9% vs. 70.0% and 68.2%, p = 0.001). Of the 38 patients who received EGFR-TKI plus crizotinib therapy, 13 patients received first-generation EGFR-TKI (i.e., gefitinib, erlotinib), 1 patient received second-generation EGFR-TKI (i.e., afatinib), and 24 patients received third-generation EGFR-TKI (i.e., osimertinib). The median age was 56 years (range, 48–65) for the patients who received EGFR-TKI plus crizotinib therapy, 57 years (range, 55–68) for patients who received crizotinib monotherapy, and 56 years (range, 50–63) for patients who received chemotherapy. The therapeutic regimens were independent of sex, age, smoking status, and mutation detection method. The concurrent EGFR mutations of the cohort were as follows: 55.7% (39/70) EGFR exon 21 L858R mutation, 40.0% (28/70) EGFR exon 19 deletion (19del), 21.4% (15/70) EGFR T790M mutation with either L858R (n = 5) or 19del (n = 10), and 50.0% (35/70) other EGFR mutations (Figure 1). Table 1 summarizes the clinicopathological characteristics of the cohort. The treatment groups had comparable clinical features including age, sex, smoking history, Eastern Cooperative Oncology Group performance status (ECOG PS), and prior EGFR-TKI therapy. However, when two groups were compared with each other, ECOG PS remained comparable between the EGFR-TKI plus crizotinib combination therapy group and crizotinib monotherapy group (p = 1) but was significantly different between EGFR-TKI plus crizotinib combination therapy group and chemotherapy group (p = 0.023) and crizotinib monotherapy group and chemotherapy group (p = 0.024).
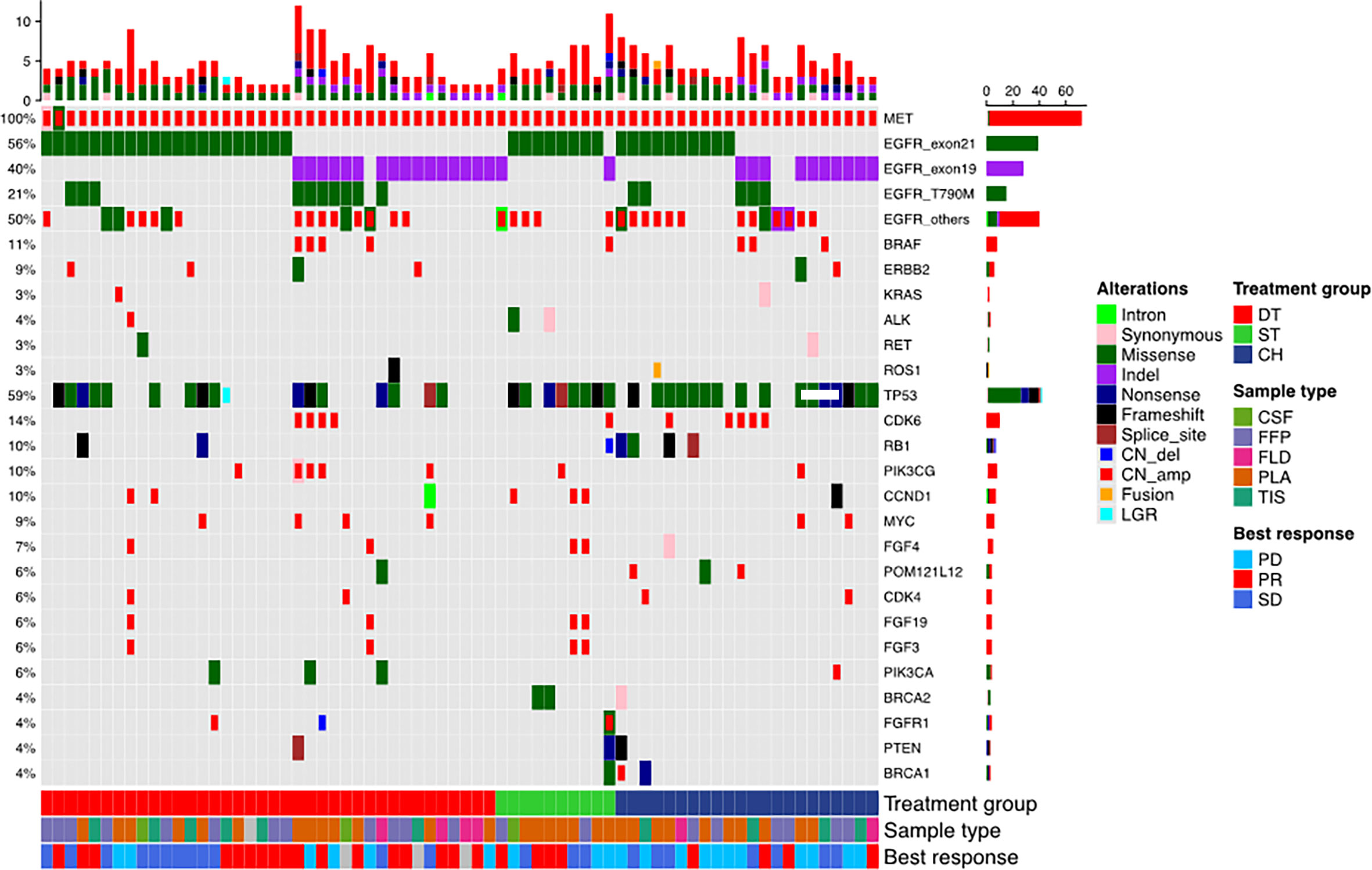
Figure 1 Mutation landscape of the cohort before receiving the three regimens: EGFR-TKI combined with crizotinib (DT), crizotinib monotherapy (ST), and chemotherapy (CH). The patient’s best responses to the treatment are also annotated at the bottom of the oncomap.
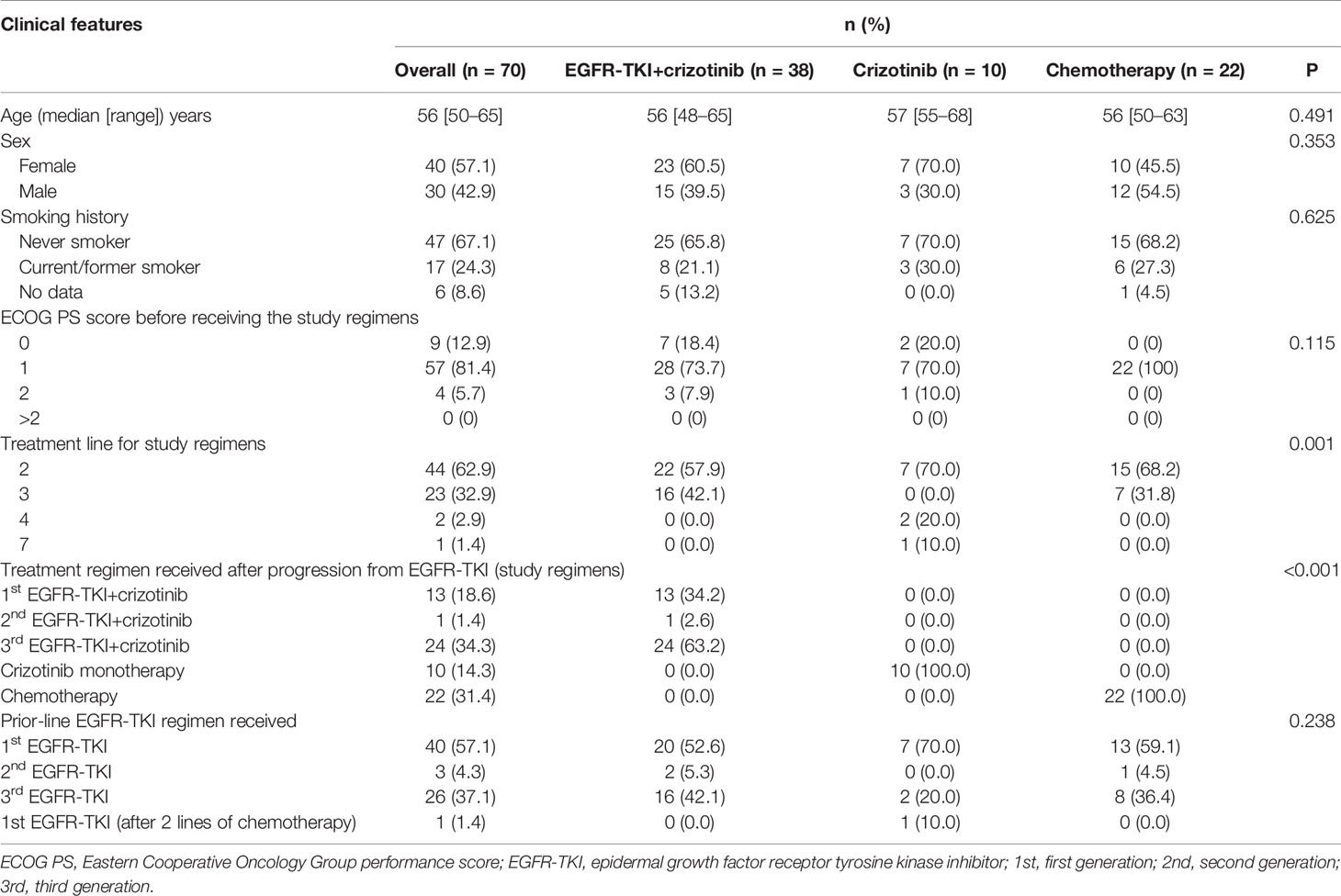
Table 1 Baseline demographic and treatment characteristics of patients with concurrent EGFR mutations and MET amplification after EGFR-TKI resistance.
Clinical Outcomes of Patients With Concurrent EGFR Mutation and MET Amplification
Among the 38 patients who received EGFR-TKI plus crizotinib combination therapy, only 35 patients were evaluable for best response. Of them, 48.6% (17/35) achieved PR and 34.3% (12/35) achieved SD, resulting in an ORR of 48.6% and DCR of 82.9%. Only 17.1% (6/35) did not benefit from the EGFR-TKI plus crizotinib. Of the 10 patients who received crizotinib monotherapy, 40.0% (4/10) and 30.0% (3/10) each achieved PR and SD, and 30% (3/10) had no clinical benefit, resulting in an ORR of 40.0% and a DCR of 70.0%. Of the 22 patients who received chemotherapy regimen, 18.2% (4/22) achieved PR, 31.8% (7/22) achieved SD, and 50.0% (11/22) had no clinical benefit, resulting in an ORR of 18.2% and a DCR of 50.0%. As compared to patients who received chemotherapy, patients who received EGFR-TKI plus crizotinib had significantly better ORR (p = 0.026) and DCR (p = 0.016) but was not statistically different from that of the patients who received crizotinib monotherapy (Figure 2A).
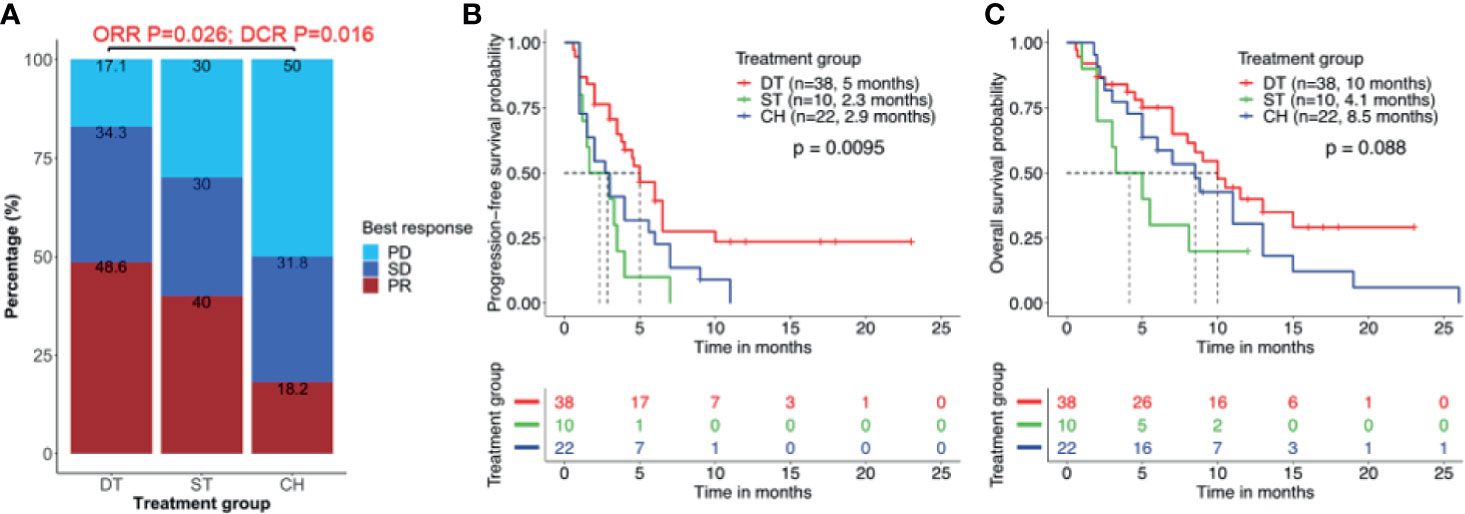
Figure 2 Clinical outcomes are better with combined therapy of EGFR-TKI and crizotinib. (A) Overall response rate (ORR) and disease control rate (DCR) were significantly higher in patients who received EGFR-TKI with crizotinib combination therapy (DT) than crizotinib monotherapy (ST) or chemotherapy (CH). (B, C) Combination therapy results in significantly longer median real-world progression-free survival (PFS) (B) but no statistically significant benefit in real-world overall survival (C). Tick marks denote censored patients.
Patients who received EGFR-TKI plus crizotinib had significantly longer median rwPFS as compared to those who received crizotinib monotherapy [5.0 vs. 2.3 months; p = 0.007; HR = 0.88, 95% CI (0.42, 2.42), adjp = 0.036] and chemotherapy regimen (5.0 vs. 2.9 months; p = 0.038; HR = 0.72, 95% CI (0.32, 2.06), adjp = 0.024] (Figure 2B). However, patients who received EGFR-TKI plus crizotinib had a trend of longer OS, with a median OS of 10.0 months but did not reach statistical significance [vs. crizotinib p = 0.058; HR = 0.65, 95% CI (0.48, 1.92), adjp = 0.17; vs. chemotherapy p = 0.095; HR = 0.59, 95% CI (0.35, 1.8), adjp = 0.20; Figure 2C). Median OS was 4.1 months for those who received crizotinib monotherapy and 8.5 months for those who received chemotherapy. The ORR (p = 0.22), DCR (p = 0.45), rwPFS (p = 0.30, adjp = 0.96), and OS (p = 0.21, adjp = 0.66) were comparable between patients who received crizotinib monotherapy and chemotherapy.
Molecular Factors Affecting Survival Outcomes
Furthermore, we studied the impact of other molecular factors on the survival outcome from the three regimens. Numerous studies have implicated the detection of concurrent TP53 mutations in poorer survival outcomes with EGFR-TKI and crizotinib therapy (31–35). Concurrent TP53 mutation was detected in 58.5% of our cohort and the most frequent co-occurring mutation. TP53 mutation was detected in 44.7% (17/38) of patients who received EGFR-TKI plus crizotinib, 80.0% (8/10) of patients who received crizotinib monotherapy, and 72.7% (16/22) of patients who received chemotherapy. Among the patients with concurrent TP53 mutation, those who received EGFR-TKI plus crizotinib (n = 17) had a significantly longer rwPFS than those who received crizotinib monotherapy (n = 8) (6.0 vs. 2.3 months, p = 0.007, adjp = 0.208) or chemotherapy (n = 16) (6.0 vs. 2.9 months, p = 0.016, adjp = 0.048) (Figure 3A). Patients with concurrent TP53 mutation who received EGFR-TKI plus crizotinib (n = 17) had a trend of longer OS but statistically comparable with the patients who received crizotinib monotherapy (n = 8) (11.5 vs. 4.1 months, p = 0.077, adjp = 0.599) or chemotherapy (n = 16) (11.5 vs. 8.5 months, p = 0.074, adjp = 0.162) (Figure 3B). Median rwPFS (p = 0.55, adjp = 0.85) and OS (p = 0.49, adjp = 0.99) were comparable for patients with concurrent TP53 mutations who received crizotinib monotherapy and chemotherapy.
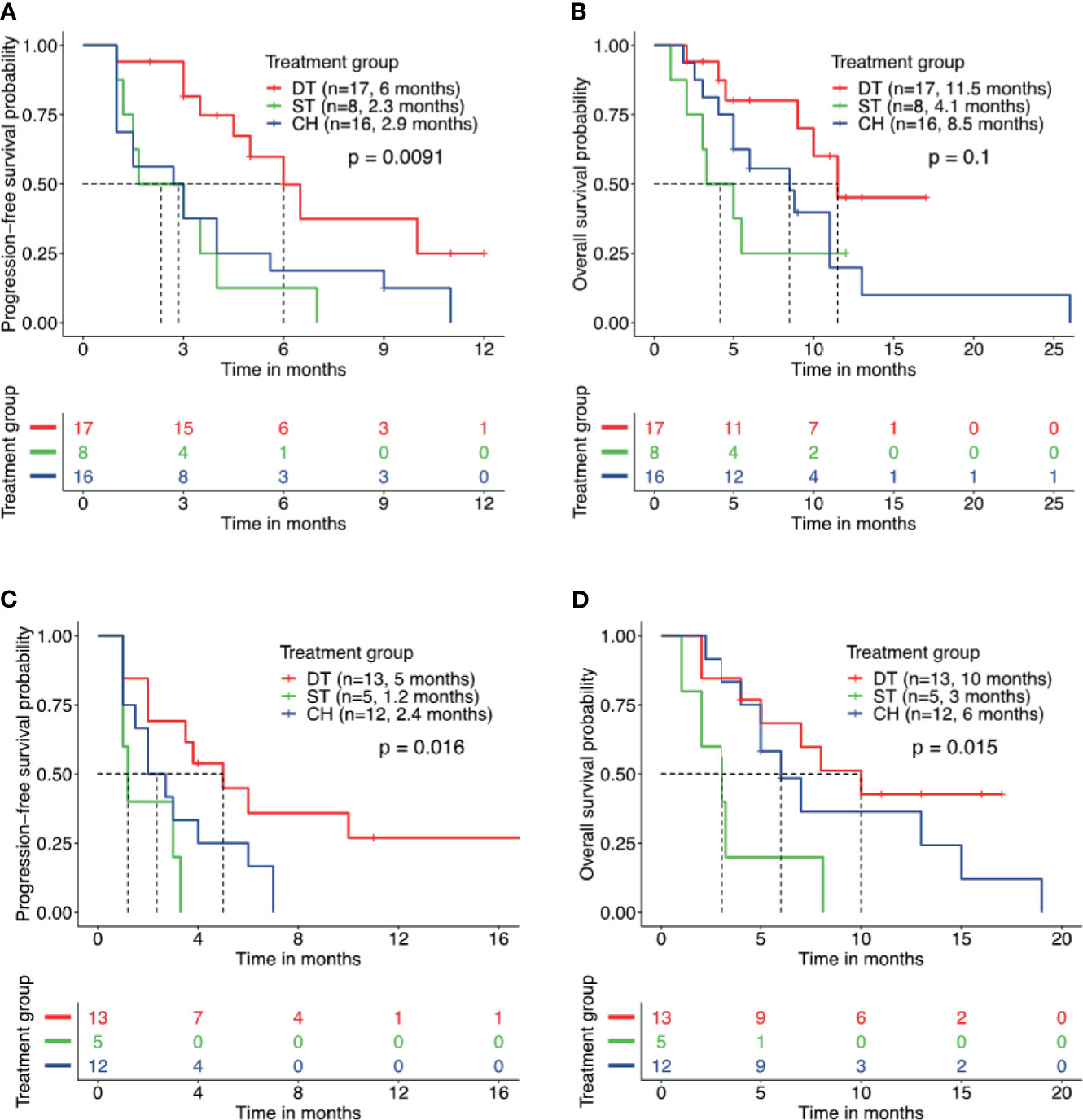
Figure 3 Patients with concurrent TP53 mutation and EGFR amplification benefit from combined therapy of EGFR-TKI and crizotinib. Kaplan–Meier curves for progression-free survival (A, C) and overall survival (B, D) demonstrate the survival benefit of patients with concurrent TP53 mutation and EGFR amplification who received combined therapy of EGFR-TKI and crizotinib (DT) than crizotinib monotherapy (ST) or chemotherapy (CH). Tick marks denote censored patients.
In addition to concurrent TP53 mutation status, the prognostic effect of EGFR amplification was also investigated. Concurrent EGFR amplification was detected in 42.9% (30/70) of the cohort and the second most frequently co-occurring mutation. Among the patients with concurrent EGFR amplification, those who received EGFR-TKI plus crizotinib (n = 13) had a significantly longer rwPFS (5.0 vs. 1.2 months, p = 0.017, adjp = 0.046) and OS (10.0 vs. 3.0 months, p = 0.02, adjp = 0.019) than those who received crizotinib monotherapy (n = 5) (Figures 3C, D). Meanwhile, patients with concurrent EGFR amplification who received EGFR-TKI plus crizotinib (n = 13) had no statistical difference in rwPFS (5.0 vs. 2.4 months, p = 0.06, adjp = 0.094) and OS (10.0 vs. 6.0 months, p = 0.35, adjp = 0.44) than patients who received chemotherapy (n = 12) (Figures 3C, D). Taken together, these data indicate that concurrent TP53 mutations and EGFR amplifications that are known to affect the prognosis of patients with EGFR-mutant NSCLC did not affect the survival outcomes of EGFR-TKI plus crizotinib combination therapy.
Exploring the Underlying Acquired Resistance Mechanism in the EGFR-TKI Plus Crizotinib Cohort
We further analyzed the molecular mechanism of acquired resistance developed during EGFR-TKI plus crizotinib by comparing the mutation profile before receiving the combination therapy and after disease progression. Among the 38 patients who received EGFR-TKI plus crizotinib, 9 patients (23.7%) submitted rebiopsy samples for NGS-based genomic testing after progression from the combination therapy. Figure 4 illustrates the mutation profile at baseline (Figure 4A), at progression (Figure 4B), and the overlap of mutation profiles at baseline and progression to highlight the mutations retained, lost, or acquired at progression (Figure 4C). Table 2 summarizes the molecular mechanisms of acquired resistance and the detailed clinical information of the nine evaluable patients. After progression, MET amplification was undetected in 66.7% (6/9) of the patients. EGFR-dependent acquired resistance mechanisms detected from the cohort included EGFR T790M (n = 2), EGFR L718Q (n = 1), and EGFR S645C (n = 1). Secondary mutation MET D1228H was detected in a patient. EGFR/MET-independent bypass mechanisms detected from our cohort included BRAF V600E (n = 1), NRAS Q61H (n = 1), KRAS amplification (n = 1), ERBB2 amplification (n = 1), CDK4 amplification (n = 1), and MYC amplification (n = 1). The two patients with unknown mechanisms of resistance had undetected MET amplification.
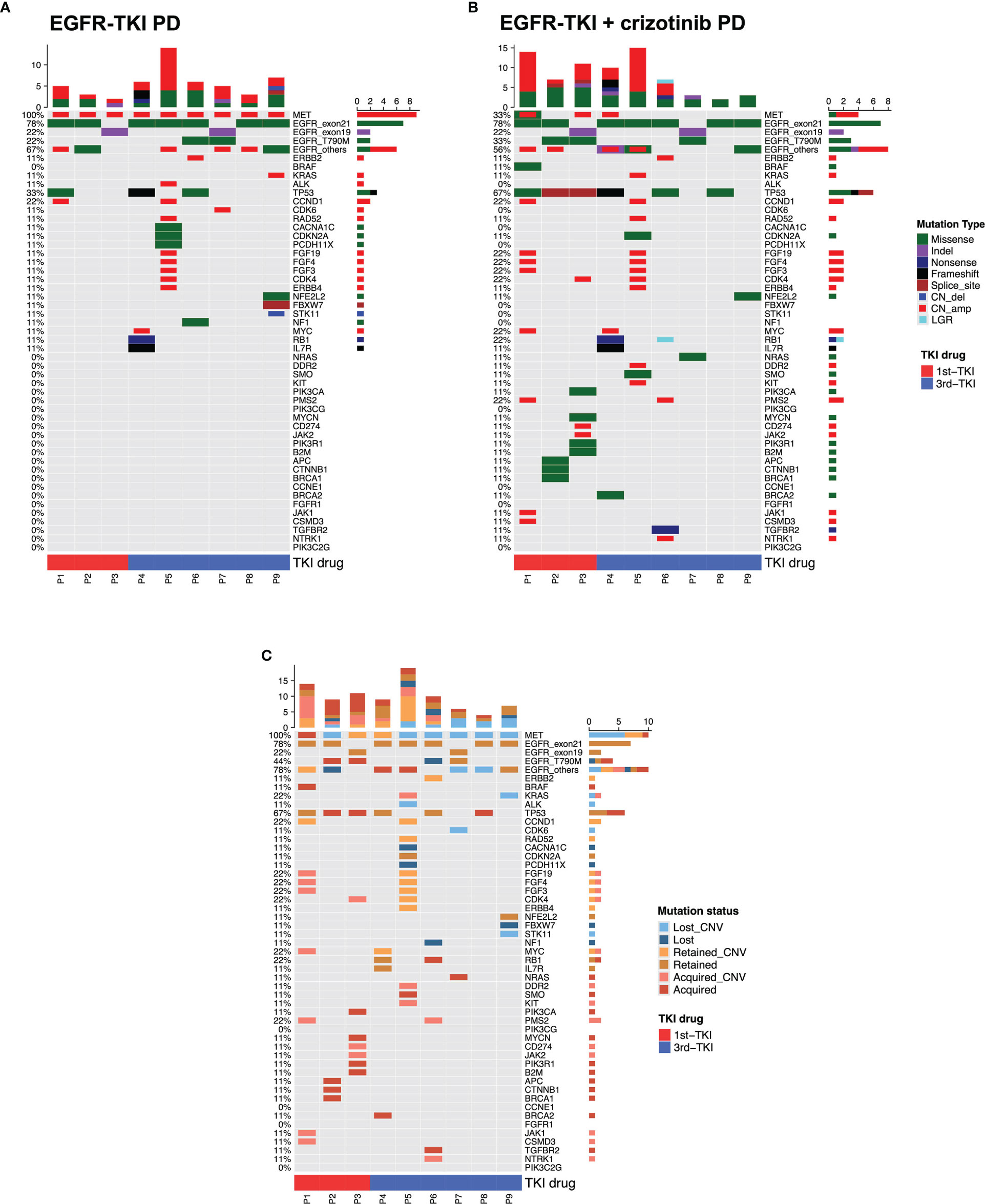
Figure 4 Mutation landscape of the nine evaluable patients after progression from EGFR-TKI with crizotinib. The oncomaps summarize the mutation profile that indicates the specific mutation types of the nine patients before receiving the combination therapy (at progression from EGFR-TKI) (A) and at progression from the combination therapy (B). (C) This oncomap integrates the mutation profile before receiving the combination therapy and at progression to indicate the mutations retained, acquired (undetected at baseline), and lost (undetected at progression).
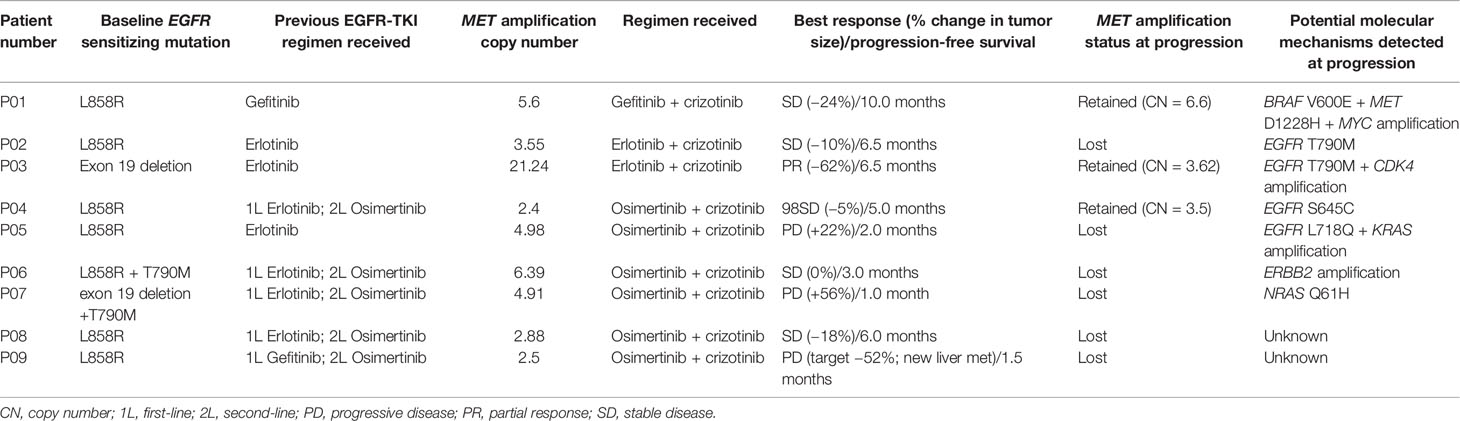
Table 2 Detailed treatment regimen and molecular mechanisms of acquired resistance to EGFR-TKI and crizotinib combination therapy of the 9 evaluable patients.
Safety and Toxicity Profiles
Table 3 summarizes the treatment-related toxicities. The most frequently reported treatment-related toxicities were neutropenia (n = 16, 22.9%) and fatigue (n = 13, 18.6%). Of the 38 patients who received EGFR-TKI combined with crizotinib, grade 1–2 neutropenia (n = 5), grade 1–3 elevated transaminase (n = 4), grade 1–2 vomiting (n = 4), grade 1–2 diarrhea (n = 4), and grade 1–3 rash (n = 4) were the most frequent toxicity. Among the 10 patients who received crizotinib monotherapy, the most frequent toxicities were grade 1–2 vomiting (n = 3) and diarrhea (n = 2). Of the 22 patients who received chemotherapy regimen, grade 1–3 neutropenia (n = 10) and fatigue (n = 8) were the most frequent toxicity. Grade 3 or above treatment-related toxicities were observed in six patients (15.8%) who received EGFR-TKI plus crizotinib and three patients (13.6%) who received chemotherapy (p = 0.591). No grade IV, fatal, or unexpected adverse events were observed in our cohort.
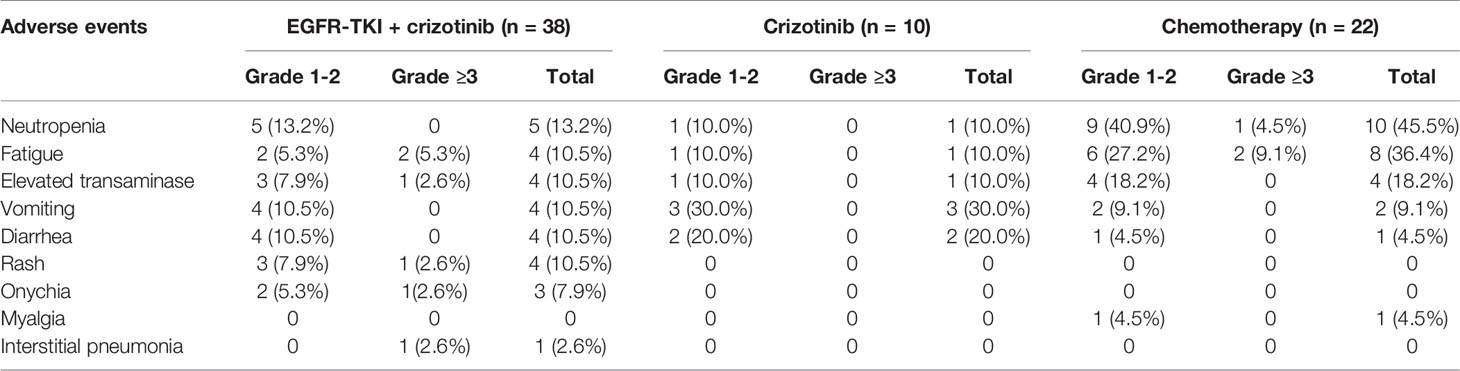
Table 3 Adverse events in each treatment group.
Case Vignette
A 45-year old man with 60 pack-year smoking history (P05, Table 2) was diagnosed with EGFR L858R-mutant stage IV lung adenocarcinoma and received a combination of osimertinib and crizotinib after acquiring MET amplification with an absolute CN of 4.98 at disease progression from first-line erlotinib therapy. The combination therapy was well tolerated except for grade 2 vomiting. It is interesting that his primary lung lesion achieved a dramatic response within 4 weeks of the combination therapy and remained responsive for 3 months (chest CT images, Supplementary Figure S1) despite the development of new metastatic lesions in the liver (abdominal CT images, Supplementary Figure S1), which led to rapid disease progression and eventual demise on January 2018. At progression, NGS analysis of the liver lesions identified EGFR L718Q, KRAS CN amplification, and the absence of MET amplification, suggesting that inhibitor resistance in the liver lesions might be mediated by both EGFR-dependent and EGFR-independent mechanisms (Supplementary Figure S1).
Discussion
The clinical success of EGFR-TKI is consistently limited by the acquisition of resistance, which inevitably occurs in almost all patients within 1 year of EGFR-TKI therapy. MET amplification-mediated resistance to EGFR-TKI occurs in approximately 20% of patients who progress on various generations of EGFR-TKI (8, 9, 15). Until now, there is still no consensus on the optimal subsequent-line treatment regimen for patients who acquired MET amplification-mediated EGFR-TKI resistance. The efficacy of crizotinib either alone or in combination with any EGFR-TKI has been explored among patients who were detected with MET amplification after progression from EGFR-TKI treatment (16, 19, 20, 22, 36, 37). Due to the limited sample size, the data in these studies were inadequate to strongly support the standard clinical use of combination therapies targeting both EGFR and MET after MET amplification-mediated EGFR-TKI resistance. To shed light on which treatment regimen is the most effective in this subset of patients, we retrospectively analyzed the clinical outcomes of 70 patients with EGFR-mutant NSCLC who acquired MET amplification during EGFR-TKI therapy and received any of the following three treatment regimens: crizotinib in combination with any EGFR-TKI (n = 38), crizotinib monotherapy (n = 10), or chemotherapy regimen (n = 22). Our study revealed a significantly higher real-world ORR, DCR, and PFS and a trend of longer OS for patients who received the combination therapy of EGFR-TKI and crizotinib than those who received either crizotinib monotherapy or chemotherapy regimen, providing clinical evidence that the combined inhibition of EGFR and MET provides clinical benefit to these patients after EGFR-TKI progression. The superior clinical outcome for the combination therapy and the similar clinical outcomes for crizotinib monotherapy and chemotherapy suggested four important points. First, NGS-based analysis of samples obtained after EGFR-TKI progression is instrumental in identifying concurrent actionable mutations to improve clinical outcomes. Second, MET amplification status identified by NGS could potentially serve as a predictive biomarker for response to crizotinib therapy after EGFR-TKI progression. Third, despite developing EGFR-TKI resistance, the tumor/s of patients detected with concurrent EGFR mutation and MET amplification at disease progression have acquired “addiction” to MET but remains “addicted” to EGFR. Fourth, the continued use of EGFR-TKI is necessary to target the remaining EGFR-addicted clones to improve clinical outcomes. To the best of our knowledge, our study is the first to include the largest cohort to compare the three subsequent-line treatment regimens after EGFR-TKI progression in the real-world setting.
Despite the clinical benefit of EGFR-TKI combined with crizotinib, these clinical responses appeared to be non-durable and heterogeneous. We speculate that the differences in MET copy number and inherent genetic heterogeneity of tumors might underlie the heterogeneous and short-lived clinical outcomes observed in our cohort. Consistent with our findings, another study reported the heterogeneous responses in patients with marked shrinkage of the primary lesion but continued progression of other metastatic lesions (20). MET amplification may also not be mutually exclusive from other molecular pathways that mediate EGFR-TKI resistance and could contribute to short-lived and heterogeneous responses of patients who receive MET-TKI after progression from EGFR-TKI therapy (20).
In addition to crizotinib, the potential effectiveness of more selective MET-TKIs, including capmatinib, savolitinib, and tepotinib, combined with any EGFR-TKIs in the management of patients with EGFR-mutant, MET-amplified NSCLC has been demonstrated in several prospective clinical studies (21, 24–26, 28). Capmatinib combined with gefitinib was shown in a phase Ib/II study to be effective in patients with EGFR-mutated, MET-amplified/overexpressed NSCLC after progressing from prior EGFR-TKI therapy, with an ORR of 47% among patients with fluorescence in situ hybridization-based MET CN of ≥6 (24). Moreover, osimertinib and savolitinib combination therapy showed promising results for patients with MET-driven resistance to either first- or second-generation EGFR-TKIs without T790M, with an ORR of 64% and a median PFS of 9.1 months (25, 26). Moreover, the efficacy of savolitinib in combination with either gefitinib (31% PR, 16/51) or osimertinib (40% PR, 19/47) was demonstrated in patients with EGFR-mutant NSCLC with concurrent MET amplification after progression on first-line EGFR-TKI treatment (27). Among the patients with MET-amplified NSCLC included in the INSIGHT study, ORR was higher among those who received tepotinib plus gefitinib than chemotherapy [66.7% (12/18) vs 42.9% (3/7)] (28). With the growing number of clinical evidence on the superiority of EGFR-TKI and MET-TKI combination therapy over chemotherapy, we anticipate the consensus on the use of the combination therapy as the standard of care for the management of NSCLC after MET amplification-mediated resistance to EGFR-TKI.
Although mechanisms of acquired resistance to EGFR-TKI monotherapy and MET-TKI monotherapy have been well-elucidated (9, 10, 21, 26, 38–44), the mechanism mediating resistance to the combination therapy of EGFR-TKI and MET-TKI remains unclear. As expected, acquired mutations observed in our cohort were on- and off-target resistance mechanisms previously reported for monotherapy of EGFR-TKI and crizotinib. Acquisition of missense mutations in MET D1228, including D1228H, was implicated as one of the on-target resistance mechanisms to crizotinib therapy that can potentially be reversed by next-generation MET-TKIs (45). An in vitro study demonstrated that EGFR S645C can promote tumor formation and is less sensitive to erlotinib (39). EGFR L718Q has been reported as one of the rare resistance mechanisms to osimertinib therapy in an EGFR L858R/T790M-positive lung adenocarcinoma patient, which might be sensitive to afatinib (46, 47). EGFR-independent resistance mechanisms, including copy number amplifications in ERBB2, genes involved in cell cycle regulation such as CDK4, and mutations in genes involved in MAPK/PIK3 signaling, such as BRAF, KRAS, and NRAS, have been reported (41). A study identified NRAS Q61K as an acquired resistance mechanism to erlotinib in EGFR L858R-mutant cell line (38). MYC amplification was implicated in primary crizotinib resistance in ALK-rearranged NSCLC (44). KRAS amplification was implicated as the mechanism of resistance from crizotinib treatment in MET exon 14 mutant-NSCLC in a study using patient tumor-derived cells and xenografts, which was effectively targeted by dual MET/PI3K inhibition (42). Nonetheless, these acquired resistance mechanisms suggest that rebiopsy at progression is necessary to identify potentially actionable mutations for therapeutic decisions. These results also indicate that current therapeutic strategies for reversing acquired resistance to EGFR-TKI or crizotinib are applicable after progression from the EGFR-TKI and crizotinib combination therapy.
Our study has several limitations. First, it is a retrospective, non-randomized study conducted in two institutions from the same city, which could result in patient selection bias. Second, MET amplification status of our cohort was only determined using DNA NGS and lacked data on standard MET amplification or overexpression status assessed using fluorescence in situ hybridization or immunohistochemistry. A multicenter collaboration is required to further establish our findings.
In conclusion, our findings provide real-world clinical evidence that the simultaneous inhibition of EGFR and MET by the combined use of EGFR-TKI and crizotinib is generally tolerable and provides benefit to patients with EGFR-mutant NSCLC who acquired MET amplification-mediated EGFR-TKI resistance. Our findings add to the accumulating evidence of the potential of combination therapy with EGFR-TKIs and MET-TKIs as the optimal subsequent-line therapeutic strategy for this subset of patients.
Data Availability Statement
The datasets presented in this study can be found in online repository. The names of the repository and accession number can be found below: National Omics Data Encyclopedia (NODE), accession number OEP002682. The dataset can be accessed through the URL: http://www.biosino.org/node/project/detail/OEP002682.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Ethics Committee of Hunan Cancer Hospital (approval number: 2021-19). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
LL, JQ, PD, and NY conceived and designed the study. HY, CZ, WJ, CH, and LL provided patient information. JT and SZ collected the data. LL and JQ analyzed the data. PD and NY supervised the project and acquired financial support. All the authors contributed to manuscript writing, editing, and approving the manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81802278), Huilan Public Welfare-Hansoh Pharma Lung Cancer Precision Medicine Research Fund (No. HL-HS2020-10), and the Natural Science Foundation of Hunan Province (No. 2019JJ50361). The funders had no role in the conceptualization, design, data collection, analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Drs. Analyn Lizaso, Ting Hou, and Zhiqiu Chen of Burning Rock Biotech for their support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.722039/full#supplementary-material
References
1. Barlesi F, Mazieres J, Merlio JP, Debieuvre D, Mosser J, Lena H, et al. Routine Molecular Profiling of Patients With Advanced Non-Small-Cell Lung Cancer: Results of a 1-Year Nationwide Programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet (2016) 387(10026):1415–26. doi: 10.1016/S0140-6736(16)00004-0
2. Xia N, An J, Jiang QQ, Li M, Tan J, Hu CP. Analysis of EGFR, EML4-ALK, KRAS, and C-MET Mutations in Chinese Lung Adenocarcinoma Patients. Exp Lung Res (2013) 39(8):328–35. doi: 10.3109/01902148.2013.819535
3. Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, et al. Phase III Study of Afatinib or Cisplatin Plus Pemetrexed in Patients With Metastatic Lung Adenocarcinoma With EGFR Mutations. J Clin Oncol (2013) 31(27):3327–34. doi: 10.1200/JCO.2012.44.2806
4. Wu YL, Sequist LV, Tan EH, Geater SL, Orlov S, Zhang L, et al. Afatinib as First-Line Treatment of Older Patients With EGFR Mutation-Positive Non-Small-Cell Lung Cancer: Subgroup Analyses of the LUX-Lung 3, LUX-Lung 6, and LUX-Lung 7 Trials. Clin Lung Cancer (2018) 19(4):e465–e79. doi: 10.1016/j.cllc.2018.03.009
5. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or Carboplatin-Paclitaxel in Pulmonary Adenocarcinoma. N Engl J Med (2009) 361(10):947–57. doi: 10.1056/NEJMoa0810699
6. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or Chemotherapy for Non-Small-Cell Lung Cancer With Mutated EGFR. N Engl J Med (2010) 362(25):2380–8. doi: 10.1056/NEJMoa0909530
7. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci Transl Med (2011) 3(75):75ra26. doi: 10.1126/scitranslmed.3002003
8. Arcila ME, Oxnard GR, Nafa K, Riely GJ, Solomon SB, Zakowski MF, et al. Rebiopsy of Lung Cancer Patients With Acquired Resistance to EGFR Inhibitors and Enhanced Detection of the T790M Mutation Using a Locked Nucleic Acid-Based Assay. Clin Cancer Res (2011) 17(5):1169–80. doi: 10.1158/1078-0432.CCR-10-2277
9. Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR-TKI Therapy in 155 Patients With EGFR-Mutant Lung Cancers. Clin Cancer Res (2013) 19(8):2240–7. doi: 10.1158/1078-0432.CCR-12-2246
10. Liu Q, Yu S, Zhao W, Qin S, Chu Q, Wu K. EGFR-TKIs Resistance via EGFR-Independent Signaling Pathways. Mol Cancer (2018) 17(1):53. doi: 10.1186/s12943-018-0793-1
11. Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, et al. AZD9291 in EGFR Inhibitor-Resistant Non-Small-Cell Lung Cancer. N Engl J Med (2015) 372(18):1689–99. doi: 10.1056/NEJMoa1411817
12. Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, et al. Novel Mutant-Selective EGFR Kinase Inhibitors Against EGFR T790M. Nature (2009) 462(7276):1070–4. doi: 10.1038/nature08622
13. Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L, et al. Increased MET Gene Copy Number Negatively Affects Survival of Surgically Resected Non-Small-Cell Lung Cancer Patients. J Clin Oncol (2009) 27(10):1667–74. doi: 10.1200/JCO.2008.19.1635
14. Schildhaus HU, Schultheis AM, Ruschoff J, Binot E, Merkelbach-Bruse S, Fassunke J, et al. MET Amplification Status in Therapy-Naive Adeno- and Squamous Cell Carcinomas of the Lung. Clin Cancer Res (2015) 21(4):907–15. doi: 10.1158/1078-0432.CCR-14-0450
15. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science (2007) 316(5827):1039–43. doi: 10.1126/science.1141478
16. Baldacci S, Mazieres J, Tomasini P, Girard N, Guisier F, Audigier-Valette C, et al. Outcome of EGFR-Mutated NSCLC Patients With MET-Driven Resistance to EGFR Tyrosine Kinase Inhibitors. Oncotarget (2017) 8(62):105103–14. doi: 10.18632/oncotarget.21707
17. Matsubara D, Ishikawa S, Sachiko O, Aburatani H, Fukayama M, Niki T. Co-Activation of Epidermal Growth Factor Receptor and C-MET Defines a Distinct Subset of Lung Adenocarcinomas. Am J Pathol (2010) 177(5):2191–204. doi: 10.2353/ajpath.2010.100217
18. Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, et al. Preexistence and Clonal Selection of MET Amplification in EGFR Mutant NSCLC. Cancer Cell (2010) 17(1):77–88. doi: 10.1016/j.ccr.2009.11.022
19. Wang W, Wang H, Lu P, Yu Z, Xu C, Zhuang W, et al. Crizotinib With or Without an EGFR-TKI in Treating EGFR-Mutant NSCLC Patients With Acquired MET Amplification After Failure of EGFR-TKI Therapy: A Multicenter Retrospective Study. J Transl Med (2019) 17(1):52. doi: 10.1186/s12967-019-1803-9
20. van Veggel B, de Langen AJ, Hashemi S, Monkhorst K, Rosenberg EH, Heideman DAM, et al. Crizotinib Treatment for Patients With EGFR Mutation Positive NSCLC That Acquire cMET Amplification After EGFR TKI Therapy Results in Short-Lived and Heterogeneous Responses. Lung Cancer (2018) 124:130–4. doi: 10.1016/j.lungcan.2018.07.030
21. Li A, Yang JJ, Zhang XC, Zhang Z, Su J, Gou LY, et al. Acquired MET Y1248H and D1246N Mutations Mediate Resistance to MET Inhibitors in Non-Small Cell Lung Cancer. Clin Cancer Res (2017) 23(16):4929–37. doi: 10.1158/1078-0432.CCR-16-3273
22. Wang Y, Tian P, Xia L, Li L, Han R, Zhu M, et al. The Clinical Efficacy of Combinatorial Therapy of EGFR-TKI and Crizotinib in Overcoming MET Amplification-Mediated Resistance From Prior EGFR-TKI Therapy. Lung Cancer (2020) 146:165–73. doi: 10.1016/j.lungcan.2020.06.003
23. Cheng JT, Yao YH, Gao YE, Zhang SL, Chen HJ, Wang Z, et al. Integrated Histological and Molecular Analyses of Rebiopsy Samples at Osimertinib Progression Improve Post-Progression Survivals: A Single-Center Retrospective Study. Lung Cancer (2020) 150:97–106. doi: 10.1016/j.lungcan.2020.10.010
24. Wu YL, Zhang L, Kim DW, Liu X, Lee DH, Yang JC, et al. Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib After Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients With EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer. J Clin Oncol (2018) 36(31):3101–9. doi: 10.1200/JCO.2018.77.7326
25. Gainor JF, Niederst MJ, Lennerz JK, Dagogo-Jack I, Stevens S, Shaw AT, et al. Dramatic Response to Combination Erlotinib and Crizotinib in a Patient With Advanced, EGFR-Mutant Lung Cancer Harboring De Novo MET Amplification. J Thorac Oncol (2016) 11(7):e83–5. doi: 10.1016/j.jtho.2016.02.021
26. Bahcall M, Sim T, Paweletz CP, Patel JD, Alden RS, Kuang Y, et al. Acquired METD1228V Mutation and Resistance to MET Inhibition in Lung Cancer. Cancer Discov (2016) 6(12):1334–41. doi: 10.1158/2159-8290.CD-16-0686
27. Sequist LV, Han JY, Ahn MJ, Cho BC, Yu H, Kim SW, et al. Osimertinib Plus Savolitinib in Patients With EGFR Mutation-Positive, MET-Amplified, Non-Small-Cell Lung Cancer After Progression on EGFR Tyrosine Kinase Inhibitors: Interim Results From a Multicentre, Open-Label, Phase 1b Study. Lancet Oncol (2020) 21(3):373–86. doi: 10.1016/S1470-2045(19)30785-5
28. Wu YL, Cheng Y, Zhou J, Lu S, Zhang Y, Zhao J, et al. Tepotinib Plus Gefitinib in Patients With EGFR-Mutant Non-Small-Cell Lung Cancer With MET Overexpression or MET Amplification and Acquired Resistance to Previous EGFR Inhibitor (INSIGHT Study): An Open-Label, Phase 1b/2, Multicentre, Randomised Trial. Lancet Respir Med (2020) 8(11):1132–43. doi: 10.1016/S2213-2600(20)30154-5
29. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New Response Evaluation Criteria in Solid Tumours: Revised RECIST Guideline (Version 1.1). Eur J Cancer (2009) 45(2):228–47. doi: 10.1016/j.ejca.2008.10.026
30. Mao X, Zhang Z, Zheng X, Xie F, Duan F, Jiang L, et al. Capture-Based Targeted Ultradeep Sequencing in Paired Tissue and Plasma Samples Demonstrates Differential Subclonal ctDNA-Releasing Capability in Advanced Lung Cancer. J Thorac Oncol (2017) 12(4):663–72. doi: 10.1016/j.jtho.2016.11.2235
31. Scoccianti C, Vesin A, Martel G, Olivier M, Brambilla E, Timsit JF, et al. Prognostic Value of TP53, KRAS and EGFR Mutations in Nonsmall Cell Lung Cancer: The EUELC Cohort. Eur Respir J (2012) 40(1):177–84. doi: 10.1183/09031936.00097311
32. Canale M, Petracci E, Delmonte A, Chiadini E, Dazzi C, Papi M, et al. Impact of TP53 Mutations on Outcome in EGFR-Mutated Patients Treated With First-Line Tyrosine Kinase Inhibitors. Clin Cancer Res (2017) 23(9):2195–202. doi: 10.1158/1078-0432.CCR-16-0966
33. Hong S, Gao F, Fu S, Wang Y, Fang W, Huang Y, et al. Concomitant Genetic Alterations With Response to Treatment and Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Patients With EGFR-Mutant Advanced Non-Small Cell Lung Cancer. JAMA Oncol (2018) 4(5):739–42. doi: 10.1001/jamaoncol.2018.0049
34. Qin K, Hou H, Liang Y, Zhang X. Prognostic Value of TP53 Concurrent Mutations for EGFR- TKIs and ALK-TKIs Based Targeted Therapy in Advanced Non-Small Cell Lung Cancer: A Meta-Analysis. BMC Cancer (2020) 20(1):328. doi: 10.1186/s12885-020-06805-5
35. Li XM, Li WF, Lin JT, Yan HH, Tu HY, Chen HJ, et al. Predictive and Prognostic Potential of TP53 in Patients With Advanced Non-Small-Cell Lung Cancer Treated With EGFR-TKI: Analysis of a Phase III Randomized Clinical Trial (CTONG 0901). Clin Lung Cancer (2021) 22(2):100–9.e3. doi: 10.1016/j.cllc.2020.11.001
36. Ou SI, Agarwal N, Ali SM. High MET Amplification Level as a Resistance Mechanism to Osimertinib (AZD9291) in a Patient That Symptomatically Responded to Crizotinib Treatment Post-Osimertinib Progression. Lung Cancer (2016) 98:59–61. doi: 10.1016/j.lungcan.2016.05.015
37. Wang Y, Li L, Han R, Jiao L, Zheng J, He Y. Clinical Analysis by Next-Generation Sequencing for NSCLC Patients With MET Amplification Resistant to Osimertinib. Lung Cancer (2018) 118:105–10. doi: 10.1016/j.lungcan.2018.02.007
38. Ohashi K, Sequist LV, Arcila ME, Moran T, Chmielecki J, Lin YL, et al. Lung Cancers With Acquired Resistance to EGFR Inhibitors Occasionally Harbor BRAF Gene Mutations But Lack Mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci USA (2012) 109(31):E2127–33. doi: 10.1073/pnas.1203530109
39. Berger AH, Brooks AN, Wu X, Shrestha Y, Chouinard C, Piccioni F, et al. High-Throughput Phenotyping of Lung Cancer Somatic Mutations. Cancer Cell (2016) 30(2):214–28. doi: 10.1016/j.ccell.2016.06.022
40. Zhang Y, Yin J, Peng F. Acquired Resistance to Crizotinib in Advanced Lung Adenocarcinoma With MET Exon 14 Skipping. Lung Cancer (2017) 113:69–71. doi: 10.1016/j.lungcan.2017.09.006
41. Nagano T, Tachihara M, Nishimura Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells (2018) 7(11):212. doi: 10.3390/cells7110212
42. Bahcall M, Awad MM, Sholl LM, Wilson FH, Xu M, Wang S, et al. Amplification of Wild-Type KRAS Imparts Resistance to Crizotinib in MET Exon 14 Mutant Non-Small Cell Lung Cancer. Clin Cancer Res (2018) 24(23):5963–76. doi: 10.1158/1078-0432.CCR-18-0876
43. Ricordel C, Friboulet L, Facchinetti F, Soria JC. Molecular Mechanisms of Acquired Resistance to Third-Generation EGFR-TKIs in EGFR T790M-Mutant Lung Cancer. Ann Oncol (2019) 30(5):858. doi: 10.1093/annonc/mdy222
44. Rihawi K, Alfieri R, Fiorentino M, Fontana F, Capizzi E, Cavazzoni A, et al. MYC Amplification as a Potential Mechanism of Primary Resistance to Crizotinib in ALK-Rearranged Non-Small Cell Lung Cancer: A Brief Report. Transl Oncol (2019) 12(1):116–21. doi: 10.1016/j.tranon.2018.09.013
45. Yap TA, Popat S. Targeting MET Exon 14 Skipping Alterations: Has Lung Cancer MET Its Match? J Thorac Oncol (2017) 12(1):12–4. doi: 10.1016/j.jtho.2016.10.019
46. Bersanelli M, Minari R, Bordi P, Gnetti L, Bozzetti C, Squadrilli A, et al. L718Q Mutation as New Mechanism of Acquired Resistance to AZD9291 in EGFR-Mutated NSCLC. J Thorac Oncol (2016) 11(10):e121–3. doi: 10.1016/j.jtho.2016.05.019
Keywords: EGFR mutation, MET amplification, EGFR-TKI and crizotinib combination, NSCLC, EGFR-TKI resistance
Citation: Liu L, Qu J, Heng J, Zhou C, Xiong Y, Yang H, Jiang W, Zeng L, Zhu S, Zhang Y, Tan J, Hu C, Deng P and Yang N (2021) A Large Real-World Study on the Effectiveness of the Combined Inhibition of EGFR and MET in EGFR-Mutant Non-Small-Cell Lung Cancer After Development of EGFR-TKI Resistance. Front. Oncol. 11:722039. doi: 10.3389/fonc.2021.722039
Received: 08 June 2021; Accepted: 23 August 2021;
Published: 01 October 2021.
Edited by:
Timothy F. Burns, University of Pittsburgh, United StatesReviewed by:
Kamini Singh, Memorial Sloan Kettering Cancer Center, United StatesMu Hao, Peking Union Medical College Hospital (CAMS), China
Copyright © 2021 Liu, Qu, Heng, Zhou, Xiong, Yang, Jiang, Zeng, Zhu, Zhang, Tan, Hu, Deng and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pengbo Deng, eW9ndXJ0MTAxNUAxNjMuY29t; Nong Yang, eWFuZ25vbmdAaG5jYS5vcmcuY24=
†These authors have contributed equally to this work